

Highlights
-
•
The full genome sequence of a bat hantavirus was determined.
-
•
It provides reference data for determining bat hantavirus’ full genomes.
-
•
Genomic analysis shows that it is distantly related to all known bat hantaviruses.
Keywords: Bat, Hantavirus, Full genome
Abstract
Hantaviruses cause life-threatening diseases in human worldwide. Rodents, insectivores and bats are known hantaviral reservoirs, but lack of complete genomic sequences of bat-borne hantaviruses impedes phylogenetic and evolutionary comparison with those of rodents and insectivores. Here, a novel bat-borne hantavirus, Laibin virus (LBV), has been identified in a black-bearded tomb bat in China. The complete genomic sequence shows that LBV is only distantly related to all previously known bat-borne hantaviruses.
Hantaviruses, responsible for hantavirus cardiopulmonary syndrome (HCPS) in the Americas and hemorrhagic fever with renal syndrome (HFRS) in Asia and Europe, are major life-threatening pathogens in human (Schmaljohn and Nichol, 2007). About 60,000–150,000 cases of HFRS are reported yearly worldwide with more than 90% in Asia, mostly in China with 29 of its 31 provinces being affected (Jonsson et al., 2010, Zhang et al., 2010, Kariwa et al., 2007).
Rodents are natural reservoirs of hantaviruses (Schmaljohn and Nichol, 2007, Jonsson et al., 2010, Zhang et al., 2010); however, recent studies have demonstrated that the natural hosts also include insectivores and bats (Zhang, 2014, Guo et al., 2013, Weiss et al., 2012). Bats are important reservoirs of many emerging deadly viral pathogens, such as filoviruses, lyssaviruses, and SARS-related coronaviruses (Calisher et al., 2006). Genetically divergent bat-borne hantaviruses have recently been identified in Africa (Weiss et al., 2012, Sumibcay et al., 2012) and Asia (Guo et al., 2013, Zhang, 2014, Gu et al., 2014), but very limited sequences are available, with none having complete sequences of all three gene segments (Table 1 ). Lack of complete genomic sequences impairs the phylogenetic and evolutionary comparison of bat-borne hantaviruses with those of human, rodents and insectivores. Here, we report the complete genomic sequence of a novel hantavirus identified from a South China bat.
Table 1.
Nucleotide sequences of all bat hantaviruses from GenBank and their identity (%) with LBV.a
| Virus | Country (Province) | Bat species | Gene | Position at LBV genome | Length (nt) | nt Identity with LBV |
|---|---|---|---|---|---|---|
| LQUV | China (Zhejiang) | Rhinolophus sinicus | S | Complete | 1,563 | 50.6 |
| M | Complete | 3,618 | 52.6 | |||
| L | 3,005–3,328 | 324 | 69.1 | |||
| HUPV | China (Hubei) | Pipistrellus abramus | S | 522–1,935 | 1,115 | 46.4 |
| L | 2,945–3,288 | 343 | 69.8 | |||
| XSV | Vietnam (Tuyên Quang) | Hipposideros pomona | S | Complete | 1,752 | 60.8 |
| M | 1,956–2,618 | 663 | 71.2 | |||
| L | 2,540–3,699 | 1,160 | 75.3 | |||
| MOUV | Côte d’Ivoire | Neoromicia nanus | L | 1,892–3,582 | 1,691 | 71.2 |
| MGBV | Sierra Leone | Nycteris hispida | L | 2,953–3,370 | 414 | 71.5 |
As part of a survey of bat-borne pathogens, thirty-two insectivorous black-bearded tomb bats (Taphozous melanopogon) were captured in July 2012 in Laibin city, Guangxi province, China. The bat species was morphologically identified by an experienced zoologist and further confirmed by PCR of its mitochondrial cytochrome b gene sequence (Wang et al., 2003). Intestines with contents, and lungs were collected and the samples of each tissue were pooled for viral metagenomic analysis as described elsewhere (He et al., 2013b). Among sequences annotated to mammal viruses, 250 identical reads with an average length of 135 nucleotides (nt) were found to exhibit 70% nt identity with the S gene of Dobrava-Belgrade hantavirus (DOBV) (Nemirov et al., 1999). To further screen for hantaviruses, total RNA was extracted from each sample using a QIAamp RNeasy Mini Kit (Qiagen), then subjected to hantavirus-specific RT-PCR using published primers (Klempa et al., 2006). Hantavirus was identified in one bat lung tissue, and designated Laibin virus (LBV).
To obtain the full genome of LBV, primers were designed according to bat-borne hantavirus sequences retrieved from GenBank (Table S). The end sequences of each segment were obtained by using conserved terminal nt sequences as primers. Overlapping amplicons were obtained by using a Fast HiFidelity PCR Kit (Tiangen), then assembled into three full gene segments. The complete sequences of S, M and L gene segments were 1,935, 3,908 and 6,531 nt in length, encoding 427, 1,127 and 2,145 aa proteins respectively (GenBank accession numbers: KM102247 and KM102249). Notably, LBV exhibited the longest M segment, 107 nt longer in the 3′ non-coding region than the largest M segment previously reported (3,801 nt; El Moro Canyon hantavirus RM-97, accession number U26828). The three segments were aligned with reported sequences using Clustal W (Version 2.0). Since most of the bat-borne hantavirus sequences were incomplete (Table 1), phylogenetic analyses were conducted based on partial sequences of the three gene segments (Fig. 1 ). The resulting topologies were similar to that described previously (Guo et al., 2013), which divided all sequences into four clades, and all bat-borne hantaviruses were clustered together. Further sequence comparison showed that the three gene segments of LBV had 46.4–75.3% nucleotide identity with other bat-borne hantaviruses (Table 1) and 44.6–67.4% with hantaviruses from humans, rats, mice, shrews and moles (Table 2 ). These results characterize LBV as being a novel bat-borne hantavirus exhibiting highest nucleotide identities with the three gene segments of Vietnamese XSV (60.8–75.3%) (Table 1).
Fig. 1.
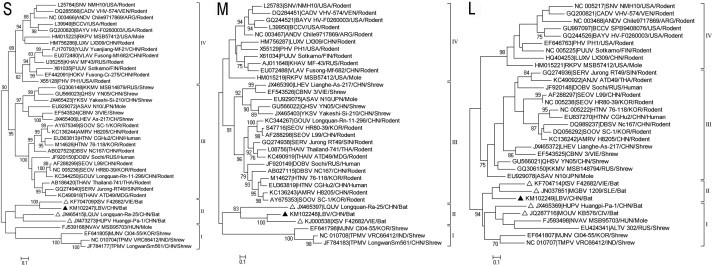
Phylogenetic trees were constructed according to partial S (positions 500–1458), M (positions 1985–2647) and L (positions 2948–3284) stretches of the hantaviral genomic segments, selected from the limited availability of sequences of most known bat-borne hantaviruses (see Table 1), fragment positions refer to complete sequences of HTNV strain 76–118 (Genbank accession Nos: NC_005218, NC_005219, NC_005222). MEGA5.0 was used to construct the phylogenetic trees with the maximum-likelihood method. Numbers above or below branches are bootstrap values of 1000 replicates. Filled triangles indicate the sequences of LBV, while open triangles indicate the sequences of other known bat hantaviruses. The scale bars indicate nucleotide substitutions per site. The accession number is prefixed to the name of each virus strain used.
Table 2.
Nucleotide and amino acid sequence identities (%) of complete sequences of three genome segments between LBV and other representative hantaviruses.a
| Hantavirus | S |
M |
L |
|||
|---|---|---|---|---|---|---|
| nt | aa | nt | aa | nt | aa | |
| HTNV/CHN/Human | 48.4 | 52.5 | 49.7 | 45.8 | NA | NA |
| DOBV/RUS/Human | 49.9 | 53.0 | 49.4 | 45.8 | 63.4 | 64.6 |
| SEOV/CHN/Rodent | 52.4 | 51.4 | 49.3 | 46.2 | 63.8 | 64.2 |
| SNV/USA/Rodent | 50.2 | 52.9 | 52.3 | 48.4 | 63.1 | 64.8 |
| PUUV/USA/Rodent | 51.9 | 53.0 | 52.6 | 46.2 | 66.2 | 65.6 |
| PHV/USA/Rodent | 48.5 | 52.1 | 52.8 | 47.5 | 64.0 | 64.8 |
| THAIV/MDG/Rodent | 53.0 | 52.8 | 48.9 | 46.0 | 64.1 | 64.4 |
| ASAV/JPN/Mole | 51.4 | 51.3 | 50.5 | 45.8 | NA | NA |
| NVAV/HUN/Mole | 58.3 | 59.6 | NA | NA | 67.4 | 68.1 |
| CBNV/VIE/Shrew | 56.1 | 53.0 | 50.4 | 45.3 | 64.0 | 64.6 |
| MJNV/KOR/Shrew | 45.5 | 46.4 | 47.6 | 42.3 | 64.2 | 64.8 |
| TPMV/IND/Shrew | 44.6 | 44.4 | 47.9 | 42.4 | 63.8 | 65.6 |
HTNV, Hantaan virus CGHu2 (EU363813, EU363819); DOBV, Dobrava-Belgrade virusAp/Sochi/hu (JF920150, JF920149, JF920148); SEOV, Seoul virus L99 (AF288299, AF288298, AF288297); SNV, Sin Nombre virus NMH10 (NC_005216, NC_005215, NC_005217); PUUV, Puumala virus Sotkamo (NC_005224, NC_005223, NC_005225); PHV, Prospect Hill virus PH-1 (X55128, X55129, EF646763); THAIV, Anjozorobe virus Em/MDG/2009/ATD49 (KC490918, KC490919, KC490922); ASAV, Asama virus N10 (EU929072, EU929075); NVAV, Nova virusMSB95703 (FJ539168, FJ593498); CBNV, Cao Bang virus 3 (EF543524, EF543526, EF543525); MJNV, Imjin virus CI04-55 (EF641805, EF641799, EF641807); TMPV, Thottapalayam virus VRC66412 (NC_010704, NC_010708, NC_010707); NA, not available.
Five blind passages in Vero-E6 cells failed to isolate the virus from the positive lung sample. However, a single viral particle of about 100 nm in diameter was observed in lung tissue by electron microscopy (EM) using previously described methods (He et al., 2013a) although no grid-like pattern typical of hantaviruses (Schmaljohn and Nichol, 2007) was seen (Fig. S). In support of the conclusion that the EM sample contained LBV, specific RT-PCR above was performed and revealed specific amplification of an LBV gene fragment.
Apart from the present report, five hantaviruses have been identified in bats worldwide; however, their complete genomic sequences have not been reported (Table 1). In present study a novel bat-borne hantavirus has been identified and its full genomic sequence has been determined. Comparison with available sequences shows that LBV is distantly related to all known bat-borne hantaviruses (Table 1), but that it is closer to Vietnamese XSV (60.8–75.3% nt identity) than to the two other Chinese strains LQUV and HUPV. Laibin city, however, is closer to Vietnamese Tuyên Quang province (the isolation site of XSV near the China-Vietnam border) than to Longquan city in Zhejiang province (LQUV) and Huangpi city in Hubei province (HUPV). Insufficient sequence data prevents a comprehensive analysis of the genetic diversities of the bat-borne hantaviruses from Côte d’Ivoire and Sierra Leone, but partial sequence analysis suggests that bat-borne hantaviruses have evolved worldwide within an independent and diverse phylogroup (Fig. 1). The successful determination of the complete sequences of the LBV gene segments may provide reference data for determining the complete genomic sequences of other bat-borne hantaviruses.
Divergent bat-borne hantaviruses have been found in a broad spectrum of bat species (Table 1) which emphasizes the importance of bats as natural worldwide reservoirs of these viruses and the need to investigate further the potential role of bats in transmission of hantaviruses to humans and other animals.
Acknowledgments
Bat experimental studies were reviewed and approved by the Military Veterinary Institute, Academy of Military Medical Sciences, China (permit number: JSY-DW-2010-02). This work was supported by the National Natural Science Foundation of China-Yunnan Province Joint Fund (U1036601), National “973” Program (Grant No. 2012CB722501) and National “863” Program (Grant No. 2012AA022006) to C. Tu.
Footnotes
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.meegid.2015.01.018.
Appendix A. Supplementary data
This document contains table and figure.
References
- Calisher C.H., Childs J.E., Field H.E., Holmes K.V., Schountz T. Bats: important reservoir hosts of emerging viruses. Clin. Microbiol. Rev. 2006;19:531–545. doi: 10.1128/CMR.00017-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu S.H., Lim B.K., Kadjo B., Arai S., Kim J., Nicolas V., Lalis A., Denys C., Cook J.A., Dominguez S.R., Holmes K.V., Urushadze L., Sidamonidze K., Putkaradze D., Kuzmin I.V., Kosoy M.Y., Song J., Yanagihara R. Molecular phylogeny of hantaviruses harbored by insectivorous bats in Côte d’Ivoire and Vietnam. Viruses. 2014;6:1897–1910. doi: 10.3390/v6051897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W.P., Lin X.D., Wang W., Tian J.H., Cong M.L., Zhang H.L., Wang M.R., Zhou R.H., Wang J.B., Li M.H., Xu J.G., Holmes E.C., Zhang Y.Z. Phylogeny and origins of hantaviruses harbored by bats, insectivores, and rodents. PLoS Pathog. 2013;9:e1003159. doi: 10.1371/journal.ppat.1003159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He B., Fan Q.S., Yang F.L., Hu T.S., Qiu W., Feng Y., Li Z.S., Li Y.Y., Zhang F.Q., Guo H.C., Zou X.H., Tu C.C. Hepatitis virus in long-fingered bats, Myanmar. Emerg. Infect. Dis. 2013;19:638–640. doi: 10.3201/eid1904.121655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He B., Li Z.S., Yang F.L., Zheng J.F., Feng Y., Guo H.C., Li Y.Y., Wang Y.Y., Su N., Zhang F.Q., Fan Q.S., Tu C.C. Virome profiling of bats from Myanmar by metagenomic analysis of tissue samples reveals more novel mammalian viruses. PLoS ONE. 2013;8:e61950. doi: 10.1371/journal.pone.0061950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson C.B., Figueiredo L.T.M., Vapalahti O. A global perspective on hantavirus ecology, epidemiology, and disease. Clin. Microbiol. Rev. 2010;23:412–441. doi: 10.1128/CMR.00062-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kariwa H., Yoshimatsu K., Arikawa J. Hantavirus infection in East Asia. Comp. Immunol. Microbiol. Infect. Dis. 2007;30:341–356. doi: 10.1016/j.cimid.2007.05.011. [DOI] [PubMed] [Google Scholar]
- Klempa B., Fichet-Calvet E., Lecompte E., Auste B., Aniskin V., Meisel H., Denys C., Koivogui L., Meulen T., Krüger D.H. Hantavirus in African wood mouse, Guinea. Emerg. Infect. Dis. 2006;12:838–840. doi: 10.3201/eid1205.051487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemirov K., Vapalahti O., Lundkvist Å., Vasilenko V., Golovljova I., Plyusnina A., Niemimaa J., Laakkonen J., Henttonen H., Vaheri A., Plyusnin A. Isolation and characterization of Dobrava hantavirus carried by the striped field mouse (Apodemus agrarius) in Estonia. J. Gen. Virol. 1999;80:371–379. doi: 10.1099/0022-1317-80-2-371. [DOI] [PubMed] [Google Scholar]
- Schmaljohn C.S., Nichol S.T. Bunyaviridae. In: Knipe D.M., Howley P.M., editors. Fields Virology. fifth ed. Lippincott, Williams & Wilkins; Philadelphia: 2007. pp. 1741–1789. [Google Scholar]
- Sumibcay L., Kadjo B., Gu S.H., Kang H.J., Lim B.K., Cook J.A., Song J.W., Yanagihara R. Divergent lineage of a novel hantavirus in the banana pipistrelle (Neoromicia nanus) in Côte d’Ivoire. Virol. J. 2012;9:34–39. doi: 10.1186/1743-422X-9-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H., Liang B., Feng J., Sheng L.X., Zhang S.Y. Molecular phylogenetic of hipposiderids (Chiroptera: Hipposideridae) and rhinolophids (Chiroptera: Rhinolophidae) in China based on mitochondrial cytochrome b sequences. Folia Zool. 2003;52:259–268. [Google Scholar]
- Weiss S., Witkowski P.T., Auste B., Nowak K., Weber N., Fahr J., Mombouli J.V., Wolfe N.D., Drexler J.F., Drosten C., Klempa B., Leendertz F.H., Krüger D.H. Hantavirus in bat, Sierra Leone. Emerg. Infect. Dis. 2012;18:159–161. doi: 10.3201/eid1801.111026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.Z. Discovery of hantaviruses in bats and insectivores and the evolution of the genus Hantavirus. Virus Res. 2014;187:15–21. doi: 10.1016/j.virusres.2013.12.035. [DOI] [PubMed] [Google Scholar]
- Zhang Y.Z., Zou Y., Fu Z.F., Plyusnin A. Hantavirus infections in humans and animals, China. Emerg. Infect. Dis. 2010;16:1195–1203. doi: 10.3201/eid1608.090470. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This document contains table and figure.