

Abstract
Rhinoviruses are the main cause of the common cold and precipitate the majority of asthma exacerbations. RT-PCR followed by internal probe hybridisation or Southern blotting, or nested PCRs are currently the most sensitive methods for their identification. However, none of the published techniques can differentiate satisfactorily rhinoviruses from other picornaviruses. Examination of the restriction maps of sequenced rhinoviruses, revealed a highly conserved BglI restriction site (GCCnnnnnGGC), located exactly in the middle of the 380-bp amplicon generated with the OL26–OL27 primer pair, which has been used extensively in the past to identify picornaviruses. Such a site was either not present, or positioned differently in other picornaviruses of known sequence. It was, therefore, considered that digestion of rhinovirus amplicons with this enzyme would result in two equal length fragments, generating a single 190-bp band in gel electrophoresis. In contrast, either one undigested 380-bp band or a double-band pattern would appear in amplicons from other picornaviruses. To test this hypothesis, Bgl digestions of OL26–OL27 amplicons from cultured and wild-type rhinoviruses, whose identity was confirmed by acid lability, as well as from echo, polio and coxsackie viruses were carried out. All rhinovirus samples were digested successfully generating single bands. Among the other picornaviruses, only 6.6% presented a single band pattern, while the rest were as predicted from the model. With a sensitivity of 100% and a specificity over 90%, the method described, which is rapid and remarkably easy to perform, can be used to distinguish rhinoviruses from other picornaviruses to a considerable extent.
Keywords: BglI, Enterovirus, Picornavirus, PCR, Restriction enzyme, RFLP, Rhinovirus
1. Introduction
Rhinoviruses are the cause of around 60% of upper respiratory infections (Papadopoulos and Johnston, 1998). Although a connection between upper respiratory infections and asthma exacerbations has long been recognised, its importance was underestimated until recently, when it was demonstrated that in the community, 85% of asthma attacks in children (Johnston et al., 1995) and 44% in adults (Nicholson et al., 1993) are precipitated by upper respiratory infections. One of the main reasons leading to this observation has been the increased sensitivity offered by the use of the polymerase chain reaction (PCR) for detecting rhinovirus infections. Standard cell culture-based methods are time-consuming and relatively insensitive for a number of viruses, especially rhinoviruses and coronaviruses (Pattemore et al., 1992), therefore several groups are undertaking currently the task of improving and evaluating PCR-based techniques for these viruses.
Rhinoviruses are members of the Picornaviridae family and exhibit considerable antigenic variability, consisting of over 100 different serotypes. As no important differences in clinical presentation are known to exist between these serotypes, conserved areas of the rhinovirus genome near the 5′ untranslated end have been used most frequently as primers (Mori and Clewley, 1994). Unfortunately, most of these areas are also conserved between the other members of the picornavirus family, most notably enteroviruses. To differentiate rhinoviruses from other picornaviruses several approaches have been suggested, including probe hybridisation, PCR-ELISA, nested PCR and sequencing of PCR amplicons (Ireland et al., 1993, Johnston et al., 1993, Mori and Clewley, 1994, Freymuth et al., 1997). However, the use of these methods increases technical demands in terms of both time and cost, without offering, in most cases, an ideal specificity.
Examination of the restriction maps of sequenced rhinoviruses, revealed a highly conserved BglI restriction site (GCCnnnnnGGC), located in the middle of the 380-bp amplicon generated with the OL26–OL27 primer pair, which has been used extensively in the past to identify picornavirus in culture and clinical samples (Gama et al., 1989, Papadopoulos et al., 1999, Nicholson et al., 1997). Such a site was either not present, or positioned differently in other picornaviruses of known sequence (Table 1 ). Therefore, restriction fragment length polymorphism (RFLP) of picornavirus amplicons using BglI, would result in two equal length fragments in rhinoviruses, generating a single 190-bp band in gel electrophoresis. In contrast either one undigested 380-bp band or a double-band pattern would appear in amplicons from other picornaviruses. To test this hypothesis, OL26–OL27 amplicons from 18 cultured and 32 wild-type rhinoviruses whose identity was confirmed by acid stability, as well as amplicons from 34 enteroviruses were digested with BglI. The method was compared with a semi-nested RT-PCR approach (Ireland et al., 1993) for the differentiation of these viruses.
Table 1.
Characteristics of BglI digestion of OL26–OL27 amplicons from sequenced human picornavirusesa
| OL26 | OL27 | Size | Bgl position | Fragments | |
|---|---|---|---|---|---|
| Rhinovirus 1a | 84–99 | 456–471 | 387 | 279 | 191/192 |
| 1b | 170–185 | 542–557 | 387 | 365 | 191/192 |
| 2 | 164–179 | 532–547 | 383 | 357 | 189/190 |
| 9 | 164–179 | 532–547 | 383 | 357 | 189/190 |
| 14 | 182–197 | 551–566 | 384 | 376 | 190/190 |
| 16 | 166–181 | 536–551 | 385 | 361 | 191/190 |
| 85 | 164–179 | 533–548 | 384 | 357 | 189/191 |
| 89 | 168–183 | 540–555 | 387 | 363 | 191/192 |
| Polio 1 | 165–180 | 541–556 | 391 | – | 391 |
| 2 | 168–183 | 545–560 | 392 | – | 392 |
| 3 | 167–182 | 544–559 | 392 | – | 392 |
| Cox.A9 | 170–185 | 546–561 | 391 | 384 | 177/203 |
| A16 | 174–189 | 550–565 | 391 | – | 391 |
| A21 | 164–179 | 539–554 | 390 | – | 390 |
| A24 | 171–186 | 548–563 | 392 | – | 392 |
| B1 | 167–182 | 543–558 | 391 | – | 391 |
| B3 | 168–183 | 544–559 | 391 | – | 391 |
| B4 | 170–185 | 546–561 | 391 | 384 | 177/203 |
| B5 | 172–187 | 546–561 | 389 | – | 389 |
| Echo 1 | 53–68 | 429–444 | 391 | – | 391 |
| 2 | 53–68 | 429–444 | 391 | – | 391 |
| 3 | 53–68 | 429–444 | 391 | – | 391 |
| 4 | 53–68 | 429–444 | 391 | 267 | 177/203 |
| 5 | 53–68 | 429–444 | 391 | – | 391 |
| 8 | 53–68 | 427–442 | 389 | – | 389 |
| 9 | 169–184 | 543–558 | 389 | 382 | 176/209 |
| 11 | 171–186 | 547–562 | 391 | – | 391 |
| 12 | 168–183 | 544–559 | 391 | – | 391 |
| 25 | 168–183 | 544–559 | 391 | – | 391 |
Position of OL26 and OL27 primers, size of generated amplicon, position of BglI restriction site and resulting fragments after digestion, in currently sequenced rhinoviruses and enteroviruses (sequence source: The Picornavirus Sequence Database. http://www.iah.bbsrc.ac.uk/virus/Picornaviridae/SequenceDatabase/).
2. Materials and methods
2.1. Viruses
Human rhinoviruses 1a, 1b, 2, 3, 5, 7, 9, 10, 14, 16, 30, 41, 44, 49, 50, 58, 70 and 89 were obtained from the MRC Common Cold Unit, Salisbury, UK, the Public Health Laboratory Service, London, UK, the National Institute for Biological Standards, Herts, UK, and the ATCC, Rockville, MD, USA. The identity of the viruses has been confirmed by neutralisation with specific antisera (ATCC). Thirty-two nasal aspirates of subjects with common colds and/or asthma exacerbations from a previous study (Johnston et al., 1995) , in which rhinovirus infection had been confirmed by culture and RT-PCR were retrieved from storage at −70°C.
Thirty enterovirus strains, including human polioviruses 1, 2 and 3 (two different isolates for each serotype), coxsackie viruses A8, A13, A18, A20, A21, A24, B1, B2, B3, B5 and B6, echoviruses 3, 5, 6, 7, 9, 11, 18, 19, 26, 30 and 32, and enteroviruses (EV) 68 and 69, were either ATCC prototype strains or were isolated and characterised at Wessex Public Health Laboratory Service, Southampton General Hospital, UK. Four additional untyped enteroviruses from the latter source, identified by tissue culture but not reacting with any of the anti-enterovirus antibodies used for typing were also included. These strains had been characterised as enteroviruses based on a qualitative acid stability assay whose outcome criterion was growth of the virus in culture after acid treatment, irrespective of titre differential.
2.2. Acid stability and titration assays
The standard assay for the differentiation between rhinoviruses and enteroviruses was used (Johnston and Tyrell, 1995). Briefly, samples were incubated at pH 3 and 7 in parallel, for 1 h at 4°C. The acid-treated sample was brought subsequently back to neutral with 0.1 N sodium hydroxide and the two samples were titrated in parallel. For the titration assay, Ohio HeLa cells, obtained from the MRC Common Cold Unit were seeded 2 days prior to infection in 96-well plates, reaching 60–70% confluence at the time of infection. Eagle’s MEM supplemented with 4% fetal calf serum, 2 mM l-glutamine, 10 mM Hepes (Gibco, Uxbridge, UK), 0.088% sodium bicarbonate, 30 mM magnesium chloride, 0.13% tryptose phosphate broth, and 40 μg/ml gentamycin (Sigma, Poole, UK) was used. Cultures were carried out in a humidified, 5% CO2 incubator at 33°C. Titrations were undertaken in quadruplicate, by logarithmic dilutions of the viruses to 1:10−9. Titration plates were inspected microscopically daily for up to 7 days for the development of cytopathic effect (CPE), after which they were fixed and stained by 5% formaldehyde, 5% ethanol, 0.1% crystal violet in PBS, and the endpoint titre was read, defined as the highest dilution at which CPE was detected in half of the wells (TCID50) and expressed as the inverse logarithm of this dilution. Acid treatment of rhinoviruses results in at least a 100-fold decrease in titre, while enteroviruses are minimally if at all affected.
2.3. RT-PCR
RT-PCR of Ohio-HeLa cell lysates was carried out as described recently (Johnston et al., 1993), with minor modifications. Briefly, to extract RNA 10 μl of sample were diluted 1:5 in ultra high quality water (UHQ) followed by the addition of an equal volume of Trizol™ reagent (Gibco) and 1:10 volume of chloroform. RNA was precipitated with isopropyl alcohol, washed with 80% ethanol, vacuum dried and resuspended in UHQ. Reverse transcription was conducted in a 50 mM Tris–HCl, 75 mM KCl, 3 mM MgCl2 buffer, with 10 mM DTT, 0.4 mM dNTPs, 0.5 μg random hexamer primers (Promega, Southampton, UK) and 100 U of reverse transcriptase (Superscript™, Gibco). The mixture was incubated at 37°C for 60 min to yield single-strand cDNA.
The PCR was carried out in a total volume of 50 μl with 10 μl of cDNA, 10 mM Tris–HCl, 50 mM KCl, 0.1% Triton X-100 buffer with 1.5 mM Mg2+, 0.2 mM dNTPs, 4.25 U Taq DNA polymerase (Promega) and 1.5 μM of primers OL27 (5′-CGG ACA CCC AAA GTA G-3′) and OL26 (5′-GCA CTT CTG TTT CCC C-3′) (Oswel DNA Service, University of Southampton), which are complementary to the antisense RNA in the 5′ non-coding region of picornaviruses at positions shown in Table 1. The thermal cycle consisted of denaturation at 94°C for 30 s, annealing at 50°C for 30 s and extension at 72°C for 2 min, for 32 cycles, including a post-PCR extension step at 72°C for 4 min. A 383–392-bp amplicon is generated, visualised by ethidium bromide staining after electrophoresis on a 2% agarose gel.
2.4. BglI digestion
Amplicons generated with the above RT-PCR were digested with BglI, by adding the enzyme in 5 μl of the buffer provided by the manufacturer to 10 μl of the amplicon solution, prior to gel electrophoresis. Time-course (5 min to 4 h at 37°C) and dose response (0.1–10 U per reaction) experiments were carried out to identify the optimal conditions for the digestion. Electrophoresis was carried out in 2 and 4% agarose, as well as in 6% polyacrylamide gels and standard ethidium bromide staining was used for visualisation. To ensure reproducibility experiments were repeated at least three times.
2.5. Rhinovirus-specific semi-nested PCR
A second, semi-nested PCR round with primers reported to be specific for rhinoviruses (Ireland et al., 1993) was carried out as a comparison assay. For this reaction, the first round PCR product was diluted 1:105 and 10 μl of this dilution were used in a total volume of 50 μl with 10 mM Tris–HCl, 50 mM KCl, 0.1% Triton X-100 buffer, 1.5 mM Mg2+, 0.2 mM dNTPs, 2 U Taq DNA polymerase (Qiagen) and 1 μM of primers OL200 (5′-GGC AGC CAC GCA GGC T-3′) and OL26 (5′-GCA CTT CTG TTT CCC C-3′) (Oswel). After an initial denaturation step at 94°C for 2 min, the thermal cycle was: 94°C for 20 s, 55°C for 20 s, 72° for 30 s, for 25 cycles, followed by a post-PCR extension at 72°C for 10 min.
3. Results
3.1. Optimisation of BglI digestion
BglI digestion of susceptible amplicons was observed even after 5 min of incubation, was complete after 15 min, while incubation periods of over 4 h had no effects on non-susceptible enterovirus amplicons (not shown). Four units of the enzyme completely digested even the strongest bands in 15 min at 37°C. While 4% agarose and 6% polyacrylamide gels offer superior separating capacity, the standard 2% agarose gel could adequately separate bands with ∼25 bp difference in length, while fragments with 1–2-bp size difference appear as a single band (Fig. 1 ).
Fig. 1.
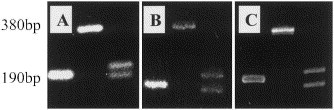
Rhinovirus and enterovirus RFLP patterns in commonly used gels. Comparison of 2% agarose (A), 4% agarose (B), and 6% polyacrylamide (C) gels for the separation of digestion fragments generated by BglI. The single 190-bp band in lane 1 is characteristic of rhinoviruses. Enteroviruses are either not digested, as in lane 2 (poliovirus 1, 380 bp) or produce a double band pattern (lane 3, coxsackie A21, 180+200 bp). While 4% agarose and 6% acrylamide have higher separating capacities, the double band pattern is easily recognised, even in the standard 2% agarose gel.
3.2. RFLP of picornavirus serotypes
All rhinovirus serotypes, as well as all wild-type rhinoviruses, were digested by BglI, producing a single ∼190-bp band, in agreement with the sequence data. Among enteroviruses only two (echovirus 32 and coxsackie B6) were digested producing a single 190-bp band, while the rest were either not digested (20/30) or produced a double (∼175 bp, 200 bp) band (8/30), as predicted from the model (Fig. 2 ). More specifically, all polioviruses (six isolates) were not digested by BglI. Four out of 11 coxsackie virus isolates produced a double band pattern, while six remained undigested. Echoviruses also produced more undigested (7/11) than double-band (3/11) forms. Finally, EV 68 was not digested while EV69 produced a double band pattern.
Fig. 2.
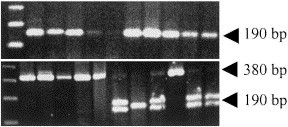
Differentiation of rhinoviruses from enteroviruses by RFLP analysis. Digestion of picornavirus amplicons generated by the OL26–OL27 primer pair, with BglI. Representative gels. Upper panel, lanes 2–11: wild-type rhinoviruses. Complete digestion in the middle of the 380-bp amplicon results in a single 190-bp band in all cases. Lower panel, lanes 2–12: poliovirus 3-isolate (A); poliovirus 3-isolate (B); poliovirus 1, poliovirus 2, enterovirus 68, coxsackie virus B5, echovirus 32, echovirus 19, coxsackie virus B3, echovirus 9, coxsackie virus A21. The absence or different placement of the BglI restriction site results in either undigested 380-bp bands or in an easily recognisable double 180+200-bp band pattern. One of two exceptions (echovirus 32) producing a single band, is shown in lane 8. Lane 1 in both panels is a 123-bp DNA ladder (Gibco).
Interestingly all four non-typed enteroviruses produced a single band pattern. When acid stability was carried out, it was shown that they were in fact acid-labile viruses, i.e. rhinoviruses.
In the examined samples, the sensitivity of RFLP for the identification of rhinoviruses in OL26–OL27 amplicons was 100%, while specificity was 92.4%.
3.3. Comparison with semi-nested PCR
The rhinovirus-specific semi-nested PCR, used for comparison was able to detect all rhinovirus samples. All coxsackie and echoviruses were negative; however, polioviruses 1, 2 and 3, were positive. In agreement with the digestion protocol, the four non-typed ‘enteroviruses’ were also positive, identifying them as rhinoviruses.
4. Discussion
The specific diagnosis of respiratory pathogens is of considerable importance for the understanding of the epidemiology and pathogenesis of the common cold and asthma exacerbations. Rhinoviruses are the most common agents related to these conditions (Johnston et al., 1995, Nicholson et al., 1997). With more than 100 characterised serotypes, their significant genetic variability leaves little choice for selection of conserved primer pairs for their identification by RT-PCR, and sequences from the 5′ untranslated end of their genome have been used in most cases. Unfortunately, these primers cannot differentiate rhinoviruses from other picornaviruses, especially enteroviruses which may in some cases produce similar symptomatology.
To address this problem, RFLP analysis was carried out in amplicons generated by a primer pair which has been extensively used for the identification of rhinoviruses both in culture and in clinical samples. The BglI restriction site, identified in maps of cloned rhinoviruses, is ideally placed in the middle of the amplicon in rhinoviruses, while it is either not present or differently positioned in enteroviruses (Table 1), resulting in easily identifiable patterns after standard gel electrophoresis. When 84 rhinovirus and enterovirus serotypes, as well as community-derived wild type viruses were tested, the theoretical prediction was confirmed in all but two cases. Interestingly, the number of enterovirus strains producing a double-band pattern was somewhat higher (∼26%) than that anticipated from the currently available sequences, where less than 20% include a BglI site. Furthermore, in some instances this pattern was obtained from enterovirus serotypes that, according to their sequence, should not possess such a site. This could possibly be attributed to an increased genetic variability of that area in enteroviruses, in contrast to the highly conserved sequence in rhinoviruses. Nevertheless this variability does not seem to affect the ability of the method to differentiate rhinoviruses from enteroviruses, since the different positioning of the BglI site is the crucial factor.
Definite practical advantages are associated with RFLP analysis. The procedure is extremely easy to perform and can be incorporated in the RT-PCR protocol as an additional step before electrophoresis. Digestion time is negligible; however, it can be extended according to convenience, when, e.g. a large number of samples have to be processed, without adversely affecting the result. The cost is low, while using lower doses and extending digestion time could possibly minimise it. Finally, by using a well characterised set of primers no extra optimisation is required, while screening can be carried out in existing stored amplicons from a considerable number of previous studies.
In contrast, alternative approaches, such as internal probe hybridisation (Johnston et al., 1993), nested PCR (Ireland et al., 1993) or sequencing of PCR amplicons (Mori and Clewley, 1994) are considerably more demanding in terms of cost, time and effort. Furthermore, the specificity of some of these methods is not ideal: the semi-nested RT-PCR we used as a comparison assay failed to differentiate polioviruses from rhinoviruses.
The main drawback of the digestion assay is that it does not increase the sensitivity of the initial RT-PCR. Nevertheless, the OL26–OL27 PCR is quite sensitive and it is not certain that further increase in sensitivity would result in a substantial improvement. As we were not able to test all members of the picornavirus family the full usefulness of the method remains to be determined. However, the 100% negative predictive value observed in the experiments indicates that the described method could be useful as a screening assay for human rhinoviruses.
An interesting finding of this study was the identification of four rhinovirus strains that were identified previously as enteroviruses by a qualitative acid lability test. In that assay, viral growth after acid treatment, irrespective of titre differential, was the criterion for their identification as enteroviruses. However, these strains did not react with any of the antibodies used for enterovirus typing. Both the digestion assay and the semi-nested PCR indicated that these strains were actually rhinoviruses, and this was confirmed by quantitative acid lability testing. This implies that optimised PCR-based methodologies, in addition to sensitivity, could before long attain the specificity required to substitute traditional assays, which are still used as a ‘gold standard’.
While the importance of rhinoviruses for the induction of upper respiratory infections and asthma exacerbations has been increasingly appreciated during the past few years, many questions regarding the epidemiology and pathogenicity of these viruses remain to be answered. The assay described for their differentiation from other picornaviruses could prove useful in this respect.
References
- Freymuth F., Vabret A., Galateau-Salle F., Ferey J., Eugene G., Petitjean J., Gennetay E., Brouard J., Jokik M., Duhamel J.F., Guillois B. Detection of respiratory syncytial virus, parainfluenzavirus 3, adenovirus and rhinovirus sequences in respiratory tract of infants by polymerase chain reaction and hybridization. Clin. Diagn. Virol. 1997;8:31–40. doi: 10.1016/s0928-0197(97)00060-3. [DOI] [PubMed] [Google Scholar]
- Gama R.E., Horsnell P.R., Hughes P.J., North C., Bruce C.B., al-Nakib W., Stanway G. Amplification of rhinovirus specific nucleic acids from clinical samples using the polymerase chain reaction. J. Med. Virol. 1989;28:73–77. doi: 10.1002/jmv.1890280204. [DOI] [PubMed] [Google Scholar]
- Ireland D.C., Kent J., Nicholson K.G. Improved detection of rhinoviruses in nasal and throat swabs by seminested RT-PCR. J. Med. Virol. 1993;40:96–101. doi: 10.1002/jmv.1890400204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston S.L., Tyrell D.A.J. Rhinoviruses. In: Lennette E.H., Lennette D.A., Lennette E.T., editors. Diagnostic Procedures for Viral, Rickettsial and Chlamydial Infections. 7. American Public Health Association; Washington: 1995. pp. 553–563. [Google Scholar]
- Johnston S.L., Sanderson G., Pattemore P.K., Smith S., Bardin P.G., Bruce C.B., Lambden P.R., Tyrrell D.A.J., Holgate S.T. Use of polymerase chain reaction for diagnosis of picornavirus infection in subjects with and without respiratory symptoms. J. Clin. Microbiol. 1993;31:111–117. doi: 10.1128/jcm.31.1.111-117.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston S.L., Pattemore P.K., Sanderson G., Smith S., Lampe F., Josephs L., Symington P., O’Toole S., Myint S.H., Tyrrell D.A.J., Holgate S.T. Community study of role of viral infections in exacerbations of asthma in 9–11 year old children [see comments] Br. Med. J. 1995;310:1225–1228. doi: 10.1136/bmj.310.6989.1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori J., Clewley J.P. Polymerase chain reaction and sequencing for typing rhinovirus RNA. J. Med. Virol. 1994;44:323–329. doi: 10.1002/jmv.1890440403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson K.G., Kent J., Ireland D.C. Respiratory viruses and exacerbations of asthma in adults [see comments] Br. Med. J. 1993;307:982–986. doi: 10.1136/bmj.307.6910.982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson K.G., Kent J., Hammersley V., Cancio E. Acute viral infections of upper respiratory tract in elderly people living in the community: comparative, prospective, population based study of disease burden. Br. Med. J. 1997;315:1060–1064. doi: 10.1136/bmj.315.7115.1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos, N.G., Johnston, S.L., 1998. Rhinoviruses. In: Zuckerman, A., Banatvala, P.J. (Eds.), Principles and Practice of Clinical Virology, 4th ed. Wiley, Chichester (in press).
- Papadopoulos, N.G., Sanderson, G., Hunter, J., Johnston, S.L., 1999. Rhinoviruses replicate effectively at lower airway temperatures. J. Med. Virol. 58, 100–104. [DOI] [PubMed]
- Pattemore P.K., Johnston S.L., Bardin P.G. Viruses as precipitants of asthma symptoms. I. Epidemiology. Clin. Exp. Allergy. 1992;22:325–336. doi: 10.1111/j.1365-2222.1992.tb03094.x. [DOI] [PMC free article] [PubMed] [Google Scholar]