

Abstract
Culture and serotyping of human enteroviruses by fluorescence microscopy are time-consuming and labor-intensive. Flow cytometry has the potential of being more rapid, sensitive, and objective but has not been used for these purposes in a clinical laboratory. Primary rhesus monkey kidney (PMK) cells were inoculated with several enterovirus serotypes and stained with enterovirus-specific antibodies for flow cytometry and indirect fluorescence antibody testing (IFA). Kinetic studies of coxsackievirus B1 and echovirus 30 infection of PMK cells were performed on days 1–4 after inoculation. Flow cytometry results for echovirus 6, 9, 11, and 30 and coxsackievirus B1 correlated with IFA in all cases. Coxsackievirus B1 and echovirus 30 infections were detected 1 day earlier by flow cytometry than IFA. Flow cytometry can be effectively used for detecting enterovirus-infected cells in a clinical laboratory with the advantages of better quantitation of low levels of infection and earlier detection of virally infected cells in culture systems.
Keywords: Enterovirus, Flow cytometry, Serotype
1. Introduction
Human enteroviruses, members of the Picornaviridae family, are among the most common human pathogens. They cause a broad spectrum of human disease, including myocarditis, hand-foot-and-mouth disease, meningoencephalitis, and paralytic myelitis (Romero, 2007). At least 68 serotypes have been defined. Periodic enterovirus outbreaks occur and are a major public health concern (Khetsuriani et al., 2006). While most enteroviral infections are not life-threatening, certain serotypes (e.g., polioviruses, enterovirus 71) can cause devastating neurological damage and death. Poliovirus outbreaks still occur in developing nations despite eradication efforts. Oral poliovirus vaccine-derived strains are an increasing cause of viral paralytic syndrome and must be differentiated from wild-type poliovirus and other causes of paralytic myelitis such as West Nile virus, coxsackievirus A7, and enterovirus 70/71 (Muir et al., 1998). Hence, the capacity to classify human enteroviruses in clinical specimens is imperative to disease prevention, treatment, and prognosis.
Historically, serotyping enteroviruses has been accomplished by a combination of indirect fluorescence antibody testing (IFA) and viral neutralization assays using anti-sera pools provided by the World Health Organization (WHO). These techniques are time-consuming, labor-intensive, and technically demanding. The WHO no longer produces anti-sera for viral neutralization studies, which now must be obtained from the few remaining sources such as the National Institute of Public Health and the Environment (RIVM, The Netherlands). Flow cytometry is a potential alternative to IFA for serotyping clinical isolates and provides quantitative information with less inter-observer variability. With flow cytometry, hundreds of cells can be analyzed in a short period of time (seconds) with increased sensitivity to detection of fluorescence signals due to the use of lasers and photomultiplier tubes, and has the ability for automation. Despite these advantages over IFA, no studies have focused on the use of flow cytometry for serotyping enterovirus prior to this study.
2. Materials and methods
2.1. Enterovirus isolates
Isolates were obtained from a frozen repository and had been previously cultured and serotyped using serum neutralization studies in the clinical virology laboratory at Associated Regional and University Pathologists (ARUP) Laboratories as previously described (She et al., 2006). All enteroviral isolates were stored at −70 °C prior to use in this study.
2.2. Preparation of cells for flow cytometric analysis
For each serotype, 5–10 shell vials containing a confluent layer of primary rhesus monkey kidney (PMK) (ViroMed Laboratories, Minnetonka, MN) were inoculated with 100 μL echovirus 6, echovirus 9, echovirus 11, echovirus 30, coxsackievirus B1, or rhinovirus 7 in 1 mL minimum essential medium (MEM) with 2% fetal bovine serum (FBS) and centrifuged for 15 min at 3000 × g. Specific viral inocula were not determined. Uninoculated cells were included as negative controls in each study. Cells were incubated at 37 °C and were harvested for staining and analysis as follows. The media were aspirated and cells were washed three times in phosphate buffered saline (PBS). Cells were overlaid with 100 μL TrypLE Trypsin Replacement (Invitrogen, Carlsbad, CA) for 30 s before addition of 1 mL MEM with 2% FBS. The cells were then transferred to a 1.5 mL microcentrifuge tube and centrifuged for 5 min at 300 × g. If cells were off the monolayer due to cytopathic effect (CPE), the overlying media was additionally saved and centrifuged. The cell pellet was washed in 500 μL PBS, centrifuged for 5 min at 300 × g, and resuspended in 100 μL PBS.
The Fix & Perm Cell Permeabilization kit (Invitrogen) was used for viral staining following the manufactures directions. Briefly, cells were transferred to a 5 mL tube and incubated with 100 μL Reagent A for 15 min at room temperature. The cells were then washed with 3 mL PBS with 5% FBS, centrifuged for 5 min at 300 × g, and resuspended in 100 μL Reagent B (permeabilization medium) and one drop (approximately 50 μL) of primary antibody which included pan-enterovirus (Diagnostic Hybrids Inc. (DHI), Athens, OH), normal mouse IgG (isotype control), coxsackievirus B blend, poliovirus blend, enterovirus blend, and echovirus blend antibodies (Millipore, Billerica, MA) in all cases. For echovirus 30, echovirus 30 antibody was tested and for echovirus 9, individual antibodies directed against echovirus 4, 6, 9, 11, and 30 were tested. For coxsackievirus B1, individual antibodies directed against coxsackievirus B1 through B6 were tested. After 20 min incubation at room temperature in the dark, the cells were washed with 3 mL PBS with 5% FBS, centrifuged for 5 min at 300 × g, and resuspended in 100 μL Reagent B and one drop of secondary antibody (fluorescein isothiocyanate (FITC)-conjugated mouse monoclonal antibody; DHI or Millipore). The cells were incubated for 15 min at room temperature in the dark and washed and centrifuged as above. The cells were resuspended in 500 μL PBS containing 1% paraformaldehyde and analyzed by flow cytometry immediately. All antibodies used were monoclonal antibodies or blends of monoclonal antibodies.
2.3. Flow cytometry and data analysis
Flow cytometer instrument setup was performed daily using Flow-Check and Flow-Set fluorospheres (Beckman Coulter, Miami, FL). The stained samples were analyzed using FC 500 flow cytometer (Beckman Coulter, Miami, FL), creating listmode files containing at least 5000 events. Listmode files were analyzed using CXP (Beckman Coulter, Miami, FL) and FlowJo (Tree Star, Ashland, OR) software.
2.4. Indirect fluorescent antibody (IFA) studies
Cells were prepared for traditional IFA staining using the trypsinized and washed cells prepared as described above. The 100 μL cell suspension was spotted to 4- or 8-well slides, dried on a slide warmer set at 37 °C, then fixed in cold acetone for 10 min. One drop of primary antibody was placed on each well and slides were incubated at 37 °C in the dark for 30 min. Slides were washed with PBS and distilled water and air-dried. One drop of FITC-conjugated secondary antibody was placed on each well and slides were incubated at 37 °C in the dark for 30 min. After a rinse with PBS and distilled water, a coverslip was placed on each slide using mounting media (DHI) and examined under a fluorescent microscope for positive, fluorescent green staining of the cells. Uninoculated cells stained with enterovirus-specific antibodies and enterovirus-infected cells stained with normal mouse antibody were included as controls.
3. Results
3.1. Flow cytometric analysis of infected PMK cells
For initial feasibility studies, flow cytometric analysis was performed on PMK cells that showed CPE following infection with echovirus 6, echovirus 9, echovirus 11, echovirus 30, or coxsackievirus B1. The percentages of infected cells were quantified by first gating on the forward and side scatter signals to help eliminate cell debris and non-cellular events that may be present. Negative control levels were determined using isotype control antibodies, which gave similar results to unstained cells or cells stained with antibodies directed against other (irrelevant) enterovirus serotypes. The infected cells represented those gated cellular events staining above negative control levels using virus specific antibodies. As shown in Fig. 1 , coxsackievirus B1-infected cells demonstrated positive staining for pan-enterovirus, coxsackievirus B blend, and coxsackievirus B1 antibodies and were negative for isotype control, echovirus blend, enterovirus blend, poliovirus blend, and coxsackievirus B2, B3, B4, B5, and B6 antibodies. Representative data shown in Fig. 2 demonstrate that echovirus 9-infected cells were positive for echoblend and echovirus 9-specific antibodies and negative for echovirus 4, 6, 11, and 30 individual specific antibodies. The thresholds used in Fig. 1, Fig. 2 were based on subjective criteria in which a natural break between positive and negative cells could be illustrated, allowing for easy comparison between infected cells and negative controls. PMK cells infected with echovirus 6, echovirus 11, or echovirus 30 demonstrated positive staining for pan-enterovirus and echovirus blend antibodies and were negative for isotype control, enterovirus blend, coxsackievirus B blend, and poliovirus blend antibodies (not shown). Echovirus 30-infected cells were additionally positive for echovirus 30-specific antibody. Rhinovirus 7-infected cells stained similarly to uninoculated cells for pan-enterovirus, echovirus blend, coxsackievirus B blend, and isotype antibodies. In all cases, IFA demonstrated qualitatively similar results compared to the flow cytometry determined percentages.
Fig. 1.
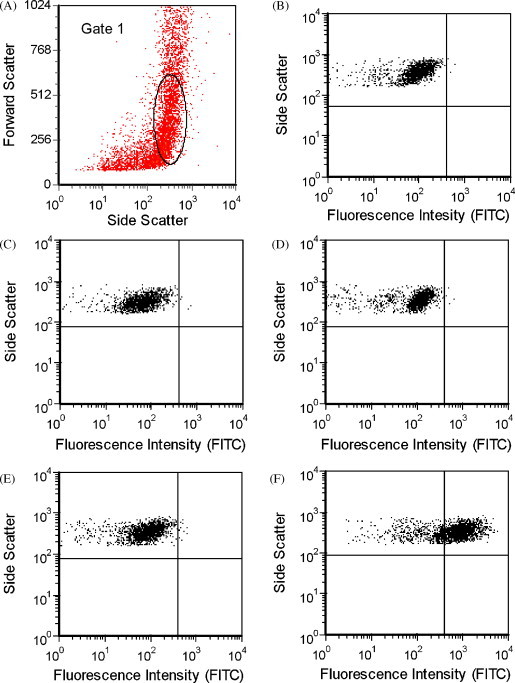
Uninfected PMK cells and coxsackievirus B1-infected PMK cells were harvested 4 days after virus was inoculated for flow cytometric evaluation. (A) Cellular events were selected for fluorescence analysis based on forward and side scatter as shown (Gate 1). Similar patterns were observed for (B) uninfected PMK cells stained with coxsackievirus B blend antibodies (0.21% positive) and (C) coxsackievirus B1-infected PMK cells stained with isotype control (0.25% positive), (D) echovirus blend (0.24% positive), or (E) coxsackievirus B6 antibody (0.60% positive). (F) The majority (71.52%) of coxsackievirus B1-infected PMK cells stained positively with coxsackievirus B blend antibody above the negative control threshold level.
Fig. 2.
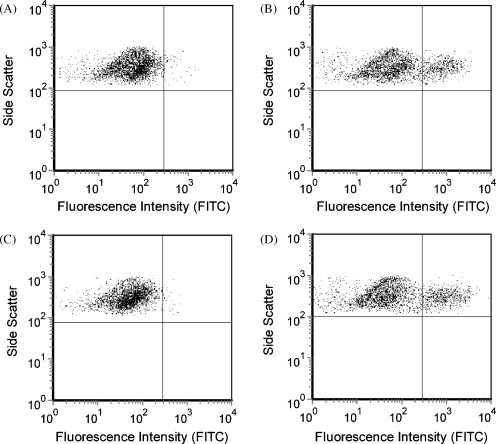
Uninfected PMK cells and echovirus 9-infected PMK cells were harvested 1 day after inoculation for flow cytometric evaluation. (A) Uninfected PMK cells stained with echovirus blend antibodies; (B) echovirus 9-infected PMK cells stained with echovirus blend antibody; (C) echovirus 4 specific antibody; (D) echovirus 9 specific antibody. Plots (B) and (D) show positively staining populations (19.0% and 22.3%, respectively) above negative controls (plots A and C).
3.2. Kinetic studies of antibody staining
PMK cell shell vials were inoculated with echovirus 30 or coxsackievirus B1 and analyzed by flow cytometry at 24, 48, 72, and 96 h after inoculation. Cells were examined for CPE and by IFA in parallel to flow cytometric testing. For coxsackievirus B1, flow cytometry and IFA detected pan-enterovirus, coxsackievirus blend, and coxsackievirus B1 antibody-stained cells 1 day after inoculation, while IFA became positive 2 days after inoculation (Fig. 3 and Table 1 ). The thresholds used in Fig. 3 were set to allow for one or no positive events in the uninfected cell control. Positive events were considered those staining above the threshold level. Similar results were obtained with echovirus 30, in that PMK cells with detectable antibody staining for pan-enterovirus or echovirus blend were found by flow cytometry 2 days after cells were inoculated, whereas IFA detected positive-staining cells by the same antibodies 3 days after inoculation. In both echovirus 30- and coxsackievirus B1-infected cells, CPE became evident at the same time IFA was positive. Different antibodies (pan-enterovirus, coxsackievirus B blend, and coxsackievirus B1 antibodies for coxsackievirus B1-infected cells and pan-enterovirus and echovirus blend antibodies for echovirus 30-infected cells) were used to monitor over time enterovirus infection in PMK shell vial cells and all gave similar results.
Fig. 3.
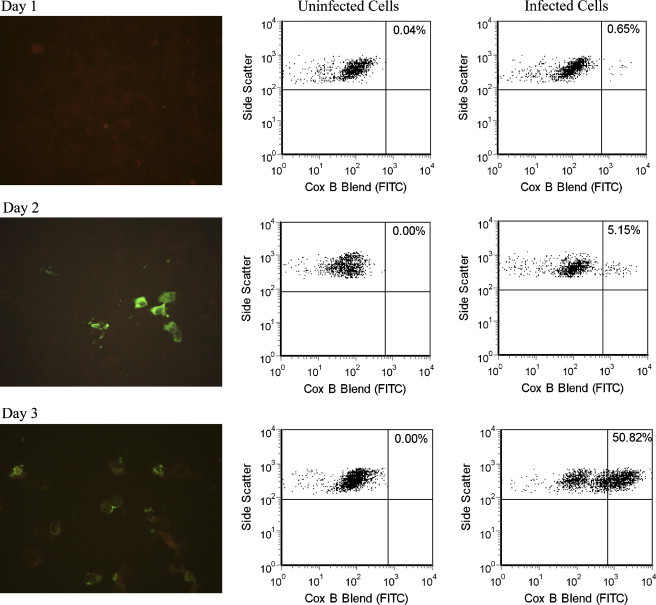
Comparison of IFA (left column) and flow cytometry analysis of uninfected PMK cells (center column) and PMK cells days 1–3 after inoculation with coxsackievirus B1 (right column). IFA showed detectable fluorescent cells with apple green cytoplasmic staining at day 2, while flow cytometry was able to detect a small number of cells with bright staining on day 1. Negative cells, counterstained with Evans Blue, are dull red on IFA.
Table 1.
Kinetic study of coxsackievirus B1-infected PMK cells as demonstrated by flow cytometric analysis, IFA, and observation of CPE. Data for flow cytometry and IFA are from cells stained with coxsackievirus B blend antibody. Flow cytometry detected positively staining, infected cells 1 day prior to IFA and appearance of CPE.
| Flow cytometry—% gated cells staining positively | IFA—semi-quantitative measurementa | CPE—semi-quantitative measurementb | |
|---|---|---|---|
| Day 1 | 0.65 | 0 | 0 |
| Day 2 | 5.2 | 1–2+ | 1–2+ |
| Day 3 | 50.8 | 3+ | 3+ |
| Day 4 | 46.9 | 4+ | 4+ |
0 = no staining observed; 1+ = 1–10% of cells show positive staining; 2+ = 11–30% of cells show positive staining; 3+ = 31–70% of cells show positive staining; 4+ = >70% of cells show positive staining.
0 = no CPE observed; 1+ = 1–10% of cells show CPE; 2+ = 11–30% of cells show CPE; 3+ = 31–70% of cells show CPE; 4+ = >70% of cells show CPE.
4. Discussion
While the applications of flow cytometry are many and varied, it has not been well-studied for detection of enterovirus infection from culture. Previous studies have focused on using flow cytometry to quantitate poliovirus infection in neuronal cells (Daley et al., 2005), characterize enterovirus binding to host cell surfaces (Freistadt and Eberle, 2006, Mbida et al., 1991, Triantafilou et al., 2001), confirm cytomegalovirus infection of tissue culture cells with a genetically engineered fluorescence reporter system (Kung et al., 2000), and serotype human immunodeficiency virus type 1 (Zolla-Pazner et al., 1995). This is the first report of a clinical application for flow cytometry in detecting enterovirus from culture.
In this study, flow cytometry proved to be a sensitive method for detecting fluorescently stained enterovirus-infected cells. For both echovirus 30- and coxsackievirus B1-infected PMK cells, flow cytometry was able to detect infection 1 day before the viral infection became detectable by IFA. This is likely due to the ability of flow cytometry to quickly analyze a larger number of cells than is routinely examined by IFA. It was also able to quantitate the level of infection even when there are low numbers of virus-infected cells. The capacity to quantitate allowed for monitoring enterovirus infection in PMK cells over a 4-day period using different antibodies which gave similar results. The coxsackievirus B1 strain used in this study was highly efficient at infecting PMK cells compared to the echovirus 30 strain. As the predilection of different enterovirus serotypes and even strains within a serotype for different cells lines is well-known (She et al., 2006), it is not surprising that a different rate of infection between the two serotypes used in this study was observed. False positives did not occur by flow cytometric analysis in that infected cells were negative after staining with isotype control antibodies and antibodies directed against other enterovirus serotypes. This is not surprising as the specificity of flow cytometry in general is related to the particular antibodies employed, and therefore, should be similar to microscopy based detection methods using the same antibodies. Also, uninoculated cells did not demonstrate positive staining with any of the antibodies used. Study of additional human enterovirus serotypes using this method is warranted.
Typing of enteroviruses by sequence analysis has emerged as an accurate surrogate for conventional serotyping methods (Oberste et al., 2000). Because highly variable regions are targeted by genotyping studies, degenerate primer sequences and/or nested designs are necessary to ensure adequate sensitivity. Use of an antibody-based method, such as flow cytometry and IFA, may have the advantage of better consistency in identifying enterovirus serotypes. Flow cytometry can also be potentially faster and less expensive than molecular methods for viral typing.
Although flow cytometric analysis was shown to be highly sensitive, the current method was cumbersome and not optimized for performance in a clinical laboratory. Shell vials were used to facilitate infection of cells with centrifugation, as is commonly done in the clinical virology laboratory. It is possible that a more efficient tissue culture system could be employed, such as use of multiple well plates (e.g., 96-well plate format). In this way the need for handling numerous shell vials per specimen is obviated. Others have found the use of centrifuged multiple well plates combined with blind immunoperoxidase staining for enterovirus to be both rapid and sensitive (Bourlet et al., 1998, Terletskaia-Ladwig et al., 2008) and this method can potentially be performed using fluorescent antibody staining followed by flow cytometric analysis instead. Because the PMK cells grow as a surface monolayer, trypsinization was used to create an optimal single cell suspension (Grogan and Collins, 1990). Another potential option is the use of lymphocytic or monocytic cell lines which have been described for cultivation of a limited number of enterovirus serotypes (Okada et al., 1987, Skarsvik et al., 2006, Vuorinen et al., 1999, Vuorinen et al., 1994). Cultivation of enterovirus in such a manner would facilitate interrogation by flow cytometry and may be studied in the future. Other cell lines that are routinely used in the clinical laboratory for isolation of enterovirus, e.g., RD, MRC-5, and BGM, should also be investigated. Directly labeled antibodies are not commercially available for enteroviruses but would be useful in streamlining the protocol described. Multicolor flow cytometry could be used to simultaneously monitor infections of different cell types and/or viruses from a single culture. Thus, modifications of the method described in this study could make the use of flow cytometry considerably more practical and automated for routine use in clinical microbiology laboratories.
5. Conclusions
Flow cytometry can be effectively used for detecting enterovirus-infected cells in a laboratory setting. Its potential advantages over fluorescent microscopy include better quantitation of low levels of infection and earlier detection of virally infected cells in culture systems. These could lead to faster laboratory identification of pathogenic viruses. The use of flow cytometry on direct clinical specimens was not explored but could be studied in the future on cellular specimens such as those from the respiratory tract. Prospective application of this method to patient isolates is still needed to confirm its validity. The analyses in this study were limited to enterovirus, but the methods that were tested and discussed can be applied to a wide array of other clinically important viruses. Adenovirus, emerging coronaviruses, and influenza virus, including highly pathogenic strains, affect the public health and in each case, knowledge of serotype provides important strain information and epidemiologic data. Additional studies are still needed to extend the methods presented to other viruses and optimize detection strategies.
Acknowledgments
This work was supported by a grant from the CAP Foundation, NIH grant 5R21DE017136 (DWB), and the ARUP Institute for Clinical and Experimental Pathology.
References
- Bourlet T., Gharbi J., Omar S., Aouni M., Pozzetto B. Comparison of a rapid culture method combining an immunoperoxidase test and a group specific anti-VP1 monoclonal antibody with conventional virus isolation techniques for routine detection of enteroviruses in stools. J. Med. Virol. 1998;54:204–209. doi: 10.1002/(sici)1096-9071(199803)54:3<204::aid-jmv11>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- Daley J.K., Gechman L.A., Skipworth J., Rall G.F. Poliovirus replication and spread in primary neuron cultures. Virology. 2005;340:10–20. doi: 10.1016/j.virol.2005.05.032. [DOI] [PubMed] [Google Scholar]
- Freistadt M.S., Eberle K.E. Fluorescent poliovirus for flow cytometric cell surface binding studies. J. Virol. Methods. 2006;134:1–7. doi: 10.1016/j.jviromet.2005.08.011. [DOI] [PubMed] [Google Scholar]
- Grogan W.L., Collins J.M. Marcel Dekker; New York: 1990. Guide to Flow Cytometry Methods. [Google Scholar]
- Khetsuriani N., Lamonte-Fowlkes A., Oberst S., Pallansch M.A. Enterovirus surveillance—United States, 1970–2005. MMWR Surveill. Summ. 2006;55:1–20. [PubMed] [Google Scholar]
- Kung S.H., Wang Y.C., Lin C.H., Kuo R.L., Liu W.T. Rapid diagnosis and quantification of herpes simplex virus with a green fluorescent protein reporter system. J. Virol. Methods. 2000;90:205–212. doi: 10.1016/s0166-0934(00)00234-2. [DOI] [PubMed] [Google Scholar]
- Mbida A.D., Pozzetto B., Sabido O., Akono Y., Grattard F., Habib M., Gaudin O.G. Competition binding studies with biotinylated echovirus 11 in cytofluorimetry analysis. J. Virol. Methods. 1991;35:169–176. doi: 10.1016/0166-0934(91)90132-j. [DOI] [PubMed] [Google Scholar]
- Muir P., Kammerer U., Korn K., Mulders M.N., Poyry T., Weissbrich B., Kandolf R., Cleator G.M., van Loon A.M. Molecular typing of enteroviruses: current status and future requirements. The European Union Concerted Action on Virus Meningitis and Encephalitis. Clin. Microbiol. Rev. 1998;11:202–227. doi: 10.1128/cmr.11.1.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberste M.S., Maher K., Flemister M.R., Marchetti G., Kilpatrick D.R., Pallansch M.A. Comparison of classic and molecular approaches for the identification of untypeable enteroviruses. J. Clin. Microbiol. 2000;38:1170–1174. doi: 10.1128/jcm.38.3.1170-1174.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada Y., Toda G., Oka H., Nomoto A., Yoshikura H. Poliovirus infection of established human blood cell lines: relationship between the differentiation stage and susceptibility of cell killing. Virology. 1987;156:238–245. doi: 10.1016/0042-6822(87)90403-x. [DOI] [PubMed] [Google Scholar]
- Romero J.R. Enteroviruses and parechoviruses. In: Murray P.R., Baron E.J., Jorgensen J.H., Landry M.L., Pfaller M.A., editors. vol. 2. ASM Press; Washington, DC: 2007. pp. 1392–1404. (Manual of Clinical Microbiology). [Google Scholar]
- She R.C., Crist G., Billetdeaux E., Langer J., Petti C.A. Comparison of multiple shell vial cell lines for isolation of enteroviruses: a national perspective. J. Clin. Virol. 2006;37:151–155. doi: 10.1016/j.jcv.2006.06.009. [DOI] [PubMed] [Google Scholar]
- Skarsvik S., Puranen J., Honkanen J., Roivainen M., Ilonen J., Holmberg H., Ludvigsson J., Vaarala O. Decreased in vitro type 1 immune response against coxsackie virus B4 in children with type 1 diabetes. Diabetes. 2006;55:996–1003. doi: 10.2337/diabetes.55.04.06.db05-0630. [DOI] [PubMed] [Google Scholar]
- Terletskaia-Ladwig E., Meier S., Hahn R., Leinmuller M., Schneider F., Enders M. A convenient rapid culture assay for the detection of enteroviruses in clinical samples: comparison with conventional cell culture and RT-PCR. J. Med. Microbiol. 2008;57:1000–1006. doi: 10.1099/jmm.0.47799-0. [DOI] [PubMed] [Google Scholar]
- Triantafilou M., Wilson K.M., Triantafilou K. Identification of Echovirus 1 and coxsackievirus A9 receptor molecules via a novel flow cytometric quantification method. Cytometry. 2001;43:279–289. doi: 10.1002/1097-0320(20010401)43:4<279::aid-cyto1060>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- Vuorinen T., Vainionpaa R., Heino J., Hyypia T. Enterovirus receptors and virus replication in human leukocytes. J. Gen. Virol. 1999;80(Pt 4):921–927. doi: 10.1099/0022-1317-80-4-921. [DOI] [PubMed] [Google Scholar]
- Vuorinen T., Vainionpaa R., Kettinen H., Hyypia T. Coxsackievirus B3 infection in human leukocytes and lymphoid cell lines. Blood. 1994;84:823–829. [PubMed] [Google Scholar]
- Zolla-Pazner S., O’Leary J., Burda S., Gorny M.K., Kim M., Mascola J., McCutchan F. Serotyping of primary human immunodeficiency virus type 1 isolates from diverse geographic locations by flow cytometry. J. Virol. 1995;69:3807–3815. doi: 10.1128/jvi.69.6.3807-3815.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]