

Abstract
Background
Respiratory viruses induce asthma exacerbations and airway hyperresponsiveness (AHR). Atopy is an important risk factor for asthma persistence.
Objective
We sought to evaluate whether atopy is a risk factor for prolonged AHR after upper respiratory tract infections (URIs).
Methods
Twenty-five children (13 atopic and 12 nonatopic children) with intermittent virus-induced asthma were studied. Clinical evaluation, skin prick tests, methacholine bronchoprovocation, questionnaires, and a nasal wash specimen were obtained at baseline. For 9 months, subjects completed diary cards with respiratory symptoms. During their first reported cold, a nasal wash specimen was obtained. Methacholine provocation was performed 10 days and 5, 7, 9, and 11 weeks later. In case a new cold developed, the provocation schedule was followed from the beginning.
Results
Viruses were detected in 17 (68%) of 25 patients during their first cold, with rhinovirus being most commonly identified (82%). AHR increased significantly 10 days after the URI, equally in both groups (P = .67), and remained so up to the fifth week. Duration of AHR in subjects experiencing a single URI ranged from 5 to 11 weeks, without a significant difference between groups. In the duration of the study, atopic children experienced more colds and asthma exacerbations than nonatopic children. Thus for duration of AHR, significant prolongation was noted in the atopic group when assessed cumulatively.
Conclusion
In asthmatic children the duration of AHR after a single natural cold is 5 to 11 weeks. However, an increased rate of symptomatic cold and asthma episodes in atopic children is associated with considerable cumulative prolongation of AHR, which might help explain the role of atopy as a risk factor for asthma persistence.
Key words: Asthma, airway hyperresponsiveness, atopy
Abbreviations used: AHR, Airway hyperresponsiveness; NW, Nasal wash; SPT, Skin prick test; URI, Upper respiratory tract infection
Asthma is characterized by abnormalities in lung function, variable airway obstruction, and airway hyperresponsiveness (AHR)1; among others, a significant correlation exists between the degree of AHR and both clinical severity and medication needs for asthma.2 A number of stimuli, including respiratory viruses, are able to induce AHR.3 Increased AHR to histamine in healthy subjects after upper respiratory tract infections (URIs) lasting up to 6 weeks was observed more than 20 years ago.4 Human experimental infections with human rhinoviruses confirmed that increased AHR to nonspecific stimuli is observed for up to 4 weeks after a viral infection in allergic subjects.5, 6, 7 Furthermore, AHR after viral infections has been documented in animal models of paramyxovirus, respiratory syncytial virus, and influenza virus infections.8, 9, 10 Most asthma exacerbations in children are associated with URIs, attributed in their majority to rhinoviruses.11, 12 Virus-induced AHR is increased in atopic individuals compared with in healthy control subjects.5, 13 Moreover, atopy is one of the strongest risk factors for the development and persistence of asthma, especially in childhood.14 More than 60% of asthmatic children are atopic, whereas the presence of atopy at the age of 12 months increases 3 to 4 times the risk for asthma persistence during later childhood and adulthood.15, 16 There are suggestions that genes responsible for atopy and AHR act together to develop the full asthma phenotype.17 Furthermore, it has been proposed that the defective epithelial repair cycle, which is characteristic of asthma and strongly correlates to AHR, is amplified by exposure to TH2 cytokines.18 Nevertheless, our current understanding of the mechanisms connecting atopy, AHR, and asthma is still incomplete. The classical model of sensitization and exposure to particular allergens19 can only partly explain the epidemiologic observations. More recently, we and others have shown that the response of atopic asthmatic individuals to rhinovirus infection is defective, with a suboptimal TH1 response and a shift toward a TH2 phenotype.20, 21 Such a response might lead to incomplete viral clearance and inflammation persistence, potentially perpetuating AHR.22
On the basis of the above, we hypothesized that atopy could be a risk factor for prolongation of AHR after respiratory viral infections. The aim of this study was to prospectively evaluate the duration of AHR in children with asthma after naturally occurring respiratory infections and assess the role of atopy as a risk factor in this respect.
Methods
Patients
Twenty-five children (16 boys), 7 to 12 years of age, with intermittent asthma and a history of disease exacerbations attributable to URI only, were recruited for the study. Asthma diagnosis was based on history, 12% or greater improvement in FEV1 after bronchodilation, and AHR (PC20 ≤ 16 mg/mL). Classification of asthma severity was performed according to the Global Initiative for Asthma.23 Atopy was evaluated by using skin prick tests (SPTs) to a panel of 18 locally relevant allergens performed outside the pollen season. Exclusion criteria were a history of seasonal or allergen-driven symptoms, current or previous use of specific immunotherapy, use of inhaled steroids within the previous 2 months, recent (<2 months) URI, and chronic conditions potentially affecting airway responsiveness.24 All parents provided written consent, and the study was approved by the hospital's ethics committee.
Study design
A prospective case-control design with 9 months' follow-up (September-June) was used. Clinical evaluation, IgE determination, SPT, and methacholine bronchial provocation were performed at baseline. Questionnaires and a nasal wash (NW) specimen were also obtained. Patients were grouped according to the presence of atopy (with n = 13). Children (or their parents) were asked to prospectively complete diary cards with upper and lower respiratory tract symptoms.11 During the first reported URI, judged either subjectively or by means of increased daily symptom scores (≥4), an NW was performed. Methacholine provocation was performed 10 days, and 5, 7, 9, and 11 weeks later. In case a new cold developed within this time period, the above provocation schedule was followed from the beginning. Bronchodilators were used as relievers; in case of persistence or deterioration of asthma symptoms, subjects were instructed to receive systemic steroids, although these were not needed on any occasion.
Methacholine provocation
Methacholine provocation was performed with the 2-minute breathing dosing protocol.25 The aerosols were generated with a nebulizer output of 0.13 mL/min (Air Dynaval Taema). The procedure was discontinued if FEV1 decreased 20% or greater from baseline values or when a 16 mg/mL concentration had been administered.26 Spirometry was performed according to the American Thoracic Society guidelines.27
Virus detection
RT-PCR was used for detection of viral RNA in NW samples, as previously described.28 PCR reactions were performed for rhinoviruses; enteroviruses; respiratory syncytial virus; coronaviruses OC43 and 229E; influenza viruses A and B; parainfluenza viruses 1, 2, and 3; adenoviruses; Chlamydia pneumoniae; and Mycoplasma pneumoniae.29, 30
Statistical analysis
Comparisons of baseline characteristics were performed with the Fisher exact test for categoric variables and t tests for continuous variables. A common cold and an asthma exacerbation were defined as an increase of the relevant symptom score totals over the personal median value for 2 days or more. The duration of each episode was defined as the time from symptom initiation to return to the median value. Severity of each was defined as the maximum symptom score. The Wilcoxon signed-rank test and the Mann-Whitney U test were used to compare independent and paired outcome variables, respectively. The time to PC20 restoration was displayed in Kaplan-Mayer curves compared with the log-rank test. P values of less than .05 were considered significant.
Results
Baseline characteristics
Table I shows the demographic, socioeconomic, and disease characteristics of the 2 groups, as obtained at baseline. Atopic asthmatic children had statistically significantly higher levels of total IgE (P = .04). A trend toward more cases of positive family history of atopy was also noted in the atopic group (P = .08). In respect to all remaining characteristics, the groups were homogeneous, with no significant differences when statistically compared. Baseline spirometric values did not differ between the 2 groups (Table I).
Table I.
Patient characteristics: Comparison of baseline characteristics between atopic and nonatopic asthmatic children included in the study
| Baseline characteristics | Nonatopic (n = 12) | Atopic (n = 13) | P value |
|---|---|---|---|
| Age (y) | 10.4 ± 1.8 | 10.3 ± 1.2 | NS |
| Sex (boys) | 50% | 77% | NS |
| IgE (mg/dL) | 78.9 ± 14.5 | 339.4 ± 117.1 | .04 |
| Residence (urban) | 66% | 54% | NS |
| Density (persons per room) | 1.5 | 1.2 | NS |
| Pet ownership | 12% | 13% | NS |
| Exposure to tobacco smoke | 75% | 62% | NS |
| School type (public) | 83% | 85% | NS |
| Paternal education level (secondary education) | 73% | 69% | NS |
| Maternal education level (secondary education) | 58% | 54% | NS |
| Father or mother with atopy | 60% | 85% | .08 |
| Colds in the previous year (n) | 3.5 | 3.6 | NS |
| Asthma exacerbations in the previous year (n) | 3.3 | 3.3 | NS |
| Emergency visits for asthma in the previous year (n) | 2.3 | 2.7 | NS |
| Hospitalizations for asthma in the previous year (n) | 0.1 | 0 | NS |
| FVC (% mean) ± SD | 92.9 ± 11.3 | 88.5 ± 9.3 | NS |
| FEV1 (% mean) ± SD | 81.4 ± 6.7 | 78.2 ± 7.4 | NS |
FVC, Forced vital capacity.
Detection of respiratory viruses with PCR
All NW specimens obtained at baseline were negative for the presence of respiratory viruses. PCR revealed the presence of a virus in 17 (68%) of 25 patients during their subsequent first URI. The most commonly identified virus was rhinovirus (14/17 [82%]). Adenovirus was found in 4 (23%) of 17 positive samples, whereas one child had both rhinovirus and adenovirus. Virus identification rates did not differ significantly between groups (nonatopic, 58%; atopic, 77%; P = .34).
Duration of AHR
All subjects completed the study. Methacholine responsiveness at baseline was slightly, but not significantly, higher in atopic individuals (time = 0, atopic PC20 = 5.9 ± 1.1 mg/mL, nonatopic PC20 = 8.4 ± 1.5 mg/mL; P = .18). All subjects experienced at least one natural cold within the study period. AHR increased significantly 10 days after the first reported URI, equally in both groups (time = 10th day, atopic PC20 = 2.3 ± 1.1 mg/mL, nonatopic PC20 = 2.6 ± 0.9 mg/mL; P = .002 in comparison with their respective baseline values in both cases and P = .67 between groups). This increase remained statistically significant up to the fifth week after the onset of the cold, progressively decreasing, although still without statistically significant differences, between the groups (time = fifth week, atopic PC20 = 3.7 ± 1.3 mg/mL, nonatopic PC20 = 4.7 ± 1.3 mg/mL; P = .005 and P = .021, respectively, in comparison with baseline and P = .29 between groups).
The above analysis was performed in subjects with no additional URI during that period (time = 0, n = 25; time = 10 days, n = 24; time = 5 weeks, n = 23). After the fifth week, several children experienced additional colds, and therefore provocations were rescheduled according to the study design; comparisons were not done on fixed time points. Eleven weeks after the initial URI, 10 children (3 atopic and 7 nonatopic children) had no further cold, and their methacholine responsiveness had returned to their respective baseline value. The mean duration of AHR in these subjects was 7.0 ± 2.0 weeks (median, 5 weeks; range, 5-11 weeks) for the atopic children and 7.3 ± 1.0 weeks (median, 7 weeks; range, 5-11 weeks) for the nonatopic children. In addition, using a simple linear regression model based on AHR values from the 10th day on and including all subjects with a single URI, the predicted time for AHR to return to its respective baseline value was 5.6 to 8.9 weeks for the atopic children and 6.7 to 10.2 weeks for the nonatopic children ( Fig 1).
Fig 1.
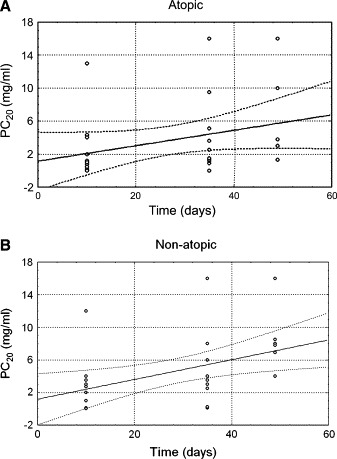
Linear regression model of AHR changes from the 10th day to the seventh week after a single URI in atopic (A) and nonatopic (B) children with intermittent virus-induced asthma. A significant and time-dependent AHR decrease is observed in both cases. The predicted time for AHR to return to baseline did not differ between the groups.
However, important differences, as shown in Fig 2, were observed when the cumulative duration of AHR was assessed prospectively in the complete cohort. All 12 nonatopic asthmatic children returned to their baseline PC20 values by day 120, after the first reported URI, whereas only 9 (67%) of 13 of the atopic children returned to their baseline PC20 value by day 200 (P = .0068), showing that in assessing the natural history of the disease, increased airway responsiveness, possibly caused by repetitive colds, is considerably more prolonged in atopic asthmatic children and might in fact last more than 6 months.
Fig 2.
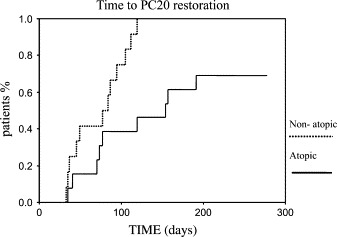
Kaplan-Meyer diagram of PC20 return to its respective baseline value after one or more naturally occurring URIs during a 9-month period in atopic and nonatopic asthmatic children. The difference is significant (P = .0068, survival analysis).
Common cold and asthma exacerbation characteristics
Atopic children had generally more disease episodes than nonatopic children ( Fig 3). The average number of colds that each subject experienced during the whole study period was marginally higher in the atopic group (atopic group, 4.6 ± 0.4; nonatopic group, 3.5 ± 0.4; P = .06). The average duration of each cold did not differ between groups (atopic group, 10.4 ± 1.8 days; nonatopic group, 9.4 ± 1.3 days; P = .81), as was the case for cold severity (atopic group, 2.2 ± 0.2; nonatopic group, 2.9 ± 0.3; P = .12).
Fig 3.
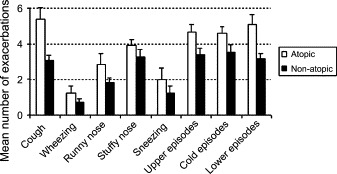
Upper respiratory tract symptom exacerbations, colds, and asthma exacerbations experienced during a 9-month period in atopic (open bars) and nonatopic (filled bars) children with intermittent asthma. ∗P < .05, #P = .06.
Atopic asthmatic children experienced significantly more exacerbations during the study period (5.1 ± 0.6 vs 3.2 ± 0.3 of the nonatopic children, P = .01). There were no differences in the duration or severity of asthma exacerbations between the groups (duration: atopic group, 8.5 ± 0.9 days; nonatopic group, 11.4 ± 2.3 days; P = .65; severity: atopic group, 4.4 ± 0.8; nonatopic group, 4.6 ± 0.9; P = .60).
No statistically significant differences were found in either the duration or severity of symptom scores between patients with positive and negative virus identification (data not shown).
All atopic children had positive SPT responses to common pollen allergens (with a wheal of ≥3 mm).31 One atopic child also presented with a positive SPT response to mites. To address the question of whether prolonged AHR in atopic children might be affected by exposure to allergens to which they were sensitized, we compared the characteristics of asthma exacerbations during the pollen season (March-June) and the cold season (October-January). There were no differences in any such characteristics within the nonatopic group ( Table II). However, within the atopic group, the number of asthma exacerbations was significantly higher during the cold season, suggesting that exposure to allergens could not account for additional disease burden in these patients (Table II).
Table II.
Seasonal distribution of asthma exacerbation characteristics in atopic and nonatopic asthmatic children
| Number | Duration (d) | Severity (index) | |
|---|---|---|---|
| Nonatopic | |||
| Winter | 1.50 (0.55) | 10.17 (15.21) | 1.43 (0.55) |
| Spring | 2.00 (1.26) | 4.67 (1.63) | 1.10 (0.33) |
| P value | .52 | .79 | .29 |
| Atopic | |||
| Winter | 2.67 (1.50) | 9.25 (4.80) | 1.52 (0.53) |
| Spring | 1.42 (0.67) | 7.08 (4.89) | 1.31 (0.35) |
| P value | .02 | .15 | .24 |
Reported values are presented as means (SD).
Winter, October through January; spring, March through June.
Discussion
This is the first study to prospectively evaluate the duration of AHR in children after naturally occurring colds for an extended period of time. Two major findings are reported: (1) the duration of postviral nonspecific AHR is considerably more prolonged than previously thought, and (2) the duration of AHR after a single cold is the same in atopic and nonatopic children; however, an increased number of symptomatic colds cumulatively lead the former to prolonged AHR.
Most previous studies assessing the duration of AHR after viral infections have used human rhinovirus experimental infections, which usually result in mild-to-moderate colds and very mild, if any, asthma exacerbations.32 Furthermore, fixed time points for up to 8 weeks after infection have been used for outcome evaluation.7, 33 In this respect these studies excluded, to a considerable extent, the possibility of interference from additional infections but on the other hand could not evaluate the duration of virus-induced AHR in real-life conditions. In our study, when the duration of AHR was assessed after a single infection, either in the subgroup of subjects who did not have additional infection for up to 11 weeks, or by means of a simple regression model, it was 7 weeks on average, ranging from 5 to 11 weeks. This time span is still longer than previously reported in human subjects (10 days to 6 weeks)4, 34 and comparable with time spans reported studies prospectively evaluating postviral AHR in experimental animals (2-22 weeks).8, 10, 35, 36 Nevertheless, when AHR was assessed in the long term after multiple colds, a considerable proportion of atopic subjects remained hyperresponsive for considerably longer, in some cases more than 6 months. This is consistent with the notion that additional, naturally occurring colds might further prolong AHR, as recently shown in an experimental animal setting,37 and might prove important in considering the natural history of the disease, as well as management of such patients. Therefore although our hypothesis that atopy might lead to postviral AHR prolongation was shown not to be directly true because there was no difference in AHR duration after a single infection, the total duration of AHR was in fact significantly longer in atopic than nonatopic children and that was associated with an increased number of colds, as well as asthma episodes.
Patient characteristics did not differ between the groups at baseline; it cannot be excluded, however, that subclinical inflammation might have been greater in atopic subjects, which were slightly, but not significantly, more hyperresponsive at baseline.
Several studies have shown that viral infection induces greater changes in nonspecific AHR in patients with respiratory allergy than in healthy control subjects.5, 7, 38, 39 Accordingly, it has been assumed that after a viral infection, the response to allergen exposure might be exaggerated, a notion elegantly confirmed by using segmental bronchial provocations with allergen.40, 41 Furthermore, clinical studies have shown synergy between viral infection and allergen exposure in sensitized individuals.42, 43 Nevertheless, other studies have shown that allergen exposure does not necessarily augment virus-mediated responses.44, 45 This might also be the case in the atopic population of our study, who, even though they were sensitized to pollen allergens, had more symptoms during the fall and winter months rather than during the pollen season. This was not completely unexpected because all our subjects were selected for a postinfectious asthma phenotype during the 2 previous years, apparently reporting symptoms only after colds and not after allergen exposure. Certainly, the possibility that unidentified allergens might have contributed to some exacerbations cannot be completely excluded. However, the fact that the identified allergens did not seem to influence the outcome, although the number of colds and subsequent asthma exacerbations during the study was higher in atopic children, suggests that an increased susceptibility to viral infection might be a more plausible mechanism for prolongation of AHR in that group. Previous evidence suggests that the immune response to rhinovirus is defective in atopic asthmatic individuals, with reduced IFN-γ production,20, 46 which might be associated with increased susceptibility to symptomatic virus infections. In fact, rhinovirus-induced IFN-γ production is strongly associated with AHR in patients with asthma.21
There is a longstanding speculation that allergic subjects, asthmatic subjects, or both have more respiratory infections than the healthy population.47 Most studies have compared respiratory symptoms after URI in atopic and healthy individuals. In a recent longitudinal cohort study, Corne et al48 showed that subjects with atopic asthma are not at greater risk of a rhinovirus infection than healthy individuals but have more frequent, severe, and longer-lasting lower respiratory tract symptoms.48 On the other hand, even within an atopic population, the outcome of an experimental rhinovirus infection is closely associated with levels of TH1 and TH2 cytokines.46 It is possible that any effects of atopy on the susceptibility to respiratory viral infections might differ between adults and children, in which cytokine responses, including IFN-γ responses, do not mature before late childhood.49, 50 Furthermore, recent evidence suggests that primary epithelial cells derived from atopic subjects are more susceptible to ex vivo rhinovirus infection than cells from healthy control subjects.51
A weakness of the present study is that PCR viral identification was performed only for the first reported cold and not for subsequent colds. Nevertheless, it is without doubt that respiratory viral infections are the cause of the common cold, whereas the association of colds with subsequent asthma exacerbations is also well established.11, 12
The degree of airway responsiveness is indicative of asthma severity and counts as an indirect marker of airway inflammation.2, 52 In this respect prolongation of virus-induced AHR might well reflect persistent airway inflammation after multiple insults. Perpetuation of subclinical airway inflammation could have a substantial effect on the risk of asthma persistence, relapse later in life, or both, providing a possible mechanism for the well-established role of atopy as a major risk factor for the persistence of asthma and AHR from childhood to adulthood.53, 54
In conclusion, the duration of AHR in children with intermittent virus-induced asthma ranges from 5 to 11 weeks after a single infection but might be considerably prolonged with repeated infections. Atopic subjects present with more symptomatic colds and thus cumulatively have significantly more prolonged AHR. Prolongation of virus-induced AHR in atopic subjects might help explain the well-established role of atopy as a risk factor for asthma persistence.
Athens, Greece
Footnotes
Disclosure of potential conflict of interest: N. Papadopoulos has received grants–research support from GlaxoSmithKline and AstraZeneca. P. Saxoni-Papageorgiou has received grants–research support from Novartis and Schering-Plough. All other authors—none disclosed.
References
- 1.Lowe L., Custovic M.S., Woodcock A. Childhood asthma. Curr Allergy Asthma Rep. 2004;4:159–165. doi: 10.1007/s11882-004-0062-9. [DOI] [PubMed] [Google Scholar]
- 2.Weiss S.T., Van Natta M.L., Zeiger R.S. Relationship between increased airway responsiveness and asthma severity in the childhood asthma management program. Am J Respir Crit Care Med. 2000;162:50–56. doi: 10.1164/ajrccm.162.1.9811005. [DOI] [PubMed] [Google Scholar]
- 3.Louis-Philippe B. Physiopathology of airway hyperresponsiveness. Curr Allergy Asthma Rep. 2003;3:166–171. doi: 10.1007/s11882-003-0030-9. [DOI] [PubMed] [Google Scholar]
- 4.Empey D.W., Laitinen L.A., Jacobs L., Gold W.M., Nadel J.A. Mechanisms of bronchial hyperreactivity in normal subjects after upper respiratory tract infection. Am Rev Respir Dis. 1976;113:131–139. doi: 10.1164/arrd.1976.113.2.131. [DOI] [PubMed] [Google Scholar]
- 5.Gern J.E., Calhoun W., Swenson C., Shen G., Busse W.W. Rhinovirus infection preferentially increases lower airway responsiveness in allergic subjects. Am Respir Crit Care Med. 1997;155:1872–1876. doi: 10.1164/ajrccm.155.6.9196088. [DOI] [PubMed] [Google Scholar]
- 6.Grunberg K., Kuijpers E.A., de Klerk E.P., de Gouw H.W., Kroes A.C., Dick E.C. Effects of experimental rhinovirus 16 infection on airway hyperresponsiveness to bradykinin in asthmatic subjects in vivo. Am J Respir Crit Care Med. 1997;155:833–838. doi: 10.1164/ajrccm.155.3.9117013. [DOI] [PubMed] [Google Scholar]
- 7.Cheung D., Dick E.C., Timmers M.C., de Klerk E.P., Spaan W.J., Sterk P.J. Rhinovirus inhalation causes long-lasting excessive airway narrowing in response to methacholine in asthmatic subjects in vivo. Am J Respir Crit Care Med. 1995;152:1490–1496. doi: 10.1164/ajrccm.152.5.7582282. [DOI] [PubMed] [Google Scholar]
- 8.Jafri H.S., Chavez-Bueno S., Mejias A., Gomez A.M., Rios A.M., Nassi S.S. Respiratory syncytial virus induces pneumonia, cytokine response, airway obstruction, and chronic inflammatory infiltrates associated with long-term airway hyperresponsiveness in mice. J Infect Dis. 2004;189:1856–1865. doi: 10.1086/386372. [DOI] [PubMed] [Google Scholar]
- 9.Folkerts G., Busse W.W., Nijkamp F.P., Sorkness R., Gern J.E. Virus induced airway hyperresponsiveness and asthma. Am J Respir Crit Care Med. 1998;157:1708–1720. doi: 10.1164/ajrccm.157.6.9707163. [DOI] [PubMed] [Google Scholar]
- 10.Walter M.J., Morton J.D., Kajiwara N., Agapov E., Holtzman M.J. Viral induction of a chronic asthma phenotype and genetic segregation from the acute response. J Clin Invest. 2002;110:165–175. doi: 10.1172/JCI14345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Johnston S.L., Pattemore P.K., Sanderson G., Smith S., Lampe F., Josephs L. Community study of role of viral infections in exacerbations of asthma in 9-11 year old children. BMJ. 1995;310:1225–1229. doi: 10.1136/bmj.310.6989.1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Papadopoulos N.G., Psarras S., Manoussakis E., Saxoni-Papageorgiou P. The role of respiratory viruses in the origin and exacerbations of asthma. Curr Opin Allergy Clin Immunol. 2003;3:39–44. doi: 10.1097/00130832-200302000-00007. [DOI] [PubMed] [Google Scholar]
- 13.Bardin P.G., Sanderson G., Robinson B.S., Holgate S.T., Tyrrell D.A. Experimental rhinovirus infection in volunteers. Eur Respir J. 1996;9:2250–2255. doi: 10.1183/09031936.96.09112250. [DOI] [PubMed] [Google Scholar]
- 14.Christodoulopoulos P., Cameron L., Nakamura Y., Lemiere C., Muro S., Dugas M. TH2 cytokine-associated transcription factors in atopic and nonatopic asthma: evidence for differential signal transducer and activator of transcription 6 expression. J Allergy Clin Immunol. 2001;107:586–591. doi: 10.1067/mai.2001.114883. [DOI] [PubMed] [Google Scholar]
- 15.Martinez F.D. Links between pediatric and adult asthma. J Allergy Clin Immunol. 2001;107(suppl):S449–S455. doi: 10.1067/mai.2001.114993. [DOI] [PubMed] [Google Scholar]
- 16.Warner J.O. Bronchial hyperresponsiveness, atopy, airway inflammation and asthma. Pediatr Allergy Immunol. 1998;9:55–56. doi: 10.1111/j.1399-3038.1998.tb00304.x. [DOI] [PubMed] [Google Scholar]
- 17.Postma D.S., Koppelman G.H., Meyers D.A. The genetics of atopy and airway hyperresponsiveness. Am J Respir Crit Care Med. 2000;162(suppl):S118–S123. doi: 10.1164/ajrccm.162.supplement_2.ras-13. [DOI] [PubMed] [Google Scholar]
- 18.Davies D.E., Wicks J., Powell R.M., Puddicombe S.M., Holgate S.T. Airway remodeling in asthma: new insights. J Allergy Clin Immunol. 2003;111:215–226. doi: 10.1067/mai.2003.128. [DOI] [PubMed] [Google Scholar]
- 19.Nelson H.S., Szefler S.J., Jacobs J., Huss K., Shapiro G., Sternberg A.L. The relationships among environmental allergen sensitization, allergen exposure, pulmonary function, and bronchial hyperresponsiveness in the Childhood Asthma Management Program. J Allergy Clin Immunol. 1999;104:775–785. doi: 10.1016/s0091-6749(99)70287-3. [DOI] [PubMed] [Google Scholar]
- 20.Papadopoulos N.G., Stanciu L.A., Papi A., Holgate S.T., Johnston S.L. A defective type 1 response to rhinovirus in atopic asthma. Thorax. 2002;57:328–332. doi: 10.1136/thorax.57.4.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brooks G.D., Buchta K.A., Swenson C.A., Gern J.E., Busse W.W. Rhinovirus-induced interferon-γ and airway responsiveness in asthma. Am J Respir Crit Care Med. 2003;168:1091–1094. doi: 10.1164/rccm.200306-737OC. [DOI] [PubMed] [Google Scholar]
- 22.Papadopoulos N.G., Stanciu L.A., Papi A., Holgate S.T., Johnston S.L. Rhinovirus-induced alterations on peripheral blood mononuclear cell phenotype and costimulatory molecule expression in normal and atopic asthmatic subjects. Clin Exp Allergy. 2002;32:537–542. doi: 10.1046/j.0954-7894.2002.01313.x. [DOI] [PubMed] [Google Scholar]
- 23.National Institutes of Health, National Heart, Lung and Blood Institute; Bethesda (MD): 2002. Global Strategy for Asthma Management and Prevention. Publication no. 02-3659. [Google Scholar]
- 24.Cain H. Bronchoprovocation testing. Clin Chest Med. 2001;22:651–659. doi: 10.1016/s0272-5231(05)70058-7. [DOI] [PubMed] [Google Scholar]
- 25.American Thoracic Society Guidelines for methacholine and exercise challenge testing-1999. Am J Respir Crit Care Med. 2000;161:309–329. doi: 10.1164/ajrccm.161.1.ats11-99. [DOI] [PubMed] [Google Scholar]
- 26.Cockcroft D.W. How best to measure airway responsiveness. Am J Respir Crit Care Med. 2001;163:1514–1515. doi: 10.1164/ajrccm.163.7.2103055b. [DOI] [PubMed] [Google Scholar]
- 27.American Thoracic Society. Standarization of spirometry: 1994 update. Am J Respir Crit Care Med. 1995;152:1107–1136. doi: 10.1164/ajrccm.152.3.7663792. [DOI] [PubMed] [Google Scholar]
- 28.Papadopoulos N.G., Sanderson G., Hunter J., Johnston S.L. Rhinovirus replicate effectively at lower airway temperatures. J Med Virol. 1999;58:100–104. doi: 10.1002/(sici)1096-9071(199905)58:1<100::aid-jmv16>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 29.Papadopoulos N.G., Hunter J., Sanderson G., Meyer J., Johnston S.L. Rhinovirus identification by BglI digestion of picornavirus RT-PCR amplicons. J Virol Methods. 1999;80:179–185. doi: 10.1016/S0166-0934(99)00045-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Papadopoulos N.G., Moustaki M., Tsolia M., Bossios A., Astra E., Prezerakou A. Association of rhinovirus infection with increased disease severity in acute bronchiolitis. Am Respir Crit Care Med. 2002;165:1285–1289. doi: 10.1164/rccm.200112-118BC. [DOI] [PubMed] [Google Scholar]
- 31.Sub-Committee on Skin Tests of the European Academy of Allergology and Clinical Immunology. Skin tests used in type I allergy testing [position paper]. Allergy 1989;44 Suppl 10:1-59. [PubMed]
- 32.Skoner D.P., Doyle W.J., Seroky J., Fireman P. Lower airway responses to influenza A virus in healthy allergic and nonallergic subjects. Am J Respir Crit Care Med. 1996;154:661–664. doi: 10.1164/ajrccm.154.3.8810602. [DOI] [PubMed] [Google Scholar]
- 33.Fleming H.E., Little F.F., Schnurr D., Avila P.C., Wong H., Liu J. Rhinovirus-16 colds in healthy and in asthmatic subjects: similar changes in upper and lower airways. Am J Respir Crit Care Med. 1999;160:100–108. doi: 10.1164/ajrccm.160.1.9808074. [DOI] [PubMed] [Google Scholar]
- 34.Grunberg K., Timmers M.C., Smits H.H., de Klerk E.P., Dick E.C., Spaan W.J. Effect of experimental rhinovirus 16 colds on airway hyperresponsiveness to histamine and interleukin-8 in nasal lavage in asthmatic subjects in vivo. Clin Exp Allergy. 1997;27:36–45. doi: 10.1111/j.1365-2222.1997.tb00670.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schwarze J., Gelfand E.W. Respiratory viral infections as promoters of allergic sensitization and asthma in animal models. Eur Respir J. 2002;19:341–349. doi: 10.1183/09031936.02.00254302. [DOI] [PubMed] [Google Scholar]
- 36.Folkerts G., Nijkamp F.P. Virus-induced airway hyperresponsiveness. Role of inflammatory cells and mediators. Am J Respir Crit Care Med. 1995;151:1666–1674. doi: 10.1164/ajrccm.151.5.7735631. [DOI] [PubMed] [Google Scholar]
- 37.Matsuse H., Behera A.K., Kumar M., Rabb H., Lockey R.F., Mohapatra S.S. Recurrent respiratory syncytial virus infections in allergen-sensitized mice lead to persistent airway inflammation and hyperresponsiveness. J Immunol. 2000;164:6583–6592. doi: 10.4049/jimmunol.164.12.6583. [DOI] [PubMed] [Google Scholar]
- 38.Trigg C.J., Nicholson K.G., Wang J.H., Ireland D.C., Jordan S., Duddle J.M. Bronchial inflammation and the common cold: a comparison of atopic and non-atopic individuals. Clin Exp Allergy. 1996;26:665–676. doi: 10.1111/j.1365-2222.1996.tb00593.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sears M.R., Burrows B., Herbison G.P., Holdaway M.D., Flannery E.M. Atopy in childhood. II. Relationship to airway responsiveness, hay fever and asthma. Clin Exp Allergy. 1993;23:949–956. doi: 10.1111/j.1365-2222.1993.tb00280.x. [DOI] [PubMed] [Google Scholar]
- 40.Lemanske R.F., Dick E.C., Swenson C.A., Vrtis R.F., Buse W.W. Rhinovirus upper respiratory tract infection increases airway hyperreactivity and late asthmatic reactions. J Clin Invest. 1989;83:1–10. doi: 10.1172/JCI113843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Calhoun W.J., Dick E.C., Schwartz L.B., Busse W.W. A common cold virus, rhinovirus 16, potentiates airway inflammation after segmental antigen bronchoprovocation in allergic subjects. J Clin Invest. 1994;94:2200–2208. doi: 10.1172/JCI117581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Green R.M., Custovic A., Sanderson G., Hunter J., Johnston S.L., Woodcock A. Synergism between allergens and viruses and risk of hospital admission with asthma: case-control study. BMJ. 2002;324:763–768. doi: 10.1136/bmj.324.7340.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Papadopoulos N.G., Psarros F., Manoussakis E., Hatzipsalti M., Bossios A., Syrigou E. Association of respiratory viral infections with disease exacerbations in patients with allergen-driven seasonal asthma and hay fever. Allergy. 2002;57:295. [abstract] [Google Scholar]
- 44.Avila P.C. Interactions between allergic inflammation and respiratory viral infections. J Allergy Clin Immunol. 2000;106:829–831. doi: 10.1067/mai.2000.111027. [DOI] [PubMed] [Google Scholar]
- 45.de Kluijver J., Evertse C.E., Sont J.K., Schrumpf J.A., van Zeijl-van der Ham C.J., Dick C.R. Are rhinovirus-induced airway responses in asthma aggravated by chronic allergen exposure? Am J Respir Crit Care Med. 2003;168:1174–1180. doi: 10.1164/rccm.200212-1520OC. [DOI] [PubMed] [Google Scholar]
- 46.Parry D.E., Busse W.W., Sukow K.A., Dick C.R., Swenson C., Gern J.E. Rhinovirus-induced PBMC responses and outcome of experimental infection in allergic subjects. J Allergy Clin Immunol. 2000;105:692–698. doi: 10.1067/mai.2000.104785. [DOI] [PubMed] [Google Scholar]
- 47.Busse W.W., Gern J.E. Do allergies protect against the effects of a rhinovirus cold? J Allergy Clin Immunol. 2000;105:889–891. doi: 10.1067/mai.2000.106378. [DOI] [PubMed] [Google Scholar]
- 48.Corne J.M., Marshall C., Smith S., Schreiber J., Sanderson G., Holgate S.T. Frequency, severity, and duration of rhinovirus infections in asthmatic and non-asthmatic individuals: a longitudinal cohort study. Lancet. 2002;359:831–834. doi: 10.1016/S0140-6736(02)07953-9. [DOI] [PubMed] [Google Scholar]
- 49.Prescott S.L. Allergy: when does it begin and where will it end? Allergy. 2003;58:864–867. doi: 10.1034/j.1398-9995.2003.00231.x. [DOI] [PubMed] [Google Scholar]
- 50.Smart J.M., Kemp A.S. Ontogeny of T-helper 1 and T-helper 2 cytokine production in childhood. Pediatr Allergy Immunol. 2001;12:181–187. doi: 10.1034/j.1399-3038.2001.012004181.x. [DOI] [PubMed] [Google Scholar]
- 51.Wark P.A., Johnston S.L., Bucchieri F., Powell R., Puddicombe S., Laza-Stanca V. Asthmatic bronchial epithelial cells have a deficient innate immune response to infection with rhinovirus. J Exp Med. 2005;201:937–947. doi: 10.1084/jem.20041901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Boulet L.P. Asymptomatic airway hyperresponsiveness: a curiosity or an opportunity to prevent asthma? Am J Respir Crit Care Med. 2003;167:371–378. doi: 10.1164/rccm.200111-084PP. [DOI] [PubMed] [Google Scholar]
- 53.Arruda L.K., Sole D., Baena-Cagnani C.E., Naspitz C.K. Risk factors for asthma and atopy. Curr Opin Allergy Clin Immunol. 2005;5:153–159. doi: 10.1097/01.all.0000162308.89857.6c. [DOI] [PubMed] [Google Scholar]
- 54.Van den Toorn L.M., Overbeek S.E., de Jongste J.C., Leman K., Hoogsteden H.C., Prins J.B. Airway inflammation is present during clinical remission of atopic asthma. Am J Respir Crit Care Med. 2001;164:2107–2113. doi: 10.1164/ajrccm.164.11.2006165. [DOI] [PubMed] [Google Scholar]