

Abstract
Human noroviruses (NoVs) are a major cause of gastroenteritis worldwide. It is thought that, similar to other RNA viruses, high mutation rates allow NoVs to evolve fast and to undergo rapid immune escape at the population level. However, the rate and spectrum of spontaneous mutations of human NoVs have not been quantified previously. Here, we analyzed the intra-patient diversity of the NoV capsid by carrying out RT-PCR and ultra-deep sequencing with 100,000-fold coverage of 16 stool samples from symptomatic patients. This revealed the presence of low-frequency sequences carrying large numbers of U-to-C or A-to-G base transitions, suggesting a role for hyper-mutation in NoV diversity. To more directly test for hyper-mutation, we performed transfection assays in which the production of mutations was restricted to a single cell infection cycle. This confirmed the presence of sequences with multiple U-to-C/A-to-G transitions, and suggested that hyper-mutation contributed a large fraction of the total NoV spontaneous mutation rate. The type of changes produced and their sequence context are compatible with ADAR-mediated editing of the viral RNA.
Keywords: Hyper-mutation, Next-generation sequencing, Norovirus, RNA virus
Highlights
-
•
Norovirus U-to-C hyper-mutants are present in patient samples.
-
•
Analysis of hyper-mutants in cell culture suggests ADAR-mediated RNA edition.
-
•
Hyper-mutation may contribute to norovirus diversity and evolution.
1. Introduction
Noroviruses (NoVs) are one of the most common causes of foodborne viral gastroenteritis, infecting over 250 million people worldwide every year. Symptoms typically last 24 to 48 h, but complications can occur in immunocompromised patients, resulting in an estimated 200,000 deaths per year mainly among elderly people and young children in developing countries (Patel et al., 2008, Robilotti et al., 2015). NoVs are positive-stranded RNA viruses belonging to the family Caliciviridae and, similar to other RNA viruses, they exhibit extremely high levels of genetic diversity (Debbink et al., 2012). NoVs have evolved into seven highly divergent genogroups (GI–GVII), which are in turn divided into genotypes. The prototypic Norwalk virus belongs to genotype GI.1, but GII.4 has become the most prevalent genotype in the last decades, being responsible for the majority of outbreaks (White, 2014). The most variable NoV genome regions are located in the surface-exposed P2 domain of the capsid (VP1) protein, which determines antibody escape (Lindesmith et al., 2008, White, 2014). Differences in genetic diversity and evolution rates among NoV genotypes have been attributed to multiple factors, including random genetic drift, receptor usage, the structural plasticity of the VP1 protein, and replication fidelity (Bull and White, 2011, Donaldson et al., 2010). However, and despite its purported importance for evolution, immune escape, and the development of efficient control strategies, the rate of spontaneous mutation of human NoVs has not been experimentally determined.
The genetic diversity of RNA viruses is ultimately driven by their extremely high rates of spontaneous mutation, which are orders of magnitude higher than those of DNA-based microorganisms and range from 10− 6 to 10− 4 per nucleotide per round of copying (Lauring et al., 2013, Sanjuán et al., 2010). Such high mutation rates are commonly attributed to the low replication fidelity of RNA virus polymerases, since these lack 3′ exonuclease activity in all viral families examined except coronaviruses (Smith and Denison, 2013, Ulferts and Ziebuhr, 2011). However, editing of the viral genome by host-encoded proteins is another possible source of mutations. Double-stranded RNA-specific adenosine deaminases (ADAR) have been suggested to edit the genome of a variety of negative-stranded RNA viruses, including measles virus (Cattaneo et al., 1988), human parainfluenza virus (Murphy et al., 1991), respiratory syncytial virus (Martínez and Melero, 2002), lymphocytic choriomeningitis virus (Zahn et al., 2007), and Rift Valley fever virus (Suspène et al., 2008), and the apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like 3 cytidine deaminase family (APOBEC3) is known to edit HIV-1 (Desimmie et al., 2014, Moris et al., 2014, Santa-Marta et al., 2013), hepatitis B virus (Suspène et al., 2005), papillomaviruses (Vartanian et al., 2008), and herpesviruses (Suspène et al. 2011). In HIV-1, 98% of spontaneous mutations in vivo are produced by APOBEC3, whereas only 2% are attributable to the viral reverse transcriptase (Cuevas et al., 2015), but the relative contribution of the viral polymerase and host-mediated editing is unknown for most other viruses.
Here, we have analyzed the intra-patient genetic diversity of a region of the NoV capsid VP1 by performing ultra-deep sequencing of stool samples obtained from infected patients. Unexpectedly, we found a small number of hyper-mutated sequences carrying large numbers of A-to-G or U-to-C base transitions that were not attributable to sequencing errors. In this sense, since the per-base error rate of the employed technology is ca. 1/1000 (Jünemann et al., 2013), it is extremely improbable to find multiple mutations in a single read. The natural genetic diversity of NoVs has been studied previously within individual patients (Nilsson et al., 2003, Obara et al., 2008, Vega et al., 2014), within defined outbreaks (Dingle, 2004, Holzknecht et al., 2015, Sasaki et al., 2006), or at larger geographic and temporal scales (Bodhidatta et al., 2015, Carlsson et al., 2009, Cotten et al., 2014, Kobayashi et al., 2015, Vega et al., 2014). However, this diversity depends on multiple factors other than spontaneous mutation rates, including natural selection, the number of replication rounds elapsed and random genetic drift, among others. To discard these confounders and focus on spontaneous mutations, we used a cell culture system in which human cells are transfected with an infectious cDNA clone (Asanaka et al., 2005, Katayama et al., 2014). Since these cells do not support viral attachment and entry, this system restricts viral replication to a single infection cycle. Whereas this is generally viewed as a limitation, single-cycle viral replication was convenient for the purpose of mutation rate estimation, because it allowed us to minimize the effects of selection and other evolutionary factors. This approach allowed us to observe hundreds of sequences carrying multiple U-to-C or A-to-G substitutions each, suggesting that a large fraction of all spontaneous mutations corresponds to hyper-mutation events. Based on the sequence context of the observed changes, we propose that NoV hyper-mutation might be driven by ADAR-mediated editing of the viral genomic RNA of either polarity during replication.
2. Methods
2.1. RT-PCR of stool samples
Viral RNA was extracted from 20% stool suspensions in PBS using the Trizol LS reagent (Invitrogen), eluted in diethyl pyrocarbonate-treated water containing RNasin (Promega) and stored at − 70 °C. RT was performed using Superscript III (Invitrogen) and random hexamers for 10 min at 25 °C, 45 min at 50 °C and 15 min at 70 °C. PCR was done with Phusion High-Fidelity DNA polymerase following manufacturer's recommendations and specific primers degenerated either for purines or pyrimidines at a final concentration of 200 μM. For VP1 region 1, two pairs of primers with different degeneration were used: primers 5′-AyGAAGAyGGCGyCGAGyGACG-3′ (forward, nucleotides 5085–5106 in accession JX459908) and 5′-GGrrrrTTTGGTGGGrCTGCTGC-3′ (reverse, nucleotides 5448–5470 in accession JX459908) were designed to account for U-to-C mutations in plus-strand RNA, and primers 5′-rTGrrGrTGGCGTCGrGTGrCG-3′ (forward) and 5′-GGAAAAyyyGGyGGGACyGCyGC-3′ (reverse) were designed to account for A-to-G mutations in plus-strand RNA. For region 2, the degenerate primer pairs were 5′-CAAGAyyCCCCAyyCCyyyGG-3′ (forward, nucleotides 5803–5823 in accession JX459908) and 5′-GGrTGrCrCCGrCTGGGGTG-3′ (reverse, nucleotides 6233–6252 in accession JX459908), and 5′-CrrGrTTCCCCrTTCCTTTGG-3′ (forward) and 5′-GGAyGACACCGACyGGGGyG-3′ (reverse), respectively. PCR conditions were 98 °C 30 s, 35 cycles of 98 °C 10 s, 68 °C 30 s, 72 °C 1 min, and a final elongation step at 72 °C 5 min.
2.2. Transfection assays
A previously described Norwalk virus infectious cDNA clone (Asanaka et al., 2005) was obtained after an MTA with Dr. M. K. Estes (Baylor College of Medicine, Houston), cloned in Escherichia coli by the heat shock method, and purified by midiprep using the PureLink HiPure Plasmid Midiprep kit (Invitrogen). Human embryonic kidney cells HEK293 were obtained from the American Tissue Culture Collection (ATCC CRL-11,268) and cultured in DMEM F12 (Dulbecco's modified Eagle medium) supplemented with 10% FBS and antibiotics at 37 °C under 5% CO2. Norovirus was recovered from the cDNA clone as described previously (Asanaka et al., 2005). Briefly, approximately 105 HEK293 cells (80% confluence) were infected with a recombinant vaccinia virus expressing bacteriophage T7 RNA polymerase at a multiplicity of infection of 10 plaque-forming units per cell and, after 1 h incubation, the inoculum was washed and cells were transfected with 0.5 μg of the infectious cDNA clone using Lipofectamine LTX Reagent (Invitrogen), following manufacturer's instructions. After 5 h incubation at 37 °C, vaccinia replication was inhibited with 25 μg/mL AraC (arabinofuranosyl cytidine) and cells were incubated for 48 h. A plasmid containing a green-fluorescent-protein (GFP) was transfected under the same conditions as a control.
2.3. RT-PCR of NoV RNA extracted from HEK293 cells
After 48 h incubation, RNA was extracted from transfected cultures using TRIzol (Invitrogen) followed by chloroform and isopropanol purification, and washed with 75% ethanol. In order to digest any remaining DNA, samples were treated with 1 U/μg RNase-free DNase I (Thermo Scientific) for 30 min at 37 °C. DNase I was heat-inactivated (10 min at 65 °C) and the RNA was column-purified using NucleoSpin RNA Clean-up XS kit (Macherey-Nagel). Purified RNA was reverse-transcribed using Accuscript High Fidelity Reverse Transcriptase (Agilent Technologies) and a sequence-specific primer. Negative-strand RNA was reverse-transcribed using the following primer 5′-ATTACTCTCTGTGCACTGTCTG-3′ (nucleotides 4790–4811 in accession NC_001959), whereas positive-strand RNA was reverse-transcribed using primer 5′-CAGTGTAGAAGAGGCTGTTGAA-3′ (nucleotides 7501–7522 in accession NC_001959). Reverse transcription conditions used were 42 °C for 60 min, followed by 70 °C at 15 min. The VP1 gene was then PCR-amplified using Phusion High Fidelity DNA polymerase (New England Biolabs) and primers 5′-GACGCyACAyCAAGCGyGG-3′ (forward, nucleotides 5376–5394 in accession NC_001959) and 5′-CTCrTGTTrCCrrCCCrrCC-3′ (reverse, nucleotides 5661–5680 in accession NC_001959). The PCR conditions used were 98 °C 30 s, 35 cycles at 98 °C 10 s, 59 °C 30 s, and 72 °C 1 min, and a final extension at 72 °C 10 min. Controls were carried out in which the PCR was performed without RT step to ensure that no remaining DNA from the infectious clone was amplified. To control for strand-specific amplification, transfection supernatants were cleaned by centrifugation at 16,000 × g, 15 min, 4 °C to separate free virions containing plus-strand genomes from cellular pellets, used for RNA extraction, and the RT step was performed with the same primer used for amplification of minus strands. As expected, these controls did not yield any visible PCR product. To obtain a shorter product for Illumina sequencing, a secondary PCR of the indicated size was done with the following cycling conditions: 98 °C 30 s, 40 cycles of 98 °C 10 s, 60 °C 30s, 72 °C 1 min, and a final extension at 72 °C 5 min.
2.4. Illumina sequencing
PCR products were sequenced in an Illumina Miseq machine using paired-end libraries. The quality of the run was first evaluated with FastQC software 0.10.1 (http://www.bioinformatics.babraham.ac.uk/projects/fastqc). For clinical samples, a base calling pipeline was run to define a consensus reference sequence for each sample. To do this, Illumina adapters and PCR primers were cut with Cutadapt software (Marcel, 2011), fastq files were trimmed using Prinseq-lite version 0.20.4 (Schmieder and Edwards, 2011), mapping was done using the Mem algorithm from Bwa 0.7.12 (http://arxiv.org/abs/1303.3997), SAM files were converted to BAM format, sorted and indexed using SAMtools software package (Li et al., 2009), and sequence variants relative to a common reference were called with VarScan 2.3.7 (Koboldt et al., 2013) using SAMtools mpileup data as input. For each sample, nucleotide changes detected at a frequency higher than 0.5 were used to construct the sample-specific reference sequence. For subsequent steps, paired-end Illumina reads were merged using PANDAseq (Masella et al., 2012) and an initial trimming of these merged fastq files was performed with Prinseq-lite version 0.20.4. Trimmed fastq files were converted into fasta and standalone blast pair-wise alignments (Camacho et al., 2009) were obtained to map reads and to obtain the number of mutations relative to the reference sequence of the sample. Since the number of reads was variable, 100,000 reads were randomly chosen for each PCR. To obtain a refined set of mutated reads, a final quality filter was applied, such that only reads with average Phred quality score higher than 28 for the specific mutated positions were considered. This filter removed less than 5% of the original reads in all samples. Specific Shell, Python, Perl and R scripts were written for these analyses.
2.5. Molecular cloning and Sanger sequencing of PCR products
PCR products were gel-purified and cloned using CloneJET PCR cloning kit (Thermo Scientific) in E. coli by the heat shock method. Transformant colonies were PCR-amplified using Taq DNA polymerase and CloneJET-specific primers (forward 5′-CGACTCACTATAGGGAGAGCGGC-3′; reverse 5′-AAGAACATCGATTTTCCATGGCAG-3′) under the following conditions: 95 °C 5 min, 35 cycles of 95 °C 30 s, 60 °C 30 s, 72 °C 2 min, and a final extension at 72 °C 5 min. Colony PCR products were column-purified and sequenced by the Sanger method. Sequence chromatograms were analyzed using the Staden software (http://staden.sourceforge.net).
3. Results
3.1. Ultra-deep sequencing reveals NoV hyper-mutants in clinical samples
We used 16 stool samples from patients acutely infected with NoV GII.4 to amplify by RT-PCR a 386-base region encompassing nucleotides 1 to 386 of the VP1 gene (reference sequence: GenBank JX459908; Fig. 1A). The RT-PCR was successful in 11/16 samples. Of these, eight belonged to newborns or children under the age of three, two to adults, and for one sample there was no available age information. We performed paired-end Illumina sequencing of these PCR products with 100,000-fold coverage (i.e. 100,000 reads per patient). For three patients, approximately one in 6000 reads contained large numbers of U-to-C or A-to-G base transitions (12–27 reads with 5–30 such mutations out of approx. 100,000 total reads; Table 1 ). Reads with less than five mutations of such type were not considered as hyper-mutants. Although the error rate of Illumina sequencing precludes analysis of low-frequency polymorphisms, it provides a powerful approach for detecting hyper-mutants. In the most mutated read, 30 of the 86 U residues were substituted for C, a pattern that cannot be explained by sequencing error. Interestingly, we found both U-to-C and A-to-G hyper-mutations, but these did not occur in the same reads. To extend our analysis, we set out to amplify by RT-PCR another region encompassing nucleotides 719 to 1168 of VP1 (450 bases, although only the 409 bases excluding primer regions were considered for subsequent analysis), which maps to the hypervariable domain P2. In two out of the three samples showing hyper-mutation in the first region, we also found hyper-mutated reads in the second region, with a maximum of 37 mutations in a single read. Furthermore, one sample which failed to amplify for the first region also yielded hyper-mutated reads in the second region (Table 1). Two of the samples showing hyper-mutation belonged to newborns, whereas the other two belonged to adults, with no significant association between age and hyper-mutation at this low sample size (Fisher's exact test, P = 0.547). Viruses carrying large numbers of mutations should not be viable and, thus, their population frequency should be strongly reduced by the action of purifying selection. Therefore, albeit very rare, hyper-mutants may reflect a relevant mutational process in NoV.
Fig. 1.

NoV genetic map, regions sequenced, and setup of transfection assays. A. In the NoV genetic map, the VP1 capsid gene is shown in red. Molecular clones encompassing the entire VP1 gene were sequenced by the Sanger method. Illumina sequencing was used to analyze smaller regions mapping to the S domain of VP1 and the hyper-variable domain P2 (dark red bars). B. An infectious cDNA clone was transfected in HEK293 cells previously infected with a recombinant vaccinia virus expressing T7 RNA polymerase, allowing for transcription of plus-strand NoV genomic RNA. A primer annealing to minus-strand copies was used for RT-PCR amplification and sequencing. Colored circles represent mutations/variants.
Table 1.
Hyper-mutant (HM) sequences found in stool samples after RT-PCR of two VP1 regions.
| Sample | Region 1 (341 bases) |
Region 2 (409 bases) |
||||||
|---|---|---|---|---|---|---|---|---|
| Total readsa | HM type | HM reads | Mutations/read (mean, min, max) | Total readsa | HM type | HM reads | Mutations/read (mean, min, max) | |
| 3106 | 99,176 | A-to-G | 12 | 7.2 (5–10) | 96,415 | U-to-C | 39 | 5.5 (5–7) |
| 3142 | 97,757 | U-to-C | 27 | 10.6 (5–30) | N/Ab | – | – | – |
| 3210 | N/Ab | – | – | – | 96,770 | U-to-C | 13 | 5.6 (5–6) |
| 3213 | 98,967 | A-to-G | 13 | 7.8 (5–12) | 95,936 | A-to-G | 8 | 17.5 (6–37) |
Reads with an average base quality (Sanger-scaled Phred) score Q > 28 at mutated positions.
RT-PCR failed.
3.2. Massive hyper-mutation in a transfection system
To minimize the effects of selection, we transfected a Norwalk virus infectious cDNA clone into HEK293 cells expressing the T7 RNA polymerase from a recombinant vaccinia virus (ATCC VR-2153). As previously described, this system supports NoV transcription, replication and encapsidation (Fig. 1B), but does not allow released virions to initiate a second infection cycle because HEK293T cells are not a natural cell target for the virus (Asanaka et al., 2005). After 48 h incubation, total RNA was extracted from cells, residual DNA was removed with DNAse I, a specific primer annealing to the minus-strand of the VP1 capsid gene was used for reverse transcription, and high-fidelity PCR amplification of a region encompassing positions 19 to 323 of the VP1 gene (305 bases, although only the 266 bases excluding primer regions were considered for subsequent analysis) was carried out. For each of three independent transfection assays, we subjected the PCR products to paired-end Illumina sequencing with the same coverage as above. Comparison of Illumina reads with the sequence of the infectious cDNA clone (reference sequence: GenBank NC_001959) revealed hundreds of sequences with multiple U-to-C substitutions. Some examples of U-to-C hyper-mutants are shown in Fig. 2 . To objectively define hyper-mutated sequences, we analyzed the distribution of the number of U-to-C transitions among the 100,000 reads obtained for each replicate assay. The distribution clearly deviated from a Poisson model of rare random events, showing an excess of sequences with high mutation counts (Fig. 3A). Based on this, we defined hyper-mutated sequences as those carrying five or more mutations in the 266-base region studied. However, the data also showed that hyper-mutation was not an all-or-nothing process and that the number of mutations per read varied continuously. Regarding U-to-C substitutions, we found 1444 hyper-mutated reads (266, 481 and 697 reads for assays 1, 2 and 3, respectively), meaning that approximately one in every 200 reads (0.48%) contained U-to-C hyper-mutations (Table 2 ). These carried 11,612 total U-to-C substitutions, the number of mutations per read varying from 5 to 31 out of the 88 U residues contained in the 266-base fragment. Since sequences were derived from minus-strand RNA, U-to-C substitutions in the reference (plus-strand) genome sequence indicate that the negative-strand template RNA contained A-to-G substitutions. Ultra-deep sequencing also revealed some A-to-G hyper-mutated sequences in two of the three assays (indicating U-to-C changes in the negative-strand template), but these were 17 times less frequent (i.e. 1444/83 = 17.4) than the former (Table 2). A-to-G hyper-mutants may be a result of plus-strand carry-over amplification during RT-PCR or, alternatively, they may represent a different mutational process.
Fig. 2.

Distribution of U-to-C mutations along a VP1 region in sequences derived from transfected HEK293 cells. The alignment on top shows two examples of highly mutated reads from each transfection assay. The heat map below indicates, for each nucleotide site, the total number of deep-sequencing reads carrying a U-to-C mutation (see color legend).
Fig. 3.
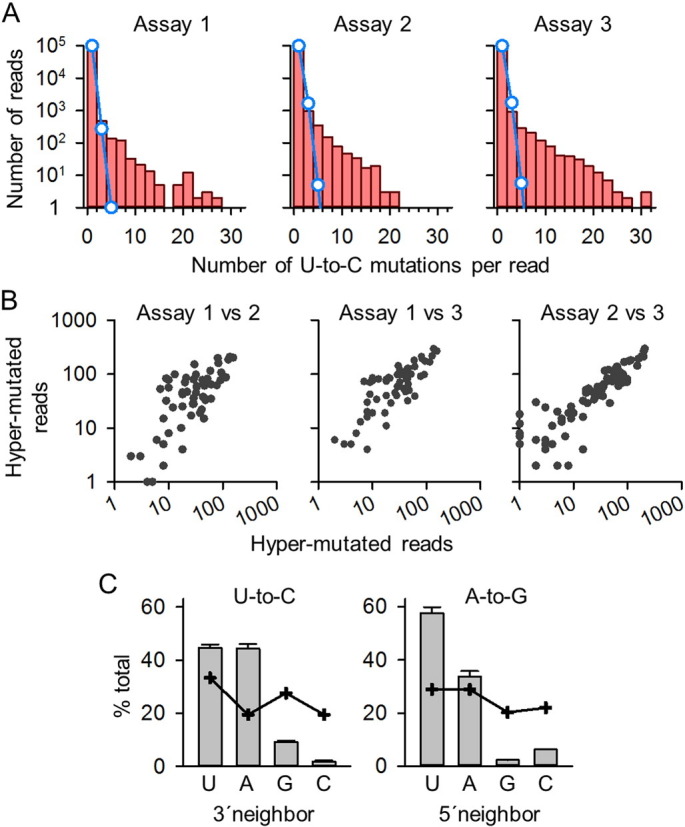
Analysis of hyper-mutation patterns. A. Distribution of the number of U-to-C mutations per deep sequencing read in each of the three transfection assays. The red histograms show the observed counts and the blue line indicates the counts expected from a Poisson model of rare random events. The single parameter of the Poisson distribution was calibrated using the number of reads carrying zero or one mutations. The strong deviation between observed and expected counts shows that sequence reads carrying multiple mutations were more frequent than expected from the Poisson model. Based on this, hyper-mutated reads were defined as those carrying five or more mutations. B. Reproducibility of U-to-C mutation frequency in three transfection assays. In the graphs, each data point corresponds to an U-containing nucleotide site, and the number of times a U-to-C mutation was observed in deep-sequencing reads is plotted for each pair of transfection assays (also represented in the heat map of Fig. 2). From left to right, Spearman correlations were 0.860, 0.855, and 0.955 (p < 10− 12 in all cases). C. Neighbor base preferences for U-to-C and A-to-G hyper-mutation. The histograms show the frequency of U, A, G, and C among 3´neighbors of U-to-C mutations (left), and the frequency of U, A, G, and C among 5´neighbors of A-to-G mutations (right). The crossed lines indicate these same frequencies among non-mutated bases (null expectation). The error bars indicate the SEM frequency from three transfection assays.
Table 2.
Hyper-mutant (HM) VP1 sequences (bases 38-303) derived from transfected HEK293 cells.
| Assay | NGS valid readsa | U-to-C HM |
A-to-G HM |
||||
|---|---|---|---|---|---|---|---|
| Number of reads | Mutations/read (mean, min, max) | Mutation rateb | Number of reads | Mutations/read (mean, min, max) | Mutation rateb | ||
| 1 | 99,982 | 266 | 8.7 (5–28) | 8.7 × 10− 5 | 67 | 7.8 (5–14) | 2.0 × 10− 5 |
| 2 | 99,917 | 481 | 8.0 (5–20) | 1.4 × 10− 4 | 0 | NA | 0 |
| 3 | 99,973 | 697 | 9.2 (5–31) | 2.1 × 10− 4 | 16 | 7.1 (5–18) | 4.3 × 10− 6 |
Reads with an average base quality (Sanger-scaled Phred) score Q > 28 at mutated positions.
Mutation rates were estimated by dividing, for a giving assay, the total number of mutations by the product of reads times the length of the PCR product (266 bases).
3.3. Reproducible effect of sequence context on hyper-mutation
Analysis of the location of mutations revealed a widespread distribution along the 266-base VP1 region. Although all of the 88 U residues showed at least one U-to-C mutation at this high sequencing depth, mutation frequencies varied strongly across sites, the pattern of variation being highly reproducible between the three biological replicates (pairwise Spearman p > 0.850, p < 10− 12; Fig. 3B). A major determinant of the frequency of U-to-C mutation was the identity of the 3′ neighboring base. Among the 11,612 U-to-C changes observed, the 3′ neighbor was U in 5228 cases, A in 5053 cases, G in 1098 cases, and C in only 233 cases. These counts clearly deviated from those of 3′ neighbors of non-mutated bases (chi-square test: p < 10− 12; Fig. 3C). After correcting for base composition, the 3′ neighbor preferences for U-to-C hyper-mutation were A > U > G > C. Interestingly, A-to-G hyper-mutated sequences showed a marked bias in the 5′ neighboring base such that, among the 638 total A-to-G mutations, the 5′ neighbor was U in 379 cases, A in 204 cases, G in 14 cases, and C in 41 cases (p < 10− 12; Fig. 3C). Therefore, A-to-G hyper-mutation had 5′ neighbor base preferences (U > A > C > G) which are exactly the reverse complement of those for U-to-C hyper-mutation. This strongly suggests a common biochemical process underlying both U-to-C and A-to-G mutations, the type of change observed depending on whether hyper-mutation occurred in the minus or plus RNA strand, respectively.
3.4. Contribution of hyper-mutation to the total NoV rate of spontaneous mutation
Based on the above data, the per-base probability of a U-to-C substitution due to hyper-mutation was (1.5 ± 0.4) × 10− 4, a value within the typical range of RNA virus rates of spontaneous mutation (Lauring et al., 2013, Sanjuán et al., 2010). To ascertain the contribution of hyper-mutation events to the total NoV mutation rate, we sought to estimate the total mutation rate from the above single-cycle transfection assays. Since the Illumina per-read accuracy is not high enough to reliably infer individual base substitutions at such low frequencies, we performed classical molecular cloning followed by Sanger sequencing. Using RNA extracts from the above transfections, we amplified by high-fidelity RT-PCR the entire VP1 gene and obtained 64 molecular clones. In total, we found 21 base substitutions in 136,032 bases, giving a mutation rate estimate of 1.5 × 10− 4 per nucleotide per cell infection (Table 3 ), a value nearly identical to the hyper-mutation rate inferred by Illumina sequencing. Furthermore, of the 21 mutations 18 were U-to-C base transitions found in a single, hyper-mutated clone. Removing this single clone, the estimated mutation rate was 2.2 × 10− 5, a value seven times lower than the estimated hyper-mutation rate.
Table 3.
Mutations found by Sanger sequencing in NoV VP1 molecular clones derived from transfected HEK293 cells.
| Total | Without hyper-mutant | |
|---|---|---|
| VP1 clones sequenced | 64 | 63 |
| Total bases sequenced | 136,032 | 133,901 |
| Total mutations | 21 | 3 |
| U-to-C substitutions | 18 | 0 |
| Total mutation frequency | 1.5 × 10− 4 | 2.2 × 10− 5 |
4. Discussion
Our results reveal that a large fraction of NoV spontaneous mutations is constituted by U-to-C and A-to-G substitutions occurring as bouts of mutations in the same RNA molecule. We argue that, depending on whether the hyper-mutation takes place in the minus or plus strand, U-to-C or A-to-G changes are observed, respectively, in the (plus strand) genomic RNA. A likely mechanism underlying these A-to-G mutations is ADAR, which edits adenosines to inosines that subsequently base-pair with cytosines (Samuel, 2011, Valente and Nishikura, 2005). A hallmark of ADAR 1 and 2 is that editing is more likely when the 5′ neighbor of the editable base is A or U and, more precisely, the neighbor base preferences have been shown to be U > A > C > G (Dawson et al., 2004, Kuttan and Bass, 2012, Lehmann and Bass, 2000, Polson and Bass, 1994). Our sequence analysis shows these same preferences, thus supporting the involvement of ADAR in NoV hyper-mutation. Previous work has shown or suggested ADAR-mediated hyper-mutation in several viruses, but these were negative-strand viruses as opposed to NoVs (Samuel, 2011). Hyper-mutation should be carried out by the interferon-inducible p150 isoform of ADAR1, since this is the only ADAR form located in the cytoplasm (George et al., 2011) where NoVs replicate. ADAR uses double-stranded RNA as a substrate and, therefore, the template RNA has to adopt a nearly perfect stem-like secondary structure or be a double-stranded replicative intermediate. The secondary structure of the NoV genomic RNA has not been solved experimentally and, although in silico RNA folding shows limited reliability for long molecules, stem-like structures are simple enough to be confidently predicted. However, the minimum free energy structure of the 266-base region encompassing VP1 nucleotides 38 to 303 predicted by the mfold algorithm (Zuker, 2003) did not show a stem-like structure. This suggests that ADAR acts on NoV double-stranded replicative intermediates. ADAR 1 is ubiquitously expressed in human tissues (Kim et al., 1994) and, although HEK293 cells express relatively low ADAR 1 levels, this activity was shown to be sufficient to edit 5% of hepatitis delta virus RNA molecules (Sato et al., 2001). In B lymphocytes, which are candidate cell targets for NoVs in vivo (Jones et al., 2014), ADAR1 and ADAR2 are more strongly expressed and have been shown to edit thousands of adenosines in cellular mRNA and long non-coding RNA (Wang et al., 2013).
A limitation of our study is that, whereas transfection assays were carried out using a cDNA clone belonging to genogroup I, viruses isolated from stool samples were all from genogroup II. However, the type of mutations produced and the neighbor base preferences were very similar in stool samples and in transfection assays. Specifically, 82.9% of the 5′ neighbors of U-to-C mutations and 72.3% of the 3′ neighbors of A-to-G mutations were A or U in clinical samples. After correcting for base composition, the resulting 3′ neighbor preferences for U-to-C mutations were A > U > G > C, whereas the 5′ preferences for A-to-G mutations were U > A > G > C in clinical samples. The similarities between the results obtained in transfection assays and in vivo support a common underlying mechanism, despite the fact that different genogroups were used for these experiments. Still, hyper-mutation was 30-fold more abundant in the transfection assays than in clinical samples. We attribute this difference to the fact that selection was mild or absent in transfection assays, whereas in stool samples (which should contain mainly free virions) we expect stronger selection against hyper-mutated genomes. Alternatively, it is possible that ADAR activity was lower in the NoV target cells in vivo than in HEK293 cells. However, B cells show extensive ADAR-mediated editing of cellular RNAs (Wang et al., 2013). Work with HIV-1 has shown that the observed levels of hyper-mutation vary depending on whether intra-cellular or virion-associated sequences are analyzed (Russell et al., 2009). APOBEC massively edits the retroviral cDNA, leading to a rate of G-to-A mutation of approximately 4 × 10− 3 per base per cell, a value two orders of magnitude higher than HIV-1 reverse transcriptase errors (Cuevas et al., 2015). In contrast, the rate observed in plasma is 44 times lower, consistent with the notion that the vast majority of APOBEC-edited HIV-1 genomes is unviable and rapidly removed by selection (Ho et al., 2013). In human hepatitis B virus, papilloma virus, herpes simplex virus 1 and Epstein–Barr virus APOBEC-edited genomes are usually found at low frequencies and their identification required a modified PCR protocol in which the lower melting temperature of A/T-rich molecules is exploited for selective amplification of hyper-mutants (Suspène et al., 2011, Suspène et al., 2005, Vartanian et al., 2008). A variant of this strategy has been devised for ADAR-edited sequences, which allowed detecting hyper-mutants in Rift Valley virus (Suspène et al., 2008). Therefore, probably with the exception of HIV-1, hyper-mutated sequences are generally rare. Selective PCR amplification is valuable for detecting these sequences but does not allow an estimation of their population frequency. As a result, few studies have determined the abundance of viral hyper-mutants in an unbiased manner. Ultra-deep sequencing provides a powerful tool for achieving this goal.
5. Conclusions
Previous work has suggested that the high genetic diversity of RNA viruses originates mainly from the low replication fidelity of their polymerases. However, our in depth analysis of NoV spontaneous mutations in clinical samples and laboratory populations supports the notion that host-driven hyper-mutation is a source of diversity comparable to or even greater than polymerase infidelity. Hyper-mutation is not necessarily an all-or-nothing process and the number of nucleotide substitutions per sequence varied extensively, suggesting that hyper-mutation may significantly contribute to NoV genetic diversity and evolution in nature. Analysis of the types of mutations produced in longitudinal studies may help elucidate this contribution.
Acknowledgments
We thank members of the Genomics facility of the University of Valencia for assistance with Illumina sequencing, and Dr. Silvia Torres for laboratory assistance, and Dr. Mary Estes for the Norwalk virus infectious cDNA clone. This work was supported by grants from the European Research Council (ERC-2011-StG-281191-VIRMUT) and the Spanish Ministerio de Economía y Competitividad (BFU2013-41329) to R.S. Illumina sequence alignments with hyper-mutated reads have been deposited in GenBank under the following accessions: KU253284–KU253322 (stool sample 3106, region 2), KU253323–KU253349 (stool sample 3142, region 1), KU253350–KU253362 (stool sample 3210, region 2), KU253363–KU253375 (stool sample 3213, region 1), KU253376–KU253383 (stool sample 3213, region 2), KU253384–KU253395 (stool sample 3106, region 1), KU289250-KU289582 (transfection 1), KU305215–KU305695 (transfection 2), and KU304502–KU305214 (transfection 3).
References
- Asanaka M., Atmar R.L., Ruvolo V., Crawford S.E., Neill F.H., Estes M.K. Replication and packaging of Norwalk virus RNA in cultured mammalian cells. Proc. Natl. Acad. Sci. U. S. A. 2005;102:10327–10332. doi: 10.1073/pnas.0408529102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodhidatta L., Abente E., Neesanant P., Nakjarung K., Sirichote P., Bunyarakyothin G., Vithayasai N., Mason C.J. Molecular epidemiology and genotype distribution of noroviruses in children in Thailand from 2004 to 2010: a multi-site study. J. Med. Virol. 2015;87:664–674. doi: 10.1002/jmv.24108. [DOI] [PubMed] [Google Scholar]
- Bull R.A., White P.A. Mechanisms of GII.4 norovirus evolution. Trends Microbiol. 2011;19:233–240. doi: 10.1016/j.tim.2011.01.002. [DOI] [PubMed] [Google Scholar]
- Camacho C., Coulouris G., Avagyan V., Ma N., Papadopoulos J., Bealer K., Madden T.L. BLAST +: architecture and applications. BMC Bioinformatics. 2009;10 doi: 10.1186/1471-2105-10-421. 421–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsson B., Lindberg A.M., Rodriguez-Diaz J., Hedlund K.O., Persson B., Svensson L. Quasispecies dynamics and molecular evolution of human norovirus capsid P region during chronic infection. J. Gen. Virol. 2009;90:432–441. doi: 10.1099/vir.0.005082-0. [DOI] [PubMed] [Google Scholar]
- Cattaneo R., Schmid A., Eschle D., Baczko K., ter Meulen V., MA B. Biased hypermutation and other genetic changes in defective measles viruses in human brain infections. Cell. 1988;55:255–265. doi: 10.1016/0092-8674(88)90048-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotten M., Petrova V., Phan M.V., Rabaa M.A., Watson S.J., Ong S.H., Kellam P., Baker S. Deep sequencing of norovirus genomes defines evolutionary patterns in an urban tropical setting. J. Virol. 2014;88:11056–11069. doi: 10.1128/JVI.01333-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuevas J.M., Geller R., Garijo R., López-Aldeguer J., Sanjuán R. Extremely high mutation rate of HIV-1 in vivo. PLoS Biol. 2015;13 doi: 10.1371/journal.pbio.1002251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson T.R., Sansam C.L., Emeson R.B. Structure and sequence determinants required for the RNA editing of ADAR2 substrates. J. Biol. Chem. 2004;279:4941–4951. doi: 10.1074/jbc.M310068200. [DOI] [PubMed] [Google Scholar]
- Debbink K., Lindesmith L.C., Donaldson E.F., Baric R.S. Norovirus immunity and the great escape. PLoS Pathog. 2012;8 doi: 10.1371/journal.ppat.1002921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desimmie B.A., Delviks-Frankenberrry K.A., Burdick R.C., Qi D., Izumi T., Pathak V.K. Multiple APOBEC3 restriction factors for HIV-1 and one Vif to rule them all. J. Mol. Biol. 2014;426:1220–1245. doi: 10.1016/j.jmb.2013.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingle K.E. Mutation in a Lordsdale norovirus epidemic strain as a potential indicator of transmission routes. J. Clin. Microbiol. 2004;42:3950–3957. doi: 10.1128/JCM.42.9.3950-3957.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson E.F., Lindesmith L.C., Lobue A.D., Baric R.S. Viral shape-shifting: norovirus evasion of the human immune system. Nat. Rev. Microbiol. 2010;8:231–241. doi: 10.1038/nrmicro2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George C.X., Gan Z., Liu Y., Samuel C.E. Adenosine deaminases acting on RNA, RNA editing, and interferon action. J. Interf. Cytokine Res. 2011;31:99–117. doi: 10.1089/jir.2010.0097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho Y.C., Shan L., Hosmane N.N., Wang J., Laskey S.B., Rosenbloom D.I., Lai J., Blankson J.N., Siliciano J.D., Siliciano R.F. Replication-competent noninduced proviruses in the latent reservoir increase barrier to HIV-1 cure. Cell. 2013;155:540–551. doi: 10.1016/j.cell.2013.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzknecht B.J., Franck K.T., Nielsen R.T., Bottiger B., Fischer T.K., Fonager J. Sequence analysis of the capsid gene during a genotype II.4 dominated norovirus season in one university hospital: identification of possible transmission routes. PLoS One. 2015;10 doi: 10.1371/journal.pone.0115331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones M.K., Watanabe M., Zhu S. Enteric bacteria promote human and mouse norovirus infection of B cells. Science. 2014;346:755–759. doi: 10.1126/science.1257147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jünemann S., Sedlazeck F.J., Prior K., Albersmeier A., John U., Kalinowski J., Mellmann A., Goesmann A., von Haeseler A., Stoye J., Harmsen D. Updating benchtop sequencing performance comparison. Nat. Biotechnol. 2013;31:294–296. doi: 10.1038/nbt.2522. [DOI] [PubMed] [Google Scholar]
- Katayama K., Murakami K., Sharp T.M., Guix S., Oka T., Takai-Todaka R., Nakanishi A., Crawford S.E., Atmar R.L., Estes M.K. Plasmid-based human norovirus reverse genetics system produces reporter-tagged progeny virus containing infectious genomic RNA. Proc. Natl. Acad. Sci. U. S. A. 2014;111:E4043–E4052. doi: 10.1073/pnas.1415096111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim U., Wang Y., Sanford T., Zeng Y., Nishikura K. Molecular cloning of cDNA for double-stranded RNA adenosine deaminase, a candidate enzyme for nuclear RNA editing. Proc. Natl. Acad. Sci. U. S. A. 1994;91:11457–11461. doi: 10.1073/pnas.91.24.11457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi M., Yoshizumi S., Kogawa S. Molecular evolution of the capsid gene in norovirus genogroup I. Sci. Rep. 2015;5:13806. doi: 10.1038/srep13806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koboldt D.C., Larson D.E., Wilson R.K. Using VarScan 2 for germline variant calling and somatic mutation detection. Curr. Protoc. Bioinformatics. 2013;44:15. doi: 10.1002/0471250953.bi1504s44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuttan A., Bass B.L. Mechanistic insights into editing-site specificity of ADARs. Proc. Natl. Acad. Sci. U. S. A. 2012;109:E3295–E3304. doi: 10.1073/pnas.1212548109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauring A.S., Frydman J., Andino R. The role of mutational robustness in RNA virus evolution. Nat. Rev. Microbiol. 2013;11:327–336. doi: 10.1038/nrmicro3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann K.A., Bass B.L. Double-stranded RNA adenosine deaminases ADAR1 and ADAR2 have overlapping specificities. Biochemistry. 2000;39:12875–12884. doi: 10.1021/bi001383g. [DOI] [PubMed] [Google Scholar]
- Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindesmith L.C., Donaldson E.F., Lobue A.D., Cannon J.L., Zheng D.P., Vinje J., Baric R.S. Mechanisms of GII.4 norovirus persistence in human populations. PLoS Med. 2008;5 doi: 10.1371/journal.pmed.0050031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcel M. Vol. 17. EMBnet J; 2011. Cutadapt Removes Adapter Sequences From High-throughput Sequencing Reads; pp. 10–12. [Google Scholar]
- Martínez I., Melero J.A. A model for the generation of multiple A to G transitions in the human respiratory syncytial virus genome: predicted RNA secondary structures as substrates for adenosine deaminases that act on RNA. J. Gen. Virol. 2002;83:1445–1455. doi: 10.1099/0022-1317-83-6-1445. [DOI] [PubMed] [Google Scholar]
- Masella A.P., Bartram A.K., Truszkowski J.M., Brown D.G., Neufeld J.D. PANDAseq: paired-end assembler for illumina sequences. BMC Bioinformatics. 2012;13 doi: 10.1186/1471-2105-13-31. 31–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moris A., Murray S., Cardinaud S. AID and APOBECs span the gap between innate and adaptive immunity. Front. Microbiol. 2014;5:534. doi: 10.3389/fmicb.2014.00534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy D.G., Dimock K., Kang C.Y. Numerous transitions in human parainfluenza virus 3 RNA recovered from persistently infected cells. Virology. 1991;181:760–763. doi: 10.1016/0042-6822(91)90913-v. [DOI] [PubMed] [Google Scholar]
- Nilsson M., Hedlund K.O., Thorhagen M., Larson G., Johansen K., Ekspong A., Svensson L. Evolution of human calicivirus RNA in vivo: accumulation of mutations in the protruding P2 domain of the capsid leads to structural changes and possibly a new phenotype. J. Virol. 2003;77:13117–13124. doi: 10.1128/JVI.77.24.13117-13124.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obara M., Hasegawa S., Iwai M., Horimoto E., Nakamura K., Kurata T., Saito N., Oe H., Takizawa T. Single base substitutions in the capsid region of the norovirus genome during viral shedding in cases of infection in areas where norovirus infection is endemic. J. Clin. Microbiol. 2008;46:3397–3403. doi: 10.1128/JCM.01932-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel M.M., Widdowson M.A., Glass R.I., Akazawa K., Vinje J., Parashar U.D. Systematic literature review of role of noroviruses in sporadic gastroenteritis. Emerg. Infect. Dis. 2008;14:1224–1231. doi: 10.3201/eid1408.071114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polson A.G., Bass B.L. Preferential selection of adenosines for modification by double-stranded RNA adenosine deaminase. EMBO J. 1994;13:5701–5711. doi: 10.1002/j.1460-2075.1994.tb06908.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robilotti E., Deresinski S., Pinsky B.A. Norovirus. Clin. Microbiol. Rev. 2015;28:134–164. doi: 10.1128/CMR.00075-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell R.A., Moore M.D., Hu W.S., Pathak V.K. APOBEC3G induces a hypermutation gradient: purifying selection at multiple steps during HIV-1 replication results in levels of G-to-A mutations that are high in DNA, intermediate in cellular viral RNA, and low in virion RNA. Retrovirology. 2009;6:16. doi: 10.1186/1742-4690-6-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel C.E. Adenosine deaminases acting on RNA (ADARs) are both antiviral and proviral. Virology. 2011;411:180–193. doi: 10.1016/j.virol.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjuán R., Nebot M.R., Chirico N., Mansky L.M., Belshaw R. Viral mutation rates. J. Virol. 2010;84:9733–9748. doi: 10.1128/JVI.00694-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santa-Marta M., de Brito P.M., Godinho-Santos A., Goncalves J. Host factors and HIV-1 replication: clinical evidence and potential therapeutic approaches. Front. Immunol. 2013;4:343. doi: 10.3389/fimmu.2013.00343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki Y., Kai A., Hayashi Y. Multiple viral infections and genomic divergence among noroviruses during an outbreak of acute gastroenteritis. J. Clin. Microbiol. 2006;44:790–797. doi: 10.1128/JCM.44.3.790-797.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato S., Wong S.K., Lazinski D.W. Hepatitis delta virus minimal substrates competent for editing by ADAR1 and ADAR2. J. Virol. 2001;75:8547–8555. doi: 10.1128/JVI.75.18.8547-8555.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmieder R., Edwards R. Quality control and preprocessing of metagenomic datasets. Bioinformatics. 2011;27:863–864. doi: 10.1093/bioinformatics/btr026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith E.C., Denison M.R. Coronaviruses as DNA wannabes: a new model for the regulation of RNA virus replication fidelity. PLoS Pathog. 2013;9 doi: 10.1371/journal.ppat.1003760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suspène R., Guetard D., Henry M., Sommer P., Wain-Hobson S., Vartanian J.P. Extensive editing of both hepatitis B virus DNA strands by APOBEC3 cytidine deaminases in vitro and in vivo. Proc. Natl. Acad. Sci. U. S. A. 2005;102:8321–8326. doi: 10.1073/pnas.0408223102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suspène R., Renard M., Henry M., Guetard D., Puyraimond-Zemmour D., Billecocq A., Bouloy M., Tangy F., Vartanian J.P., Wain-Hobson S. Inversing the natural hydrogen bonding rule to selectively amplify GC-rich ADAR-edited RNAs. Nucleic Acids Res. 2008;36 doi: 10.1093/nar/gkn295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suspène R., Aynaud M.M., Koch S., Pasdeloup D., Labetoulle M., Gaertner B., Vartanian J.P., Meyerhans A., Wain-Hobson S. Genetic editing of herpes simplex virus 1 and Epstein–Barr herpesvirus genomes by human APOBEC3 cytidine deaminases in culture and in vivo. J. Virol. 2011;85:7594–7602. doi: 10.1128/JVI.00290-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulferts R., Ziebuhr J. Nidovirus ribonucleases: structures and functions in viral replication. RNA Biol. 2011;8:295–304. doi: 10.4161/rna.8.2.15196. [DOI] [PubMed] [Google Scholar]
- Valente L., Nishikura K. ADAR gene family and A-to-I RNA editing: diverse roles in posttranscriptional gene regulation. Prog. Nucleic Acid Res. Mol. Biol. 2005;79:299–338. doi: 10.1016/S0079-6603(04)79006-6. [DOI] [PubMed] [Google Scholar]
- Vartanian J.P., Guetard D., Henry M., Wain-Hobson S. Evidence for editing of human papillomavirus DNA by APOBEC3 in benign and precancerous lesions. Science. 2008;320:230–233. doi: 10.1126/science.1153201. [DOI] [PubMed] [Google Scholar]
- Vega E., Donaldson E., Huynh J., Barclay L., Lopman B., Baric R., Chen L.F., Vinje J. RNA populations in immunocompromised patients as reservoirs for novel norovirus variants. J. Virol. 2014;88:14184–14196. doi: 10.1128/JVI.02494-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang I.X., So E., Devlin J.L., Zhao Y., Wu M., Cheung V.G. ADAR regulates RNA editing, transcript stability, and gene expression. Cell Rep. 2013;5:849–860. doi: 10.1016/j.celrep.2013.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White P.A. Evolution of norovirus. Clin. Microbiol. Infect. 2014;20:741–745. doi: 10.1111/1469-0691.12746. [DOI] [PubMed] [Google Scholar]
- Zahn R.C., Schelp I., Utermohlen O., LD v. A-to-G hypermutation in the genome of lymphocytic choriomeningitis virus. J. Virol. 2007;81:457–464. doi: 10.1128/JVI.00067-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]