

Abstract
ORF5 sequences of porcine reproductive and respiratory syndrome virus (PRRSV) were analysed to determine genetic diversity, codon usage, positive and negative selection sites and potential changes in the predicted glycoprotein 5 (GP5). A hypothetical GP5 containing all selected sites was constructed to determine its characteristics. These sequences corresponded to isolates obtained 10 years apart (1991–1995, 18 strains) and a second set (n = 46) from 2000 to 2005. Similarity to Lelystad virus (LV) decreased from 95.5% in 1991–1995 to 89.5% in 2000–2005. Three highly variable regions were found in ORF5. Codon usage was different in both sets for leucine, glutamine, serine and proline. Thus, 2000–2005 sequences used codons more similar to those present in highly expressed pig genes compared to the 1991–1995 set. Twenty four sites of positive selection and 20 sites of negative selection were found in GP5, most of them in transmembrane regions. Additional glycosylation in N37 of GP5 was common in 2000–2005 but some sequences lack a glycosylation site in N46. The hypothetical GP5 was only 88.1% similar to LV and was less hydrophobic. Taking together these results suggest that PRRSV is still adapting to pig cells.
Keywords: Porcine reproductive and respiratory virus, ORF5, Genetic diversity, Codon usage
Porcine reproductive and respiratory syndrome virus (PRRSV) belongs to the genus Arterivirus. Two different genotypes are currently known, the European one and the American one. Genetic similarity between both genotypes ranges from 50 to 70% depending on the open reading frame (ORF) examined (Meng et al., 1994). PRRSV genome is composed of nine ORFs of which ORF1a and 1b encode the viral polymerase, ORFs 2–6 encode envelope and membrane proteins (called GP2a, GP2b, GP3, GP4, GP5 and M, respectively) and ORF7 encodes the non-glycosylated viral nucleocapsid protein (Snijder and Meulenberg, 1998, Wu et al., 2005). Among the envelope proteins, GP5 seems to be one of the key viral structures. It is thought that attachment and entry to the target cells is mediated by GP5 or GP5-M heterodimers (Snijder et al., 2003). In addition, the neutralisation epitope of PRRSV is located in the middle of the GP5 ectodomain (Gonin et al., 1999, Ostrowski et al., 2002, Plagemann, 2004a, Plagemann, 2004b).
From early studies, it became evident that ORF5 was one of the most variable regions in the PRRSV genome although other parts also show a considerable degree of variability as occurs in ORF3 and in non-structural protein 2 (nsp2) (Fang et al., 2004, Oleksiewicz et al., 2000). ORF5 heterogeneity was initially reported for American strains but nowadays is evident that European isolates are diverse as well (Forsberg et al., 2002, Mateu et al., 2003, Pesch et al., 2005, Pirzadeh et al., 1998). Genetic variability observed in ORF5 is consistent with the well known fact that RNA-polymerases of RNA viruses have a relatively poor fidelity (Castro et al., 2005) and with the notion that selective pressures act favouring viral variants better fitted for spread and persistence in the target hosts. As GP5 is exposed in the viral envelope, participates in viral attachment to cells and contains a neutralisation epitope, it is a potential target for these selective pressures. Interestingly, the adaptative sites in GP5 seem not to be restricted to the known B-epitopes but also to other regions (Hanada et al., 2005).
In the present study, a large set of ORF5 sequences from Spanish PRRSV strains obtained 10 years apart were analysed to determine the changes in ORF5 and GP5 and to figure out the potential impact of those changes.
ORF5 sequences of Spanish PRRSV strains were randomly selected from isolates available in our laboratory obtained from sera of pigs of Spanish epidemiologically unrelated commercial farms or were retrieved from Spanish sequences deposited at Genbank. For PRRSV isolates, viral RNA was extracted, amplified by PCR and sequenced as described before (Mateu et al., 2003). The final set of sequences included the Lelystad virus (LV) (Genbank accession number M96262), the first Spanish isolate from 1991 (Genbank accession number X92942), a second Spanish isolate from 1991 (strain CReSA-VP21, GenBank accession number DQ009647), 15 isolates from 1991 to 1995 (Suárez et al., 1996) and 46 unrelated isolates retrieved in our laboratory from 2000 to 2005 (Genbank accession numbers AF495499–AF495502, AF495504–AF5521, DQ009625–DQ009646). For comparative purposes, one set of 16 non Spanish European-type ORF5 sequences representing strains isolated in the period 1991–1995 in several countries of Europe were also analysed. This set comprised sequences from Belgium (n = 2), Denmark (n =2), France (n = 2), Germany (n = 2), The Netherlands (n = 3), Poland (n = 2) and United Kingdom (n = 1) (GenBank accession numbers: AY035900–AY035903, AY035918, AY035919, AY035921, AY035922, AY035926, AY035927, AF378799, U40696, U40696 and M96262).
ORF5 sequences were initially aligned using ClustalW (Thompson et al., 1994) and a similarity matrix was constructed. Alignments were retrieved using Bio-edit software v.7.0.5 (available at http://www.mb.mahidol.ac.th/Downloads/Mol-Bio/Bioedit/Bioedit.htm) and entropy plots were constructed to determine genetic variability in ORF5. Entropy was calculated as where entropy H(l) is equal to the summatory of f(b,l), namely the frequency at which a given residue (b) is found at a given position (l), multiplied by the neperian logarithm of f(b,l). With this formula, the higher the value of H(l), the higher the variability at a given position. Lelystad virus was used as a reference for alignments.
Codon usage for each set of sequences was analysed using GCUA software v.1.0 (available at http://bioinf.may.ie). In a first step, relative codon usage was calculated for each set of sequences by means of the synonymous codon usage measures (RSCU) and taking into account the effective number of codons (ENC) in the gene. Then, a correspondence analysis (CA) was done in order to determine trends in the variation of codon usage. A linear regression analysis was used to evaluate correlation between codon usage bias and nucleotide composition. p-Values lower than 0.05 were considered to be significant. To ascertain the possible significance of changes in codon usage, 10 sequences of genes highly expressed in pigs were also analysed. Genes included were: creatin kinase (Genbank accession number AY754869); interferon-beta (NM_001003923), pyruvate dehydrogenase (X52990), myosin heavy chain (NM_214136); haptoglobin (AF492467), hemoglobin epsilon (NM_214447); plasminogen activator (AF364605), albumin (NM_00100528), alpha amylase (AF064742) and interleukin-1 beta (M86725).
In a subsequent step, aligned sequences were examined to determine the codons corresponding to each aminoacid in the GP5 protein. For each nucleotide position the rate of mutation (percentage of strains having a different nucleotide) was calculated compared to LV. Also, for each codon the ratio between synonymous and non-synonymous mutations (S/NS) was determined. As an initial criterion, codons where at least 25% of the examined strains had a mutation and had an S/NS higher than 3 were considered as potentially negatively selected while ratios below 0.3 were potential points for positive selection. These thresholds were set arbitrarily. In the third step, the probability that n strains from a set of N sequences shared the same mutation was calculated. For this calculation it was assumed that each sequence was epidemiologically unrelated to the others. Considering the degeneracy of the genetic code, the probability of a synonymous mutation (Ps) for a given aminoacid at a given point was calculated as Ps = (codons coding the same aminoacid-1)/60. The probability of a non-synonymous mutation was 1-Ps. The probability that n unrelated sequences shared a mutated codon encoding the same aminoacid was calculated according to a binomial distribution. Positive or negative selection at a given point was arbitrarily considered to occur when the probability was ≤1 × 10−9.
Predicted GP5 aminoacid sequences were aligned, similarities to LV were calculated and a bootstrapped phylogenetic tree was constructed using the neighbor-joining method (1000 iterations) using LV as the outgroup. An entropy plot of predicted GP5 was constructed to determine conserved and highly variable regions of the protein.
The sequence of a hypothetical strain containing all positively selected mutations was written and analysed using Bio-edit. The hypothetical GP5 was compared with other available European GP5 sequences using the Blastp utility (http://www.ncbi.nlm.nih.gov/BLAST). Finally, for this hypothetical strain transmembrane regions and N-glycosylation sites were evaluated using TMPred and NetNGlyc utilities at Expasy server (http://www.expasy.org) and the hydrophobic profile (Kyle and Dolittle method) was determined using Bio-edit.
For the Spanish 1991–1995 set of nucleotide sequences, the percentage of similarity to LV ranged from 99.1 to 87.2% (average, 95.5 ± 3.6%). For the 2000–2005 set, similarity to LV ranged from 94.9 to 81.7% (average 89.5 ± 2.8%). Entropy analysis showed three highly variable regions. The first one was located between nucleotide residues 165 and 189; the second one between residues 315 and 339 and the third one was located between residues 360 and 369. Other points of high variability were found at residues 36–39, 429–432 and 480–489. Regions located between nucleotides 99–120 and 123–165 were the less variable part.
As expected, the CA globally showed that codon usage was not significantly different in 1991–1995 and 2000–2005. However, when specific aminoacids were examined, codon usage differed among 1991–1995 sequences and those of 2000–2005 for leucine, glutamine, serine and proline (Table 1 ). Thus, predominant codon for leucine in the 1991–1995 set was CTC (RCSU = 2.08) while in 2000–2005 was TTG (RCSU = 1.99). For glutamine, the only codon present in the older set of sequences was CAA (RSCU = 2.0) while in the set 2000–2005 appeared the codon CAG (RSCU = 0.28). For serine, older strains preferentially carried AGC (RSCU = 2.16) while newer strains preferentially used TCC (RSCU = 1.46) and for proline, older strains preferentially had CCG (RSCU = 1.41) instead of the CCC codon of the newer strains (RSCU = 1.30). Results for 1991–1995 non-Spanish European-type sequences were similar. The most frequent codons used for leucine were TTG (RSCU = 2.09) and CTC (RSCU = 1.97); for glutamine CAA (RSCU = 1.81) and CAG (RSCU = 0.19), for serine AGC (RSCU = 1.87) and TCC (RSCU = 1.41); and for proline CCG (RSCU = 1.84) and CCA (RSCU = 1.05). Some differences related to the country of origin were observed. Polish strains and the British strain preferentially coded leucine with the codon CTC. The codification of glutamine with the codon CAG was only found in Belgian strains.
Table 1.
Codon usage in the ORF5 gene of Spanish sequences of porcine reproductive and respiratory syndrome strains (1991–1995 and 2000–2005)
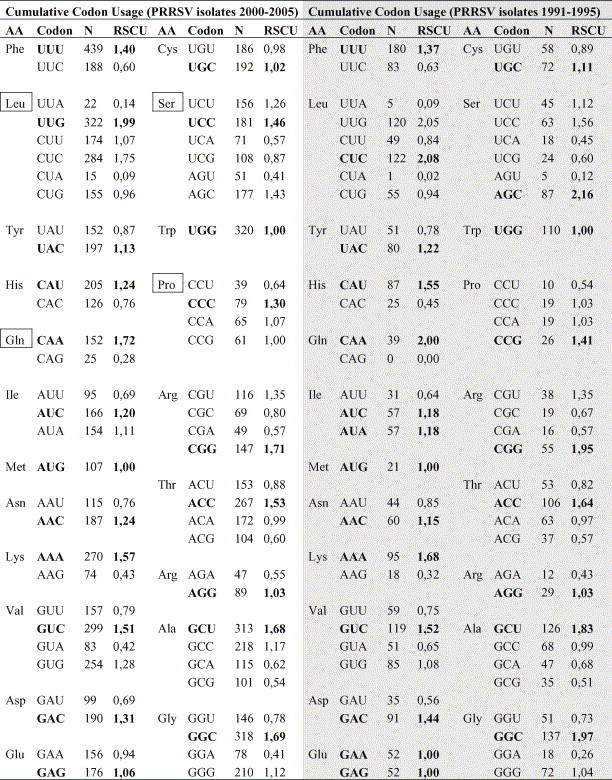
AA: aminoacid; N: number of effective codons; RSCU: synonymous codon usage measure. In bold is shown the most frequent codon for each aminoacid. Boxes show the main changes between 1991–1995 and 2000–2005 sequences.
Ten highly expressed pig genes were also analysed for codon usage. In those genes, the most commons codons for leucine, glutamine, serine and proline were TTG (RSCU = 0.81); CAG (RSCU = 1.45); TCC (RSCU = 1.36) and CCC (RSCU = 1.26).
The analysis of codons present in each sequence showed an overall S/NS of 1.41. In the set of sequences from 2000 to 2005, 24 codons showed the characteristics of a positive selection according to the calculated probabilities (Table 2 ). Interestingly, 12 strains changed Asn-46 to Asp-46, losing thus one glycosylation site. Also, 11 of those 12 strains gained a glycosylation site by changing Asp-37 to Asn-37. For the 1991–1995 set, the number of positively selected sites was seven. Another 20 codons showed the characteristics of a negative selection and were mainly distributed in three segments of the predicted protein (residues 73–89; 108–113 and 153–169) (Table 3 ). Codon usage significantly changed (p < 0.05) in 14 of these negatively selected positions between strains of 1991–1995 and those of 2000–2005 (Table 4 ).
Table 2.
Potential sites for positive selection of mutations in Spanish strains of porcine reproductive and respiratory syndrome virus GP5
| Position in GP5 | Lelystad virus | Spanish strains 2000–2005 (n = 47)a |
Spanish strains 1991–1995 (n = 17) |
||||
|---|---|---|---|---|---|---|---|
| Aminoacid | Aminoacid (strains) | NS/Sb | Probabilitya | Aminoacid (strains) | NS/Sb | Probabilitya | |
| 10 | Phenilalanine | Serine (14) | 18/3 | 1.3 × 10−20 | No change | 0/0 | N.A. |
| 12 | Threonine | Isoleucine (12) | 20/0 | 1.8 × 10−13 | No change | 0/0 | N.A. |
| 13 | Proline | Glutamine (12) | 21/18 | 1.3 × 10−18 | Leu (1) | 2/8 | 0.03 |
| Leucine (7) | 3.2 × 10−6 | ||||||
| 21 | Phenilalanine | Serine (5) | 6/0 | 7.5 × 10−9 | No change | 0/0 | N.A. |
| 32 | Alanine | Valine (15) | 19/0 | 9.6 × 10−17 | Valine (5) | 5/0 | 3.2 × 10−7 |
| 36 | Glycine | Aspartic acid (12) | 17/2 | 2.1 × 10−17 | Aspartic (2) | 3/0 | 8.0 × 10−3 |
| 37 | Aspartic acid | Asparagine (38) | 0/47 | 1.5 × 10−53 | Asparagine (5) | 9/0 | 1.5 × 10−7 |
| Serine (5) | 9.7 × 10−4 | Serine (4) | 8.9 × 10−6 | ||||
| 46 | Asparagine | Aspartic acid (12) | 15/14 | 1.5 × 10−12 | No change | 0/0 | N.A. |
| 56 | Aspartic acid | Glycine (16) | 42/0 | 6.7 × 10−11 | Glutamic acid (4) | 7/1 | 2.6 × 10−6 |
| Glutamic acid (14) | 4.2 × 10−15 | ||||||
| 60 | Serine | Asparagine (16) | 33/0 | 6.2 × 10−33 | Asparagine (1) | 2/0 | 0.03 |
| 63 | Glycine | Aspartic acid (18) | 45/0 | 1.1 × 10−20 | Aspartic acid (11) | 12/0 | 3.2 × 10−19 |
| Serine (12) | 5.7 × 10−12 | ||||||
| 96 | Glycine | Serine (13) | 15/10 | 7.8 × 10−19 | Serine (2) | 2/1 | 8.0 × 10−4 |
| 100 | Threonine | Isoleucine (11) | 24/0 | 9.1 × 10−11 | Isoleucine (3) | 5/0 | 3.4 × 10−4 |
| 101 | Alanine | Threonine (30) | 46/0 | 1.1 × 10−29 | Threonine (8) | 9/0 | 1.6 × 10−9 |
| 106 | Glycine | Lysine (16) | 26/3 | 4.8 × 10−15 | Lysine (4) | 3/1 | 3.8 × 10−7 |
| 111 | Cysteine | Serine (36) | 47/0 | 1.4 × 10−54 | Serine (15) | 15/0 | 2.1 × 10−27 |
| 116 | Alanine | Valine (17) | 26/1 | 1.5 × 10−18 | Phenilalanine (1) | 1/0 | 0.02 |
| 119 | Phenilalanine | Leucine (9) | 17/10 | 7.0 × 10−12 | Leucine (3) | 3/1 | 1.3 × 10−6 |
| 122 | Phenilalanine | Leucine (30) | 35/0 | 9.3 × 10−42 | Leucine (9) | 9/0 | 9.9 × 10−17 |
| 123 | Valine | Alanine (13) | 19/11 | 8.6 × 10−12 | No change | 0/0 | N.A. |
| 143 | Phenilalanine | Histidine (9) | 12/9 | 4.1 × 10−14 | No change | 0/0 | N.A. |
| 154 | Valine | Isoleucine (39) | 42/0 | 4.9 × 10−66 | Isoleucine (10) | 10/0 | 1.7 × 10−15 |
| 172 | Aspartic acid | Glycine (34) | 41/5 | 2.6 × 10−39 | Glycine (10) | 10/6 | 5.7 × 10−10 |
| 174 | Asparagine | Aspartic acid (35) | 43/0 | 1.7 × 10−52 | Aspartic acid (10) | 11/0 | 1.8 × 10−17 |
Probability: probability, according to a binomial distribution, that in a set of n independent sequences with a mutation, a given number of strains shared the same non-synonymous mutation. For strains detected in the period 2000–2005 only mutations with probabilities lower than 1.0 × 10−9 are shown.
NS/S: non synonymous/synonymous mutations.
Table 3.
Potential sites for negative selection of mutations in Spanish strains of porcine reproductive and respiratory syndrome virus GP5 obtained in 2000–2005
| Position in GP5 | Aminoacid | S/NSa | Probabilityb |
|---|---|---|---|
| 8 | Glycine | 20/2 | 2.0 × 10−24 |
| 73 | Proline | 17/1 | 1.3 × 10−21 |
| 79 | Leucine | 33/4 | 1.3 × 10−54 |
| 85 | Threonine | 15/4 | 9.6 × 10−17 |
| 89 | Phenilalanine | 16/3 | 3.3 × 10−26 |
| 91 | Aspartic acid | 31/4 | 3.69 × 10−51 |
| 108 | Tyrosine | 34/8 | 3.6 × 10−53 |
| 109 | Valine | 21/7 | 3.9 × 10−22 |
| 112 | Serine | 22/8 | 3.9 × 10−33 |
| 113 | Valine | 34/8 | 4.6 × 10−37 |
| 135 | Alanine | 36/5 | 8.4 × 10−42 |
| 137 | Arginine | 28/5 | 9.3 × 10−26 |
| 140 | Arginine | 25/7 | 1.9 × 10−21 |
| 153 | Arginine | 35/5 | 2.5 × 10−31 |
| 160 | Proline | 16/3 | 1.2 × 10−18 |
| 163 | Valine | 25/4 | 5.8 × 10−29 |
| 164 | Glutamic acid | 39/3 | 4.9 × 10−66 |
| 169 | Alanine | 21/7 | 3.9 × 10−22 |
| 194 | Threonine | 14/4 | 1.5 × 10−15 |
| 200 | Glutamic acid | 35/8 | 7.4 × 10−55 |
S/NS: synonymous/non-synonymous mutations.
Probability: probability, according to a binomial distribution, that in a set of n independent sequences with a mutation, a given number of strains shared a synonymous mutation.
Table 4.
Significant changes (p < 0.05) in codon usage for negatively selected sites of ORF5 of porcine reproductive and respiratory strains isolated in 1991–1995 or 2000–2005
| Position in GP5 | Aminoacids | Codons 1991–1995a | Codons 2000–2005a |
|---|---|---|---|
| 73 | Proline | CCG (18/18) | CCG (30/46) |
| CCA (15/46) | |||
| CCC (1/46) | |||
| 79 | Leucine | CTC (12/18) | CTC (11/44) |
| CTT (6/18) | CTT (33/44) | ||
| 85 | Threonine | ACA (18/18) | ACA (29/45) |
| ACG (13/45) | |||
| ACC (3/45) | |||
| 89 | Phenilalanine | TTT (18/18) | TTT (29/46) |
| TTC (17/46) | |||
| 91 | Aspartic acid | GAC (11/18) | GAC (13/45) |
| GAT (7/18) | GAT (32/45) | ||
| 109 | Valine | GTA (15/17) | GTA (20/41) |
| GTG (2/17) | GTG (21/41) | ||
| 112 | Serine | AGC (15/18) | AGC (18/40) |
| AGT (3/18) | AGT (22/40) | ||
| 135 | Alanine | GCC (7/17) | GCC (7/43) |
| GCT (10/17) | GCT (34/43) | ||
| GCG (2/43) | |||
| 140 | Arginine | CGT (13/17) | CGC (16/40) |
| CGC (4/17) | CGC (24/40) | ||
| 153 | Arginine | AGA (8/18) | AGA (8/43) |
| AGG (10/18) | AGG (35/43) | ||
| 160 | Proline | CCA (18/18) | CCA (29/45) |
| CCG (11/45) | |||
| CCC (5/45) | |||
| 163 | Valine | GTA (13/15) | GTA (18/43) |
| GTG (2/15) | GTG (16/43) | ||
| GTC (9/43) | |||
| 164 | Glutamic acid | GAA (16/18) | GAA (6/45) |
| GAG (2/18) | GAG (39/45) | ||
| 169 | Alanine | GCC (10/18) | GCC (20/41) |
| GCT (8/18) | GCT (21/41) | ||
Differences in the denominator to 18 (1991–1995 sequences) or to 46 (2000–2005 strains) reflect non-synonymous mutations.
Average similarity of predicted GP5 proteins with regards to LV was 83.8% for the 2000–2005 set and 94.4% in the 1991–1995 set. The bootstrapped tree of the predicted aminoacid sequences of GP5 a high diversity in GP5 (Fig. 1 ). Although, in general, strains from 2000 to 2005 tended to cluster together while 1991–1995 sequences were scattered along the tree, bootstrap values only supported small clusters and did not provide evidence for a clear evolutionary line between modern and older strains.
Fig. 1.

Bootstrapped tree (neighbor-joining method) of predicted aminoacid sequences of GP5 of PRRS virus. In italics (light colour) sequences from 1991 to 1995 period; in bold type, sequences from 2000 to 2005. Spanish strains 1991–1995 = Suarez, CRESA-VP21 and Olot91. Spanish Strains 2000–2005 = AF49XXX and CReSA. All other strains correspond to European non-Spanish isolates from 1991 to 1995. Bootstrap values are shown in italics close to the nodes.
The entropy analysis showed three highly variable regions located between aminoacids 56–63; 105–113 and 120–130. These segments corresponded to the parts with higher entropy values for the nucleotide sequence. The most conserved region was found between residues 38–55.
A hypothetical GP5 containing all positively and negatively selected sites was analysed to make a prediction of its characteristics. This hypothetical GP5 had a similarity of 88.1% compared to LV. As shown by BLAST comparison, the 10 sequences closest to the hypothetical GP5 (besides those included in the study) had a similarity ranging from 91 to 88% (average 89.4%). The older strain included in this set of 10 was a Spanish sequence of 1991. Interestingly, of the 24 predicted positive selection sites, 16 were present in sequences from other European countries and eight were predominant (>70% frequency) regardless of the country of origin of the PRRSV strain. Regarding negative selection sites, 19 out of 20 were present in all sequences (Fig. 2 ).
Fig. 2.

Comparison with other non-Spanish European strains of the hypothetical GP5 containing all predicted sites for positive and negative selection in Spanish PRRSV sequences. The hypothetical GP5 was BLASTed and the 10 closest non-Spanish matches were included in the alignment along with Lelystad virus GP5 (LV) and an European-type strain isolated in USA. Protein sequences are represented by its GenBank accession number followed by the country of origin (AT: Austria, DK: Denmark, IT: Italy, US: United States of America) and the year of isolation. Amino acids are presented with a one-letter code in the row corresponding to LV. (- - -) Discontinuous underlined: predicted signal peptide. (—) Continuous underlined: predicted N-glycosylation sites. ( ) Boxed segments: predicted transmembrane regions in the hypothetical GP5. (
) Boxed segments: predicted transmembrane regions in the hypothetical GP5. ( ) Negative selection sites. Positive selection sites are marked with the one-letter aminoacid code in the hypothetical GP5. Selection site: the last row shows whether or not the predicted selection sites were found in other non-Spanish European sequences. Dots (·) indicate negative selection site found in 11/11 non-Spanish European sequences. Aminoacid symbol (X) indicates positive selection site found in at least one non-Spanish European sequence. Encircled aminoacid symbol (®) indicates positive selection site found in ≥8/11 non-Spanish European sequences. (*) Predicted positive or negative selection site not found in non-Spanish European sequences.
) Negative selection sites. Positive selection sites are marked with the one-letter aminoacid code in the hypothetical GP5. Selection site: the last row shows whether or not the predicted selection sites were found in other non-Spanish European sequences. Dots (·) indicate negative selection site found in 11/11 non-Spanish European sequences. Aminoacid symbol (X) indicates positive selection site found in at least one non-Spanish European sequence. Encircled aminoacid symbol (®) indicates positive selection site found in ≥8/11 non-Spanish European sequences. (*) Predicted positive or negative selection site not found in non-Spanish European sequences.
For the hypothetical strain, the signal peptide comprised residues 1–34 (1–32 in LV); transmembrane regions were predicted to exist at the following segments: 69–90 and 108–127 (same segments in LV). Potential N-glycosylation sites were predicted at residues 37 and 53 (46 and 53 in LV). Comparison of the hydrophobicity profiles of LV and the hypothetical GP5 showed that the latter was less hydrophobic (not shown).
The study of the evolution and adaptation of viruses to their hosts is a question of relevance because provides insight on the mechanisms by which a viral variant gains prevalence in a population. A large endemic population with a high replacement rate is a suitable frame to study such phenomena. This is the case of Spain for PRRSV. Spanish pig population is the second largest in Europe with some 24 million pigs and, according to FAO statistics, imports every year about 1.2 million live pigs (http://faostat.fao.org).
The present study was conducted with two sets of PRRSV sequences, one corresponding to the period 1991–1995 and the other to 2000–2005. In this lapse of years, average similarity to LV changed from above 95% in 1991–1995 to below 90% in 2000–2005. These values suggest an increase in divergence of about 0.5% per year. If divergence increased at a constant rate and sequences from 1991 to 1995 shared an average similarity of 95% to LV, original PRRSV strains in pigs could have originated some 10 years before; namely, about 1981–1985. This is the predicted date in which PRRSV is thought to have entered the domestic pig population (Forsberg et al., 2001, Hanada et al., 2005, Plagemann, 2003).
The entropy analysis showed that this divergence arise from mutations scattered in ORF5 although hypervariable regions could be recognised. This has been described before (Pesch et al., 2005, Pirzadeh et al., 1998) and it is thought that these hypervariable regions can correspond to potentially immunogenic sites. Actually, the neutralisation epitope of GP5 is located in the middle of the ectodomain (Plagemann, 2004b) and the first hypervariable region flanked this epitope.
Codon usage was different for leucine, glutamine, serine and proline in either set of Spanish sequences. When compared with the codon usage of other early European PRRSV strains, results were similar for glutamine, serine and proline while leucine was preferentially coded with TTG as did the most recent Spanish strains. Most frequent codons for these aminoacids in 2000–2005 sequences were similar to the codon usage in some highly expressed swine genes. These results can be interpreted as a sign of either a selection or an adaptation of PRRSV to the codon usage most adequate for an efficient replication in the pig host. Also, this adaptation can have other implications. Several authors (Cook et al., 2005, Kheyar et al., 2005) have shown that optimising codon usage of arterivirus genes to that of mammalian cells results in an increase of the levels of expression of viral genes as well as increases immunogenicity of viral proteins. In the present case, our results suggest that PRRSV is still adapting to the swine host. This should be taken into account when designing attenuated vaccines because adequate levels of expression of viral proteins are important to develop strong immune responses.
The codon analysis revealed and average S/NS of 1.41. This ratio was similar to that determined by Hanada et al. (2005) for Coronaviruses but lower than reported by others (Pesch et al., 2005) for PRRSV. This high rate of non-synonymous mutations may have important implications for the design of vaccines since these variable points may constitute inefficient targets for the immune system.
The examination of positively and negatively selected sites showed 24 potential sites for positive selection and 20 for negative selection. Ten of the 24 sites for positive selection were located in transmembrane sections of the predicted GP5, a fact that suggests that many of these adaptations were not selected because of a pressure of the neutralising antibodies. In contrast, the negatively selected sites were concentrated in the last 100 aminoacids of GP5 (14/20 sites) for which no neutralising antibodies have been detected so far (Plagemann, 2004a, Plagemann, 2004b). These negatively selected sites clustered in three segments of the predicted GP5. The first two of these clusters (residues 73–89 and 108–113) corresponded to predicted transmembrane regions while the function of the third segment is still unknown. Since variability of aminoacids in those points is restricted in spite of the presence of several possible codons, it is reasonable to think that these sites are probably crucial for virus integrity or functionality.
The phylogenetic analysis of GP5 sequences did not support a clear line of evolution from older to newer Spanish strains. However, clustering was evident for some newer sequences. With the available data it is impossible to know whether the newer strains represent a PRRSV type that slowly evolved from older strains or if they represent an old type that gained predominance. Unfortunately, we were not able to obtain Spanish sequences from 1996 to 1999 and therefore we could not fill this gap; however, the analysis of 153 GP5 European-type sequences from different countries and periods yielded similar results (not shown). The closest matches to the hypothetical GP5 included strains from different countries and periods but the oldest known match was a Spanish strain from 1991 not included in this study. This fact would support the hypothesis that similarity to the hypothetical GP5 has some benefit because this profile has become predominant in Spain all over the years.
The hypothetical strain was similar to LV but had two additional characteristics that lacked in LV. The first was a change in the sites of glycosylation compared to LV. One was located at the start of the ectodomain (Asp-37) and the second was located at the known neutralisation epitope (Asp-53). This hypothetical GP5 would lose thus the glycosylation site at Asp-46. In contrast, positive selection in position 37 introduces a new glycosylation site. Previous studies suggested that the lack of this glycosylation at Asp-46 reduces virus infectivity and can be a marker of attenuation (Wissink et al., 2004). However, according to Pesch et al. (2005), glycosylation at position 46 can be found in three European-type attenuated vaccines of which only one also has a glycosylation at position 37. Several authors claimed that these additional glycosylation sites may serve to mask the key B-epitopes (Chen et al., 1998) although this has not been proven yet for PRRSV. The second fact that differentiates the hypothetical GP5 from that of LV was a lesser hydrophobic profile. The consequence would be a more exposed GP5 that could better interact with the receptors in target cells.
The present study shows that ORF5 of PRRSV has increased its genetic diversity over time. This evolution included positive and negative selections of given aminoacids in specific sites of the PRRSV genome, mainly in transmembrane segments of GP5. Also, codon usage for leucine, glutamine, serine and proline changed and in more recent sequences have more resemblance to codon usage in highly expressed pig genes suggesting that a process of adaptation to pig is taking place. These data, if further confirmed by other studies with PRRSV isolates of other countries, may be useful to understand the evolution of PRRSV as well as can be relevant for the design of new and more efficacious vaccines.
Acknowledgements
Our grateful thanks to Dr. Anna Barceló for her assistance in sequencing and to Dr. Dieter Klein for providing data for Austrian strains.
References
- Castro C., Arnold J.J., Cameron C.E. Incorporation fidelity of the viral RNA-dependent RNA polymerase: a kinetic, thermodynamic and structural perspective. Virus Res. 2005;107:141–149. doi: 10.1016/j.virusres.2004.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z., Li K., Rowland R.R., Plagemann P.G. Neuropathogenicity and susceptibility to immune response are interdependent properties of lactate dehydrogenase-elevating virus (LDV) and correlate with the number of N-linked polylactosaminoglycan chains on the ectodomain of the primary envelope glycoprotein. Adv. Exp. Med. Biol. 1998;440:583–592. doi: 10.1007/978-1-4615-5331-1_76. [DOI] [PubMed] [Google Scholar]
- Cook R.F., Cook S.J., Bolin P.S., Howe L.J., Zhou W., Montelaro R.C., Issel C.J. Genetic immunization with codon-optimized equine infectious anemia virus (EIAV) surface unit (SU) envelope protein gene sequences stimulates immune responses in ponies. Vet. Microbiol. 2005;108:23–37. doi: 10.1016/j.vetmic.2005.04.004. [DOI] [PubMed] [Google Scholar]
- Fang Y., Kim D.Y., Ropp S., Steen P., Christopher-Hennings J., Nelson E.A., Rowland R.R. Heterogeneity in Nsp2 of European-like porcine reproductive and respiratory syndrome viruses isolated in the United States. Virus Res. 2004;100:229–235. doi: 10.1016/j.virusres.2003.12.026. [DOI] [PubMed] [Google Scholar]
- Forsberg R., Oleksiewicz M.B., Petersen A.M., Hein J., Botner A., Storgaard T. A molecular clock dates the common ancestor of European-type porcine reproductive and respiratory syndrome virus at more than 10 years before the emergence of disease. Virology. 2001;289:174–179. doi: 10.1006/viro.2001.1102. [DOI] [PubMed] [Google Scholar]
- Forsberg R., Storgaard T., Nielsen H.S., Oleksiewicz M.B., Cordioli P., Sala G., Hein J., Botner A. The genetic diversity of European type PRRSV is similar to that of the North American type but is geographically skewed within Europe. Virology. 2002;299:38–47. doi: 10.1006/viro.2002.1450. [DOI] [PubMed] [Google Scholar]
- Gonin P., Pirzadeh B., Gagnon C.A., Dea S. Seroneutralization of porcine reproductive and respiratory syndrome virus correlates with antibody response to the GP5 major envelope glycoprotein. J. Vet. Diagn. Invest. 1999;11:20–26. doi: 10.1177/104063879901100103. [DOI] [PubMed] [Google Scholar]
- Hanada K., Suzuki Y., Nakane T., Hirose O., Gojobori T. The origin and evolution of porcine reproductive and respiratory syndrome viruses. Mol. Biol. E. 2005;22:1024–1031. doi: 10.1093/molbev/msi089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kheyar A., Jabrane A., Zhu C., Cleroux P., Massie B., Dea S., Gagnon C.A. Alternative codon usage of PRRS virus ORF5 gene increases eucaryotic expression of GP(5) glycoprotein and improves immune response in challenged pigs. Vaccine. 2005;23:4016–4022. doi: 10.1016/j.vaccine.2005.03.012. [DOI] [PubMed] [Google Scholar]
- Mateu E., Martin M., Vidal D. Genetic diversity and phylogenetic analysis of glycoprotein 5 of European-type porcine reproductive and respiratory virus strains in Spain. J. Gen. Virol. 2003;84:529–534. doi: 10.1099/vir.0.18478-0. [DOI] [PubMed] [Google Scholar]
- Meng X.J., Paul P.S., Halbur P.G. Molecular cloning and nucleotide sequencing of the 3′-terminal genomic RNA of the porcine reproductive and respiratory syndrome virus. J. Gen. Virol. 1994;75:1795–1801. doi: 10.1099/0022-1317-75-7-1795. [DOI] [PubMed] [Google Scholar]
- Oleksiewicz M.B., Botner A., Toft P., Grubbe T., Nielsen J., Kamstrup S., Storgaard T. Emergence of porcine reproductive and respiratory syndrome virus deletion mutants: correlation with the porcine antibody response to a hypervariable site in the ORF 3 structural glycoprotein. Virology. 2000;267:135–140. doi: 10.1006/viro.1999.0103. [DOI] [PubMed] [Google Scholar]
- Ostrowski M., Galeota J.A., Jar A.M., Platt K.B., Osorio F.A., Lopez O.J. Identification of neutralizing and nonneutralizing epitopes in the porcine reproductive and respiratory syndrome virus GP5 ectodomain. J. Virol. 2002;76:4241–4250. doi: 10.1128/JVI.76.9.4241-4250.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesch S., Meyer C., Ohlinger V.F. New insights into the genetic diversity of European porcine reproductive and respiratory syndrome virus (PRRSV) Vet. Microbiol. 2005;107:31–48. doi: 10.1016/j.vetmic.2005.01.028. [DOI] [PubMed] [Google Scholar]
- Pirzadeh B., Gagnon C.A., Dea S. Genomic and antigenic variations of porcine reproductive and respiratory syndrome virus major envelope GP5 glycoprotein. Can. J. Vet. Res. 1998;62:170–177. [PMC free article] [PubMed] [Google Scholar]
- Plagemann P.G. Porcine reproductive and respiratory virus: origin hypothesis. Emerg. Infect. Dis. 2003;9:903–905. doi: 10.3201/eid0908.030232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plagemann P.G. GP5 ectodomain epitope of porcine reproductive and respiratory syndrome virus, strain Lelystad virus. Virus Res. 2004;102:225–230. doi: 10.1016/j.virusres.2004.01.031. [DOI] [PubMed] [Google Scholar]
- Plagemann P.G. The primary GP5 neutralization epitope of North American isolates of porcine reproductive and respiratory syndrome virus. Vet. Immunol. Immunopathol. 2004;102:263–275. doi: 10.1016/j.vetimm.2004.09.011. [DOI] [PubMed] [Google Scholar]
- Suárez P., Zardoya R., Martin M.J., Prieto C., Dopazo J., Solana A., Castro J.M. Phylogenetic relationships of european strains of porcine reproductive and respiratory syndrome virus (PRRSV) inferred from DNA sequences of putative ORF-5 and ORF-7 genes. Virus Res. 1996;42:159–165. doi: 10.1016/0168-1702(95)01305-9. [DOI] [PubMed] [Google Scholar]
- Snijder E.J., Dobbe J.C., Spaan W.J. Heterodimerization of the two major envelope proteins is essential for arterivirus infectivity. J. Virol. 2003;77:97–104. doi: 10.1128/JVI.77.1.97-104.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijder E.J., Meulenberg J.J. The molecular biology of arteriviruses. J. Gen. Virol. 1998;79:961–979. doi: 10.1099/0022-1317-79-5-961. [DOI] [PubMed] [Google Scholar]
- Thompson J.D., Higgins D.G., Gibson T.J. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wissink E.H., Kroese M.V., Maneschijn-Bonsing J.G., Meulenberg J.J., van Rijn P.A., Rijsewijk F.A., Rottier P.J. Significance of the oligosaccharides of the porcine reproductive and respiratory syndrome virus glycoproteins GP2a and GP5 for infectious virus production. J. Gen. Virol. 2004;85:3715–3723. doi: 10.1099/vir.0.80402-0. [DOI] [PubMed] [Google Scholar]
- Wu, W.H., Fang. Y., Rowland. R.R., Lawson, S.R., Christopher-Hennings, J., Yoon, K.J., Nelson, E.A., 2005. The 2b protein as a minor structural component of PRRSV. Virus Res [Epub ahead of print August 10]. [DOI] [PMC free article] [PubMed]