

The application of molecular biology tools to the diagnosis of infectious disease is increasing in small animal veterinary medicine. The polymerase chain reaction (PCR), reverse transcriptase PCR (RT-PCR), analysis of restriction fragment length polymorphisms (RFLP), and others that often were developed initially for research purposes have increased knowledge of some infectious disease agents. In the process, there was rapid recognition of the diagnostic potential of these assays, and some have become available for diagnostic testing of suspected infections in dogs and cats.
The tools of molecular biology rely on biochemical properties of nucleic acids (DNA and RNA) imparted by the nucleotide composition (relative proportions of each of the four nucleotides) and the nucleotide sequence. The nucleotide composition of nucleic acids influences their denaturation (the separation of the two complementary strands that compose DNA or DNA/RNA hybrids) and hybridization properties (the ability of nucleic acid sequences to bind to each other to form double-stranded nucleic acid moieties). Nucleotide sequence also influences hybridization properties and dictates susceptibility to nucleic acids being “cut” at specific locations using restriction enzymes. This discussion is not intended to provide the details necessary to perform, or even understand all aspects of the molecular techniques presented, but rather is meant to convey an overview of some of the available techniques, the diagnostic power of these applications, some of the diagnostic limitations of these assays, and the variety of clinical applications possible with the molecular techniques.
Molecular assays for detecting infectious agents
The most widely used of the molecular tools for the diagnosis of infectious disease are the PCR and the RT-PCR. The PCR is used for initial detection of DNA and so is most useful for detection of infectious agents that contain DNA as their primary genetic material. Because the PCR is incapable of detecting RNA, in order to detect infectious agents that have RNA as their primary genetic material (many viruses for example), a copy of DNA (cDNA) can be made from the infectious agent's RNA through the process of reverse transcription (RT). Transcription normally produces a messenger RNA from a DNA template; the enzyme reverse transcriptase promotes the synthesis of a DNA molecule from an RNA template. Once the cDNA has been synthesized, a PCR can be performed subsequently, and the entire process is referred to as RT-PCR.
The PCR uses short single-stranded segments of nucleotides, called primers, the sequences of which are complimentary to DNA sequences of the intended target DNA, for example, the DNA of an infectious organism. Primers serve as the initial template upon which a new DNA molecule can be synthesized. The primers and other necessary reagents of the PCR are added to a volume of solution containing representative DNA from the sample of interest, including host DNA and DNA from the intended target of detection. The test sample can be anything that could harbor the agent of interest such as tissue, fluids such as urine or blood, stool, or others. The reaction mix is heated to separate DNA into its two strands, then cooled to allow primers to bind to complimentary regions of denatured target DNA. The reaction then is heated again in an extension step to promote the addition of nucleotides to the primer ends, thus building a “new” strand of DNA. The PCR cocktail is subjected to 25 to 40 cycles of heating and cooling to preferentially amplify target DNA segments. Amplification occurs on a geometric scale; theoretically, one copy of the target DNA sequence can be amplified to over 30 million copies in 25 cycles. The PCR products, or amplicons, are detected most commonly by using gel electrophoresis. The presence of target DNA in the test sample is suggested by observation of a specific and predicted size DNA band in the gel (Fig. 1 ).
Fig. 1.
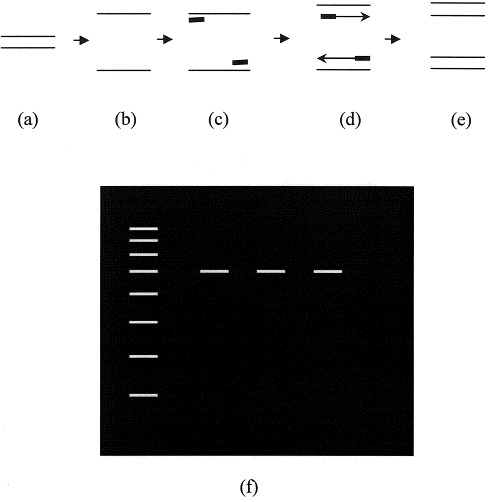
Schematic representation of the polymerase chain reaction (PCR). DNA (a) in the test sample is heated to separate the two strands (b). The reaction is cooled to allow primer (short bold lines) annealing to the target DNA (c), then heated again to allow addition of nucleotides to extend the nascent DNA molecule (d). At the end of the cycle, the target DNA has been duplicated (e). Each new DNA can then itself be a targeted molecule for the next cycle of amplification. The process is repeated for 25–40 cycles, and then products are visualized following gel electrophoresis (f). The left hand lane represents DNA markers of a known size against which the size of the amplicons is measured.
The most common clinical application of the PCR to infectious disease diagnosis in dogs and cats is the detection of a single suspected infectious agent. In these instances, some or all of the DNA sequence of the infectious agent must be known to design agent-specific primers. The PCR can be used, however, to cast a wider net in cases in which an infectious agent is suspected, but for which specific sequence information is not available, or in which the presence of an infectious disease is suspected, but one is unsure of which specific agent is present. In these situations, instead of performing the PCR with primers that are specific for a single agent, universal primers can be designed to amplify a segment of a gene that retains a high degree of sequence similarity, or homology, among a large group of related infectious organisms. Related is a relative term when used in this sense and could refer to organisms related by genus, family, or more broadly still as for example all prokaryotic bacteria. One example of this broad-based approach is illustrated by detection of the gene encoding the 16S ribosomal RNA (rRNA) of prokaryotic bacteria, a gene that maintains a high degree of homology across many genera of bacteria. If a product is amplified using these universal primers, the presence of bacterial DNA in the reaction is established. The reaction amplicons are analyzed by sequencing or other strategies, and the identity of the agent established by documenting similarities to organisms already existing in large databases.
The universal primer PCR can be the first step in design of a genus or species-specific PCR when detailed genetic information regarding the target agent is unknown. Amplicons obtained from universal primer PCR can be sequenced, with the sequence analysis then suggesting a specific genus, or perhaps a specific species, of organism. Sequences that are identified as unique to the 16S rRNA gene or other genes of the detected organism then become the foundation for design of the next set of specific primers. Thus, one has gone from casting a broad net with universal primers to the development of a focused PCR in just a few steps. Such a strategy was employed in the development of a PCR assay specific for Haemobartonella felis [1].
Although primer selection is critical for establishing PCR specificity, there are steps that should be taken to confirm the specificity of the amplicons, primarily to be sure that there are no other unknown DNA segments that share the same sequence of the primer binding sites of the target organism. Confirmation of the specificity is particularly crucial when assays first are developed or are applied to new sample types that represent a different pool of DNA than that in which the assay was developed. Among the methods that can confirm amplicon specificity, DNA hybridization, amplicon sequencing, and analysis of restriction enzyme digestion patterns are among the most commonly used. DNA hybridization uses a short DNA segment that is labeled to permit detection and which recognizes a sequence within the amplicon flanked by the primer binding sites. Detection of the probe will occur only when the probe has become bound to its complementary sequence in the amplicon and is retained through several washes that remove unbound probe. Obtaining a predicted sequence in the amplicon following sequence analysis also confirms the specificity of the amplification.
With primers designed to be very specific for the target sequence, the PCR is very specific. The PCR is typically also very sensitive because of its ability to detect very small numbers of target DNA. Modifications of the PCR have been developed to further increase the sensitivity of the PCR, typically by adding a small volume containing amplicons from a first reaction to a second PCR. The primers of the second PCR are complementary to sequences of the amplicons from the first reaction, a technique known as nested PCR. The sensitivity of any of the PCR strategies facilitates the detection of some organisms below the threshold of detection of routine microbial cultures, cytology, histology, or perhaps immunohistochemical detection.
In another modification of the PCR, the in situ PCR, the reaction is conducted on tissue samples (biopsies for example), and the detection of the amplicons achieved by means other than gel electrophoresis to allow demonstration of the target sequence in a particular tissue location. In situ PCR thus allows correlation of agent detection with the presence of a histological lesion, or localizes the agent to a particular cell type, a finding that would provide a compelling argument that the organism detected played a role in disease causation. In situ PCR can be combined with other techniques such as immunohistochemistry to more completely characterize cells or tissues in which the amplicons are detected, information that can be useful in understanding disease pathogenesis.
Polymerase chain reaction and RT-PCR are limited to diagnostic or research laboratories with the equipment and personnel trained to perform the assays. It is possible that in the future, assays relying on this technology may be available in a bedside format accessible to the practitioner or staff. Indeed, such bedside assays are already appearing in the human medical field and are considered standard approaches to the diagnosis of some infections of people [2]. In the meantime, the practitioner still has access to some of these laboratory-based assays as samples, such as tissue and fluids (blood, effusions, and others), usually are easily acquired and submitted. Any special collection or handling requirements as set forth by the laboratory should be understood before sample acquisition and submission. It is important to recognize that not all laboratories will be in a position to offer all available PCR-based assays for infectious disease diagnosis, and samples submitted to one laboratory for a given test may be sent to a different laboratory better equipped to perform the assay. Cost of the assays will vary with the laboratory and can reflect how frequently the test is performed and technical aspects of the assay.
Restriction fragment length polymorphisms
DNA is susceptible to being “cut” at restriction sites by restriction enzymes. The restriction site recognized by a given restriction enzyme is defined by DNA nucleotide sequences; thus, each restriction enzyme recognizes and cuts at a very specific nucleotide sequence. There are many restriction enzymes used for cutting DNA, and for a given restriction enzyme, multiple restriction sites may exist within a particular DNA segment from a given individual or organism. When DNA that is obtained from an organism is subjected to the action of restriction enzymes and the cut DNA subjected to gel electrophoresis, a pattern of restriction fragments of varying lengths that is unique to the organism is produced (Fig. 2 ). Variations in the lengths of fragments, known as polymorphisms, arise from differences in DNA sequence between the restriction sites. Differences in the restriction fragment lengths alter the migrating properties of the fragments during gel electrophoresis, allowing comparison between organisms. The more restriction enzymes, within reason, a given DNA sample is cut with, the more patterns there can be for analysis. Analysis of restriction fragment length polymorphisms (RFLP) often is referred to as “DNA fingerprinting,” as RFLP are typically unique to individuals or groups of closely related organisms. Comparison of polymorphisms between an unknown and a known pattern can facilitate detection of a new or variant organism related to the known agent. RFLP analysis often is combined with PCR or RT-PCR; the PCR-based assays produce an amplicon, the identity of which can be more precisely established by RFLP analysis.
Fig. 2.
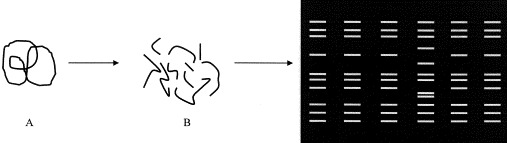
Schematic representation of restriction fragment length polymorphism (RFLP), or DNA fingerprinting, analysis. DNA (A) is cut with a restriction enzyme to produce a number of DNA fragments of varying lengths (B). The DNA fragments are separated by gel electrophoresis to produce a pattern unique to the organism and the enzyme used to cut the DNA. In this representation, six different samples were analyzed, with one of the six (in the 4th lane) clearly exhibiting a pattern different from the other five.
Compared with the PCR-based assays, analysis of RFLPs as a strictly diagnostic technique has had more limited application to the diagnosis of infectious disease in small animal medicine. RFLP is quite useful in demonstrating relatedness or divergence of infectious organisms in an individual or populations of animals. Comparison of RFLP patterns can demonstrate emergence of variant pathogens within a population, or even within the same individual in the case of persistent infections. A recent report describes by RFLP analysis the emergence of variants of Bartonella henselae within cats with chronic infection, a finding that could suggest a mechanism for persistent infection [3].
In situ hybridization
Another technique that has seen somewhat limited applications in small animals is the use of labeled genetic probes to identify the nucleic acid of an infectious agent within a particular cell or tissue type, a technique known as in situ hybridization. In situ hybridization, like the PCR-based assays, takes advantage of the fact that complementary DNA sequences will bind to each other with high affinities. Thus, a probe with a complementary sequence to a known gene can be labeled to permit easy detection, and the probe added, under appropriate conditions, to a sample with cells or tissues to see if the target nucleic acid is present. In situ hybridization thus generally requires a priori knowledge of the organism that is being sought, as the probes tend to be organism-specific. Because in situ hybridization, like in situ PCR, can associate the nucleic acid of an infectious agent with a particular cell or tissue type, in situ hybridization can be a powerful tool for elucidating cell tropism of infectious agents, often a key to the pathophysiology of infectious diseases. For example, in situ hybridization has been used to localize feline immunodeficiency virus (FIV) to particular cells in the thymus [4], canine and feline herpesvirus to a number of tissues [5], [6], and H felis on erythrocytes [7].
Representational differential analysis
Representational differential analysis (RDA) makes use of the PCR and hybridization properties of nucleic acids to “subtract” normal DNA from the total pool of DNA in a test sample [8]. When normal DNA is subtracted from the total DNA present, what remains are exogenous nucleic acid sequences such as those of infectious organisms. The exogenous sequences then can be amplified by the PCR, the amplicons analyzed by sequencing or other strategies, and the results compared with information in databases to identify relationships to known infectious organisms. The author is aware of no published studies in which RDA has been used to establish the diagnosis of an infectious disease in small animal patients. This approach has been used in people to suggest the existence of a novel herpesvirus infection considered to be a likely cause of Kaposi's sarcoma in people with HIV-1 infection, however [9].
Analysis of DNA libraries
An interesting approach to establishing the identity of unknown infectious organisms takes advantage of a host immune response and the ability to put large segments of DNA into bacterial plasmids or other organisms like yeast, which can be cultured. The cultured organisms often are referred to as expression vectors, since the proteins encoded by the inserted DNA sequence can be produced, or expressed, in detectable quantities in culture. In so doing, a library of DNA, or cDNA if starting from RNA, is generated and can be analyzed by a number of methods. With one technique, nucleic acids are collected from an individual with the suspected infectious disease, and the nucleic acid cloned, or inserted, into an expression vector. Each of the clones generated is cultured, with the clones producing proteins encoded by the inserted genes. The patient's serum then is used to screen each of the cultured clones for the production of proteins recognized by the patient's antibodies. Clones recognized by patient antibodies then are analyzed to establish a putative identity of the clone based on its similarity to other pathogens or organisms. This technique was used to establish the identity of a new hepatitis virus in people associated with transfusion-associated hepatitis [10]. To accomplish this feat, the investigators had to screen approximately one million clones to find one that produced a candidate infectious disease antigen.
Limitations of molecular assays
Like all diagnostic test results, interpretation of results generated from molecular assays needs to be considered in light of all available information about the patient, including history, physical examination findings, and results of other diagnostic tests. Although very useful in the assessment of dogs and cats for infectious diseases, the molecular assays are not without limitations. For example, the extreme sensitivity of the PCR is also its biggest limitation, as false positive results can occur with even minute contamination of test samples or reactions. Contamination can occur during sample collection or during the course of performing the PCR. It is important to know that the laboratory has taken relevant steps to eliminate contamination and to ensure that a positive result is truly positive and not a reflection of assay contamination. The extreme sensitivity of the PCR is the biggest obstacle to routine use of universal primer PCR as a diagnostic tool, as contamination with bacteria at any step has the potential to result in a false-positive reaction. Thus, universal primer PCR is better for detection of organisms in normally sterile sites, such as blood, nonepithelial tissues, or other locations from which samples can be obtained aseptically.
As sensitive as they are, the PCR or RT-PCR are nonetheless susceptible to false-negative results. The PCR-based assays may be falsely negative if there are PCR inhibitors, which may include proteins such as hemoglobin or others, if the DNA or RNA in a sample is of poor quality because of degradation, or if there are technical problems with the assay. Performing the assay on an inappropriate sample also could cause false-negative results. For example, the sensitivity of detection of canine distemper virus in one experimental study improved if whole blood, serum and cerebrospinal fluid were tested by RT-PCR, as any one of these samples from any given dog could be negative [11]. Another theoretical cause of false-negative results would be changes in an organism's nucleic acid sequences that preclude primer binding during the annealing step of the PCR. If primers do not anneal to target DNA sequences, a new DNA strand cannot be synthesized.
Most laboratories performing diagnostic PCR will include known positive and negative samples as controls for technical problems with the assay, or as a guard against false-positive results from contamination of the assay. To help further reduce false-positive results from reaction contamination, some laboratories will include a control sample that has all the components of the reaction cocktail except a nucleic acid source, replacing the test sample with water. If a product is detected in one of these reagent controls, products in any other sample must be considered as potential contaminants.
Detection of a nucleic acid of an infectious organism by one of the molecular methods does not necessarily mean that the agent is the cause of clinical disease. A molecular assay may be positive in instances where the organism is in fact not a cause of the clinical disease, as might occur with agents that cause latent infections. Likewise, a negative result would not guarantee that an infectious agent is not responsible for the clinical disease observed. A microbial toxin produced at a site distant from the tissue sampled for the assay may be responsible for the clinical disease and thus would not be detected in the molecular assays. As previously suggested, a variant of the organism not recognized by a particular primer pair would lead to a false-negative result in the face of infection. If the sample submitted was not appropriate for detection of the agent (eg, submission of tissue when blood was needed, or collection of a sample at the wrong stage of infection), the result of an assay could be negative and not reflect the true infection status of the patient. Detection of Borrelia burgdorferi was better accomplished by PCR of skin samples than blood samples because of the low level of spirochetemia associated with that infection [12].
Another limitation of the molecular assays is that, in general, they are not well-suited to assess responses to therapy. It is likely that nucleic acids persist in the host after the death of an organism, and although the duration of DNA persistence often is presumed to be less than a few weeks, the actual duration of DNA persistence following organism death is not known. One group of investigators of acute Rocky Mountain spotted fever was surprised to find that PCR results for Rickettsia rickettsii were positive for at least 8 days beyond the last positive culture [13], a combination of results that would be consistent with the persistence of DNA for a time after all detectably viable organisms had been eliminated from the host. Modifications of the PCR can provide quantitative information regarding the copy numbers (numbers of identical DNA segments in the test sample) of nucleic acid present, which in most instances would be interpreted to reflect the number of organisms actually present in the sample. Thus, a decline in copy number following a therapeutic intervention would be consistent with a therapeutic response. Such an approach could prove more helpful in assessing responses to therapy as compared with a nonquantitative assay. The RT-PCR has been suggested as more useful than PCR assays to monitor responses to therapy [14]. RNA is typically more labile than DNA. Free RNA, or RNA that is not involved in protein synthesis as might occur with the death of an infectious agent, typically is quickly destroyed by host RNAses, enzymes that destroy RNA. Thus, the argument holds that if RNA from an infectious organism is detected in an RT-PCR assay, the presence of viable organisms is implied. RT-PCR-based testing of viral load is standard for people infected with HIV-1 [15].
Clinical applications of molecular diagnostic tools
Molecular diagnostic techniques will not supplant the use of traditional methods of infectious disease diagnosis such as microbial culture, serologic assays, and microscopic detection of organisms. Molecular biological assays, however, do offer particular advantages over traditional methods of infection diagnosis in certain clinical settings. The molecular assays can also be complementary to the traditional approaches. Advantages to the molecular approaches can include more rapid confirmation of the presence or absence of a particular pathogen; the assays, especially the PCR and RT-PCR, have the potential to be completed within 24 to 48 hours of the laboratory's receipt of the sample, including time needed for sample processing. This compares favorably to the days or weeks sometimes required for cultures to be declared positive or negative. The time advantage is dependent on the turnaround times of the laboratory, however, which could reflect demand for the assay and strategies that minimize expense of the assay. Another advantage of the molecular assays is that, since recovery of viable organisms is typically not a goal of the molecular assays, there is no need to use special media for transport of many samples, which makes sample acquisition easier for most in private practice. Depending on the assay and the laboratory, some PCR assays can be cost-competitive with traditional approaches.
One of the calling cards of the techniques described previously is their ability to provide information that could be helpful in the diagnosis of organisms that cannot be cultured (eg, many viruses, some mycoplasma such as Haemobartonella felis) or organisms that are difficult or slow to grow in culture (eg, Mycobacteria). Because detection of these agents by molecular methods does not require having viable organisms, detection of their nucleic acid “footprints” can give clues to their existence in a host. As stated previously, a PCR-based assay exists for documentation of H felis infection. Likewise, PCR approaches have been used to document infection with mycobacterial agents in dogs and cats [16], [17], [18]. These assays also have been applied to the diagnosis of enteric viral infections, as PCR-based assays have been developed for pathogens such as canine and feline coronaviruses [19], [20].
Another advantage of the molecular assays is the fact that they can detect evidence of infection before an infected patient mounts a detectable antibody response. Thus, the molecular assays are attractive for establishing the diagnosis of acute infections before seroconversion, and they could obviate the need for collection of acute and convalescent samples often required for demonstration of seroconversion. Assays for Rocky Mountain spotted fever and leptospirosis have been developed [13], [21], [22], and these infections would be good examples of diseases that historically have required evidence of serologic conversion to provide evidence of infection.
Although serologic conversion can provide strong evidence of infection, there are some diseases for which antibodies exist because of vaccine-induced antibodies (eg, leptospirosis) or because natural exposure to organisms is common (eg, Toxoplasma gondii). For infections such as these, serologic assays often add supportive evidence of infection, but the confidence provided by positive serology can be slim in some clinical situations. Thus, detection of the molecular footprints in a seropositive animal would add an extra degree of confidence in a diagnosis. For example, detection of leptospiral DNA in the blood or urine of a dog with clinical signs would provide strong evidence of infection and could provide evidence of serovar-specific infection supported by antibody titers that are high against that particular serovar. These assays also have the potential to clarify whether young animals that are antibody positive to an infectious disease are infected, or are positive simply because of maternally derived antibodies. Clarification of the infection status of kittens that are positive for antibodies to FIV could be clarified by PCR assays that detect the FIV provirus in feline blood cells were one commercially available.
Another application of the molecular assays would be documentation of latent infections or animals that are carriers of infectious agents. PCR assays have documented latent herpesvirus infection in dogs [23] and clinically normal cats [24]. PCR has been used to document clinically silent ehrlichial infections in dogs 34 months after experimental inoculation [25].
Molecular assays also can differentiate between pathogenic and nonpathogenic forms of organisms or between vaccinal and field isolates of infections for which modified-live virus vaccines exist. RFLP analysis is used commonly to document the existence of pathogenic versions of bacteria such as Escherichia coli [26]. Manifestations of canine distemper virus or feline panleukopenia virus infections that follow closely on the heels of vaccination can be examined by use of the molecular techniques to determine if the clinical disease is caused by the vaccine isolate or a field isolate [27], [28].
Clinicians frequently encounter cases of animals that die or are euthanized before a definitive diagnosis is established. In some of these patients, evidence or suspicion of an infectious etiology is generated by results of necropsy and histological examination of tissues, but blood or other tissues may not be available, or are no longer suitable, for traditional diagnostic techniques such as serology and culture. An infectious etiology can be supported in some of these cases by the molecular assays, since some of them can be performed on nucleic acid extracted from paraffin-embedded tissue blocks. Thus, another advantage of the molecular assays is the ability to retrospectively analyze samples for infectious disease [5], [6].
Selection of appropriate antimicrobial therapy also can be directed by PCR-based assays and RFLP analysis. Genes in infectious organisms can encode antimicrobial resistance proteins, and these genes could be targets of detection for PCR or RFLP. Thus, resistance to a particular class of antimicrobials could be documented before the organism has been cultured and antimicrobial resistance patterns identified by more routine methods. Molecular methods are the tests of choice for detection of methicillin-resistant Staphylococcus aureus and vancomycin-resistant enterococci in people [2].
Other clinical applications that are supported by molecular diagnostic tools include the characterization of zoonotic infections. A novel chlamydial infection in a person was traced, using PCR and RFLP, to an organism harbored by the patient's cat [29]. Cat scratch disease caused by Bartonella clarridgeiae infection in a person occurred with an isolate identical to one recovered from the patient's cat [30]. These assays also have tremendous potential to define new causes of infectious disease, be it detection of variants of well-known organisms, or perhaps even novel infectious causes of well-characterized clinical syndromes that have defied etiologic description. Recently, an ehrlichial-like syndrome was observed in dogs that were negative for Ehrlichia canis by an E canis-specific PCR routinely used by the investigators. Using primers specific for the Ehrlichia genus 16S ribosomal RNA gene, the investigators were able to amplify ehrlichial DNA from the clinical patients [31]. Sequence and DNA hybridization analysis of this amplicon suggested that the dog was infected with E ruminantium, an ehrlichial agent not previously associated with clinical disease in dogs. The investigators were careful to state that detection of the DNA of this ehrlichial organism was not definitive proof that the infection was the cause of the dog's clinical disease, but their finding certainly raises that possibility.
Establishing a relationship between organisms and disease
The power of the molecular assays to uncover the existence of new organisms or new variants in ill animals raises questions about cause and effect, since in many of these cases, recovery of viable organisms may not be accomplished easily. The classic methodology of establishing the relationship between a putative infectious agent and clinical disease was to fulfill criteria established by Koch. Briefly, Koch's postulates held that a given agent was the cause of clinical disease if:
-
•
The agent was found in every case of the disease.
-
•
The agent was not found in other diseases.
-
•
The agent could be isolated and cultured, and caused disease in a new host.
A fourth postulate, that the agent could be isolated from the experimentally inoculated host, is considered an additional point of proof of infectious disease causation.
Although rigorous satisfaction of the postulates provides convincing evidence of a cause and clinical effect, the postulates break down with regards to infectious organisms that cannot be cultured, or are very difficult to culture. To account for the applications of the tools of molecular biology to the diagnosis of infectious disease, and especially with regards to discovery of novel infectious organisms, newer criteria to support disease causation have been proposed [32]. Researchers have suggested that these criteria include:
-
•
The sequence of the putative agent should be detectable in most cases of the disease.
-
•
Hosts without disease caused by the putative agent should have no, or few, copy numbers.
-
•
Resolution of the clinical disease should be associated with either a decrease in copy number or an inability to detect the agent.
-
•
Detection of the agent before onset of clinical disease, or an increasing copy number that is associated with the onset or severity of clinical disease, makes cause and effect more likely.
-
•
The properties of the putative agent inferred from its sequence or genetic relationship to other organisms should be consistent with other agents of that particular type.
-
•
If a phenotype, such as a predictable pattern of clinical signs, laboratory abnormalities, or histopathological lesions, is predictable based on the presence of the sequence or increasing copy numbers, a causal link is strengthened.
-
•
The strength of a causal link is enhanced if the sequence is detected readily in lesions, but not in normal tissues.
-
•
The ability to detect the sequence should be reproducible.
Questions arise from a consideration of these criteria when the clinician is presented with a patient or a literature description of a patient from which the nucleic acid of an infectious agent has been detected. First and foremost, the clinician needs to ask if it is likely that the infectious organism is truly responsible for the clinical signs or other abnormalities detected in the patient. In many cases, the association will be suggested because of well-established links between the infectious agent and clinical signs (eg, acute fever, petechial hemorrhage and thrombocytopenia in a dog with R rickettsii). In other cases, the link may be more difficult to discern, as for example a cat with signs of hemolytic anemia and detection of Haemobartonella by a PCR. Although it is known that Haemobartonella can cause hemolytic anemia, it is also known that there are other causes of hemolytic anemia in cats, and that many cats are asymptomatic carriers of Haemobartonella. Thus, a PCR-positive blood sample would not guarantee that hemolytic anemia was caused by Haemobartonella felis infection. Likewise, T gondii parasitemia has been documented in experimentally infected cats that show no clinical signs of infection [33]. Thus, a cat demonstrating signs of neurological or respiratory disease that was positive for T gondii on the basis of a PCR on a blood sample may or may not have clinical toxoplasmosis. The strength of disease association in such an instance would be bolstered by a positive result from a tissue lesion. Thus, as is the case with interpretation of other diagnostic tests for infectious organisms, an understanding of the biology of the infection is also important for proper interpretation of results of molecular assays.
With increasing clinical application of the PCR-based assays or other molecular approaches, there likely will be increasing reports of novel infectious organisms being associated with clinical diseases in dogs and cats. When reading such reports, it would do the reader well to recall some of the guidelines suggested above before concluding that the organism detected is the cause of a new disease. For example, there have been several recent reports of dogs with Bartonella infections detected by PCR with a diverse array of clinical diseases including granulomatous diseases and peliosis hepatis [34], [35]. These conditions have been associated with Bartonella infection through detection of Bartonella DNA in lesions. Granulomatous disease and peliosis hepatis in dogs have not yet been shown conclusively to be caused by Bartonella infections, however, as few of the criteria for establishing a cause and effect by molecular diagnostic methods have been fulfilled.
Summary
The era of diagnostic molecular biology has arrived for small animal clinicians, and it is a near certainty that assays such as the PCR and RT-PCR will become more widely available for a wider array of infectious agents. Already there is an extensive list of infectious diseases of dogs and cats that have been investigated with molecular tools. A partial list is included in box 1 .
Box 1. Partial list of infectious agents for which molecularassays have been developed and used in dogs and catsa.
Viral [36], [37], [38], [39], [40], [41], [42]
Bornavirus in dogs and cats
Canine adenovirus
Parvovirus (canine and feline)
Feline calicivirus
Rabies
Bacterial [36], [43]
Chlamydia psittaci
Helicobacter
Protozoal [44], [45], [46], [47]
Cytauxzoon felis
Leishmania in dogs and cats
Babesia species
Hepatozoon americanum
Fungal [48], [49]
Histoplasma capsulatum
Cryptococcus neoformans
aThese are infectious agents for which the molecular assays described in this article have been used to study the organism in cats and dogs. To author's knowledge, none of these assays are available for routine diagnostic tests.
An understanding of the advantages and disadvantages of the molecular techniques and some of the questions these techniques can answer for clinicians can serve practitioners well in their approach to the diagnosis of infectious diseases in dogs and cats. It is likely that additional applications of these tools to small animal medicine will become apparent as investigators use and refine them for their research purposes, or as new uses emerge from human medical applications. Clinicians also are likely to reap the benefits of this knowledge. Because samples often are acquired easily from clinical patients in most practice settings, access to these tools puts all clinicians in the group of discoverers of new, or variations of, infectious diseases and their clinical manifestations.
References
- 1.Messick J.B, Berent L.M, Cooper S.K. Development and evaluation of a PCR-based assay for detection of Haemobartonella felis in cats and differentiation of H. felis from related bacteria by restriction fragment length polymorphism analysis. J Clin Microbiol. 1998;36:462–466. doi: 10.1128/jcm.36.2.462-466.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Versalovic J, Lupski J.R. Molecular detection and genotyping of pathogens: more accurate and rapid answers. Trends Microbiol. 2002;10(10S):S15–S21. doi: 10.1016/s0966-842x(02)02438-1. [DOI] [PubMed] [Google Scholar]
- 3.Kabeya H, Maruyama S, Irei M, Takahashi R, Yamashita M, Mikami T. Genomic variations among Bartonella henselae isolates derived from naturally infected cats. Vet Microbiol. 2002;89:211–221. doi: 10.1016/s0378-1135(02)00175-x. [DOI] [PubMed] [Google Scholar]
- 4.Johnson C.M, Papadi G.P, Tompkins W.A, Sellon R.K, Orandle M.S, Bellah J.R. Biphasic thymus response by kittens inoculated with feline immunodeficiency virus during fetal development. Vet Pathol. 1998;35(3):191–201. doi: 10.1177/030098589803500304. [DOI] [PubMed] [Google Scholar]
- 5.Suchy S, Bauder B, Gelbmann W, Lohr C.V, Teifke J.P, Weissenbock H. Diagnosis of feline herpesvirus infection by immunohistochemistry, polymerase chain reaction, and in situ hybridization. J Vet Diagn Invest. 2000;12(2):186–191. doi: 10.1177/104063870001200220. [DOI] [PubMed] [Google Scholar]
- 6.Schulze C, Baumgartner W. Nested polymerase chain reaction and in situ hybridization for diagnosis of canine herpesvirus infection in puppies. Vet Pathol. 1998;35(3):209–217. doi: 10.1177/030098589803500306. [DOI] [PubMed] [Google Scholar]
- 7.Berent L.M, Messick J.B, Cooper S.K, Cusick P.K. Specific in situ hybridization of Haemobartonella felis with a DNA probe and tyramide signal amplification. Vet Pathol. 2000;37(1):47–53. doi: 10.1354/vp.37-1-47. [DOI] [PubMed] [Google Scholar]
- 8.Lisitsyn N, Lisitsyn N, Wigler M. Cloning the difference between two complex genomes. Science. 1993;259:946–951. doi: 10.1126/science.8438152. [DOI] [PubMed] [Google Scholar]
- 9.Chang Y, Cesarman E, Pessin M.S. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science. 1994;265:1865–1869. doi: 10.1126/science.7997879. [DOI] [PubMed] [Google Scholar]
- 10.Choo Q.L, Kuo G, Weiner A.J, Overby L.R, Bradley D.W, Houghton M. Isolation of a cDNA clone derived from a bloodborne non-A, non-B viral hepatitis genome. Science. 1989;244:359–364. doi: 10.1126/science.2523562. [DOI] [PubMed] [Google Scholar]
- 11.Frisk A.L, König M, Moritz A, Baumgärtner W. Detection of canine distemper virus nucleoprotein RNA by reverse transcription-PCR using serum, whole blood, and cerebrospinal fluid from dogs with distemper. J Clin Microbiol. 1999;37(11):3634–3643. doi: 10.1128/jcm.37.11.3634-3643.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Straubinger R.K. PCR-based quantification of Borrelia burgdorferi organisms in canine tissues over a 500-day postinfection period. J Clin Microbiol. 2000;38(6):2191–2199. doi: 10.1128/jcm.38.6.2191-2199.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Breitschwerdt E.B, Papich M.G, Hegarty B.C, Gilger B, Hancock S.I, Davidson M.G. Efficacy of doxycycline, azithromycin, or trovafloxacin for treatment of experimental Rocky Mountain spotted fever in dogs. Antimicrob Agents Chemother. 1999;43(4):813–821. doi: 10.1128/aac.43.4.813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Felek S, Unver A, Stich R.W, Rikihisa Y. Sensitive detection of Ehrlichia chafeensis in cell culture, blood and tick specimens by reverse-transcription-PCR. J Clin Microbiol. 2001;39(2):460–463. doi: 10.1128/JCM.39.2.460-463.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pfaller M.A. Molecular approaches to diagnosing and managing infectious diseases: practicality and costs. Emerg Infect Diseases. 2001;7(2):312–318. doi: 10.3201/eid0702.010234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hughes M.S, Ball N.W, Love D.N, Canfield P.J, Wigney D.I, Dawson D. Disseminated Mycobacterium genavense infection in a FIV-positive cat. J Feline Med Surg. 1999;1(1):23–29. doi: 10.1016/S1098-612X(99)90006-2. [DOI] [PubMed] [Google Scholar]
- 17.Appleyard G.D, Clark E.G. Histologic and genotypic characterization of a novel Mycobacterium species found in three cats. J Clin Microbiol. 2002;40(7):2425–2430. doi: 10.1128/JCM.40.7.2425-2430.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pressler B.M, Hardie E.M, Pitulle C, Hopwood R.M, Sontakke S, Breitschwerdt E.B. Isolation and identification of Mycobacterium kansasii from pleural fluid of a dog with persistent pleural effusion. J Am Vet Med Assoc. 2002;220(9):1336–1340. doi: 10.2460/javma.2002.220.1336. [DOI] [PubMed] [Google Scholar]
- 19.Pratelli A, Buonavoglia D, Martella V, Tempesta M, Lavazza A, Buonavoglia C. Diagnosis of canine coronavirus infection using nested PCR. J Virol Methods. 2000;84:91–94. doi: 10.1016/S0166-0934(99)00134-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Foley J.E, Poland A, Carlson J, Pedersen N.C. Patterns of feline coronavirus infection and fecal shedding from cats in multiple cat environments. J Am Vet Med Assoc. 1997;210(9):1307–1312. [PubMed] [Google Scholar]
- 21.Cai H.Y, Hornby G, Key D.W, Osuch M.R, Maxie M.G. Preliminary study on differentiation of Leptospira grippotyphosa and Leptospira sejroe from other common pathogenic leptospiral serovars in canine urine by polymerase chain reaction assay. J Vet Diagn Invest. 2002;14(2):164–168. doi: 10.1177/104063870201400214. [DOI] [PubMed] [Google Scholar]
- 22.Zuerner R.L, Bolin C.A. Differentiation of Leptospira interrogans isolates by IS1500 hybridization and PCR assays. J Clin Microbiol. 1997;35(10):2612–2617. doi: 10.1128/jcm.35.10.2612-2617.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miyoshi M, Ishii Y, Takiguchi M, Takada A, Yasuda J, Hashimoto A. Detection of canine herpesvirus DNA in the ganglionic neurons and the lymph node lymphocytes of latently infected dogs. J Vet Med Sci. 1999;61(4):375–379. doi: 10.1292/jvms.61.375. [DOI] [PubMed] [Google Scholar]
- 24.Burgesser K.M, Hotaling S, Schiebel A, Ashbaugh S.E, Roberts S.M, Collin J.K. Comparison of PCR, virus isolation, and indirect fluorescent antibody staining in the detection of naturally occurring feline herpesvirus infections. J Vet Diagn Invest. 1999;11(2):122–126. doi: 10.1177/104063879901100203. [DOI] [PubMed] [Google Scholar]
- 25.Harrus S, Waner T, Aizenberg I, Foley J.E, Poland A.M, Bark H. Amplification of ehrlichial DNA from dogs 34 months after infection with Ehrlichia canis. J Clin Microbiol. 1998;36(1):73–76. doi: 10.1128/jcm.36.1.73-76.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smith K.A, Kruth S, Hammermueller J, Gyles C, Wilson J.B. A case-control study of verocytotoxigenic Escherichia coli infection in cats with diarrhea. Can J Vet Res. 1998;62(2):87–92. [PMC free article] [PubMed] [Google Scholar]
- 27.Ohashi K, Iwatsuki K, Nakamura K, Mikami T, Kai C. Molecular identification of a recent type of canine distemper virus in Japan by restriction fragment length polymorphism. J Vet Med Sci. 1998;60(11):1209–1212. doi: 10.1292/jvms.60.1209. [DOI] [PubMed] [Google Scholar]
- 28.Horiuchi M, Yuri K, Soma T, Katae H, Nagasawa H, Shinagawa M. Differentiation of vaccine virus from field isolates of feline panleukopenia virus by polymerase chain reaction and restriction fragment length polymorphism analysis. Vet Microbiol. 1996;53:283–293. doi: 10.1016/s0378-1135(96)01225-4. [DOI] [PubMed] [Google Scholar]
- 29.Hartley J.C, Stevenson S, Robinson A.J, Littlewood J.D, Carder C, Cartledge J. Conjunctivitis due to Chlamydophila felis (Chlamydia psittaci feline pneumonitis agent) acquired from a cat: case report with molecular characterization of isolates from the patient and cat. J Infect. 2001;43(1):7–11. doi: 10.1053/jinf.2001.0845. [DOI] [PubMed] [Google Scholar]
- 30.Kordick D.L, Hilyard E.J, Hadfield T.L, Wilson K.H, Steigerwalt A.G, Brenner D.J. Bartonella clarridgeiae, a newly recognized zoonotic pathogen causing inoculation papules, fever, and lymphadenopathy (cat scratch disease) J Clin Microbiol. 1997;35(7):1813–1818. doi: 10.1128/jcm.35.7.1813-1818.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Allsopp M.T.E.P, Allsopp B.A. Novel Ehrlichia genotype detected in dogs in South Africa. J Clin Microbiol. 2001;39(11):4204–4207. doi: 10.1128/JCM.39.11.4204-4207.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fredericks D.N, Relman D.A. Sequence-based identification of microbial pathogens: a reconsideration of Koch's postulates. Clin Microbiol Rev. 1996;9:18–33. doi: 10.1128/cmr.9.1.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Burney D.P, Lappin M.R, Spilker M, McReynolds L. Detection of Toxoplasma gondii parasitemia in experimentally inoculated cats. J Parasitol. 1999;85(5):947–951. [PubMed] [Google Scholar]
- 34.Kitchell B.E, Fan T.M, Kordick D.L. Peliosis hepatis in a dog infected with Bartonella henselae. J Am Vet Med Assoc. 2000;216:519–523. doi: 10.2460/javma.2000.216.519. [DOI] [PubMed] [Google Scholar]
- 35.Pappalardo B.L, Brown T, Gookin J.L. Granulomatous disease associated with Bartonella infection in two dogs. J Vet Intern Med. 2000;14:37–42. doi: 10.1892/0891-6640(2000)014<0037:gdawii>2.3.co;2. [DOI] [PubMed] [Google Scholar]
- 36.Sykes J.E, Allen J.L, Studdert V.P, Browning G.F. Detection of feline calicivirus, feline herpesvirus-1 and Chlamydia psittaci mucosal swabs by multiplex RT-PCR/PCR. Vet Microbiol. 2001;81:95–108. doi: 10.1016/s0378-1135(01)00340-6. [DOI] [PubMed] [Google Scholar]
- 37.Reeves N.A, Helps C.R, Gunn-Moore D.A, Blundell C, Finnemore P.L, Pearson G.R. Natural Borna disease virus infection in cats in the United Kingdom. Vet Rec. 1998;143(19):523–526. doi: 10.1136/vr.143.19.523. [DOI] [PubMed] [Google Scholar]
- 38.Pereira C.A, Monezi T.A, Mehnert D.U, D'Angelo M, Durigon E.L. Molecular characterization of canine parvovirus in Brazil by polymerase chain reaction assay. Vet Microbiol. 2000;75(2):127–133. doi: 10.1016/s0378-1135(00)00214-5. [DOI] [PubMed] [Google Scholar]
- 39.Steinel A, Munson L, van Vuuren M, Truyen U. Genetic characterization of feline parvovirus sequences from various carnivores. J Gen Virol. 2000;81(Pt 2):345–350. doi: 10.1099/0022-1317-81-2-345. [DOI] [PubMed] [Google Scholar]
- 40.Meurs K.M, Fox P.R, Magnon A.L, Liu S, Towbin J.A. Molecular screening by polymerase chain reaction detects panleukopenia virus DNA in formalin-fixed hearts from cats with idiopathic cardiomyopathy and myocarditis. Cardiovasc Pathol. 2000;9(2):119–126. doi: 10.1016/S1054-8807(00)00031-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weissenbock H, Nowotny N, Caplazi P, Kolodziejek J, Ehrensperger F. Borna disease in a dog with lethal meningoencephalitis. J Clin Microbiol. 1998;36(7):2127–2130. doi: 10.1128/jcm.36.7.2127-2130.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ito M, Itou T, Sakai T, Santos M.F.C, Arai Y.T, Takasaki T. Detection of rabies virus RNA isolated from several species of animals in Brazil by RT-PCR. J Vet Med Sci. 2001;63(12):1309–1313. doi: 10.1292/jvms.63.1309. [DOI] [PubMed] [Google Scholar]
- 43.Foley J.E, Solnick J.V, Lapointe J.M, Jang S, Pedersen N.C. Identification of a novel enteric Helicobacter species in a kitten with severe diarrhea. J Clin Microbiol. 1998;36(4):908–912. doi: 10.1128/jcm.36.4.908-912.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Poli A, Abramo F, Barsotti P, Leva S, Gramiccia M, Ludovisi A. Feline leishmaniosis due to Leishmania infantum in Italy. Vet Parasitol. 2002;106:181–191. doi: 10.1016/s0304-4017(02)00081-x. [DOI] [PubMed] [Google Scholar]
- 45.Meinkoth J, Kocan A.A, Whitworth L, Murphy G, Fox J.C, Woods J.P. Cats surviving natural infection with Cytauxzoon felis: 18 cases (1997–1998) J Vet Intern Med. 2000;14(5):521–525. doi: 10.1892/0891-6640(2000)014<0521:csniwf>2.3.co;2. [DOI] [PubMed] [Google Scholar]
- 46.Gaskin A.A, Schantz P, Jackson J, Birkenheuer A, Tomlinson L, Gramiccia M. Visceral leishmaniasis in a New York foxhound kennel. J Vet Intern Med. 2002;16(1):34–44. doi: 10.1892/0891-6640(2002)016<0034:vliany>2.3.co;2. [DOI] [PubMed] [Google Scholar]
- 47.Inokuma H, Okuda M, Ohno K, Shimoda K, Onishi T. Analysis of the 18S rRNA gene sequence of a Hepatozoon detected in two Japanese dogs. Vet Parasitol. 2002;106(3):265–271. doi: 10.1016/s0304-4017(02)00065-1. [DOI] [PubMed] [Google Scholar]
- 48.de Medeiros Muniz M, Pizzini C.V, Peralta J.M, Reiss E, Zancope-Oliveira R.M. Genetic diversity of Histoplasma capsulatum strains isolated from soil, animals, and clinical specimens in Rio de Janeiro state, Brazil, by a PCR-based random amplified polymorphic DNA assay. J Clin Microbiol. 2001;39(12):4487–4494. doi: 10.1128/JCM.39.12.4487-4494.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kano R, Fujino Y, Takamoto N, Tsujimoto H, Hasegawa A. PCR detection of the Cryptococcus neoformans CAPS9 gene from a biopsy specimen from a case of feline cryptococcosis. J Vet Diagn Invest. 2001;13(5):439–442. doi: 10.1177/104063870101300516. [DOI] [PubMed] [Google Scholar]