

Summary
This study describes a novel non-specific universal virus detection method that permits molecular detection of viruses in biological materials containing mixtures of cells and viruses. Samples are subjected to nuclease digestion and ultracentrifugation to separate encapsidated viral nucleic acids from cellular nucleic acids. A degenerate oligonucleotide primer PCR (DOP-PCR) that has been optimized for the non-specific amplification of virus sized genomes is then employed. Virus identification is performed by sequencing of cloned DOP-PCR products followed by sequence comparison to sequences published in GenBank.
This method was used to detect a variety of DNA viruses (including HSV, VZV, SV40, AAV, and EBV) and RNA viruses (including HTLV-I, HTLV-II, influenza, and poliovirus), which were spiked into cells, constitutively expressed in cell culture, or detected in productively infected cultured cells. This novel approach was compared with a non-specific virus detection method used previously and found to be several logs more sensitive. This type of approach has potential utility in solving virus detection and discovery problems where other methods have failed.
Abbreviations: DOP-PCR, degenerate oligo primer-polymerase chain reaction; RDA, representational difference analysis; CPE, cytopathic effect; SISPA, sequence-independent single primer amplification
Keywords: Universal virus detection, Non-specific virus detection, DOP-PCR, Virus discovery, Capsid preparation
1. Introduction
The detection of unknown viruses is still one of the most challenging problems in diagnostic virology. If a known virus is suspected to be present in a biological sample, it is usually not difficult to use the known viral sequence to design specific PCR primers, and to implement a specific detection assay. However, in an era of emerging pathogens and bioterrorism (including the potential for bioterrorism agents genetically engineered to avoid detection using standard methods), improved methods for virus discovery and detection that are less dependent upon the availability of sequence information are needed. Moreover, if such methods were standardized, they could be used potentially to demonstrate (to a defined degree of sensitivity) the absence of a contaminating virus in a biological specimen with potential application to clinical or regulatory problems.
Conventional PCR methods are used frequently to identify or exclude the presence of a viral pathogen suspected to be present in a biological sample. PCR is both very sensitive and specific, especially when coupled with sequencing of PCR products. However, its reliance on specific primers complementary to the pathogen genome sequence makes conventional PCR analyses unsuitable for screening of biological samples for the presence of unknown viruses.
PCR methods using primers based on consensus sequences shared among related viruses were used to detect parainfluenza viruses 1–3 (Corne et al., 1999), to identify and detect coronaviruses (Sampath et al., 2005; van der Hoek et al., 2004), to discover new herpes viruses (VanDevanter et al., 1996) and to identify West Nile virus as the etiologic agent of human encephalitis in the recent New York outbreak (Briese et al., 1999). These primers are designed to anneal to sequence regions highly conserved among members of a virus family, such as polymerase genes. Because these regions are almost never completely conserved, these consensus primers generally include some degeneracy that permits binding to all or the most common known variants on the conserved sequence. This general approach has been automated in an innovative high throughput system (Sampath et al., 2005). However, to be successful, these methods require sufficient sequence information on a group of related viruses to permit the design of consensus primers.
Subtractive methods can identify genomic sequence differences between two related samples, and have also been used for virus discovery. Representational difference analysis (RDA) (Lisitsyn et al., 1993) involves molecular subtraction of uninfected negative control sequences from cells believed to also contain virus. Differential display also involves the comparison of amplified representational libraries of two or more different samples. RDA was used to discover HHV-8 (Chang et al., 1994) and TTV (Linnen et al., 1996), while differential display was used to first identify human metapneumovirus growing in infected cell cultures (van den Hoogen et al., 2001). Even though these methods are not limited generally by primer specificity, they require a matched uncontaminated negative control that is otherwise identical genetically to the test sample, and they are technically challenging and time consuming.
The goal of the present study was to develop sensitive molecular techniques that could be used to detect viruses rapidly in biological specimens, in the absence of any specific information about the virus and without the need for a negative sample. In order to identify viral genomes in any given biological sample, it is necessary to use methods that exclude cellular nucleic acids, which are the most abundant nucleic acids in most samples. The approach described here is based on physical separation of viral and cellular nucleic acids, followed by non-specific amplification of the purified viral nucleic acids. Encapsidation of viral nucleic acids is used as a basis for separation of viral nucleic acids from cellular nucleic acids. Viral capsids protect viral nucleic acids from digestion by nucleases, and also provide viral nucleic acids with a density that permits capsids to be separated from other cellular debris, including nucleic acids (Denniston et al., 1981). After purification of viral capsids, viral DNA and RNA are extracted (separately, in order to preserve an indication of whether a viral genome is DNA or RNA) and RNA is copied into cDNA by reverse transcription. Samples are then amplified using a non-specific DOP-PCR assay that is designed to amplify at least a portion of any viral genome. This report discusses this method and an evaluation of its sensitivity to detect viruses spiked into cell culture, present during acute infection of cell culture, or expressed constitutively in cell culture.
2. Materials and methods
2.1. Samples
Samples used to assess the sensitivity of the non-specific virus detection methods included virus stocks, Vero cells (African Green Monkey Kidney Cells) spiked with varying amounts of virus stocks, cells known to be infected chronically or latently with various viruses (either alone or spiked into uninfected Vero cells), and cells acutely infected with known DNA or RNA viruses (either alone or spiked into uninfected Vero cells). SV40 genomic DNA (obtained from Life Technologies, Gaithersburg, MD), in 10-fold dilution series, was used as a control.
2.1.1. Cells
Vero, 1A2 (Croce et al., 1979), MJ (G11) (Popovic et al., 1983), Mo T (Saxon et al., 1978), and Jiyoye (Kohn et al., 1967) cells were sourced from the American Type Culture Collection (Manassas, VA), and maintained in culture according to the instructions provided. To investigate detection of viruses expressed in cells, 1A2, MJ, MoT and Jiyoye cells were harvested and pelleted by centrifugation, resuspended in medium, counted and then diluted serially (10-fold serial dilutions containing 106 to 101 cells), using uninfected Vero cells to make up a total of 106 cells.
2.1.2. Viruses
HSV-1 (strain 17syn+) (Dr. Stephen Straus, NIH), HSV-2 (strain 333) (Dr. Gary Hayward, Johns Hopkins University), influenza type A virus (Dr. Zhiping Ye, CBER/FDA), titered stocks of simian virus 40 (SV40) (Dr. Andrew Lewis, CBER/FDA), varicella-zoster Virus (Oka strain) (Michiaki Takahashi, Osaka University, Japan), poliovirus type 2 (Sabin strain) (Dr. Konstantin Chumakov, CBER/FDA) and adeno-associated virus (AAV type 2) (Dr. Robert Kotin, NIH) were used in these experiments.
2.1.3. Cell spiking experiments
Viruses were diluted serially in cell culture medium to obtain 10-fold serial dilutions of 106 to 101 infectious units. Virus dilutions were mixed with 105 or 106 uninfected Vero cells.
2.1.4. Infected cell experiments
HSV-1 and HSV-2 were used to infect Vero cells at a multiplicity of infection (MOI) of 1. Human poliovirus type 2 was used to infect semi-confluent Vero cells at a MOI of 1. Varicella-zoster virus (VZV) was grown in MRC-5 cells. Cells were harvested when about 75% cytopathic effect (CPE) was evident. The cells were pelleted by centrifugation, resuspended in medium, and counted using a hemocytometer. Virus-infected cells were serially diluted to obtain 10-fold serial dilutions of 106 to 101 infected cells. Infected cell samples were then adjusted to a total of 106 cells using uninfected Vero cells.
2.2. Purification of viral nucleic acids
The viral nucleic acid extraction method is based on viral capsid purification techniques described previously (Denniston et al., 1981). Briefly, samples were suspended in viral buffer (30 mM Tris/HCl pH 7.5, 3.6 mM CaCl2, 5 mM Mg Acetate, 125 mM KCl and 0.5 mM EDTA), and homogenized (using a disposable tip homogenizer, Omni International, Marietta, GA) to disrupt cell and nuclear membranes. Cellular nucleic acids were digested away by treatment with nucleases DNase I (100 Kunitz Units, Sigma–Aldrich, St. Louis, MO) and RNase ONE (50–100 U, Promega, Madison, WI). Samples were then extracted with 0.4 volumes of trichloro-trifluoro-ethane (Sigma–Aldrich, St. Louis, MO). The encapsidated viral nucleic acids were recovered in the aqueous phase and then pelleted through a discontinuous glycerol gradient as described previously (Denniston et al., 1981). The capsid pellet was resuspended in viral buffer and was re-treated (as above) with DNase and RNase. Viral nucleic acids (DNA and/or RNA) were extracted from the isolated capsids using the Qiagen (Valencia, CA) Whole Blood Kit (for DNA) or the Trizol reagent (Invitrogen, Carlsbad, CA) (for RNA). In order to detect viral RNA, samples were subjected to reverse transcription using random hexamer primers and Superscript Reverse Transcriptase (Life Technologies, Gaithersburg, MD) prior to DOP-PCR.
2.3. Degenerate oligonucleotide primer polymerase chain reaction (DOP-PCR)
A method described previously for generating representational libraries of eukaryotic genomes, known as degenerate oligonucleotide primer PCR (DOP-PCR) (Telenius et al., 1992) was adapted to amplify non-specifically portions of any viral genome. DOP-PCR primer populations were designed with a short (four to six nucleotide) 3′-anchor sequence (which allows the primers to bind in consistent locations), preceded by a non-specific degenerate sequence (of six to eight nucleotides in these experiments). Immediately upstream of the non-specific degenerate sequence, each primer also contained a defined 5′-sequence of 10 nucleotides in length (Table 1 ).
Table 1.
Design and optimization of degenerate oligonucleotide primer used in the universal viral nucleic acid amplification
| Primer number | 5′ specific sequence | Ambiguous sequence | 3′-anchor sequence | Detection limit |
|---|---|---|---|---|
| 1 | CCGACTCGAG | NNNNNN | ATGTGG | 106 |
| 2 | CCGACTCGAG | INNNNNN | TGTGG | 102 |
| 3 | CCGACTCGAG | INNNNNN | TGTG | 105 |
| 4 | CCGACTCGAG | IINNNNNN | GTGG | >106 |
| 5 | CCGACTCGAG | IIINNNNNN | GTGG | 103 |
| 6 | CCGACTCGAG | NNNNNNIII | GTGG | >106 |
| 7 | CCGACTCGAG | IINNNIINNNI | TGTG | 105 |
| 8 | CCGACTCGAG | IINNNNNN | TGTG | 103 |
| 9 | CCGACTCGAG | IINNNIINNN | TGTG | >106 |
| 10 | CCGACTCGAG | IINNNIINNN | TGT | >106 |
| 11 | CCGACTCGAG | IIINNNNNN | TGG | 104 |
| 12 | CCGACTCGAG | IINNNIINNNI | TG | >106 |
| 13 | GCGCCATCAG | INNNNNN | TGTGG | 102 |
| 14 | GCCCGCTCAG | INNNNNN | TGTGG | 102 |
| 15 | CCGACTCGAG | INNNNNN | TTCTG | 102 |
| 16 | CCGACTCGAG | INNNNNN | TCTG | 105 |
| 17 | CCGACTCGAG | INNNNNN | TTCT | 105 |
| 18 | CCGACTCGAG | INNNNNN | GTCT | 105 |
| 19 | CCGACTCGAG | IINNNNNN | TTCT | >106 |
The sequence of the primer population used in most of the experiments reported in this manuscript was 5′-CCGACTCGAGINNNNNN TGTGG-3′, where N refers to an equimolar representation of all four deoxyribonucleotides, I refers to inosine, the underlined nucleotides correspond to the 3′-anchor sequence, and the italicized nucleotides correspond to the degenerate sequence. Because of the six Ns in each degenerate sequence, each reaction included a mixture of 4096 different primers. The PCR cycling program used in these experiments was: Initial denaturation for 5 min at 95 °C, followed by 5 cycles of 1 min at 94 °C, 1.5 min at 30 °C, slow ramping to 72 °C at 0.2 °C/s, and 3 min extension at 72 °C. This was then followed by 35 cycles of 1 min at 94 °C, 1 min at 55 °C, and 2 min extension at 72 °C, with the addition of 14 s per cycle to the extension step. Amplifications were performed on a BioMetra TGradient thermocycler (Biometra, Göttingen, Germany). The PCR reaction contained 1.5 mM MgCl2, 10 mM KCl, 10 mM Tris pH 8.4, 200 μM dNTP, 2.4 μM DOP primer and 2.5 U low-DNA Taq (PerkinElmer, Wellesley, MA).
2.4. Adapter ligation and sequence-independent single primer amplification (SISPA)
Adapter ligation and sequence-independent single primer amplification was carried out as described previously (Allander et al., 2001). Briefly, restriction endonuclease Csp6.1 was used to digest viral genomes (HSV and SV40, quantified by spectrophotometry after gradient purification) in a 10-fold dilution series. The DNA fragments were purified using MiniElute columns (Qiagen, Valencia, CA) after restriction digestion, and ligated to the adapter NCsp (hybridized oligonucleotides NBam24, AGG CAA CTG TGC TAT CCG AGG GAG; and NCsp11, TAC TCC CTC GG). The ligation product was used as a template for PCR, using the oligonucleotide NBam24 as a primer for 40 cycles of amplification. The resulting viral PCR amplicons were subjected to electrophoresis, gel-purified and then cloned, and 2–3 clones from each band were sequenced to confirm their origin.
2.5. Cloning and identification of PCR-amplified products
PCR amplifications were resolved by electrophoresis on a 1.2% agarose gel, excised from the gel, recovered using the Qiagen MinElute gel extraction kit and ligated to the TOPO-TA cloning vector (Invitrogen, Carlsbad, CA). The ligated products were then transformed into TOP-10 chemically competent cells (Invitrogen, Carlsbad, CA). DNA extracted from bacterial clones containing the DOP-PCR-amplified products was analyzed by restriction endonuclease digestion, and plasmids containing inserts were sequenced using T7 or T3 primer. Sequences were compared with those submitted previously to GenBank using the TFASTX program (GCG, Madison, Wisconsin).
2.6. Real-time PCR
Real-time PCR for SV40 was performed in order to evaluate the efficiency of the capsid preparation and further confirm the sensitivity of the DOP-PCR (Pal et al., 2006). The primers were: 5′-CAG GGA AGG GTT GCA AAT ATC A and 5′-GCA CAT TAG GAC TTT GGC TTT GA, and the probe was: 5′-FAM-TGT CTG CCT CAT CAA TAT TAT CAT AGG TGT GCC CA-TAMRA. ABI Universal Mastermix was used according to the manufacturer's instructions, with cycling conditions of 50 °C × 2 min, 95 °C × 10 min, followed by 40 cycles of 95 °C × 15 s and 60 °C × 1 min.
3. Results
A degenerate oligonucleotide primer PCR (DOP-PCR) method, developed originally for non-specific amplification of large genomes, was adapted to amplify viral nucleic acids non-specifically. As originally described, this method used a degenerate primer mixture (5′-CCGACTCGAGINNNNNNATGTGG-3′) that contained a six nucleotide 3′-anchor sequence (ATGTGG) (Telenius et al., 1992) (Table 1). Because the primer mixture includes a 6-nucleotide degenerate sequence (NNNNNN) adjacent to the 3′-anchor sequence, the mixture includes 4096 (46) different primers, and there is thus a small quantity of an individual primer with a 3′ end that matches at least 12 consecutive nucleotides (the 6 nucleotide degenerate sequence plus the 6 nucleotide 3′-anchor sequence) at template locations complementary to the anchor sequence. At low stringency during the first few DOP-PCR cycles, at least 12 consecutive nucleotides from the 3′ end of the primer anneal to DNA sequences on the PCR template. In subsequent cycles at higher stringency, these initial PCR products are amplified efficiently further using the same primer population.
Efficient amplification of viral genomes is generally not feasible when a 3′-anchor sequence of 6 nucleotides is used, because such a sequence appears only (on average) once every 4096 nucleotides, leading to a sensitivity of only 106 copies of the SV40 genome (Table 1, primer #1). Thus, to adapt this method for amplification of viral genomes, primers with shorter 3′-anchor sequences were designed and tested (Table 1). Primer qualifying experiments were carried out using a 10-fold dilution series of the 5.2 kb SV40 genome, in order to compare the sensitivity of different primer populations. These experiments focused on variations of 3′-anchor sequences TGTGG (primer #s 2–12) and TTCTG (primer #s 14–17), to increase the number of locations at which an anchor sequence is likely to bind to viral nucleic acid. Inosines were added at different locations of the degenerate portion of the primers in an effort to further improve binding at the 3′ end of the primer (primer #s 2–17). In experiments using a 3′-anchor sequence of four nucleotides (primer #s 3–6, 8, 9, 11, 14–17), sensitivity to detect DNA templates was reduced, probably because the primer length that matched the DNA template at its 3′ end was too short (even when combined with the 6-nucleotide degenerate sequence or with additional inosines). Sequencing of DOP-PCR products and comparison with original template sequences revealed that this method typically amplifies genome fragments that match the 3′-anchor sequence on opposite strands at both ends (with occasional mismatches), and that nucleotide matches upstream of the degenerate sequence are not required for efficient amplification (with only random [a median of 2 matches in the original viral sequence out of ten nucleotides] typically observed in this region, regardless of template). Use of different 10-nucleotide sequences did not influence sensitivity (primer #s 13 and 14), further supporting the conclusion that the 5′-sequence does not play a role in DOP-PCR primer sensitivity. The best sensitivity was obtained with a 5-nucleotide 3′-anchor sequence, in combination with one additional inosine inserted upstream of the degenerate sequence (primer #2). Consequently, an optimized primer population with these features was used in the virus detection experiments described in this manuscript.
Fig. 1 shows a comparison of the ability of the optimized DOP-PCR primer (5′-CCGACTCGAGINNNNNNTGTGG-3′) and that of adapter ligation PCR (Allander et al., 2001) to amplify a 10-fold dilution series (in water) of full-length 152 kb HSV-2 and 5.2 kb SV40 genomic DNA. Quantities of viral DNA ranging from 109 (for HSV-2) or 1010 (for SV40) copies to 102 copies (genome copy numbers were based on spectrophotometric readings of viral nucleic acids extracted directly from gradient-purified viral capsids) were subjected to amplification, and the DOP-PCR detected 100 copies of SV40 DNA and 1000 copies of HSV-2 DNA in these reactions (Fig. 1, top panels). Adapter ligation PCR (Fig. 1, lower panels) detected 109 copies (but not lower quantities) of both viral DNAs, implying a substantial advantage to amplification of viral genomes by DOP-PCR. PCR-amplified bands from both DOP-PCR and adapter ligation PCR were confirmed to be from the original viruses by gel-purification, cloning and sequencing. Analysis of sequences obtained from SISPA of 109 genome equivalents of SV40 revealed that approximately 55% of the genome was detected. Sequences ranged from 300 to 800 bp; sequences from theoretical SISPA amplicons outside of this size range were not identified in this experiment, accounting for the approximately 45% of the genome that was not obtained.
Fig. 1.
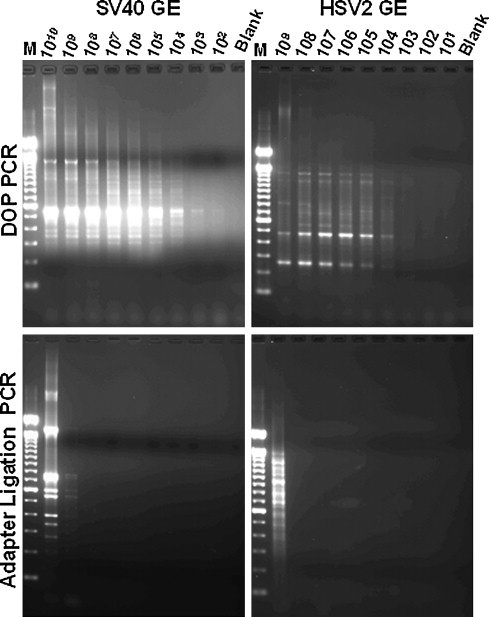
Sensitivity of viral genome detection. (A) Detection of SV40 and HSV-2 genome, using the optimized DOP-PCR primer. 10-fold serial dilutions of SV40 or HSV-2 genomic DNA were subjected to DOP-PCR amplification. Lanes: M: 100 bp ladder, numerical lane headings represent the number of genome equivalents (GE) of SV40 or HSV-2, respectively, subjected either to DOP-PCR using our optimized primer or to adapter ligation PCR. Blank: water control.
To investigate the efficiency of the capsid preparation, SV40 virus was spiked into uninfected Vero cells in a 10-fold dilution series, and subjected to capsid purification as described. Materials prior to and after capsid purification were analyzed in parallel by DOP-PCR and by real-time PCR (Fig. 2 ). The real-time PCR studies revealed that the original samples contained two to three logs more DNA copies than PFUs, and that the capsid preparation yielded a viral DNA copy number about one to one and a half logs higher than the number of PFU spiked into the Vero cells. These results imply a high efficiency of capsid preparation and nucleic acid extraction. Because a PFU in culture often represents an aggregation of viruses, it was not surprising to detect more DNA copies than PFU. Consistent with the results shown in Fig. 1, the DOP-PCR detected SV40 in samples spiked with 100 PFU of virus, which by real-time PCR corresponded to approximately 5 × 103 virus genomes after capsid purification. DOP-PCR on pre-capsid preparation material detected 105 PFU (approximately 107 copies) of SV40, confirming the value for this assay of using the capsid preparation.
Fig. 2.
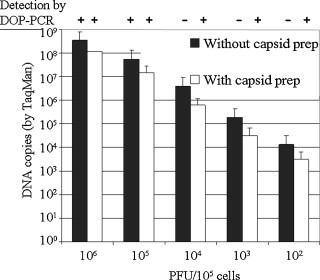
Detection of SV40 viral genome after capsid preparation using real-time PCR and DOP-PCR. Ten-fold serial dilutions of virus stock (measured in PFU) were mixed with 106 Vero cells and examined both prior to and after capsid preparation. The SV40 viral nucleic acid was extracted from the viral capsids and 1/10 of the DNA was used as template for amplified using real-time and DOP-PCR in parallel. Detected quantities of viral nucleic acid are shown for each original sample. DOP-PCR results (+ or −) are shown at the top of the graph.
The ability of the optimized primer to detect other viruses was examined in capsid preparations from biological samples (Fig. 3 ). For samples suspected to contain RNA viruses, a random-hexamer primed reverse transcription step was employed prior to DOP-PCR. Discrete amplification products were obtained using influenza type A virus (at a hemaglutination titer of 1:1, grown in allantoic fluid, lane 1), poliovirus type 2 (at 105 PFU, grown in African Green Monkey Kidney cells, lane 3), varicella-zoster virus grown in MRC-5 cells (100 PFU spiked into 106 Vero cells, lane 5), and SV40 grown in Vero cells (1000 PFU spiked into 106 Vero cells). When the reverse transcriptase step was omitted, no amplification was observed in the RNA virus samples, suggesting that the removal of cellular DNA was fairly complete (lanes 2 and 4). All discrete bands were gel-purified from lanes 1, 3, 5 and 6, cloned, sequenced and compared with sequences in GenBank. For each virus, sequences obtained from each band matched with that of the original virus, and the original virus sequence revealed the presence of the anchor sequence (TGTGG) on both strands flanking the obtained sequences. In addition, experimental results showed the same banding pattern each time a virus was studied.
Fig. 3.
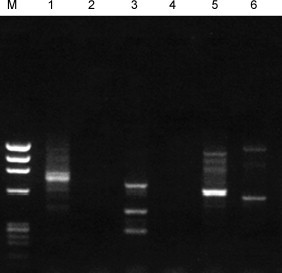
Non-specific DOP-PCR amplification of viruses. Samples in lanes 1–6 were obtained by amplification using the DOP-PCR-based method.
M: ΦX/HaeIII size markers, Lane 1: Influenza virus A (1:1 by HA), with RT step Lane 2: Influenza virus A (1:1 by HA), no RT step; Lane 3: Poliovirus (Sabin strain type 2), with RT step; Lane 4: Poliovirus (Sabin strain type 3), no RT step; Lane 5: VZV (100 PFU spiked into 106 Vero cells); Lane 6: SV40 (100–1000 PFU spiked into 106 Vero cells).
Table 2 summarizes experiments using this method to detect the DNA viruses VZV, SV40, and AAV, and the RNA viruses polio virus and influenza virus. For VZV, SV40, and AAV, the capsid purification approach, combined with the non-specific PCR technique, provided sensitivity in the range of 100–1000 infectious units in experiments where 10-fold virus dilutions were spiked into 106 Vero cells.
Table 2.
Sensitivity of non-specific detection method (using nuclease digestion, ultracentrifugation, and modified primer in DOP-PCR) to detect viruses in 106 Vero cell background
| Virus | Spiked into | Sensitivity |
|---|---|---|
| VZV | 106 cells | 100 PFU |
| SV40 | 106 cells | 1000 PFU |
| AAV | 106 cells | 1000 PFU |
| Polio | Vaccine | ∼105 IU |
| Influenza | 106 cells | 1:1 by HA |
Several cell lines known to harbor persistent viral infections were examined subsequently. Jiyoye cells, derived from a patient with African Burkitt's lymphoma (Kohn et al., 1967), contain latent EBV genomes, but have not been reported to express virus. 1A2 cells contain latent EBV genomes and produce virus during normal passage in cell culture (Croce et al., 1979). Mo T cells and MJ cells produce constitutively HTLV-2 and HTLV-1, respectively (Popovic et al., 1983, Saxon et al., 1978). One million cells from each line were tested for the presence of viruses using this method (Fig. 4 ). No specific bands were obtained on electrophoresis using material from Jiyoye cells, consistent with the presence only of latent virus in those cells. However, EBV sequences were found in 5/5 clones from the bands associated with 1A2 cells, and HTLV-1 and HTLV-2 sequences in 1/3 and 4/4 clones obtained from the bands (unique to the RT-treated samples) associated with MJ and Mo T cells, respectively.
Fig. 4.
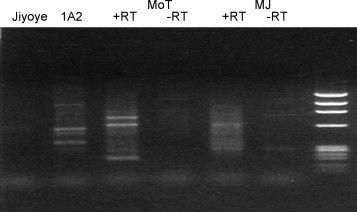
Detection of viruses in cells that constitutively produce virus. Jiyoye (EBV genome-containing), 1A2 (EBV-producing), Mo T (HTLV-2 producing) and MJ (HTLV-1 producing) cells were subjected to virus detection. The results obtained after electrophoresis of DOP-PCR products from each cell type (with or without RT) are shown in the lane corresponding to each cell type. Bands were excised from the gel, cloned, and sequenced, and in each case, the experiment revealed the starting virus. The right-most lane contains ΦX/HaeIII size markers. The experiments were repeated three times with identical results.
Many clinical samples might contain a small proportion of infected cells, against a background of infected cells. Thus, it was of interest to study further the sensitivity to detect infectious viruses growing in cell culture in controlled samples, in this context. Detection limits for viruses in cells infected acutely or persistently after dilution into uninfected Vero cells were examined (Table 3 ). In these experiments, quantities ranging from 101 to 106 infected cells were added to uninfected Vero cells to a total of 106 cells. The method was able to detect HSV-1 or Sabin strain (type 2) poliovirus from 1000 (or more) infected Vero cells against a background of 106 total cells. EBV was successfully detected in samples containing 1000 or more 1A2 cells against a background of 106 uninfected Vero cells. These data indicate that DOP-PCR can detect HSV, EBV, or poliovirus when approximately one cell per thousand is infected. The sensitivity for detection of infectious HTLV-2 appeared to be lower, but the method detected HTLV-2 sequences in 105 Mo T cells against a background of 106 total cells.
Table 3.
Sensitivity of viral nucleic acid detection in infected cells (when infected cells were diluted with uninfected Vero cells to a total of 106 cells per sample)
| Sample | Minimum number of infected cells detected when diluted into Vero cells (total sample: 106 cells) |
|---|---|
| HSV-1 infected Vero cells | 1000 |
| Poliovirus type 2 infected Vero cells | 1000 |
| 1A2 cells | 1000 |
| Mo T cells | 105 |
4. Discussion
Viruses were detected non-specifically in the presence of substantial amounts of contaminating host DNA by combining physical separation of viral nucleic acids from cellular nucleic acids with the non-specific amplification of the viral nucleic acids. This non-specific approach to amplifying viral nucleic acids was several logs more sensitive than a non-specific amplification method described previously, implying a significant advance. A variety of DNA and RNA viruses were detected non-specifically in test samples designed to mimic detection problems that involve virus particles against a background of cellular nucleic acids.
Previous studies reported viral detection methods based upon nuclease digestion of biological samples. The main difference between those and the approach presented in the current study lies in the amplification schemes. In one of the previous studies (in which a new bovine parvovirus was detected in serum), PCR primers were ligated to purified viral nucleic acids after digestion with a restriction endonuclease (SISPA) (Allander et al., 2001). The same scheme was also used to identify a new human parvovirus and to isolate new strains of human TTV (Jones et al., 2005). This approach is most suitable for detection of DNA viruses because digestion with a restriction endonuclease of cDNAs reverse-transcribed from RNA viruses would require second-strand synthesis from cDNA, which is known to be an inefficient process.
Purification of viral nucleic acids from viral capsids yields advantages and disadvantages. Because non-encapsidated viral nucleic acid is removed in the process of purification and thus not amplified, some potential sensitivity is lost. The ratio between infectious virions and total virus nucleic acid may vary for different viruses and is determined not only by viral but also by host factors. In samples with low PFU to nucleic acid ratios, capsid purification could substantially reduce the ability to detect viral nucleic acids. However, in the SV40 experiment (Fig. 2), capsid purification yielded a 3-log improvement in DOP-PCR assay sensitivity, while reducing total viral DNA by approximately one log. Although it seems likely that the purification techniques described above would be applicable to other types of samples, caution should be used in extrapolating these results to samples that may have higher PFU to nucleic acid ratios, or that may be more complex and thus interfere either with the capsid purification or the DOP-PCR. Additional validation of the capsid purification may thus be necessary to extend this method to other sample types.
A significant advantage of the capsid purification is that any viral nucleic acid amplified from capsids is more likely to be derived from infectious virus, and is therefore potentially more relevant in a clinical setting. Another advantage is the lack of need for a matched negative control (to perform molecular subtractions). A third advantage is the ability to handle very large samples; the elimination of cellular nucleic acids permits the concentration of viral nucleic acids to levels that would not be possible if cellular nucleic acids were retained. This advantage mitigates the somewhat reduced sensitivity of DOP-PCR relative to traditional PCRs, and also can compensate for the potential loss of non-encapsidated viral nucleic acid during the capsid preparation.
The non-specific PCR was based on primers that contain a short 3′-anchor sequence. PCR-assays using such primers were found to be sensitive (able to detect as few as 100 copies of viral nucleic acid for large virus genomes, and 100–1000 copies of viral nucleic acid for small genomes). The specific 3′-anchor sequence of the primer population led to a reproducible amplification pattern for each virus. Thus, the pattern of bands observed in DOP-PCR may provide some preliminary clues to the virus sequences present in a sample. When contaminating cellular nucleic acids are incompletely degraded (while the virus nucleic acids are still protected by the capsid), we hypothesize that the use of a 3′-anchor sequence may minimize the random amplification of very small cellular sequences (which are seldom long enough to contain two copies of the 3′-anchor sequence on opposite strands), helping to explain the absence of cellular nucleic acid amplification in the assay.
When performing these non-specific PCR amplifications, it is necessary to take extensive precautions to avoid PCR contamination. While specific PCRs result in false positives only if contaminating sequences that share significant homology with the used primers are present, false positive DOP-PCR products could stem from every conceivable form of nucleic acid contamination. Such contamination could not only cause delays in detection and more sequencing but could also camouflage virus sequences. In some early experiments, spurious sequences were detected due to reagent contamination with small amounts of bacterial, fungal, or algal nucleic acids, sometimes caused by residual contaminants in polymerases. However, the use of low-DNA Taq polymerase, and careful attention to the quality of other reagents minimized these problems.
The sensitivity of DOP-PCR was compared with adapter ligation followed by single-primer PCR (SISPA), and an approximately six log better sensitivity of viral genome amplification was observed for the DOP-PCR. Although the study of adapter ligation PCR did not match the previously published sensitivity of 106 copies (Allander et al., 2001) on multiple attempts, the current approach was still 3–4 logs more sensitive than the published sensitivity for SISPA. The inability to match the published figure for SISPA may reflect variability in this technique, which depends on the efficiency of additional enzymatic steps (restriction endonuclease digestion, ligation) that are difficult to standardize.
This method provides partial sequence information of viral genomes present in biological samples. These sequences can then be compared with sequences reported previously in databases such as GenBank. Although initial determination of a given sequence to be of viral origin requires the presence of reported sequences previously from related viruses in the database, even unrecognized sequences may be used to design follow-up studies of more specific PCR assays to study these sequences further, and possibly identify yet unknown viruses. The DOP-PCR could thus prove to be a useful tool for virus discovery, detection of unsuspected, genetically modified and identification of emerging viruses.
5. Conclusion
The method described in this report can be used to identify viruses either directly from biological specimens or from cell culture in cases when the virus is cultivable. This method provides an opportunity to identify viruses in samples from which detection by other methods has failed. In its current form, the method seems most likely to be applicable to samples like serum or body fluids. Although implementation of the DOP-PCR for tissue or environmental samples will require additional work and validation, this assay is able to detect viruses of very different natures, including enveloped and non-enveloped viruses, viruses with DNA or RNA genomes, with small or large genomes, and even a segmented genome, without any specific prior knowledge regarding the sequence of the genome. The non-specific amplification method described here could also be paired with other virus nucleic acid purification techniques. There is potential utility of this method for virus detection and discovery, particularly in cases where other methods are unsuccessful in identifying viruses.
Acknowledgements
The authors acknowledge the National Vaccine Program Office for its support of this work. Portions of this work were also supported by the National Institutes of Allergy and Infectious Diseases, via a grant to the Mid Atlantic Regional Center for Excellence in Biodefense and Emerging Infectious Diseases, and by the National Institutes of Allergy and Infectious Diseases, via an interagency agreement with FDA/CBER. Helpful comments are highly acknowledged from Dr. Jeffrey Cohen of the National Institutes of Allergy and Infectious Diseases and from Dr. Shuang Tang of FDA/CBER.
References
- Allander T., Emerson S.U., Engle R.E., Purcell R.H., Bukh J. A virus discovery method incorporating DNase treatment and its application to the identification of two bovine parvovirus species. Proc. Natl. Acad. Sci. U.S.A. 2001;98:11609–11614. doi: 10.1073/pnas.211424698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briese T., Jia X.Y., Huang C., Grady L.J., Lipkin W.I. Identification of a Kunjin/West Nile-like flavivirus in brains of patients with New York encephalitis. Lancet. 1999;354:1261–1262. doi: 10.1016/s0140-6736(99)04576-6. [DOI] [PubMed] [Google Scholar]
- Chang Y., Cesarman E., Pessin M.S., Lee F., Culpepper J., Knowles D.M., Moore P.S. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science. 1994;266:1865–1869. doi: 10.1126/science.7997879. [DOI] [PubMed] [Google Scholar]
- Corne J.M., Green S., Sanderson G., Caul E.O., Johnston S.L. A multiplex RT-PCR for the detection of parainfluenza viruses 1-3 in clinical samples. J Virol Methods. 1999;82:9–18. doi: 10.1016/s0166-0934(99)00073-7. [DOI] [PubMed] [Google Scholar]
- Croce C.M., Shander M., Martinis J., Cicurel L., D’Ancona G.G., Dolby T.W., Koprowski H. Chromosomal location of the genes for human immunoglobulin heavy chains. Proc. Natl. Acad. Sci. U.S.A. 1979;76:3416–3419. doi: 10.1073/pnas.76.7.3416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denniston K.J., Madden M.J., Enquist L.W., Vande Woude G. Characterization of coliphage lambda hybrids carrying DNA fragments from Herpes simplex virus type 1 defective interfering particles. Gene. 1981;15:365–378. doi: 10.1016/0378-1119(81)90180-3. [DOI] [PubMed] [Google Scholar]
- Jones M.S., Kapoor A., Lukashov V.V., Simmonds P., Hecht F., Delwart E. New DNA viruses identified in patients with acute viral infection syndrome. J. Virol. 2005;79:8230–8236. doi: 10.1128/JVI.79.13.8230-8236.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohn G., Mellman W.J., Moorhead P.S., Loftus J., Henle G. Involvement of C group chromosomes in five Burkitt lymphoma cell lines. J. Natl. Cancer Inst. 1967;38:209–222. [PubMed] [Google Scholar]
- Linnen J., Wages J., Jr., Zhang-Keck Z.Y., Fry K.E., Krawczynski K.Z., Alter H., Koonin E., Gallagher M., Alter M., Hadziyannis S., Karayiannis P., Fung K., Nakatsuji Y., Shih J.W., Young L., Piatak M., Jr., Hoover C., Fernandez J., Chen S., Zou J.C., Morris T., Hyams K.C., Ismay S., Lifson J.D., Hess G., Foung S.K., Thomas H., Bradley D., Margolis H., Kim J.P. Molecular cloning and disease association of hepatitis G virus: a transfusion-transmissible agent. Science. 1996;271:505–508. doi: 10.1126/science.271.5248.505. [DOI] [PubMed] [Google Scholar]
- Lisitsyn N., Lisitsyn N., Wigler M. Cloning the differences between two complex genomes. Science. 1993;259:946–951. doi: 10.1126/science.8438152. [DOI] [PubMed] [Google Scholar]
- Pal A., Sirota L., Maudru T., Peden K., Lewis A.M., Jr. Real-time, quantitative PCR assays for the detection of virus-specific DNA in samples with mixed populations of polyomaviruses. J. Virol. Methods. 2006;135:32–42. doi: 10.1016/j.jviromet.2006.01.018. [DOI] [PubMed] [Google Scholar]
- Popovic M., Sarin P.S., Robert-Gurroff M., Kalyanaraman V.S., Mann D., Minowada J., Gallo R.C. Isolation and transmission of human retrovirus (human t-cell leukemia virus) Science. 1983;219:856–859. doi: 10.1126/science.6600519. [DOI] [PubMed] [Google Scholar]
- Sampath R., Hofstadler S.A., Blyn L.B., Eshoo M.W., Hall T.A., Massire C., Levene H.M., Hannis J.C., Harrell P.M., Neuman B., Buchmeier M.J., Jiang Y., Ranken R., Drader J.J., Samant V., Griffey R.H., McNeil J.A., Crooke S.T., Ecker D.J. Rapid identification of emerging pathogens: coronavirus. Emerg. Infect. Dis. 2005;11:373–379. doi: 10.3201/eid1103.040629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxon A., Stevens R.H., Quan S.G., Golde D.W. Immunologic characterization of hairy cell leukemias in continuous culture. J. Immunol. 1978;120:777–782. [PubMed] [Google Scholar]
- Telenius H., Carter N.P., Bebb C.E., Nordenskjold M., Ponder B.A., Tunnacliffe A. Degenerate oligonucleotide-primed PCR: general amplification of target DNA by a single degenerate primer. Genomics. 1992;13:718–725. doi: 10.1016/0888-7543(92)90147-k. [DOI] [PubMed] [Google Scholar]
- van den Hoogen B.G., de Jong J.C., Groen J., Kuiken T., de Groot R., Fouchier R.A., Osterhaus A.D. A newly discovered human pneumovirus isolated from young children with respiratory tract disease. Nat. Med. 2001;7:719–724. doi: 10.1038/89098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanDevanter D.R., Warrener P., Bennett L., Schultz E.R., Coulter S., Garber R.L., Rose T.M. Detection and analysis of diverse herpesviral species by consensus primer PCR. J. Clin. Microbiol. 1996;34:1666–1671. doi: 10.1128/jcm.34.7.1666-1671.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]