

Highlights
-
•
Epitope-based vaccine design facilitates laboratory research and save valuable time and money.
-
•
Immunoinformatics tools were used to predict B and T-cell epitopes within the E2 protein.
-
•
Highest population coverage was recorded 84.94% in Papua New Guinea.
-
•
Docking study revealed that E2 binds with lowest possible binding energy.
-
•
Pharmacophore study suggested MAN as the most effective inhibitor of the CHIKV E2.
Abbreviations: CHIKV, Chikungunya virus; HLAs, human leukocyte antigens; BFV, Barmah Forest Virus; RRV, Ross River Viruses; ONNV, o’nyong-nyong; SFV, Semliki Forest viruses; SINV, Sindbis; ORFs, open reading frames; UniPortKB, UniProt Knowledge Base; CTL, cytotoxic T-lymphocyte; IC50, half-maximal inhibitory concentration; MHC, major histocompatibility complex; IEDB, Immune Epitope Database; TAP, transport associated proteins; SOPMA, self-optimized prediction method with alignment; GRAVY, grand average hydropathy; FAO, Food and Agriculture Organization; WHO, World Health Organization; RCSB, Research Collaboratory for Structural Bioinformatics; 3D, three dimensional; CASTp, Computed Atlas of Surface Topography of proteins; NAG, N-Acetyl-d-Glucosamine; MAN, Alpha-d-Mannose; MSE, Selenomethionine; NDG, 2-(Acetylamino)-2-Deoxy-A-d-Glucopyranose; ACToR, Aggregated Computational Toxicology Resource; admetSAR, absorption, distribution, metabolism, excretion, and toxicity Structure–Activity Relationship database
Keywords: Chikungunya virus, Vaccine, Inhibitor, HLA, Pharmacophore study
Abstract
Recent concerning facts of Chikungunya virus (CHIKV); a Togaviridae family alphavirus has proved this as a worldwide emerging threat which causes Chikungunya fever and devitalizing arthritis. Despite severe outbreaks and lack of antiviral drug, a mere progress has been made regarding to an epitope-based vaccine designed for CHIKV. In this study, we aimed to design an epitope-based vaccine that can trigger a significant immune response as well as to prognosticate inhibitor that can bind with potential drug target sites by using various immunoinformatics and docking simulation tools. Initially, whole proteome of CHIKV was retrieved from database and perused to identify the most immunogenic protein. Structural properties of the selected protein were analyzed. The capacity to induce both humoral and cell-mediated immunity by T cell and B cell were checked for the selected protein. The peptide region spanning 9 amino acids from 397 to 405 and the sequence YYYELYPTM were found as the most potential B cell and T cell epitopes respectively. This peptide could interact with as many as 19 HLAs and showed high population coverage ranging from 69.50% to 84.94%. By using in silico docking techniques the epitope was further assessed for binding against HLA molecules to verify the binding cleft interaction. In addition with this, the allergenicity of the epitopes was also evaluated. In the post therapeutic strategy, three dimensional structure was predicted along with validation and verification that resulted in molecular docking study to identify the potential drug binding sites and suitable therapeutic inhibitor against targeted protein. Finally, pharmacophore study was also performed in quest of seeing potent drug activity. However, this computational epitope-based peptide vaccine designing and target site prediction against CHIKV opens up a new horizon which may be the prospective way in Chikungunya virus research; the results require validation by in vitro and in vivo experiments.
1. Introduction
The word Chikungunya means something ‘that bends up’ which in turn refers to the distorted posture of a patient due to severe joint pain caused by Chikungunya fever (Thiboutot et al., 2010). Chikungunya fever (CHIKV) and related arthralgic symptoms are the results of Chikungunya virus infection; an arboreal alphavirus of the Togaviridae family of viruses. CHIKV is a spherical shape enveloped virus with 60–80 nm diameter and contains a positive sense single stranded linear RNA of approximately 11.8 kb (Powers and Logue, 2007).
There are about 30 species in the genus alphavirus, all are arthropod-borne viruses and they are distinguished in 7 antigenic complexes (Caglioti et al., 2013). These viruses cause encephalitis and febrile arthralgia in vertebrates including CHIKV and several other alphaviruses that are known to cause disease in humans. Mosquitoes of Aedes sp. are the primary vectors of CHIKV predominantly Aedesaegypti (Caglioti et al., 2013, Lahariya and Pradhan, 2006). But recent cases has been found to be caused by A. albopictus, much to the worry of the people due to widespread geographical distribution of A. albopictus, which was previously thought to be the secondary vector (Nougairede et al., 2013, Knudsen, 1995).
The onset of the Chikungunya virus infection is acute and clinical features generally vary (Vanlandingham et al., 2005, Schuffenecker et al., 2006). Symptoms emerge usually 4–7 days after the mosquito bite in case of humans and patient suffers from fever, headache and severe polyarthralgia followed by rash which last about 5–7 days and the disease is usually self-limiting (Robinson, 1955, Paquet et al., 2006). The arthralgia primarily affects the small joints of hand and legs that lasts a few days in acute cases but in chronic cases joint pain may persist for months and a study reported that over 12% of the patients suffer from prolonged joint symptoms (Brighton, 1984).
CHIKV infection was first recorded in 1952 in Mankonde spoken areas of Tanzania-Mozambique border. But the virus came into wide attention during its huge outbreak in the Indian Ocean Islands (Caglioti et al., 2013, Enesrink, 2006). There have been several CHIKV outbreaks since its recognition in 1952 and its reemergence in 2004 mainly in Africa and the countries in and around Indian Ocean and South-East Asia (Lahariya and Pradhan, 2006, Burt et al., 2012). In 2005 there have been outbreaks in la Reunion Island (Schuffenecker et al., 2006). Recent outbreaks from 2006 to present include Italy in 2007 (Rezza et al., 2007), France 2010 (Grandadam et al., 2011) and Madagascar (Pistonet et al., 2009). The virus reached Americas in 2013 through its outbreak in the Caribbean (Leparc-Goffart et al., 2014).
The viral genome codes for four nonstructural proteins (nsp1–4) and three structural proteins (C, E1, and E2) with two cleavage products E3 and 6k which are laid out in two open reading frames (ORFs). The 5′ ORF codes for non-structural protein precursors and 3′ ORF codes for structural proteins (Grakoui et al., 1989, Rashad et al., 2014).
The surface of CHIKV is composed of 80 trimeric spikes of envelope glycoprotein E1 and E2 heterodimers. Upon entering the acidic environment of endosomes causes dissociation of trimeric spikes of E1–E2 heterodimers and reorganization into E1 homotrimers. These trimers with the help of hydrophobic fusion trimer (fusion loop) enter the host cell and undergo refolding to form a hairpin like structure. This interaction fuses the virus and host cell membrane and the viral nucleocapsid enters the host cell. This phenomenon is mediated by low pH along with cholesterol like all other alphaviruses and virus budding also requires presence of these mediators (Rashad et al., 2014). Along with host cell proteins positive strand replicase produce 26S sub-genomic positive strand RNAs and 49S genomic RNAs (Barton et al., 1991, Shirako and Strauss, 1994). The 26S RNAs encode the structural protein precursors which are cleaved by a serine protease to produce capsid (c), pE2, 6K and E1. The C protein is thought to induce the autocatalytic activity and contain conserved regions known to have the similar activity in other alphaviruses (Grakoui et al., 1989). In addition to that a furin like protease activity cleaves the pE2 into E2 and E3 in the plasma membrane. Before that, pE2 and E1 undergoes post transcriptional modification in the golgi apparatus. CHIKV nucleocapsid has 120 dimers of C protein which after its formation in the cytoplasm, contains the viral RNA, buds out of the infected cell enveloped in host cell's lipid membrane with viral glycoprotein spikes (Rashad et al., 2014, Tang, 2012).
During infection receptor binding is carried out by E2 whereas E1 is responsible for membrane fusion. Glycoprotein E3 facilitates correct folding of E2 and dimerization with E1, also prevents the dimer from premature fusion with membranes (Rashad and Keller, 2013).
The emergence of A. albopictus as the new and prominent vector has been greatly facilitated by a mutation in the E1 envelope protein A226V that enables the CHIKV to utilize A. albopictus for its transmission into vertebrates (Ozden et al., 2008, Kumar et al., 2008). Recent repeated outbreaks in native and new regions most of which are modern and highly developed have generated consternation mainly due to the socio economic effect and the extent of the distressing symptoms of Chikungunya fever. Despite the extremity of the disease there is no specific treatment for the disease till date (Weaver et al., 2012). Recent events involving the disease once again bring up the necessity of a selective anti-viral drug or a vaccine to treat and to protect from future infection of CHIKV. But despite some improvements in the recent years, there is no approved vaccine or drug against CHIKV is available (Lee-Jah et al., 2014).
As of lately, host immune response against CHIKV gained significant concentration. Though Type I IFN and related pathways have been seen to be very important in controlling viral replication, its effects are insufficient to completely eradicate CHIKV from the system. As a result, CHIKV is detected in tissues long after IFN-α/β level returns to normal. These facts along with poor understanding of CHIKV pathogenesis endorse that adaptive immunity has a big role in complete elimination of the virus, although adaptive immunity against CHIKV is still not fully characterized (Couderc et al., 2008, Schilte et al., 2010, Labadie et al., 2010, Her et al., 2010, Werneke et al., 2011, Gardner et al., 2010, Gardner et al., 2012, Couderc et al., 2009). Adaptive immunity works through the recognition of T cell epitopes by T lymphocytes by subsequent presentation of the antigen by HLA molecules. This trigger T cell mediated cytotoxicity and activates humoral immune response. Moreover, B cell receptors (BCR) recognize specific epitopes in their linear form and with the help of helper T cells differentiate into antibody producing plasma cells. Antibodies bind to their specific antigens and triggers phagocytosis or complement pathway. Some plasma cells develop into memory B cells that ensure long lasting immunity against the virus (Janeway et al., 2001).
To completely eliminate the chance of re-infection a proper vaccine that induces both humoral and cell mediated immune response is necessary. This requirement is furthers reinforced by the fact that there has been reports of microevolution (Schuffenecker et al., 2006) in the CHIKV genome that might give the virus ability to evade humoral immunity. T-cell epitope based vaccine design technique can be summarized as the identification of immune dominant epitopes of the virus and synthesizing it to be used as vaccines to produce specific immune response (Atanas and Irini, 2013). Immunoinformatics research emphasizes on mapping immune dominant B-Cell and T-cell epitopes to facilitate laboratory research and reduce valuable time and money necessary for the job.
In the present study we have analyzed the whole proteome of the CHIKV to determine its potential immunogenic regions to predict a candidate for vaccine development along with a genome wide search to find out the most eligible drug target site and simulated inhibition of the target site by a predicted inhibitor molecule using computational methods. The goal of the study was to facilitate future laboratory based endeavors searching for complete treatment and prevention of Chikungunya virus infection.
2. Materials and methods
The flow chart representing the overall procedures of pre and post therapy for CHIKV is illustrated in Fig. 1 .
Fig. 1.

The flow chart representing the overall procedures of pre and post therapy for CHIKV. Notes: UniProtKB: UniProt Knowledgebase; CHIKV: Chikungunya virus; CTL: cytotoxic T-lymphocyte; IC50: half-maximal inhibitory concentration; MHC: major histocompatibility complex; IEDB: Immune Epitope Database; TAP: transporter of antigen presentation; 3D: three dimensional; HLA: human leukocyte antigen; HLA-B: the-major histocompatibility complex, class I, B, CASTp: Computer Atlas of Surface Topology of Proteins; OSIRIS property explorer: organic chemistry portal; ACToR: Aggregated Computational Toxicology Resource; admetSAR: a comprehensive source and free tool for assessment of chemical ADMET properties.
2.1. Sequence retrieval
The amino acid sequences of the complete proteome of Chikungunya virus except the variable strains were retrieved from UniProt Knowledge Base (UniProtKB) database in FASTA format. UniProtKB is a comprehensive source for protein sequence and annotation information as well as with accurate, consistent and rich annotation it works as the center for the collection of functional information on proteins. Different non-structural proteins (NSP) were excluded from this selection. A total available 1100 envelope (E) proteins, 204 structural (S) polyproteins, 3 capsid (C) proteins and only single coat and k protein respectively were categorized. Then the sequences were analyzed to study antigenicity, solvent accessible regions, surface accessibility, flexibility and MHC class I binding sites (The UniProt Consortium, 2014, Apweiler et al., 2004).
2.2. Highest antigenic protein uncovering
To isolate the highest antigenic protein, proteins were then submitted in the VaxiJen v2.0 Server which was used for the prediction of potent antigens and subunit vaccines with defaults parameter (Doytchinova and Flower, 2007). Plain sequence format was submitted, virus was selected as target organism. All the antigenic proteins with their respective score were then filtered in Excel. A single antigenic protein with highest antigenicity scores was selected for further evaluation.
2.3. Primary and secondary structure analysis
ProtParam tool (Colovos and Yeates, 1993a) and self-optimized prediction method with alignment (SOPMA) (Geourjon and Deleage, 1995a) of Expasy server were used to analyze different physiological and chemical properties of the selected protein. Theoretical isoelectric point (pI), molecular weight, amino acid composition, grand average hydropathicity (GRAVY), estimated half-life, extinction coefficient (Gill and Von, 1989), instability index (Guruprasad et al., 1990), aliphatic index (Ikai, 1980) of the protein were calculated using the preset parameters through protparam. Properties like solvent accessibility, transmembrane helices, globular regions, bend region, random coil and coiled-coil region were predicted by SOPMA.
3. Pre-therapy (vaccine development) against CHIKV
3.1. T cell epitope identification
Consistent predictions of CTL epitopes are very important for coherent vaccine design. The most important thing is that they can reduce the wet lab experimental effort needed to identify epitopes. For this reason, NetCTL-1.2 (Larsen et al., 2007), a web-based server designed for predicting human CTL epitopes in any given protein was used. The overall score was calculated by integrating predictions of proteasomal cleavage, TAP transport efficiency, and MHC class I affinity. The threshold value for epitope identification was set at 0.5 for our present study which have sensitivity and specificity of 0.89 and 0.94, respectively. On the basis of overall score, 5 epitopes were selected for further dry lab experimentation.
For MHC-1 binding prediction (Buus et al., 2003), the Stabilized Matrix Base Method (SMM) (Peters and Sette, 2005) was used to calculate IC50 values of peptide binding to MHC-1 molecules. For both frequent and non-frequent allele, peptide length was set to 9 amino acids earlier to the prediction. The alleles having binding affinity IC50 less than 200 nm were selected for further consideration.
Another IEDB analysis resource tool was also implemented to predict proteasomal cleavage score, TAP score, processing score, and MHC-1 binding score using SMM for each and every selected peptide (Tenzer et al., 2005).
3.2. Epitope conservancy prediction
Epitope conservancy prediction for individual epitopes was calculated using the IEDB analysis resource. Conservancy can be defined as the portion of protein sequences that restrain the epitope measured at or exceeding a specific level of identity. The epitope conservancy calculation tool was executed as a Java web-application (Bui et al., 2007).
3.3. Population coverage calculation
Population coverage for each epitope was calculated by the IEDB population coverage tool. Population coverage was predicted through an online tool based on MHC binding and/or T cell restriction data. This tool is aimed in order to determine the fraction of individuals predicted to respond to a given set of epitopes with known MHC restrictions. For every single population coverage, the tool computed the following information: (1) predicted population coverage, (2) HLA combinations recognized by the population, and (3) HLA combinations recognized by 90% of the population (PC90). All epitope and their MHC-I molecules were inserted and population coverage area selected before submission (Bui et al., 2007).
3.4. Allergenicity appraisal
The online server AllerHunter was used to predict the allergenicity of the proposed epitope for vaccine development. This server predicts allergenicity through a combinational prediction, by using both incorporation of the Food and Agriculture Organization (FAO)/World Health Organization (WHO) allergenicity assessment proposal and support vector machines (SVM)-pairwise sequence similarity. AllerHunter predicts allergen in addition to non-allergens with high specificity which makes AllerHunter is a very constructive program for allergen cross-reactivity prediction (Liao and Noble, 2003, Muh et al., 2009).
3.5. Design of the three-dimensional (3D) epitope structure
To perform docking simulation study, the top conserve YYYELYPTM epitope was inserted to PEP-FOLD (Thevenet et al., 2012) server, which is a de novo approach designed to predict peptide structures from amino acid sequences. It is worked on the basis of structural alphabet (SA) letters to explain the structural conformations of 4 successive residues, couples the predicted series of SA letters to a greedy algorithm a well as a coarse-grained force field (Maupetit et al., 2009, Maupetit et al., 2010). As an output, this server is modeled 5 proposed 3D structures. Consequently, the best model was selected to analyze the interactions with HLAs.
3.6. Docking simulation study
A docking study was performed in order to make sure the binding between HLA molecules and our finding epitope by using AutoDock Vina (Trott and Olson, 2010). For this kind of purpose, a crystal structure of the HLA-B*3501 molecule named 3 LKN was retrieved from the Research Collaboratory for Structural Bioinformatics (RCSB) protein database (Berman et al., 2000). Before conducting the docking study, the NP418 epitope of influenza, which was complexed in the binding groove of HLA-B*3501 (Gras et al., 2010), was removed by using Discovery studio (Van Joolingen et al., 2005). After the separation of the NP418 epitope of influenza, docking study between predicted epitope and prepared HLA-B*3501 was performed. Then another docking between influenza NP418 epitope and prepared HLA-B*3501 was done in order to compare with the former one.
3.7. Prediction of the B-cell epitope
Linear B-cell epitopes were predicted from the given highest immunogenic protein sequence through the B-cell epitope prediction tools of IEDB. The most significant properties for predicting B-cell epitopes are flexibility, antigenicity, surface accessibility, hydrophilicity, and linear epitope predictions (Fieser et al., 1987). We analyzed the flexibility, antigenicity, surface accessibility, hydrophilicity, and linear epitope predictions of our selected highest antigenic protein by using the Karplus and Schulz flexibility prediction (Karplus and Schulz, 1985), Kolaskar and Tongaonkar antigenicity scale (Kolaskar and Tongaonkar, 1990), Emini surface accessibility prediction (Emini et al., 1985), Parker hydrophilicity prediction (Parker et al., 1986), and Bepipred linear epitope prediction algorithms (Andersen et al., 2006), respectively of IEDB analysis resource. Several wet lab experiments revealed that the antigenic portion was situated in beta turn regions of a protein (Rini et al., 1992) for that region the Chou and Fasman beta turn (Chou and Fasman, 1978) prediction tool was used.
4. Post-therapy (drug design) against CHIKV
4.1. Homology modeling and refinement
To predict the three-dimensional (3D) structure of the selected protein Phyre2 (Protein Homology/Analogy Recognition Engine) (Kelley and Sternberg, 2009) was used. FASTA format data was inputted and intensive modeling mode was selected to generate protein model. Homology based modeling contain major local distortions which includes steric clashes, irregular H-hydrogen bonding networks and unphysical phi/psi angles which reduce the structure models usefulness for high-resolution functional analysis. ModRefiner (Dong and Yang, 2011); an algorithm for atomic-level, high-resolution protein structure refinement was used to refine homology predicted protein structure. ModRefiner refine protein structures based on a two-step atomic level energy minimization by using physical and knowledge-based force field. Main chain structures are formally constructed from initial Cα traces and side-chains are then refined with the backbone atoms. Refinement was done for several times to get better minimized protein energy. Finally, the protein was visualized by Swiss-PDB Viewer (Guex and Peitsch, 1997).
4.2. Evaluation and validation of the structure
To evaluate the accuracy and stereo-chemical properties of the predicted model PROCHECK (Laskowski et al., 1996) by Ramachandran plot analysis (Ramachandran et al., 1963) was done through “Protein structure and model assessment tools” of Swiss-model workspace. Refined PDB format of the protein was uploaded and 2.5 Å resolution value was selected. Protein structure assessment, 3D profiling of the predicted protein and model quality estimation was done by ERRAT (Colovos and Yeates, 1993b), Verify3D (Eisenberg et al., 1997) and QMEAN (Benkert et al., 1998) respectively. All the parameters were kept unchanged for the above evaluation tools.
4.3. Active site analysis
Active site analysis provides a noteworthy insight of the docking simulation study. The active binding sites of the protein were searched based on the structural association of template and the model construct with Computed Atlas of Surface Topography of proteins (CASTp) (Dundas et al., 2006) server. This was used to recognize and determine the binding sites, surface structural pockets, internal cavities of proteins and active sites, area, shape and volume of every pocket.
4.4. Protein–ligand docking
In silico docking simulation study, was carried out to recognize the inhibiting potential against envelope protein 2 (UniProt ID: T2ASQ1). Docking study was performed by Autodock Vina (Perryman et al., 2014). Before starting the docking stimulation study, envelope protein 2 was modified by adding polar hydrogen. A grid (box size: 76 × 76 × 76 Å and box center: 16.072 × 26.5007 × 3.7748 for x, y, and z, respectively) was designed in which various binding modes were generated for the most favorable bindings. The overall combined binding with envelope protein 2 (UniProtID: T2ASQ1) and NAG, MAN, MSE, NDG were obtained by using PyMOL (Seeliger and de Groot, 2010) (The PyMOL Molecular Graphics System, Version 1.5.0.4, Schrödinger, LLC).
4.5. Pharmacophore studies
The pharmacophore property of our selected ligand was carried out to use the online-based and license-agreed software. The Osiris property explorer (Zhang et al., 2013), ACToR (Aggregated Computational Toxicology Resource) and admetSAR (absorption, distribution, metabolism, excretion, and toxicity Structure–Activity Relationship database) were employed for its studies. ACToR is a set of software applications that take into one central location many types and sources of data on environmental chemicals. Presently, the ACToR database has information on chemical structure, in vitro bioassays and in vivo toxicology assays derived from more than 150 sources (Judson et al., 2008). The admetSAR is an open source, structure searchable, and updated database on a regular basis that gathers, and handles existing ADMET-associated information from the available literature. In admetSAR database, over 210,000 ADMET annotated data points for more than 96,000 unique compounds from a huge number of diverse literatures (Cheng, 2012).
5. Result
5.1. Identifying the Highest Antigenic Protein
The query for Chikungunya virus structural and non-structural protein resulted in a total of 1312 hits. All the proteins were evaluated by VaxiJen server, which generated an overall score for each protein sequence denoting their potentiality to create immune response. The protein sequence with the UniProtKB ID: T2ASQ1 attained the highest score of 0.6056 in VaxiJen analysis among all the query proteins. The protein itself is the Chikungunya virus envelope protein 2 (E2) consisting of 422 amino acids. The protein has been selected for further analysis in the present study.
5.2. Primary and secondary structure determination
The function of a protein correlates with the structural features of the protein. The ProtParam server analyzes the protein sequence and computes some parameters that decide the stability and function of the protein. Just as that secondary structural features of a protein also reveal its functional characteristics to some extent. From ProtParam generated results showed 35.42 Instability Index (II), 72.96 aliphatic index and a negative GRAVY (grand average hydropathy) of −0.497 for the protein. SOPMA calculated the secondary structural features of the protein and reported that the protein is dominated by random coils consisting 48.34%. Alpha Helix and Extended Strands formed 17.77% and 25.59% of the protein respectively. Lastly it showed Beta turns constituting 8.29%. The parameters calculated by both the tools are shown in Table 1, Table 2 respectively. The secondary structure plot is shown in Fig. 2 .
Table 1.
Different physico-chemical properties of Chikungunya envelope protein 2.
| Parameter | Value |
|---|---|
| Molecular weight | 47548.5 |
| Extinction coefficients Abs. 0.1% (=1 g/l) 1.088, assuming all pairs of Cys residues form cystines |
55,195 |
| Ext. coefficient Abs. 0.1% (=1 g/l) 1.078, assuming all Cys residues are reduced |
54,320 |
| Theoretical pI | 9.01 |
| Total number of negatively charged residues (Asp + Glu) | 38 |
| Total number of positively charged residues (Arg + Lys) | 51 |
| Instability index | 35.42 |
| Grand average of hydropathicity (GRAVY) | −0.497 |
| Aliphatic index | 72.96 |
Table 2.
Secondary structure analysis through SOPMA of Chikungunya virus envelope protein 2.
| Secondary structure | Percentage |
|---|---|
| Alpha helix (Hh) | 17.77% |
| Extended strand (Ee) | 25.59% |
| Beta turn (Tt) | 8.29% |
| Random coil (Cc) | 48.34% |
| 310 helix | 0.00% |
| π helix | 0.00% |
| Isolated β-bridge | 0.00% |
| Bend | 0.00% |
Fig. 2.

Secondary structure plot of envelope protein of Chikungunya virus. Here, helix is indicated by blue, while extended strands and beta turns are indicated by red and green, respectively. (For interpretation of the color information in this figure legend, the reader is referred to the web version of the article.)
5.3. T-cell epitope prediction
T-cell epitopes on the current protein were predicted using NetCTL server. The server identifies likely overlapping epitopes on the given protein sequence by using neural network architecture and generates a combinatorial score by predicting peptide-MHC class 1 binding based on all MHC class 1 supertypes, proteasomal C terminal cleavage and TAP transport efficiency all together. From the generated results the first five epitopes VTNHKKWQY, STKDNFNVY, VMHKKEVVL, LYPDHPTLL, and YYYELYPTM were selected on basis of their height combinatorial score.
The previously selected epitopes were found to be recognized by a range of MHC class 1 molecule according to IEDB MHC class 1 binding prediction tool. This tool is based on stabilized matrix method (SMM) and gives an output result for HLA binding affinity of the epitopes in IC50 nM unit. Binding affinity of the epitopes with the MHC-I molecules have an inverse relationship with the IC50 value. In the present study we opted for the selection of the MHC-I molecules with coupled IC50 value less than 200 nM (IC50 < 200), this ensured the selection of the MHC-1 molecules (Table 3 ) for which the selected epitopes showed higher affinity.
Table 3.
Most potential 5 T-cell epitopes with interacting MHC-1 alleles, total processing score and epitope conservancy result.
| Epitope | Interacting MHC-1 allele with an affinity, 200 (total score of proteasome score, TAP score, MHC score, processing score and MHC-1 binding) | Epitope conservancy analysis result |
|---|---|---|
| VTNHKKWQY | HLA-A*68:23 (2.02) HLA-A*32:07 (1.36) HLA-C*05:01 (1.16) HLA-C*12:03 (1.10) HLA-B*27:20 (0.87) HLA-A*30:02 (0.67) HLA-A*32:15 (0.64) HLA-B*15:17(0.55) HLA-A*80:01(0.45) |
37.29% |
| STKDNFNVY | HLA-A*68:23 (2.52) HLA-C*12:03 (2.16) HLA-A*32:07 (1.55) HLA-B*15:01 (1.03) HLA-A*32:15 (0.98) HLA-C*03:03 (0.93) HLA-A*30:02 (0.81) HLA-A*26:02 (0.80) HLA-B*15:02 (0.76) HLA-B*40:13 (0.73) HLA-C*14:02 (0.63) HLA-B*15:17 (0.59) |
45.76% |
| VMHKKEVVL | HLA-B*27:20 (1.17) HLA-A*02:50 (0.95) HLA-B*40:13 (0.86) HLA-A*32:07 (0.86) HLA-A*02:11 (0.82) HLA-A*68:23 (0.50) HLA-B*15:03 (0.49) HLA-C*14:02 (0.46) HLA-A*02:12 (0.27) HLA-C*03:03 (0.12) HLA-A*02:16 (−0.11) HLA-C*12:03 (−0.13) |
66.10% |
| LYPDHPTLL | HLA-A*02:17 (0.94) HLA-C*14:02 (0.90) HLA-A*32:07 (0.80) HLA-A*02:50 (0.64) HLA-A*68:23 (0.45) HLA-A*24:03 (0.36) HLA-C*12:03 (0.22) HLA-B*40:13 (0.14) HLA-B*15:02 (0.01) HLA-C*06:02 (−0.11) HLA-A*24:02 (−0.26) HLA-B*27:20 (−0.27) |
68.88% |
| YYYELYPTM | HLA-C*14:02 (1.31) HLA-A*02:17 (0.75) HLA-A*24:03 (0.44) HLA-C*07:02 (0.32) HLA-A*32:07 (0.21) HLA-A*68:23 (0.19) HLA-C*03:03 (0.12) HLA-B*27:20 (0.04) HLA-C*12:03 (−0.07) HLA-B*42:01 (−0.30) HLA-A*24:02 (−0.54) HLA-A*23:01 (−0.60) HLA-B*15:03 (−0.67) HLA-B*15:02 (−0.68) HLA-C*06:02 (−0.87) HLA-A*02:11 (−0.89) HLA-A*32:15 (−0.89) HLA-B*35:01 (−0.94) HLA-A*02:50 (−0.95) |
69.49% |
MHC-I processing efficiency tool of IEDB predicts an overall score for each epitope based on proteasomal cleavage efficiency, TAP transport efficiency and MHC-I binding efficiency combined. The proteasomal complex contains enzymes that digest proteins to form smaller peptides. Produced peptides are recognized by MHC class 1 molecules and MHC-1 forms a complex with the peptides. The peptide-MHC class 1 complexes are transported to the endoplasmic reticulum, a process facilitated by transport associated proteins (TAP) before being presented to the T-cells on the plasma membrane of the cell. The higher the combined score of the peptides the better they are processed for presentation and that is critical step for creating a successful immune response. The scores obtained from IEDB MHC-1 binding analysis and processing tools are summarized in Table 3.
Better immune response depends on the successful recognition of epitopes by HLA molecules with significant affinity. So, a peptide recognized by the highest number of HLA alleles has the best potential to induce a strong immune response. Among the 5 epitopes studied, one epitope has interacted with higher number of HLA alleles than the other epitopes. The 9-mer epitope YYYELYPTM showed affinity for highest 19 MHC-1 molecules including HLA-C*14:02, HLA A*02:17, HLA-A*24:03, HLA-C*07:02, HLA-A*32:07, HLA-A*68:23, HLA-C*03:03, HLA-B*27:20, HLA-C*12:03, HLA-B*42:01, HLA-A*24:02, HLA-A*23:01, HLA-B*15:03, HLA-B*15:02, HLA-C*06:02, HLA-A*02:11, HLA-A*32:15, HLA-B*35:01 and HLA-A*02:50.
5.4. Epitope conservancy prediction
Conserved epitopes can provide a more effective immunization therefore better conservancy of an epitope is expected. Epitope conservancy analysis revealed (Table 3) the epitope LYPDHPTLL to be 68.88% conserved whereas epitope YYYELYPTM scored 69.49%. Other three epitopes VTNHKKWQY, STKDNFNVY, VMHKKEVVL showed 32.29%, 45.76% and 66.10% conservancy respectively.
5.5. Prediction of population coverage
Identified optimum MHC-I binders for each epitopes were considered for the population coverage analysis of the epitopes. Population coverage calculates the percentage of people living in a specific region to be potentially responsive to the query epitopes. IEDB's population coverage analysis tool revealed highest 84.94% population coverage of the 5 epitopes in Papua New Guinea. The epitopes showed 72.87% coverage in West Africa along with 69.71% and 71.83% coverage in East Africa and Central Africa respectively. Among newer Chikungunya affected regions Italy and North America the epitopes showed 79.83% and 77.05% population coverage respectively. 82.44% cumulative population coverage for Philippines and 69.50% cumulative population coverage for South Asian region are also achieved. The results are shown in Fig. 3 .
Fig. 3.

Population coverage, based on MHC-I restriction data. Different CHIKV-affected regions were selected for evaluation of the population coverage of the proposed epitopes. Notes: In the graphs, the line (-o-) represents the cumulative percentage of population coverage of the epitopes; the bars represent the population coverage for each epitope. Abbreviations: CHIKV, Chikungunya Virus; HLA, human leukocyte antigen; MHC-I, major histocompatibility complex class I; PC90, 90% population coverage.
5.6. Allergenicity assessment
The query sequence does not meet the criteria set by the FAO/WHO evaluation scheme for cross-reactive allergen prediction. Hence, the query sequence is classified as a non-allergen by the FAO/WHO evaluation scheme. AllerHunter predicted the query sequence as a non-allergen with score of 0.04 (SE = 95.2%, SP = 59.7%).
5.7. Selection of T cell epitope
Among the five primarily selected epitopes, the epitope ‘YYYELYPTM’ was found more suitable as a vaccine candidate than other epitopes by considering its overall epitope conservancy, population coverage and by the affinity for highest number of HLA molecules.
5.8. Docking simulation study
AutoDock Vina predicted the binding models for the epitope YYYELYPTM with HLA molecules. HLA molecules are stored in protein Data Bank as crystal structures are generally complexed with other peptides or epitopes which were gained experimentally by various research works. So, here at first we retrieved HLA-B*3501 complexed with influenza NP418 epitope crystal structure. Then influenza NP418 epitope was removed from HLA molecule. After that, all the docking simulations were done with this epitope removed HLA molecule. The binding energy of predicted epitope with HLA-B*3501 receptor was found to be −7.1 kcal/mol. This binding energy was compared with the binding energy of influenza NP418 epitope to HLA B*3501 was −7.6 kcal/mol. The 3D structures shown in Fig. 4 of HLA and epitope are visualized and captured with Pymol molecular graphics system.
Fig. 4.

Docking simulation study generated by Autodock Vina. (A) Structure of our predicted epitope, “YYYELYPTM” and (B) visualization of docking results of “YYYELYPTM” with HLAB*3501.
5.9. B-cell epitope prediction
Potential B cell epitopes have several characteristics which are necessary for successful recognition by B cells. These features include hydrophilicity, surface accessibility and beta turn prediction. The query protein was scanned to identify B cell epitopes by several web based tools available in IEDB.
Kolaskar and Tongaonkar antigenicity prediction tool assessed the protein for B cell epitopes analyzing the physico-chemical properties of the amino acids and their abundance in known B cell epitopes. The tool subsequently came up with a result shown in Table 4 and Fig. 5 predicting an average antigenic propensity value of 1.032 for the protein with the maximum value of 1.284 and minimum of 0.860. The tool was primed with the threshold value of 1.00 to search for antigenically potent regions and the region from 397 to 406 a 9 mer epitope showed preferred B cell epitope characteristics.
Table 4.
Kolaskar and Tongaonkar antigenicity analysis.
| No. | Start | End | Peptide | Peptide length |
|---|---|---|---|---|
| 1 | 8 | 23 | VYKATRPYLAHCPDCG | 16 |
| 2 | 25 | 35 | GHSCHSPVALE | 11 |
| 3 | 47 | 55 | KIQVSLQIG | 9 |
| 4 | 86 | 94 | RTSLPCKIT | 9 |
| 5 | 99 | 105 | HFILARY | 7 |
| 6 | 110 | 116 | TLTVGFT | 7 |
| 7 | 120 | 137 | KISLCNKPVHHDPPVIGR | 18 |
| 8 | 147 | 159 | GKELPCSTYVQST | 13 |
| 9 | 166 | 171 | IEVHMP | 6 |
| 10 | 184 | 194 | SGNVKITVNGQ | 11 |
| 11 | 196 | 203 | VRYKCNCG | 8 |
| 12 | 216 | 230 | INNCKVDQCHAAVTN | 15 |
| 13 | 237 | 244 | NSPLVPRN | 8 |
| 14 | 253 | 268 | KIHIPFPLANVTCRVP | 16 |
| 15 | 282 | 296 | VIMLLYPDHPTLLSY | 15 |
| 16 | 313 | 327 | KKEVVLTVPTEGLEV | 15 |
| 17 | 349 | 362 | GHPHEIILYYYELY | 14 |
| 18 | 364 | 384 | TMTVVVVSVATFILLSMVGIA | 21 |
| 19 | 392 | 399 | RRRCITPY | 8 |
Fig. 5.

Kolashkar and Tongaonkar antigenicity prediction of the most antigenic protein, T2ASQ1. Notes: The x-axis and y-axis represent the sequence position and antigenic propensity, respectively. The threshold value is 1.0. The regions above the threshold are antigenic, shown in yellow. (For interpretation of the color information in this figure legend, the reader is referred to the web version of the article.)
Surface accessibility of B cell epitopes is required because hydrophilic regions are generally exposed on the surface and likely to evoke B cell immune response. The Emini surface accessibility prediction and Parker hydrophilicity prediction tools were utilized. The analyses are shown in Table 5 along with graphical rendition from both the tools in Fig. 6, Fig. 7 respectively.
Table 5.
Emini surface accessibility analysis.
| No. | Start | End | Peptide | Peptide length |
|---|---|---|---|---|
| 1 | 9 | 15 | YKATRPY | 7 |
| 2 | 36 | 42 | RIRNEAT | 7 |
| 3 | 57 | 74 | KTDDRHDWTKLRYMDNHM | 18 |
| 4 | 104 | 109 | RYQKGE | 6 |
| 5 | 138 | 148 | EKFHSRPQHGK | 11 |
| 6 | 171 | 179 | PPDTPDRTL | 9 |
| 7 | 230 | 237 | NHKKWQYN | 8 |
| 8 | 268 | 274 | PKARNPT | 7 |
| 9 | 296 | 307 | YRNMGEEPNYQE | 12 |
| 10 | 330 | 338 | GNNEPYKYW | 9 |
| 11 | 398 | 403 | PYEQKP | 6 |
Fig. 6.

Emini surface accessibility prediction of the most antigenic protein, T2ASQ1. Notes: The x-axis and y-axis represent the sequence position and surface probability, respectively. The threshold value is 1.000. The regions above the threshold are antigenic, shown in yellow. (For interpretation of the color information in this figure legend, the reader is referred to the web version of the article.)
Fig. 7.

Parker hydrophilicity prediction of the most antigenic protein, T2ASQ1. Notes: The x-axis and y-axis represent the position and score, respectively. The threshold is 1.673. The regions having beta turns in the protein are shown in yellow color, above the threshold value. (For interpretation of the color information in this figure legend, the reader is referred to the web version of the article.)
The beta turns in a protein are generally surface accessible and hydrophilic in nature. Chou and Fasman Beta turn prediction (Fig. 8 ) was done for the protein to locate the beta turn regions in the query protein as beta turns have a significant effect in inducing antigenicity. Produced results identified a region from 394 to 406 with constant predicted B turn region.
Fig. 8.

Chou and Fasman beta-turn prediction of the most antigenic protein, T2ASQ1. Notes: The x-axis and y-axis represent the position and score, respectively. The threshold is 1.001. The regions having beta turns in the protein are shown in yellow color, above the threshold value. (For interpretation of the color information in this figure legend, the reader is referred to the web version of the article.)
Experimental data showed that the region of a peptide that interacts with the antibody tends to be flexible. Karplus Schulz flexibility prediction tool identified the flexible regions on the query protein. The region from 397 to 405 is considerably the most favorable region in the flexibility prediction analysis. Results are shown in Fig. 9 .
Fig. 9.

Karplus and Schulz flexibility prediction of the most antigenic protein, T2ASQ1. Notes: The x-axis and y-axis represent the position and score, respectively. The threshold is 1.0. The flexible regions of the protein are shown in yellow color, above the threshold value. (For interpretation of the color information in this figure legend, the reader is referred to the web version of the article.)
Bepipred is a machine learning process based on Hidden-Markov model, a tool to determine Linear B cell epitopes. This tool was utilized to eliminate the fact that single scale amino acid propensity profile cannot reliably predict antigenic epitopes every time and to obtain a better result from the epitope prediction tools than the receiver operating characteristics (ROC) plot. The Bepipred predicted epitopes on the protein are shown in Table 6 and Fig. 10 .
Table 6.
Bepipred linear epitope prediction.
| No. | Start | End | Peptide | Peptide length |
|---|---|---|---|---|
| 1 | 1 | 6 | STKDNF | 6 |
| 2 | 20 | 29 | PDCGEGHSCH | 10 |
| 3 | 39 | 45 | NEATDGT | 7 |
| 4 | 57 | 63 | KTDDRHD | 7 |
| 5 | 73 | 79 | HMPTNAK | 7 |
| 6 | 107 | 110 | KGET | 4 |
| 7 | 116 | 116 | T | 1 |
| 8 | 127 | 136 | PVHHDPPVIG | 10 |
| 9 | 140 | 151 | FHSRPQHGKELP | 12 |
| 10 | 155 | 186 | YVQSTAATTEEIEVHMPPDTPDRTLMSQQSGN | 32 |
| 11 | 202 | 211 | CGGSNEGQTI | 10 |
| 12 | 233 | 253 | KWQYNSPLVPRNAELGDRKGK | 21 |
| 13 | 268 | 278 | PKARNPTVTYG | 11 |
| 14 | 298 | 307 | NMGEEPNYQE | 10 |
| 15 | 322 | 350 | TEGLEVTWGNNEPYKYWPQLSTNGTAHGH | 29 |
| 16 | 397 | 406 | TPYEQKPGAT | 10 |
Fig. 10.

Bepipred linear epitope prediction of the most antigenic protein, T2ASQ1. Notes: The x-axis and y-axis represent the position and score, respectively. The threshold is 0.35. The regions having beta turns are shown in yellow. The highest peak region indicates the most potent B-cell epitope. (For interpretation of the color information in this figure legend, the reader is referred to the web version of the article.)
After cross processing all the data derived from the previous B cell epitope prediction tools, the region from 397 to 405 amino acids is found to be the best capable region for inducing B cell response.
5.10. Model building and refinement
A three dimensional structure of the Chikungunya envelope protein E2 was predicted using the Phyre2 server. The tertiary structure of a protein depicts its molecular basis of function and interaction. As homology modeling tools often generate models with some local distortion, the MODELLER generated model was refined with ModRefiner to obtain a more stereo-chemically accurate model. Refined model showed that most of the residues of the protein (>90%) is in the most favored region. The refined model was used to carry out subsequent analysis. A Swiss-PDB generated view of the 3D model is displayed in Fig. 11 .
Fig. 11.

Swiss-PDB generated image of the Chikungunya virus envelope protein 2.
5.11. Model verification and validation
The predicted model in the current study was verified to measure its accuracy comparing it with high resolution models with a several structure validation tools. PROCHECK performed an overall analysis of the model and delivered the Ramachandran plot shown in Fig. 12 and Table 7 . ERRAT analysis obtained results are presented in Fig. 13 . Another verification tool Verify3D that generates an environmental profile graph for a given protein while QMEAN server was used for the verification of protein model.
Fig. 12.

Ramachandran plot analysis of T2ASQ1. Here, red region indicates favored region, yellow region for allowed and light yellow shows generously allowed region and white for disallowed region. Phi and psi angels determine torsion angels. (For interpretation of the color information in this figure legend, the reader is referred to the web version of the article.)
Table 7.
Ramachandran plot of envelope protein 2 from Chikungunya virus.
| Ramachandran plot statistics | CHIKV E2 |
|
|---|---|---|
| Residue | % | |
| Residues in the most favored regions [A,B,L] | 339 | 93.4 |
| Residues in the additional allowed regions [a,b,l,p] | 21 | 5.8 |
| Residues in the generously allowed regions [a,b,l,p] | 3 | 0.8 |
| Residues in the disallowed regions [xx] | 0 | 0.0 |
| Number of non-glycine and non-proline residues | 363 | 100.0 |
| Number of end residues (excl. Gly and Pro) | 2 | |
| Number of glycine residues | 27 | |
| Number of proline residues | 30 | |
| Total number of residues | 422 | |
Fig. 13.

ERRAT generated result of CHIKV E2 where 95% indicates rejection limit.
5.12. Active site prediction
The active sites of the Chikungunya virus envelope protein 2 is shown in Fig. 14 derived from CASTp server. The calculated results revealed that amino acid position 13–341 is predicted to be conserved with the active site. The server predicted the best active site with an area of 593.2 and formed with 1063.2 amino acid residues.
Fig. 14.

Active site analysis. (A) Active site information by CASTp. Green color shows the active site position from 13 to 241 with the beta-sheet in between them. (B) The table shows the area and the volume for different active sites of Chikungunya virus E2 and the best active site remains in an area of 593.2 and a volume of 1063.2 amino acid. (C) The 3D structure of best active site. (For interpretation of the color information in this figure legend, the reader is referred to the web version of the article.)
5.13. Protein–ligand docking
AutoDock Vina delivered results with complete docking records. The log file is in Table 8 . Calculation of the root mean square deviation (RMSD) between co-ordinates of the atoms and formation of clusters based on RMSD values computed the resemblance of the docked structures. The most favorable docking is considered to be the conformation with the lowest binding energy. From the attained results binding energy for N-Acetyl-d-Glucosamine [NAG] was −4.8 kcal/mol, for Alpha-d-Manose [MAN] −5.9 kcal/mol, for Selenomethionine [MSE] −3.0 kcal/mol, for 2-(Acetyleamino)-2-Deoxy-A-d-Glucopyranose [NDG] −4.8 kcal/mol. The PyMol generated figures for the binding complexes are in Fig. 15 .
Table 8.
Ligands information for docking study and protein–ligand interaction.
| Ligands | Identifiers | Formula | Molecular weight | Binding energy (kcal/mol) | Chemical structure |
|---|---|---|---|---|---|
| N-Acetyl-d-Glucosamine [NAG] | 2-(Acetylamino)-2-deoxy-beta-d-glucopyranose N-[(2R,3R,4R,5S,6R)-6-(hydroxymethyl)-2,4,5-tris(oxidanyl)oxan-3-yl]ethanamide |
C8H15NO6 | 221.21 g/mol | −4.8 | 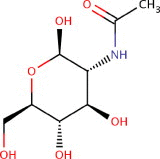 |
| Alpha-d-Mannose [MAN] | Alpha-d-mannopyranose (2S,3S,4S,5S,6R)-6-(hydroxymethyl)oxane-2,3,4,5-tetrol |
C6H12O6 | 180.16 g/mol | −5.9 | 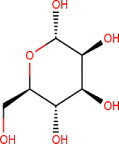 |
| Selenomethionine [MSE] | (2S)-2-Amino-4-(methylselanyl)butanoic acid (2S)-2-amino-4-methylselanyl-butanoic acid |
C5H11NO2Se | 196.11 g/mol | −3.0 | 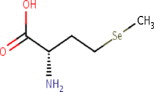 |
| 2-(Acetylamino)-2-Deoxy-A-d-Glucopyranose [NDG] | 2-(Acetylamino)-2-deoxy-alpha-d-glucopyranose N-[(2S,3R,4R,5S,6R)-2,4,5-trihydroxy-6-(hydroxymethyl)oxan-3-yl]ethanamide |
C8H15NO6 | 221.21 g/mol | −4.8 | 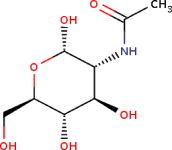 |
Fig. 15.

Overall binding between CHIKV E2 and (A) NAG; (B) MAN; (C) MSE; (D) NDG.
5.14. Pharmacophore analysis
The results from OSIRIS property explorer, ACToR and admetSAR have been tabulated to analyze its drug likeness, drug score, and turmeric, mutagenic and structural polarity (Table 9 ) which is quite satisfactory for the adequacy as a possible drug candidate.
Table 9.
Pharmacophore properties of selected ligand.
| Ligand | Pharmacophore | Evaluation/score |
|---|---|---|
| Alpha-d-mannose [MAN] | Drug likeness | −3.78 |
| Drug score | 0.5 | |
| Bioaccumulative | No | |
| Meets human health criteria (hazard) | No | |
| Solubility | 0.25 | |
| The polar surface area | 110.3 | |
| Persistent | No | |
| Substance category | Organic | |
| Inherently toxic | No | |
| Tumeric | No indication | |
| Irritant | No indication | |
| Mutagenic | No indication | |
| Reproductive effect | No indication |
6. Discussion
Diseases are global scale burden. As newer viruses are affecting humans more frequently than ever, vaccine development within a short time to keep up with the rising viral attacks has become crucial (Marshall, 2004, De Groot and Rappuoli, 2004, Korber et al., 2006).
In recent years next generation sequencing and advanced genomics and proteomic technologies brought about a significant change in computational immunology. With the abundance of data available like never before, newer immunoinformatics tools are being developed that are being used to get a head start in developing vaccines with a better understanding of immune response of human body against a multitude of organisms (Fauci, 2006, Purcell et al., 2007). In a very contrasting scenario a very little has been done in case of Chikungunya virus. RNA viruses (e.g. Chikungunya virus) are more likely to mutate than other DNA viruses. Such a mutation in the envelope protein of the CHIKV virus brought it to the developed part of the world where despite of its benign nature it is long lasting debilitating symptoms caused serious concerns owing to socio-economic effects (Weaver, 2014). As a result, recently a surge of activity principally concentrated on developing vaccine for Chikungunya virus has started but yet with no outcome.
In the present study we tried to identify major immunogenic epitopes on Chikungunya viral proteins using immune informatics tools and predict a vaccine along with novel inhibitors of Chikungunya virus envelope protein E2.
Traditional vaccination approaches are based on complete pathogen either live attenuated or inactivated. Among the major problems these vaccines brought are crucial safety concerns, because those pathogens being used for immunization may become activated and cause infection. Moreover due to genetic variation of pathogen strains around the world vaccines are likely to lose their efficacy in different regions or for a specific population. But novel vaccine approaches like DNA vaccines and epitope based vaccines have the potential to overcome these barriers for these vaccines can create more effective, specific, strong and long lasting immune response with minimal structure and devoid of all the undesired effects (Arnon, 2006). On top of that neutralizing peptide based vaccine design has been successfully suggested by using in silico approach against rhinovirus (Lapelosa et al., 2009) dengue virus (Chakraborty et al., 2010), Human coronaviruses (Sharmin and Islam, 2014), Hepatitis C virus (Idrees and Ashfaq, 2013), Saint Louis encephalitis virus (Hasan et al., 2013). Such studies regarding those viruses established immunoinformatics study.
Meanwhile, though most of the epitope based vaccines trigger the B-cell epitopes, T cell epitope based vaccines designs are inspired owing to a stronger CD8+ T-cell mediated immune response of the host cell to the infected T cells (Klavinskis et al., 1989). Moreover, antigenic drifts might render humoral immunity and antigenic memory response non-effective and the pathogens pass through. T cell based vaccines are free of this limitation and along with a strong B cell epitope, a multi epitope vaccine is a good prospect which can trigger both cell mediated and humoral immune response (Trainor et al., 2007).
A T cell epitope is considered strong and potent if it is well conserved among the sequenced CHIKV E2 proteins in the database. The predicted T-cell epitope of the current study YYYELYPTM is calculated to be 69.49% conserved. It is the most conserved epitope among the 5 most potential epitopes selected from NetCTL T-cell epitope analysis. In epitope based vaccine development a highly conserved epitope is expected to deliver a broader protection across different strains. Moreover, as RNA viruses like CHIKV are more likely to mutate due to a lack of proof reading activity of RNA polymerase, a vaccine candidate epitope must come from the portion of the protein showing much conservancy that will ensure an effective long lasting immunization (Trainor et al., 2007).
To endorse the selected epitope YYYELYPTM, the result from MHC-I and epitope interaction tool suggested that this epitope interacts with the highest number of HLA alleles. It was found that YYYELYPTM interacts with 19 HLA-A, HLA-B and HLA-C alleles in total. Analyzing similar data from other study showed this specific high affinity binding is absolutely desired because the efficiency of an epitope vaccine greatly relies on the precise interaction between epitope and HLA alleles (Chakraborty et al., 2010). This specific high affinity binding is absolutely desired because the efficiency of an epitope vaccine greatly relies on the precise interaction between epitope and HLA alleles. Those HLA alleles, for which YYYELYPTM showed affinity, were searched for population coverage. The search was primarily concentrated on the CHIKV endemic regions. Highest population coverage was recorded in Papua New Guinea; one of the recent CHIKV affected regions, which is of more interest because outbreak in Papua New Guinea was identified, caused by a mutated CHIKV. 69.71–72.87% coverage was recorded in different regions of endemic Africa. A significant percentage of coverage is attained in the most recent CHIKV outbreak regions Italy and North America. In addition to that the epitope is declared as non-allergen, an unquestionable feature a vaccine must have. These results implicate that the vaccine would be effective for a huge population throughout a wide geographical region.
The epitope was docked and compared with a control to measure its docking efficacy with a specific HLA allele HLA-B*3501. The results are concluding that this epitope can bind as efficiently as the control considered in the study. This computational analysis confirms the epitopes affinity for the MHC-I molecules and upholds its position as a novel vaccine candidate.
Chikungunya virus envelope protein (E2) was also searched for B cell epitopes as B cell epitopes can induce both primary and secondary immunity. Several tools from IEDB database generated results analyzing the protein based on the key characteristics of B cell epitopes. We cross referenced all the data and the region from 397 to 405 found to be predicted as potent B cell epitope by Bepipred tool. The region is composed of beta turns and flexible. The region is surface accessible and hydrophilic comparatively than other regions and proved to be antigenic. The 9-mer epitope TPYEQKPGA is the most favorable as B cell epitope as per predicted results.
As the chronic stage of the CHIKV infection can last for months to years along with severe polyarthralgia its debilitating effects call for a therapeutic agent that can minimize or completely eliminate the chronic symptoms. Moreover, for complete safeguard against CHIKV infection a universal drug is compulsory. If the effectiveness of the vaccine is reduced due to mutation in the virus the drug may work alongside to diminish the symptoms faster. In another scenario, in case of any sudden Chikungunya outbreak in a non-vaccinated area vaccination would become obsolete. Only post-therapeutic measure or drug would be effective until vaccine kicks in. In the present study along with pre therapy vaccine development we also predicted the active sites on the highest antigenic protein of the Chikungunya virus and subsequently predicted the novel inhibitors of the protein (Queyriaux et al., 2008).
As suggested by VaxiJen analysis the envelope protein 2 of CHIKV appeared to be the eligible drug target. A valuable insight was achieved from the primary and secondary analysis of the protein from ProtParam and SOPMA tools. The protein is found to be stable in vitro with below 40 instability index negative GRAVY and, higher aliphatic index. The abundance coiled region in SOPMA generated results implicated the higher conservancy and stability of the protein (Guruprasad et al., 1990, Gasteiger et al., 2005, Geourjon and Deleage, 1995b).
The three dimensional structure generated by Phyre2 server was acceptable. But homology modeling algorithms often generate models that lack desired quality because of significant local distortions, including steric clashes, unphysical phi/psi angles and irregular hydrogen bonding networks which restrict its use for high resolution functional analysis. Structure refinement overcomes this problem to some extent (Dong and Yang, 2011). Our predicted model was refined with the help of ModRefiner and the resultant structure had 93.4% of its residues laid out in the most favorable core region and 5.8% in additional allowed region with only 0.8% of the residues in generously allowed regions. These parameters are confirmation of a good quality model.
Our predicted structure scored significantly well in structure validation analysis. Verify 3D matches a 3D structure profile with its amino acid sequence. Generally quality structures score higher in this analysis. This result ranges from −1 (bad) to +1 (good) so 0.73 for our predicted protein speaks of its good environmental profile (Gardner et al., 2012). ERRAT generated results showed 75.676 quality factors for the protein model, this value is below the 95% rejection limit (Bowie et al., 1991).
The geometrical aspect of the model was computed by QMEAN scoring function with a composite function of six different structural descriptors. To estimate the absolute quality of the model the query model is compared to high resolution X-ray structures of same size by QMEAN server which reflects the structure's “degree of nativeness”. But membrane proteins expectantly get lower Z-score owing to their physico-chemical properties than other soluble proteins. As a result a modest score was received from the QMEAN server analysis for the predicted protein model (Benkert et al., 1998, Benkert et al., 2009a, Benkert et al., 2009b, Benkert et al., 2011, Hasan et al., 2014a, Hasan et al., 2014b).
CASTp server generated docking reports of active sites on CHIKV E2. AutoDockVina reported the most favorable bindings of the CHIKV E2 with N-Acetyl-d-Glucosamine [NAG], Alpha-d-Mannose [MAN], Selenomethionine [MSE], 2-(Acetylamino)-2-Deoxy-A-d-Glucopyranose [NDG]. Among them Alpha-d-Mannose [MAN] forms the cluster with E2 in the lowest possible binding energy conformation.
The results from OSIRIS property explorer, ACToR and admetSAR have been tabulated to analyze its drug likeness, drug score, turmeric, mutagenic and structural polarity which are quite satisfactory for the adequacy as a possible drug candidate. MAN may act as a potent safe drug candidate against CHKV as there have no probable tumorigenic as well as irritant tendency.
From the current study we have suggested a potent T cell epitope along with a B cell epitope which can effectively be used for the development of multi peptide vaccine to induce a complete immune response against Chikungunya virus. In addition to that Alpha-d-Mannose [MAN] is found to be the most effective inhibitor of the CHIKV envelope protein 2. Although this study emphasizes on the algorithms available, it is important to verify all the criteria in vitro to assess the immunogenicity and recognize the epitopes of a protein. Both in vivo and in vitro analysis are required along with this in silico study and to determine the binding affinity binding chip assay may be useful.
7. Conclusion
As the present study to identify epitope binding HLA alleles was based on computational tools, there may be more HLA alleles that can recognize the epitopes or may be the predicted epitope might not show the same affinity as predicted computationally in the experimental settings. CHIKV envelope protein 2 is distinctively associated with viral maturation, multiplication and infection. It establishes CHIKV E2 as an interesting drug target site. But, to validate the findings of the current study extensive laboratory based techniques are required. Nonetheless these finding will serve the purpose of the ground data for such kind of study in future.
Conflict of interests
Authors disclose no potential conflict of interests.
Funding sources
There is no funding source.
Author's contributions
M.A.H. has made substantial contributions to conception and design, acquisition of data, analysis and interpretation of data. M.A.K. and A.D. carried out the molecular genetic studies, participated in the sequence alignment and drafted the manuscript. M.H.H.M. worked for computational analysis. M.U.H. conceived of the study, and participated in its design and coordination and helped to draft the manuscript. All authors read and approved the final manuscript.
Acknowledgements
We cordially thank Adnan Mannan, Assistant Professor of the Department of Genetic Engineering and Biotechnology, University of Chittagong, for his suggestions and inspiration during our research proceedings.
References
- Andersen P.H., Nielsen M., Lund O. Prediction of residues in discontinuous B-cell epitopes using protein 3D structures. Protein Sci. 2006;15(11):2558–2567. doi: 10.1110/ps.062405906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apweiler R., Bairoch A., Wu C.H. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 2004;32(1):D115–D119. doi: 10.1093/nar/gkh131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnon R. A novel approach to vaccine design—epitope-based vaccines. FEBS J. 2006;273:33–34. [Google Scholar]
- Atanas P., Irini D. T-cell epitope vaccine design by immunoinformatics. Open Biol. 2013;3:120139. doi: 10.1098/rsob.120139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton D.J., Sawicki S.G., Sawicki D.L. Solubilization and immunoprecipitation of alphavirus replication complexes. J. Virol. 1991;65:1496–1506. doi: 10.1128/jvi.65.3.1496-1506.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benkert P., Tosatto, Schomburg D. QMEAN, a comprehensive scoring function for model quality assessment. Proteins Struct. Funct. Bioinform. 1998;71(1):261–277. doi: 10.1002/prot.21715. [DOI] [PubMed] [Google Scholar]
- Benkert P., Schwede T., Tosatto S.C. QMEANclust: estimation of protein model quality by combining a composite scoring function with structural density information. BMC Struct. Biol. 2009;20(9):35. doi: 10.1186/1472-6807-9-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benkert P., Kunzli M., Schwede T. QMEAN server for protein model quality estimation. Nucleic Acids Res. 2009;1(37):W510–W514. doi: 10.1093/nar/gkp322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benkert P., Biasini M., Schwede T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics. 2011;27(3):343–350. doi: 10.1093/bioinformatics/btq662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman H.M., Westbrook J., Feng Z., Gilliland G., Bhat T.N. The protein data bank. Nucleic Acids Res. 2000;28(1):235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowie J.U., Luthy R., Eisenberg D. A method to identify protein sequences that fold into a known three-dimensional structure. Science. 1991;253(5016):164–170. doi: 10.1126/science.1853201. [DOI] [PubMed] [Google Scholar]
- Brighton S.W. Chloroquine phosphate treatment of chronic Chikungunya arthritis: an open pilot study. S. Afr. Med. J. 1984;66:217–218. [PubMed] [Google Scholar]
- Bui H.H., Sidney J., Li W., Fusseder N., Sette A. Development of an epitope conservancy analysis tool to facilitate the design of epitope-based diagnostics and vaccines. BMC Bioinform. 2007;8(1):361. doi: 10.1186/1471-2105-8-361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burt F.J., Rolph M.S., Rulli N.E., Mahalingam S., Heise M.T. Chikungunya: a re-emerging virus. Lancet. 2012;379(9816):662–671. doi: 10.1016/S0140-6736(11)60281-X. [DOI] [PubMed] [Google Scholar]
- Buus S., Lauemoller S.L., Worning P., Kesmir C., Frimurer T.S. Sensitive quantitative predictions of peptide-MHC binding by a ‘Query by Committee’ artificial neural network approach. Tissue Antigens. 2003;62(5):378–384. doi: 10.1034/j.1399-0039.2003.00112.x. [DOI] [PubMed] [Google Scholar]
- Caglioti C., Lalle E., Castilletti C., Carletti F., Capobianchi M.R. Chikungunya virus infection: an overview. New Microbiol. 2013;36(3):211–227. [PubMed] [Google Scholar]
- Chakraborty S., Chakravorty R., Ahmed M. A computational approach for identification of epitopes in dengue virus envelope protein: a step towards designing a universal dengue vaccine targeting endemic regions. In Silico Biol. 2010;10(5–6):235–246. doi: 10.3233/ISB-2010-0435. [DOI] [PubMed] [Google Scholar]
- Cheng Feixiong. admetSAR: a comprehensive source and free tool for assessment of chemical ADMET properties. J. Chem. Inform. Model. 2012;52(11):3099–3105. doi: 10.1021/ci300367a. [DOI] [PubMed] [Google Scholar]
- Chou P.Y., Fasman G.D. Prediction of the secondary structure of proteins from their amino acid sequence. Adv. Enzymol. Relat. Areas Mol. Biol. 1978;47:45–148. doi: 10.1002/9780470122921.ch2. [DOI] [PubMed] [Google Scholar]
- Colovos C., Yeates T.O. Verification of protein structures: patterns of nonbonded atomic interactions. Protein Sci. 1993;2:1511–1519. doi: 10.1002/pro.5560020916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colovos C., Yeates T.O. Verification of protein structures, patterns of nonbonded atomic interactions. Protein Sci. 1993;2(9):1511–1519. doi: 10.1002/pro.5560020916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couderc T., Chrétien F., Schilte C., Disson O., Brigitte M., Guivel-Benhassine F., Touret Y., Barau G., Cayet N., Schuffenecker I. A mouse model for Chikungunya: young age and inefficient type-I interferon signaling are risk factors for severe disease. PLoS Pathog. 2008;4:e29. doi: 10.1371/journal.ppat.0040029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couderc T. Prophylaxis and therapy for Chikungunya virus infection. J. Infect. Dis. 2009;200:516–523. doi: 10.1086/600381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Groot A.S., Rappuoli R. Genome-derived vaccines. Expert Rev. Vaccines. 2004;3(1):59–76. doi: 10.1586/14760584.3.1.59. [DOI] [PubMed] [Google Scholar]
- Dong X., Yang Z. Improving the physical realism and structural accuracy of protein models by a two-step atomic-level energy minimization. Biophys. J. 2011;101:2525–2534. doi: 10.1016/j.bpj.2011.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doytchinova I.A., Flower D.R. VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 2007;8:4. doi: 10.1186/1471-2105-8-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dundas J., Ouyang Z., Tseng J., Binkowski A., Turpaz Y. CASTp, computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res. 2006;34:116–118. doi: 10.1093/nar/gkl282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg D., Luthy R., Bowie J.U. VERIFY3D, assessment of protein models with three-dimensional profiles. Methods Enzymol. 1997;277:396–404. doi: 10.1016/s0076-6879(97)77022-8. [DOI] [PubMed] [Google Scholar]
- Emini E.A., Hughes J.V., Perlow D.S., Boger J. Induction of hepatitis A virus-neutralizing antibody by a virus-specific synthetic peptide. J. Virol. 1985;55(3):836–839. doi: 10.1128/jvi.55.3.836-839.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enesrink M. Infectious diseases: massive outbreak draws fresh attention to little-known virus. Science. 2006;311:1085. doi: 10.1126/science.311.5764.1085a. [DOI] [PubMed] [Google Scholar]
- Fauci A.S. Emerging and re-emerging infectious diseases: influenza as a prototype of the host–pathogen balancing act. Cell. 2006;124(4):665–670. doi: 10.1016/j.cell.2006.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fieser T.M., John A., Tainer H. Influence of protein flexibility and peptide conformation on reactivity of monoclonal anti-peptide antibodies with a protein α-helix. Proc. Natl. Acad. Sci. U.S.A. 1987;84(23):8568–8572. doi: 10.1073/pnas.84.23.8568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner J., Anraku I., Le T.T., Larcher T., Major L., Roques P., Schroder W.A., Higgs S., Suhrbier A. Chikungunya virus arthritis in adult wild-type mice. J. Virol. 2010;84:8021–8032. doi: 10.1128/JVI.02603-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner C.L., Burke C.W., Higgs S.T., Klimstra W.B., Ryman K.D. Interferon-alpha/beta deficiency greatly exacerbates arthritogenic disease in mice infected with wild-type Chikungunya virus but not with the cell culture-adapted live-attenuated 181/25 vaccine candidate. Virology. 2012;425:103–112. doi: 10.1016/j.virol.2011.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasteiger E., Hoogland C., Gattiker A., Duvaud S., Wilkins M.R. The Proteomics Protocols Handbook. 2005. Protein identification and analysis tools on the ExPASy Server; pp. 571–607. [Google Scholar]
- Geourjon C., Deleage G. SOPMA: significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Comput. Appl. Biosci. 1995;11(6):681–684. doi: 10.1093/bioinformatics/11.6.681. [DOI] [PubMed] [Google Scholar]
- Geourjon C., Deleage G. SOPMA, significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Comput. Appl. Biosci. 1995;11(6):681–684. doi: 10.1093/bioinformatics/11.6.681. [DOI] [PubMed] [Google Scholar]
- Gill S.C., Von H.P. Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 1989;182(2):319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- Grakoui A., Levis R., Raju R., Huang H.V., Rice C.M. Cis-acting mutation in the Sindbis virus junction region which affects subgenomicrna-synthesis. J. Virol. 1989;63:5216–5227. doi: 10.1128/jvi.63.12.5216-5227.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandadam M., Caro V., Plumet S., Thiberge J.M., Souares Y. Chikungunya virus, southeastern France. Emerg. Infect. Dis. 2011;17(5):910–913. doi: 10.3201/eid1705.101873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gras S., Kedzierski L., Valkenburg S.A. Cross reactive CD8+ T-cell immunity between the pandemic H1N1-2009 and H1 N1-1918 influenza A viruses. Proc. Natl. Acad. Sci. U.S.A. 2010;107(28):12599–12604. doi: 10.1073/pnas.1007270107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guex N., Peitsch M.C. SWISS-MODEL and the Swiss-PdbViewer, an environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- Guruprasad K., Reddy B.V., Pandit M.W. Correlation between stability of a protein and its dipeptide composition, a novel approach for predicting in vivo stability of a protein from its primary sequence. Protein Eng. 1990;4(2):155–161. doi: 10.1093/protein/4.2.155. [DOI] [PubMed] [Google Scholar]
- Hasan M.A., Hossain M., Alam M.J. A computational assay to design an epitope-based peptide vaccine against Saint Louis encephalitis virus. Bioinform. Biol. Insights. 2013;7:347–355. doi: 10.4137/BBI.S13402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasan M.A., Alauddin S.M., Al-Amin M., Nur S.M., Mannan A. In silico molecular characterization of cysteine protease YopT from Yersinia pestis by homology modeling and binding site identification. Drug Target Insights. 2014;13(8):1–9. doi: 10.4137/DTI.S13529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasan M.A., Mazumder M.H.H., Khan M.A., Hossain M.U., Chowdhury A.S.M.H.K. Molecular characterization of legionellosis drug target candidate enzyme phosphoglucosamine mutase from Legionella pneumophila (strain Paris): an in silico approach. Genomics Inform. 2014;12(4):268–275. doi: 10.5808/GI.2014.12.4.268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Her Z., Malleret B., Chan M., Ong E.K., Wong S.C., Kwek D.J., Tolou H., Lin R.T., Tambyah P.A., Rénia L., Ng L.F. Active infection of human blood monocytes by Chikungunya virus triggers an innate immune response. J. Immunol. 2010;184:5903–5913. doi: 10.4049/jimmunol.0904181. [DOI] [PubMed] [Google Scholar]
- Idrees S., Ashfaq U.A. Structural analysis and epitope prediction of HCV E1 protein isolated in Pakistan: an in-silico approach. Virol. J. 2013;10:113. doi: 10.1186/1743-422X-10-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikai A. Thermostability and aliphatic index of globular proteins. J. Biochem. 1980;88(6):1895–1898. [PubMed] [Google Scholar]
- Janeway C.A., Travers P., Walport M., Shlomchik M.J. 5th ed. Garland Science; New York/London: 2001. Immunobiology. ISBN 0-8153-4101-6. [Google Scholar]
- Judson R., Richard A., Dix D., Houck K., Elloumi F. ACToR—aggregated computational toxicology resource. Toxicol. Appl. Pharmacol. 2008;233(1):7–13. doi: 10.1016/j.taap.2007.12.037. [DOI] [PubMed] [Google Scholar]
- Karplus P.A., Schulz G.E. Prediction of chain flexibility in proteins. Naturwissenschaften. 1985;72:212–213. [Google Scholar]
- Kelley L.A., Sternberg M.J. Protein structure prediction on the web: a case study using the Phyre server. Nat. Protoc. 2009;4:363–371. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- Klavinskis L.S., Whitton J.L., Oldstone M.B. Molecularly engineered vaccine which expresses an immunodominant T-cell epitope induces cytotoxic T lymphocytes that confer protection from lethal virus infection. J. Virol. 1989;63(10):4311–4316. doi: 10.1128/jvi.63.10.4311-4316.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudsen A.B. Global distribution and continuing spread of Aedes albopictus. Parassitologia. 1995;37:91–97. [PubMed] [Google Scholar]
- Kolaskar A.S., Tongaonkar P.C. A semi-empirical method for prediction of anti-genic determinants on protein antigens. FEBS Lett. 1990;276(1–2):172–174. doi: 10.1016/0014-5793(90)80535-q. [DOI] [PubMed] [Google Scholar]
- Korber B., LaBute M., Yusim K. Immunoinformatics comes of age. PLoS Comput. Biol. 2006;2(6):e71. doi: 10.1371/journal.pcbi.0020071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar N.P., Joseph R., Kamaraj T., Jambulingam P. A226V mutation in virus during the 2007 Chikungunya outbreak in Kerala, India. J. Gen. Virol. 2008;89:1945–1948. doi: 10.1099/vir.0.83628-0. [DOI] [PubMed] [Google Scholar]
- Labadie K., Larcher T., Joubert C., Mannioui A., Delache B., Brochard P., Guigand L., Dubreil L., Lebon P., Verrier B. Chikungunya disease in nonhuman primates involves long-term viral persistence in macrophages. J. Clin. Invest. 2010;120:894–906. doi: 10.1172/JCI40104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahariya C., Pradhan S.K. Emergence of Chikungunya virus in Indian subcontinent after 32 years: a review. J. Vector Borne Dis. 2006;43(4):151–160. [PubMed] [Google Scholar]
- Lapelosa M., Gallicchio E., Arnold G.F., Arnold E., Levy R.M. In silico vaccine design based on molecular simulations of rhinovirus chimeras presenting HIV-1 gp41 epitopes. J. Mol. Biol. 2009;385(2):675–691. doi: 10.1016/j.jmb.2008.10.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen M.V., Lundegaard C., Lamberth K., Buus S., Lund O. Large-scale validation of methods for cytotoxic T-lymphocyte epitope prediction. BMC Bioinform. 2007;8:424. doi: 10.1186/1471-2105-8-424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski R.A., Rullmannn J.A., MacArthur M.W., Kaptein R., Thornton J.M. AQUA and PROCHECK-NMR, programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR. 1996;8:477–486. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- Lee-Jah C., Kimberly A.D., Floreliz H.M., Jamie G.S., Sandra S. Safety and tolerability of Chikungunya virus-like particle vaccine in healthy adults: a phase 1 dose-escalation trial. Lancet. 2014;14:61185–61195. doi: 10.1016/S0140-6736(14)61185-5. [DOI] [PubMed] [Google Scholar]
- Leparc-Goffart I., Nougairede A., Cassadou S., Prat C., de Lamballerie X. Chikungunya in the Americas. Lancet. 2014;383:514. doi: 10.1016/S0140-6736(14)60185-9. [DOI] [PubMed] [Google Scholar]
- Liao L., Noble W.S. Combining pairwise sequence similarity and support vector machines for detecting remote protein evolutionary and structural relationships. J. Comput. Biol. 2003;10(6):857–868. doi: 10.1089/106652703322756113. [DOI] [PubMed] [Google Scholar]
- Marshall S.J. Developing countries face double burden of disease. Bull. World Health Organ. 2004;82(7):556. [PMC free article] [PubMed] [Google Scholar]
- Maupetit J., Derreumaux P., Tufféry P. PEP-FOLD: an online resource for de novo peptide structure prediction. Nucleic Acids Res. 2009;37(Web Server issue):W498–W503. doi: 10.1093/nar/gkp323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maupetit J., Derreumaux P., Tuffery P. A fast and accurate method for large-scale de novo peptide structure prediction. J. Comput. Chem. 2010;31:726–738. doi: 10.1002/jcc.21365. [DOI] [PubMed] [Google Scholar]
- Muh H.C., Tong J.C., Tammi M.T. AllerHunter: a SVM-pairwise system for assessment of allergenicity and allergic cross-reactivity in proteins. PLoS ONE. 2009;4(6):e5861. doi: 10.1371/journal.pone.0005861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nougairede A., De Fabritus L., Aubry F., Gould E.A., Holmes E.C. Random codon re-encoding induces stable reduction of replicative fitness of Chikungunya virus in primate and mosquito cells. PLoS Pathog. 2013;9(2):e1003172. doi: 10.1371/journal.ppat.1003172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozden S., Lucas-Hourani M., Ceccaldi P.E., Basak A., Valentine M. Inhibition of Chikungunya virus infection in cultured human muscle cells by furin inhibitors: impairment of the maturation of the E2 surface glycoprotein. J. Biol. Chem. 2008;283:21899–21908. doi: 10.1074/jbc.M802444200. [DOI] [PubMed] [Google Scholar]
- Paquet C., Quatresous I., Solet J.L., Sissoko D., Renault P. Chikungunya outbreak in reunion: epidemiology and surveillance. Euro Surveill. 2006;11:2. doi: 10.2807/esw.11.05.02891-en. [DOI] [PubMed] [Google Scholar]
- Parker J.M., Guo D., Hodges R.S. New hydrophilicity scale derived from high-performance liquid chromatography peptide retention data: correlation of predicted surface residues with antigenicity and X-ray-derived accessible sites. Biochemistry. 1986;25(19):5425–5432. doi: 10.1021/bi00367a013. [DOI] [PubMed] [Google Scholar]
- Perryman A.L., Santiago D.N., Forli S., Santos-Martins D., Olson A.J. Virtual screening with AutoDock Vina and the common pharmacophore engine of a low diversity library of fragments and hits against the three allosteric sites of HIV integrase: participation in the SAMPL4 protein-ligand binding challenge. J. Comput. Aided Mol. Des. 2014;28(4):429–441. doi: 10.1007/s10822-014-9709-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters B., Sette A. Generating quantitative models describing the sequence specificity of biological processes with the stabilized matrix method. BMC Bioinform. 2005;6:132. doi: 10.1186/1471-2105-6-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pistonet T., Ezzedine K., Schuffenecher I., Receveur M.C., Malvy D. An imported case of Chikungunya fever from Madagascar: use of the sentinel traveller for detecting emerging arboviral infections in tropical and European countries. Travel Med. Infect. Dis. 2009;7:52–54. doi: 10.1016/j.tmaid.2008.10.001. [DOI] [PubMed] [Google Scholar]
- Powers A.M., Logue C.H. Changing patterns of Chikungunya virus: re-emergence of a zoonotic arbovirus. J. Gen. Virol. 2007;88(9):2363–2377. doi: 10.1099/vir.0.82858-0. [DOI] [PubMed] [Google Scholar]
- Purcell A.W., McCluskey J., Rossjohn J. More than one reason to rethink the use of peptides in vaccine design. Nat. Rev. Drug Discov. 2007;6(5):404–414. doi: 10.1038/nrd2224. [DOI] [PubMed] [Google Scholar]
- Queyriaux B., Simon F., Grandadam M., Michel R., Tolou H. Clinical burden of Chikungunya virus infection. Lancet Infect. Dis. 2008;8(1):2–3. doi: 10.1016/S1473-3099(07)70294-3. [DOI] [PubMed] [Google Scholar]
- Ramachandran G.N., Ramakrishnan C., Sasisekharan V. Stereochemistry of polypeptide chain configurations. J. Mol. Biol. 1963;7:95–99. doi: 10.1016/s0022-2836(63)80023-6. [DOI] [PubMed] [Google Scholar]
- Rashad A.A., Keller P.A. Structure based design towards the identification of novel binding sites and inhibitors for the Chikungunya virus envelope proteins. J. Mol. Graph. Modell. 2013;44:241–252. doi: 10.1016/j.jmgm.2013.07.001. [DOI] [PubMed] [Google Scholar]
- Rashad A.A., Mahalingam S., Keller P.A. Chikungunya virus: emerging targets and new opportunities for medicinal chemistry. J. Med. Chem. 2014;57(4):1147–1166. doi: 10.1021/jm400460d. [DOI] [PubMed] [Google Scholar]
- Rezza G., Nicoletti L., Angelini R., Romi R., Finarelli A.C. Infection with Chikungunya virus in Italy: an outbreak in a temperate region. Lancet. 2007;370:1840–1846. doi: 10.1016/S0140-6736(07)61779-6. [DOI] [PubMed] [Google Scholar]
- Rini J.M., Schulze-Gahmen U., Wilson I.A. Structural evidence for induced fit as a mechanism for antibody–antigen recognition. Science. 1992;255(5047):959–965. doi: 10.1126/science.1546293. [DOI] [PubMed] [Google Scholar]
- Robinson M.C. An epidemic of virus disease in southern province, Tanganyika territory, in 1952–53. Trans. R. Soc. Trop. Med. Hyg. 1955;49(1):28–32. doi: 10.1016/0035-9203(55)90080-8. [DOI] [PubMed] [Google Scholar]
- Schilte C., Couderc T., Chretien F., Sourisseau M., Gangneux N., Guivel-Benhassine F., Kraxner A., Tschopp J., Higgs S., Michault A. Type I IFN controls Chikungunya virus via its action on nonhematopoietic cells. J. Exp. Med. 2010;207:429–442. doi: 10.1084/jem.20090851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuffenecker I., Iteman I., Michault A., Murri S., Frangeul L. Genome microevolution of Chikungunya viruses causing the Indian Ocean outbreak. PLoS Med. 2006;3(7):e263. doi: 10.1371/journal.pmed.0030263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeliger D., de Groot B.L. Ligand docking and binding site analysis with PyMOL and Autodock/Vina. J. Comput. Aided Mol. Des. 2010;24:417–422. doi: 10.1007/s10822-010-9352-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharmin, Islam A highly conserved WDYPKCDRA epitope in the RNA directed RNA polymerase of human coronaviruses can be used as epitope-based universal vaccine design. BMC Bioinform. 2014;15:161. doi: 10.1186/1471-2105-15-161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirako Y., Strauss J.H. Regulation of Sindbis virus RNA replication: uncleaved P123 and nsP4 function in minus-strand RNA synthesis, whereas cleaved products from P123 are required for efficient plus-strand RNA synthesis. J. Virol. 1994;68:1874–1885. doi: 10.1128/jvi.68.3.1874-1885.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang B.L. The cell biology of Chikungunya virus infection. Cell Microbiol. 2012;14:1354–1363. doi: 10.1111/j.1462-5822.2012.01825.x. [DOI] [PubMed] [Google Scholar]
- Tenzer S., Peters B., Bulik S., Schoor O., Lemmel E. Modeling the MHC class I pathway by combining predictions of proteasomal cleavage, TAP transport and MHC class I binding. Cell. Mol. Life Sci. 2005;62:1025–1037. doi: 10.1007/s00018-005-4528-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The UniProt Consortium Activities at the Universal Protein Resource (UniProt) Nucleic Acids Res. 2014;42(D1):D191–D198. doi: 10.1093/nar/gkt1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thevenet P., Shen Y., Maupetit J., Guyon F., Derreumaux P. PEP-FOLD: an updated de novo structure prediction server for both linear and disulfide bonded cyclic peptides. Nucleic Acids Res. 2012;40:W288–W293. doi: 10.1093/nar/gks419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiboutot M.M., Kannan S., Kawalekar O.U., Shedlock D.J., Khan A.S. Chikungunya: a potentially emerging epidemic? PLoS Negl. Trop. Dis. 2010;4(4):e623. doi: 10.1371/journal.pntd.0000623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trainor N.B., Crill W.D., Roberson J.A., Chang G.J. Mutation analysis of the fusion domain region of St. Louis encephalitis virus envelope protein. Virology. 2007;360(2):398–406. doi: 10.1016/j.virol.2006.10.033. [DOI] [PubMed] [Google Scholar]
- Trott O., Olson A.J. AutoDockVina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Joolingen W.R., Jong T.D., Lazonder A.W., Savelsbergh E.R., Manlove S. Co-Lab: research and development of an online learning environment for collaborative scientific discovery learning. Comput. Hum. Behav. 2005;21(4):671–688. [Google Scholar]
- Vanlandingham D.L., Hong C., Klingler K., Tsetsarkin K., McElroy K.L. Differential infectivities of o’nyong-nyong and Chikungunya virus isolates in Anopheles gambiae and Aedesaegypti mosquitoes. Am. J. Trop. Med. Hyg. 2005;72(5):616–621. [PubMed] [Google Scholar]
- Weaver S.C. Arrival of Chikungunya virus in the new world: prospects for spread and impact on public health. PLoS Negl. Trop. Dis. 2014;8(6):e2921. doi: 10.1371/journal.pntd.0002921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver S.C., Osorio J.E., Livengood J.A., Chen R., Stinchcomb D.T. Chikungunya virus and prospects for a vaccine. Expert Rev. Vaccines. 2012;11:1087–1101. doi: 10.1586/erv.12.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werneke S.W., Schilte C., Rohatgi A., Monte K.J., Michault A., Arenzana-Seisdedos F., Vanlandingham D.L., Higgs S., Fontanet A., Albert M.L., Lenschow D.J. ISG15 is critical in the control of Chikungunya virus infection independent of UbE1L mediated conjugation. PLoS Pathog. 2011;7:e1002322. doi: 10.1371/journal.ppat.1002322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang E., Tian H., Xu S., Yu X., Xu Q. Iron-catalyzed direct synthesis of imines from amines or alcohols and amines via aerobic oxidative reactions under air. Org. Lett. 2013;15(11):2704–2707. doi: 10.1021/ol4010118. [DOI] [PubMed] [Google Scholar]