

Abstract
This review focuses on bioinformatics technologies employed by the EU-sponsored multidisciplinary VIZIER consortium (Comparative Structural Genomics of Viral Enzymes Involved in Replication, FP6 Project: 2004-511960, active from 1 November 2004 to 30 April 2009), to achieve its goals. From the management of the information flow of the project, to bioinformatics-mediated selection of RNA viruses and prediction of protein targets, to the analysis of 3D protein structures and antiviral compounds, these technologies provided a communication framework and integrated solutions for steady and timely advancement of the project. RNA viruses form a large class of major pathogens that affect humans and domestic animals. Such RNA viruses as HIV, Influenza virus and Hepatitis C virus are of prime medical concern today, but the identities of viruses that will threaten human population tomorrow are far from certain. To contain outbreaks of common or newly emerging infections, prototype drugs against viruses representing the Virus Universe must be developed. This concept was championed by the VIZIER project which brought together experts in diverse fields to produce a concerted and sustained effort for identifying and validating targets for antivirus therapy in dozens of RNA virus lineages.
Keywords: VIZIER, VaZyMolO, VirAliS, Xtrack, EDBase
1. Introduction
This review describes the development and application of bioinformatics by a multidisciplinary network of researchers faced with the challenge of dissecting the proteome of RNA viruses in a structure-based search for antivirals over a span of approximately 5 years.
The European Union supported a consortium of researchers, known as VIZIER, who proposed to target the replicative enzymes of RNA viruses for antivirus therapy (Coutard et al., 2008). RNA viruses form the largest virus class, including major pathogens of humans and domestic animals (Moya et al., 2004). The underlying VIZIER concept champions a proactive approach to combating virus infections. At its core, it envisions that prototype drugs against viruses representing the Virus Universe must be developed and put on the shelf to become immediately available when they are needed most: to contain an outbreak of an “old” or newly emerging infection. To develop this broad range of drugs, targets for antiviral therapy must be identified in all major virus lineages. This is a huge undertaking, whose scale is determined by the known diversity of viruses and the complexity of virus proteomes. As the number of known viruses, closely tracked by the number of sequenced genomes, expands exponentially in time (Belshaw et al., 2009), so do the resources necessary to develop drugs, should each virus to be targeted.
These relationships and associated challenges were recognized early in the VIZIER project. To address them, a specialized bioinformatics section was included in the consortium. This section had three missions which it pursued in close contact with partners from other sections. Firstly, to develop and manage a “traditional” information component of the project that brought together researchers active in bioinformatics, virology, protein production, crystallography, enzymology, and drug development and testing. To this end, versatile and easy-to-use software tools and databases had to be implemented. Secondly, to provide the consortium with recommendations regarding RNA viruses and targets to be studied. This latter broad effort was complemented by the contributions of partners from other sections who selected viruses and designed targets using different approaches and often based on their expertise with a particular group of viruses or proteins. Thirdly, to develop software tools assisting with specialized tasks such as protein production and structure determination, and the dissemination of these results to other members of the consortium.
For target prediction and domain design (Carugo et al., 2007), two software platforms were used: VaZyMolO (Ferron et al., 2005) and VirAliS (Gorbalenya et al., unpublished). The former provided a friendly WEB-based interface to traditional bioinformatics tools used for comparative sequence analysis, e.g. BLAST, allowing the user to exploit existing annotations from VaZyMolO or VIZIER Databases and other public resources in order to derive information related to a sequence query. Every participant in the consortium could use VaZyMolO as a shared platform to design targets for a selected virus. The second platform, Viralis, was used for expert-based predictions across a broad range of viruses. The interaction of researchers with the Viralis platform was typically managed through a strictly defined protocol which included submission of virus genomes for analysis and receipt of predictions through e-mail.
Managing the information flow and predicting targets requires specialized technologies that were brought to the project and further developed by teams involved in the bioinformatics section. For organizing the information component of the project, including management of accumulated data, we considered using initially a specialized Laboratory Information Management System (LIMS). However, a delay with its development and implementation, as well as a lack of motivation to use it, prompted a search for alternative solutions. As a result, VIZIER Targets Database (see below) took over some major functionalities of data management (Fig. 1 ). It was also connected to the Xtrack software platform (Harris and Jones, 2002) which provided tools and an environment for the analysis of 3D structures. This database became a hub for protein crystallization and structures solved by the consortium. Xtrack, connected to EDBase – a server for 3D structure quality assessment – were also used to distribute the targets within the consortium and coordinate work in the crystallography section of VIZIER.
Fig. 1.
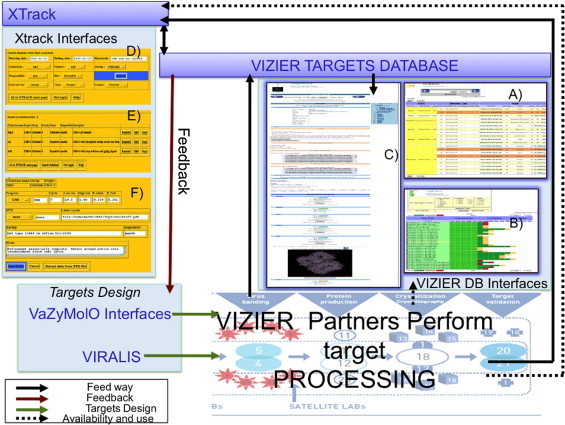
VIZIER targets data pathway. Information stored into the VIZIER Targets Database can be accessed through the three VIZIER Targets DB interfaces including: (A) Targets Data; (B) Targets Status; (C) Target Focus. Xtrack provides also three Interfaces for users that are (D)–(F). Both VIZIER and Xtrack databases are inter-linked. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of the article.)
Some characteristics of the software platforms listed above and associated activities are briefly reviewed below. Other results with a major bioinformatics component are summarized in separate reviews elsewhere in this issue of Antiviral Research.
2. VIZIER Targets Database
The VIZIER consortium gathers European scientists coming from different fields of biology, including bioinformatics, virology, protein production, crystallography, enzymology and drug development (Coutard et al., 2008). In order to enable technical and scientific communication inside the consortium, a website was devised. It served as a common portal to access the core and different specialized databases. The core database, named VIZIER Targets DB, was conceived, implemented and interfaced for managing all VIZIER-related information. Through VIZIER Targets DB, project members easily retrieved data for any target, including its status in the processing pipeline.
Three online interfaces, all empowered with search capabilities, are available to retrieve information at different levels:
-
-
The “Targets Data Interface” displays, in a tabular format, the information related to selected targets. This information may include the virus strain used to generate target DNA and amino-acid sequences, the primers’ sequences for DNA amplification, as well as the description of the last step in the protein processing (Fig. 1(A)). The complete table can be re-arranged by ranking column values, and lines can be highlighted to facilitate comparison. This interface enables a fast checking of the processing progress for a set of proteins. When several closely related constructs are designed the interface provides a snapshot comparing constructs. It assists the user with assessing the impact, on protein production, of any variation in constructs, e.g. tag position, and amino-acid deletion, insertion, or replacement. For example, this type of comparison has been used to identify a crystallizable domain of an Astrovirus protease (Speroni et al., 2009).
-
-
The “Targets Status Interface” displays the status of the processed targets, from PCR production to structure determination using color coding (Fig. 1(B)). The status at each stage can be completed (green), ongoing (orange) or aborted (red). It also indicates the partners in charge of each production step. Using this interface, a comparative view of targets that have passed a production step (cloning, production, crystallization) can be retrieved easily. For example, one search request was sufficient to find what protein from “flavivirus” was “at least crystallized”, as illustrated in Fig. 3 of Coutard et al. (2008).
-
-
The VIZIER “Targets Focus Interface” was developed to present all data for each target on a single page (Fig. 1(C)). While the two previous interfaces allow one to get a comparative view of data and status within a set of targets, this interface focuses on a specific target and displays the technical information available in the database (sequencing of the expression clone, production file, etc.). Since the production of a given protein can be done in different laboratories for different purposes, the original production file can be shared through this interface, or as a PDF file exported from the ELN electronic laboratory book (Contur Software). In addition to the data provided by the VIZIER partner, data from other resources have been integrated, including computer-generated data such as the isoelectric point, the molecular weight and the amino-acid composition of the current target. PDB files related to the target, whenever available can be downloaded and can also be displayed using a Jmol applet launched from the page. Links to additional information on other resources are also displayed (e.g. Xtrack, ICTV DB, GenBank (Benson et al., 2008), PDB (Berman et al., 2007)). This and the other two target DB interfaces were used during the project as a “basic LIMS” without requiring any specific program download.
-
-
This extended use of the VIZIER targets DB was facilitated by a barcode-like nomenclature for constructs that was developed by the consortium. Specifically, a name was assigned to each construct submitted to the DB; for many targets the initial name assignment was made upon target prediction by Viralis (see Section 4.2). The name consists of several parts which are organized in the following order from left to right: (i) the two-digit VIZIER Partner number, (ii) virus abbreviation, (iii) strain designation, (iv) two-digit construct number, (v) protein abbreviation, (vi) function abbreviation, (vii) two-digit target number, (viii) purification tag and its position in the construct. For example, VZ10SV-AY694184-11-3DL-RdRp-01HC is the name of the 3DL protein of the Sapovirus (SV), strain AY694184 provided by partner 10 (VZ10). It is the 11th construct (-11-) of the first predicted target (01) for the RNA-dependent RNA polymerase (RdRp) domain that was cloned fused with His tag at the C-terminus (HC). A search for targets in the VIZIER targets DB can be done on each of the 8 parts of the name.
Target submission and updates of the target's progress processing can be done by any partner using a single or batch procedure. The single procedure is usually required when a single target has to be submitted or updated. The batch mode, based on a pre-filled Excel file, enables the submission or an update for multiple targets at once.
Two thousand four hundred and two different constructs were submitted to the VIZIER Targets Database during the project. They could be identified using a keywords search or, alternatively, BLAST-mediated comparison (Altschul et al., 1997) initiated with a sequence query. A BLAST engine was installed and interfaced on the VIZIER web-server to run against the protein, nucleic acid, and genomic sequences corresponding to the VIZIER targets and against the proteins of the PDB. The BLAST search was routinely used for checking the uniqueness of a prospective target before starting its processing in an effort to avoid undesirable redundancy in the project. Also, BLAST-mediated searching of PDB helped with identifying unique targets whose structures had not already been solved elsewhere.
To facilitate browsing over all information available at the website, a communication protocol between the different VIZIER Targets DB interfaces has been designed and implemented. It enabled access to a specific target focus page from the two other interfaces or from a VIZIER BLAST results page. Following the same approach, the VIZIER Targets DB has also been linked to external resources and particularly to the Xtrack Database dedicated to VIZIER Targets. Since Xtrack was designed to explore, generate and archive crystallographic data, by linking to this resource, we simply and elegantly complement the VIZIER Targets DB.
The VIZIER Targets Database and its interfaces held a central position in the VIZIER project by acting as a reference repository allowing one to browse among the whole set of targets and their data (Fig. 1). The targets data pathway starts with VaZyMolO interfaces and Viralis that were used to design the targets that entered into the VIZIER pipeline. The results and related information of each processing step performed in the VIZIER pipeline were submitted in the VIZIER Targets Database by the corresponding project members. Specific information could also be added into Xtrack. All accumulated information could be accessed and the target processing could be followed easily by all project members using the dedicated interfaces. The VIZIER Targets DB played an important role for providing feedback from sections downstream in the pipeline to researchers who designed the target constructs (Fig. 1).
3. Identifying domain targets for antivirus therapy
3.1. Virus selection
The VIZIER strategy is to study viruses that comprehensively represent the RNA Virus Universe. Due to practical considerations, the project was focused on human and other mammalian viruses. In total, viruses that belong to 8 families/groups of positive-stranded RNA viruses without a DNA stage in their life cycle (ssRNA+) viruses, 3 families/groups of negative-stranded RNA (ssRNA−) viruses, and 4 families/groups of double-stranded (dsRNA) viruses have been analyzed to different degrees using VaZyMolO and Viralis platforms (see below).
The selection of viruses to be characterized by VIZIER was based on several considerations, including virus host and diversity. It was made largely before the project started in close cooperation between virologists and bioinformaticians to include representatives of most major lineages of RNA viruses. For some large virus groups, notably Picornaviruses, with numerous human viruses, a rational selection of a reasonable number of viruses presented a challenge and was extended during the project. A detailed phylogenetic analysis of human enteroviruses (a subset of the Picornaviridae) that form several clusters of closely related viruses (HEV-A, HEV-B, HEV-C and HEV-D) provides an example of how this challenge was addressed (Fig. 2 ). From four human enteroviruses, the cluster HEV-C is of particular clinical importance. It includes three serotypes of poliovirus (PV), a significant human pathogen, and 11 serotypes of coxsackie A virus (C-CAV), a benign human pathogen. Using rooted phylogenetic analysis, it was demonstrated that PV was likely to have originated from an C-CAV-like ancestor (Jiang et al., 2007). Characterization of chimeras and recombinants between PVs and C-CAVs provided support for this evolutionary reconstruction. Based on these observations, it was further proposed that diverse C-CAVs, currently circulating in human population, form a potential reservoir for a new PV-like agent that may emerge. A new pathogenic virus could evolve once PV has been eradicated, after PV vaccination has stopped and once the human population has become immunologically naïve with respect to anti-PV antibodies (Gromeier et al., 1999). This reasoning provided the basis for the decision to include C-CAVs on the list of viruses analyzed by VIZIER.
Fig. 2.
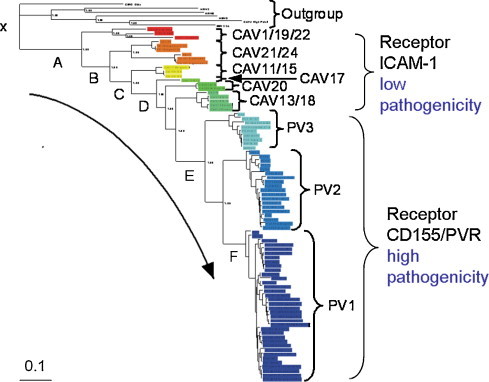
Rooted phylogenetic analysis of C cluster Human Enteroviruses for the structural part of polyprotein. Figure was modified from Fig. 1 in (Jiang et al., 2007).
3.2. Domain target prediction
VIZIER was primarily focused on enzymes of genome replication and expression, many of which, e.g. RNA-dependent RNA polymerase (RdRp) and proteases, are essential for virus viability and are expected to be excellent targets. Other replicative proteins, either non-essential or poorly characterized, were also studied by VIZIER on two major grounds: (1) their analysis may add to our knowledge base of the essential enzymes, and (2) targeting these domains with drugs may produce a dominant-negative effect that will suppress virus growth (Crowder and Kirkegaard, 2005). Because of these considerations, the bioinformatics section aimed at producing comprehensive delineation of protein domains that were used in the project as a starting point on the long road to drug development.
The prediction of domain boundaries is critical for domain identification. There are several approaches to tackle domain boundary prediction for protein sequences, which can roughly be classified as either ab initio or homology-based; they can also be combined in a common framework (Yoo et al., 2008, Liu and Rost, 2004). Using the former approach, multi-domain proteins can be split into domains using different statistics accumulated about the domain borders in proteins and largely ignoring homology relationships. These statistics commonly take into account distributions of structurally disordered regions, secondary structure elements, solvent accessibility and amino-acid properties across globular domains and inter-domain junctions, as well as domain size variation, among others (Bryson et al., 2007, Dovidchenko et al., 2007, Suyama and Ohara, 2003, Dumontier et al., 2005). The second approach explores the homology relationships between proteins that commonly have different sizes and may include two or more domains (Sim et al., 2005, George and Heringa, 2002). The underlying idea is to align as many homologs as possible to identify those N- and C-termini that, when combined, would be separated by the shortest distance; by implication they would flank the smallest common domain for the analyzed set. Among parameters that are of critical importance for the success of this approach are the sensitivity and accuracy of methods used to identify and align homologs in the protein family. The predictive power of all these methods has improved over the years but remains limited in relation to the actual number of domains in proteins and the precise location of domain boundaries (Dovidchenko et al., 2007, Bryson et al., 2007).
RNA viruses have the smallest genomes among all living forms (in the narrow range of ∼3.0–33.0 kb). They are hierarchically inter-connected in a network of evolutionary relationships that could include also very distantly related cellular homologs (Gorbalenya, 1995). Consequently, the RNA virus proteome, although not fully dissected, is dominated by proteins of just a few families (Gorbalenya and Koonin, 1993a), making the homology approach especially powerful in domain delineation.
RNA viruses tend to encode one or two large multi-domain polyproteins that are assembled into functional complexes. These include major replicative enzymes that either readily operate in this form or are processed further to smaller products. In RNA viruses whose polyproteins are proteolytically processed to mature proteins, cleavage sites tend to be highly conserved in and between viruses (Kitamura et al., 1981, Gorbalenya and Snijder, 1996). These sites can be described by characteristic sequence signatures that proved to be invaluable for delineating domains with the authentic termini, including viruses from poorly characterized families (Gorbalenya et al., 1989, Gorbalenya, 2001, Ziebuhr et al., 2001). Neural network predictors have been developed for the identification of a subset of these sites that are recognized by 3C proteases of some picornaviruses (Blom et al., 1996) and 3C-like proteases of coronaviruses (Kiemer et al., 2004) and they can assist in the domain identification. In many other viruses that employ sites with deviant sequence signatures, which could also be relatively poorly conserved, these predictors may not work so well, leaving the site identification to experts.
It was shown that the authentic terminus could be essential for functioning of some key replicative enzymes of RNA viruses, e.g. PV RNA-dependent RNA polymerase (RdRp) (Gohara et al., 1999). Consistent with this observation, characterization of terminally modified derivatives of viral enzymes may miss structural features that could be critical for designing antivirus drugs (Thompson and Peersen, 2004, Hansen et al., 1997). Because of these considerations, defining targets with authentic termini (boundaries) was considered to be beneficial in the VIZIER project. Occasionally such targets may be terminally modified (truncated or extended) for practical reasons, e.g. to address difficulties with cloning, protein expression and purification, crystallization, or structure determination.
At the start of the Vizier project it was estimated that an “average” RNA virus genome may encode 4–6 replicative domains. In line with these estimates, Viralis has been used for predicting approximately 790 original and refined targets for more than a hundred viruses that were submitted for analysis (Fig. 3 ). Among predicted domains are diverse and distantly related RdRps (Kamer and Argos, 1984), helicases of three superfamilies (HEL1, HEL2 and HEL3) (Gorbalenya and Koonin, 1993b), diverse proteases employing papain-like and chymotrypsin-like folds (CHL-Pro and PL-Pro, respectively) (Gorbalenya et al., 1989a, Gorbalenya et al., 1989b, Gorbalenya et al., 1991), diverse methyltransferases targeting 2′O and N7 atoms in RNA substrates (OMT and NMT, respectively) (Rozanov et al., 1992, Koonin, 1993, Ferron et al., 2002, Feder et al., 2003), 3′-to-5′ exoribonuclease (ExoN) (Snijder et al., 2003), uridylate-specific endoribonuclease (NendoU) (Snijder et al., 2003), acyltransferase (AT) (Hughes and Stanway, 2000), cyclic phosphodiesterase (CPD) (Mazumder et al., 2002, Snijder et al., 2003), diverse Zn-binding domains (Zc) (Gorbalenya, 1992, Gorbalenya et al., 2006), ubiquitin-like domains (Ub) (Serrano et al., 2007, Ratia et al., 2006), proteins covalently bound to virus RNA (VPg) (Gorbalenya and Koonin, 1993a, Paul et al., 1998) and adenosine diphosphate-ribose 1″-phosphatase (ADRP) (Snijder et al., 2003). Also a large number of poorly characterized domains that were recognized as unique for phylogenetically compact groups of viruses, e.g. group 2a of coronaviruses (g2aUD) (Gorbalenya, unpublished observations; Snijder et al., 2003, Neuman et al., 2008), were predicted. Numerous targets represented by various combinations of adjacent domains were also delineated. For a considerable number of viruses, the originally predicted targets were refined after feedback from colleagues about their experience with cloning, protein expression, crystallization and structure solving, and by accommodating newly published results from other laboratories.
Fig. 3.
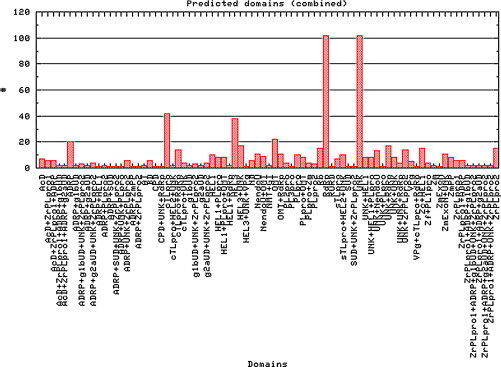
Separate domains and their combinations (targets) predicted for RNA viruses with Viralis.
4. Software platforms for RNA virus bioinformatics
4.1. VaZyMolO
The VaZyMolO (Viral enZyme Module lOcalization) is a database of modular annotations on viral proteins (Ferron et al., 2005) that is available as a stand-alone resource and a web-based portal at http://www.vazymolo.org. It aims at defining protein modules suitable for purification and crystallization; it could be useful for structural genomics on viral replication. Proteins are annotated using tools to define amino-acid composition, and conduct hydrophobic clusters analysis, secondary structure prediction, homology modeling using solved structures and data mining on biochemistry (function and motifs, active sites, cleavage sites, etc.).
For the VIZIER project, specific interfaces to this database have been developed (available to project members as private pages from http://www.vazymolo.org), and the database itself was reshaped and completed by additional content such as feedback information from experimental results as well as genomic information to facilitate the browsing within the database interfaces. These VaZyMolo Interfaces constitute a web access point to the VaZyMolO database and allows the use of the database content as a tool for target design by identifying modularity within viral proteins (disorder, hydrophobic parts, linkers, etc.).
This online access is provided through three major interfaces including VaZyMolO Browser, Browser Focus and Blast and Tools:
-
-
The VaZyMolO Browser interface allows navigation through the data available in the VaZyMolO database by using a search module and sort function capabilities. Thus it facilitates the identification of domains of interest present in browsed proteomes.
-
-
The VaZyMolO Browser Focus interface deals with organism, genomic, proteomic and modularity information. It is available by selecting an organism or a specific protein on the VaZyMolO Browser interface. It allows the user to zoom in on information related to the selected organism (virus) and to map an overview for each CDS on the viral genome or, for a VaZyMolO domain, on the corresponding proteins. Diverse data including the amino-acid sequences, computed molecular weight, isoelectric point, and the presence of homologous domains in other proteins can be accessed (Fig. 4 ).
-
-
The VaZyMolO Blast and Tools provides an interface to programs that use a sequence as the input for launching different analyses:
-
(a)
BLASTX and BLASTP mediate comparison of a query sequence against the VaZyMolO database to identify sequence regions having similarities with predicted domains in VaZyMolO.
-
(b)
BLAST mediates query comparison against two public databases, PDB and Merops (Rawlings et al., 2008), to retrieve information about known structures and proteins with known peptidase functions, respectively.
-
(c)
External tools such as HCA (hydrophobic cluster analysis), TMHMM (Prediction of transmembrane helices in proteins) (Krogh et al., 2001), are also available from this interface. These tools allow the user to refine or check the provided domain predictions.
Fig. 4.
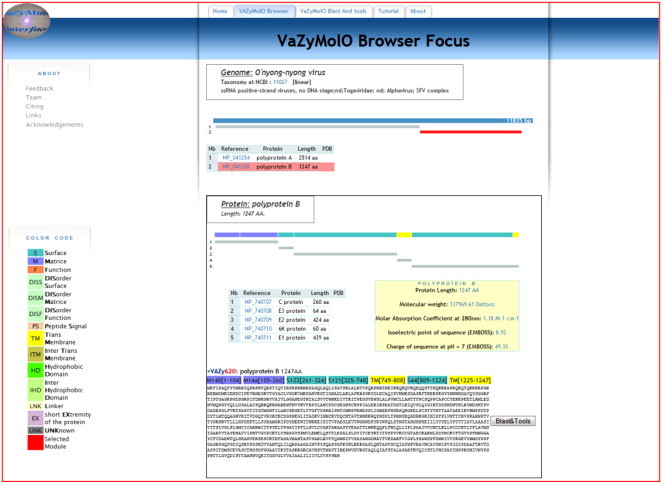
The VaZyMolO Browser Focus Interface. It allows browsing from genomic information to a protein's module details.
Since the prediction methods are partly based on similarity searches, the efficiency of VaZyMolO depends partly on the number of sequences available in the database. Indeed, the larger the sequence database is, the more reliable may be the results. The current version of the VaZyMolO database contains proteins derived from the fully sequenced virus genomes available in GenBank on 1 January 2008 and amounts to 656 virus sequences. Compared to the original Vazymolo launched in 2005 by Ferron and colleagues (Ferron et al., 2005), not only is the number of sequences four times larger, but the virus families are more diverse (see Table 1 ). Other RNA viruses of plant or insect origins, which can share some similarities with mammalian viruses, are now available in the VaZyMolO; ds RNA viruses remain to be included.
Table 1.
Virus entries in VaZyMolO: impact of the Vizier project.a.
| Class | No. | Current no. |
|---|---|---|
| Family | Before the VIZIER project | |
| ssRNA negative-strand viruses | 64 | 111 |
| Bornaviridae | 1 | 1 |
| Filoviridae | 3 | 4 |
| Paramyxoviridae | 27 | 36 |
| Rhabdoviridae | 12 | 22 |
| Arenaviridae | 3 | 13 |
| Bunyaviridae | 9 | 23 |
| Orthomyxoviridae | 6 | 12 |
| ssRNA positive-strand viruses | 50 | 545 |
| Arteriviridae | 4 | 4 |
| Coronaviridae | 8 | 9 |
| Flaviviridae | 32 | 45 |
| Narnaviridae | 6 | 8 |
| Picornaviridae | 0 | 38 |
| Flexiviridae | 0 | 64 |
| Comoviridae | 0 | 18 |
| Sequiviridae | 0 | 3 |
| Luteoviridae | 0 | 18 |
| Tombusviridae | 0 | 41 |
| Caliciviridae | 0 | 16 |
| Potyviridae | 0 | 66 |
| Astroviridae | 0 | 6 |
| Hepeviridae | 0 | 1 |
| No Family | 0 | 104 |
| Tetraviridae | 0 | 4 |
| Bromoviridae | 0 | 25 |
| Marnaviridae | 0 | 1 |
| Tymoviridae | 0 | 14 |
| Closteroviridae | 0 | 19 |
| Barnaviridae | 0 | 1 |
| Nodaviridae | 0 | 9 |
| Idae | 0 | 1 |
| Dicistroviridae | 0 | 14 |
| Togaviridae | 0 | 16 |
The numbers in the right column indicate the current number of entries of selected RNA virus families described in VaZyMolO.
To expand sources of annotation beyond those retrieved through similarity-based searches, a new tool identifying disorder regions in proteins was devised. It is a metaserver producing a consensus by analyzing the results generated by different existing tools. It improves the speed and accuracy of defining disordered domains for the annotation procedure. This program is named MeDor (Metaserver of Disorder) (Lieutaud et al., 2008). It provides a graphical interface with a unified view of the output of multiple disorder predictors (Fig. 5 ). It allows fast, simultaneous analysis of a query sequence by multiple predictors and easy comparison of the results provided by the latter. It also enables standardized access to disorder predictors and allows meaningful comparisons among various query sequences. Regions of interest to be removed can then be manually highlighted and their sequences can be retrieved. The MeDor program was added to the tools available from the VaZyMolO Blast and Tools interface to enhance the capabilities of this interface. A stand-alone version of the program is available online for downloading.
Fig. 5.
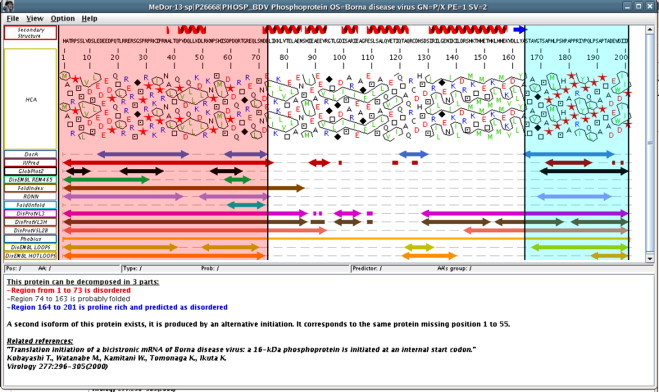
The MeDor program's output for phosphoprotein P24 of the Borna disease virus (accession number P26668). The highlighted regions in red and blue, and the textual comments constitute annotations made by a user. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of the article.)
VaZyMolO, therefore, now constitutes an integrated tool for target design, based on similarity searches and further improved by new annotations integrating feedback generated by VIZIER experiments.
4.2. Viralis
The Viralis software platform was developed to assist handling of RNA virus genomes, building alignments, and to facilitate diverse analyses of sequence-based information. It is mainly written in Perl/Tcl and includes in-house genome and alignment MySQL relational databases (VDB-GS and VDB-GA, respectively) and integrated bioinformatics tools for comparative sequence analysis that are accessed from specialized modules. An original XML protocol was developed in Viralis for inter-module data transfer including information about biopolymer sequences, alignments, secondary structure and accompanying annotation. These modules provide access also to local copies of the public genome and protein databases including GenBank (Benson et al., 2008), UniProt (Bairoch et al., 2009), PFAM (Finn et al., 2008), CDD (Marchler-Bauer et al., 2009) and PDB (Berman et al., 2007). The databases are regularly updated and can be searched using diverse programs, including HMMER (Eddy, 1996), BLAST (Altschul et al., 1997), and COMPASS (Sadreyev and Grishin, 2003), to retrieve sequences that are subsequently aligned using ClustalX (Thompson et al., 1997), T-Coffee (Notredame et al., 2000, Simossis et al., 2005), MUSCLE (Edgar, 2004), or Dialign (Morgenstern, 2004) programs. Alignments can be analyzed to predict disorder regions using FoldUnfold (Galzitskaya et al., 2006) and RONN (Yang et al., 2005), and to map secondary structure elements identified in the tertiary structures of proteins (Kabsch and Sander, 1983) included in the alignment. Also the PROSITE database (Hulo et al., 2008) assists with the identification of sequence signatures characteristic for different protein families. Using these instruments, experts build and analyze alignments to produce functional and structural assignments mainly through transfer of knowledge from characterized proteins to their homologs. This annotation transfer is currently expert-mediated; in future, it could be made in a statistically rigorous fashion. As a step in this direction, comparative analysis of various annotation transfer statistics, based on the likelihood ratio criterion, has been conducted (Leontovich et al., 2008).
The VIZIER users interacted with Viralis through a dedicated page (Fig. 6 ) of the Viralis website written in PHP. It mediates sequence upload for expert analysis and access to up-to-date status of processing of all submitted queries and a database of predicted targets. To initiate query processing, the user was required to define a virus family of the uploaded sequence. Two ways of sequence uploading to Viralis were provided: by copy-and-paste or through retrieving a publicly available sequence from GenBank/RefSeq using their identifiers (gi, accession number, accession number with version, locus name). Each submitted sequence was assigned a unique name tag encoding names of submitting partner and virus to be studied, according to a convention developed by the VIZIER consortium (see Section 2). After a sequence was annotated in Viralis, tentative targets were sent from the Domain Prediction Response (DPR) module to the user who requested predictions (Fig. 7 ). Each target was selected from a pre-compiled domain list to initiate a response with the DPR module. At this stage, a target was assigned with a unique tag that was based on the respective sequence tag extended to include name abbreviations for protein and domain, and target version (two digits). The target response file included amino-acid and nucleotide sequences of the target flanked by extra sequences that are clearly highlighted and numbered. This format allowed the target recipient to verify the identity of the target and readily design primers for cloning. The DPR module provided also a mechanism for canceling target predictions by sending a notification to the target recipient. In practise, it was used mainly to correct target name assignments. The VIZIER users had ready access to all submitted sequences and received targets through the progress monitoring table at the Viralis WEB site (Fig. 6).
Fig. 6.
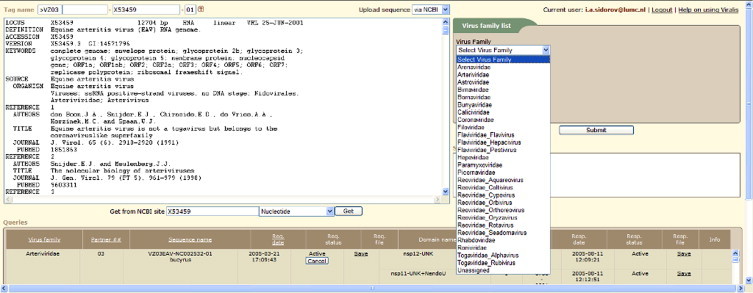
Viralis Web interface to handle user queries and access targets predicted in RNA viruses.
Fig. 7.
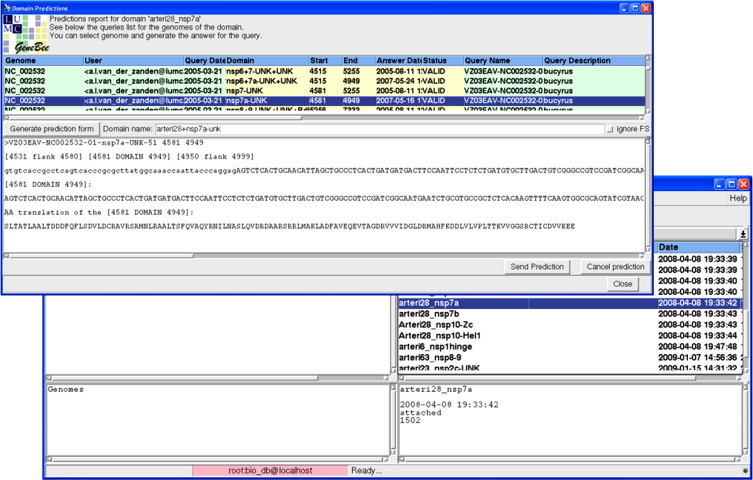
Viralis Domain Prediction Response module interface.
Virus analysis by Viralis was conducted in the context of publicly available genome sequences for respective virus families. To ensure that VDB-GS is up-to-date, an original procedure was developed for virus genome retrieval from GenBank (Sidorov, Samborskiy and Gorbalenya, in preparation). It is based on several sequence characteristics, including genome conservation and length, and protein domain organization. When combined, these characteristics can uniquely identify genomes of a particular family. The procedure is essentially annotation-independent, virtually eliminating a possible negative effect of incomplete or erroneous entry annotations on genomes retrieval. A web site, provisionally called SARGENS (http://veb.lumc.nl/SARGENS), was also designed using Perl to provide public access to regularly updated databases of complete genomes of selected RNA virus families. They are available for downloading in the Fasta format and their GenBank records can be accessed through links at the SARGENS web page.
In VIZIER and outside of the project, the results of bioinformatics analyses are often communicated as a multiple sequence alignment or tree whose preparation may be a cumbersome process that involves replacement of sequence ID with names informative to humans. To facilitate this conversion, Sidorov et al. (2009) have developed a sequence name annotation-based designer (SNAD) (http://veb.lumc.nl/SNAD/); its functionalities were also incorporated into the Viralis platform. The SNAD web site provides a user-friendly interface to a versatile tool that works with identifiers presented as a plain list or in diverse formats of multiple sequence alignments and trees. It can convert sequence IDs into names using sequence annotation from several public databases according to a name template that controls the structure and content of the name. The user can choose from a set of predefined templates or design a new template using a versatile template-building facility. An example of SNAD-mediated conversion using a virus tree as input and one of the predefined templates is illustrated in Fig. 8 . This tool facilitates communication and knowledge dissemination about genome-based data that dominate output in virus-related research.
Fig. 8.
ID-to-name conversion in tree of alphaviruses by SNAD.
5. Laboratory Without Walls (LW2)
5.1. Why is this needed within VIZIER?
VIZIER includes more than 20 laboratories, of which eight are actively involved in trying to determine protein structures using X-ray crystallography. Systems were needed to ensure interactions between the crystallographic laboratories, and between structural biologists and virologists. The Laboratory Without Walls concept (LW2) is our attempt to achieve this difficult goal. With the correct access privileges, a virologist in Leiden would be able to access the crystallographic information for a particular target that is being evaluated and generated by the crystallography group in Pavia, for example. At the same time, the target allocation committee may require access to the latest set of new targets from the protein production pipeline, and the necessary information that would be required to assign a particular target to a particular laboratory. The LW2 system, therefore, is needed for coordination as well as collaboration between VIZIER laboratories. It is achieved with a web-based front end to a database system Xtrack/EDBase whose essential components are outlined in Fig. 9 .
Fig. 9.
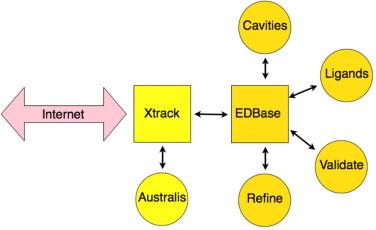
Xtrack/EDBase components and interactions.
5.2. Xtrack
Xtrack is a Laboratory Notebook System (LNS), a simplified Laboratory Information Management System (LIMS) that allows users to keep track of crystallographic data from protein expression through to crystallization, data collection and processing (Harris and Jones, 2002). It is written in PHP and accesses a Postgres database through SQL commands. Xtrack was originally designed to be extremely simple to use by beginners and infrequent users, and we have tried to keep this philosophy in our more recent updates and developments. The deposited information is arranged around a collection; an entity associated with a set of crystallographic data associated with a particular structure. Collections in turn are split up into more detailed pages that may be related to the target, protein expression, crystallization, data collection, structure solution, refinement and validation; see Harris and Jones (2002) for more details. Access to the database is controlled by user accounts and passwords. All Xtrack users must be registered and assigned to one or more groups. The user may then read and modify data only within those groups, but all members within a particular group are implicitly trusted, and may modify any group-related data. In the Vizier implementation, each user belongs to two groups – one representing their laboratory, and the pseudo-group ‘Vizier’ which has only read access to other groups’ data.
Xtrack has been further developed for purposes that are specific to VIZIER's needs. One vital aspect of our project is the allocation of targets to the crystallographic laboratories. Since this target-related information is kept in Marseille and we had no desire to duplicate this, we have built bi-directional connections between the databases. The main target list page in Xtrack has a column labelled ‘submission’, containing names like VZ04MODV-Q8QL64-01-UNK-MT-01HN. These codes are the links to the Marseille VIZIER database entry for that target. At the bottom of that VIZIER database page is a link back to Xtrack. The implementation is through PHP's libcurl package, which uses a secure connection protocol. The connection is also restricted to the IP address of our two hosts. With this information in hand, new targets are given an initial allocation code POOL before being assigned to a crystallographic laboratory. Every night, a script is run to carry out a BLAST search (Altschul et al., 1990) of all targets within Xtrack amongst themselves and against the deposited structures at the Protein Data Bank (PDB). It then becomes relatively straightforward for the target allocation committee to see the most closely related structures that are being worked on within VIZIER and outside the consortium. The crystallographic laboratories can also use this information to decide on their internal priority for a particular target. Fig. 10 , shows an Xtrack listing for one such target, VZ93, a methyltransferase from Modoc virus (Jansson et al., 2009).
Fig. 10.
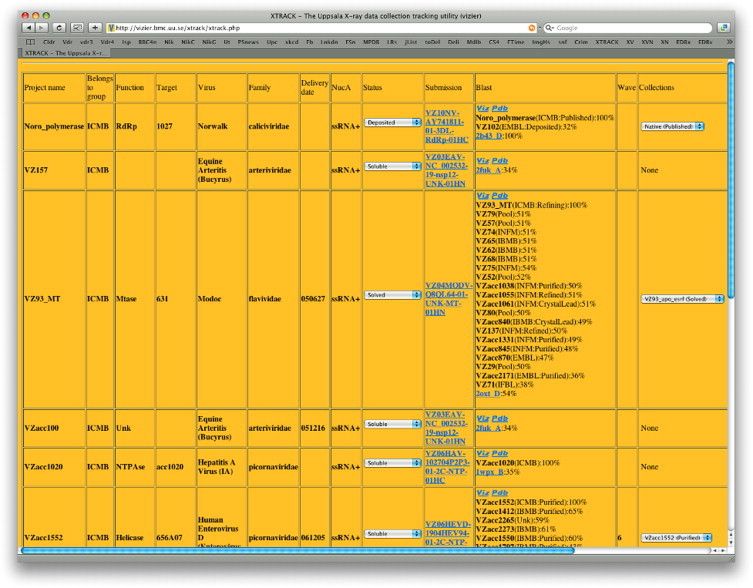
Xtrack project listing, centred on target VZ93. Some fields are clipped to the right of the image.
This target was one of the first such enzymes produced within VIZIER, but as the project progressed a number of closely related targets became available. The interested consortium member can click on a button to see a superposition of the most closely related 3D structures, generated by our AuStrAliS structural supposition server. AuStrAliS is a web-based server that finds structural alignments between protein chains. It can either be run from a stand-alone interface, or from the Xtrack-PDB BLAST hit page. It performs pairwise alignments between selected structures, and then superimposes the molecules for viewing either in a Java viewer (Jmol) within the browser window, or by downloading a package that can be read into O (Jones et al., 1991). It is powered by LSQMAN for making the detailed superpositions (Kleywegt and Jones, 1997a, Kleywegt and Jones, 1997b).
5.3. EDBase
The starting point for this system was the Uppsala Electron-Density Server, EDS (Kleywegt et al., 2004). EDS is a service for evaluating the electron density and model quality of crystal structures deposited in the PDB. It consists of a number of modules written in C, Perl, Fortran and Java, and is again accessed through the web. The LW2 implementation of EDS is known as EDBase, and is activated directly from Xtrack. EDBase, Fig. 11a, is a utility that allows crystallographers to upload their experimental data and molecular models to our server, and to then request calculation of maps and statistics, along with a series of quality factors that summarize the state of their current model. These include the usual crystallographic R-factor, as well as a number of quality control coefficients that have been developed in Uppsala (Kleywegt and Jones, 1995, Kleywegt and Jones, 1997a, Kleywegt and Jones, 1997b) such as the average real-space correlation coefficient (Jones et al., 1991), and the number of Ramachandran outliers (Kleywegt and Jones, 1996a). If calculations have already been made on previous models, then those quality factors that have become worse in the latest model are flagged in red. At this point the user can also choose to perform a refinement on the uploaded model within EDBase. In fact they can choose to perform several different refinements using different weighting parameters, and then see which quality factors improve and which deteriorate with the different refinement strategies.
Fig. 11.
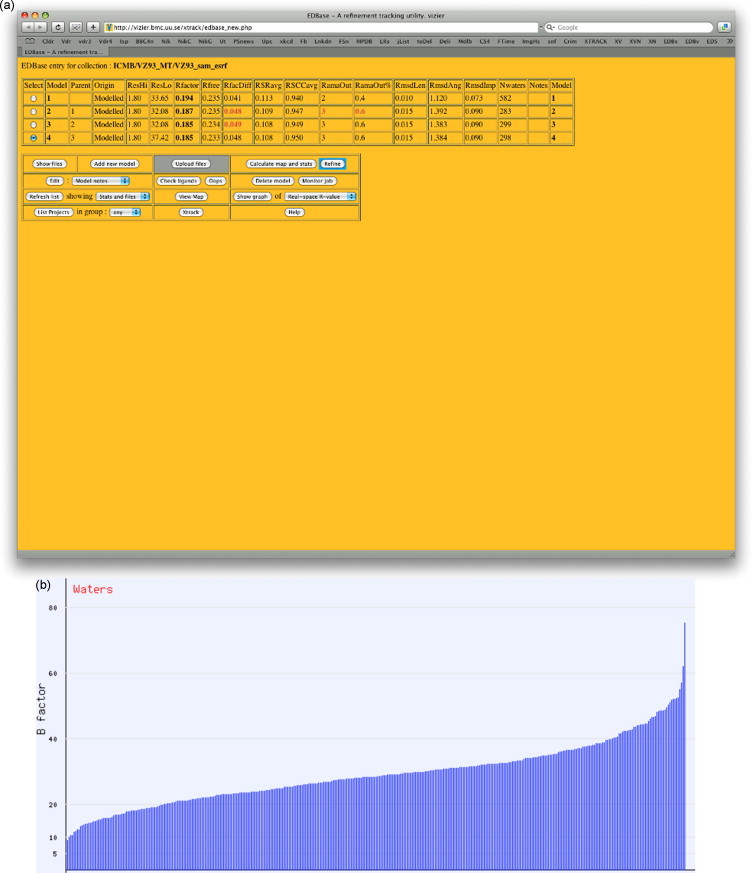
Access to EDBase for VZ93 example. (a) Four models (with goodness-of-fit indicators) are stored in the database and (b) solvent B-factor distribution after refinement.
Two crystallographic refinement systems are available with EDBase, REFMAC5 (Murshudov et al., 1997) which is part of the CCP4 package (Collaborative Computational Project No. 4, 1994), and Buster-TNT (Blanc et al., 2004) via the Autobuster script. Autobuster, in turn, has scripts for adding additional waters at any stage in the refinement.
EDBase also provides access to the water-adding functionality in O version 12 (TAJ, to be published). These tools provide a more quantitative approach to deciding the level at which electron-density maps (especially maps made with amplitudes of type |Fo| − |Fc|) should be inspected, and how to decide on the addition of new water molecules during crystallographic refinement. Such decisions are made in terms of the average carbonyl oxygen of the structure under refinement, using a 3D profile that is specific to this structure and crystallographic dataset. If an EDBase user elects to select water molecules based on a low fraction (e.g. 0.5), more waters will be selected which after crystallographic refinement will show a gradual increase in temperature factor. For structures solved to suitably high resolution, such a plot of increasing B-factors will often show a hockey-stick profile, Fig. 11b, that clearly indicates where waters should be deleted from the model. O's use of carbonyl oxygen profiles conveniently allows the user to recognize peaks that are not water molecules but ions or larger entities. The water_electron function in O will normalize the ‘water’ peak into electrons in an attempt to clarify the nature of the atom/group. False positives may occur due to model errors, non-spherical density, and/or low resolution.
EDBase is also equipped with a connection to the Oops program (Kleywegt and Jones, 1996b), which will generate a series of O macros that will step the user through suspect regions of a protein structure by jumping to residues that have unusual statistics. This data and information is available to the EDBase user for downloading to a local computer for offline work and analysis with O (Jones et al., 1991). Numerous residue-based goodness of fit indicators are provided as an aid to identifying problem areas in the structure.
A number of other novel features have been added to EDBase that provide functionality that is otherwise difficult to achieve. ValLigURL, Fig. 12 , is a ligand-searching tool for the validation of ligand structures (Kleywegt and Harris, 2007). The server scans the PDB for a particular ligand, and then compares the geometries of the hits with that of the given ligand structure. As well as a geometric comparison, the structures can also be shown superimposed in the Jmol molecular viewer, and a SMILES representation of the ligand is generated that can be used to search for previously unpublished ligand structures. This service is intended to indicate anomalies in the geometry of refined ligands, by highlighting differences from published structures of the same ligand, and also to aid in identification of novel ligand conformations. It has connections to several external ligand servers:
-
(1)
BabelWeb/ChemDB (Chen et al., 2005) is used to generate the canonical SMILES representation of the ligand so that the user can take this away to their favorite similarity search engine.
-
(2)
Direct links are offered to PDBeChem's searches for substructures and superstructures that are found in the PDB (Golovin et al., 2004). From the results pages of these searches, PDBe offers links to further pages about similar ligands, including full chemical information, and a list of the PDB entries containing them.
-
(3)
There are connections to Relibase's similarity searches (Hendlich et al., 2003). This service now requires a cost-free registration to Relibase before the VaLigURL link can be used. The user can then get access to a list of similar ligands, and links to the PDB structures containing them.
-
(4)
The connection to SuperLigands (Michalsky et al., 2005) not only finds similar structures, but can superimpose them, and display the result using Chime.
-
(5)
The link to BindingDB (Liu et al., 2007) gives access to many more parameters for the ligand.
Fig. 12.
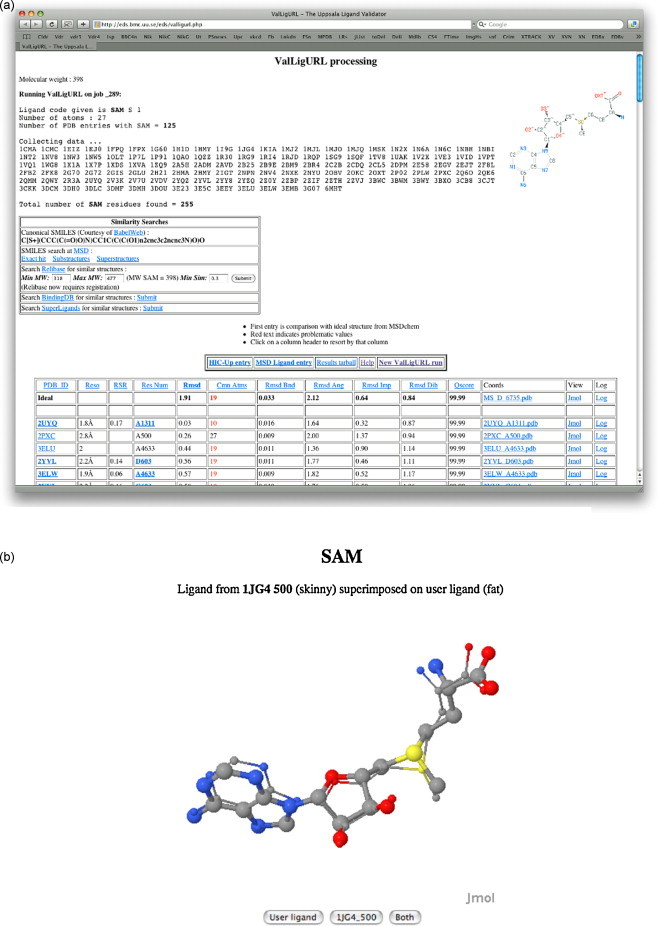
(a) Access to ValLigURL for the VZ95 example. (b) JMol superposition of SAM (s-adenosylmethionine) to PDB entry 1JG4.
CavitySearch is a service that allows users to upload their protein coordinates and then calculate and visualize pockets, tunnels and other cavities. It is available directly from the model listings in the VIZIER EDBase server, or as a stand-alone server. Two algorithms are available – the VOIDOO algorithm (Kleywegt and Jones, 1994) that identifies buried cavities, and the Delaney algorithm (Delaney, 1992) that identifies pockets that connect with the surface, and the user can choose either to show all cavities or only the largest. Probe size, grid size, and minimum and maximum cavity sizes for inclusion can all be set by the user. The calculated cavities are output as net (so-called ‘chicken-wire’) surfaces that can either be viewed directly in the AstexViewer (Hartshorn, 2002), or can be downloaded as a compressed tar-ball file together with startup and macro files for easy viewing within O.
6. Concluding remarks and perspectives
The scale of the VIZIER project, including its broad RNA-virus-wide scope, multidisciplinary approach, and the involvement of over 20 partners from different countries, presented unique opportunities and considerable challenges. Informatics and, notably bioinformatics, were brought to the project to improve inter-partner communication and coordination, to equip the consortium with tools for large-scale and timely analysis of sequences and structures, and to provide recommendations regarding target domain definition. Expectations and their execution were regularly debated at consortium meetings as the project progressed. These discussions raised awareness concerning cultural differences integral to such a broad enterprise, helped build bridges and develop practical solutions to produce inter-partner synergy. Each of the four major software platforms instrumental in the project's success, the VIZIER Target DB, VaZyMolO, Viralis and Xtrack/EDBase, was advanced with new tools, and their databases were updated. They were used to predict and analyze hundreds of domain targets, and conduct other analyses that led, particularly, to the development of a novel concept of virus emergence. To date, VaZyMolO database is publicly accessible without any restrictions and Xtrack and SNAD codes are freely available to the virology and the structural biology communities.
What is the next step? Although the VIZIER project is over, reaching its ultimate goal – to put prototype drugs on the shelf for RNA viruses of all major lineages – remains a work in progress. As is evident from studies conducted in and outside VIZIER, solving the three-dimensional structures of proteins – a crucial step toward developing prototype drugs – proved to be very difficult for RNA viruses, particularly ssRNA− viruses. Thus predicting new domains and refining old predictions remains a part of any sustainable effort to dissect the RNA virus proteome. Suitable target constructs need to be predicted for new viruses that are being identified literally every day, and at an ever increasing pace. With virus numbers soon counted in many orders of magnitude, the selection of those prototypes worth more detailed investigations becomes largely a genomics-based process for which bioinformatics-mediated frameworks need to be developed.
The VIZIER project has undoubtedly contributed to building bridges between our virology and structural biology communities. The informatics/bioinformatics described here has played an important role in forming and developing these interactions, be it through web-based tools, databases or stand-alone programs designed within the project. These tools have not only helped in the daily follow-up of the project, but they have considerably simplified inter-partner communications and the establishment of a common language that contributed to the project culture. Other information-related challenges abound downstream in the pipeline. Most of the laboratories involved in drug testing in VIZIER, for example, have their own pre-existing drug or chemical library databases. These are served with tools describing, managing, and retrieving information about small organic chemical molecules that have a drug potential. It becomes vital to integrate these drug databases into a network that could be used to produce, in a common format, data about structural, chemical, antiviral, pharmacological and toxicological properties related to viruses and targets, and to access all related information acquired on emerging viruses. With these challenges, any successor of VIZIER is sure to become an information-based project if it is to succeed.
7. Availability
Xtrack are freely available to all interested parties. SNAD code is freely available to academics. VaZyMolO database is freely accessible through web site. The AstexViewer software is used by permission and includes code developed by Astex Technology Limited, UK. Jmol is an open-source Java viewer for displaying chemical structures in 3D, http://www.jmol.org/.
Acknowledgements
The authors wish to thank all VIZIER colleagues with whom they collaborated during the project for their advice, questions and patience. The Viralis team (AEG, AAK, DVS, IAS, AML) thanks for collaboration their present and former colleagues at Leiden and Moscow including I. V. Antonov, B. Brandt, J. Faase, A. V. Golovin, C. Lauber, E. P. Nikitina, A. V. Nikonov, V. K. Nikolaev, D. A. Reshetov, M. N. Rozanov, V. A. Sorokin. VaZyMolO, Viralis and Xtrack/EDBase have been developed with partial support from EU IP Project VIZIER (CT 2004-511960). The Viralis development and usage were also partially supported by grants from the Netherlands Bioinformatics Center (BioRangeSP3.2.2) and the Collaborative Agreement in Bioinformatics between LUMC and MSU (CRDF GAP1473).
Contributor Information
Alexander E. Gorbalenya, Email: A.E.Gorbalenya@lumc.nl.
Bruno Coutard, Email: Bruno.coutard@afmb.univ-mrs.fr.
T. Alwyn Jones, Email: alwyn@xray.bmc.uu.se.
References
- Altschul S.F., Madden T.L., Schaffer A.A., Zhang J., Zhang Z., Miller W., Lipman D.J. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Research. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bairoch A., UniProt Consortium, Bougueleret L., Altairac S., Amendolia V., Auchincloss A., Argoud-Puy G., Axelsen K., Baratin D., Blatter M.C., Boeckmann B., Bolleman J., Bollondi L., Boutet E., Quintaje S.B., Breuza L., Bridge A., Decastro E., Ciapina L., Coral D., Coudert E., Cusin I., Delbard G., Dornevil D., Roggli P.D., Duvaud S., Estreicher A., Famiglietti L., Feuermann M., Gehant S., Farriol-Mathis N., Ferro S., Gasteiger E., Gateau A., Gerritsen V., Gos A., Gruaz-Gumowski N., Hinz U., Hulo C., Hulo N., James J., Jimenez S., Jungo F., Junker V., Kappler T., Keller G., Lachaize C., Lane-Guermonprez L., Langendijk-Genevaux P., Lara V., Lemercier P., Le Saux V., Lieberherr D., Lima T.D., Mangold V., Martin X., Masson P., Michoud K., Moinat M., Morgat A., Mottaz A., Paesano S., Pedruzzi I., Phan I., Pilbout S., Pillet V., Poux S., Pozzato M., Redaschi N., Reynaud S., Rivoire C., Roechert B., Schneider M., Sigrist C., Sonesson K., Staehli S., Stutz A., Sundaram S., Tognolli M., Verbregue L., Veuthey A.L., Yip L., Zuletta L., Apweiler R., Alam-Faruque Y., Antunes R., Barrell D., Binns D., Bower L., Browne P., Chan W.M., Dimmer E., Eberhardt R., Fedotov A., Foulger R., Garavelli J., Golin R., Horne A., Huntley R., Jacobsen J., Kleen M., Kersey P., Laiho K., Leinonen R., Legge D., Lin Q., Magrane M., Martin M.J., O’Donovan C., Orchard S., O’Rourke J., Patient S., Pruess M., Sitnov A., Stanley E., Corbett M., di Martino G., Donnelly M., Luo J., van Rensburg P., Wu C., Arighi C., Arminski L., Barker W., Chen Y.X., Hu Z.Z., Hua H.K., Huang H.Z., Mazumder R., McGarvey P., Natale D.A., Nikolskaya A., Petrova N., Suzek B.E., Vasudevan S., Vinayaka C.R., Yeh L.S., Zhang J. The Universal Protein Resource (UniProt) 2009. Nucleic Acids Research. 2009;37:D169–D174. doi: 10.1093/nar/gkn664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belshaw R., de Oliveira T., Markowitz S., Rambaut A. The RNA virus database. Nucleic Acids Research. 2009;37:D431–D435. doi: 10.1093/nar/gkn729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson D.A., Karsch-Mizrachi I., Lipman D.J., Ostell J., Wheeler D.L. GenBank. Nucleic Acids Research. 2008;36:D25–D30. doi: 10.1093/nar/gkm929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman H., Henrick K., Nakamura H., Markley J.L. The worldwide Protein Data Bank (wwPDB): ensuring a single, uniform archive of PDB data. Nucleic Acids Research. 2007;35:D301–D303. doi: 10.1093/nar/gkl971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc E., Roversi P., Vonrhein C., Flensburg C., Lea S.M., Bricogne G. Refinement of severely incomplete structures with maximum likelihood in BUSTER-TNT. Acta Cryst. 2004;D60:2210–2221. doi: 10.1107/S0907444904016427. [DOI] [PubMed] [Google Scholar]
- Blom N., Hansen J., Blaas D., Brunak S. Cleavage site analysis in picornaviral polyproteins: discovering cellular targets by neural networks. Protein Science. 1996;5:2203–2216. doi: 10.1002/pro.5560051107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryson K., Cozzetto D., Jones D.T. Computer-assisted protein domain boundary prediction using the Dom-Pred server. Current Protein & Peptide Science. 2007;8:181–188. doi: 10.2174/138920307780363415. [DOI] [PubMed] [Google Scholar]
- Carugo O., Djinovic-Carugo K., Gorbalenya A.E., Tucker P. Workshop on the definition of protein domains and their likelihood of crystallization – editorial. Current Protein & Peptide Science. 2007;8:119–120. [Google Scholar]
- Chen J., Swamidass S.J., Bruand J., Baldi P. ChemDB: a public database of small molecules and related chemoinformatics resources. Bioinformatics. 2005;21:4133–4139. doi: 10.1093/bioinformatics/bti683. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project, N. 4 The CCP4 suite: programs for protein crystallography. Acta Crystallographica. 1994;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- Coutard B., Gorbalenya A.E., Snijder E.J., Leontovich A.M., Poupon A., de Lamballerie X., Charrel R., Gould E.A., Gunther S., Norder H., Klempa B., Bourhy H., Rohayem J., L’hermite E., Nordlund P., Stuart D.I., Owens R.J., Grimes J.M., Tucker P.A., Bolognesi M., Mattevi A., Coll M., Jones T.A., Aqvist J., Unge T., Hilgenfeld R., Bricogne G., Neyts J., La Colla P., Puerstinger G., Gonzalez J.P., Leroy E., Cambillau C., Romette J.L., Canard B. The VIZIER project: preparedness against pathogenic RNA viruses. Antiviral Research. 2008;78:37–46. doi: 10.1016/j.antiviral.2007.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowder S., Kirkegaard K. Trans-dominant inhibition of RNA viral replication can slow growth of drug-resistant viruses. Nature Genetics. 2005;37:701–709. doi: 10.1038/ng1583. [DOI] [PubMed] [Google Scholar]
- Delaney J.S. Finding and filling protein cavities using cellular logic operations. J. Mol. Graphics. 1992;10:174. doi: 10.1016/0263-7855(92)80052-f. [DOI] [PubMed] [Google Scholar]
- Dovidchenko N.V., Lobanov M.Y., Galzitskaya O.V. Prediction of number and position of domain boundaries in multi-domain proteins by use of amino acid sequence alone. Current Protein & Peptide Science. 2007;8:189–195. doi: 10.2174/138920307780363460. [DOI] [PubMed] [Google Scholar]
- Dumontier M., Yao R., Feldman H.J., Hogue C.W.V. Armadillo: domain boundary prediction by amino acid composition. Journal of Molecular Biology. 2005;350:1061–1073. doi: 10.1016/j.jmb.2005.05.037. [DOI] [PubMed] [Google Scholar]
- Eddy S.R. Hidden Markov models. Current Opinion in Structural Biology. 1996;6:361–365. doi: 10.1016/s0959-440x(96)80056-x. [DOI] [PubMed] [Google Scholar]
- Edgar R.C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feder M., Pas J., Wyrwicz L.S., Bujnicki J.M. Molecular phylogenetics of the RrmJ/fibrillarin superfamily of ribose 2′-O-methyltransferases. Gene. 2003;302:129–138. doi: 10.1016/s0378-1119(02)01097-1. [DOI] [PubMed] [Google Scholar]
- Ferron F., Longhi S., Henrissat B., Canard B. Viral RNA-polymerases – a predicted 2′-O-ribose methyltransferase domain shared by all Mononegavirales. Trends in Biochemical Sciences. 2002;27:222–224. doi: 10.1016/s0968-0004(02)02091-1. [DOI] [PubMed] [Google Scholar]
- Ferron F., Rancurel C., Longhi S., Cambillau C., Henrissat B., Canard B. VaZyMolO: a tool to define and classify modularity in viral proteins. Journal of General Virology. 2005;86:743–749. doi: 10.1099/vir.0.80590-0. [DOI] [PubMed] [Google Scholar]
- Finn R.D., Tate J., Mistry J., Coggill P.C., Sammut S.J., Hotz H.R., Ceric G., Forslund K., Eddy S.R., Sonnhammer E.L., Bateman A. The Pfam protein families database. Nucleic Acids Research. 2008;36:D281–D288. doi: 10.1093/nar/gkm960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galzitskaya O.V., Garbuzynskiy S.O., Lobanov M.Y. FoldUnfold: web server for the prediction of disordered regions in protein chain. Bioinformatics. 2006;22:2948–2949. doi: 10.1093/bioinformatics/btl504. [DOI] [PubMed] [Google Scholar]
- George R.A., Heringa J. SnapDRAGON: a method to delineate protein structural domains from sequence data. Journal of Molecular Biology. 2002;316:839–851. doi: 10.1006/jmbi.2001.5387. [DOI] [PubMed] [Google Scholar]
- Gohara D.W., Ha C.S., Kumar S., Ghosh B., Arnold J.J., Wisniewski T.J., Cameron C.E. Production of “authentic” poliovirus RNA-dependent RNA polymerase (3D(pol)) by ubiquitin-protease-mediated cleavage in Escherichia coli. Protein Expression and Purification. 1999;17:128–138. doi: 10.1006/prep.1999.1100. [DOI] [PubMed] [Google Scholar]
- Golovin A., Oldfield T.J., Tate J.G., Velankar S., Barton G.J., Boutselakis H., Dimitropoulos D., Fillon J., Hussain A., Ionides J.M.C., John M., Keller P.A., Krissinel E., McNeil P., Naim A., Newman R., Pajon A., Pineda J., Rachedi A., Copeland J., Sitnov A., Sobhany S., Suarez-Uruena A., Swaminathan G.J., Tagari M., Tromm S., Vranken W., Henrick K. E-MSD: an integrated data resource for bioinformatics. Nucleic Acids Research. 2004;32(Database issue):D211–D216. doi: 10.1093/nar/gkh078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbalenya A.E. Host-related sequences in RNA virus genomes. Seminars in Virology. 1992;3:359–371. [Google Scholar]
- Gorbalenya A.E. Origin of RNA viral genomes; approaching the problem by comparative sequence analysis. In: Gibbs A.J., Calisher C.H., Garcia-Arenal F., editors. Molecular Basis of Virus Evolution. Cambridge University Press; Cambridge, UK: 1995. pp. 49–66. [Google Scholar]
- Gorbalenya A.E. Big nidovirus genome – when count and order of domains matter. Advances in Experimental Medicine and Biology. 2001;494:1–17. [PubMed] [Google Scholar]
- Gorbalenya A.E., Donchenko A.P., Blinov V.M., Koonin E.V. Cysteine proteases of positive strand RNA viruses and chymotrypsin-like serine proteases: a distinct protein superfamily with a common structural fold. FEBS Letters. 1989;243:103–114. doi: 10.1016/0014-5793(89)80109-7. [DOI] [PubMed] [Google Scholar]
- Gorbalenya A.E., Donchenko A.P., Koonin E.V., Blinov V.M. N-Terminal domains of putative helicases of flavi- and pestiviruses may be serine proteases. Nucleic Acids Research. 1989;17:3889–3897. doi: 10.1093/nar/17.10.3889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbalenya A.E., Enjuanes L., Ziebuhr J., Snijder E.J. Nidovirales: evolving the largest RNA virus genome. Virus Research. 2006;117:17–37. doi: 10.1016/j.virusres.2006.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbalenya A.E., Koonin E.V. Comparative analysis of the amino acid sequences of the key enzymes of the replication and expression of positive-strand RNA viruses. Validity of the approach and functional and evolutionary implications. Soviet Science Reviews D: Physicochemical Biology. 1993;11:1–84. [Google Scholar]
- Gorbalenya A.E., Koonin E.V. Helicases: amino acid sequence comparisons and structure–function relationships. Current Opinion in Structural Biology. 1993;3:419–429. [Google Scholar]
- Gorbalenya A.E., Koonin E.V., Donchenko A.P., Blinov V.M. Coronavirus genome: prediction of putative functional domains in the non-structural polyprotein by comparative amino acid sequence analysis. Nucleic Acids Research. 1989;17:4847–4861. doi: 10.1093/nar/17.12.4847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbalenya A.E., Koonin E.V., Lai M.M.C. Putative papain-related thiol proteases of positive-strand RNA viruses. FEBS Letters. 1991;288:201–205. doi: 10.1016/0014-5793(91)81034-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbalenya A.E., Snijder E.J. Viral cysteine proteinases. Perspectives in Drug Discovery and Design. 1996;6:64–86. doi: 10.1007/BF02174046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gromeier M., Wimmer E., Gorbalenya A.E. Genetics, pathogenesis and evolution of picornaviruses. In: Domingo E., Webster R.G., Holland J.J., editors. Origin and Evolution of Viruses. Academic Press; San Diego: 1999. pp. 287–343. [Google Scholar]
- Hansen J.L., Long A.M., Schultz S.C. Structure of the RNA-dependent RNA polymerase of poliovirus. Structure (Cambridge) 1997;5:1109–1122. doi: 10.1016/s0969-2126(97)00261-x. [DOI] [PubMed] [Google Scholar]
- Harris M., Jones T.A. Xtrack – a web-based crystallographic notebook. Acta Crystallographica D: Biological Crystallography. 2002;58:1889–1891. doi: 10.1107/s0907444902012696. [DOI] [PubMed] [Google Scholar]
- Hartshorn M.J. AstexViewer: a visualisation aid for structure-based drug design. Journal of Computer-Aided Molecular Design. 2002;16:871–881. doi: 10.1023/a:1023813504011. [DOI] [PubMed] [Google Scholar]
- Hendlich M., Bergner A., Gunther J., Klebe G. Relibase: design and development of a database for comprehensive analysis of protein–ligand interactions. Journal of Molecular Biology. 2003;326:607–620. doi: 10.1016/s0022-2836(02)01408-0. [DOI] [PubMed] [Google Scholar]
- Hughes P.J., Stanway G. The 2A proteins of three diverse picornaviruses are related to each other and to the H-rev107 family of proteins involved in the control of cell proliferation. Journal of General Virology. 2000;81:201–207. doi: 10.1099/0022-1317-81-1-201. [DOI] [PubMed] [Google Scholar]
- Hulo N., Bairoch A., Bulliard V., Cerutti L., Cuche B.A., de Castro E., Lachaize C., Langendijk-Genevaux P.S., Sigrist C.J. The 20 years of PROSITE. Nucleic Acids Research. 2008;36:D245–D249. doi: 10.1093/nar/gkm977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansson A.M., Jakobsson E., Johansson P., Lantez V., Coutard B., de Lamballerie X., Unge T., Jones T.A. Structure of the methyltransferase domain from the Modoc virus, a flavivirus with no known vector. Acta Crystallographica. 2009;D65:1–10. doi: 10.1107/S0907444909017260. [DOI] [PubMed] [Google Scholar]
- Jiang P., Faase J.A.J., Toyoda H., Paul A., Wimmer E., Gorbalenya A.E. Evidence for emergence of diverse polioviruses from C-cluster coxsackie A viruses and implications for global poliovirus eradication. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:9457–9462. doi: 10.1073/pnas.0700451104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones T.A., Zou J.Y., Cowan S.W., Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Cryst. 1991;A47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- Kabsch W., Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- Kamer G., Argos P. Primary structural comparison of RNA-dependent polymerases from plant, animal and bacterial-viruses. Nucleic Acids Research. 1984;12:7269–7282. doi: 10.1093/nar/12.18.7269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiemer L., Lund O., Brunak S., Blom N. Coronavirus 3CLpro proteinase cleavage sites: possible relevance to SARS virus pathology. BMC Bioinformatics. 2004;5:72. doi: 10.1186/1471-2105-5-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura N., Semler B.L., Rothberg P.G., Larsen G.R., Adler C.J., Dorner A.J., Emini E.A., Hanecak R., Lee J.J., Vanderwerf S., Anderson C.W., Wimmer E. Primary structure, gene organization and polypeptide expression of poliovirus RNA. Nature. 1981;291:547–553. doi: 10.1038/291547a0. [DOI] [PubMed] [Google Scholar]
- Kleywegt G.J., Harris M.R. ValLigURL: a server for ligand-structure comparison and validation. Acta Crystallographica D. 2007;63:935–938. doi: 10.1107/S090744490703315X. [DOI] [PubMed] [Google Scholar]
- Kleywegt G.J., Harris M.R., Zou J.Y., Taylor T.C., Wahlby A., Jones T.A. The Uppsala electron-density server. Acta Crystallographica D: Biological Crystallography. 2004;60:2240–2249. doi: 10.1107/S0907444904013253. [DOI] [PubMed] [Google Scholar]
- Kleywegt G.J., Jones T.A. Detection, delineation, measurement and display of cavities in macromolecular structures. Acta Crystallographica. 1994;D50:178–185. doi: 10.1107/S0907444993011333. [DOI] [PubMed] [Google Scholar]
- Kleywegt G.J., Jones T.A. Where freedom is given, liberties are taken. Structure. 1995;3:535–540. doi: 10.1016/s0969-2126(01)00187-3. [DOI] [PubMed] [Google Scholar]
- Kleywegt G.J., Jones T.A. Efficient rebuilding of protein structures. Acta Crystallographica. 1996;D52:829–832. doi: 10.1107/S0907444996001783. [DOI] [PubMed] [Google Scholar]
- Kleywegt G.J., Jones T.A. Phi/Psi-chology: Ramachandran revisited. Structure. 1996;4:1395–1400. doi: 10.1016/s0969-2126(96)00147-5. [DOI] [PubMed] [Google Scholar]
- Kleywegt G.J., Jones T.A. Detecting folding motifs and similarities in protein structures. Methods in Enzymology. 1997;277:525–545. doi: 10.1016/s0076-6879(97)77029-0. [DOI] [PubMed] [Google Scholar]
- Kleywegt G.J., Jones T.A. Model-building and refinement practice. Methods in Enzymology. 1997;277:208–230. doi: 10.1016/s0076-6879(97)77013-7. [DOI] [PubMed] [Google Scholar]
- Koonin E.V. Computer-assisted identification of a putative methyltransferase domain in NS5 protein of flaviviruses and λ2 protein of reovirus. Journal of General Virology. 1993;74:733–740. doi: 10.1099/0022-1317-74-4-733. [DOI] [PubMed] [Google Scholar]
- Krogh A., Larsson B., von Heijne G., Sonnhammer E.L.L. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. Journal of Molecular Biology. 2001;305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- Leontovich A.M., Tokmachev K.Y., van Houwelingen H.C. The comparative analysis of statistics, based on the likelihood ratio criterion, in the automated annotation problem. BMC Bioinformatics. 2008;9:31. doi: 10.1186/1471-2105-9-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieutaud P., Canard B., Longhi S. MeDor: a metaserver for predicting protein disorder. BMC Genomics. 2008;9(Suppl. 2):S25. doi: 10.1186/1471-2164-9-S2-S25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T., Lin Y. BindingDB: a web-accessible database of experimentally determined protein–ligand binding affinities. Nucleic Acids Research. 2007;35(Database issue):D198–201. doi: 10.1093/nar/gkl999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.F., Rost B. Sequence-based prediction of protein domains. Nucleic Acids Research. 2004;32:3522–3530. doi: 10.1093/nar/gkh684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchler-Bauer A., Anderson J.B., Chitsaz F., Derbyshire M.K., DeWeese-Scott C., Fong J.H., Geer L.Y., Geer R.C., Gonzales N.R., Gwadz M., He S., Hurwitz D.I., Jackson J.D., Ke Z., Lanczycki C.J., Liebert C.A., Liu C., Lu F., Lu S., Marchler G.H., Mullokandov M., Song J.S., Tasneem A., Thanki N., Yamashita R.A., Zhang D., Zhang N., Bryant S.H. CDD: specific functional annotation with the Conserved Domain Database. Nucleic Acids Research. 2009;37:D205–D210. doi: 10.1093/nar/gkn845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazumder R., Iyer L.M., Vasudevan S., Aravind L. Detection of novel members, structure–function analysis and evolutionary classification of the 2H phosphoesterase superfamily. Nucleic Acids Research. 2002;30:5229–5243. doi: 10.1093/nar/gkf645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalsky E., Dunkel M., Goede A., Preissner R. SuperLigands – a database of ligand structures derived from the Protein Data Bank. BMC Bioinformatics. 2005;6:122. doi: 10.1186/1471-2105-6-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgenstern B. DIALIGN: multiple DNA and protein sequence alignment at BiBiServ. Nucleic Acids Research. 2004;32:W33–W36. doi: 10.1093/nar/gkh373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moya A., Holmes E.C., Gonzalez-Candelas F. The population genetics and evolutionary epidemiology of RNA viruses. Nature Reviews Microbiology. 2004;2:279–288. doi: 10.1038/nrmicro863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov G.N., Vagin A.A., Dodson E.J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallographica. 1997;D53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Neuman B.W., Joseph J.S., Saikatendu K.S., Serrano P., Chatterjee A., Johnson M.A., Liao L., Klaus J.P., Yates J.R., Wuethrich K., Stevens R.C., Buchmeier M.J., Kuhn P. Proteornics analysis unravels the functional repertoire of coronavirus nonstructural protein 3. Journal of Virology. 2008;82:5279–5294. doi: 10.1128/JVI.02631-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notredame C., Higgins D.G., Heringa J. T-Coffee: a novel method for fast and accurate multiple sequence alignment. Journal of Molecular Biology. 2000;302:205–217. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- Paul A.V., van Boom J.H., Filippov D., Wimmer E. Protein-primed RNA synthesis by purified poliovirus RNA polymerase. Nature. 1998;393:280–284. doi: 10.1038/30529. [DOI] [PubMed] [Google Scholar]
- Ratia K., Saikatendu K.S., Santarsiero B.D., Barretto N., Baker S.C., Stevens R.C., Mesecar A.D. Severe acute respiratory syndrome coronavirus papain-like protease: structure of a viral deubiquitinating enzyme. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:5717–5722. doi: 10.1073/pnas.0510851103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawlings N.D., Morton F.R., Kok C.Y., Kong J., Barrett A.J. MEROPS: the peptidase database. Nucleic Acids Research. 2008;36:D320–D325. doi: 10.1093/nar/gkm954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozanov M.N., Koonin E.V., Gorbalenya A.E. Conservation of the putative methyltransferase domain: a hallmark of the ‘Sindbis-like’ supergroup of positive-strand RNA viruses. Journal of General Virology. 1992;73:2129–2134. doi: 10.1099/0022-1317-73-8-2129. [DOI] [PubMed] [Google Scholar]
- Sadreyev R., Grishin N. COMPASS: a tool for comparison of multiple protein alignments with assessment of statistical significance. Journal of Molecular Biology. 2003;326:317–336. doi: 10.1016/s0022-2836(02)01371-2. [DOI] [PubMed] [Google Scholar]
- Serrano P., Johnson M.A., Almeida M.S., Horst R., Herrmann T., Joseph J.S., Neuman B.W., Subramanian V., Saikatendu K.S., Buchmeier M.J., Stevens R.C., Kuhn P., Wuthrich K. Nuclear magnetic resonance structure of the N-terminal domain of nonstructural protein 3 from the severe acute respiratory syndrome coronavirus. Journal of Virology. 2007;81:12049–12060. doi: 10.1128/JVI.00969-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidorov I.A., Reshetov D.A., Gorbalenya A.E. SNAD: sequence name annotation-based designer. BMC Bioinformatics. 2009;10:251. doi: 10.1186/1471-2105-10-251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sim J., Kim S.Y., Lee J. PPRODO: prediction of protein domain boundaries using neural networks. Proteins-Structure Function and Bioinformatics. 2005;59:627–632. doi: 10.1002/prot.20442. [DOI] [PubMed] [Google Scholar]
- Simossis V.A., Kleinjung J., Heringa J. Homology-extended sequence alignment. Nucleic Acids Research. 2005;33:816–824. doi: 10.1093/nar/gki233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijder E.J., Bredenbeek P.J., Dobbe J.C., Thiel V., Ziebuhr J., Poon L.L.M., Guan Y., Rozanov M., Spaan W.J.M., Gorbalenya A.E. Unique and conserved features of genome and proteome of SARS-coronavirus, an early split-off from the coronavirus group 2 lineage. Journal of Molecular Biology. 2003;331:991–1004. doi: 10.1016/S0022-2836(03)00865-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suyama M, Ohara O. DomCut: prediction of inter-domain linker regions in amino acid sequences. Bioinformatics. 2003;19:673–674. doi: 10.1093/bioinformatics/btg031. [DOI] [PubMed] [Google Scholar]
- Thompson A.A., Peersen O.B. Structural basis for proteolysis-dependent activation of the poliovirus RNA-dependent RNA polymerase. EMBO Journal. 2004;23:3462–3471. doi: 10.1038/sj.emboj.7600357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson J.D., Gibson T.J., Plewniak F., Jeanmougin F., Higgins D.G. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Research. 1997;25:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z.R., Thomson R., McNeil P., Esnouf R.M. RONN: the bio-basis function neural network technique applied to the detection of natively disordered regions in proteins. Bioinformatics. 2005;21:3369–3376. doi: 10.1093/bioinformatics/bti534. [DOI] [PubMed] [Google Scholar]
- Yoo P.D., Sikder A.R., Zhou B.B., Zomaya A.Y. Improved general regression network for protein domain boundary prediction. BMC Bioinformatics. 2008;9:S12. doi: 10.1186/1471-2105-9-S1-S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziebuhr J., Thiel V., Gorbalenya A.E. The autocatalytic release of a putative RNA virus transcription factor from its polyprotein precursor involves two paralogous papain-like proteases that cleave the same peptide bond. Journal of Biological Chemistry. 2001;276:33220–33232. doi: 10.1074/jbc.M104097200. [DOI] [PMC free article] [PubMed] [Google Scholar]