

Abstract
Background
Acute exacerbation (AE) is currently established as a distinct condition with acute deterioration of respiratory status in idiopathic pulmonary fibrosis (IPF). Recently, several studies have reported that AE also occurred in interstitial pneumonias other than IPF, such as collagen vascular disease-associated interstitial pneumonia (CVD-IP). However, the incidence of AE in CVD-IP and its clinical characteristics remain to be fully determined. This study was conducted to elucidate cumulative incidence of AE in CVD-IP and its clinical features.
Methods
We reviewed 83 biopsy-proven CVD-IP patients, estimated cumulative incidence of AE, and examined its clinical characteristics.
Results
Among 83 CVD-IP patients, six patients with a mean age of 65.7 years developed AE (overall incidence, 7.2%; 1-year incidence, 1.25%). Underlying CVDs included rheumatoid arthritis (RA) (n = 5; overall incidence, 20.0%) and primary Sjögren syndrome (n = 1; overall incidence, 5.9%). Patients with AE showed acute respiratory deterioration with severe hypoxemia (mean PaO2/FiO2 ratio, 131). Radiologically, ground-glass opacity was superimposed on the underlying reticular abnormalities. Preexisting histological patterns included three usual interstitial pneumonia (UIP) and two non-specific interstitial pneumonia (NSIP). Five (83.3%) of six patients died of respiratory failure despite intensive therapy. Univariate Cox's proportional hazards analysis showed that age and RA diagnosis were significantly associated with AE. Multivariate Cox's proportional hazards analysis indicated that age was an independent significant factor predicting AE.
Conclusions
These data suggest that AE can occur in CVD-IP, and this condition is closely similar to that of IPF with poor prognosis. AE is most common in RA, and associated with higher ages.
Keywords: Collagen vascular disease, Interstitial pneumonia, Acute exacerbation
Abbreviations: ADM, amyopathic dermatomyositis; CVD, collagen vascular disease; DM, dermatomyositis; MCTD, mixed connective tissue disease; NSIP, non-specific interstitial pneumonia; PM, polymyositis; pSS, primary Sjögren syndrome; RA, rheumatoid arthritis; SLE, systemic lupus; SSc, systemic sclerosis; UIP, usual interstitial pneumonia
Introduction
In idiopathic pulmonary fibrosis (IPF), acute exacerbation (AE) has been increasingly recognized as an acute, often fatal, clinical incident, which occurs during the course of this disease.1, 2, 3, 4, 5, 6 Much effort has been devoted to characterize this condition, and most recently the Idiopathic Pulmonary Fibrosis Clinical Research Network (IPF net) have proposed a definition and criteria for AE of IPF.6 In IPF, AE is characterized by rapid worsening of respiratory symptoms, acute deterioration of hypoxemia, newly developed bilateral ground-glass opacity and/or consolidation on chest radiographs or computed tomography (CT) scans, and the absence of other alternative causes, such as infection, heart failure and embolism.5, 6 Histologically, diffuse alveolar damage (DAD) is superimposed on the underlying fibrotic pattern of usual interstitial pneumonia (UIP) in most patients with AE of IPF.1, 2, 7, 8
Recently, AE has also been shown to occur in interstitial pneumonias other than IPF, such as idiopathic non-specific interstitial pneumonia (NSIP) and collagen vascular disease-associated interstitial pneumonia (CVD-IP).9, 10, 11, 12, 13, 14 Parambil et al. have reported AE in three of 18 patients with primary Sjögren syndrome-associated interstitial pneumonia (pSS-IP).11 More recently, Park et al. have described this condition in four of 93 patients with CVD-IP.13 To date, however, the cumulative reported number of CVD-IP patients with AE is small, and the incidence of this condition in CVD-IP, and its clinical characteristics, remain to be fully determined. Thus, the present study was conducted to estimate cumulative incidence of AE in CVD-IP, and to establish its clinical features.
Patients and methods
Patients
The study population included 83 consecutive patients (35 males and 48 females) with CVD-IP who underwent open or thoracoscopic lung biopsy in our facilities from 1987 to 2007. Each CVD was diagnosed according to established criteria.15, 16, 17, 18, 19, 20 A diagnosis of amyopathic dermatomyositis (ADM) was based on modified Euwer's criteria in our previous study21: (1) characteristic dermatological manifestations of classic dermatomyositis (DM), including a heliotrope rash and Gottoron's papules; (2) no muscle weakness; (3) no increase in serum muscle enzymes during the observation period. AE was defined using the criteria recently proposed by the IPF net, with slight modification for adaptation to CVD-IP6: (1) previous diagnosis of CVD-IP; (2) unexplained worsening or development of dyspnea within 30 days; (3) high-resolution CT (HRCT) with new bilateral ground-glass abnormality and/or consolidation superimposed on a background reticular or honeycomb pattern; (4) no evidence of pulmonary infection by negative respiratory culture, including endotracheal aspirate or bronchoalveolar lavage, and serological test results for respiratory pathogens; and (5) exclusion of alternative causes, such as left heart failure, pulmonary embolism, and an identifiable cause of acute lung injury. Patients with AE needed to meet all five criteria. The study protocol was approved by the Ethical Committee of the Hamamatsu University School of Medicine.
Data collection
Clinical data were obtained from medical records. Laboratory findings and pulmonary function tests at the time of surgical lung biopsy and AE, and outcome were also recorded.
Pathological review
Biopsy specimens were obtained from at least two sites. Lung biopsy specimens were independently reviewed by three pathologists (T.V.C, Y.N., S.I.) who were unaware of the clinical or physiological findings. When classification differed between the pathologists (kappa coefficient of agreement = 0.40), a consensus opinion on the overall histopathological pattern was reached. Histological classification was based on the previously published criteria for idiopathic interstitial pneumonias from the American Thoracic Society (ATS)/European Respiratory Society (ERS) consensus statement.22 A histological pattern that could not be placed in any patterns described in the consensus statement was categorized as unclassifiable interstitial pneumonia. Sections stained with hematoxylin–eosin were reviewed from all cases, and sections in which elastic tissue was stained were reviewed in selected cases.
HRCT
HRCT findings were reviewed by a radiologist (Y.T.) and two pulmonologists (T.S., N.I.), without knowledge of any clinical data. HRCT examination of the lungs was performed on 1.0- or 1.5-mm thick sections to evaluate radiographic abnormalities.
Bronchoalveolar lavage
BAL findings were retrospectively reviewed. BAL was performed as described previously.23 Briefly, a fiberoptic bronchoscope was passed transorally and wedged in a segmental or subsegmental bronchus of the middle lobe. Three 50-ml aliquots of sterile 0.9% saline were instilled and the returns were gently aspirated through the side channel of the bronchoscope. BAL fluid (BALF) was centrifuged at 800g for 10 min to obtain the cellular components. The total cell count was determined using a hemocytometer and a differential cell count was taken on Giemsa-stained cytocentrifuged preparations.
Statistical analysis
For two group comparisons involving binary data, we used either the chi-square test or Fisher's exact test, depending on expected frequencies. The cumulative incidence of AE in CVD-IP patients was estimated using Kaplan–Meier curves. The overall incidence was calculated by dividing the number of patients developing AE during the observation periods by the total number of patients. Cox proportional hazards regression analysis was used to identify significant variables associated with AE. Statistical analyses were performed using JMP Start Statistics (SAS Institute Inc., NC, USA). The p value <0.05 was considered significant. All data are expressed as a mean ± SD.
Results
Incidence of AE in CVD-IP
The clinical characteristics of 83 CVD-IP patients proven by surgical lung biopsy at the initial presentation are shown in Table 1 . During the observation period, six patients (7.2%) developed AE with a 1-year incidence of 1.25% (Table 2 ; Fig. 1 ). There were four males and two females. Among the underlying CVDs, AE was found in rheumatoid arthritis (RA) and primary Sjögren syndrome (pSS) (Table 2). The overall and 1-year incidence of AE were highest in RA (20.0 and 2.58%, respectively), followed by pSS (5.9 and 1.05%, respectively). No AE occurred in patients with CVDs other than RA and pSS (Table 2). With regard to histological patterns, patients with UIP showed higher frequency of AE than those with NSIP (overall incidence, 10.0 vs. 4.0%; 1-year incidence, 2.00 vs. 0.70%, respectively) (Table 2). There was no significant difference in the observation periods for the underlying CVDs, or histological patterns (data not shown).
Table 1.
Clinical characteristics of CVD-IP at initial presentation.
| Number of patients or values | |
|---|---|
| Sex, male/female | 35/48 |
| Age, years | 58.7 ± 9.5a |
| Smoking habit | |
| Current/ex-smoker/never | 24/6/53 |
| Underlying collagen vascular diseases | |
| RA | 25 |
| PM/DM/ADM | 21 |
| pSS | 17 |
| SSc | 13 |
| SLE | 4 |
| MCTD | 2 |
| Adult Still's disease | 1 |
| Histological pattern | |
| NSIP | 50 (60.3%) |
| UIP | 30 (36.1%) |
| Unclassifiable | 3 (3.6%) |
| Observation periods, years | 6.0 ± 5.6 |
| Laboratory data | |
| %VC (n = 83) | 82.9 ± 17.9 |
| DLco, % (n = 78) | 75.0 ± 25.6 |
| PaO2 on room air, Torr (n = 83) | 79.8 ± 9.8 |
| KL-6, U/ml (n = 63) | 1003 ± 1642 |
| Treatment for interstitial pneumonia | |
| Corticosteroid, % | 65.0 |
| Immunosuppressant, % | 22.9 |
RA, rheumatoid arthritis; SSc, systemic sclerosis; pSS, primary Sjögren syndrome; PM, polymyositis; DM, dermatomyositis; ADM, amyopathic dermatomyositis; SLE, systemic lupus erythematosus; MCTD, mixed connective tissue disease; NSIP, non-specific interstitial pneumonia; UIP, usual interstitial pneumonia.
mean ± SD.
Table 2.
Incidence of AE in CVD-IP and its association with histological patterns.
| Total (n = 83) | RA (n = 25) | pSS (n = 17) | SSc (n = 13) | PM/DM/ADM (n = 21) | SLE (n = 4) | Othersa (n = 3) | |
|---|---|---|---|---|---|---|---|
| No. of patients developing AE | 6 (7.2%) | 5 (20.0%) | 1 (5.9%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) |
| Histologic patterns | |||||||
| UIP | 3/30b (10.0%) | 2/10 (20.0%) | 1/10 (10.0%) | 0/5 (0%) | 0/3 (0%) | 0/2 (0%) | |
| NSIP | 2/50 (4.0%) | 2/13 (15.4%) | 0/7 (0%) | 0/8 (0%) | 0/18 (0%) | 0/2 (0%) | 0/2 (0%) |
| Unclassifiable | 1/3 (33.3%) | 1/2 (50.0%) | 0/1 (0%) | ||||
AE, acute exacerbation.
Others include MCTD and adult Still's disease.
Number of patients developing AE / total number of patients showing each histological pattern.
Figure 1.

Cumulative incidence of AE in CVD-IP.
Clinical features
The clinical characteristics of six patients with AE of CVD-IP are listed in Table 3 . The age at the onset of AE was 65.7 ± 5.3 years. All patients presented with rapidly deteriorating respiratory symptoms, such as dyspnea and cough, within 30 days (18.3 ± 8.3 days). Four patients (66.7%) had fever, and fine crackles were present in all patients. The duration between diagnosis of interstitial pneumonia and onset of AE varied widely, with a mean of 66.5 months. One patient (Case 6) developed AE after surgical lung biopsy. In all six patients, the activity of the underlying CVD was stable before the onset of AE. Low-dose corticosteroids were given to one patient, and there were no alterations of medications within 3 months before the onset of AE in any patients.
Table 3.
Clinical characteristics of patients with AE of CVD-IP.
| No. | Underlying disease | Age (years), sex | Smoking status | Histological pattern | Duration between diagnosis of IP and AE, months | Activity of underlying CVD at AE | Treatment before AE | Treatment for AE | Outcome |
|---|---|---|---|---|---|---|---|---|---|
| 1 | RA | 57, female | Former | UIP | 99 | Stable | Bucillamine | MPSL, PSL CPA, CsA | Died |
| 2 | RA | 63, male | Current | UIP | 86 | Stable | Mizoribine | MPSL, PSL CPA | Died |
| 3 | RA | 65, male | Former | NSIP | 129 | Stable | PSL (10 mg/day) | PSL | Died |
| 4 | RA | 68, male | Former | NSIP | 31 | Stable | Bucillamine | MPSL, PSL CPA | Died |
| 5 | RA | 69, male | Current | Unclassifiable | 52 | Stable | Bucillamine | MPSL, PSL | Survived |
| 6 | pSS | 72, female | Never | UIP | 2 | Stable | None | MPSL, PSL CPA, CsA | Died |
IP, interstitial pneumonia; AE, acute exacerbation; PSL, prednisolone; MPSL, methylprednisolone; CPA, cyclophosphamide; CsA, cyclosporine.
Laboratory and BAL findings
All patients had severe hypoxemia on admission for AE. The P/F ratio (PaO2/FiO2 ratio) was 131 ± 78 (Fig. 2 ). Serum levels of C-reactive protein (CRP) and lactate hydroxygenase (LDH) were markedly increased in all patients (CRP, 20.1 ± 9.4 mg/ml; LDH, 571 ± 249 IU/L) (Fig. 2). Additionally, an elevation of serum KL-6 (normal range, <500 U/ml), a marker of interstitial pneumonia, was found in all five patients tested (1926 ± 1045 U/ml) (Fig. 2). BAL was performed in one patient (Case 4), and this analysis showed an increase in total cell counts, as well as neutrophilia (total cell count, 2.5 × 106 cells/ml BAL fluid; neutrophils, 64.0%).
Figure 2.

Laboratory data of patients with CVD-IP at the onset of AE. Horizontal bars indicate mean values. P/F ratio, PaO2/FiO2 ratio.
Microbiological examination
Microbiological studies of BAL fluid (n = 1), endotracheal aspirates (n = 5), sputum (n = 6) and blood (n = 6) revealed no infectious agents, including bacteria, mycobacteria, fungi or Pneumocystis jiroveci. The results of serological studies for atypical pathogens, such as Mycoplasma or Chlamydophila spp., were negative in all patients. In three patients, serum titers of viruses, including influenza viruses, parainfluenza viruses, respiratory syncytial viruses, rhinovirus, and coronaviruses, were measured, and no significant elevation of any virus titer was found. Three patients underwent a test for urinary antigens of Legionella spp. (Binax Now, Binax, Scarborough, ME, USA), which revealed a negative result in all patients.
HRCT and histological findings
The most conspicuous finding of HRCT at the onset of AE was newly developed bilateral ground-glass opacity (100%), with or without areas of consolidation, superimposed on the bibasilar reticular abnormalities (Fig. 3 ). Patchy air space consolidation was present in five patients (83.3%). Four patients with AE underwent contrast-material enhanced CT, which revealed no evidence of pulmonary embolism. As a result of severe respiratory failure on admission, the second surgical lung biopsy could not be performed at the onset of AE in any patients. Autopsy was performed in one patient (Case 4), which showed a DAD pattern superimposed on the background of NSIP (Fig. 4 ).
Figure 3.
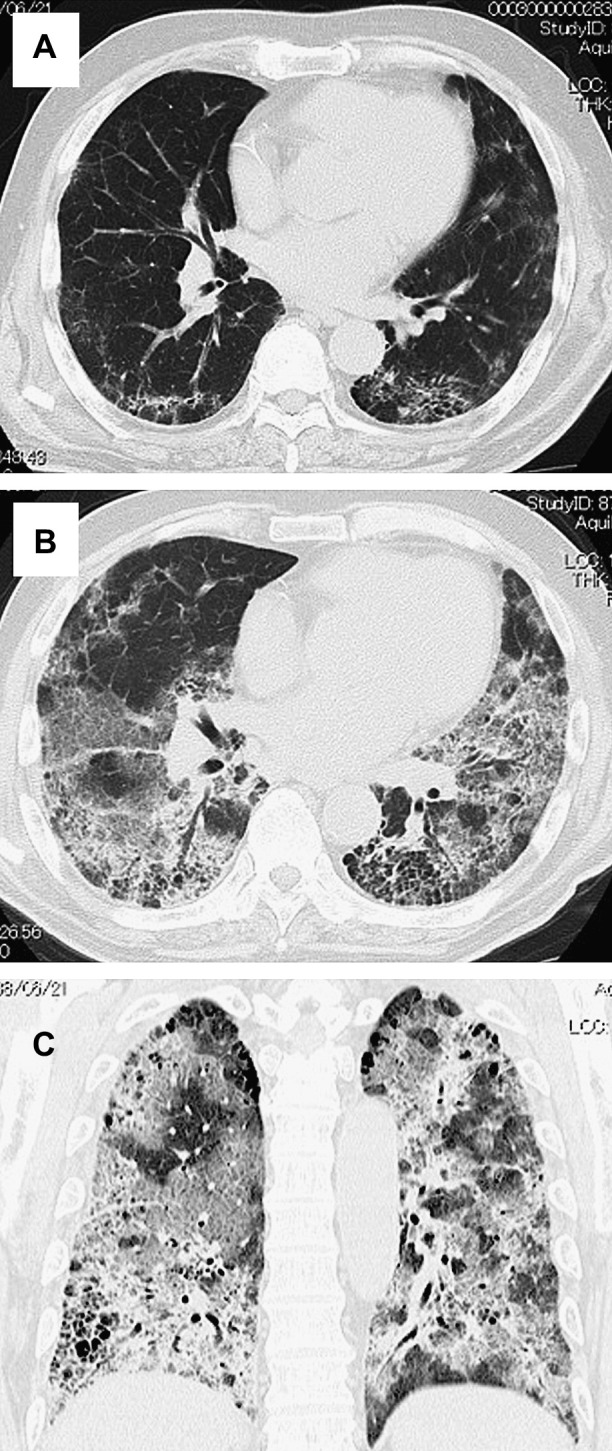
HRCT of AE of CVD-IP (Case 5). (A) Three months before AE. Transverse thin-section CT of the lower lobe shows mild reticular and cystic opacities in subpleural areas of the lung. (B and C) Transverse and coronal thin-section CT of the lower lobe at the onset of AE reveal superimposition of ground-glass opacity spreading to both lungs.
Figure 4.
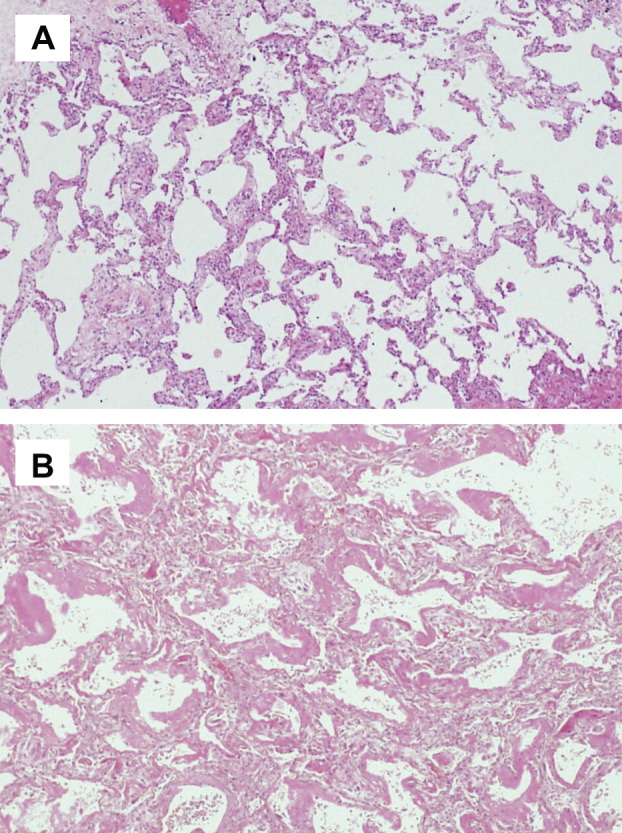
Histopathology of AE of CVD-IP (Case 6). (A) Specimen from surgical lung biopsy obtained before the onset of AE shows temporary uniform fibrosis of the alveolar wall, with mild infiltration of mononuclear cells (fibrotic NSIP) (hematoxylin–eosin, ×200). (B) Autopsy specimen reveals scattered hyaline membranes lining thickened alveolar septa, with interstitial edema (hematoxylin–eosin, ×400).
Treatment and outcome
Intravenous bolus of methylprednisolone, 1000 mg once a day for 3 days, was given to all patients, followed by 1 mg/kg/day prednisolone (Table 3). Four patients were intravenously administered 500–750 mg/m2 cyclophosphamide. In addition, two patients received 2–3 mg/kg/day cyclosporine. Despite these intensive anti-inflammatory and immunosuppressive therapies, all patients required mechanical ventilation, and five (83.3%) of them died of respiratory failure. Among these five patients, four died within 40 days after the onset of AE (26.0 ± 11.6 days). One patient survived (Case 5); his respiratory status had worsened before the onset of AE, and he received supplemental oxygen therapy after discharge.
Associated factors with AE development
To determine the associated factors with AE development at the initial diagnosis, we first assessed the valuables by univariate Cox's proportional hazards regression models. The variables included sex, age, smoking history, CVD diagnosis, histologic patterns, pulmonary function test results, laboratory findings, and arterial blood gas analysis at the initial diagnosis. Among the valuables, age and RA diagnosis were significantly associated with AE development (Hazard Ratio 1.181 and 2.536, p = 0.0037 and p = 0.0484, respectively) (Table 3). Moreover, multivariate Cox's proportional hazards regression models, including those two valuables, showed that age was an independent significant factor for AE development (Hazard Ratio 1.221, p = 0.0052) (Table 4, Table 5 ).
Table 4.
Associated factors for AE development: univariate Cox proportional hazards models.
| Variables | Hazard ratio | 95% CI |
p Value | |
|---|---|---|---|---|
| Lower | Upper | |||
| Age, years | 1.181 | 1.050 | 1.374 | 0.0037 |
| Sex, female | 0.764 | 0.283 | 1.744 | 0.5281 |
| Smoking habit, yes | 2.336 | 0.898 | 10.33 | 0.0844 |
| CVD, RA | 2.536 | 1.006 | 11.16 | 0.0484 |
| Histology, NSIP | 0.545 | 0.203 | 1.234 | 0.1452 |
| VC, % | 0.984 | 0.942 | 1.029 | 0.4681 |
| DLco, % | 1.054 | 0.975 | 1.208 | 0.1824 |
| PaO2, Torr | 1.008 | 0.924 | 1.111 | 0.8585 |
| KL-6, U/ml | 1.000 | 0.997 | 1.000 | 0.9314 |
AE, acute exacerbation; CI, confidence interval; CVD, collagen vascular diseases; RA, rheumatoid arthritis; UIP, usual interstitial pneumonia.
Table 5.
Associated factors for AE development: multivariate Cox proportional hazards models.
| Variables | Hazard ratio | 95% CI |
p Value | |
|---|---|---|---|---|
| Lower | Upper | |||
| Age, years | 1.221 | 1.054 | 1.495 | 0.0052 |
| CVD, RA | 2.473 | 0.930 | 11.21 | 0.0716 |
AE, acute exacerbation; CI, confidence interval; CVD, collagen vascular diseases; RA, rheumatoid arthritis.
Discussion
The present study reviewed 83 patients with biopsy-proven CVD-IP, and demonstrated AE in six patients (7.2%) during the observation period. The incidence of AE in CVD-IP was confirmed as 7.2% overall and 1.25% at 1 year, and this condition occurred mostly in patients with RA. The clinical characteristics of AE of CVD-IP were similar to those of IPF with high mortality (83.3%). These data suggest that AE can occur in CVD-IP with poor prognosis.
Recently, it has been recognized that IPF patients present with acute clinical deterioration, termed as AE of IPF.1, 2, 3, 4, 5, 6 According to the IPF net criteria, with slight modification, we found six patients with AE in our series of 83 CVD-IP patients undergoing surgical lung biopsy. Typically, these six patients had acute respiratory deterioration within 30 days. Their HRCT scan revealed bilateral ground-glass opacity with or without air space consolidation superimposed on preexisting reticular abnormality. The results of extensive microbiological studies, such as trachea aspirates and BAL, were negative. They responded poorly to intensive therapy with high mortality rate. Collectively, the features of our CVD-IP patients closely fitted those of AE of IPF in the literature.1, 2, 3, 4, 5, 6 To date, there have been a few studies about AE in series of CVD-IP patients.9, 11, 13 Recent studies by Parambil et al. and Park et al. have independently demonstrated AE of CVD-IP in three of 18 biopsy-proven pSS-IP patients and in four of 93 biopsy-proven CVD-IP patients, respectively.11, 13 Additionally, Parambil et al. have examined nine CVD-IP patients with a DAD pattern on surgical lung biopsy without known etiology, and found that six of them had preexisting interstitial pneumonia.9 Collectively, these results confirmed that CVD-IP patients can develop AE.
The incidence of AE in IPF remains unknown. The reported incidence varies widely, because there are differences in the definition of AE, as well as differences in the populations studied. However, several prospective randomized control or retrospective longitudinal cohort studies have reported the likely incidence. Kim et al. indicated a 1-year incidence of 8.5% in a retrospective longitudinal cohort of 147 patients with biopsy-proven IPF.4 In a randomized controlled trial of pirfenidone, Azuma et al. reported a 9-month incidence of 14.3% in 36 placebo-treated patients.24 Taken together, the incidence of AE of IPF was estimated to be in the range of 5–19% per year.5 In CVD-IP, the present study revealed an overall incidence of 7.2%, with a mean 6.0 years follow-up, and a 1-year incidence of 1.25%. Compared with the reported incidence of AE of IPF, the incidence of CVD-IP appears to be lower. Among the underlying CVD, AE was found exclusively in patients with RA and pSS. Five of our six CVD-IP patients with AE had RA (overall incidence, 20.0%; 1-year incidence, 2.58%), and the other patient had pSS. Consistent with our observations, Park et al. described four CVD-IP patients who developed AE, and found that most of them (75%) had RA,13 although they did not mention the overall number for each CVD-IP. Collectively, these data suggest that patients with CVD-IP develop AE less frequently than those with IPF, and among CVD-IP, this condition is most common in RA.
Histologically, three of our six CVD-IP patients with AE had a UIP pattern on surgical lung biopsy, and two had an NSIP pattern, and the other had unclassifiable interstitial pneumonia. Originally, AE was reported to occur in UIP, but recent studies have described this condition in interstitial pneumonia with patterns other than UIP, such as the NSIP pattern.10, 11, 12, 13, 14 So far, there are few data about the histology of AE in CVD-IP. Parambil et al. have recently shown that acute respiratory deterioration in CVD-IP represented DAD pattern on the underlying chronic fibrotic processes, such as UIP and NSIP.9 As a result of severe respiratory failure, the second surgical lung biopsy at the onset of AE could not be performed in our patients, but an autopsy of one patient with NSIP revealed a DAD pattern superimposed on a background of an NSIP pattern (Fig. 3). Collectively, these results suggest that AE occurs in either UIP or NSIP patterns of CVD-IP.
Radiological findings of our CVD-IP patients were comparable to those of AE in IPF, as described previously.6, 14, 25 We found new, extensive ground-glass opacity mostly with patchy air space consolidation, superimposed on background reticular abnormality. In laboratory findings, in addition to severe hypoxemia, marked elevations of serum CRP and LDH levels were observed. Interestingly, the serum levels of KL-6, a marker of interstitial pneumonia, were strikingly increased in all patients tested at the onset of AE. Similarly, in IPF, the serum levels of KL-6 were shown to be elevated at AE, which suggests that this can be used as a marker of this condition.26 A few data are available on BAL findings of AE, and neutrophilia in BAL fluid has been reported.2, 4, 8 We performed BAL in only one patient, and marked neutrophilia was noted.
Little is known about the causative factors of AE of IPF. Several studies have suggested that surgical lung biopsy may cause AE.10, 27 In agreement with those observations, AE occurred immediately after surgical lung biopsy in one patient with pSS-IP. In the remaining five patients, however, the causative factors of this condition were not determined. Interestingly, the activity of the underlying CVD was stable in all patients at the onset of AE. This suggests that development of AE is not associated with deterioration of the underlying CVD, and can occur in patients in a stable condition. In addition, no data are available about the predictive factors for AE at the initial diagnosis. We found that age and RA diagnosis were significantly associated with AE using by univariate Cox's proportional hazards regression models. In addition, we demonstrated that age remained significant in multivariate Cox's proportional hazards regression models, suggesting that CVD-IP patients with higher ages are more likely to develop AE.
Despite intensive anti-inflammatory and immunosuppressive therapies, the outcome associated with AE of CVD-IP was poor in our patients. All patients were treated with high-dose corticosteroids, and four of them also received immunosuppressive agents, such as cyclophosphamide and cyclosporine. However, five (83.3%) of our six patients died of respiratory failure. This poor outcome of our CVD-IP patients was similar to that in AE of IPF.3, 4, 5, 6 Consistent with our observations, all four patients with AE of CVD-IP reported by Park et al. died.13 These data suggest that AE of CVD-IP is characterized by poor prognosis, which is comparable to that of IPF.
There are several limitations to the present study. First, this was a retrospective study, so there were selection and recall biases. Second, the study population was restricted to biopsy-proven cases, and may not accurately reflect the general population of CVD-IP patients. Third, although this study included a relatively large proportion of patients with AE in CVD-IP, the sample size was still too small to determine the precise incidence and clinical characteristics of this condition. Future studies will be required in a large number of patients to confirm our observations.
In conclusion, the present study demonstrated that AE occurred in CVD-IP with poor prognosis, and this condition in CVD-IP closely resembled AE of IPF. In addition, AE is most common in RA, and significantly associated with higher ages. Although AE occurs less frequently in CVD-IP than in IPF, attention should be made on this distinct condition of CVD-IP.
Conflict of interest statement
None of the authors has declared any conflict of interest related to this work.
References
- 1.Kondoh Y., Taniguchi H., Kawabata Y., Yokoi T., Suzuki K., Takagi K. Acute exacerbation in idiopathic pulmonary fibrosis. Analysis of clinical and pathologic findings in three cases. Chest. 1993;103:1808–1812. doi: 10.1378/chest.103.6.1808. [DOI] [PubMed] [Google Scholar]
- 2.Ambrosini V., Cancellieri A., Chilosi M. Acute exacerbation of idiopathic pulmonary fibrosis: report of a series. Eur Respir J. 2003;22:821–826. doi: 10.1183/09031936.03.00022703. [DOI] [PubMed] [Google Scholar]
- 3.Martinez F.J., Safrin S., Weycker D. The clinical course of patients with idiopathic pulmonary fibrosis. Ann Intern Med. 2005;142:963–967. doi: 10.7326/0003-4819-142-12_part_1-200506210-00005. [DOI] [PubMed] [Google Scholar]
- 4.Kim D.S., Park J.H., Park B.K., Lee J.S., Nicholson A.G., Colby T. Acute exacerbation of idiopathic pulmonary fibrosis: frequency and clinical features. Eur Respir J. 2006;27:143–150. doi: 10.1183/09031936.06.00114004. [DOI] [PubMed] [Google Scholar]
- 5.Hyzy R., Huang S., Myers J., Flaherty K., Martinez F. Acute exacerbation of idiopathic pulmonary fibrosis. Chest. 2007;132:1652–1658. doi: 10.1378/chest.07-0299. [DOI] [PubMed] [Google Scholar]
- 6.Collard H.R., Moore B.B., Flaherty K.R. Acute exacerbations of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2007;176:636–643. doi: 10.1164/rccm.200703-463PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rice A.J., Wells A.U., Bouros D. Terminal diffuse alveolar damage in relation to interstitial pneumonias. An autopsy study. Am J Clin Pathol. 2003;119:709–714. doi: 10.1309/UVAR-MDY8-FE9F-JDKU. [DOI] [PubMed] [Google Scholar]
- 8.Parambil J.G., Myers J.L., Ryu J.H. Histopathologic features and outcome of patients with acute exacerbation of idiopathic pulmonary fibrosis undergoing surgical lung biopsy. Chest. 2005;128:3310–3315. doi: 10.1378/chest.128.5.3310. [DOI] [PubMed] [Google Scholar]
- 9.Parambil J.G., Myers J.L., Ryu J.H. Diffuse alveolar damage: uncommon manifestation of pulmonary involvement in patients with connective tissue diseases. Chest. 2006;130:553–558. doi: 10.1378/chest.130.2.553. [DOI] [PubMed] [Google Scholar]
- 10.Kondoh Y., Taniguchi H., Kitaichi M. Acute exacerbation of interstitial pneumonia following surgical lung biopsy. Respir Med. 2006;100:1753–1759. doi: 10.1016/j.rmed.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 11.Parambil J.G., Myers J.L., Lindell R.M., Matteson E.L., Ryu J.H. Interstitial lung disease in primary Sjogren syndrome. Chest. 2006;130:1489–1495. doi: 10.1378/chest.130.5.1489. [DOI] [PubMed] [Google Scholar]
- 12.Churg A., Muller N.L., Silva C.I., Wright J.L. Acute exacerbation (acute lung injury of unknown cause) in UIP and other forms of fibrotic interstitial pneumonias. Am J Surg Pathol. 2007;31:277–284. doi: 10.1097/01.pas.0000213341.70852.9d. [DOI] [PubMed] [Google Scholar]
- 13.Park I.N., Kim D.S., Shim T.S. Acute exacerbation of interstitial pneumonia other than idiopathic pulmonary fibrosis. Chest. 2007;132:214–220. doi: 10.1378/chest.07-0323. [DOI] [PubMed] [Google Scholar]
- 14.Silva C.I., Muller N.L., Fujimoto K. Acute exacerbation of chronic interstitial pneumonia: high-resolution computed tomography and pathologic findings. J Thorac Imaging. 2007;22:221–229. doi: 10.1097/01.rti.0000213588.52343.13. [DOI] [PubMed] [Google Scholar]
- 15.Arnett F.C., Edworthy S.M., Bloch D.A. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988;31:315–324. doi: 10.1002/art.1780310302. [DOI] [PubMed] [Google Scholar]
- 16.Preliminary criteria for the classification of systemic sclerosis (scleroderma). Subcommittee for scleroderma criteria of the American Rheumatism Association Diagnostic and Therapeutic Criteria Committee. Arthritis Rheum. 1980;23:581–590. doi: 10.1002/art.1780230510. [DOI] [PubMed] [Google Scholar]
- 17.Smolen J.S., Steiner G. Mixed connective tissue disease: to be or not to be? Arthritis Rheum. 1998;41:768–777. doi: 10.1002/1529-0131(199805)41:5<768::AID-ART3>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 18.Tan E.M., Cohen A.S., Fries J.F. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1982;25:1271–1277. doi: 10.1002/art.1780251101. [DOI] [PubMed] [Google Scholar]
- 19.Bohan A., Peter J.B. Polymyositis and dermatomyositis (first of two parts) N Engl J Med. 1975;292:344–347. doi: 10.1056/NEJM197502132920706. [DOI] [PubMed] [Google Scholar]
- 20.Yamaguchi M., Ohta A., Tsunematsu T. Preliminary criteria for classification of adult Still's disease. J Rheumatol. 1992;19:424–430. [PubMed] [Google Scholar]
- 21.Suda T., Fujisawa T., Enomoto N. Interstitial lung diseases associated with amyopathic dermatomyositis. Eur Respir J. 2006;28:1005–1012. doi: 10.1183/09031936.06.00038806. [DOI] [PubMed] [Google Scholar]
- 22.American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. This joint statement of the American Thoracic Society (ATS), and the European Respiratory Society (ERS) was adopted by the ATS board of directors, June 2001 and by the ERS Executive Committee, June 2001. Am J Respir Crit Care Med. 2002;165:277–304. doi: 10.1164/ajrccm.165.2.ats01. [DOI] [PubMed] [Google Scholar]
- 23.Suda T., Sato A., Ida M., Gemma H., Hayakawa H., Chida K. Hypersensitivity pneumonitis associated with home ultrasonic humidifiers. Chest. 1995;107:711–717. doi: 10.1378/chest.107.3.711. [DOI] [PubMed] [Google Scholar]
- 24.Azuma A., Nukiwa T., Tsuboi E. Double-blind, placebo-controlled trial of pirfenidone in patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2005;171:1040–1047. doi: 10.1164/rccm.200404-571OC. [DOI] [PubMed] [Google Scholar]
- 25.Akira M., Hamada H., Sakatani M., Kobayashi C., Nishioka M., Yamamoto S. CT findings during phase of accelerated deterioration in patients with idiopathic pulmonary fibrosis. AJR Am J Roentgenol. 1997;168:79–83. doi: 10.2214/ajr.168.1.8976924. [DOI] [PubMed] [Google Scholar]
- 26.Yokoyama A., Kohno N., Hamada H. Circulating KL-6 predicts the outcome of rapidly progressive idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 1998;158:1680–1684. doi: 10.1164/ajrccm.158.5.9803115. [DOI] [PubMed] [Google Scholar]
- 27.Yuksel M., Ozyurtkan M.O., Bostanci K., Ahiskali R., Kodalli N. Acute exacerbation of interstitial fibrosis after pulmonary resection. Ann Thorac Surg. 2006;82:336–338. doi: 10.1016/j.athoracsur.2005.09.036. [DOI] [PubMed] [Google Scholar]