

Abstract
The ability of porcine reproductive and respiratory syndrome virus (PRRSV) to establish a persistent infection is the principal contributing factor to the world-wide spread of the disease. Several studies have documented the course of viral infection in postnatally infected pigs; however, very little is known regarding sites of virus replication during persistent infection of pigs exposed to PRRSV in utero. In this study, virus replication and PRRSV-specific antibody were followed for several hundred days in a group of pigs derived from three sows infected at 90 days of gestation with PRRSV isolate VR-2332. Eighty-four percent of pigs were born viremic with a mortality of 54% within 21 days after birth. At approximately 60 days sera from pigs were negative for virus by virus isolation. Analysis of virus replication in the tissues of pigs randomly sacrificed between 63 and 132 days showed no evidence of virus in lung and other non-lymphoid organs. However, virus was easily recovered from tonsil and lymph nodes and in situ hybridization identified these tissues as sites of virus replication. Even though replication was at a low level, virus was easily transmitted to sentinel pigs. By 260 days pigs became seronegative and did not transmit virus to sentinel pigs. Sacrifice of remaining pigs after 300 days showed no evidence of virus in blood and tissues. This study shows that congenital PRRSV-infected pigs can support virus replication for an extended period during which virus replication is primarily restricted to tonsil and lymph nodes.
Keywords: Porcine reproductive and respiratory syndrome virus (PRRSV), Persistence, Congenital infection, Pig-viruses
1. Introduction
Porcine reproductive and respiratory syndrome (PRRS) is caused by an enveloped positive-stranded RNA virus, PRRSV, belonging to the family Arteriviridae (Wensvoort et al., 1991, Benfield et al., 1992, Nelsen et al., 1999). Other members of the arterivirus group include lactate dehydrogenase-elevating virus (LDV) of mice, equine arteritis virus (EAV), and simian hemorrhagic fever virus (SHFV; for review see Plagemann, 1996). The arteriviruses, toroviruses, and coronaviruses are members of a single order, Nidovirales (Cavanagh, 1997). Arteriviruses structurally resemble togaviruses, but similar to coronaviruses, replicate via a nested 3′-co-terminal set of subgenomic mRNAs that possesses a common leader and a poly-A tail (Snijder and Mulenberg, 1998). The arteriviruses exhibit several important properties relevant to the study of viral pathogenesis, including cytopathic replication in macrophages, the capacity to establish and maintain an asymptomatic persistent infection, as well as cause severe and fatal disease (Plagemann, 1996).
The outcome following experimental PRRSV infection is dependent on the age and reproductive status of the pig, as well as the isolate used for infection (reviewed in Rossow, 1998). Infection of adult pigs usually produces a nonfatal disease, characterized by mild flu-like symptoms, a transient elevation in temperature and inappetance. The reproductive form of PRRS occurs following the infection of pregnant gilts or sows. Accordingly, virus-induced reproductive failure can present clinically as increased regular and delayed returns to estrus and not-in-pig sows, as well as abortions, mummified fetuses, stillbirths and weak-born pigs. (Collins et al., 1992, Christianson et al., 1993, Mengeling et al., 1994, Benfield et al., 1999, Rossow et al., 1999). Surviving neonates exhibit the severest form of respiratory disease with mortality often reaching 100% within 3 weeks after birth. Besides interstitial pneumonia, viral lesions in infected neonates can be found in primary and secondary lymphoid organs, including bone marrow, thymus, lymph node, spleen and tonsil, and non-lymphoid organs, such as brain, heart, kidney, and stomach (Rossow et al., 1994, Rossow et al., 1996, Rossow, 1998, Feng et al., 2001). The complex pathology following exposure to PRRSV in utero represents a unique form of the disease referred to as congenital PRRS.
Another outcome following PRRSV infection is persistently infected swine herds, a property that has contributed to the world-wide spread of the disease (Albina, 1997, Chung et al., 1997, Duan et al., 1997, Wills et al., 1997, Allende et al., 2000, Bierk et al., 2001). It is assumed that persistence is maintained by the ability of PRRSV to evade host defenses and establish a long-term asymptomatic infection within individual pigs. Even though PRRSV-specific antibody appears as early as 5 days post-infection and is followed by serum neutralizing activity and cell-mediated immunity (Bautista and Molitor, 1997, Bautista and Molitor, 1999, Rowland et al., 1999, Rowland et al., 2001) persistently infected pigs can continue to shed virus. The mechanistic basis for PRRSV persistence is not known. Similar to LDV, PRRSV may sustain replication in a subpopulation of renewable macrophages, while avoiding host-defenses (Plagemann et al., 1995). Another possibility is the restriction of virus replication to immunoprivileged sites, such as the male reproductive tract (Christopher-Hennings et al., 1995, Sur et al., 1997), which is observed during infection with other arteriviruses, such as EAV and LDV (Holyoak et al., 1993, Anderson et al., 1995). In previous work characterizing quasispecies evolution during infection, we proposed that antigenic drift in the ectodomain of GP5 may lead to the selection of clones that are resistant to neutralizing antibody or change the tropism of the virus for a different population of cells (Rowland et al., 1999).
Previous experimental studies designed to understand PRRSV persistence have characterized virus replication during PRRSV infection of postnatal pigs. These studies indicate that virus is present in the blood for approximately 20–30 days after infection; however, in a significant number of animals, virus can be detected in tonsil for up to 150 days and by RT-PCR in semen for 90 days after infection (Wills et al., 1997, Christopher-Hennings et al., 1995, Allende et al., 2000). More recently Wills et al. (2003) reported that six of 28 pigs, infected at 35 days after birth, were positive for the presence of virus at 225 days. However, the ability of these long-term infected pigs to shed virus was not determined. Very little is known regarding the course of virus replication in pigs exposed to virus in utero. The purpose of this study was to further understand persistent infection in congenitally infected pigs by characterizing the course of clinical disease, sites of virus replication and the capacity to transmit virus in a group of pigs exposed to PRRSV in utero.
2. Materials and methods
2.1. Virus, cells and animals
Three pregnant sows at 85–90 days gestation were infected with PRRSV isolate VR-2332 (Benfield et al., 1992, Collins et al., 1992) passage 5, which was plaque-purified and propagated on MARC-145 cells, a subclone of the MA-104 cell line (Kim et al., 1993). Five milliliters of virus, at a concentration of 104 TCID50/ml, was divided into two doses and administered into each nare with a 5 cc syringe. The sows were monitored daily for signs of PRRSV infection and allowed to farrow naturally. Umbilical cord and blood samples were collected from newborn pigs at birth and prior to nursing. Blood was then collected from surviving pigs at weekly intervals for the duration of the study. Neonates were monitored daily for clinical signs. Pigs that succumbed to acute PRRSV infection were necropsied and tissues evaluated for pathology and virus replication. At 19 days all surviving pigs were weaned and placed together in a single isolation room in the animal care facility.
2.2. Measurements of antibody and virus neutralizing activity
PRRSV-specific antibody in serum was measured using the “old format” HerdChek® PRRS ELISA (IDEXX) according to the manufacturer’s instructions. This assay was done by personnel at an accredited diagnostic laboratory (Animal Disease Research and Diagnostic Laboratory at South Dakota State University). An S/P ratio greater than 0.39 was considered seropositive. PRRSV neutralizing activity in serum was measured according to Rowland et al. (1999). Serial 1:2 dilutions of serum were prepared in culture medium on 96 well plates. An equal volume of the VR-2332 PRRSV, at a concentration of 2000 TCID50/ml, was added to each sample and incubated for 1 h at 37 °C then transferred to a 96-well plate containing confluent MARC-145 cells. After 3 days the plates were fixed in 80% acetone and infected cells detected with FITC-labeled anti-nucleocapsid monoclonal antibody, SDOW-17 (Nelson et al., 1993) diluted in PBS with 5% FBS. Neutralizing activity was reported as the log of the highest serum dilution that prevented the replication of PRRSV in MARC-145 cells (negative for SDOW-17 staining).
2.3. Virus isolation from serum and tissues
Virus isolation was performed by incubating dilutions of serum or tissue homogenate on 96-well plates containing MARC-145 cells. For virus isolation from tissues, approximately 10 g of tissue was homogenized in 5 ml of Hanks balanced salt solution and centrifuged at 500×g for 20 min to remove debris. Dilutions (1:10 and 1:40) of cleared homogenate or serum were incubated on 96-well plates of MARC-145 cells for 3 days. Plates were then fixed in 80% acetone and stained with FITC-SDOW-17.
2.4. RT-PCR for the detection of viral RNA
Nested RT-PCR amplification of ORF 7, described by Christopher-Hennings et al. (1995), was used for the detection of viral RNA in serum and tissues. Samples were homogenized in an equal volume of lysis buffer (4 M guanidinium thiocyanate, 25 mM sodium citrate [pH.7], 0.5% Sarkosyl [N-lauryl sarcosine] 0.1 M 2-mercaptoethanol). Five hundred microliters of lysate was added to 250 ul of phenol and 250 ul chloroform and mixed. The aqueous phase was removed, the RNA precipitated with ethanol and then reconstituted in 30 ul of distilled water. One microliter of RNA was reverse transcribed using MuLV reverse transcriptase and the outer antisense PCR primer. The outer sense and antisense primers for PCR amplification of a 484 bp product incorporating the entire ORF7 region were 5′-TCGTGTTGGGTGGCAGAAAAGC-3′ and 5′-GCCATTCACCACACATTCTTCC-3′, respectively. The outer primers were incorporated into a reaction mixture containing cDNA, 2 mM MgCl2, 1× PCR buffer and Taq polymerase. The product was subjected to a second round of amplification using the nested sense and antisense primers, 5′-CCAGATGCTGGGTAAGATCATC-3′ and 5′-CAGTGTAACTTATCCTCCCTGA-3′, respectively. The second round of amplification incorporated 3 mM MgCl2, 1× PCR buffer and Taq polymerase. Forty cycles of amplification were performed for each primer pair. All PCR reactions incorporated a 94 °C denaturing step (25 s), a 58 °C annealing step (25 s), and a 74 °C polymerization step (25 s). The reaction products were electrophoresed on an agarose gel and the final 236 bp product detected with ethidium bromide.
2.5. In situ hybridization
In situ hybridization for the identification of cells supporting virus replication was done according to Lawson et al. (1997) using a modification of the procedure originally described by Anderson et al. (1995). Tissues were fixed in 10% neutral buffered formalin for 4 h at room temperature and embedded in paraffin. Thin sections were mounted on Denhardt’s solution-treated slides and paraffin removed with xylene. After pretreatment, the tissue sections were hybridized with a [35S] dCTP labeled random-primed probe complementary to a 484 nucleotide region covering all of ORF 7. Slides were hybridized for 72 h at 42 °C, washed, dipped in Kodak NTB-2 autoradiographic emulsion, exposed for approximately 3 days, developed and counterstained with Mayer’s hematoxylin and eosin Y. A positive control included tissues from pigs experimentally infected with PRRSV. Negative controls included tissues from uninfected pigs and tissues from experimentally infected pigs hybridized with a probe prepared using cDNA from LDV ORF7 region.
3. Results
3.1. Clinical disease
Three pregnant sows were infected with VR-2332 at 90 days gestation. Clinical signs in sows were minimal, consisting of inappetance in two of the three animals, which began at 3–4 days post-inoculation and lasted approximately 2 days. All sows seroconverted and farrowed naturally at 114 days of gestation. Outcomes for the 35 piglets from the three infected sows are summarized in Table 1 . Seven pigs were stillborn and due to the degraded condition of the tissues were not subjected to further study. Analysis of virus and antibody in blood and/or umbilical cords from the 28 live neonates showed that at least 20 or 74% were virus isolation (VI) or RT-PCR positive for PRRSV at the time of farrowing indicating that in utero infection was successful. The eight neonates that were identified as PRRSV-negative at the time of birth became VI-positive by 7 dpf. Neonates as a group exhibited signs consistent with PRRSV infection, with pronounced dyspnea being the most common symptom. About half of the pigs exhibited more severe disease signs, including ocular edema and inappetance. Microhistology showed PRRSV-induced pathological changes in the lungs and other organs of acutely infected symptomatic pigs, including interstitial pneumonia with the infiltration of proteinaceous debris from the interstitial space into the alveoli. Lymph nodes were generally enlarged with germinal center hypertrophy, hyperplasia and necrosis. Lesions in the brain (encephalitis) and heart (myocarditis) and stomach of some pigs were also observed.
Table 1.
Summary of pigs exposed to VR-2332 in utero
| Number of gilts infected at 95 days gestation | 3 |
| Number of pigs farrowed | 35 |
| Number of stillborn pigs | 7 (20%) |
| Number of pigs born live | 28 (80%) |
| Number of live pigs born viremic | 20 (74%)a |
| Mortality 1–21 dpfb | 15 (54%) |
| Mortality from PRRSV after 21 days | 0 |
| Pigs euthanized between 63 and 132 dpf | 9 |
| Number of pigs maintained after 132 dpf | 4 |
Percentage based on the analysis of 24 blood samples recovered from piglets at birth.
Days post-farrowing.
There was a 54% mortality between 1 and 21 dpf. Only one pig in the early mortality group was confirmed to have died from a non-PRRS-related cause. Pigs that survived beyond 21 dpf eventually recovered and showed no external signs of acute infection and there were no PRRSV-related deaths after 21 dpf. For the purpose of comparison, we included a litter from a single mock-infected sow. The 10 piglets, born at the same time as the PRRSV pigs, appeared normal and showed no mortality within 21 dpf.
3.2. The umbilical cord as a site of virus replication
In the developing fetus, there are two potential sources of virus, which can cause fetal damage. The first source is virus replication within the tissues and organs of the developing fetus. Another source is virus replication in the accessory tissues, such as the umbilical cord and placenta. In this study, we were able to obtain 24 umbilical cords of piglets born to infected sows, including 18 umbilical cords from infected newborns and 6 umbilical cords from non-viremic pigs born to infected sows. Virus isolation showed all umbilical cords from infected neonates to be positive for PRRSV. Neonates that were VI or RT-PCR negative for PRRSV in serum were also negative for PRRSV in umbilical cord tissues. The recovery of virus from umbilical cords presented the possibility that virus was isolated from circulating blood and not from cells supporting virus replication. In situ hybridization, for the detection of cells possessing viral RNA, was performed on a single microscopic cross-section of each umbilical cord. We identified PRRSV RNA-positive cells in 10 of the 15 tissues cross-sections from VI-positive umbilical cords, indicating the presence of infected cells. A representative example of PRRSV RNA-positive cells in an umbilical cord cross-section is shown in Fig. 3A. Positive cells were generally found within the smooth muscle regions of umbilical cord tissues. As a negative control, in situ hybridization was performed on umbilical cord sections from six uninfected pigs. All six were negative for the presence of cells containing viral RNA. Even though cells supporting virus replication were easily identified in umbilical cords of infected piglets, histology at both gross and microscopic levels revealed no virus-associated lesions.
Fig. 3.
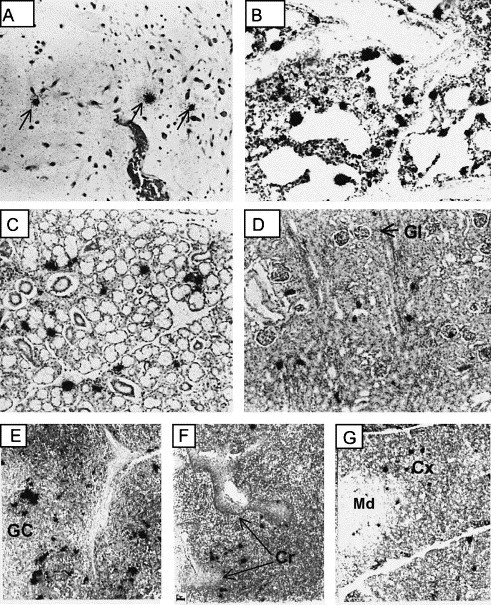
Cells supporting PRRSV replication in tissues during acute infection. Representative photomicrographs showing in situ hybridization of PRRSV RNA during the first 21 dpf. (A) umbilical cord, (B) lung with interstitial pneumonia, (C) salivary gland, (D) kidney, (E) lymph node, (F) tonsil and (G) thymus. Arrows in (A) identify representative hybridization signals. Photomicrographs taken at low magnification. Key: Gl, glomerulus; GC, germinal center; Cr, crypt; Md, medulla; Cx, cortex.
3.3. The association between timing of in utero infection and mortality
Based on the presence of virus and PRRSV-specific antibody in 24 pre-suckling serum samples, newborn piglets could be placed into one of the three groups. The first group contained four pigs (16%) that were VI-negative and seronegative. We assume that these neonates were not infected in utero, but were infected immediately prior to farrowing or immediately after farrowing as a result of contact with the dam or infected litter mates. The second group contained 13 (54%) pigs that were VI-positive for PRRSV, but seronegative. And finally, the third group was composed of seven (30%) neonates that were both VI-positive and seropositive. Each litter contained a mixture of pigs that could be placed in all three groups, indicating that fetuses were infected at different times in the same sow. Since pigs are immunocompetent at around 70 days of gestation and there is no passive transfer of antibody from the mother to the fetus (Tizard, 1996) we can conclude that infected neonates that were born seropositive were infected earlier than those that were VI-positive and seronegative.
Based on the infection of fetuses at different times in utero, we proposed that the earlier a fetus is infected the greater the probability of early mortality after birth. To test this hypothesis, neonates were divided into two groups. The first group was composed of the 13 piglets that died within 21 dpf and showed symptoms of severe PRRS. (One pig succumbed from other than PRRSV infection and was eliminated from the analysis.) The second group included the 10 remaining pigs, which recovered and became asymptomatic. In the long-term survivor group only one serum sample was identified as positive for PRRSV antibody, as indicated by an ELISA S/P ratio greater than 0.39 (Fig. 1 ). In pigs that died prior to 21 days, 6 of 13 pigs were seropositive for PRRSV. Chi-square statistical analysis of these data showed a statistically significant association between the presence of PRRSV antibody and mortality prior to 21 dpf (P=0.036).
Fig. 1.
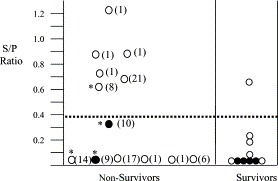
Correlation of PRRSV-specific antibody at the time of birth with outcome. The data show PRRSV-specific antibody levels and viral status in pre-suckling serum samples obtained from 23 newborn pigs. The non-survivors were those pigs that succumbed within 21 days after farrowing. The survivors were those that recovered from acute PRRSV infection. The horizontal line identifies the 0.39 S/P ratio used as the cut-off for determining if a sample was positive or negative for PRRSV antibody. Samples that were positive or negative for virus are presented by open and closed circles, respectively. The number in parentheses identifies the day after farrowing on which that pig died. Asterisks indicate samples from pigs that were infected with S. suis. For Chi-square analysis the Proc Freq statement was utilized in SAS 6.12 to determine the Chi-square statistic. The association between the presence of antibody and mortality was significant at the P=0.036 level.
It should be noted that in the non-survivor group, 2 of the 13 pigs were VI-negative and seronegative at birth. These two piglets, and two others in the non-survivor group, exhibited clinical features consistent with Streptococcus sp. infection, including swollen joints filled with pus, recruitment of PMNs into inflammatory lesions, and the presence of streptococci in lung and other tissues. Streptococcus suis was isolated from tissues of all four pigs.
3.4. Viremia and antibody response
The presence of virus in the blood was determined by VI, and later when serum samples became VI-negative, by RT-PCR. The analysis of PRRSV-specific immune responses included measurements of total anti-PRRSV activity by ELISA and virus neutralizing activity. These data, collected over a period of 300 days after birth, are graphically summarized in Fig. 2 . By 7 dpf, all pigs were VI-positive in serum, with the percentage of VI-positive serum samples declining at about 21 dpf. The decline in the number of VI-positive serum samples was associated with the appearances of peak antibody levels and virus neutralizing activity. The disappearance of virus from the blood also correlated with the recovery of pigs from acute disease. The last VI- and RT-PCR-positive serum samples were obtained at 48 and 78 dpf, respectively. Pigs that attained a VI-negative status remained VI-negative throughout the remainder of the study. However, it should be noted that pigs that became PCR negative did not always stay negative. Periodically we found serum samples between 78 and 228 dpf, which were PCR-positive, even though the previous and subsequent samples taken from the same animal were negative by PCR. These intermittent PCR-positive results can best be explained by the presence of a small amount of viral RNA, but at or near the lower detection limits of the PCR assay (data not shown). There were no positive RT-PCR serum samples after 228 dpf.
Fig. 2.
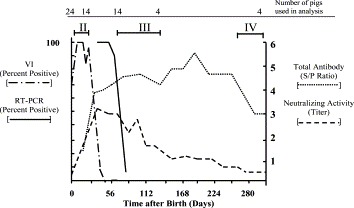
Antibody and viremia during PRRSV infection. PRRSV-specific antibody responses are presented as total ELISA antibody (S/P ratio) and virus neutralizing activity (titer). Data are presented as mean values. The presence of virus in serum was determined by VI on MARC-145 cells or RT-PCR of ORF7 RNA as described in Section 2. The results are presented as the percentage of pigs that were positive for virus or viral RNA. The number of samples used to calculate each data point are indicated at the top. Data after 132 dpf was obtained from a group of four pigs retained for long-term study. The Roman numerals show periods of infection during which virus replication and pathology were assessed in tissues.
Mean PRRSV antibody peaked at approximately 28 dpf with ELISA S/P ratios ranging between 2.60 and 4.80 (n=13), well above the 0.39 cut-off that identified a serum sample as seropositive. Since pigs were suckling prior to 19 days, we assume that passive antibody from the mother was contributing to the humoral response. After 28 days, antibody gradually declined and by 260 dpf the remaining four pigs had attained a seronegative status (S/P ratio<0.4) and remained seronegative throughout the remainder of the study. Virus neutralizing activity in serum was first detected in one pig at 7 dpf (neutralizing titer=4). By 56 dpf all pigs showed detectable neutralizing activity. Since these measurements were made several weeks after weaning, we presume that virus neutralizing activity was generated by the immune system of the congenitally infected pigs. At their peak, between days 56 and 260, neutralizing titers ranged between 1:16 and 1:256 (Fig. 2). This relatively low amount of neutralizing activity was not unexpected and is typical of the neutralizing response during infection of older pigs (Yoon et al., 1995, Wills et al., 1997). Interestingly, virus neutralizing activity was detected in sera well beyond 260 days, even though all pigs were seronegative for PRRSV by the IDEXX ELISA test (S/P ratios below 0.40). The source of the virus neutralizing activity in serum was not determined. However, the presence of neutralizing activity in “seronegative” sera can be explained based on the different sensitivities of the assays used to measure antibody and neutralizing activity. For example, we cannot rule out the possibility that pigs possessing an ELISA S/P ratio less than 0.4 have some PRRSV antibody, but at amounts below the confidence limits of the assay.
3.5. Sites of virus replication
The analysis of virus replication in tissues of congenitally infected pigs focused on three periods of infection, identified by Roman numerals II, III and IV in Fig. 2. Each period was unique in terms of clinical disease signs, viremia, serology and pathology. The first period, indicated by Roman numeral II in Fig. 2, represented the acute, symptomatic stage of infection, which covered the first 21 days after birth. The second period, identified by Roman numeral III focused on a period when pigs were asymptomatic for clinical disease signs and negative in serum for PRRSV by VI and largely negative in serum by RT-PCR, but all pigs were still seropositive. And finally, we assessed virus replication in a group of four infected pigs, which had become seronegative (Roman numeral IV).
During acute infection (Roman numeral II, Fig. 2) PRRSV was easily isolated from all lymphoid and non-lymphoid tissues examined (lung, heart, aorta, kidney, liver, testes, salivary gland, intestine, brain, stomach, thymus, spleen, tonsil, and lymph nodes). In situ hybridization identified cells containing viral RNA in the same tissues. Fig. 3 B–D shows representative examples of RNA-positive cells in lung, salivary gland and kidney. These three tissues represent sources of PRRSV shedding by oral–nasal and urinary secretions. Also shown are representative examples of virus replication in cells of lymph node, tonsil and thymus (Fig. 3E–G). In the tonsil, cells supporting virus replication were in close proximity to the tonsilar crypts. These data are consistent with our previous study, identifying multiple sites of virus replication in gnotobiotic piglets at 21 days after experimental infection with VR-2332 (Lawson et al., 1997).
Roman numeral III in Fig. 2 shows the next period of infection covered by our study. During this stage, PRRSV-specific antibody was declining, but all pigs were seropositive and possessed virus neutralizing activity at a serum dilution of at least 1:16. There were no overt clinical signs of acute PRRSV infection and all pigs were VI-negative in serum. Virus isolation and RT-PCR were performed on lymphoid and non-lymphoid tissues from nine of the asymptomatic pigs, sacrificed at random between 63 and 132 dpf. VI and RT-PCR results for lung, mandibular lymph node, mesenteric lymph node, tonsil and serum are presented in Table 2 . In contrast to the acutely infected pigs, all lung and non-lymphoid tissue samples were negative for virus and viral RNA. The absence of detectable amounts of virus in lungs was based on negative results for VI, RT-PCR and in situ hybridization assays of tissue samples from each lung quadrant. Virus isolation, RT-PCR and in situ hybridization also failed to identify virus or viral RNA in kidney, bladder, heart, aorta, liver, intestines, stomach, testes/ovaries, nasal turbinates, and salivary glands (data not shown). Positive VI and RT-PCR results were frequently obtained from homogenates of tonsil (seven of nine pigs) and mandibular lymph nodes (eight of nine pigs; Table 2). Mesenteric, aortic, cervical, tracheal, medial iliac lymph nodes also yielded virus and/or viral RNA, but at much lower frequencies (see Table 2 for mesenteric lymph node data). Virus or viral RNA was not detected in spleen or thymus (data not shown). Consistent with the VI and RT-PCR results, in situ hybridization identified PRRSV RNA-positive cells in tonsil and mandibular lymph nodes. PRRSV RNA-positive cells in a mandibular lymph node at 63 dpf and tonsil at 132 dpf are shown in Fig. 4 . Only a few RNA-positive cells could be identified within a given tissue section. Therefore, the locations of RNA-positive positive cells were confirmed by performing hybridization of adjacent tissue sections (Fig. 4). All pigs sacrificed during this time possessed detectable amounts of virus neutralizing activity. Together these data show that PRRSV replication during asymptomatic infection is absent from lung and other non-lymphoid tissues and is primarily restricted to tonsil and lymph nodes, even in the presence of humoral immunity.
Table 2.
RT-PCR and virus isolation from sera and tissues of seropositive, VI-negative PRRSV-infected pigs
| Pig no. | Grpa | dpfb | Antibodyc | RT-PCR (virus isolation)d |
||||
| Serume | Lung | TNSL | MDLN | MSLN | ||||
| 186 | 1 | 63 | 3.87 (16) | + (35) | − (−) | NDf (+) | + (−) | ND (−) |
| 189 | 2 | 75 | 2.09 (16) | − (20) | − (−) | ND (−) | + (−) | ND (+) |
| 62 | 2 | 75 | 4.22 (16) | − (28) | − (−) | + (+) | + (−) | ND (−) |
| 199 | 1 | 91 | 2.87 (64) | − (41) | − (−) | − (−) | + (−) | + (−) |
| 67 | 3 | 92 | 2.02 (16) | − (28) | − (−) | ND (+) | ND (+) | − (−) |
| 16 | 1 | 104 | 4.13 (32) | − (34) | − (−) | + (−) | + (−) | − (−) |
| 72 | 2 | 104 | 3.83 (256) | − (48) | − (−) | ND (+) | + (−) | + (−) |
| 69 | 2 | 132 | 2.31 (16) | − (34) | − (−) | + (+) | − (−) | − (−) |
| 71 | 2 | 132 | 1.57 (16) | − (34) | − (−) | + (−) | + (−) | − (−) |
Group designation based on virus and serology at the time of birth. Grp 1, virus-negative and seronegative; Grp 2, virus-positive and seronegative; Grp 3, virus-positive and seropositive.
Days post-farrowing on which the pig was sacrificed.
PRRSV antibody status, ELISA response and neutralizing titer (in parenthesis) at the time of sacrifice.
RT-PCR of ORF7 and virus isolation (in parenthesis) were performed on serum, lung, tonsil (TNSL), mandibular lymph node (MDLN) and mesenteric lymph node (MSLN).
The number in parentheses identifies the last day that a positive VI was obtained.
Not determined.
Fig. 4.
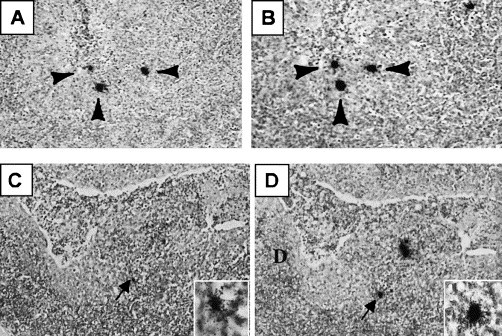
Identification of cells supporting PRRSV replication in secondary lymphoid organs during asymptomatic infection. Photomicrographs, taken at low magnification, showing in situ hybridization results in adjacent thin sections of a lymph node (A and B) at 63 dpf and a tonsil (C and D) at 132 dpf. The arrows identify RNA-positive cells that are present in both sections. Inset is a higher magnification showing the hybridization signal.
To investigate the significance of this low level of virus replication in asymptomatic pigs, we studied the ability of congenital PRRS pigs to transmit virus. Age-matched PRRSV seronegative sentinel pigs were introduced into the PRRSV-infected group at 64 dpf (2 sentinels and 12 principals), 84 dpf (2 sentinels and 10 principals), 98 dpf (1 sentinel, 8 principals) and 112 dpf (1 sentinel, 6 principals). Sentinel pigs were allowed to commingle with the infected herd for 1 week and then removed to separate isolation rooms for observation and assessment of infection by VI and serology. All sentinel pigs were VI-positive in serum within 1 week after introduction and seroconverted a week later. These results indicate that under natural conditions of normal pig-to-pig contact PRRSV is efficiently transmitted to naïve pigs even though virus replication is low.
Congenital PRRS pigs that became seronegative (see Roman numeral IV in Fig. 2) were the subject of the last part of our study. By 260 dpf, all sera from four remaining infected pigs were VI-negative, RT-PCR negative and ELISA seronegative for PRRSV antibody (S/P ratio less than 0.4). To determine if PRRSV pigs were capable of shedding virus, four age-matched PRRSV seronegative sentinel pigs were introduced at 260 dpf and maintained in continual contact with the PRRSV pigs for at least 56 days. Commingling culminated in the mating of each PRRSV pig to an aged-matched sentinel. Two litters were obtained from the three PRRSV females. The adult female pigs and litters were sacrificed and assessed for PRRSV replication. All three female pigs and associated litters were negative for PRRSV antibody and viral RNA as indicated by negative results for in situ hybridization and by performing RT-PCR in lungs, lymph nodes and tonsils. The single sentinel female pig and litter obtained after mating to the seronegative PRRSV boar were also negative for PRRSV. Since previous work identified long-term virus shedding in semen (Christopher-Hennings et al., 1995), we also performed RT-PCR and in situ hybridization on testes and male accessory reproductive organs of the remaining male pig. There was no evidence of virus replication in any of the male reproductive tissues.
4. Discussion
The results from this study show that congenital PRRSV infection can be divided into distinct stages, unique in terms of serology, virology and clinical disease. The first stage covers the period of in utero exposure to virus. These data demonstrate that not all fetuses are infected at the same time and some fetuses may escape infection altogether. Furthermore, the positive association between the presence of antibody at birth with early mortality indicates that the earlier a fetus is infected the more severe the clinical disease (Fig. 1). Since it takes 1–2 weeks to develop antibody, we assume that seropositive fetuses were infected 1–2 weeks after infection of the sow or approximately 1–2 weeks prior to birth. As a whole, these data show that the placenta is a barrier to PRRSV, but this barrier does break down with time. There are at least two sources of virus-related damage that can account for early mortality in the antibody-positive fetuses. The first is the formation of PRRSV lesions in fetal organs. However, the target organs in the fetus may be different from those in the postnatally infected pig. For instance, porcine alveolar macrophages, which support virus replication to high levels in postnatal pigs are absent from the fetal lung. In preliminary studies, we found relatively large amounts of virus in the thymus from fetuses obtained 1–2 weeks prior to birth (data not shown). Feng et al. (2001) observed thymic lesions in congenital PRRSV pigs sacrificed at 12 dpf. The possible effect of PRRSV infection on fetal/neonatal primary immune organs has obvious implications in the increased susceptibility of congenitally infected pigs to secondary infections. A second source of fetal damage is through the disruption of support tissues, such as placenta and umbilical cord. Lager and Halbur (1996) reported lesions in umbilical cord, which appeared to be sufficient to cause anoxia in the developing fetus. Even though umbilical cord was identified as a sight of virus replication in this study, we were not able to identify PRRSV-related lesions. The absence of umbilical cord lesions may reflect the different isolate used in this study.
Even though immune abnormalities represent a hallmark of PRRSV infection, congenital infection did not appear to adversely affect the capacity of surviving pigs to develop an antibody response to PRRSV later in life. As a group, the antibody levels in congenital PRRS pigs peaked at about 28 days after birth (Fig. 2), which is similar to the time-course appearance of antibody in pigs infected several weeks postnatally (Yoon et al., 1995, Allende et al., 2000). Congenital PRRS pigs were able to mount a prolonged neutralizing antibody response at levels typically found in experimentally infected older pigs (Yoon et al., 1995). These results indicate that fetuses exposed to PRRSV during late gestation do not become immunotolerant to the virus later in life.
A unique aspect of this study was the thorough analysis of virus replication in pig tissues during asymptomatic infection (Roman numeral III, Fig. 2). This stage of congenital PRRSV infection was characterized as the period of time when pigs no longer exhibited clinical signs of acute disease, were VI-negative in serum, but still seropositive. In this study it was represented by a group of nine pigs sacrificed between 63 and 132 days after birth. Perhaps, the most interesting finding was the identification of virus in tonsil and mandibular lymph node, but not in lung and other non-lymphoid organs. These results are in agreement with studies identifying tonsil as a source of virus during persistent infection (Wills et al., 1997, Allende et al., 2000, Horter et al., 2002), but contradict other studies identifying pulmonary macrophages as the source of virus replication in persistently infected pigs (Mengeling et al., 1996, Duan et al., 1997). Our conclusion that virus is absent from the lungs in persistently infected pigs is based on a thorough analysis of virus replication, including the use of VI, RT-PCR and in situ hybridization. Continuous virus replication in regional lymph nodes accounts for the efficient transmission via oral–nasal secretions and in semen during persistent infection (Christopher-Hennings et al., 1998). The basis for an eventual change in organ tropism, from multiple sites of PRRSV replication to preferential replication in tonsil and lymph nodes, is not known. In previous work we identified the emergence of a virus sub-population during persistent infection, which possessed a mutation in the ectodomain of ORF5, the major envelope glycoprotein. This mutation may increase the tropism of PRRSV for replication in a macrophage subpopulation that resides in tonsils and lymph nodes.
We followed virus and antibody in four congenital PRRSV pigs for more than a year. At the time of necropsy these pigs were seronegative for PRRSV (S/P ELISA ratio less than 0.4) and showed no evidence of virus replication, as determined by the absence of viral RNA in serum and in tissues. In this study we were not able to determine exactly when virus disappeared from these pigs. However, the last RT-PCR positive serum sample was obtained at 228 dpf and sentinel pigs did not become infected when introduced at 260 dpf. The failure of sentinel pigs to become infected after an extended period of intimate contact with the congenital PRRSV-infected pigs indicated that PRRSV replication was not reactivated.
The results from this study show that congenital PRRS pigs can shed virus for at least 112 dpf and perhaps for as long at 250 dpf, which support the conclusions of a recent study by Wills et al. (2003). The isolation of virus from lymph nodes at 132 dpf in this study is longer than the isolation of infectious virus at 84 days from pigs infected at 1 month of age (Allende et al., 2000), but is similar to length of time reported by Wills et al. (2003). Because of viral strain differences, it is difficult to assess the impact of congenital infection on virus replication, shedding and persistence by making comparisons with other published studies performed in older pigs. However, in agreement with Allende et al. (2001) PRRSV replication does not establish a steady-state equilibrium in the manner of other persistent arteriviruses, such as LDV or SHFV, but gradually declines over time, with the lymphoid organs as the last vestige of virus replication. The presence of an extended period of ongoing PRRSV replication in pigs with substantial anti-PRRSV humoral and cell-mediated immune responses suggests that the eventual elimination of the virus from congenital PRRS pigs may ultimately reflect the disappearance of PRRSV-permissive cells with age.
5. Conclusion
Based on our analysis of clinical disease, PRRSV-specific antibody and virus replication, congenital PRRSV infection can be divided into four distinct stages. Stage I covers the period of in utero exposure to virus during which time fetuses are infected at different times and some fetuses remain uninfected. The positive association between the presence of PRRSV antibody at birth with early mortality indicates that the earlier a fetus is infected the more severe the clinical disease outcome. Stage II is the period of acute infection. During this time pigs show signs of acute PRRSV. In this study virus was isolated from all tissues, and all tissues contained cells supporting virus replication. Stage III is a period when pigs no longer exhibit clinical disease signs, are VI-negative and largely RT-PCR negative in serum and possess peak levels of virus neutralizing activity. Even though virus replication is relatively low, persistently infected pigs can efficiently transmit virus to naive pigs. Stage IV of congenital PRRSV infection is viral clearance.
Acknowledgements
This work was supported by the USDA National Research Initiative for Competitive Grants Program Grants # 95-37204-2233 and # 97-35204-5071, National Pork Producer Council Grant # 1795, National Science Foundation Grant # OSR-9108773, South Dakota Agricultural Experiment Station and the South Dakota Future Fund. We thank Curt Nelson and Scott Kistler for their excellent assistance in the care and welfare of the animals.
References
- Albina E. Epidemiology of porcine reproductive and respiratory syndrome (PRRS): an overview. Vet. Microbiol. 1997;55:309–316. doi: 10.1016/s0378-1135(96)01322-3. [DOI] [PubMed] [Google Scholar]
- Allende R, Laegreid W.W, Kutish G.F, Galeota J.A, Wills R.W, Osario F.A. Porcine reproductive and respiratory syndrome virus: description of persistence in individual pigs upon experimental infection. J. Virol. 2000;74:10834–10837. doi: 10.1128/jvi.74.22.10834-10837.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson G.W, Rowland R.R.R, Even C, Palmer G.A, Plagemann P.G.W. Lactate dehydrogenase-elevating virus persists in liver, spleen, lymph nodes and testis and results in accumulation of viral RNA in germinal centers concomitant with the polyclonal activation of B cells. J. Virol. 1995;69:5177–5185. doi: 10.1128/jvi.69.8.5177-5185.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bautista E.M, Molitor T.W. Cell-mediated immunity to porcine reproductive and respiratory syndrome virus. Viral Immunol. 1997;10:83–94. doi: 10.1089/vim.1997.10.83. [DOI] [PubMed] [Google Scholar]
- Bautista E.M, Molitor T.W. IFN gamma inhibits porcine reproductive and respiratory syndrome virus replication in macrophages. Arch. Virol. 1999;144:1191–1200. doi: 10.1007/s007050050578. [DOI] [PubMed] [Google Scholar]
- Benfield D.A, Nelson E.A, Collins J.E, Harris L, Goyal S.M, Robison D, Christianson W.T, Morrison R.B, Gorcyca D, Chladek D. Characterization of swine infertility and respiratory syndrome (SIRS) virus (Isolate ATCC VR-2332) J. Vet. Diagn. Invest. 1992;4:127–133. doi: 10.1177/104063879200400202. [DOI] [PubMed] [Google Scholar]
- Benfield, D.A., Collins, J.E., Dee, S.A., Halbur, P.G., Joo, H.S., Lager, K.M., Mengeling, W.L., Murtaugh, M.P., Rossow, K.D., Stevenson, G.W., Zimmerman, J.J., 1999. Porcine reproductive and respiratory syndrome. In: Straw, B., D’Allaire, S., Mengeling, W.L., Taylor, D.J. (Eds.), Diseases of Swine, 8th ed. Iowa State University Press, Ames, IA, Chapter 18, pp. 201–232.
- Bierk M.D, Dee S.A, Rossow K.D, Collins J.E, Guedes M.I, Pijoan C, Molitor T.W. Diagnostic investigation of chronic porcine reproductive and respiratory syndrome virus in a breeding herd of pigs. Vet. Rec. 2001;148:687–690. doi: 10.1136/vr.148.22.687. [DOI] [PubMed] [Google Scholar]
- Cavanagh D. Nidovirales: a new order comprising Coronaviridae and Arteriviridae. Arch. Virol. 1997;142:629–633. [PubMed] [Google Scholar]
- Christianson W.T, Choi C.S, Collins J.E, Molitor T.W, Morrison R.B, Joo H.S. Pathogenesis of porcine reproductive and respiratory syndrome virus infection in mid gestation sows and fetuses. Can. J. Vet. Res. 1993;57:262–268. [PMC free article] [PubMed] [Google Scholar]
- Christopher-Hennings J, Nelson E.A, Hines R.J, Nelson J.K, Swenson S.L, Zimmerman J.J, Chase C.L, Yaeger M.J, Benfield D.A. Persistence of porcine reproductive and respiratory syndrome virus in serum and semen of adult boars. J. Vet. Diagn. Invest. 1995;7:456–464. doi: 10.1177/104063879500700406. [DOI] [PubMed] [Google Scholar]
- Christopher-Hennings J, Nelson E.A, Nelson J.K, Rossow K.D, Shivers J.L, Yaeger M.J, Chase C.C, Garduno R.A, Collins J.E, Benfield D.A. Identification of porcine reproductive and respiratory syndrome virus in semen and tissues from vasectomized and nonvasectomized boars. Vet. Pathol. 1998;35:260–267. doi: 10.1177/030098589803500404. [DOI] [PubMed] [Google Scholar]
- Chung W.B, Lin M.W, Chang W.F, Hsu M, Yang P.C. Persistence of porcine reproductive and respiratory syndrome virus in intensive farrow-to-finish pig herds. Can. J. Vet. Res. 1997;61:292–298. [PMC free article] [PubMed] [Google Scholar]
- Collins J.E, Benfield D.A, Christianson W.T, Harris L, Hennings J.C, Shaw D.P, Goyal S.M, Gorcyca D, Chladek D, McCullough S, Morrison R.B, Joo H.S. Isolation of swine infertility and respiratory syndrome virus (Isolate ATCC VR-2332) in North America and experimental reproduction of the disease in gnotobiotic pigs. J. Vet. Diagn. Invest. 1992;4:117–126. doi: 10.1177/104063879200400201. [DOI] [PubMed] [Google Scholar]
- Duan X, Nauwynck H.J, Pensaert M.B. Virus quantification and identification of cellular targets in the lungs lymphoid tissues of pigs at different time intervals after inoculation with porcine reproductive and respiratory syndrome virus (PRRSV) Vet. Microbiol. 1997;56:9–19. doi: 10.1016/S0378-1135(96)01347-8. [DOI] [PubMed] [Google Scholar]
- Feng W, Laster S.M, Tompkins M, Brown T, Xu J.S, Altier C, Gomez W, Benfield D, McCaw M.B. In utero infection by porcine reproductive and respiratory syndrome virus is sufficient to increase susceptibility of piglets to challenge by Streptococcus suis Type II. J. Virol. 2001;75:4889–4895. doi: 10.1128/JVI.75.10.4889-4895.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holyoak G.R, Little T.V, McCollam W.H, Timoney P.J. Relationship between onset of puberty and establishment of persistent infection with equine arteritis virus in the experimentally infected colt. J. Comp. Pathol. 1993;109:29–46. doi: 10.1016/S0021-9975(08)80238-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horter D.C, Pogranichniy R.M, Chang C.C, Evans R.B, Yoon K.J, Zimmerman J.J. Characterization of the carrier state in porcine reproductive and respiratory syndrome virus infection. Vet. Microbiol. 2002;86:213–228. doi: 10.1016/s0378-1135(02)00013-5. [DOI] [PubMed] [Google Scholar]
- Kim H.S, Kwang J, Yoon I.J, Joo H.S, Frey M.L. Enhanced replication of porcine reproductive and respiratory syndrome (PRRS) virus in a homogeneous subpopulation of MA-104 cell line. Arch. Virol. 1993;133:477–483. doi: 10.1007/BF01313785. [DOI] [PubMed] [Google Scholar]
- Lager K.M, Halbur P.G. Gross and microscopic lesions in porcine fetuses infected with porcine reproductive and respiratory syndrome virus. J. Vet. Diagn. Invest. 1996;8:275–282. doi: 10.1177/104063879600800301. [DOI] [PubMed] [Google Scholar]
- Lawson S.R, Rossow K.D, Collins J.E, Benfield D.A, Rowland R.R.R. Porcine reproductive and respiratory syndrome virus infection of gnotobiotic pigs: sites of virus replication and co-localization with MAC-387-positive cells 21 days post-infection. Virus Res. 1997;51:105–113. doi: 10.1016/s0168-1702(97)00086-5. [DOI] [PubMed] [Google Scholar]
- Mengeling W.L, Lager K.M, Vorwald A.C. Temporal characterization of transplacental infection of porcine fetuses with porcine reproductive and respiratory syndrome virus. Am. J. Vet. Res. 1994;55:1391–1398. [PubMed] [Google Scholar]
- Mengeling W.L, Vorwald A.C, Lager K.M, Brockmeier S.L. Diagnosis of porcine reproductive and respiratory syndrome using infected macrophages from live pigs. Vet. Microbiol. 1996;49:105–115. doi: 10.1016/0378-1135(95)00173-5. [DOI] [PubMed] [Google Scholar]
- Nelsen C.J, Murtaugh M, Faaberg K.S. Porcine reproductive and respiratory syndrome virus comparison: divergent evolution on two continents. J. Virol. 1999;73:270–280. doi: 10.1128/jvi.73.1.270-280.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson E.A, Christopher-Hennings J, Drew T, Wensvoort G, Collins J.E, Benfield D.A. Differentiation of United States and European isolates of porcine reproductive and respiratory syndrome (PRRS) virus using monoclonal antibodies. J. Clin. Microbiol. 1993;31:3184–3189. doi: 10.1128/jcm.31.12.3184-3189.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plagemann, P.G.W., 1996. Lactate dehydrogenase-elevating virus and related viruses. In: Fields, B. (Ed.), Fields Virology, 3rd ed. Lippincott-Raven, Philadelphia, PA, pp. 1105–1120.
- Plagemann P.G.W, Rowland R.R.R, Even C, Faaberg K.S. Lactate dehydrogenase-elevating virus: an ideal persistent virus? Springer Semin. Immunopathol. 1995;17:167–186. doi: 10.1007/BF00196164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossow K.D. Porcine reproductive and respiratory syndrome. Vet. Pathol. 1998;35:1–20. doi: 10.1177/030098589803500101. [DOI] [PubMed] [Google Scholar]
- Rossow K.D, Morrison R.B, Goyal S.M, Singh G.S, Collins J.E. Lymph node lesions in neonatal pigs congenitally exposed to porcine reproductive and respiratory syndrome virus. J. Vet. Diagn. Invest. 1994;6:368–371. doi: 10.1177/104063879400600316. [DOI] [PubMed] [Google Scholar]
- Rossow K.D, Laube K.L, Goyal S.M, Collins J.E. Fetal microscopic lesions in porcine reproductive and respiratory syndrome virus-induced abortion. Vet. Pathol. 1996;33:95–99. doi: 10.1177/030098589603300115. [DOI] [PubMed] [Google Scholar]
- Rossow K.D, Shivers J.L, Yeske P.E, Polson D.D, Rowland R.R.R, Lawson S.R, Murtaugh M.P, Nelson E.A, Collins J.E. Porcine reproductive and respiratory syndrome virus infection in neonatal pigs characterised by marked neurovirulence. Vet. Rec. 1999;144:444–448. doi: 10.1136/vr.144.16.444. [DOI] [PubMed] [Google Scholar]
- Rowland R.R.R, Steffen M, Ackerman T, Benfield D.A. The evolution of porcine reproductive and respiratory syndrome virus: quasispecies and emergence of a virus subpopulation during infection of pigs with VR-2332. Virology. 1999;259:262–266. doi: 10.1006/viro.1999.9789. [DOI] [PubMed] [Google Scholar]
- Rowland R.R.R, Kim T.S, Robinson B, Stefanick J, Guanghua L, Lawson S.R, Benfield D.A. Inhibition of porcine reproductive and respiratory syndrome virus by interferon-gamma and recovery of virus replication with 2 aminopurine. Archiv. Virol. 2001;146:539–555. doi: 10.1007/s007050170161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijder E.J, Mulenberg J.M. The molecular biology of arteriviruses. J. Gen. Virol. 1998;79:961–979. doi: 10.1099/0022-1317-79-5-961. [DOI] [PubMed] [Google Scholar]
- Sur J.H, Doster A.R, Christian J.S, Galeota J.A, Wills R.W, Zimmerman J.J, Osorio F.A. Porcine reproductive and respiratory syndrome virus replicates in testicular germ cells, alters spermatogenesis, and induces germ cell death by apoptosis. J. Virol. 1997;71:9170–9179. doi: 10.1128/jvi.71.12.9170-9179.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tizard, I.R., 1996. Immunity in the fetus and newborn. In: Veterinary Immunology: An Introduction. Saunders, Philadelphia, PA, Chapter 19, pp. 237–254.
- Wensvoort G, Terpstra C, Pol J.M, ter Laak E.A, Bloemrad M, deKluyer E.P, Kragten C, van Buiten L, den Besten A, Wagenaar F, Broekhuijsen J.M, Moonen P.L.J.M, Zetstra T, de Boer E.A, Tibben H.J, de Jong M.F, van’t Veld P, Groenland G.J.R, van Gennep J.A, Voets M.T.H, Verheijden J.H.M, Braamskamp J. Mystery swine disease in The Netherlands: the isolation of Lelystad virus. Vet. Quart. 1991;13:121–130. doi: 10.1080/01652176.1991.9694296. [DOI] [PubMed] [Google Scholar]
- Wills R.W, Zimmerman J.J, Yoon K.J, Swenson S.L, McGinley M.J, Hill H.T, Platt K.B, Christopher-Hennings J, Nelson E.A. Porcine reproductive and respiratory syndrome virus: a persistent infection. Vet. Microbiol. 1997;55:231–240. doi: 10.1016/s0378-1135(96)01337-5. [DOI] [PubMed] [Google Scholar]
- Wills R.W, Doster A.R, Galeota J.A, Sur J.H, Osorio F.A. Duration of infection and proportion of pigs persistently infected with porcine reproductive and respiratory syndrome virus. J. Clin. Microbiol. 2003;41:58–62. doi: 10.1128/JCM.41.1.58-62.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon K.J, Zimmerman J.J, Swenson S.L, McGinley M.J, Eernisse K.A, Brevik A, Rhinehart L, Frey M.L, Hill H.T, Platt K. Characterization of the humoral immune response to porcine reproductive and respiratory syndrome (PRRS) virus infection. J. Vet. Diagn. Invest. 1995;7:305–312. doi: 10.1177/104063879500700302. [DOI] [PubMed] [Google Scholar]