

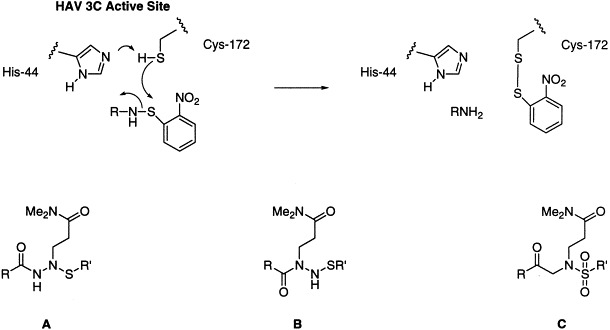
Abstract
Hepatitis A virus (HAV) 3C proteinase is a picornaviral cysteine proteinase that is essential for cleavage of the initially synthesized viral polyprotein precursor to mature fragments and is therefore required for viral replication in vivo. Since the enzyme generally recognizes peptide substrates with l-glutamine at the P1 site, four types of analogues having an azaglutamine residue were chemically synthesized: hydrazo-o-nitrophenylsulfenamides A (e.g. 16); frame-shifted hydrazo-o-nitrophenylsulfenamides B (e.g. 25–28); the azaglutamine sulfonamides C (e.g. 7, 8, 11, 12); and haloacetyl azaglutamine analogues 2 and 3. Testing of these compounds for inhibition of the HAV 3C proteinase employed a C24S mutant in which the non-essential surface cysteine was replaced with serine and which displays identical catalytic parameters to the wild-type enzyme. Sulfenamide 16 (type A) showed no significant inhibition. Sulfenamide 27 (type B) had an IC50 of ca 100 μM and gave time-dependent inactivation of the enzyme due to disulfide bond formation with the active site cysteine thiol, as demonstrated by electrospray mass spectrometry. Sulfonamide 8 (type C) was a weak competitive inhibitor with an IC50 of approximately 75 μM. The haloacetyl azaglutamine analogues 2 and 3 were time-dependent irreversible inactivators of HAV 3C proteinase with rate constants kobs/[I] of 680 M−1 s−1 and 870 M−1 s−1, respectively, and were shown to alkylate the active site thiol. ©
Keywords: Cysteine proteinase, enzyme inhibitors, azapeptides, antiviral agents
1. Introduction
The family of picornaviruses has more than 200 known members, divided into six genera, and contains pathogens such as poliovirus, human rhinovirus (HRV), encephalornyocarditis virus, foot and mouth disease virus and hepatitis A virus (HAV).1, 2, 3, 4These viruses possess a positive single-stranded RNA genome whose translation in mammalian cells produces a large protein product. The co- and post-translational processing of this initial polyprotein relies on highly specific and organized cleavage by viral cysteine proteinases, which are attractive targets for development of therapeutic agents.4, 5Among these, the 3C cysteine proteinases have been the subject of extensive structural investigation, with crystal structures reported for HAV 3C,6, 7HRV 3C,[8]and poliovirus 3C proteins.[9]Analogous 3C proteinases have also been detected in human and avian coronaviruses.10, 11These essential viral enzymes are especially appealing as targets for pharmaceutical intervention because their unique active site geometry[1]makes them highly specific in their choice of substrates and cleavage sites that are absent in non-infected mammalian cells. In addition, the active site residues appear to be relatively invariant in rhinovirus, where large numbers of serotypes (>100) make vaccine development difficult.[12]
Structurally the 3C proteinases have topologies that resemble the two domain β-barrel fold of the chymotrypsin-like serine proteinases.1, 5, 6, 7, 8The active site cysteine (Cys-172 in HAV 3C) is in close proximity to a histidine residue (His-44 in HAV 3C) that acts as a general base to form the nucleophilic thiolate anion. Substrate recognition, by analogy to the chymotrypsin family, is believed to involve anti-parallel binding of the backbone of the peptide to the proteinase and the interaction of the side chains with the enzyme in several key subsites. Hydrogen-bonding between the carbonyl oxygen of the side chain of the P1 glutamine of the substrate (or peptidic inhibitor) and a histidine at the S1 subsite (His-191 in HAV 3C) is probably one of the key recognition events (1, 5–8). The wild type HAV 3C has 219 amino acids with a molecular weight of 24 kilodaltons and exists as an active monomer. For peptide substrates mimicking the 2B/2C junction of the large precursor polyprotein, the k cat of this enzyme is typically about 1.8 s−1 with a K m of 2.1 mM at a pH of 7.5.[5]
In the search for therapeutic leads, enzyme inhibition studies on HAV 3C and HRV 3C proteins have employed a large variety of compounds, including peptide aldehydes,13, 14, 15peptide fluoromethyl ketones,[16]β-lactams,[17]isatins,[18]homophthalimides,[19]vinylogous esters and sulfones,12, 20, 21and halomethyl carbonyl compounds.22, 23Some of these compounds do show in vitro antiviral activity in cell culture,12, 15, 16, 18, 19, 20, 21but toxicity can be a problem for substances that are generally reactive with thiols.[18]
In the present study we examine a number of sulfur-containing azaglutamine derivatives as potential inhibitors of HAV 3C proteinase. Incorporation of hydrazino functionality into peptide backbones (azapeptides) is well-established for generation of proteinase inhibitors,24, 25including cysteine proteinases.[26]Recently such analogues have been shown to act as active site titrants for cysteine proteinase,[27]and certain symmetrical bis-hydrazides are extremely potent nanomolar inhibitors of cathepsin K, a cysteine proteinase involved in bone-remodeling.28, 29In one study on HRV 3C proteinase, an N-bromoacetyl azaglutamine dipeptide analogue 1 was shown to have an IC50 of approximately 48 nM and to be a time-dependent irreversible inactivator with a k inact/K i value >2500 M−1 s−1.[22]However, in this case the inhibitory activity depends on the highly reactive bromoacetyl moiety, which is likely to react non-specifically with a variety of thiols present in mammalian cells. The amide character of sulfenamides and the use of aryl sulfenamides as amino acid protecting groups which are removable by thiols under acidic conditions (Scheme. 1 ),30, 31, 32suggested that such functionality could display sufficiently low intrinsic reactivity and still form a disulfide with the active site thiol (Cys-172) of HAV 3C cysteine proteinase. Thus sulfenamide azaglutamine analogues of type A and B, as well as sulfonamide derivatives C, were synthesized and investigated for inhibition of this enzyme. Although the sulfonamides C would not be expected to react with the active site cysteine, they have some potential to bind to active site residues, and they also provide a new sulfur-containing aza analogue motif for investigation. The haloacetyl azaglutamine analogues 2 and 3 were also prepared for comparison. In each case the side chain amide functionality bears two methyls since such groups are well-accepted by the HAV 3C enzyme and hinder interactions with possible reactive intermediates, especially at the P1 carbonyl site.13, 16, 33

Scheme. 1.
2. Results and Discussion
2.1. Synthesis and testing of peptidosulfonamides C
Examination of structure C indicates that condensation of the known α-bromoketone 4 34, 35with the anion derived from sulfonamide 5 or 6 of β-alanine dimethylamide using the procedure of Jones[36]would afford the dipeptide analogues 7 or 8, respectively (Scheme. 2 ). The required sulfonamides are readily available by deprotection of N-Boc-β-alanine dimethylamide[37]followed by reaction with the appropriate sulfonyl chloride. As expected, the condensation[36]with the bromoketone proceeds readily, and can be followed by hydrogenolysis to remove the Cbz group in quantitative yield and form the corresponding amino compounds 9 and 10. Studies on sequence recognition by HAV 3C proteinase using model peptides suggests that the P4 residue should be a hydrophobic amino acid such as l-leucine, and that the P2 and P3 residues can be l-alanine.[38]This idea is supported by inhibition of the enzyme at nanomolar levels by aldehyde and fluoromethylketone peptide analogues that have such sequences.13, 16Thus coupling of 9 or 10 to N-acetyl-l-leucinyl-l-alanine proceeds readily with ethyl chloroformate to generate the target tetrapeptide azaglutamine sulfonamide derivatives 11 and 12.
Scheme. 2.
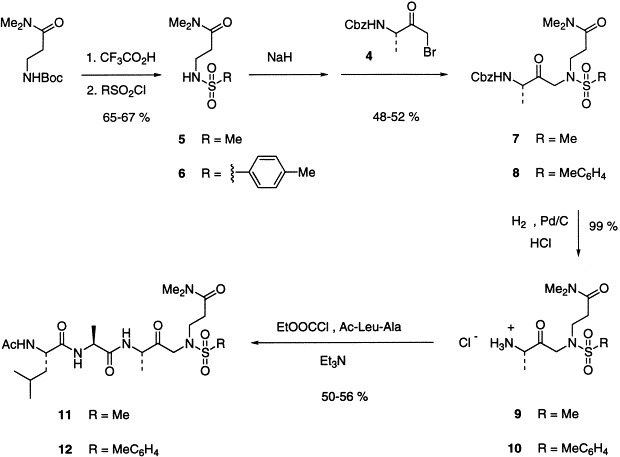
Assay of the sulfonamides 7, 8, 11, and 12 for inhibition of HAV 3C proteinase employed a C24S mutant in which the non-essential surface cysteine was replaced with serine and which displays catalytic parameters indistinguishable from the wild-type enzyme.16, 39All of these compounds are weak competitive inhibitors, with 8 being the most potent with an IC50 value of 75 μM. This is significantly lower than the K m for an ideal hexapeptide substrate (2.1 mM), but not potent enough to warrant extensive study in view of the nanomolar inhibition achieved with a peptide aldehyde[13]and a fluoromethyl ketone.[16]The other sulfonamides had IC50 values ≥100 μM.
2.2. Synthesis and testing of sulfenamide azapeptides and haloacetyl azapeptides
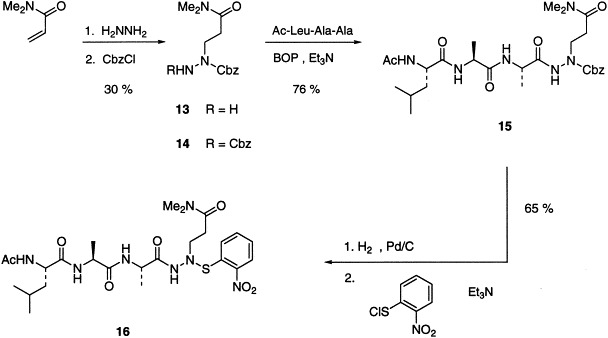
For the synthesis of potential inhibitors with the structural framework A, Michael addition of hydrazine to N,N-dimethylacrylamide followed by protection of the more nucleophilic secondary nitrogen with benzyl chloroformate generates 13 along with the corresponding bisacylated product 14 (Scheme. 3 ). Coupling of the free primary nitrogen with the tripeptide, N-acetyl-l-leucinyl-l-alanyl-l-alanine proceeds smoothly to give 15. Hydrogenolytic deprotection liberates the alkylated nitrogen, which then immediately reacts with o-nitrosulfenyl chloride to afford the target sulfenamide 16. During these transformations it became clear that in the azaglutamine series, compounds which have amide character at only one nitrogen are prone to facile decomposition and should be protected at the other basic nitrogen as quickly as possible. Disappointingly, 16 shows no significant inhibition (IC50>1 mM) of HAV 3C proteinase despite the presence of the P4-P1 backbone. Not only is there no detectable reaction with the inhibitor, but the level of competitive binding at the active site is also very low. Presumably the aromatic sulfenamide moiety is too bulky to be easily accommodated in the enzyme site which binds the P1′ substrate residue. Other sulfenamides were prepared wherein the o-nitrophenyl group is replaced with methyl or trifluoroethyl, but these compounds display very poor stability, especially in water, and were not pursued further.
Scheme. 3.
The preparation of structural type B again begins with Michael addition of hydrazine to N,N-dimethylacrylamide, this time with protection of the central nitrogen with a tert-butoxycarbonyl group to generate 17 (Scheme. 4 ). Reaction with benzyl chloroformate affords 18, which can then be deprotected with trifluoroacetic acid to liberate the secondary nitrogen and permit its acylation with a variety of protected amino acids and peptides. Such condensations afford azaglutamine derivatives 19–22. Because of the relative instability of the intermediate having the free basic nitrogen and solubility problems with the N-acetyl-l-leucinyl-l-alanyl-l-alanine tripeptide, it proved advantageous to form tetrapeptide analogues via a two stage coupling. Thus compounds 21 and 22 were deprotected and coupled to the dipeptide, N-acetyl-l-leucinyl-l-alanine, to give 23 and 24, respectively. Hydrogenolytic removal of the Cbz groups of 19, 20, 23, and 24 followed by immediate reaction with o-nitrosulfenyl chloride yields the corresponding azaglutamine sulfenamide derivatives 25–28, respectively. These compounds all have a hydrazo nitrogen at the nominal position of the substrate P1 carbonyl and sulfur at the P1′ nitrogen site.
Scheme. 4.

Assay of the type B compounds (i.e. 25–28) for inhibition of HAV 3C proteinase shows better binding than with the type A analogue 16, presumably because the frame shift on the peptide backbone allows the bulky o-nitrophenyl group to occupy a site further down the P′ binding region in the enzyme active site. However, these substances still give only modest levels of inhibition with IC50 values of 0.4 to 0.5 mM, with the exception of the tetrapeptide analogue 27, which has an initial IC50 of ca 100 μM. Electrospray mass spectrometry of the C24S HAV 3C enzyme inhibited with 27 for a prolonged period (≥3 h) does show formation of an enzyme-nitrophenyl disulfide, with the protein molecular ion peak shifting to higher mass by 154±1 units (−H+C6H4NO2S). The relatively poor competition with substrate may again be due to unfavorable interactions of the aromatic portion of the inhibitor with the enzyme active site. Although the sulfenamide azapeptides are not as potent inhibitors of HAV 3C proteinase as the peptidic aldehydes13, 14, 15or fluoromethyl ketone,[16]they represent new structural types that are reasonably accessible and stable, and may be useful for inhibition of other cysteine proteinases. Although additional thiols were not added to assay mixtures in the present study, and there is a possibility of non-specific reaction of compounds B and C with free thiols in vivo, removal of o-nitrophenylsulfenyl groups by extraneous thiols generally requires harsh conditions that include strong acid (e.g. p-toluenesulfonic acid) with high concentrations of thiol.30, 31, 32Cell culture studies could help to determine whether such sulfenamide functionalities are likely to be benign and stable in vivo.
Peptidic halomethyl carbonyl compounds are potent inactivators of 3C proteinases, 16, 22, 23and as mentioned above, the bromoacetyl azaglutamine derivative 1 has been reported as an irreversible inhibitor of the HRV 3C enzyme.[22]The availability of precursors for aza analogues from our preceding work encouraged the construction of chloroacetyl and bromoacetyl azaglutamine derivatives 2 and 3, respectively (Scheme. 5 ). Thus reductive deprotection of 15 as in Scheme. 3, followed by immediate acylation with chloroacetyl chloride or bromoacetyl bromide affords the target compounds directly. As expected, both 2 and 3 show potent time-dependent irreversible inhibition of HAV 3C proteinase with rate constants k obs/[I] of 680 M−1 s−1 and 870 M−1 s−1, respectively. Electrospray mass spectrometry of the enzyme-inhibitor complexes show addition of 470 mass units to the molecular ion and suggest that in each case there is displacement of halogen by the active site thiol of Cys-172. This is in accord with the reaction of HAV 3C with a tetrapeptide fluoromethyl ketone which has been studied by NMR spectrometry,[16]as well as with the displacement of halogen from iodoacetyl peptides by the active site thiol.23, 40Our compounds are somewhat less active against the HAV 3C enzyme than 1 is with the HRV 3C proteinase (k inact/K i value>2500 M−1 s−1).[22]The reasons for this are uncertain, but most likely reflect differences between the proximal subsite interactions (P1 and P2) between the two enzymes with their cognate inhibitors. Possibly the presence of two methyl groups in the glutamine analogue side chain also has some effect. Moreover, it is clear that recognition of the peptide moiety in inhibitors 2 and 3 is crucial because the inherently more reactive compound, N-iodoacetamide, has a second order rate constant for inactivation of only about 3.1 M−1 s−1.[23]
Scheme. 5.

In conclusion, the present work provides synthetic access to four types of aza amino acid peptide analogues. Although some show no binding (structural type A) or only weak initial inhibitory activity (structural types B and C) toward the hepatitis A virus 3C proteinase, the haloacetyl azaglutamine tetrapeptides 2 and 3 as well as the sulfenamide azaglutamine derivative 27 irreversibly react with the active site thiol (Cys-172). It is not unreasonable to expect that such substrate analogues may be more potent and useful for other types of cysteine proteinases. Current studies with other types of inhibitors of picornaviral proteinases will be reported shortly.
3. Experimental
3.1. General methods and enzyme assays
Most general procedures and instrumentation have been previously described.[41]Preparation, purification and assays of HAV 3C (C24S) protein with and without inhibitors were done as previously reported.13, 16Purity of the enzyme samples was greater than 90% as determined by SDS-PAGE analysis. Proteinase concentrations were determined spectrophotometrically using ε280=1.2 mg mL−1. Enzyme activity was monitored using the trinitrobenzene sulfonate (TNBS) assay as previously described.13, 16, 38Reaction mixtures were incubated in reaction buffer at 20 °C. Aliquots (10 μL) were removed from the reaction mixture at timed intervals and peptide hydrolysis was quenched with 50 μL of 0.25 M sodium borate, pH 10. A solution (12.5 μL) of freshly prepared 0.14 M TNBS (Johnson–Matthey, Ward Hill, MA) in 0.25 M sodium borate solution was added to the quenched reaction mixture and incubated for 10 min at 20 °C. The color was stabilized by adding 200 μL of 3.5 mM Na2SO3, 0.2 M KH2PO4. The concentration of free amine generated during peptide hydrolysis was determined by measuring the absorbance at 405 nM using a microtitre plate reader (Biorad, Richmond, CA). Proteinase inactivation was quantitatively evaluated by progress curve analysis as previously described.[13]The extent of peptide proteolysis (release of α-amino groups) was monitored using the TNBS assay. The concentrations of potential inhibitors were varied from 0.5 to 500 μM; substrate (Ac-ELRTQSFS-amide) concentration was 2 mM and HAV-3C proteinase (C24S mutant) concentration was 0.07 μM. Enzyme was dialyzed against reaction buffer to remove DTT immediately prior to use. Reactions were initiated with enzyme and absorbances were converted into μmoles of product using a glycine standard curve. All determinations were performed in triplicate with different enzyme and inhibitor preparations. Progress curves were fitted using least squares non-linear regression analysis using Mac Curve Fit 1.0.7, (K. Raner):
| P=νo(1−e−kt)k |
where ν o is the initial velocity and k is the apparent first order rate constant (k obs) for the inactivation process. The weighted average of parameter estimates from a minimum of three individual experiments was used to calculate reported rate constants.
General peptide synthesis and purification procedures have been described earlier,[16]and employed a Rainin PS-3 solid-phase peptide synthesizer using standard Fmoc chemistry on Wang Resin. All peptides were purified by reverse-phase HPLC (C-18, 5×25 cm, Vydac, 2%/min linear gradient of 0.1% TFA/water adding 0.1% TFA/acetonitrile). Peptide structures were verified by NMR and mass spectrometry. In cases where restricted rotation around amide bonds generates conformers detectable by NMR spectrometry, high temperature studies were done to demonstrate equilibria and coalescence of resonances. Mass spectrometry of enzyme-inhibitor complexes employed instrumentation and methodology specified in preceding publications.13, 23
3.1.1. 3-[N1-(Chloroacetyl)-N2-(acetyl-l-leucyl-l-alanyl-l-alanyl)hydrazino]l-N,N-(dimethyl)propanamide (2)
To a solution of Cbz-hydrazino derivative 15 (56.2 mg, 0.10 mmol) in methanol (10 mL) under argon was added 10% palladium on charcoal catalyst (10 mg). The mixture was stirred under an atmosphere of hydrogen until gas absorption ceased. The catalyst was removed by filtration through a column of Celite and the filtrate was concentrated in vacuo to give the deprotected hydrazino derivative (42.8 mg, quantitative). To a solution of this (42.8 mg, 0.1 mmol) in CH2Cl2 (5 mL) at −10 °C was added triethylamine (30.1 μL, 0.2 mmol) and chloroacetyl chloride (12.5 μL, 0.15 mmol). After removal of the cooling bath, the solution was stirred at room temperature for 1 h and then concentrated in vacuo. The crude product was purified by HPLC (linear gradient elution over 20 min of 0.1% TFA in acetonitrile and 0.1% TFA in water, from 20% to 40%, t R 9.2 min) to give 2 (22.7 mg, 45%) as a white powder. Spectral characterization indicated a mixture of conformers (conformer A: conformer B, 3:1): mp 133–143 °C (dec); IR (μscope) 3282, 2956, 2937, 2871, 1641, 1631, 1529, 1447, 1402, 1369 cm−1; 1H NMR (360 MHz, CD3OD) (conformer A) δ 4.40–4.00 (m, 5H, α-CH Leu, 2 α-CH Ala and COCH2Cl), 4.00–3.50 (brs, 2H, NCH2), 3.03 (s, 3H, NCH3), 2.90 (s, 3H, NCH3), 2.67 (t, 2H, J=7.3 Hz, COCH2), 1.97 (s, 3H, COCH3), 1.75–1.60 (m, 1H, CH Leu), 1.60–1.50 (m, 2H, CH2 Leu), 1.42 (d, 3H, J=7.2 Hz, CH3 Ala), 1.36 (d, 3H, J=7.2 Hz, CH3 Ala), 0.96 (d, 3H, J=6.5 Hz, CH3 Leu), 0.92 (d, 3H, J=6.5 Hz, CH3 Leu); (conformer B) δ 4.40–4.00 (m, 5H, α-CH Leu, 2 α-CH Ala, and CH2Cl), 4.00–3.50 (brs, 2H, NCH2), 3.03 (s, 3H, NCH3), 2.90 (s, 3H, NCH3), 2.67 (t, 2H, J=7.3 Hz, COCH2), 1.96 (s, 3H, COCH3), 1.75–1.60 (m, 1H, CH Leu), 1.60–1.50 (m, 2H, CH2 Leu), 1.43 (d, 3H, J=7.2 Hz, CH3 Ala), 1.35 (d, 3H, J=7.2 Hz, CH3 Ala), 0.96 (d, 3H, J=6.5 Hz, CH3 Leu), 0.92 (d, 3H, J=6.5 Hz, CH3 Leu); 13C NMR (75 MHz, CD3OD) (conformer A) δ 175.20, 175.02, 174.40, 173.66, 172.87, 170.32, 53.67, 50.43, 49.71, 46.51, 42.66, 41.69, 37.68, 35.69, 31.79, 25.90, 23.39, 22.44, 21.97, 17.65, 16.89; (conformer B) δ 175.20, 175.02, 174.40, 173.66, 172.87, 170.32, 53.34, 50.70, 50.03, 46.57, 42.70, 41.75, 37.68, 35.69, 31.72, 25.90, 23.48, 22.44, 21.86, 17.32, 16.89; MS (FAB) 505.2 (47) (MH+).
3.1.2. 3-[N1-(Bromoacetyl)-N2-(acetyl-l-leucyl-l-alanyl-l-alanyl)hydrazino]-N,N-(dimethyl)propanamide (3)
The procedure used for the preparation of 2, with Cbz-hydrazino derivative 15 (58.2 mg, 0.10 mmol) and 10% palladium on charcoal catalyst (10 mg) in methanol (10 mL), followed by triethylamine (30.1 μL, 0.2 mmol) and bromoacetyl bromide (13 μL, 0.15 mmol) in CH2Cl2 (5 mL) gave the crude product 3. Purification by HPLC (linear gradient elution over 20 min of 0.1% TFA in acetonitrile and 0.1% TFA in water, from 20% to 40%, t R 9.8 min) gave pure 3 (21.9 mg, 40%) as a white powder. Spectral characterization indicated a mixture of conformers (conformer A:conformer B, 3:1): mp 81–90 °C (dec); IR (μscope) 3283, 2956, 2935, 2871, 1645, 1537, 1448, 1402, 1370 cm−1; 1H NMR (360 MHz, CD3OD) (conformer A) δ 4.40–4.20 (m, 3H, α-CH Leu and 2 α-CH Ala), 4.20–3.50 (m, 4 H, COCH2Br and NCH2), 3.04 (s, 3H, NCH3), 2.86 (s, 3H, NCH3), 2.75–2.60 (brs, 2H, COCH2),1.97 (s, 3H, COCH3), 1.76–1.60 (m, 1H, CH Leu), 1.60–1.48 (in, 2H, CH2 Leu), 1.42 (d, 3H, J=7.2 Hz, CH3 Ala), 1.36 (d, 3H, J=7.2 Hz, CH3 Ala), 0.96 (d, 3H, J=6.5 Hz, CH3 Leu), 0.92 (d, 3H, J=6.5 Hz, CH3 Leu); (conformer B) δ 4.40–4.20 (m, 3H, α-CH Leu and 2 α-CH Ala), 4.20–3.50 (m, 4 H, CH2Br and NCH2), 3.04 (s, 3H, NCH3), 2.86 (s, 3H, NCH3), 2.75–2.60 (brs, 2H, COCH2), 1.98 (s, 3H, COCH3), 1.76–1.60 (m, 1H, CH Leu), 1.60–1.48 (m, 2H, CH2 Leu), 1.41 (d, 3H, J=7.2 Hz, CH3 Ala), 1.37 (d, 3H, J=7.2 Hz, CH3 Ala), 0.96 (d, 3H, J=6.5 Hz, CH3 Leu), 0.92 (d, 3H, J=6.5 Hz, CH3 Leu); 13C NMR (75 MHz, CD3OD) (conformer A) δ 175.14, 174.98, 174.36, 173.63, 172.86, 170.52, 53.64, 50.38, 49.97, 46.52, 42.68, 41.71, 37.72, 35.72, 31.73, 25.91, 23.41, 22.46, 21.99, 17.71, 16.96; (conformer B) δ 175.14, 174.98, 174.36, 173.63, 172.86, 170.52, 53.40, 50.63, 49.86, 46.58, 42.68, 41.71, 37.72, 35.72, 31.66, 25.91, 23.50, 22.46, 21.88, 17.45, 16.96; MS (FAB) 549.0 (27) (MH+).
3.1.3. 3-(Methylsulfonylamino)-N,N-(dimethyl)propanamide (5)
Trifluoroacetic acid (11.6 mL, 150 mmol) was added dropwise to a stirred solution of N-t-Boc-β-Ala-NMe2 [37](2.16 g, 10 mmol) in dry distilled CH2Cl2 (11.6 mL) under argon at room temperature. The reaction mixture was stirred at room temperature for 2 h. The solvent was removed in vacuo and the residue was dried overnight under high vacuum to generate the known[42]β-alanine dimethylamide as a trifluoroacetate salt (2.30 g, quantitative): IR (CH2Cl2 cast) 2948, 1679, 1639, 1503, 1408, 1203, 1174 cm−1; 1H NMR (360 MHz, CD2Cl2) δ 7.90–7.60 (brs, 3H, NH3 +), 3.35–3.25 (m, 2H, CH2N), 2.98 (s, 3H, NCH3), 2.91 (s, 3H, NCH3), 2.74 (t, 2H, J=5.4 Hz, COCH2); 13C NMR (75 MHz, CD2Cl2) δ 171.57 (CON), 161.3 (q, COO−), 116.3 (q, CF3), 37.21 (NCH3), 36.90 (CH2NH3 +), 35.50 (NCH3), 29.57 (CH2); MS (FAB) 116.1 (100) (MH+-CF3COO−).
Triethylamine (0.90 mL, 6.00 mmol) was added dropwise to a stirred solution of this amine salt (1.15 g, 5.00 mmol) in dry CH2Cl2 (20 mL) under argon at 0 °C. The mixture was stirred at 0 °C for 5 min, and then methanesulfonyl chloride (0.39 mL, 5.00 mmol) and triethylamine (0.90 mL, 6.00 mmol) were added dropwise. The reaction mixture was stirred at 0 °C for 2 h, and then concentrated in vacuo. EtOAc (150 mL) was added to the residue, and the precipitate was removed by filtration. The filtrate was washed with water (20 mL), saturated aq NaHCO3 (20 mL) and brine (50 mL), and dried (Na2SO4). The solvent was removed in vacuo to give an oil. Purification by flash chromatography (ethyl acetate) gave the sulfonamide 5 (0.65 g, 67%) as an oil: IR (CH2Cl2 cast) 3200, 2932, 1632, 1403, 1316, 1146 cm−1; 1H NMR (360 MHz, CDCl3) δ 5.52–5.37 (brs, 1H, CONH), 3.40–3.27 (m, 2H, CH2N), 2.97 (s, 3H, NCH3), 2.92 (s, 3H, SO2CH3), 2.91 (s, 3H, NCH3), 2.59 (t, 2H, J=5.1 Hz, COCH2); 13C NMR (75 MHz, CD2Cl2) δ 172.95 (CON), 39.72 (SO2CH3), 39.06 (CH2N), 36.87 (NCH3), 35.12 (NCH3), 33.40 (COCH2); HRMS calcd for C6H14N2O3S 194.0803, found 194.0782.
3.1.4. 3-(p-Methylphenylsulfonylamino)-N,N-(dimethyl)propanamide (6)
The procedure for preparation of 5 was employed to react amine salt (1.15 g, 5.00 mmol) obtained by deprotection of Boc-β-alanine dimethylamide,[37]triethylamine (1.8 mL, 12.00 mmol) and p-toluenesulfonyl chloride (0.95 g, 5.00 mmol) to give the previously reported[43]sulfonamide 6 (0.88 g, 65%) as an oil: IR (CH2Cl2 cast) 3180, 2927, 1631, 1495, 1401, 1326, 1158, 1093 cm−1; 1H NMR (360 MHz, CD2Cl2) δ 7.75–7.70 (m, 2H, ArH), 7.36–7.31 (m, 2H, ArH), 5.80–5.65 (brs, 1H, CONH), 3.20–3.09 (m, 2H, CH2N), 2.87 (s, 3H, NCH3), 2.86 (s, 3H, NCH3), 2.46 (t, 2H, J=5.5 Hz, COCH2), 2.42 (s, 3H, CH3); 13C NMR (75 MHz, CD2Cl2) δ 171.25 (CON), 143.67 (C, Ar), 137.74 (C, Ar), 129.96 (CH, Ar), 127.18 (CH, Ar), 39.33 (NCH2), 37.03 (NCH3), 35.20 (NCH3), 32.92 (COCH2), 21.50 (CH3); HRMS calcd for C12H18N2O3S 270.1038, found 270.1034.
3.1.5. (3S)-{N3-(Benzyloxycarbonyl)-N1-[3′-(N,N-dimethylamino)-3′-oxopropyl]-N1-(methylsulfonyl)}-1,3-diaminobutan-2-one (7)
To a stirred solution of sulfonamide 5 (0.82 g, 4.2 mmol) in anhydrous DMF (10 mL) under argon at 0 °C, was added NaH (60% dispersion in mineral oil) (0.237 g, 5.9 mmol) in small portions, and the mixture was stirred at 0 °C for an additional 30 min. The resulting yellow anion solution was added dropwise over a 30 min period to a stirred solution of bromo compound 4 34, 35(1.51 g, 5.0 mmol) in DMF (5 mL). After 1 h at room temperature, the solvent was removed in vacuo, and the residue was dissolved in CH2Cl2, and washed with 1% aq HCl (10 mL), 5% aq NaHCO3 (10 mL) and brine (10 mL). The organic phase was dried (Na2SO4) and concentrated in vacuo to give an oil. Purification by flash chromatography (ethyl acetate: petroleum spirit, 1:1) gave the sulfonamide 7 (0.83 g, 48%) as an oil: IR (CH2Cl2 cast) 3315, 3032, 2935, 1715, 1637, 1522, 1454, 1328, 1248, 1146 cm−1; 1H NMR (360 MHz, CDCl3) δ 7.45–7.29 (m, 5H, ArH), 5.61–5.45 (brs, 1H, CONH), 5.12 (s, 2H, OCH2), 4.47 (s, 2H, CH2), 4.40–4.25 (m, 1H, CH), 3.60–3.40 (m, 2H, CH2N), 2.97 (s, 3H, NCH3), 2.94 (s, 3H, SO2CH3), 2.88 (s, 3H, NCH3), 2.70–2.50 (m, 2H, COCH2), 1.35 (d, 3H, J=7.1 Hz, CH3); 13C NMR (75 MHz, CD2Cl2) δ 206.43 (CO), 170.98 (CON), 156.19 (OCON), 136.94 (C, Ar), 128.75 (CH, Ar), 128.38 (CH, Ar), 128.25 (CH, Ar), 67.08 (OCH2), 55.16 (CH2), 53.96 (CH), 44.95 (NCH2), 39.42 (SO2CH3), 37.16 (NCH3), 35.34 (NCH3), 34.06 (COCH2), 16.92 (CH3); HRMS cald for C18H27N3O6S 413.1621, found 413.1632.
3.1.6. (3S)-{N3-(Benzyloxycarbonyl)-N1-[3′-(N,N-dimethylamino)-3′-oxopropyl]-N1-(p-methylphenylsulfonyl)}-1,3-diaminobutan-2-one (8)
The procedure used for the preparation of 7, was employed to condense sulfonamide 6 (1.00 g, 3.7 mmol) and bromomethyl ketone 4 34, 35(1.33 g, 4.44 mmol) to give 8 (0.94 g, 52%) as a waxy solid: IR (CH2Cl2 cast) 3295, 3032, 2935, 1714, 1638, 1519, 1497, 1401, 1336, 1248, 1156, 1093 cm−1; 1H NMR (360 MHz, CD2Cl2) δ 7.67 (d, 2H, J=8.2 Hz, ArH), 7.41–7.20 (m, 7 H, ArH), 5.65 (d, 1H, J=6.9 Hz, CONH), 5.09 (s, 2H, OCH2), 4.46–4.28 (m, 3H, CH and COCH2N), 3.41 (t, 2H, J=6.3 Hz, CH2N), 2.91 (s, 3H, NCH3), 2.82 (s, 3H, NCH3), 2.67–2.50 (m, 2H, COCH2), 2.42 (s, 3H, ArCH3), 1.32 (d, 3H, J=7.1 Hz, CH3); 13C NMR (75 MHz, CD2Cl2) δ 204.93 (CO), 171.05 (CON), 156.13 (OCON), 144.04 (C, Ar), 137.09 (C, Ar), 130.00 (CH, Ar), 128.86 (CH, Ar), 128.44 (CH, Ar), 128.32 (CH, Ar), 127.59 (CH, Ar), 115.93 (C, Ar), 67.11 (OCH2), 55.86 (COCH2N), 53.81 (CH), 45.69 (NCH2), 37.19 (NCH3), 35.37 (NCH3), 33.96 (COCH2), 21.62 (Ar CH3), 17.30 (CH3); HRMS calcd for C24H32N3O6S 490.2012, found 490.2001.
3.1.7. (3S)-{N1-[3′-(N,N-Dimethylamino)-3′-oxopropyl]-N1-(methyl-sulfonyl)}-1,3-diaminobutan-2-one hydrochloride (9)
To a solution of N-Cbz sulfonamide 7 (0.580 g, 1.40 mmol) in methanol-HCl (40 mL) under argon was added 10% palladium on charcoal catalyst (58 mg). The mixture was stirred under an atmosphere of hydrogen until gas absorption ceased. The catalyst was removed by filtration through a column of Celite, and the filtrate was concentrated in vacuo to give deprotected sulfonamide 9 (0.443 g, quantitative) as a glassy solid: IR (MeOH cast) 3440, 2921, 1737, 1619, 1325, 1145 cm−1; 1H NMR (300 MHz, CD3OD) δ 4.55 (d, 1H, J=19.3 Hz, CHHA), 4.41 (d, 1H, J=19.3 Hz, CHBH), 4.25 (q, 1H, J=7.2 Hz, CH), 3.53 (t, 2H, J=6.4 Hz, CH2), 3.01 (s, 3H, SO2CH3), 2.94 (s, 3H, NCH3), 2.87 (s, 3H, NCH3), 2.74 (d, 3H, J=6.4 Hz, COCH2), 1.53 (d, 3H, J=7.2 Hz, CH3); 13C NMR (75 MHz, CD3OD) δ 204.14, 173.57, 55.35, 54.04, 46.17, 39.28, 38.32, 36.38, 34.12, 15.76; MS (FAB) 279.9 (33) (MH+-HCl).
3.1.8. (3S)-{N1-[3′-(N,N-Dimethylamino)-3′-oxopropyl]-N1-(p-methylphenylsulfonyl)}-1,3-diaminobutan-2-one hydrochloride (10)
The procedure used for the preparation of 9, was adapted to convert N-Cbz sulfonamide 8 (0.500 g, 1.02 mmol) with 10% palladium on charcoal (50 mg) in MeOH-HCl (40 mL) to the title compound 10 (0.400 g, quantitative) as a glassy solid: IR (MeOH cast) 3440, 2927, 1742, 1621, 1598, 1494, 1334, 1156 cm−1; 1H NMR (300 MHz, CD3OD) δ 7.69 (d, 1H, J=8.3 Hz, ArH), 7.35 (d, 1H, J=8.3 Hz, ArH), 4.36 (s, 2H, COCH2N), 4.25 (q, 1H, J=7.3 Hz, CH), 3.38 (t, 2H, J=6.4 Hz, NCH2), 2.92 (s, 3H, NCH3), 2.82 (s, 3H, NCH3), 2.67–2.58 (m, 2H, COCH2), 2.37 (s, 3H, ArCH3), 1.48 (d, 3H, J=7.3 Hz, CH3); 13C NMR (75 MHz, CD3OD) δ 204.25, 173.54, 143.55, 137.64, 129.89, 127.28, 55.27, 54.01, 45.96, 37.24, 35.27, 32.92, 21.58, 16.20; MS (FAB) 356.0 (97) (MH+-HCl).
3.1.9. (3S)-{N3-(Acetyl-l-leucyl-l-alanyl)-N1-[3′-(N,N-dimethylamino)-3′-oxopropyl]-N1-(methylsulfonyl)}-1,3-diaminobutan-2-one (11)
A solution of the dipeptide Ac-Leu-Ala-OH (0.216 g, 0.89 mmol) in dry THF (10 mL) under argon at 0 °C was treated with ethyl chloroformate (90 μL, 0.89 mmol) and triethylamine (0.13 mL, 0.91 mmol) and stirred for 20 min. A solution of sulfonamido amine hydrochloride 9 (0.280 g, 0.89 mmol) in THF (5 mL), and triethylamine (0.13 mL, 0.91 mmol) were added, and the reaction mixture was stirred for 30 min. It was then allowed to warm to room temperature and stirred overnight. To the mixture was added EtOAc (40 mL), and the organic phase was washed with saturated aq NaHCO3 (20 mL) and brine (20 mL), dried (Na2SO4) and concentrated in vacuo to give the crude product 11. Purification by HPLC (linear gradient elution over 25 min of acetonitrile and water, from 20% to 35%, t R 10.4 min) gave 11 (0.224 g, 50%) as a white solid: mp 122–124 °C; IR (MeOH cast) 3287, 2935, 1731, 1650, 1643, 1547, 1327, 1202, 1147 cm−1; 1H NMR (300 MHz, CD3OD) δ 4.45–4.15 (m, 5H, α-CH Leu, 2 α-CH Ala, and COCH2N), 3.46 (t, 2H, J=7.0 Hz, NCH2), 3.06 (s, 3H, SO2CH3), 2.96 (s, 3H, NCH3), 2.89 (s, 3H, NCH3), 2.69 (d, 3H, J=7.0 Hz, COCH2), 1.97 (s, 3H, COCH3), 1.72–1.59 (m, 1H, CH Leu), 1.58–1.50 (m, 2H, CH2 Leu), 1.37 (d, 3H, J=7.2 Hz, CH3 Ala), 1.33 (d, 3H, J=7.2 Hz, CH3 Ala), 0.96 (d, 3H, J=6.5 Hz, CH3 Leu), 0.92 (d, 3H, J=6.5 Hz, CH3 Leu); 13C NMR (100 MHz, CD3OD) δ 207.20, 175.37, 175.32, 173.83, 173.28, 55.22, 54.12, 54.02, 50.65, 46.19, 40.14, 39.49, 38.86, 36.38, 34.12, 25.67, 22.80, 22.39, 21.97, 19.39, 15.76; MS (FAB) 506.3 (80) (MH+).
3.1.10. (3S)-{N3-(Acetyl-l-leucyl-l-alanyl)-N1-[3′-(N,N-dimethylamino)-3′-oxopropyl]-N1-(p-methylphenylsulfonyl)}-1,3-diaminobutan-2-one (12)
The procedure used for the preparation of 11, was used to condense dipeptide Ac-Leu-Ala-OH (0.156 g, 0.64 mmol) and sulfonamido amine hydrochloride 10 (0.250 g, 0.64 mmol) in the presence of ethyl chloroformate (62 μL, 0.64 mmol) and triethylamine (0.178 mL, 1.28 mmol). Purification by HPLC (linear gradient elution over 20 min of acetonitrile and water, from 30% to 50%, t R 15.8 min) gave the title compound 12 (0.208 g, 56%) as a solid. Spectral characterization showed a mixture of conformers (conformer A:conformer B, 3:1): mp 145–147 °C; IR (MeOH cast) 3381, 2957, 1734, 1651, 1644, 1575, 1538, 1455, 1335, 1202, 1157 cm−1; 1H NMR (360 MHz, CD3OD) (conformer A) δ 7.77–7.69 (m, 2H, ArH), 7.37 (d, 2H, J=8.4 Hz, ArH), 4.41–4.15 (m, 5H, α-CH Leu, 2 α-CH Ala, and COCH2N), 3.50–3.30 (m, 2H, NCH2), 2.99 (s, 3H, NCH3), 2.87 (s, 3H, NCH3), 2.65 (d, 2H, J=7.3 Hz, COCH2), 2.42 (s, 3H, ArCH3), 1.94 (s, 3H, COCH3), 1.70–1.58 (m, 1H, CH Leu), 1.58–1.50 (m, 2H, CH2 Leu), 1.37 (d, 3H, J=7.3 Hz, CH3 Ala), 1.32 (d, 3H, J=7.2Hz, CH3 Ala), 0.97 (d, 3H, J=6.5 Hz, CH3 Leu), 0.93 (d, 3H, J=6.5 Hz, CH3 Leu); (conformer B) δ 7.77–7.69 (m, 2H, ArH), 7.37 (d, 2H, J=8.4 Hz, ArH), 4.41–4.15 (m, 5H, α-CH Leu, 2 α-CH Ala, and COCH2N), 3.50–3.30 (m, 2H, NCH2), 2.99 (s, 3H, NCH3), 2.87 (s, 3H, NCH3), 2.65 (d, 2H, J=7.3 Hz, COCH2), 2.42 (s, 3H, ArCH3), 1.94 (s, 3H, COCH3), 1.70–1.58 (m, 1H, CH Leu), 1.58–1.50 (m, 2H, CH2 Leu), 1.36 (d, 3H, J=7.3 Hz, CH3 Ala), 1.29 (d, 3H, J=7.2 Hz, CH3 Ala), 0.96 (d, 3H, J=6.5 Hz, CH3 Leu), 0.92 (d, 3H, J=6.5 Hz, CH3 Leu); 13C NMR (100 MHz, CD3OD) (conformer A) δ 206.04, 175.17, 173.74, 173.56, 173.10, 145.00, 138.13, 130.75, 128.53, 55.91, 54.01, 53.93, 50.57, 46.54, 41.36, 37.64, 35.64, 33.84, 25.88, 23.18, 22.46, 22.30, 21.46, 17.50, 15.80; (conformer B) δ 206.13, 175.17, 174.97, 173.74, 173.10, 145.00, 138.13, 130.75, 128.53, 55.91, 54.01, 53.93, 50.57, 46.54, 41.36, 37.64, 35.64, 33.97, 25.88, 23.18, 22.46, 22.30, 21.46, 17.50, 15.90; MS (FAB) 582.2 (52) (MH+).
3.1.11. 3-[N1-(Benzyloxycarbonyl)hydrazino]-N,N-(dimethyl)propanamide (13)
N,N-Dimethylacrylamide (4.96 g, 50 mmol) was added dropwise to a solution of hydrazine monohydrate (2.50 g, 50 mmol) in MeOH (30 mL). The reaction mixture was stirred at room temperature for 2 h, diluted with MeOH (30 mL), dried (Na2SO4), and then concentrated in vacua to give crude 3-hydrazino-N,N-(dimethyl)propanamide (6.5 g). A stirred solution of this compound (2.62 g, 20 mmol) in a mixture of diethyl ether (30 mL), water (15 mL) and 1 N NaOH (20 mL) was cooled in an ice-bath. Benzyl chloroformate (2.8 mL, 20 mmol) was added and stirring was continued in the ice-bath for 1 h. The precipitate was filtered, the filtrate was concentrated in vacuo to 20 mL, and then extracted with ethyl acetate (3×80 mL). The combined organic extracts were washed with brine (20 mL), dried (Na2SO4) and the solvent was concentrated in vacua to give an oil. Purification by flash chromatography (ethyl acetate:methanol, 95:5) gave the desired 13 (1.59 g, 30%) as a white solid: mp 48–50 °C; IR (CH2Cl2 cast) 3334, 3215, 3031, 2941, 1699, 1643, 1586, 1497, 1411, 1200, 1109 cm−1; 1H NMR (300 MHz, CD2Cl2) δ 7.40–7.24 (m, 5H, ArH), 5.12 (s, 2H, CH2O), 4.22–4.07 (brs, 2H, NH2), 3.69 (t, 2H, J=7.2 Hz, NCH2), 2.94 (s, 3H, NCH3), 2.86 (s, 3H, NCH3), 2.60 (t, 2H, J=7.2 Hz, CH2CO); 13C NMR (75 MHz, CD2Cl2) δ 171.05, 155.11, 137.14, 128.69, 128.25, 128.15, 67.57, 47.36, 37.17, 35.07, 31.70; HRMS calcd for C13H19N3O3 265.1426, found 265.1432. Anal. calcd for C13H19N3O3: C, 58.85; H, 7.22; N, 15.84. Found: C, 58.89; H, 7.01; N, 15.73.
3.1.12. 3-[N1-(Benzyloxycarbonyl)-N2-(acetyl-l-leucyl-l-alanyl-l-alanyl)hydrazino]-N,N-(dimethyl)propanamide (15)
To a solution of tripeptide Ac-Leu-Ala-Ala-OH (0.315 g, 1 mmol), Cbz hydrazino derivative 13 (0.265 g, 1 mmol) and benzotriazolyl-N-oxytris(dimethylamino)phosphonium hexafluorophosphate (BOP) (0.442 g, 1 mmol) in DMF (10 mL) was added triethylamine (0.139 mL, 1 mmol). The mixture was stirred at room temperature for 6 h, and then concentrated in vacuo to give an oil. Purification by HPLC (linear gradient elution over 20 min of acetonitrile and water, from 20% to 40%, t R 13.8 min) gave 15 (0.428 g, 76%) as a white powder: mp 141–142 °C; IR (CH2Cl2 cast) 3267, 3038, 2955, 2931, 1721, 1628, 1500, 1449, 1116 cm−1; 1H NMR (360 MHz, CD3OD) δ 7.40–7.20 (brs, 5H, ArH), 5.12 (s, 2H, OCH2), 4.40–4.20 (m, 3H, α-CH), 3.86–3.65 (m, 2H, NCH2), 3.04 (s, 3H, NCH3), 2.86 (s, 3H, NCH3), 2.67 (t, 2H, J=7.3 Hz, COCH2), 1.95 (s, 3H, CH3), 1.72–1.58 (in, 1H, CH Leu), 1.57–1.50 (m, 2H, CH2), 1.48–1.20 (m, 6 H, CH3), 0.96 (d, 3H, J=6.5 Hz, CH3), 0.92 (d, 3H, J=6.5 Hz, CH3); 13C NMR (75 MHz, CD3OD) δ 175.21, 174.71, 174.11, 173.46, 173.06, 157.01, 137.48, 129.51, 129.22, 129.05, 69.08, 53.84, 50.56, 49.84, 47.37, 41.5 1, 37.77, 35.64, 32.16, 25.91, 23.22, 22.39, 22.24, 17.54, 17.38; MS (FAB) 563.4 (34) (MH+).
3.1.13. 3-[N1-(o-Nitrophenylsulfenyl)-N2-(acetyl-l-leucyl-l-alanyl-l-alanyl)hydrazino]-N,N-(dimethyl)propanamide (16)
To a solution of Cbz derivative 15 (56.2 mg, 0.10 mmol) in methanol (10 mL) under argon was added 10% palladium on charcoal catalyst (10 mg). The mixture was stirred under an atmosphere of hydrogen until gas absorption ceased. The catalyst was removed by filtration through a column of Celite, and the filtrate was concentrated in vacuo to give the unstable deprotected hydrazine (42.8 mg, quantitative). To a stirred solution of this crude material (42.8 mg, 0.10 mmol) and o-nitrophenylsulfenyl chloride (18.9 mg, 0.10 mmol) in DMF (1 mL) under argon, at −10 °C, was added triethylamine (14 μL, 0.10 mmol). After 30 min at −10 °C, the reaction mixture was allowed to warm to room temperature and was stirred for 1 h. The solution was then concentrated in vacuo. Purification by HPLC (linear gradient elution over 20 min of acetonitrile and water, from 25% to 40%, t R 10.6 min) gave the title compound 16 (37.8 mg, 65%) as a yellow powder. Spectral characterization indicated a mixture of conformers (conformer A:conformer B, 5:6): mp 133–145 °C (dec); IR (μscope) 3278, 3268, 3070, 2956, 2935, 2872, 1651, 1628, 1593, 1512, 1450, 1335 cm−1; 1H NMR (360 MHz, CD3OD) (conformer A) δ 8.52 (d, 1H, J=8.5 Hz, ArH), 8.26 (dd, 1H, J=8.5, 1.0 Hz, ArH), 7.76–7.65 (m, 1H, ArH), 7.40–7.30 (m, 1H, ArH), 4.38–4.13 (m, 3H, α-CH Leu and 2 α-CH Ala), 3.76–3.60 (m, 2H, NCH2), 2.98 (s, 3H, NCH3), 2.89 (s, 3H, NCH3), 2.77–2.58 (m, 2H, COCH2), 1.98 (s, 3H, COCH3), 1.76–1.62 (m, 1H, CH Leu), 1.62–1.50 (m, 2H, CH2 Leu), 1.41–1.24 (m, 6 H, 2 CH3 Leu), 1.00–0.82 (m, 6 H, 2 CH3 Ala); (conformer B) δ 8.43 (d, 1H, J=8.5 Hz, ArH), 8.26 (dd, 1H, J=8.5, 1.0 Hz, ArH), 7.76–7.65 (m, 1H, ArH), 7.40–7.30 (m, 1H, ArH), 4.38–4.13 (m, 3H, α-CH Leu and 2 α-CH Ala), 3.76–3.60 (m, 2H, NCH2), 2.98 (s, 3H, NCH3), 2.89 (s, 3H, NCH3), 2.77–2.58 (m, 2H, COCH2), 1.97 (s, 3H, COCH3), 1.76–1.62 (m, 1H, CH Leu), 1.62–1.50 (m, 2H, CH2 Leu), 1.41–1.24 (m, 6 H, 2 CH3 Leu), 1.00–0.82 (m, 6 H, 2 CH3 Ala); 13C NMR (75 MHz, CD3OD) (conformer A) δ 175.10, 174.65, 173.68, 173.57, 173.13, 143.91, 143.10, 135.34, 127.26, 126.51, 126.37, 57.89, 53.57, 50.55, 49.60, 41.68, 37.84, 35.82, 32.74, 25.90, 23.44, 22.45, 22.02, 17.65, 17.28; (conformer B) δ 175.13, 174.77, 173.76, 173.68, 173.13, 143.91, 143.10, 135.34, 127.06, 126.51, 126.37, 57.89, 53.46, 50.78, 49.60, 41.68, 37.84, 35.82, 32.74, 25.90, 23.40, 22.53, 21.92, 17.56, 17.51; MS (FAB) 582.1 (25) (MH+).
3.1.14. 3-[N1-(tert-Butyloxycarbonyl)hydrazino]-N,N-(dimethyl)propanamide (17)
A stirred solution of the crude 3-hydrazino-N,N-(dimethyl)propanamide (2.62 g, 20 mmol), which was obtained as in the preparation of 13, in a mixture of dioxane (30 mL), water (15 mL) and 1 N NaOH (20 mL) was cooled in an ice-bath. Di-tert-butyl pyrocarbonate (4.8 g, 22 mmol) was added and stirring was continued at room temperature for 1 h. The precipitate was filtered, the filtrate was concentrated in vacuo to 25 mL, and then extracted with ethyl acetate (3×80 mL). The combined organic extracts were washed with brine (20 mL), dried (Na2SO4) and concentrated in vacuo to give an oil. Purification by flash chromatography (ethyl acetate:methanol, 95:5) gave unstable propanamide 17 (2.25 g, 53%) as a colorless oil: IR (CH2Cl2 cast) 3334, 3217, 2975, 2932, 1694, 1645, 1397, 1365, 1168 cm−1; 1H NMR (360 MHz, CDCl3) δ 4.38–4.20 (brs, 2H, NH2), 3.66 (t, 2H, J=7.0 Hz, NCH2), 2.98 (s, 3H, NCH3), 2.89 (s, 3H, NCH3), 2.60 (t, 2H, J=7.0 Hz, COCH2), 1.42 (s, 9 H, (CH3)3C); 13C NMR (75 MHz, CDCl3) δ 170.54, 155.80, 79.62, 46.20, 36.60, 34.49, 30.99, 27.73; HRMS calcd for C10H21N3O3 231.1583, found 231.1582.
3.1.15. 3-[N1-(tert-Butyloxycarbonyl)-N2-(benzyloxycarbonyl)hydrazino]-N,N-(dimethyl)propanamide (18)
To a suspension of Boc-propanamide 17 (2.31 g, 10 mmol) and anhydrous potassium carbonate (6.91 g, 50 mmol) in dry THF (20 mL), was added benzyl chloroformate (1.88 g, 11 mmol) with vigorous stirring. The mixture was heated to 40–50 °C for 2 h. The precipitate of KHCO3 and K2CO3 was removed by filtration. The filtrate was concentrated in vacuo. The residue was dissolved in ethyl acetate (100 mL), washed with 1 N NaHCO3 (20 mL), water (20 mL) and brine (20 mL), dried (Na2SO4) and concentrated in vacuo to give an oil. Purification by flash chromatography (ethyl acetate) gave 18 (3.2 g, 87%) as a colorless oil: IR (CH2Cl2 cast) 3251, 2976, 2934, 1741, 1709, 1632, 1497, 1400, 1366, 1257, 1209, 1159 cm−1; 1H NMR (360 MHz, CD2Cl2) δ 7.38–7.30 (m, 5H, ArH), 7.22–7.05 (brs, 1H, CONH), 5.14 (s, 2H, CH2O), 3.70 (t, 2H, J=6.7 Hz, NCH2), 2.96 (s, 3H, NCH3), 2.87 (s, 3H, NCH3), 2.61 (t, 2H, J=6.7 Hz, CH2CO), 1.40 (s, 9 H, C(CH3)3); 13C NMR (75 MHz, CD2Cl2) δ 171.33, 156.54, 155.11, 136.65, 128.84, 128.53, 128.38, 81.50, 67.60, 47.62, 37.36, 35.30, 31.99,28.27; MS (FAB) 366.0 (28) (MH+).
3.1.16. 3-[N1-(Acetyl-d-alanyl)-N2-(benzyloxycarbonyl)hydrazino]-N,N-(dimethyl)propanamide (19)
To a stirred solution of Boc derivative 18 (1.10 g, 3 mmol) in dry CH2Cl2 (10 mL) under argon, was added trifluoroacetic acid (10 mL). The solution was stirred at room temperature for 2 h and was then concentrated in vacuo. Toluene (10 mL) was added, and the mixture was concentrated in vacuo. The residue was dried in vacuo overnight to give the crude unstable hydrazino compound. This was dissolved in dry THF (3 mL), and triethylamine (1.57 mL, 10.5 mmol) was added to form the amine solution A. A solution of N-acetyl-d-alanine (0.393 g, 3.0 mmol) in dry THF (10 mL) was cooled to −5 °C with stirring under argon. Triethylamine (0.45 mL, 3.0 mmol) and ethyl chloroformate (0.29 mL, 3.0 mmol) were added. The mixture was stirred for 20 min at −5 °C, then the amine solution A was added. After 30 min at −5 °C, the reaction mixture was allowed to warm to room temperature and stirred for 2 h. The precipitate was filtered and the filtrate was concentrated in vacuo. The residue was dissolved in ethyl acetate (100 mL), washed with said aq NH4Cl (15 mL), 1 N NaHCO3 (15 mL) and brine (15 mL), dried (Na2SO4) and concentrated in vacua to give an oil. Purification by flash chromatography (ethyl acetate:methanol, 95:5) gave 19 (0.91 g, 80%) as a colorless oil: IR (CH2Cl2 cast) 3282, 2936, 1738, 1645, 1500, 1455, 1259, 1189 cm−1; 1H NMR (360 MHz, CD3OD) δ 7.42–7.25 (m, 5H, ArH), 5.18 (s, 2H, CH2O), 4.80–4.70 (m, 1H, CH), 4.10–3.90 (brs, 1H, NCHAH), 3.70–3.40 (brs, 1H, NCHHB), 2.99 (s, 3H, NCH3), 2.88 (s, 3H, NCH3), 2.80–2.40 (brs, 2H, CH2CO), 1.90 (s, 3H, CH3), 1.30–1.00 (brs, 3H, CH3); 13C NMR (75 MHz, CD2Cl2) δ 175.08, 171.84, 169.38, 156.82, 136.17, 128.91, 128.70, 128.50, 68.21, 46.18, 45.85, 37.44, 35.47, 31.62, 23.25, 18.52; MS (FAB) 379.0 (28) (MH+).
3.1.17. 3-[N1-(Acetyl-l-alanyl)-N2-(benzyloxycarbonyl)hydrazino]-N,N-(dimethyl)propanamide (20)
The procedure used for the preparation of 19 was adapted to prepare the trifluoroacetate salt (1.14 g, 3 mmol) and condense it with N-acetyl-l-alanine (0.393 g, 3 mmol) to give 20 (0.86 g, 76%) as a gum: IR (CH2Cl2 cast) 3284, 2937, 1738, 1646, 1500, 1455, 1259, 1189 cm−1; 1H NMR (360 MHz, CD2Cl2) δ 8.64–8.48 (brs, 1H, N-NH), 7.47–7.28 (m, 5H, ArH), 6.50–6.30 (brs, 1H, CONH), 5.18 (s, 2H, CH2O), 4.92–4.78 (s, 1H, CH), 4.25–4.00 (brs, 1H, NCHHB), 3.50–3.10 (brs, 1H, NCHAH), 2.95 (s, 3H, NCH3), 2.87 (s, 3H, NCH3), 2.80–2.40 (brs, 2H, CH2CO), 1.90 (s, 3H, CH3), 1.21 (d, 3H, J=6.6 Hz, CH3); 13C NMR (75 MHz, CD2Cl2) δ 175.05, 171.89, 169.36, 156.86, 136.16, 128.92, 128.71, 128.50, 68.23, 46.23, 45.86, 37.45 35.48, 31.66, 23.29, 18.60; MS (FAB) 379.2 (65) (MH+).
3.1.18. 3-[N1-(tert-Butyloxycarbonyl-l-alanyl)-N2-(benzyloxycarbonyl)hydrazino]-N,N-(dimethyl)propanamide (21)
The above procedure for the preparation of 19, was adapted to prepare and condense the trifluoroacetate salt (0.376 g, 1 mmol) and N-t-Boc-l-alanine (0.131 g, 1 mmol) to give 21 (0.350 g, 80%) as a colorless oil: IR (CH2Cl2 cast) 3242, 2976, 2934, 1742, 1712, 1670, 1650, 1499, 1454, 1246, 1167 cm−1; 1H NMR (300 MHz, CD2Cl2) δ 8.55–8.25 (s, 1H, N-NH), 7.45–7.25 (m, 5H, ArH), 5.30–5.10 (m, 3H, CH2O, CONH), 4.70–4.50 (s, 1H, CH), 4.35–4.00 (s, 1H, NCHHB), 3.40–3.00 (s, 1H, NCHAH), 2.93 (s, 3H, NCH3), 2.88 (s, 3H, NCH3), 2.86–2.30 (m, 2H, CH2CO), 1.40 (s, 9 H, C(CH3)3), 1.19 (d, 3H, J=6.8 Hz, CH3); 13C NMR (75 MHz, CD2Cl2) δ 175.31, 172.09, 156.93, 155.21, 136.23, 128.94, 128.72, 128.50, 79.50, 68.22, 46.85, 46.31, 37.49, 35.51, 31.77, 28.45, 18.81; MS (FAB) 437.1 (59) (MH+).
3.1.19. 3-[N1-(tert-Butyloxycarbonyl-β-alanyl)-N2-(benzyloxycarbonyl)hydrazino]-N,N-(dimethyl)propanamide (22)
The procedure used for the preparation of 19, was adapted to prepare and convert the trifluoroacetate salt (2.93 g, 7.8 mmol) and N-t-Boc-β-alanine (1.47 g, 7.8 mmol) to 22 (2.14 g, 63%) as an oil: IR (CH2Cl2 cast) 3248, 2975, 2933, 1738, 1711, 1670, 1651, 1635, 1500, 1402, 1247, 1172 cm−1; 1H NMR (300 MHz, CD2Cl2) δ 8.50–8.30 (s, 1H, N-NH), 7.50–7.25 (m, 5H, ArH), 5.30–5.10 (s, 3H, CH2O, CONH), 4.30–4.05 (brs, 1H, NCHHB), 3.40–3.10 (m, 3H, NCHAH, NCH2), 2.95 (s, 3H, NCH3), 2.88 (s, 3H, NCH3), 2.86–2.34 (m, 4 H, 2 CH2CO), 1.40 (s, 9 H, C(CH3)3); 13C NMR (100 MHz, CD2Cl2) δ 174.32, 171.94, 156.45, 156.08, 136.27, 128.87, 128.63, 128.40, 78.96, 67.91, 45.77, 37.36, 36.33, 35.40, 33.05, 31.91, 28.45; MS (FAB) 437.2 (9) (MH+).
3.1.20. 3-[N1-(Acetyl-l-leucyl-l-alanyl-l-alanyl)-N2-(benzyloxycarbonyl)hydrazino]-N,N-(dimethyl)propanamide (23)
The above procedure for the preparation of 19, was adapted to convert 21 (0.130 g, 0.3 mmol) and Ac-Leu-Ala-OH (73.2 mg, 0.3 mmol) to the crude product. This was purified by HPLC (linear gradient elution over 20 min of acetonitrile and water, from 20% to 40%, t R 12.8 min) to give 23 (0.118 g, 70%) as a white solid: mp 183–184 °C; IR (CH2Cl2 cast) 3280, 2955, 2934, 1741, 1634, 1537, 1455, 1412, 1243 cm−1; 1H NMR (300 MHz, CD3OD) δ 7.43–7.25 (m, 5H, ArH), 5.15 (s, 2H, CH2O), 4.80–4.70 (m, 1H, CH), 4.40–4.20 (m, 2H, 2 CH), 4.10–3.80 (brs, 1H, NCHBH), 3.70–3.40 (brs, 1H, NCHAH), 2.97 (s, 3H, NCH3), 2.88 (s, 3H, NCH3), 2.70–2.50 (brs, 2H, CH2CO), 1.96 (s, 3H, CH3), 1.72–1.57 (m, 1H, CH), 1.57–1.45 (m, 2H, CH2), 1.30–1.10 (m, 6 H, 2 CH3), 0.95 (d, 3H, J=6.3 Hz, CH3), 0.91 (d, 3H, J=6.3 Hz, CH3); 13C NMR (75 MHz, CD3OD) δ 176.22, 174.71, 174.05, 173.31, 172.96, 157.61, 137.44, 129.60, 129.36, 129.16, 68.77, 53.16, 50.10, 46.73, 46.44, 41.90, 37.64, 35.67, 31.80, 25.91, 23.42, 22.40, 21.98, 17.91; MS (FAB) 563.1 (16) (MH+).
3.1.21. 3-[N1-(Acetyl-l-leucyl-l-alanyl-β-alanyl)-N2-(benzyloxycarbonyl)hydrazino]-N,N-(dimethyl)propanamide (24)
The above procedure for the preparation of 19, was adapted to convert 22 (0.130 g, 0.3 mmol) and Ac-Leu-Ala-OH (73.2 mg, 0.3 mmol) to the crude product. This was purified by HPLC (linear gradient elution over 20 min of acetonitrile and water, from 20% to 40%, t R 14.2 min) to give 24 (0.110 g, 65%) as a white solid: mp 156–158 °C; IR (CH2Cl2 cast) 3289, 2956, 2934, 1738, 1644, 1538, 1451, 1412, 1243 cm−1; 1H NMR (300 MHz, CD2Cl2) δ 8.85–8.50 (brs, 1H, N-NH), 7.43–7.30 (m, 5H, ArH), 7.30–7.15 (brs, 1H, CONH), 7.15–7.00 (s, 1H, CONH), 6.85–6.50 (brs, 1H, CONH), 5.16 (s, 2H, CH2O), 4.55–4.30 (m, 2H, 2 CH), 4.30–4.00 (s, 1H, NCHBH), 3.60–3.20 (m, 3H, NCHAH, NCH2), 2.93 (s, 3H, NCH3), 2.86 (s, 3H, NCH3), 2.86–2.00 (m, 4 H, 2 CH2CO), 1.95 (s, 3H, CH3), 1.75–1.40 (m, 3H, CH and CH2), 1.35–1.25 (d, 3H, J=7.0 Hz, CH3), 0.91 (d, 3H, J=6.3 Hz, CH3), 0.89 (d, 3H, J=6.3 Hz, CH3); 13C NMR (75 MHz, CD2Cl2) δ 174.25, 172.72, 172.40, 172.00, 170.80, 156.66, 136.33, 128.95, 128.71, 128.43, 68.03, 52.47, 49.40, 45.64, 41.67, 37.47, 35.69, 35.49, 32.31, 31.86, 25.16, 23.18, 23.14, 22.06, 18.5 1; MS (FAB) 563.2 (38) (MH+).
3.1.22. 3-[N1-(Acetyl-d-alanyl)-N2-(o-nitrophenylsulfenyl)hydrazino]-N,N-(dimethyl)propanamide (25)
To a solution of Cbz-hydrazino derivative 19 (0.378 g, 1 mmol) in methanol (20 mL) under argon was added 10% palladium on charcoal catalyst (38 mg). The mixture was stirred under an atmosphere of hydrogen until absorption of gas ceased. The catalyst was removed by filtration through a column of Celite and the filtrate was concentrated in vacuo to give 3-[N′-(acetyl-d-alanyl)hydrazinol-N,N-(dimethyl)propanamide (0.244 g, quantitative) as a colorless oil: IR (CH2Cl2 cast) 3314, 3214, 2934, 1636, 1542 cm−1; 1H NMR (360 MHz, CD3OD) δ 5.24 (q, 1H, J=7.0 Hz, CH), 3.86–3.60 (m, 2H, NCH2), 3.04 (s, 3H, NCH3), 2.91 (s, 3H, NCH3), 2.72 (t, 2H, J=6.9 Hz, CH2CO), 1.95 (s, 3H, CH3), 1.27 (d, 3H, J=7.0 Hz, CH3); 13C NMR (75 MHz, CD3OD) δ 176.60, 173.55, 172.70, 48.38, 47.18, 37.73, 35.67, 31.74, 22.42, 17.54; MS (FAB) 245.3 (100) (MH+). Anal. calcd for C10H20N4O3: C, 49.17; H, 8.25; N, 22.93. Found: C, 48.86; H, 8.52; N, 22.62.
To a stirred solution of this intermediate (81.1 mg, 0.33 mmol) and o-nitrophenylsulfenyl chloride (62.9 mg, 0.33 mmol) in DMF (2 mL) under argon, at −10 °C, was added triethylamine (50 μL, 0.33 mmol). After 30 min at −10 °C, the reaction mixture was allowed to warm to room temperature, and was then stirred for 1 h. The solution was concentrated in vacuo. Purification by HPLC (linear gradient elution over 20 min of acetonitrile and water, from 25% to 45%, flow rate 15.0 mL min−1, t R 11.6 min) gave the title compound 25 (0.108 g, 82%) as a yellow powder. Spectral characterization indicated a mixture of conformers (conformer A:conformer B, 2:1): mp 52–63 °C (dec); IR (CH2Cl2 cast) 3283, 3209, 2934, 1641, 1592, 1565, 1510, 1337, 735 cm−1; 1H NMR (360 MHz, CD2Cl2) (conformer A) δ 8.33–8.20 (m, 3H, N-NH, ArH), 7.80 (ddd, 1H, J=7.7, 7.1, 1.3 Hz, ArH), 7.38–7.28 (m, 1H, ArH), 6.41 (d, 1H, J=6.5 Hz, CONH), 5.15–5.05 (m, 1H, CH), 3.88–3.77 (m, 2H, NCH2), 2.95 (s, 3H, NCH3), 2.92 (s, 3H, NCH3), 2.75–2.67 (m, 2H, CH2CO), 1.88 (s, 3H, COCH3), 1.17 (d, 3H, J=6.8 Hz, CH3 Ala); (conformer B) δ 8.33–8.20 (m, 3H, N-NH, ArH), 7.69 (ddd, 1H, J=7.7, 7.1, 1.3 Hz, ArH), 7.38–7.28 (m, 1H ArH), 6.58 (d, 1H, J=6.5 Hz, CONH), 4.97–4.87 (m, 1H, CH), 3.88–3.77 (m, 2H, NCH2), 2.88 (s, 3H, NCH3), 2.85 (s, 3H, NCH3), 2.82–2.74 (m, 2H, CH2CO), 1.95 (s, 3H, COCH3), 1.30 (d, 3H, J=6.8 Hz, CH3 Ala); 13C NMR (75 MHz, CD3OD) (conformer A) δ 174.40, 172.29, 169.37, 143.05, 141.31, 134.74, 126.51, 125.96, 125.82, 45.94, 45.79, 37.54, 35.59, 32.43, 23.18, 18.69; (conformer B) δ 173.09, 170.63, 169.58, 142.54, 141.31, 134.50, 125.92, 125.82, 125.46, 45.66, 45.19, 37.15, 35.29, 31.70, 23.18, 18.69; MS (FAB) 398.0 (21) (MH+). Anal. calcd for C16 H 23N5O5S: C, 48.35; H, 5.83; N, 17.62. Found: C, 48.11; H, 5.56; N, 17.32.
3.1.23. 3-[N1-(Acetyl-l-alanyl)-N2-(o-nitrophenylsulfenyl)hydrazino]-N,N-(dimethyl)propanamide (26)
The above procedure for the preparation of 25 was used with Cbz-hydrazino derivative 20 (0.378 g, 1 mmol) to give the intermediate 3-[N 1-(acetyl-lL-alanyl)hydrazino]-N,N-(dimethyl)propanamide (0.243 g, quantitative) as a colorless oil: IR (CH2Cl2 cast) 3312, 3213, 2933, 1633, 1541 cm−1; 1H NMR (300 MHz, CD3OD) δ 5.24 (q, 1H, J=7.0 Hz, CH), 3.80–3.60 (m, 2H, NCH2), 3.04 (s, 3H, NCH3), 2.91 (s, 3H, NCH3), 2.72 (t, 2H, J=6.9 Hz, CH2CO), 1.94 (s, 3H, CH3), 1.27 (d, 3H, J=7.0 Hz, CH3); 13C NMR (75 MHz, CD3OD) δ 176.59, 173.55, 172.69, 48.15, 47.18, 37.74, 35.67, 31.74, 22.43, 17.54; MS (FAB) 245.2 (100) (MH+).
This compound (0.100 g, 0.41 mmol), o-nitrophenylsulfenyl chloride (77.7 mg, 0.41 mmol) and triethylamine (57.2 μL, 0.41 mmol) were reacted as above to give the crude product. This was purified by HPLC (linear gradient elution over 20 min of acetonitrile and water, from 25% to 45%, t R 10.1 min) to give the title compound 26 (0.120 g, 74%) as a yellow powder. Spectral characterization indicated a mixture of conformers (conformer A:conformer B, 2:1): mp 58–70 °C (dec); IR (CH2Cl2 cast) 3284, 3209, 2934, 1642, 1592, 1565, 1510, 1337, 735 cm−1; 1H NMR (300 MHz, CD2Cl2) (conformer A) δ 8.34–8.20 (m, 3H, N-NH, ArH), 7.78 (ddd, 1H, J=7.7, 7.2, 1.3 Hz, ArH), 7.40–7.28 (m, 1H, ArH), 6.34 (d, 1H, J=7.30 Hz, CONH), 5.15–5.00 (m, 1H, CH), 3.90–3.77 (m, 2H, NCH2), 2.95 (s, 3H, NCH3), 2.92 (s, 3H, NCH3), 2.71 (t, 2H, J=5.5 Hz, CH2CO), 1.88 (s, 3H, COCH3), 1.16 (d, 3H, J=6.8 Hz, CH3); (conformer B) δ 8.34–8.20 (m, 3H, N-NH, ArH), 7.69 (ddd, 1H, J=7.7, 7.2, 1.3 Hz, ArH), 7.40–7.28 (m, 1H, ArH), 6.48 (d, 1H, J=7.10 Hz, CONH), 4.98–4.85 (m, 1H, CH), 3.90–3.77 (m, 2H, NCH2), 2.88 (s, 3H, NCH3), 2.86 (s, 3H, NCH3), 2.80 (t, 2H, J=5.5 Hz, CH2CO), 1.95 (s, 3H, COCH3), 1.30 (d, 3H, J=6.8 Hz, CH3); 13C NMR (75 MHz, CD3OD) (conformer A) δ 174.36, 172.33, 169.40, 143.05, 141.28, 134.75, 126.51, 125.97, 125.83, 45.94, 45.84, 37.57, 35.61, 32.46, 23.20, 18.69; (conformer B) δ 173.05, 170.07, 169.62, 143.05, 142.50, 134.50, 125.97, 125.83, 125.44, 45.68, 45.14, 37.16, 35.31, 31.69, 23.20,18.69; MS (FAB) 398.2 (17) (MH+).
3.1.24. 3-[N1-(Acetyl-l-leucyl-l-alanyl-l-alanyl)-N2-(o-nitrophenyl-sulfenyl)hydrazino]-N,N-(dimethyl)propanamide (27)
The procedure used for the preparation of 16, with Cbz-hydrazino derivative 23 (58.2 mg, 0.10 mmol) and 10% palladium on charcoal catalyst (10 mg) in methanol (10 mL), followed by o-nitrophenylsulfenyl chloride (18.9 mg, 0.10 mmol) gave the crude product 27. Purification by HPLC (linear gradient elution over 20 min of acetonitrile and water, from 25% to 40%, t R 15.6 min) gave the title compound 27 (29.1 mg, 50%) as a yellow powder. Spectral characterization indicated a mixture of conformers (conformer A:conformer B, 4: 5): mp 94–105 °C (dec); IR (CH2Cl2 cast) 3284, 3071, 2956, 2934, 1637, 1592, 1512, 1449, 133 8, 1305 cm−1; 1H NMR (300 MHz, CD3OD) (conformer A) δ 8.39 (d, 1H, J=8.0 Hz, ArH), 8.25 (d, 1H, J=8.4 Hz, ArH), 7.85–7.70 (m, 1H, ArH), 7.42–7.28 (m, 1H, ArH), 5.11–5.00 (m, 1H, NCHCON-N), 4.43–4.28 (m, 2H, α-CH Leu and α-CH Ala), 3.90–3.68 (m, 2H, NCH2), 2.96 (s, 3H, NCH3), 2.88 (s, 3H, NCH3), 2.90–2.80 (m, 2H, COCH2), 1.98 (s, 3H, COCH3), 1.76–1.58 (m, 1H, CH Leu), 1.58–1.46 (m, 2H, CH2 Leu), 1.37–1.18 (m, 6 H, 2 CH3 Ala), 0.95 (d, 3H, J=6.5 Hz, CH3 Leu), 0.91 (d, 3H, J=6.5 Hz, CH3 Leu); (conformer B) δ 8.25 (d, 2H, J=8.4 Hz, ArH), 7.85–7.70 (m, 1H, ArH), 7.42–7.28 (m, 1H, ArH), 4.86–4.76 (m, 1H, NCHCON-N), 4.43–4.28 (m, 2H, α-CH Leu and α-CH Ala), 3.90–3.68 (m, 2H, NCH2), 3.01 (s, 3H, NCH3), 2.91 (s, 3H, NCH3), 2.80–2.70 (m, 2H, COCH2), 1.98 (s, 3H, COCH3), 1.76–1.58 (m, 1H, CH Leu), 1.58–1.46 (m, 2H, CH2 Leu), 1.37–1.18 (m, 6 H, 2 CH3 Ala), 0.95 (d, 3H, J=6.5 Hz, CH3 Leu), 0.91 (d, 3H, J=6.5 Hz, CH3 Leu); 13C NMR (75 MHz, CD3OD) (conformer A) δ 175.91, 174.66, 174.15, 173.35, 172.65, 143.98, 142.13, 135.55, 135.34, 127.17, 126.55, 53.46, 50.03, 46.89, 46.73, 41.67, 37.65, 35.70, 32.50, 29 25.91, 23.28, 22.43, 22.15, 17.97, 17.78; (conformer B) δ 175.91, 174.66, 174.15, 173.35, 173.25, 144.35, 142.13, 135.55, 135.34, 127.17, 126.55, 53.18, 50.03, 46.89, 46.51, 41.86, 37.76, 35.70, 32.26, 25.91, 23.44, 22.41, 21.96, 17.95, 17.62; MS (FAB) 582.3 (4) (MH+).
3.1.25. 3-[N1-(Acetyl-l-leucyl-l-alanyl-β-alanyl)-N2-(o-nitrophenyl-sulfenyl)hydrazino]-N,N-(dimethyl)propanamide (28)
The procedure used for the preparation of 16, was employed with Cbz-hydrazino derivative 24 (58.2 mg, 0.10 mmol) and 10% palladium on charcoal catalyst (10 mg) in methanol (10 mL), followed by o-nitrophenylsulfenyl chloride (18.9 mg, 0.10 mmol) to give the crude product 28. Purification by HPLC (linear gradient elution over 20 min of acetonitrile and water, from 25% to 40%, t R 17.5 min) gave the title compound 28 (35 mg, 60%) as a yellow powder. Spectral characterization indicated a mixture of conformers (conformer A:conformer B, 2:1): mp 66–78 °C (dec); IR (CH2Cl2 cast) 3288, 3064, 2955, 2935, 1646, 1592, 1541, 1511, 1448, 1337 cm−1; 1H NMR (300 MHz, CD3OD) (conformer A) δ 8.38 (dd, 1H, J=8.3, 1.1 Hz, ArH), 8.26 (dd, 1 H, J=8.4, 1.3 Hz, ArH), 7.86–7.72 (m, 1H, ArH), 7.45–7.30 (m, 1H, ArH), 4.35–4.17 (m, 2H, α-CH Leu and α-CH Ala), 3.86–3.70 (m, 2H, N-NCH2), 3.54–3.37 (m, 2H, CONCH2), 3.00 (s, 3H, NCH3), 2.89 (s, 3H, NCH3), 2.82–2.65 (m, 4 H, 2 COCH2), 1.98 (s, 3H, COCH3 0.72–1.58 (m, 1H, CH Leu), 1.58–1.50 (m, 2H, CH2 Leu), 1.33 (d, 3H, J=7.3 Hz, CH3 Ala), 0.96 (d, 3H, J=6.4 Hz, CH3 Leu), 0.92 (d, 3H, J=6.4 Hz, CH3 Leu); (conformer B) δ 8.28 (dd, 1H, J=8.3 Hz, 1.1 Hz, ArH), 8.26 (dd, 1H, J=8.4 Hz, 1.3 Hz, ArH), 7.86–7.72 (m, 1H, ArH), 7.45–7.30 (m, 1H, ArH), 4.35–4.17 (m, 2H, α-CH Leu and α-CH Ala), 3.86–3.70 (m, 2H, N-NCH2), 3.54–3.37 (m, 2H, CONCH2), 2.90 (s, 3H, NCH3), 2.86 (s, 3H, NCH3), 2.82–2.65 (m, 4 H, 2 COCH2), 1.96 (s, 3H, COCH3), 1.72–1.58 (m, 1H, CH Leu), 1.58–1.50 (m, 2H, CH2 Leu), 1.33 (d, 3H, J=7.3 Hz, CH3 Ala), 0.96 (d, 3H, J=6.4 Hz, CH3 Leu), 0.92 (d, 3H, J=6.4 Hz, CH3 Leu); 13C NMR (75 MHz, CD3OD) (conformer A) δ 175.23, 174.96, 174.83, 173.72, 172.94, 144.32, 142.55, 135.54, 127.11, 126.56, 126.49, 53.90, 50.49, 46.17, 41.44, 37.76, 36.77, 35.68, 33.78, 32.25, 25.91, 23.22, 22.47, 22.22, 17.81; (conformer B) δ 175.23, 175.10, 174.83, 173.38, 172.61, 144.22, 143.97, 135.34, 127.11, 126.56, 126.49, 54.02, 50.49, 46.54, 41.44, 37.53, 36.84, 35.68, 33.13, 32.43, 25.91, 23.22, 22.47, 22.22, 17.81; MS (FAB) 582.3 (22) (MH+). Anal. calcd for C25H39N7O7S: C, 51.62; H, 6.76; N, 16.86. Found: C, 51.25; H, 6.83; N, 16.51.
Acknowledgements
The authors would like to thank: Drs. Michael N. James and Ernst M. Bergmann (Biochemistry Department, University of Alberta) for many helpful discussions and suggestions. Colin Luo and Shirley Shechosky are gratefully acknowledged for extensive assistance with enzyme assays. These investigations were supported by the US National Institutes of Health (Grant AI 38249) and the Natural Sciences and Engineering Research Council of Canada.
References
- 1.(a) Bergmann, E. M.; James, M. N. G. In Proteinases of Infectious Agents, Dunn, B., Ed.; Academic Press: San Diego, CA, 1998, in press. (b) Gorbalenya, A. E; Snijder, E. J. Perspectives in Drug Discovery and Design1996, 6, 64–86. [DOI] [PMC free article] [PubMed]
- 2.Rueckert, R. R. In Fields Virology; Fields, B. N.; Knipe, D. M.; Howley, P. M.; Channock, R. M.; Melnick, J. L.; Monath, T. P.; Roizmann, B.; Straus, S. E., Eds.; Lippincott-Raven Publishers: Philadelphia, PA, 1997.
- 3.Ryan, M. D.; Flint, M. J. Gen. Virol.1997, 78, 699–723. [DOI] [PubMed]
- 4.(a) Krausslich, H. G.; Wimmer, E. Annu. Rev. Biochem.1988, 57, 701–754. (b) Rasnick, D. Perspectives in Drug Discovery and Design1996, 6, 47–63. [DOI] [PubMed]
- 5.Malcolm, B. A. Protein Sci.1995, 4, 1439–1445. [DOI] [PMC free article] [PubMed]
- 6.Allaire, M.; Chernaia, M. M.; Malcolm, B. A.; James, M. N. G. Nature1994, 369, 72–76. [DOI] [PubMed]
- 7.Bergmann, E. M.; Mosimann, S. C.; Chernaia, M. M.; Malcolm, B. A.; James, M. N. G. J. Virol.1997, 71, 2436–2448. [DOI] [PMC free article] [PubMed]
- 8.Matthews, D. A.; Smith, W. W.; Ferre, R. A.; Condon, B.; Budahazi, G.; Sisson, W.; Villafranca, J. E.; Janson, C. A.; McElroy, H. E.; Gribskov, C. L.; Worland, S. Cell1994, 77, 761–771. [DOI] [PubMed]
- 9.Mosimann, S. C.; Cherney, M. M.; Sia, S.; Plotch, S.; James, M. N. G. J. Mol. Biol.1997, 273, 1032–1047. [DOI] [PubMed]
- 10.Ng, L. F. P.; Liu, D. X. Virology, 1998, 243, 388–395. [DOI] [PMC free article] [PubMed]
- 11.Ziebuhr, L; Heusipp, G.; Siddell, S. G. J.Virology1997; 71, 3992–3997. [DOI] [PMC free article] [PubMed]
- 12.Dragovich, P. S.; Webber, S. E.; Babine; R. E.; Fuhrman; S. A.; Patick; A. K.; Matthews; D. A.; Lee, C. A.; Reich, S. H.; Prins; T. L.; Marakovits, J. T.; Littlefield, E. S.; Zhou, R.; Tikhe, L.; Ford, C. E.; Wallace, M. B.; Meador, J. W. III; Ferre, R. A.; Brown, E. L.; Binford, S. L.; Harr, J. E. V.; DeLisle, D. M.; Worland, S. T. J. Med. Chem.1998, 41, 2806–2818, and references therein. [DOI] [PubMed]
- 13.Malcolm, B. A.; Lowe, C.; Shechosky, S.; McKay, R. T.; Yang, C. C.; Shah, V. L.; Simon, R. L.; Vederas, J. C. and Santi, D. V. Biochemistry1995, 34, 8172–8179. [DOI] [PubMed]
- 14.Kaldor, S. W.; Hammond, M.; Dressman, B. A.; Labus, J. M.; Chadwell, F. W.; Kline, A. D.; Heirnz, B. A. Bioorg. Med. Chem. Lett.1995, 5, 2021–2026.
- 15.Shepherd, T. A.; Cox, G. A.; McKinney, E.; Tang, L.; Wakulchik, M.; Zimmerman, R. E.; Villarreal, E. C. Bioorg. Med. Chem. Lett.1996, 6, 2893–2896.
- 16.Morris, T. S.; Frormann, S.; Shechosky, S.; Lowe, C.; Lall, M. S.; Gauss-Müller, V.; Plurcell, R. H.; Emerson, S. U.; Vederas, J. C.; Malcolm, B. A. Bioorg. Med. Chem.1997,5, 797–807. [DOI] [PubMed]
- 17.Skiles, J. W.; McNeil, D. Tetrahedron1990, 31, 7277–7280.
- 18.Webber, S. E.; Tikhe, J.; Worland, S. T.; Fuhrman, S. A.; Hendrickson, T. R.; Matthews, D. A.; Love, R. A.; Patick, A. K.; Meador, J. W.; Ferre, R. A.; Brown, E. L.; DeLisle, D. M.; Ford, C. E.; Binford, S. L. J. Med. Chem.1996, 39, 5072–5082. [DOI] [PubMed]
- 19.Jungheim, L. N.; Cohen, J. D.; Johnson, R. B.; Villarreal, E. C.; Wakulchik, M.; Loncharich, R. L.; Wang, Q. M. Bioorg. Med. Chem. Lett.1997, 7, 1589–1594.
- 20.Kong, J.; Venkatraman, S.; Furness, K.; Nimkar, S.; Shepherd, T. A.; Wang, Q. M.; Aubé, J.; Hanzlik, R. P. J. Med. Chem.1998, 41, 2579–2587. [DOI] [PubMed]
- 21.Dragovich, P. S.; Webber, S. E.; Babine; R. E.; Fuhrman; S. A.; Patick,; A. K.; Matthews,; D. A.; Reich, S. H.; Marakovits, J. T.; Prins; T. J.; Zhou, R.; Tikhe, J.; Littlefield, E. S.; Bleckman, T. M.; Wallace, M. B.; Little, T. L.; Ford, C. E.; Meador, J. W. III; Ferre, R. A.; Brown, E. L.; Binford, S. L.; DeLisle, D. M.; Worland, S. T. J. Med. Chem.1998, 41, 2819–2834. [DOI] [PubMed]
- 22.Sham, H. L.; Rosenbrook, W.; Kati, W.; Betebenner, D. A.; Wideburg, N. E.; Saldivar, A.; Plattner, J. J.; Norbeck, D. W. J. Chem. Soc., Perkin Trans. 11995, 1081–1082.
- 23.McKendrick, J. E.; Frormann, S.; Luo, C.; Semchuck, P.; Vederas, J. C.; Malcolm, B. A. Int. J. Mass. Spectrom.1998, in press.
- 24.For discussion and leading references to azapeptides see: Han, H.; Janda, K. D. J. Am. Chem. Soc.1996, 118, 2539–2544.
- 25.For an example of an orally-available azapeptide inhibitor of HIV-1 protease see: Fässler, A.; Bold, G.; Capraro, H.-G.; Cozens, R.; Mestan, J.; Poncioni, B.; Rösel, J.; Tintelnot-Blomley, M.; Lang, M. J. Med. Chem.1996, 39, 3203–3216. [DOI] [PubMed]
- 26.Abeles; R. H.; Magrath, J. J. Med. Chem.1992, 35, 4279–4283. [DOI] [PubMed]
- 27.Xing, R.; Hanzlik, R. P. J. Med. Chem.1998, 41, 1344–1351. [DOI] [PubMed]
- 28.Thompson, S. K.; Halbert, S. M.; Bossard, M. J.; Tornaszek, T. A.; Levy, M. A.; Zhao, B.; Smith, W. W.; Abdel-Meguid, S. S.; Janson, C. A.; D'Alessio, K. J.; McQueney, M. S.; Amegadzie, B. Y.; Hanning, C. R.; DesJarlais, R. L.; Briand, J.; Sarkar, S. K.; Huddleston, M. J.; Ijames, C. F.; Carr, S. A.; Garnes, K. T.; Shu, A.; Heys, J. R.; Bradbeer, J.; Zembryki, D.; Lee-Rykaczewski, L.; James, I. E.; Lark, M. W.; Drake, F. K.; Gowen, M.; Gleason, J. G.; Veber, D. F. Proc. Natl. Acad. Sci.1997, 94, 14249–14254. [DOI] [PMC free article] [PubMed]
- 29.For related work using amino ketones see: Yamashita, D. S.; Smith, W. W.; Zhao, B.; Janson, C. A.; Tomaszek, T. A.; Bossard, M. J.; Levy, M. A.; Oh, H. J.; Carr, T. J.; Thompson, S. K.; Ijames, C. F.; Carr, S. A.; McQueney, M.; D'Alessio, K. L.; Amegadzie, B. Y.; Hanning, C. R.; Abdel-Meguid, S.; DesJarlais, R. L.; Gleason, J. G.; Veber, D. F. J. Am. Chem. Soc.1997, 119, 11351–11352.
- 30.Greene, T. W.; Wuts, P. G. M. Protective Groups in Organic Synthesis, 2nd ed.; Wiley: New York, 1991; pp 377–378.
- 31.Kice, J. L.; Kutateladze, A. G. J. Org. Chem.1992, 57, 3298–3303.
- 32.Pu, Y.; Lowe, C.; Sailer, M.; Vederas, J. C. J. Org. Chem.1994, 59, 3642–3655.
- 33.A preliminary portion of this work has been mentioned in a recent review but without experimental detail: Frormann, S.; Huang, Y.; Lall, M.; Lowe, C.; Vederas, J. C. In Recent Advances in the Chemistry of Anti-Infective Agents, Bentley, P. H.; O'Hanlon, P. J.; Eds., Royal Society of Chemistry: Cambridge, UK, 1997: pp 298–315.
- 34.Wood, J. L.; Jeong, S.; Salcedo, A.; Jenkins, J. J. Org. Chem.1995, 60, 286–287.
- 35.Albeck, A.; Persky, R. Tetrahedron1994, 50, 6333–6346.
- 36.Jones, C. D. J. Org. Chem.1972, 37, 3624–3625.
- 37.Dado, G. P.; Gellman, S. H. J. Am. Chem. Soc.1994, 116, 1054–1062.
- 38.Jewell, D. A.; Swietnicki, W.; Dunn, B. M.; Malcolm, B. A. Biochemistry1992, 31, 7862–7869. [DOI] [PubMed]
- 39.Malcolm, B. A.; Chin, S. M.; Jewell, D. A.; Stratton-Thomas, J. R.; Thudium, K. B.; Ralston, R.; Rosenberg, S. Biochemistry1992, 31, 3358–3363. [DOI] [PubMed]
- 40.A crystal structure of HAV 3C with N-iodoacetyl-Val-Phe-NH2, has been solved by Michael N. James and Ernst M. Bergmann (Biochemistry Department, University of Alberta). Details of the crystallization methodology and solution of the crystal structure will be reported elsewhere.
- 41.Witter, D. J.; Vederas, J. C. J. Org. Chem.1996, 61, 2613–2623. [DOI] [PubMed]
- 42.Lettieri, G.; Brancaccio, G.; Larizza, A. Boll. Chim. Farm.1981, 12, 308–310. [PubMed]
- 43.Molander, G. A.; Stengel, P. J. Tetrahedron1997, 53, 8887–8912.