

Highlights
-
•
Novel bufavirus was detected in Miniopterus schreibersii bats in Hungary.
-
•
This is the first time any parvovirus was detected in European bats.
-
•
Novel bat bufaviruses were closely related to human bufaviruses.
-
•
A possible intragenic recombination event was detected within bufaviruses.
Keywords: Bat, Bufavirus, Parvovirus, Hungary, Metagenomics, Viruses
Abstract
Bats are important hosts of many viruses and in several cases they may serve as natural reservoirs even for viruses with zoonotic potential worldwide, including Europe. However, they also serve as natural reservoir for other virus groups with important evolutionary relevance and yet unknown zoonotic potential. We performed viral metagenomic analyses on Miniopterus schreibersii bat fecal samples. As a result, a novel parvovirus was detected for the first time in European bats. Although, bufavirus was recently discovered as a novel human infecting parvovirus, here we report sequence data of the first bufavirus from European bats related to human bufaviruses. Based on our sequence data a possible intragenic recombination event was detected within bufaviruses which may serves as an important milestone in their evolution.
Bats are the most diverse and species-rich taxa among vertebrates, with over 1250 species from all continents, except Antarctica (Teeling et al., 2005). After the SARS pandemic in 2002–2003, bats and their role in the ecology of emerging viruses became the objective of epidemiological investigations. They have been recognized for the natural reservoir hosts of many viruses with zoonotic potential (coronaviruses, paramyxoviruses, orthoreoviruses) (Kohl and Kurth, 2014) or viruses with important evolutionary relevance (Orthomyxoviruses, Hepadnaviruses, Hepaciviruses) (Tong et al., 2013, Drexler et al., 2013, Quan et al., 2013).
The family Parvoviridae comprises non-enveloped viruses with a single-stranded DNA genome of approximately 5 kb. The subfamily Densovirinae includes only arthropod-infecting viruses, while Parvovirinae contains human and other vertebrate-infecting viruses. The Parvovirinae subfamily is currently subdivided into 8 genera (Cotmore et al., 2014). Among DNA viruses, parvoviruses exhibit the highest mutation rate, almost comparable to that possessed by RNA viruses, furthermore they are extremely prone to recombination (Decaro et al., 2009, Shackelton et al., 2007). Parvoviruses are associated with a wide spectrum of acute and chronic diseases in humans and animals, with at least five known groups (Dependoviruses, Parvovirus B19, Human bocaviruses, Parv4 viruses and recently described Bufaviruses) infecting humans (Brown, 2010, Phan et al., 2012).
The discovery of bufavirus in 2012 from fecal samples of children with acute diarrhea revealed a novel human-infecting parvovirus (Phan et al., 2012). Since then bufavirus has also been detected in Bhutan, Finland and the Netherlands from human samples (Yahiro et al., 2014, Väisänen et al., 2014, Smits et al., 2014). The evolutionary origin of this novel virus is still unknown. Here we report the first parvovirus sequence data from European bats related to human bufaviruses. Phylogenetic data demonstrate the potential relatedness between bat-origin parvoviruses and primate protoparvoviruses.
Fecal samples were collected from a single Miniopterus schreibersii bat colony, at a cave in Szársomlyó mountain, Hungary (GPS coordinates 45°51′17N; 18°24′40E), 2013. Sample collection was performed as described previously (Kemenesi et al., 2015). All bat species in Europe are strictly protected under the Flora, Fauna, Habitat Guidelines of the European Union (92/43/EEC) and the Agreement on the Conservation of Populations of European Bats (http://www.eurobats.org). Invasive bat sampling is prohibited, hence we collected faecal samples and all animal handling processes were conducted by a trained chiropterologist with appropriate license for safe handling of bats.
After homogenization, samples were centrifuged at 15,000g for 10 min. DNA was extracted from 200 μl of supernatant using GeneJet Viral DNA and RNA Purification Kit (Thermo Scientific) following the manufacturer’s instructions. Samples were subjected to random primed PCR and semiconductor sequencing using the Ion Torrent PGM platform. Library preparation, size selection of library DNA, emulsion PCR, templated Ion Sphere bead enrichment and sequencing, bioinformatics analysis was carried out as described in detail elsewhere (Mihalov-Kovács et al., 2014).
In one sample parvovirus sequences were detected. These consensus sequences were 250 and 127 nt long and were represented by 24 and 2 sequence reads, respectively. The identified viral sequences were used for further primer design, using Oligo Explorer v1.2 software. In addition, PCR primers (BParV-F: 5′-ATC TGG GGC TTA TTC ATT TGG-3′ and BParV-R: 5′-CGG GTT ATG TCC TTG TTG TAT-3′) were designed for routine screening and 5 out of the total 13 samples were found to be positive. The expected amplicon size for screening PCR was approximately 230 nt long. Another set of primers (BParV Long-F: 5′-AAA TGG ATG CTT GGT CAT CCA-3′ and BParV Long-R: 5′-TTC CTT TGA AGA CCA TGG TTC-3′) were designed to obtain a larger fragment of the genome using Phusion High-Fidelity DNA Polymerase (Thermo Scientific), according to the manufacturer’s instructions. Sequences were further edited and aligned using GeneDoc 3.2 and ClustalX 2.0, respectively. The phylogenetic trees were constructed with MEGA v5.0 software using the Maximum-Likelihood method, based on the General Time Reversible model (GTR + G + I). For amino acid analyses 352 amino acid long sequence of NS1 region was analyzed using Neighbor-joining method with Poisson model. Number of bootstraps for simulations was 1000. Similarity plot analysis was performed with SimPlot v3.5.1 software using the Kimura 2 parameter model with a window size of 500 nt and a step size of 20 nt (Lole et al., 1999).
A 3438 nt long fragment of the genome (encompassing a 975 nt of the NS1 region and a 2013 nt segment of the VP1/VP2 genes) was obtained and used further in genetic analyses (Fig. 1 ). In addition to the NS1 and the VP1/VP2 regions, a smaller ORF, encoding the putative middle protein (PMP) was also identified with a length of 450 nucleotides. Phospholipase A2 motif of the VP1 region was identified, which is typically present in most of the parvoviruses, but absent in other viral families. A Walker loop motif (GXXXXGK T/S) was identified in the NS1 region as GPASTGKS in our sequences, same as described previously in human bufaviruses (Phan et al., 2012, Yahiro et al., 2014). The nucleotide sequence identities with the reference Chinese bat parvovirus strains fell between 77% and 81%. On the other hand, nucleotide sequence similarity with viruses in the Parvovirinae family ranged between 45% and 47%, while the amino acid sequence identity ranged between 35% and 42%. The nucleotide and amino acid identity between bat bufavirus (BtBV) strains and those detected from human patient samples are shown in Table 1 .
Fig. 1.
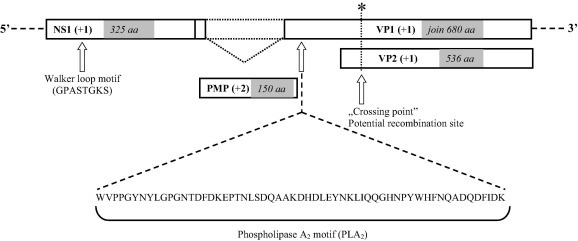
Genome organization of novel bat bufaviruses. A Walker loop motif (GPASTGKS) in the NS1 region and the phospholipase A2 (PLA2) motif of the VP1 region were identified. A potential intragenic recombination site of the VP region is shown.
Table 1.
Nucleotide and amino acid identities between bat bufavirus (BtBV) strains and those detected from human patient samples.
|
KR078343 (BtBV/V3/HUN/2013) |
KR078344 (BtBV/V7/HUN/2013) |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Total |
NS1 |
VP1 |
Total |
NS1 |
VP1 |
|||||||
| nt | aa | nt | aa | nt | aa | nt | aa | nt | aa | nt | aa | |
| KC154060 | 77 | 61 | 80 | 66 | 76 | 59 | 77 | 61 | 80 | 66 | 76 | 58 |
| KC154061 | 81 | 67 | 84 | 71 | 81 | 68 | 81 | 67 | 84 | 71 | 81 | 68 |
| JX027295 | 65 | 44 | 63 | 44 | 68 | 49 | 65 | 44 | 63 | 44 | 68 | 49 |
| JX027296 | 64 | 44 | 63 | 44 | 68 | 49 | 65 | 44 | 63 | 44 | 68 | 49 |
| JX027297 | 63 | 43 | 62 | 42 | 67 | 47 | 63 | 44 | 62 | 42 | 67 | 48 |
| KM580348 | 63 | 43 | 63 | 43 | 66 | 47 | 63 | 44 | 63 | 43 | 66 | 47 |
| JX627576 | 64 | 43 | 64 | 43 | 67 | 47 | 64 | 44 | 64 | 43 | 67 | 48 |
Based on the phylogenetic analyses (Fig. 2 A), the novel BtBVs (KR078343-“BtBV/V3/HUN/2013” and KR078344-“BtBV/V7/HUN/2013”) were closely related to human bufaviruses belonging to the Protoparvovirus genus and Primate protoparvovirus 1 species. Hungarian BtBV sequences were highly similar to those viruses detected previously from bats in China (unpublished data, GenBank accession numbers: KC154061 and KC154060), which therefore are also related to bufavirus sequences. Although the Hungarian viruses were closely related to bufaviruses, they formed a separate monophyletic branch (Fig. 2B and C). Phylogeny revealed that bat-related bufaviruses might have had common unknown ancestors with bufaviruses identified from humans so far. In a recent publication, a novel parvovirus (WUHARV) was described in nonhuman primates, which were most closely related to bufavirus 2 genotype (Handley et al., 2012). Based on recombination analyses (Fig. 3 ), an intragenic recombination event was detected in the viral protein segment, which assumes that this nonhuman primate bufavirus might be a result of past recombination event between bat and human bufaviruses.
Fig. 2.
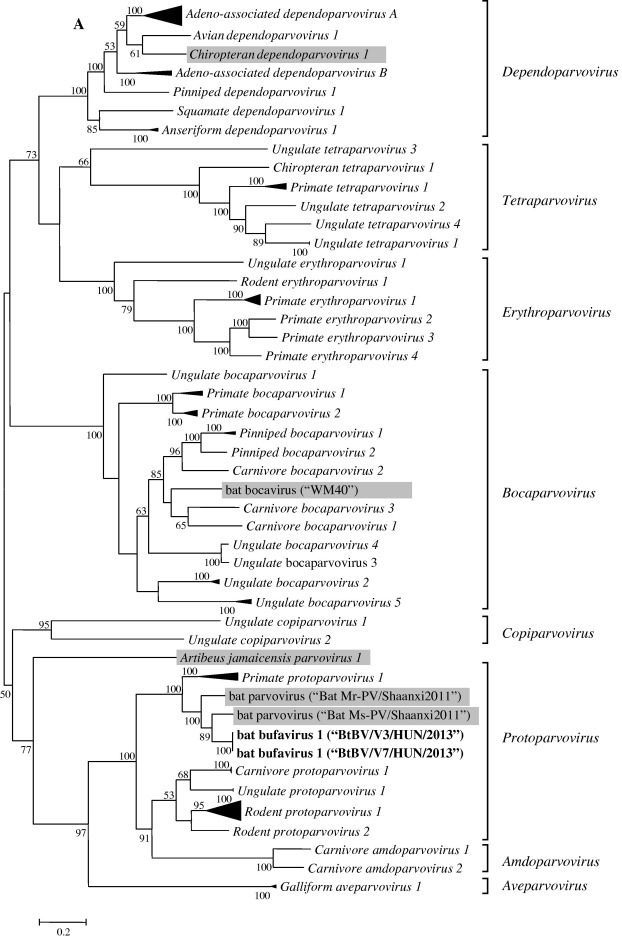
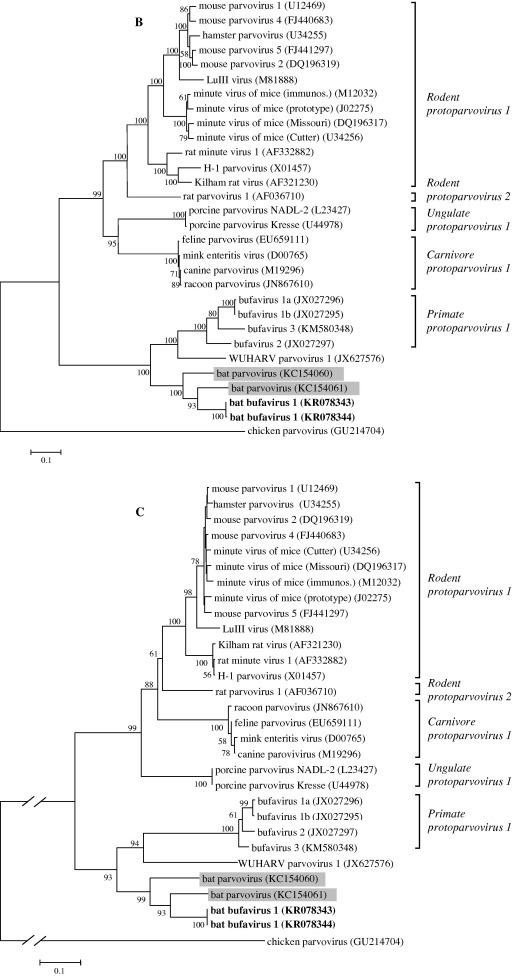
Nucleotide (A and B) and amino acid (C) based phylogenetic analyses of the Parvoviridae family (A) and Protoparvovirus genus (B and C) along with the novel BtBV strains detected in Hungary. Phylogenetic trees were constructed with MEGA v5.0 software using Maximum-Likelihood method with the General Time Reversible model (GTR + G + I) based on nucleic acid sequences of a 3394 bp coding region of the genome. For amino acid analyses, 352 amino acid long sequence of NS1 region was analyzed using Neighbor-joining method with Poisson model. Number of bootstraps for simulations was 1000. Bat-derived parvoviruses detected previously by others are indicated in gray. Hungarian BtBV strains identified in this study are marked in bold face.
Fig. 3.
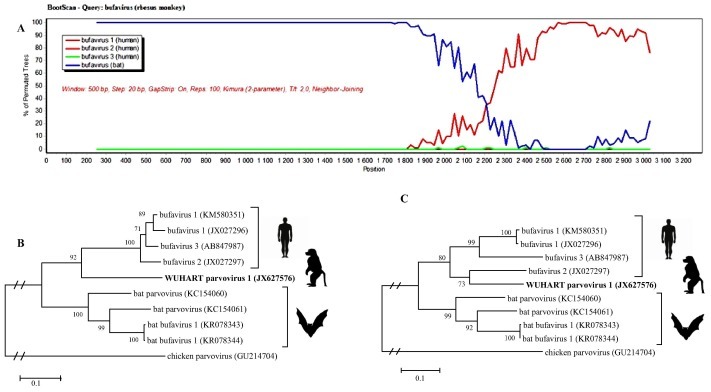
Recombination analyses within bufaviruses. A Simplot (A) analysis revealed significant recombination between the existing bat (blue line) and human (red line) bufavirus strains. Phylogenetic analyses performed with sequence data before (B) and after (C) the hypothetic crossing-point verified the possible recombination event.
Viral metagenomic analyses of M. schreibersii bats provide the first sequence data of bat parvoviruses from Europe with phylogenetic relatedness to the recently described human-infecting bufaviruses. The discovery of this novel BtBV along with the detailed molecular and recombination analyses expands our knowledge on the evolutionary history of bufaviruses and widens the list of human infecting viruses with possible bat origin. The shared evolutionary history between primate and bat bufaviruses seems to be interesting; however, the zoonotic transmission potential and the possible health risk posed by the newly described virus remains to be determined. High mutation rate and ability to recombination may facilitate fast evolution of bufaviruses, which might be quaint enough for investigate with further molecular characterization of both human and animal related bufavirus genomes.
Acknowledgements
Research activity of Ferenc Jakab was supported by the TÁMOP 4.2.4. A/2-11-1-2012 0001 – National Excellence Program Elaborating and operating an inland student and researcher personal support system. The project was subsidized by the European Union and co-financed by the European Social Fund. Krisztián Bányai was supported by the “Momentum program”. This study was approved by The National Inspectorate for Environment, Nature and Water (No. #14/2138-7/2011). The present scientific contribution is dedicated to the 650th anniversary of the foundation of the University of Pécs, Hungary.
References
- Brown K.E. The expanding range of parvoviruses which infect humans. Rev. Med. Virol. 2010;20:231–244. doi: 10.1002/rmv.648. [DOI] [PubMed] [Google Scholar]
- Cotmore S.F., Agbandje-McKenna M., Chiorini J.A., Mukha D.V., Pintel D.J., Qiu J., Soderlund-Venermo M., Tattersall P., Tijssen P. The family Parvoviridae. Arch. Virol. 2014;159:1239–1247. doi: 10.1007/s00705-013-1914-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decaro N., Desario C., Parisi A., Martella V., Lorusso A., Miccolupo A., Mari V., Colaianni M.L., Cavalli A., Di Trani L., Buonavoglia C. Genetic analysis of canine parvovirus type 2c. Virology. 2009;385:5–10. doi: 10.1016/j.virol.2008.12.016. [DOI] [PubMed] [Google Scholar]
- Drexler J.F., Geipel A., König A., Corman V.M., van Riel D., Leijten L.M., Bremer C.M., Rasche A., Cottontail V.M. Bats carry pathogenic hepadnaviruses antigenically related to hepatitis B virus and capable of infecting human hepatocytes. Proc. Natl. Acad. Sci. 2013;110:16151–16156. doi: 10.1073/pnas.1308049110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handley S.A., Thackray L.B., Zhao G., Presti R., Miller A.D., Droit L., Abbink P., Maxfield L.F., Kambal A. Pathogenic simian immunodeficiency virus infection is associated with expansion of the enteric virome. Cell. 2012;151:253–266. doi: 10.1016/j.cell.2012.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemenesi G., Zhang D., Marton S., Dallos B., Görföl T., Estók P., Boldogh S., Kurucz K., Oldal M. Genetic characterization of a novel picornavirus detected in Miniopterus schreibersii bats. J. Gen. Virol. 2015;96:815–821. doi: 10.1099/jgv.0.000028. [DOI] [PubMed] [Google Scholar]
- Kohl C., Kurth A. European bats as carriers of viruses with zoonotic potential. Viruses. 2014;6:3110–3128. doi: 10.3390/v6083110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lole K.S., Bollinger R.C., Paranjape R.S., Gadkari D., Kulkarni S.S., Novak N.G., Ingersoll R., Sheppard H., Ray S.C. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J. Virol. 1999;73:152–160. doi: 10.1128/jvi.73.1.152-160.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihalov-Kovács E., Fehér E., Martella V., Bányai K., Farkas S.L. The fecal virome of domesticated animals. Virusdisease. 2014;25:150–157. doi: 10.1007/s13337-014-0192-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan T.G., Vo N.P., Bonkoungou I.J., Kapoor A., Barro N., O’Ryan M., Kapusinszky B., Wang C., Delwart E. Acute diarrhea in West African children: diverse enteric viruses and a novel parvovirus genus. J. Virol. 2012;86:11024–11030. doi: 10.1128/JVI.01427-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quan P.L., Firth C., Conte J.M., Williams S.H., Zambrana-Torrelio C.M., Ellison J.A., Gilbert A.T., Kuzmin I.V. Bats are a major natural reservoir for hepaciviruses and pegiviruses. Proc. Natl. Acad. Sci. 2013;110:8194–8199. doi: 10.1073/pnas.1303037110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shackelton L.A., Hoelzer K., Parrish C.R., Holmes E.C. Comparative analysis reveals frequent recombination in the parvoviruses. J. Gen. Virol. 2007;88:3294–3301. doi: 10.1099/vir.0.83255-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smits S.L., Schapendonk C.M., van Beek J., Vennema H., Schürch A.C., Schipper D., Bodewes R., Haagmans B.L., Osterhaus A.D. New viruses in idiopathic human diarrhea cases, the Netherlands. Emerg. Infect. Dis. 2014;20:1218–1222. doi: 10.3201/eid2007.140190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teeling E.C., Springer M.S., Madsen O., Bates P., O’brien S.J., Murphy W.J. A molecular phylogeny for bats illuminates biogeography and the fossil record. Science. 2005;307:580–584. doi: 10.1126/science.1105113. [DOI] [PubMed] [Google Scholar]
- Tong S., Zhu X., Li Y., Shi M., Zhang J., Bourgeois M., Yang H., Chen X., Recuenco S. New world bats harbor diverse influenza A viruses. PLoS Pathog. 2013;9:e1003657. doi: 10.1371/journal.ppat.1003657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Väisänen E., Kuisma I., Phan T.G., Delwart E., Lappalainen M., Tarkka E., Hedman K., Söderlund-Venermo M. Bufavirus in feces of patients with gastroenteritis, Finland. Emerg. Infect. Dis. 2014;20:1077–1080. doi: 10.3201/eid2006.131674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yahiro T., Wangchuk S., Tshering K., Bandhari P., Zangmo S., Dorji T., Tshering K., Matsumoto T., Nishizono A. Novel human bufavirus genotype 3 in children with severe diarrhea, Bhutan. Emerg. Infect. Dis. 2014;20:1037–1039. doi: 10.3201/eid2006.131430. [DOI] [PMC free article] [PubMed] [Google Scholar]