

Abstract
Objective:
To report a novel mutation in GHR and to characterize a novel mechanism of nonclassical growth hormone insensitivity.
Context:
Laron syndrome (LS) is a well-described disorder of growth hormone insensitivity due to mutations in the growth hormone receptor (GHR) that leads to short stature. Biochemically, LS patients classically have elevated levels of growth hormone (GH), but low levels of insulin-like growth factor (IGF)-1, IGF binding protein (IGFBP)-3 and GH binding protein (GHBP).
Design:
Case presentation with in vitro functional studies.
Patients:
A young male Caucasian child with short stature was found to have growth hormone insensitivity manifested by elevated levels of GH and GHBP.
Measurements:
Growth hormone stimulation tests revealed baseline GH level of 20.9 μg/L and maximum stimulated GH level of 52.7 μg/L and GHBP level of 4868 pmol/L. GHR gene sequencing revealed a novel heterozygous nonsense mutation (c.800G > A, p.Trp267*) in the transmembrane domain of the receptor. Immunoblot analysis of transfected GHR p.Trp267* in HEK293 revealed inhibition of GH-induced STAT5 signalling that was overcome with increasing doses of recombinant human GH.
Results:
Using an in vitro model, we show that elevated levels of GHBP inhibit the action of GH. Furthermore, our studies demonstrate that this inhibition by GHBP can be overcome by increasing doses of recombinant human GH.
Conclusions:
To our knowledge, this is the first study to demonstrate in vitro that elevated levels of GHBP attenuate the effect of GH and inhibit GH-induced signalling, thereby leading to short stature. Though this inhibition was overcome in vitro with supraphysiologic doses of GH, significantly above endogenously available GH, it remains to be seen whether such an effect can be replicated in vivo.
Keywords: GHR, growth hormone, growth hormone insensitivity, growth hormone receptor, hormone replacement, paediatric endocrinology, pituitary, short stature
1 |. INTRODUCTION
Laron syndrome (LS) is a rare genetic disorder resulting from bi-allelic (and occasionally mono-allelic) mutation(s) in GHR, the gene encoding the growth hormone receptor.1,2 Loss of growth hormone receptor (GHR) function results in primary growth hormone insensitivity (GHI) resulting in typical clinical characteristics including short stature, atypical facies, obesity and delayed bone age.1,3 Biochemically, LS classically presents with low levels of circulating insulin-like growth factor (IGF)-1, IGF binding protein (IGFBP)-3, GH binding protein (GHBP) and acid-labile subunit (ALS) while circulating GH levels remain normal or even elevated.4–6 While cases of classic LS are due to bi-allelic mutations in the extracellular domain of GHR leading to low levels of GHBP, rare heterozygous mutations of GHR can lead to a dominant-negative effect resulting in a milder GHI syndrome.6–10
Growth hormone (GH) exerts its action via GHR which is encoded on chromosome 5p13.1–12 and consists of 9 introns and 10 exons. Exons 2–7 encode the extracellular portion of GHR, exon 8 is responsible for the transmembrane portion of GHR that anchors the receptor, and exons 9 and 10 encode the intracellular regions of GHR.1,7 GHR is a homodimeric cytokine receptor that lacks intrinsic kinase activity and relies on Janus kinase 2 (JAK2) to induce signal transducer and activator of transcription (STAT)-5b pathway.11 GHBP is the soluble extracellular domain of GHR that results from proteolytic cleavage of the extracellular ligand-binding domain of GHR and thereby binds GH while in circulation.6,12 Physiologic GHBP regulation is complex and remains to be fully understood although GHBP levels have been shown to be affected by age, body mass index, insulin and leptin concentrations, as well as by pathologic states such as insulin dependent diabetes mellitus, hypothyroidism, chronic kidney disease and malnutrition.12 To date, approximately 100 mutations have been described in human GHR leading to short stature.13 GHR mutations that result from aberrant splicing of exons 8 and 9 that encode the transmembrane and intracellular domains of GHR, respectively, have been implicated in previously reported cases of short stature with elevated GHBP.8,11,14,15
Here, we describe a male Caucasian child referred for severe postnatal growth failure who was found to have a novel heterozygous mutation in GHR resulting in elevated levels of GHBP. We describe the first nonsense mutation in the transmembrane domain (exon 8) and, in an in vitro system, demonstrate that the mutation results in increased release of the extracellular portion of GHR (ie GHBP) into the extracellular space. We show that the released GHBP acts as an inhibitor of growth hormone action, and, further, that this inhibition may be dampened with increased dosing of supplementary GH. Our results thus provide evidence for a new mechanism of dominant-negative effects of GHR defects. This may have important potential therapeutic ramifications for patients with this class of mutation.
2 |. PATIENT AND METHODS
2.1 |. Patient
A 23-month-old Caucasian male was referred to the paediatric endocrine clinic for severe short stature. He was born at 37 weeks gestation with a reported birthweight of 2200 g (−2.7 SDS) and birth length of 43.2 cm (−3.5 SDS). Mother’s pregnancy and postnatal history were unremarkable. The patient’s past medical and surgical histories were otherwise unremarkable. There was no reported significant history of short stature in the extended family. Mother’s adult height was 152 cm (−1.7 SDS) and father’s adult height was 185 cm (1.3 SDS) with a calculated mid-parental target height of 175 cm (−0.16 SDS). Consanguinity was denied. Developmental milestones were normal, and he was reported to be an active and otherwise healthy toddler. On presentation at 23 months, his weight was 10.6 kg (−1.0 SDS), and his height was 75.8 cm (−3.6 SDS). On physical examination, he was noted to have a mildly prominent forehead, but the rest of the examination was otherwise normal. His extremities were proportional. Screening laboratory evaluation revealed a slightly low plasma glucose of 3.33 mmol/L and an otherwise normal comprehensive metabolic panel, normal thyroid function tests (TSH 3.63 mIU/L, free T4 12.9 pmol/L), coeliac panel (tissue transglutaminase IgA antibody undetectable, IgA 0.44 g/L) and urinalysis. Skeletal survey was normal. IGF-1 measured by chemi-luminescence immunoassay was 65 μg/L (normal 16–134 μg/L), and IGFBP3 measured by immunoassay was 2.7 mg/L (normal 0.7–3.6 mg/L). Dual-agent GH stimulation test with clonidine and arginine revealed a baseline value of 20.9 μg/L with a supraphysiological response of 32.5 μg/L at 60 minutes and 52.7 μg/L at 120 minutes. The patient’s GHBP was elevated at 4868 pmol/L (normal 267–1638 pmol/L). The patient’s mother’s IGF-1 level was 332.9 μg/L (normal 75–265 μg/L), and her GHBP was elevated at 5977 pmol/L (normal 440–4260 pmol/L). No paternal or additional family evaluation was available. GHR gene sequencing subsequently revealed a heterozygous variant, c.800G > A, p.Trp267* (PreventionGenetics, Wisconsin). Deletion/Duplication analysis of the GHR gene was negative (PreventionGenetics). Table 1 summarizes key clinical and biochemical features.
TABLE 1.
Summary of clinical and biochemical status at presentation
| Age | 23 mo |
| Sex | Male |
| Ethnicity | Caucasian |
| Weight, kg (Z-score, CDC) | 10.6 (−1.6) |
| Height, cm (Z-score, CDC) | 75.8 (−3.1) |
| Ethnicity | Caucasian |
| Gestational age at birth | 37 wks |
| Birthweight, kg (Z-score, Fenton) | 2.2 (−2.7) |
| Birth length, cm (Z-score, Fenton) | 43.2 (−3.5) |
| Mid-parental height, cm (Z-score) | 175 (−0.3) |
| Biochemistry* | |
| IGF-1, μg/L (RR) | 65 (16–134) |
| IGFBP-3, mg/L (RR) | 2.7 (0.7–3.6) |
| GHBP, pmol/L (RR) | 4868 (267–1638) |
| GH basal, μg/L | 20.9 |
| GH stimulated, 1-h, μg/L | 32.5 |
| GH stimulated, 2-h, μg/L | 52.7 |
| Genetics evaluation | |
| Mutation in GHR | Heterozygous, Nonsense, c.800G > A, p.Trp267* |
See text for additional results.
At a follow-up visit at 3 years of age, his growth velocity was 6.9 cm/y (Figure 1). The patient was started on recombinant human IGF-1 (rhIGF-1) therapy at a dose of 0.04 mg/kg twice daily at the age of 3 years and 7 months, and titrated to a maximum of 0.05 mg/kg twice daily. The patient stopped therapy after 6 months. His annualized growth velocity on rhIGF-1, however, did not change likely due to suboptimal rhIGF-1 dosing (Figure 1).
FIGURE 1.
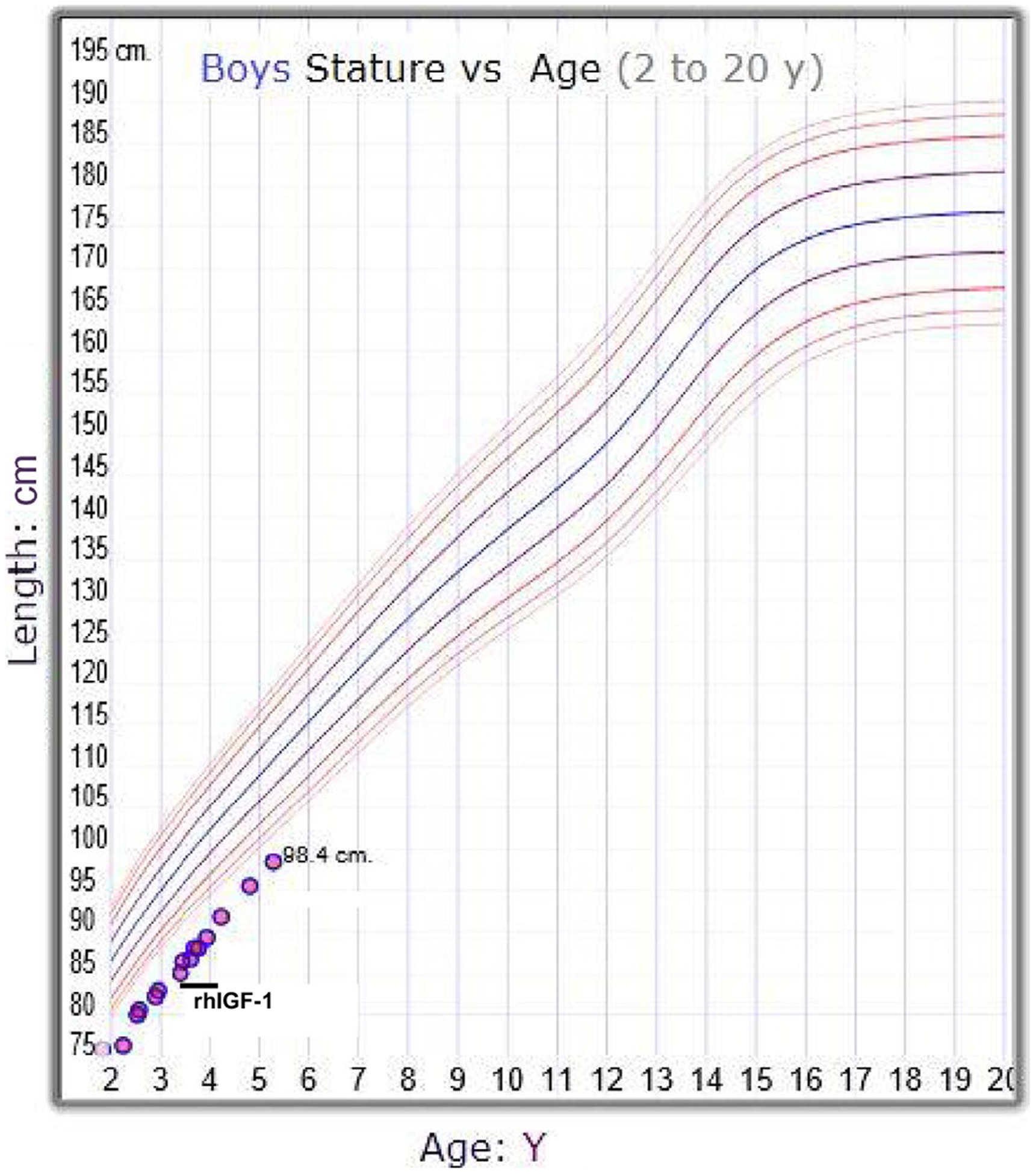
The CDC growth chart (male) of the patient carrying the heterozygous GHR mutation. The time frame of rhIGF-1 treatment is indicated on the growth curve. CDC, Centers for Disease Control and Prevention
2.2 |. Sample procurement and consenting
Informed consent was obtained from the family (patient P1; mother) to participate and provide samples (DNA, whole blood), in compliance with the Institutional Review Board at Cincinnati Children’s Hospital Medical Center (CCHMC). Genomic DNA was isolated from peripheral blood of the patient and the mother as previously described.16
Targeted Sanger sequencing of the GHR coding exons (NM_000163.4), exons 2–10, was performed as previously described.16
2.3 |. Regenerating the GHR variants for functional studies
The plasmid pcDNA carrying the full-length human GHR (kind gift from Dr Richard Ross) was used as template to re-generate the variant c.800G > A. The variant was generated using the QuikChange II XL Site-Directed Mutagenesis Kit from Agilent technologies, following the manufacturer’s protocol. The primers for the site-directed mutagenesis were designed based on modified human GHR cDNA sequences, where 22 nucleotides within exon 8 were substituted without altering the encoded amino acid residues17 to permit stable maintenance of human GHR cDNA in Escherichia coli. Forward and corresponding reverse primers, c.800G > A nucleotide change is in bold: 5′-TACTTTCCATAGTTACTCATCATC. Underlined nucleotides are substituted according to Wang and Woods.17
2.4 |. Cell culture and transfection
HEK293 cells were maintained in Dulbecco’s Modified Eagle medium (DMEM) supplemented with 10% foetal bovine serum (FBS) at 37°C in 5% CO2. For in vitro functional analyses, HEK293 cells were transfected with the vector (pcDNA3.1), wild-type (WT), the mutant GHR variant or co-transfected with both variants in a 1:1 ratio, using Polyjet transfection reagent (SignaGen Laboratories). After 24 hours of transfection, the cells were serum starved in serum-free media (SFM, DMEM supplemented with 0.1% bovine serum albumin, BSA) for 48 hours then treated with rhGH (NutropinAQ, Genetech) for 20 minutes and conditioned media (supernatant) collected for analysis. In some experiments, fresh SFM were added after serum starvation prior to rhGH treatment at time points indicated. All transfection experiments were performed at least three independent times.
2.5 |. Immunoblot analysis
Transfected cells were treated with rhGH for 20 minutes as indicated; cell lysates were prepared and processed as previously described.18 Protein concentrations of the lysates or cell culture supernatants were determined by the Bradford assay, and equal quantities of protein were loaded onto a 10% sodium dodecyl sulphate-polyacrylamide gel electrophoresis gel. After electrophoresis, the proteins were transferred onto nitrocellulose membranes, blocked using 5% BSA or 5% nonfat milk in 1× tris (hydroxymethyl) aminomethane-buffered saline with Tween 20 (TBST) and incubated with the appropriate primary antibodies. Following incubation, the blots probed with the horseradish peroxidase–conjugated secondary antibodies were washed in 1× TBST and developed using the super signal maximum sensitivity detection system (Thermo Fisher Scientific; catalog no. 37075). Primary antibodies employed were: anti-GHBP (BioVision Incorporated; 6660–30T; 1:1000 dilution), anti-GHR (Santa Cruz Technology, sc-137185; 1:500 dilution), anti-STAT5B (Boster Biological Technology; PA1841; 1:2000 dilution) and anti-pY-STAT5 (Cell Signaling Technology; 9351; 1:1000 dilution).
3 |. RESULTS
3.1 |. A novel nonsense mutation in exon 8 of the GHR gene is identified
Gene sequencing of the patient’s GHR was performed and confirmed that the proband carried a novel heterozygous nonsense mutation (NM_000163.4, c.800G > A, p.Trp267*; Figure 2). This variant localizes to exon 8 of GHR which encodes the transmembrane domain of GHR (residues 264–288) and the c.800G > A leads to a nonsense conversion of tryptophan at position 267. This leads to a predicted premature truncation of the protein with a preserved extracellular domain. No other variant was identified. This variant was inherited from the patient’s mother (Figure 2).
FIGURE 2.
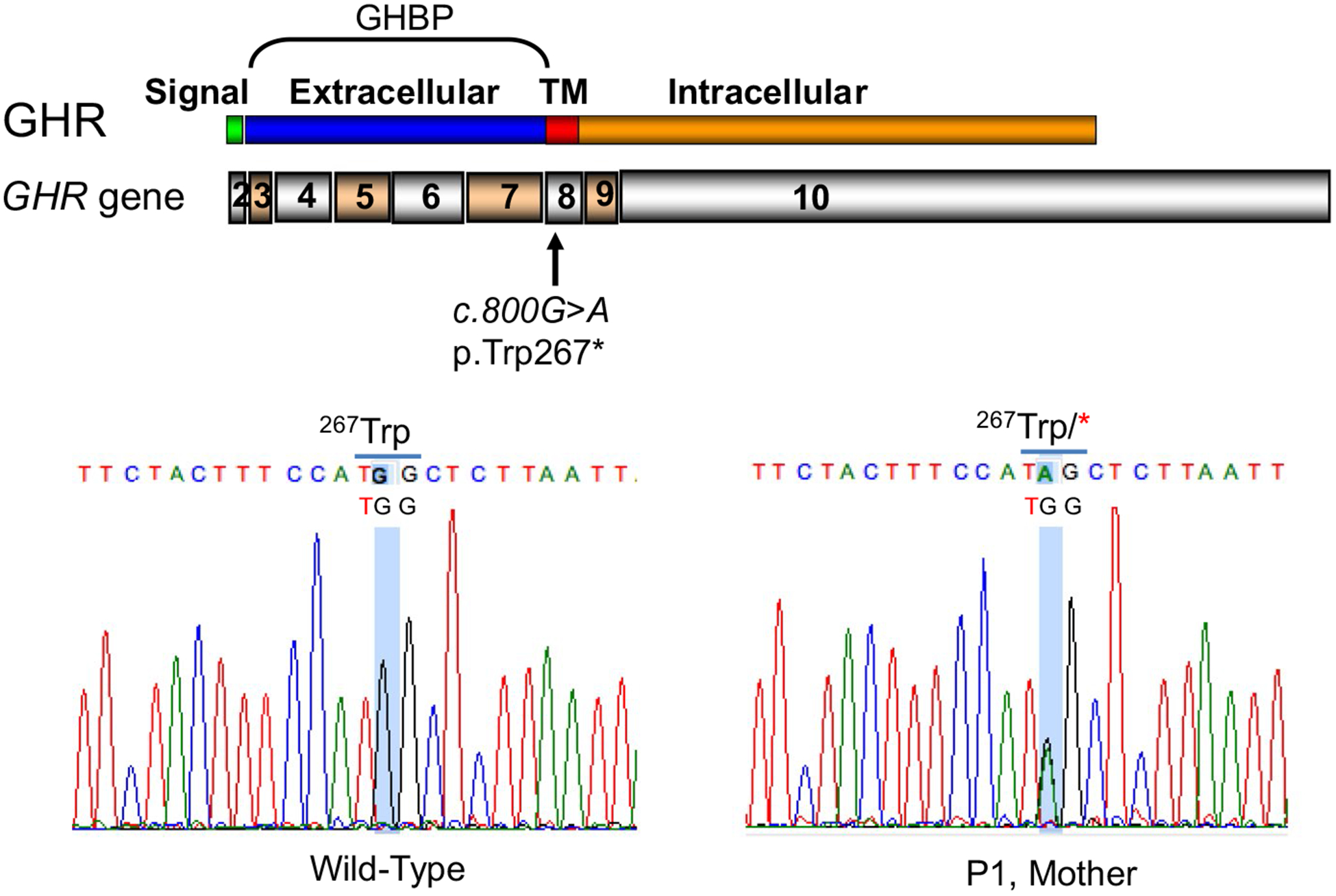
Identification of heterozygous GHR c.800G > A (p.Trp267*) in exon 8. Schematic of the GHR gene, exons 2–10, encoding for GHR domains as indicated. TM, transmembrane. Electropherograms indicating heterozygous ic.800G > A identified in the patient and his mother
3.2 |. Secreted GHR p.Trp267* inhibits GH-induced STAT5 signalling
An expressed truncated GHR c.800G > A, p.Trp267* is predicted to be unable to anchor to the cell surface. To test this hypothesis, thereby providing an explanation for the elevated GHBP in serum samples from both the patient and the mother, the variant was regenerated and evaluated in HEK293 reconstitution system. It is of note that the human GHR cDNA template used for regeneration carries 22 nucleotide substitutions within exon 8 without altering the amino acid residue sequences.17 Unlike other mammalian growth hormone receptor cDNAs (eg rodent, rabbit and porcine), these substitutions were necessary for stable maintenance of human GHR cDNA in E coli for molecular manipulation.17
In reconstituted HEK293 cells, transfection and analysis of regenerated GHR p.Trp267*, and wild-type GHR (schema shown in Figure 3A) demonstrated that both the GHR-WT and the truncated GHR p.Trp267* were well expressed after 48 hours in SFM (serum-free media), with both GHR variants detectable in cell lysates. Only the GHR p.Trp267*, however, was robustly detected in media conditioned by transfected cells (CM, conditioned media; Figure 3B). These results supported the secretion of the truncated GHR variant into the extracellular milieu. Further, when co-expressed with GHR-WT, mimicking the in vivo heterozygous state of the patient, presence of the GHR p.Trp267* (GHBP) significantly blunted responsiveness to rhGH, based on tyrosine-phosphorylated STAT5 (pY-STAT5) as the read-out (Figure 3B). Replacing the 48 hours CM post-transfection with fresh SFM, suggested that it is the secreted GHR p.Trp267* which appears to be affecting GH-induced effects, since GH response was not blunted until the accumulation of secreted GHR p.Trp267* (Figure 3B).
FIGURE 3.
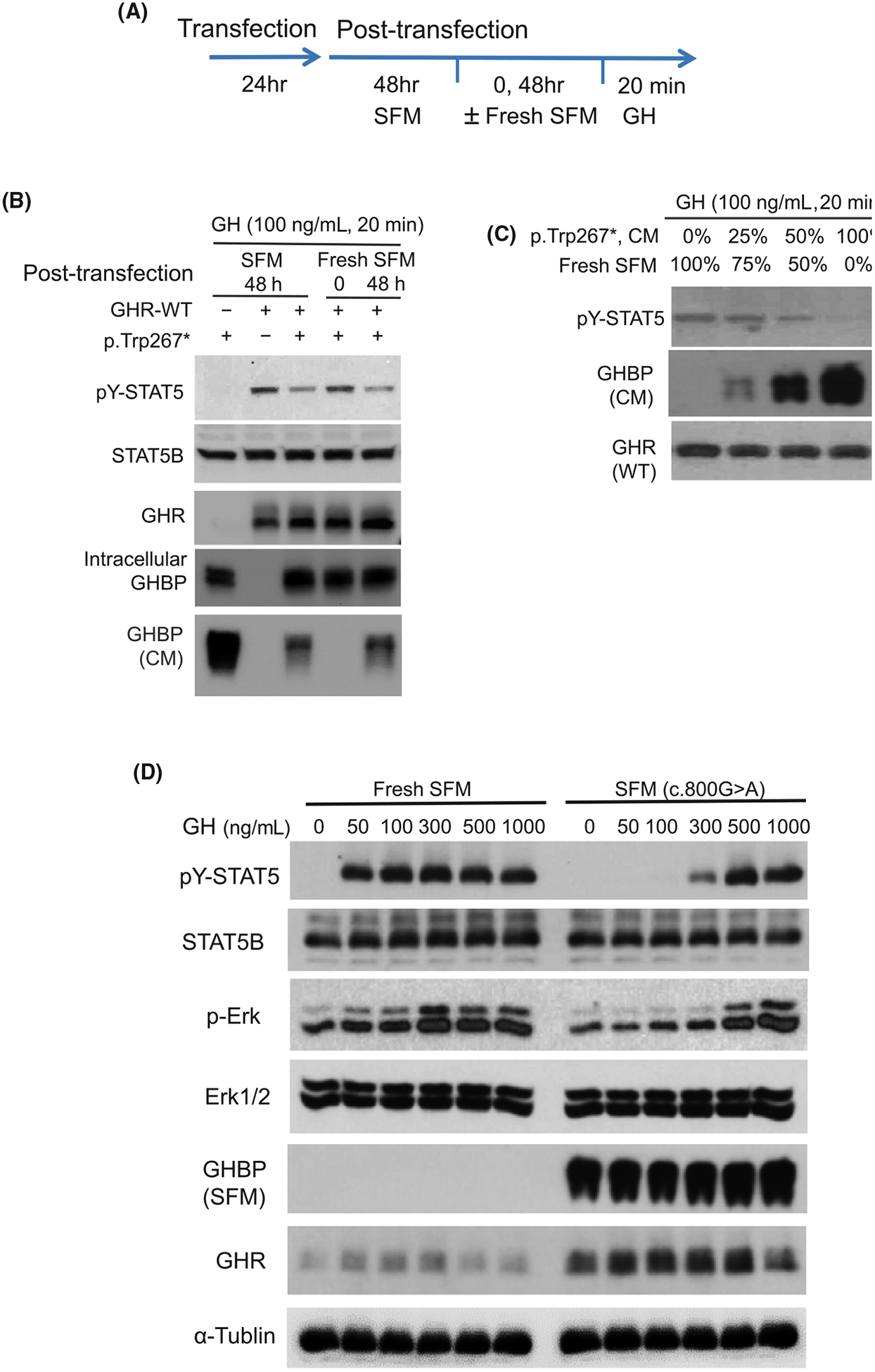
Transient transfected GHR p.Trp267* in HEK293 is secreted and inhibits GH-induced STAT5 activation. A, Schematic of transfection and post-transfection procedures, In some experiments, the 48 h post-transfection serum-free media (SFM) was removed and replaced with fresh SFM (indicated as ‘Fresh SFM’) for 0 or 48 h, prior to GH treatment (100 ng/mL). B, Immunoblot analysis of GH-treated transfected HEK293 cell lysates. GHBP in conditioned media (CM) were also analysed. Proteins immuno-detected are as indicated. C, Effect on GH-induced STAT5 signalling in the presence of increasing proportion of CM carrying GHR p.Trp267* mixed with Fresh SFM. D, Effects on GH-induced STAT5 signalling with increasing dose of GH
To further confirm that it is the secreted GHR p.Trp267* which was interfering with GH-induced pY-STAT5, HEK293 cells co-transfected with GHR-WT + GHR p.Trp267* were exposed to increasing proportion of 48-hour fresh SFM conditioned by GHR p.Trp267* (Figure 3C). A dose-dependent blunting of GH-induced signalling was observed with increasing proportion of GHR p.Trp267* CM, which coincided with increasingly higher levels of GHBP.
We next asked whether increasing rhGH dosing (50–1000 ng/mL) can overcome the inhibitory effects of excess secreted GHR p.Trp267*. GH doses as low as 50 ng/mL induced pY-STAT5 in cell lysates when 48-hour CM was replaced with fresh SFM lacking GHR p.Trp267* (Figure 3D). In the presence of GHR p.Trp267*, GH dosing of at least 300 ng/mL was necessary for detection of pY-STAT5 response (Figure 3D). Collectively, these results suggest that an appropriate dose of rhGH may be able to override the inhibitory effects of excess GHBP, though this effect may be dependent on the amount of excess GHBP.
4 |. DISCUSSION AND CONCLUSION
We present the first GHR transmembrane domain mutation, a novel heterozygous nonsense mutation, in a 23-month-old Caucasian boy who was referred for severe short stature. The c.800G > A variant in exon 8 of GHR generates a premature stop codon, p.Trp267*, within the transmembrane domain of GHR. The resulting truncated GHR peptide consists only of the extracellular domain and 4 residues of the 24 amino acid residues transmembrane domain. A GHR lacking the transmembrane and intracellular domains would be unable to form either dimeric GHR receptors or to anchor to the cell surface. The net effect is secretion of the extracellular domain of GHR, explaining the markedly elevated serum GHBP concentrations detected in both the patient and his mother. Importantly, we demonstrated in vitro that the accumulation of secreted GHR p.Trp267*, detected as GHBP, could inhibit GH-induced signalling, suggesting that abnormally high concentrations of circulating GHBP can act as a ‘sink’ for GH with negative impacts on GH actions. As previously reported, baseline endogenous levels of GH are elevated in individuals with growth hormone insensitivity,10 likely in response to GHR defects and attenuated downstream IGF1 signalling. In this case, the compensatory elevation of GH appears inadequate to overcome the dampening effects of GHBP. We, therefore, propose that the heterozygous GHR p.Trp267* mutation, despite the presence of one wild-type copy of the GHR gene, has a unique dominant-negative effect in which elevated levels of released GHBP inhibit the effect of GH-induced actions, leading to short stature.
Heterozygous GHR splicing mutations resulting in excision of exon 8 associated with supraphysiological levels of serum GHBP and short stature have been previously reported, including by our group.8,11,15,19 We had speculated that increasing concentrations of circulating GHBP could reduce the availability of GH for interactions with the cell membrane-anchored GHR8 and, indeed, our in vitro reconstitution studies with GHR p.Trp267* provided evidence that increasing GHBP in the conditioned media correlated with reduced GH-induced cell signalling. Further, we demonstrated that this inhibitory effect may be partially reversed by increasing the dosage of GH treatment. The striking ability of increased GH dosages to restore GH signalling, at least in vitro, suggests a potential novel therapeutic strategy for our patient and other patients with similar mechanisms of GH resistance. We, therefore, recommend measuring serum GHBP in patients with elevated GH levels on stimulation testing. As previously noted, GHBP levels may aid in identifying the aetiology of GHI.12 Furthermore, our results suggest that supraphysiologic level of rhGH may overcome certain cases of GH insensitivity.
It is noteworthy that the mother, from whom our patient inherited the GHR p.Trp267* variant, was not frankly short, although her height did fall in the lower end of the normal range. This is comparable to previous reports,8,11,15,19 and consistent with the clinical heterogeneity of patients with dominant-negative mutations in GHR.9 Both the patient and his mother, moreover, had normal or slightly elevated levels of IGF-1. Although the aetiology of the mother’s elevated IGF-1 is presently unknown, the implication is that circulating IGF-1 levels may not always accurately reflect GH action in vivo.
To date, there have only been seven dominant-negative, heterozygous GHR mutations described though there are likely many undiagnosed cases.9 Importantly, patients with dominant-negative mutations may present with milder phenotypes compared to patients with classical homozygous autosomal-recessive GHI, and therefore be characterized as having idiopathic short stature or may be missed during routine clinical work. A robust GH stimulation test in the background of normal IGF-1 can be an important clue of a nonclassical GHI phenotype, as it was for our present case.
Our patient only received low-dose rhIGF-1 therapy resulting in incrementally improved growth velocity from 6.9 cm/y prior to treatment to 7.8 cm/y during the treatment period. However, noncompliance was an issue. As previously noted, rhIGF-1 may only transiently increase growth velocity due to a negative feedback mechanism that inhibits GH secretion.9 In light of our in vitro studies, recombinant human GH (rhGH) therapy may be an option to overcome the inhibitory effect of elevated GHBP levels.
In summary, we report the first pathological GHR mutation in the transmembrane domain identified in a 23-month-old male patient with short stature. The heterozygous GHR p.Trp267* nonsense mutation exerted a negative effect through a novel dominant-negative mechanism, in which the excess truncated GHR p.Trp267* secreted into the extracellular milieu and detected as GHBP inhibited GH actions. The milder clinical presentation of our patient, furthermore, supports the notion that patients with dominant-negative GHR mutations could be missed during routine clinical workup. We recommend evaluation of serum GHBP in cases where the measured serum GH, IGF-1 and IGFBP-3 are uncharacteristically normal. Optimal therapy in cases of elevated GHBP associated with nonclassical GHI and short stature could include higher rhGH dosing, with or without rhIGF-1, as has been demonstrated for other dominant-negative GHR mutations.9
Acknowledgments
Vivian Hwa and Sowmya Krishnan co-senior authors. Research reported in this publication was supported by the National Institutes of Health under Award Numbers NIH NICHHC R01HD078592 and R21HD098417 to Vivian Hwa, and UG1OD024950 and U54GM104938 to Sowmya Krishnan. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
REFERENCES
- 1.Backeljauw PF, Chernausek SD. The insulin-like growth factors and growth disorders of childhood. Endocrinol Metab Clin North Am. 2012;41(2):265–282, v. [DOI] [PubMed] [Google Scholar]
- 2.Sanchez JE, Perera E, Baumbach L, Cleveland WW. Growth hormone receptor mutations in children with idiopathic short stature. J Clin Endocrinol Metab. 1998;83(11):4079–4083. [DOI] [PubMed] [Google Scholar]
- 3.Laron Z, Kauli R. Fifty seven years of follow-up of the Israeli cohort of Laron Syndrome patients-From discovery to treatment. Growth Horm IGF Res. 2016;28:53–56. [DOI] [PubMed] [Google Scholar]
- 4.Laron Z. Insulin-like growth factor-I treatment of children with Laron syndrome (primary growth hormone insensitivity). Pediatr Endocrinol Rev. 2008;5(3):766–771. [PubMed] [Google Scholar]
- 5.Savage MO, Attie KM, David A, Metherell LA, Clark AJ, Camacho-Hubner C. Endocrine assessment, molecular characterization and treatment of growth hormone insensitivity disorders. Nat Clin Pract Endocrinol Metab. 2006;2(7):395–407. [DOI] [PubMed] [Google Scholar]
- 6.David A, Metherell LA, Clark AJ, Camacho-Hubner C, Savage MO. Diagnostic and therapeutic advances in growth hormone insensitivity. Endocrinol Metab Clin North Am. 2005;34(3):581–595, viii. [DOI] [PubMed] [Google Scholar]
- 7.Ross RJ. The GH receptor and GH insensitivity. Growth Horm IGF Res. 1999;9:42–46; discussion 45–46. [DOI] [PubMed] [Google Scholar]
- 8.Aalbers AM, Chin D, Pratt KL, et al. Extreme elevation of serum growth hormone-binding protein concentrations resulting from a novel heterozygous splice site mutation of the growth hormone receptor gene. Horm Res. 2009;71(5):276–284. [DOI] [PubMed] [Google Scholar]
- 9.Vairamani K, Merjaneh L, Casano-Sancho P, et al. Novel dominant-negative GH receptor mutations expands the spectrum of GHI and IGF-I deficiency. J Endocr Soc. 2017;1(4):345–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Storr HL, Chatterjee S, Metherell LA, et al. Nonclassical GH insensitivity: characterization of mild abnormalities of GH action. Endocr Rev. 2019;40(2):476–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Iida K, Takahashi Y, Kaji H, et al. Growth hormone (GH) insensitivity syndrome with high serum GH-binding protein levels caused by a heterozygous splice site mutation of the GH receptor gene producing a lack of intracellular domain. J Clin Endocrinol Metab. 1998;83(2):531–537. [DOI] [PubMed] [Google Scholar]
- 12.Schilbach K, Bidlingmaier M. Growth hormone binding protein – Physiological and analytical aspects. Best Pract Res Clin Endocrinol Metab. 2015;29(5):671–683. [DOI] [PubMed] [Google Scholar]
- 13.Lin S, Li C, Li C, Zhang X. Growth hormone receptor mutations related to individual dwarfism. Int J Mol Sci. 2018;19(5):1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klammt J, Shen S, Kiess W, et al. Clinical and biochemical consequences of an intragenic growth hormone receptor (GHR) deletion in a large Chinese pedigree. Clin Endocrinol. 2015;82(3):453–461. [DOI] [PubMed] [Google Scholar]
- 15.Woods KA, Fraser NC, Postel-Vinay MC, Savage MO, Clark AJ. A homozygous splice site mutation affecting the intracellular domain of the growth hormone (GH) receptor resulting in Laron syndrome with elevated GH-binding protein. J Clin Endocrinol Metab. 1996;81(5):1686–1690. [DOI] [PubMed] [Google Scholar]
- 16.Aisenberg J, Auyeung V, Pedro HF, et al. Atypical GH insensitivity syndrome and severe insulin-like growth factor-I deficiency resulting from compound heterozygous mutations of the GH receptor, including a novel frameshift mutation affecting the intracellular domain. Horm Res Paediatr. 2010;74(6):406–411. [DOI] [PubMed] [Google Scholar]
- 17.Wang YD, Wood WI. Amino acids of the human growth hormone receptor that are required for proliferation and Jak-STAT signaling. Mol Endocrinol. 1995;9(3):303–311. [DOI] [PubMed] [Google Scholar]
- 18.Derr MA, Aisenberg J, Fang P, Tenenbaum-Rakover Y, Rosenfeld RG, Hwa V. The growth hormone receptor (GHR) c.899dupC mutation functions as a dominant negative: insights into the pathophysiology of intracellular GHR defects. J Clin Endocrinol Metab. 2011;96(11):E1896–E1904. [DOI] [PubMed] [Google Scholar]
- 19.Silbergeld A, Dastot F, Klinger B, et al. Intronic mutation in the growth hormone (GH) receptor gene from a girl with Laron syndrome and extremely high serum GH binding protein: extended phenotypic study in a very large pedigree. J Pediatr Endocrinol Metab. 1997;10(3):265–274. [DOI] [PubMed] [Google Scholar]