

Abstract
A multiplex RT-nested PCR has been developed to detect and differentiate the closely related prawn viruses, gill-associated virus (GAV) from Australia and yellow head virus (YHV) from Thailand. RT-PCR using primers to conserved sequences in the ORF1b gene amplified a 794 bp region of either GAV or YHV. Nested PCR using a conserved sense primer and either a GAV- or YHV-specific antisense primer to a divergent sequence differentially amplified a 277 bp region of the primary PCR amplicon. Multiplexing the YHV antisense primer with a GAV antisense primer to another divergent sequence allowed the viruses to be distinguished in a single nested PCR. Nested PCR enhanced detection sensitivity between 100- and 1000-fold and GAV or YHV RNA was detectable in ∼10 fg lymphoid organ total RNA. The multiplex RT-nested PCR was also able to co-detect GAV and YHV RNA mixed over a wide range of concentrations to simulate potential dual-infection states. The robustness of the test was examined using RNA samples from Penaeus monodon prawns infected either chronically or acutely with GAV or YHV and collected at different locations in Eastern Australia and Thailand between 1994 and 1998. GAV- (406 bp) or YHV-specific (277 bp) amplicons were differentially generated in all cases, including five YHV RNA samples in which no primary RT-PCR amplicon was detected. Sequence analysis of GAV and YHV PCR amplicons identified minor variations in the regions targeted by the virus-specific antisense primers. However, none occurred at positions that critically affected the PCR.
Keywords: Yellow head virus, Gill-associated virus, Multiplex RT-PCR, Penaeus monodon, Penaeid shrimp, Prawn
1. Introduction
Yellow head virus (YHV) from Thailand and gill-associated virus (GAV) from Australia have recently been classified in new taxa (genus Okavirus, family Roniviridae) within the Nidovirales (Cowley et al., 2000b, Cowley and Walker, 2002, Sittidilokratna et al., 2002, Mayo, 2002). The rod-shaped, enveloped virions of YHV and GAV are morphologically indistinguishable and each virus can be highly pathogenic for the black tiger prawn (Penaeus monodon) (Boonyaratpalin et al., 1993, Chantanachookin et al., 1993, Lu et al., 1995, Spann et al., 1997). They also cause similar histopathology in diseased prawns and in situ hybridisation (ISH) and transmission electron microscopy (TEM) have detected infection in lymphoid organ (LO), gills, haemocytes, cuticular epithelium, nerves, eyes and in connective tissues of head organs and abdominal muscle (Boonyaratpalin et al., 1993, Chantanachookin et al., 1993, Lu et al., 1995, Spann et al., 1997, Tang and Lightner, 1999, Tang et al., 2002, Smith, 2000, Callinan et al., 2003). A chronic GAV infection state also occurs in which virus, originally described as lymphoid organ virus (LOV), is primarily observed by ISH and TEM in foci of hypertrophied cells or ‘spheroids’ within the LO of apparently healthy prawns (Spann et al., 1995). Chronic infections are highly prevalent in P. monodon captured from the wild or farmed along the Pacific coast of Queensland, Australia (Spann et al., 1995, Spann et al., 2003, Cowley et al., 2000a, Walker et al., 2001). There is also evidence of chronic GAV infection in healthy P. monodon broodstock and postlarvae in some regions of southeast Asia (Phan, 2001; T.W. Flegel, personal communication) and YHV infections have been described in healthy P. monodon farmed in Thailand (Pasharawipas et al., 1997).
Penaeid prawn species from Hawaii and the western hemisphere, including Penaeus stylirostris, P. aztecus, P. duorarum, P. setiferus and Penaeus vannamei, are susceptible to experimental infection with YHV (Lu et al., 1994, Lightner et al., 1998), and species farmed commercially in Australia, including P. esculentus, P. merguiensis and Penaeus japonicus, are susceptible to GAV (Spann et al., 2000). However, some age/size and species-related differences have been noted in their susceptibility to disease. Histopathology consistent with YHV infection has also been reported in P. japonicus cultured in Taiwan that displayed typical signs of severe white spot syndrome virus (WSSV) disease and appeared to be co-infected (Wang et al., 1996). In Australia, RT-nested PCR has detected GAV in low levels in healthy P. esculentus co-cultivated with P. monodon (Walker et al., 2001). There is no other published evidence of natural YHV or GAV infection in prawn species other than P. monodon although there are reports that small shrimp such as Palaemon styliferus and krill (Acetes spp.) (Flegel et al., 1997) and crabs (e.g. Scylla serrata) may carry these viruses. Nevertheless, the ability of YHV and GAV to cause disease experimentally suggests these viruses represent a significant threat to cultivated prawns.
Antibody (Lu et al., 1996, Nadala et al., 1997, Nadala and Loh, 2000, Sithigorngul et al., 2000, Sithigorngul et al., 2002, Soowannayan et al., 2003), ISH (Tang and Lightner, 1999, Tang et al., 2002, Spann et al., 2003), conventional RT-PCR (Wongteerasupaya et al., 1997, Tang and Lightner, 1999, Cowley et al., 1999, Cowley et al., 2000a) and real-time RT-PCR (Dhar et al., 2002) methods have been used to detect either YHV or GAV in clinical samples. Of the antibody-based diagnostic techniques, immuno-histochemistry using monoclonal antibodies to a surface glycoprotein and the nucleocapsid protein of YHV (Sithigorngul et al., 2000, Sithigorngul et al., 2002) has recently allowed the detection and distinction of YHV and GAV infections (Soowannayan et al., 2003). ISH methods using DNA probes targeted to regions in the ORF1b gene have also allow cross-detection of YHV and GAV (Tang et al., 2002). RT-PCR tests reported to date have not considered genotypic sequence differences but have cross-detected YHV and GAV by chance (Cowley et al., 1999, Cowley et al., 2000a). To avoid the need for separate PCR tests to recognise YHV and GAV infections definitely, we describe here a sensitive multiplex RT-nested PCR that exploits genome sequence variations between reference Thai YHV and Australian GAV isolates to co-detect and distinguish these viruses.
2. Materials and methods
2.1. Prawn samples
Healthy P. monodon infected chronically with GAV (Spann et al., 1995) and moribund prawns displaying characteristic clinical signs, histopathology and TEM evidence of acute GAV infection (Spann et al., 1997) were obtained from hatcheries and farms in northeast and southeast Queensland between February 1996 and March 1998. Experimental infections with the reference GAV isolate, which was derived from a farm disease outbreak in northeast Queensland in 1996, were conducted as described previously (Spann et al., 1997). Prawn tissues were collected at 5–7 days post-infection when cumulative mortalities had reached ∼40%. P. monodon were also infected experimentally with a reference YHV isolate by injecting the tail muscle with haemolymph containing YHV (Wongteerasupaya et al., 1995, Wongteerasupaya et al., 1997). Lymphoid organs and haemolymph were collected from YHV-infected prawns 3–5 days post-infection. Prawns from farm outbreaks of YHV disease were collected from different locations in Thailand between 1994 and 1998. The origins of prawns used as sources of GAV and YHV RNA samples are listed in Table 1 .
Table 1.
Origin of GAV- and YHV-infected Penaeus monodon tested in the multiplex RT-nested PCR
| Number | Date | Location | Source | Description | Tissue |
| GAV#1 | 14-10-97 | Jacobs Well, SEQ | Cairns | H, I, B, N | LO |
| GAV#2 | 13-10-97 | Jacobs Well, SEQ | Innisfail | H, I, B, N | LO |
| GAV#3 | 25-09-97 | Cleveland, SEQ | Innisfail | R, I, B, N | LO |
| GAV#4 | 05-03-98 | Innisfail, NQ | Innisfail | W, I, B, N | LO |
| GAV#5 | 27-03-98 | Cardwell, NQ | Townsville | F, B,I , N | LO |
| GAV#6 | 12-02-96 | Mourilyan, NQ | NQ | F, P, M | LO |
| GAV#7 | 06-03-97 | Bundaberg, SEQ | NQ | F, P, M | LO |
| GAV#8 | 18-03-97 | Jacobs Well, SEQ | NQ | F, P, M | LO |
| GAV#9 | 17-04-96 | Jacobs Well, SEQ | NQ | F, P, M | Ceph |
| GAV#10 | 16-04-96 | Jacobs Well, SEQ | NQ | F, P, M | Ceph |
| YHV#1 | NK-96 | NK | CP | F, P, M | Hae |
| YHV#2 | 13-10-95 | Rayong | AAHRI | R, F, P, M | Gill/hae |
| YHV#3 | 01-02-95 | Surattanee | NK | R, F, I, M | Gill/hae |
| YHV#4 | 29-10-94 | Songkhla | CP | R, F, P | Gill/hae |
| YHV#5 | 18-01-96 | Chachuengsao | AAHRI | R, F, P, M | Gill/hae |
| YHV#6 | NK-97 | Bangpakong | CP | R, F, P, M | Gill/hae |
| YHV#7 | 16-12-97 | NK | CP | F, I, M | Hae |
| YHV#8 | 17-03-98 | Banpo | CP | H, P | Ceph |
| YHV#9 | 17-03-98 | Klongsuan | CP | H, P | Ceph |
All GAV-infected prawns were derived from wild broodstock sourced from Cairns, Innisfail or Townsville in north Queensland, Australia. All YHV-infected prawns were collected from locations in Thailand. Description: codes are F, farm stock; H, hatchery stocks of wild broodstock; W, wild broodstock collected directly from a trawler; R, research facility stock; B, broodstock; I, individual; P, pool of 2–10 prawns; M, mortalities in stock at time of collection and N, no obvious disease symptoms. GAV#6; YHV#1, reference isolates; NQ, north Queensland; SEQ, southeast Queensland; NK, not known. Tissues: gill, lymphoid organ (LO), haemolymph (hae), whole cephalothorax (ceph). CP: Shrimp Culture Research Centre of Charoen Pokphand Co. Ltd., Thailand. AAHRI: The Aquatic Animal Health Research Institute, Thailand.
2.2. RNA isolation
For RNA isolation, whole head homogenates (Spann et al., 1997), haemolymph, gill or LO tissue were collected using sterilised instruments and either processed immediately or snap frozen on dry ice and stored in liquid nitrogen until used. Gill or LO tissue was homogenised in TRIzol–LS™ (Invitrogen) using a pellet pestle. Haemolymph and 0.22 μm filtered head homogenates were dissolved directly in TRIzol–LS™ and RNA was isolated according to the manufacturer’s instructions. RNA was resuspended in DEPC–water, heated at 55 °C for 5 min, quantified by spectrophotometry (A260 nm) and stored at −70 °C. All reagents and tissue samples were handled in a separate laboratory using a laminar flow cabinet and aerosol-resistant barrier pipette tips to prevent contamination with PCR products.
2.3. Primers
The sequences of primers used for generic RT-PCR amplification of GAV and YHV (GY) and specific nested PCR amplification of either GAV (G) or YHV (Y) are listed: GY1 (5′-GACATCACTCCAGACAACATCTG-3′); GY2 (5′-CATCTGTCCAGAAGGCGTCTATGA-3′); G3 (5′-GCGTTCCTTTGTGAGCATAAATGA-3′); Y3 (5′-ACGCTCTGTGACAAGCATGAAGTT-3′); GY4 (5′-GTGAAGTCCATGTGTGTGAGACG-3′); GY5 (5′-GAGCTGGAATTCAGTGAGAGAACA-3′); and G6 (5′-GTAGTAGAGACGAGTGACACCTAT-3′). The length (23–24 nt) and melting temperatures (T m±4 °C) of all primers were closely matched. The generic GY2 nested PCR primer was designed with a slightly higher T m to compensate for a single base mismatch with the reference GAV sequence (Cowley et al., 2000b). All oligonucleotides were synthesised using an Oligo-1000 DNA Synthesiser (Beckman). RT-PCR with primer pair GY1–GY4 was expected to generate a 794 bp amplicon. Semi-nested and nested PCRs were designed to generate amplicons of 293 bp (primer pair GY1–G3 or GY1–Y3), 277 bp (primer pair GY2–G3 or GY2–Y3) and 406 bp (primer pair GY2–G6).
2.4. RT-PCR
Total RNA (100 ng) and 35 pmol primer GY5 were incubated at 70 °C for 10 min in 6 μl DEPC–water and chilled on ice. cDNA was synthesised by adding 2 μl Superscript II buffer × 5 (Invitrogen), 1 μl 100 mM DTT and 0.5 μl 10 mM dNTP mix and incubation at 42 °C for 2 min prior to adding 0.5 μl 200 U/μl Superscript II reverse transcriptase (Invitrogen) and further incubation at 42 °C for 1 h. Reverse transcriptase was inactivated at 70 °C for 15 min and the reaction quenched on ice. For PCR, 1 μl cDNA reaction was amplified in 50 μl containing 1×Taq buffer (10 mM Tris–HCl pH 9.0, 50 mM KCl, 0.1% Triton X-100), 1.5 mM MgCl2, 35 pmol each primer GY1 and GY4, 200 μM each dNTP and 2.5 U Taq polymerase (promega). The reaction mixture was overlaid with 50 μl liquid paraffin and heated at 85 °C for 3 min prior to adding cDNA (Chou et al., 1992). DNA was amplified by 35 cycles of 95 °C/30 s, 66 °C/30 s, 72 °C/45 s followed by 72 °C/7 min final extension using either a FTS-960 (Corbett Research) or PCR Sprint (Hybaid) thermal cycler. All PCRs utilised 0.5 ml thin-walled tubes. PCR products (10 μl) were resolved in 2% agarose–TAE gels containing 0.5 μg/ml ethidium bromide (Sambrook et al., 1989).
2.5. Differential nested PCRs
An aliquot (typically 0.5 μl) of the RT-PCR was amplified by nested PCR using a 50 μl reaction volume and primer sets GY2–G3, GY2–Y3 or GY2–Y3/G6. Semi-nested PCR reactions utilizing the primer pairs GY1–G3 or GY1–Y3 were also evaluated. Nested PCR conditions were as for the first PCR except that the extension time was reduced to 30 s. To increase sensitivity in cases when the nested PCR was negative, the amplification was repeated using up to 10 μl of the RT-PCR. Aliquots (10 μl) of the nested PCR were resolved in 2% agarose–TAE gels as above.
To determine the detection sensitivity of the RT-nested PCR for GAV and YHV, serial 10-fold dilutions of LO total RNA were prepared in DEPC–water containing 10 ng/μl LO total RNA from an uninfected P. japonicus to maintain a constant amount of RNA in the cDNA synthesis reaction. Aliquots (1 μl) of diluted RNA were reverse transcribed using primer GY5 and amplified by PCR (primer pair GY1–GY4) and nested PCR using various primer sets (i.e. GY1–G3, GY1–Y3, GY2–G3, GY2–Y3 or GY2–Y3/G6). To simulate dual GAV/YHV infections, 10-fold dilutions of one virus RNA were prepared using a diluent comprising either 100 or 1 ng/μl of LO total RNA from prawns infected with the other virus. To avoid cross-contamination, all PCR mixes were prepared in a laminar flow cabinet using aerosol-resistant tips and in a different laboratory to that used to analyze PCR products.
2.6. Sequencing
Sequences in the G3/Y3 and G6 differential primer regions were determined for RT-PCR products amplified from all 10 GAV and the eight YHV field RNA samples. PCR products (794 bp) amplified with primer pair GY1–GY4 were purified using QIAquick™ columns (Qiagen). Both DNA strands of GAV and YHV PCR amplicons were sequenced using primers GY1, GY4, Y3 or G6, Big Dye™ V2 reagent (Applied Biosystems Inc., ABI) and automated ABI Model-377 apparatus (ABI) at the Australian Genome Research Facility, University of Queensland. The sequence in the GY4 and GY5 primer regions of YHV RNA samples was determined using PCR amplicons generated with other primers (Hodgson et al., unpublished). Nucleotide sequence chromatograms were analysed using SeqEd 3.1 (ABI).
3. Results
3.1. Selection of generic and specific GAV and YHV primers
Overall, the nucleotide sequences of the ORF1b genes of reference isolates of GAV from Australia and YHV from Thailand (Tang and Lightner, 1999, Cowley et al., 1999, Sittidilokratna et al., 2002) display 19.5% divergence and the majority of differences occur in codon ‘wobble’ positions not altering the pp1ab coding sequence. However, some localised regions display higher levels of variation and virus-specific primers used in the differential RT-nested PCR were targeted to two of these sites. Regions targeted by the generic and specific primers are shown in Fig. 1 . The site targeted by cDNA primer GY5 was conserved in three reported YHV sequences (Wongteerasupaya et al., 1997, Tang and Lightner, 1999, Sittidilokratna et al., 2002) and GAV (Cowley et al., 1999). GAV and YHV sequences at the site targeted by virus-specific primers G3 and Y3 varied at 12/24 nt positions associated with three aa changes, including one at the primer 3′-end that was divergent at all three codon positions. GAV-specific primer G6 was targeted to a sequence that diverged at 10/24 nt positions, of which three occurred at the primer 3′-end and were associated with one aa change. The extent of sequence variation was expected to restrict annealing of these primers to the non-homologous DNA template and mismatching at their 3′-termini was included as two or more mismatches at this position severely abrogates PCR amplification (Kwok et al., 1990).
Fig. 1.
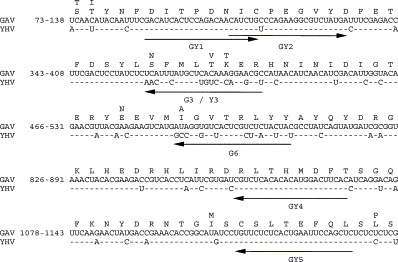
GAV and YHV nucleotide and amino acid sequences in the ORF1b gene region targeted by primers used in the multiplex RT-nested PCR. YHV sequence differences are indicated above and below the GAV sequences and the primer targets are highlighted (arrows). Numbering is taken from a GAV–YHV sequence comparison reported previously (Cowley et al., 1999).
3.2. RT-PCR to detect GAV or YHV
From cDNA synthesised using antisense primer GY5, PCR using the conserved primer pair GY1–GY4 amplified a 794 bp product from both the GAV and YHV (Fig. 2a ). The PCR was tested at three annealing temperatures (64, 66 and 68 °C) and 66 °C was selected for subsequent amplifications, as the amplicon yield was slightly greater than at 68 °C. At the selected temperature there was also an absence of very minor products seen at 64 °C when higher viral RNA template levels were amplified (data not shown).
Fig. 2.
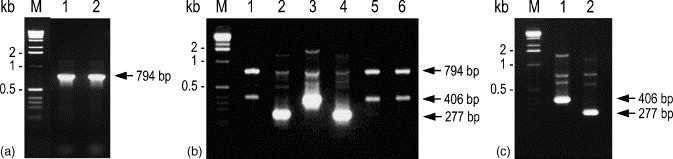
Amplification of GAV and YHV RNA by RT-PCR followed by nested PCR with various primer combinations. (a) PCR amplification (primer pair GY1–GY4) of a 794 bp product from cDNA synthesised from reference GAV (lane 1) and YHV (lane 2) RNA using primer GY5. (b) Nested PCR amplification of RT-PCR products from GAV (lanes 1–3) and YHV (lanes 4–6) using primer pairs GY2–Y3 (lanes 1 and 4), GY2–G3 (lanes 2 and 5) and GY2–G6 (lanes 3 and 6). (c) Nested PCR amplification of a 406 bp GAV-specific product (lane 1) or 277 bp YHV-specific product (lane 2) using the multiplexed primer set GY2–Y3/G6. PCR products (10 μl) were resolved in a 2% agarose–TAE gel containing 0.5 μg/ml ethidium bromide. M=1 kb DNA ladder (Invitrogen).
3.3. Nested PCRs to differentiate GAV from YHV
Initially, separate nested PCRs were tested using a conserved sense primer (GY2) in conjunction with an antisense primer specific to either GAV (G3) or YHV (Y3) (see Fig. 1). Primer pairs GY2–G3 and GY2–Y3 amplified only a 277 bp fragment of the 794 bp RT-PCR amplicons of GAV and YHV, respectively (Fig. 2a and b). Nested PCR using primer GY2 in combination with GAV-specific antisense primer G6 similarly amplified only a 406 bp fragment of GAV (Fig. 2b). When the virus-specific nested PCR did not proceed due to mismatching of the antisense primer to the template, elevated levels of the 794 bp GY1–GY4 amplicon and a ∼400 bp non-specific product were detected (Fig. 2b, lanes 1, 5 and 6). Semi-nested PCRs in which primer GY1 was substituted for GY2 produced comparable results to those shown in Fig. 2b except the amplicon (293 bp) was longer (data not shown).
A multiplexed nested PCR using primer GY2 in combination with both YHV- (Y3) and GAV-specific (G6) antisense primers was tested for its ability to differentiate the viruses in a single reaction. Nested PCR amplicons of a size expected for YHV (277 bp) or GAV (406 bp) were generated only from primary PCR amplicons derived from YHV or GAV, respectively (Fig. 2c). Minor amounts of the primary 794 bp amplicon and of two other products that differed in size between GAV and YHV were also detected.
3.4. Sensitivity of the RT-nested PCR
The sensitivity limits of the RT-PCR and nested PCR were examined using serial 10-fold dilutions of LO total RNA from P. monodon infected experimentally with GAV or YHV at levels sufficient to give good yields of the primary 794 bp PCR amplicon (Fig. 3 ). The RT-PCR sensitivity limit was ∼1 pg RNA (amplicon barely visible) for either GAV or YHV. The sensitivity limits of nested PCRs using primer pairs GY2–G3 or GY2–G6 for GAV and GY2–Y3 for YHV were identical (10 fg RNA) and at least 100-fold greater than that of the primary PCR. Semi-nested PCRs employing primer GY1 instead of GY2 increased sensitivity to the same extent (data not shown).
Fig. 3.
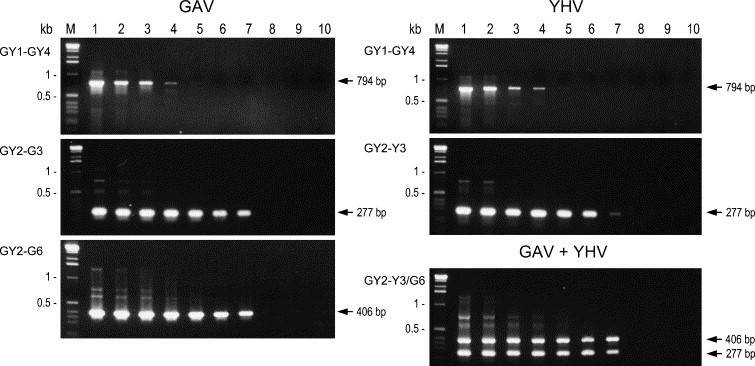
Detection limits of the RT-PCR and nested PCR primer combinations using titrations of LO total RNA from P. monodon infected with the reference GAV or YHV isolates. cDNA synthesised using primer GY5 and serial 10-fold RNA dilutions (100 ng/μl to 1 fg/μl, lanes 1–9, respectively) diluted in 10 ng/μl P. japonicus LO RNA, which was also used as a negative control (lane 10), was amplified by PCR using primer pair GY1–GY4. Nested PCRs were performed using primer pairs GY2–G3 and GY2–G6 for GAV and GY2–Y3 for YHV. Multiplex nested PCR (primer set GY2–Y3/G6) was also performed using 1:1 mixtures of the GAV and YHV RT-PCRs. PCR products (10 μl) were resolved as in Fig. 2. M=1 kb DNA ladder.
To determine whether the presence of the two virus-specific antisense primers (Y3 and G6) might affect the detection sensitivity, the multiplex nested PCR (primer set GY2–Y3/G6) was tested using 1:1 mixtures of the primary PCRs of the GAV and YHV RNA dilution series (Fig. 3). YHV-specific (277 bp) and GAV-specific (406 bp) products were co-amplified to the same detection limit (10 fg RNA) obtained in the nested PCRs (GY2–Y3 or GY2–G6) employing each antisense primer alone.
3.5. RT-nested PCR differentiation of field samples of GAV and YHV
Five RNA samples from healthy, wild P. monodon broodstock chronically infected with GAV, five samples from moribund farmed prawns infected acutely with GAV and nine samples from moribund farmed prawns acutely infected with YHV were tested using the multiplex RT-nested PCR (Fig. 4 ). The RNA originated from tissues of either individual prawns or pools of 2–10 prawns. Prawns were collected at different locations in eastern Australia or Thailand between 1994 and 1998 (Table 1). Among the GAV samples, less primary (794 bp) PCR amplicon was generated from the two cephalothorax homogenates than from LO tissue (Fig. 4a). In contrast, a high yield of 794 bp amplicon occurred only in sample YHV#1 and none was detected in five of the YHV RNA samples. Multiplex nested PCR (primer set GY2–Y3/G6) generated a 406 bp amplicon in all 10 GAV samples and a 277 bp amplicon from all YHV samples except YHV#8 (Fig. 4b). Moreover, comparable yields of the 277 bp amplicon were generated from the former YHV samples irrespective of whether a primary PCR amplicon was detected. For the five YHV samples (#3, #5–#8) in which little or no primary PCR amplicon was detected, the PCR was repeated using 2 μl instead of 1 μl cDNA. Increasing the input cDNA enhanced the 794 bp amplicon yield only for sample YHV#6 (data not shown). However, 277 bp nested PCR amplicon yields were enhanced visibly for most samples and an amplicon was detected for the previously negative YHV#8 (Fig. 4c). The yield of YHV#8 amplicon increased further when either 5 or 10 μl of the primary PCR was employed in the nested PCR.
Fig. 4.
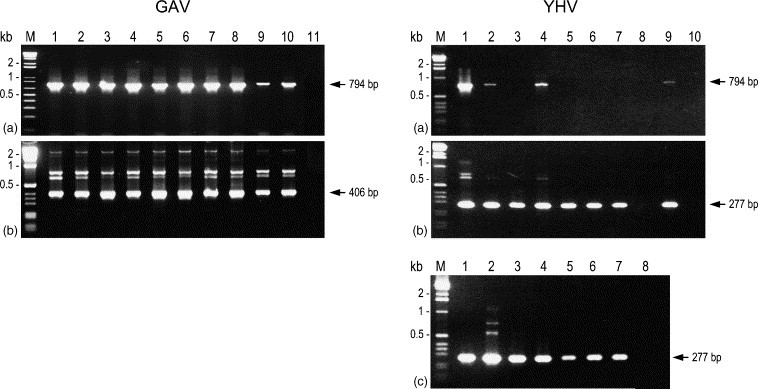
Detection of RNA from different GAV and YHV samples using the multiplex RT-nested PCR. The origin of P. monodon from which RNA was isolated is described in Table 1. Isolates GAV#1 to GAV#10 correspond to GAV lanes 1–10, respectively, and isolates YHV#1 to YHV#9 correspond to YHV lanes 1–9, respectively. GAV lane 11 and YHV lane 10 were negative controls comprising LO RNA from uninfected P. japonicus. (a) cDNA synthesised from 100 ng RNA using primer GY5 amplified by PCR using the primer pair GY1–GY4. (b) RT-PCR products amplified by nested PCR using the multiplex primer set GY2-Y3/G6. (c) For selected YHV samples (#3 (lane 1), #5 (lane 2), #6 (lane 3), #7 (lane 4) and #8 (lanes 5–7)) which generated little or no 794 bp PCR amplicon, the multiplex nested PCR was repeated using RT-PCRs performed using 2 μl instead of 1 μl input cDNA. cDNA from P. japonicus RNA was used as a negative control (lane 8). For isolate YHV#8, 5 μl (lane 6) and 10 μl (lane 7) of the primary PCR were also amplified by nested PCR. PCR products (10 μl) were resolved as in Fig. 2. M=1 kb or 1 kb plus DNA ladder (Invitrogen).
3.6. PCR co-detection of GAV and YHV RNA mixed at various ratios
To simulate prawn co-infections with GAV and YHV at various infection levels, a 10-fold dilution series of reference YHV RNA was prepared in a diluent comprising 100 ng/μl of the reference GAV RNA. A reciprocal GAV RNA dilution series was prepared similarly in a diluent comprising YHV RNA and thus all samples contained at least 100 ng/μl RNA. Using cDNA prepared from selected dilutions, comparable yields of primary PCR amplicon (794 bp) were generated (data not shown). However, GAV-specific (406 bp) and YHV-specific (277 bp) amplicons were co-detected in the multiplex nested PCR only to a limit equal to a major:minor viral RNA ratio of 1:10−2 (100:1 ng/μl) when either GAV or YHV represented the minor RNA species (Fig. 5a , lanes 4–6). Overexposure of the gel image detected very low levels of amplicon of the minor RNA in the YHV and GAV dilutions containing 10 pg/μl of the minor RNA (data not shown). In any case, this was equivalent to a 103 to 105-fold reduction in the detection limit of either virus RNA compared to that obtained (i.e. 10 fg/μl) when diluted in uninfected P. japonicus RNA (see Fig. 3). The test was repeated using 10-fold RNA dilutions prepared as above in a diluent containing 1 ng/μl rather than 100 ng/μl of either YHV or GAV RNA (Fig. 5b). Barely detectable but comparable amounts of the 794 bp RT-PCR amplicon were generated from all samples (data not show). Compared to Fig. 5a, the nested PCR detection limit of the minor GAV or YHV RNAs increased significantly to include dilutions containing a major:minor RNA species ratio of 1:10−4 (i.e. 1 ng/μl:100 fg/μl).
Fig. 5.

Multiplex RT-nested PCR amplification of GAV and YHV RNA mixed at various ratios to simulate dual-infection states. RNA was mixed 1:1 at concentrations of either (a) 100 ng/μl (lane 5) or (b) 1 ng/μl (lane 6) and serial 10-fold dilutions of virus RNA were prepared in a diluent of reciprocal virus RNA at these two concentrations. (a) YHV RNA amounts 100 fg, 10 pg, 100 pg, 1 ng (lanes 1–4, respectively) and 100 ng (lanes 5–9), GAV RNA amounts 100 ng (lanes 1–5) and 1 ng, 100 pg, 10 pg, 100 fg, (lanes 6–9, respectively) and P. japonicus negative control RNA (lane 10). (b) YHV RNA amounts 10 fg, 100 fg, 1 pg, 10 pg, 100 pg (lanes 1–5, respectively) and 1 ng (lanes 6–11), GAV RNA amounts 1 ng (lanes 1–6) and 100 pg, 10 pg, 1 pg, 100 fg, 10 fg (lanes 7–11, respectively). In both tests, cDNA synthesised using primer GY5 was amplified by PCR (primer pair GY2–GY4) followed by nested PCR with the multiplex primer set GY2–Y3/G6. PCR products (10 μl) were resolved as in Fig. 2. M=1 kb DNA ladder.
3.7. Sequence differences among GAV and YHV RNA samples
Primary PCR amplicons from all 10 GAV isolates including the reference isolate were sequenced to identify any nucleotide variations in the G3/Y3 and G6 nested-PCR primer regions (Fig. 6 ). In the 682 nt region immediately downstream of primer GY1, a maximum of 1.8% (12/682 nt) divergence was detected among the different GAV isolates. All nine field samples varied from the reference GAV#6 isolate (Cowley et al., 1999, Cowley et al., 2000b) at a silent position (U→C) 6 nt from the 3′-end of primer G3 and at a silent position (U→C) 15 nt from the 3′-end of primer G6. Six GAV isolates also varied at a silent position (U→C) 12 nt from the end of G6. Sample GAV#4 varied at a position (U→C) 1 nt from the 3′-end of primer G6 that would cause an amino acid substitution. The sequences also provided information in the GY2 primer target downstream of the region that overlapped primer GY1. Six of the GAV samples varied at a silent position (A→G) 15 nt from the 3′-end of GY2 (data not shown).
Fig. 6.

Nucleotide sequences of genome regions targeted by primers G3/Y3 and G6 in the nine GAV and eight YHV field samples compared to the reference isolates (GAV#6 and YHV#1). The GAV#6 amino acid sequence is shown, as are differences between this sequence and that of YHV#1. Nucleotide variations to the GAV#6 sequence are shown (bold), as are variations (bold) to the YHV#1 sequence.
For seven of the eight YHV field samples, sequence analysis of RT-PCR amplicons (0.7 kb) generated for other purposes (Hodgson et al., unpublished) identified no nucleotide variations, compared to the reference YHV#1 (Tang and Lightner, 1999, Sittidilokratna et al., 2002), in regions targeted by primers GY4 and GY5. For all eight YHV field samples, similar analysis of semi-nested PCR amplicons generated using the primer pair GY2–GY4 identified a maximum divergence of 2.1% (15/730 nt) compared to the reference sequence. In each amplicon, most variations were due to the coexistence of two nucleotides (most commonly U/C=Y or A/G=R) in relatively equal amounts. Variations from the reference YHV isolate in the G3/Y3 and G6 primer targets were only detected in four field samples. In samples YHV#3 and #7, a U/C double nucleotide occurred at a silent position either 14 nt from the 3′-end of primer G3/Y3 while a U/C also occurred 12 nt from the 3′-end of primer G6 in samples YHV#4 and #5.
4. Discussion
GAV from Australia and YHV from Thailand are viral pathogens that pose significant disease threats to the culture of P. monodon in countries that primarily farm this species. The viruses are morphologically indentical, induce similar histopathology (Boonyaratpalin et al., 1993, Chantanachookin et al., 1993, Spann et al., 1997), have similar tissue tropism (Tang and Lightner, 1999, Tang et al., 2002, Spann et al., 2003) and are readily cross-detected by ISH (Tang et al., 2002). However, the level of nucleotide sequence variation in the ORF1b (19.5%) (Cowley et al., 1999, Sittidilokratna et al., 2002) and ORF3 (25%) genes (Jitrapakdee et al., 2003) and antigenic variations detected with monoclonal antibodies (Soowannayan et al., 2003) indicate that GAV and YHV represent distinct geographic topotypes. Moreover, mortalities accumulate more rapidly in P. monodon infected experimentally with YHV (Boonyaratpalin et al., 1993, Spann et al., 1997) and, while YHV can induce cephalothorax yellowing in diseased P. monodon, whole body reddening has been reported for GAV (Spann et al., 1997). Nevertheless, these differences are subjective and/or observed inconsistently and would be unreliable in distinguishing between infections with the two viruses.
We describe the development and assessment of an RT-nested PCR to co-detect and distinguish GAV from YHV. The RT and primary PCR steps employ primers to conserved ORF1b gene sequences to amplify either virus, while the nested PCR employs multiplexed virus-specific primers to distinguish the viruses based on amplicon size. The two virus-specific primers incorporated 10–12/24 mismatches, including three at their 3′-termini associated with amino acids shifts. The number and positioning of the mismatches precluded amplification of the non-homologous sequence as two or more substitutions at the 3′-end of a primer have been shown previously to severely inhibit PCR (Sommer and Tautz, 1989, Kwok et al., 1990). In addition, the 23–24 mer PCR primers were used at an annealing temperature (66 °C) that would ensure specificity but accommodate minor sequence variations known to occur among different GAV and YHV isolates. As these crustacean okaviruses encode an RNA-dependent RNA polymerase (Cowley et al., 2000b, Sittidilokratna et al., 2002, Cowley and Walker, 2002) most closely related to those of coronaviruses and arteriviruses, it is likely that it will lack proofreading ability and establish virus quasispecies populations that are a source of genetic drift (see reviews Steinhauer and Holland, 1987, Holland et al., 1992).
Initial tests using non-multiplexed nested PCRs showed that amplicons were generated only when the GAV-specific (G3 or G6) or YHV-specific (Y3) antisense primer matched the virus template. However, in tests in which these did not match, the primary PCR (794 bp) amplicon was more evident, probably as the result of carry over of the GY1–GY4 primers. A non-specific product (∼400 bp) of similar size to the 406 bp GY2–G6 primer amplicon was also apparent in these nested PCRs. However, this DNA did not occur in the multiplexed nested PCR incorporating both YHV- (Y3) and GAV-specific (G6) antisense primers in which its production was quenched by the preferential amplification of virus template. Multiplexing of the antisense primers targeted to different divergent sequences also readily distinguished GAV (406 bp) from YHV (277 bp) in a single nested PCR on the basis of amplicon size.
Nested PCR extended the RT-PCR detection limit 100–1000-fold. Although we made no attempt to relate this directly to template copies, viral RNA was detectable in ∼10 fg LO total RNA isolated from heavily infected P. monodon. Moreover, the detection limit was unaffected by primer multiplexing and was comparable to that of a GAV RT-nested PCR targeting a downstream region of the ORF1b gene (Cowley et al., 2000a). For diagnostic purposes, this additional sensitivity is extremely useful in detecting unapparent infections in healthy prawns when virus levels are often below the detection threshold of one-step RT-PCR (Cowley et al., 2000a, Walker et al., 2001).
The multiplex RT-nested PCR allowed co-detection of GAV and YHV in LO total RNA mixed at various ratios to simulate dual-infection states. This was examined as chronic GAV infection is highly prevalent in healthy wild and farmed P. monodon in eastern Australia (Spann et al., 1995, Cowley et al., 2000a, Walker et al., 2001) and so any introduction of exotic YHV into this population would likely involve superinfection of GAV-infected prawns. However, when GAV RNA was diluted in 100 ng/μl YHV RNA and visa versa, the nested PCR detection limit of the minor viral RNA species (10 pg–1 ng) was found to be quenched 103 to 105-fold compared to that (10 fg) observed in the absence of competitor. We do not know to what extent the RT and/or PCR steps contributed to this anomaly. However, the effect was concentration-dependent. When 1 ng/μl rather than 100 ng/μl competitor RNA was used, the detection limit of the minor viral RNAs (100 fg) approached that obtained without competitor. Although there is obviously some dependence on virus loads, the nested PCR theoretically has the capability to co-detect and distinguish GAV from YHV in dual-infected prawns when relative RNA template numbers range between 1:1 and 1:104.
GAV and YHV samples from P. monodon collected at different locations in eastern Australia and Thailand over a 2–4 year period were distinguished using the multiplex RT-PCR. All YHV samples were from diseased prawns while GAV samples were obtained from both healthy and diseased animals. There was some variability in amplicon yields among the YHV samples compared to GAV and these appear to be due primarily to the tissue type selected for RNA isolation. YHV RNA samples were mainly from haemolymph while LO was mostly used for GAV. YHV was not detected in one sample using the standard conditions. However, by increasing the cDNA quantity used in the primary PCR, and primary PCR quantity used in the nested PCR, a YHV amplicon was detected. The use of a published RT-nested PCR method has shown that nested PCR is often required to detect low levels of GAV in the LO of healthy, chronically-infected P. monodon (Cowley et al., 2000a and unpublished, Walker et al., 2001). Moreover, LO and gills appear to be a better source of GAV than haemocytes when infection levels are low (Cowley et al., 2000a). Unfortunately we were unable to determine whether this is also the case in the YHV-infected prawns as LO or separate gill tissues were not available for testing.
PCR amplicons from the GAV and YHV samples were sequenced to identify any nucleotide variations in the genome regions targeted by virus-specific nested PCR primers G3/Y3 and G6. Variations of up to 1.8% for GAV and 2.1% for YHV were detected between the reference (Cowley et al., 1999, Cowley et al., 2000b, Sittidilokratna et al., 2002) and field samples. These primarily occurred at codon wobble positions and, among the YHV samples, many were due to the presence of two different nucleotides in relatively equal amounts. This was not unexpected as most YHV RNA samples came from tissues pooled from 5–10 prawns and it is likely that individuals contained minor genotypic variants. In all YHV samples, sequences targeted by primers GY4 and GY5 were conserved. However, single variations due to co-infections occurred in either the Y3 or G6 nested PCR primer targets in 4 samples. Among the GAV samples, 1–3 nt variations were detected in sequences targeted by these nested PCR primers. However, these variations, including one which caused an A:C mismatch 1 nt from the 3′-end of primer G6, did not adversely affect the performance of the PCR. This is consistent with data demonstrating that single primer-template mismatches (other than A:G, G:A, C:C or A:A mismatches at the primer 3′-terminus) are unlikely to be deleterious to PCR (Sommer and Tautz, 1989, Kwok et al., 1990). In all, the absence of substantial sequence variations in the ORF1b gene region targeted by the multiplex RT-nested PCR suggests the test should be quite robust in distinguishing the Thailand YHV genotype from the GAV genotype that occurs commonly in healthy and diseased P. monodon from Eastern Australia.
Potential mechanisms for the inadvertent introduction of major prawn viral pathogens such as YHV, WSSV and Taura syndrome virus (TSV) into regions previously free of these agents are well documented (Lightner et al., 1997, Nunan et al., 1998). Of primary concern is the translocation of frozen uncooked prawns or live broodstock and seed carrying unapparent virus infections. The sequence divergence between Thai YHV and Australian GAV isolates suggests that introductions of these or genetically related viruses, could be detected and traced to the source of the introduction. The multiplex RT-nested PCR described here provides a rapid and sensitive initial tool to co-detect and differentiate these viruses. However, we are currently obtaining P. monodon samples from other locations in Australia and Asia to determine the extent of genetic diversity among YH-complex viruses present in different prawn populations (Walker et al., 2001, Soowannayan et al., 2003). Such epidemiological data will help establish whether these viruses are endemic or have been recently introduced through movements of brooders or postlarvae. Moreover, sequence information on different YH-complex viruses may allow development of a modified multiplexed RT-PCR test to co-detect and distinguish other genotypic variants of YHV/GAV.
Acknowledgements
We thank CSIRO Marine Research and Queensland prawn farm and hatchery operators for supplying prawns. This research was supported in part by the Australian Centre for International Agricultural Research, the National Centre for Genetic Engineering and Biotechnology of Thailand and the Thailand Research Fund.
References
- Boonyaratpalin S., Supamattaya K., Kasornchandra J., Direkbusaracom S., Aekpanithanpong U., Cantanachookin C. Non-occluded baculo-like virus, the causative agent of yellow-head disease in the black tiger shrimp (Penaeus monodon) Fish Patho. 1993;l28:103–109. [Google Scholar]
- Callinan R.B., Jiang L., Smith P.T., Soowannayan C. Fatal, virus-associated peripheral neuropathy and retinopathy in farmed Penaeus monodon in eastern Australia. I. Pathol. Dis. Aquat. Org. 2003;53:181–193. doi: 10.3354/dao053181. [DOI] [PubMed] [Google Scholar]
- Chantanachookin C., Boonyaratpalin S., Kasornchandra J., Sataporn D., Ekpanithanpong U., Supamataya K., Sriurairatana S., Flegel T.W. Histology and ultrastructure reveal a new granulomas-like virus in Penaeus monodon affected by yellow-head disease. Dis. Aquat. Org. 1993;17:145–157. [Google Scholar]
- Chou Q., Russell M., Birch D.E., Raymond J., Bloch W. Prevention of pre-PCR mispriming and primer dimerisation improves low-copy number amplifications. Nucl. Acids Res. 1992;20:1717–1723. doi: 10.1093/nar/20.7.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowley J.A., Walker P.J. The complete genome sequence of gill-associated virus of Penaeus monodon prawns indicates a gene organisation unique among nidoviruses. Arch. Virol. 2002;147:1977–1987. doi: 10.1007/s00705-002-0847-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowley J.A., Dimmock C.M., Spann K.M., Walker P.J. Detection of Australian gill-associated virus (GAV) and lymphoid organ virus (LOV) of Penaeus monodon by RT-nested PCR. Dis. Aquat. Org. 2000;39:159–167. doi: 10.3354/dao039159. [DOI] [PubMed] [Google Scholar]
- Cowley J.A., Dimmock C.M., Spann K.M., Walker P.J. Gill-associated virus of Penaeus monodon prawns: an invertebrate virus with ORF1a and ORF1b genes related to arteri- and corona-viruses. J. Gen. Virol. 2000;81:1473–1484. doi: 10.1099/0022-1317-81-6-1473. [DOI] [PubMed] [Google Scholar]
- Cowley J.A., Dimmock C.M., Wongteerasupaya C., Boonsaeng V., Panyim S., Walker P.J. Yellow head virus from Thailand and gill-associated virus from Australia are closely related but distinct viruses. Dis. Aquat. Org. 1999;36:153–175. doi: 10.3354/dao036153. [DOI] [PubMed] [Google Scholar]
- Dhar A.K., Roux M.M., Klimpel K.R. Quantitative assay for measuring the Taura syndrome virus and yellow head virus load in shrimp by real-time RT-PCR using SYBR Green chemistry. J. Virol. Meth. 2002;104:69–82. doi: 10.1016/S0166-0934(02)00042-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flegel, T.W., Sriurairatana, S., Wongteerasupaya, C., Boonsaeng, V., Panyim, S., Withyachumnarnkul, B., 1997. Progress in characterization and control of yellow-head virus of Penaeus monodon. In: Flegel, T.W., Menasveta, P., Paisarnrat, S. (Eds.), Shrimp biotechnology in Thailand. National Center for Genetic Engineering and Biotechnology, Bangkok, pp. 71–78.
- Holland J.J., De La Torre J.C., Steinhauer D.A. RNA virus populations as quasispecies. Curr. Top. Microbiol. Immunol. 1992;176:1–20. doi: 10.1007/978-3-642-77011-1_1. [DOI] [PubMed] [Google Scholar]
- Jitrapakdee S., Unajak S., Sittidilokratna N., Hodgson R.A.J., Cowley J.A., Walker P.J., Panyim S., Boonsaeng V. Identification and analysis of gp116 and gp64 structural glycoproteins of yellow head nidovirus of Penaeus monodon shrimp. J. Gen. Virol. 2003;84:863–873. doi: 10.1099/vir.0.18811-0. [DOI] [PubMed] [Google Scholar]
- Kwok S., Kellogg D.E., McKinney N., Spasic D., Goda L., Levenson C., Sninsky J.J. Effects of primer-template mismatches on the polymerase chain reaction: human immunodeficiency virus type 1 model studies. Nucl. Acids Res. 1990;18:999–1005. doi: 10.1093/nar/18.4.999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lightner D.V., Hasson K.W., White B.L., Redman R.M. Experimental infection of western hemisphere Penaeid shrimp with Asian white spot syndrome virus and Asian yellow head virus. J. Aquat. Anim. Health. 1998;10:271–281. [Google Scholar]
- Lightner D.V., Redman R.M., Poulos B.T., Nunan L.M., Mari J.L., Hasson K.W. Risk of spread of penaeid shrimp viruses in the Americas by the international movement of live and frozen shrimp. Rev. Sci. Tech. 1997;16:146–160. doi: 10.20506/rst.16.1.1010. [DOI] [PubMed] [Google Scholar]
- Lu Y., Tapay L.M., Brock J.A., Loh P.C. Infection of the yellow head baculo-like virus (YBV) in two species of penaeid shrimp, Penaeus stylirostris (Stimpson) and Penaeus vannamei (Boone) J. Fish Dis. 1994;17:649–656. [Google Scholar]
- Lu Y., Tapay L.M., Loh P.C. Development of a nitrocellulose-enzyme immunoassay for the detection of yellow-head from penaeid shrimp. J. Fish Dis. 1996;19:9–13. [Google Scholar]
- Lu Y., Tapay L.M., Loh P.C., Brock J.A., Gose R.B. Distribution of yellow-head virus in selected tissues and organs of penaeid shrimp Penaeus vannamei. Dis. Aquat. Org. 1995;23:67–70. [Google Scholar]
- Mayo M.A. A summary of taxonomic changes recently approved by the ICTV. Arch. Virol. 2002;147:1655–1656. doi: 10.1007/s007050200039. [DOI] [PubMed] [Google Scholar]
- Nadala E.C.B., Tapay L.M., Cao S., Loh P.C. Detection of yellowhead virus and Chinese baculovirus in penaeid shrimp by the western blot technique. J. Virol. Methods. 1997;69:39–44. doi: 10.1016/s0166-0934(97)00136-5. [DOI] [PubMed] [Google Scholar]
- Nadala E.C.B., Loh P.C. Dot-blot nitrocellulose enzyme immunoassays for the detection of white-spot virus and yellow-head virus of penaeid shrimp. J. Virol. Methods. 2000;84:175–179. doi: 10.1016/s0166-0934(99)00140-8. [DOI] [PubMed] [Google Scholar]
- Nunan L.M., Poulos B.T., Lightner D.V. The detection of white spot syndrome virus (WSSV) and yellow head virus (YHV) in imported commodity shrimp. Aquaculture. 1998;160:19–30. [Google Scholar]
- Phan, T.T.N., 2001. Prevalence and co-prevalence of white spot syndrome virus (WSSV) and yellow head complex viruses (YHV-complex) in cultured giant tiger prawn (Penaeus monodon) in Vietnam. M.Sc. thesis, University of Queensland.
- Pasharawipas, T., Flegel, T.W., Sriurairatana, S., Morrison, D.J., 1997. Latent yellow-head infections in Penaeus monodon and implications regarding tolerance. In: Flegel, T.W., Menasveta, P., Paisarnrat, S. (Eds.), Shrimp Biotechnology in Thailand. National Center for Genetic Engineering and Biotechnology, Bangkok, pp. 45–53.
- Sambrook, J., Fritsch, E.F., Maniatis, T., 1989. Molecular Cloning: A Laboratory Manual, second ed. Cold Spring Harbor Laboratory Press, New York.
- Sithigorngul P., Chauychuwong P., Sithigorngul W., Longyant S., Chaivisuthangkura P., Menasveta P. Development of a monoclonal antibody specific to yellow head virus (YHV) from Penaeus monodon. Dis. Aquat. Org. 2000;42:27–34. doi: 10.3354/dao042027. [DOI] [PubMed] [Google Scholar]
- Sithigorngul P., Rukpratanporn S., Longyant S., Chaivisuthangkura P., Sithigorngul W., Menasveta P. Monoclonal antibodies specific to yellow-head virus (YHV) of Penaeus monodon. Dis. Aquat. Org. 2002;49:71–76. doi: 10.3354/dao049071. [DOI] [PubMed] [Google Scholar]
- Sittidilokratna N., Hodgson R.A.J., Cowley J.A., Jitrapakdee S., Boonsaeng V., Panyim S., Walker P.J. Complete ORF1b-gene sequence indicates yellow head virus is an invertebrate nidovirus. Dis. Aquat. Org. 2002;50:87–93. doi: 10.3354/dao050087. [DOI] [PubMed] [Google Scholar]
- Smith P.T. Diseases of the eye of farmed shrimp Penaeus monodon. Dis. Aquat. Org. 2000;43:159–173. doi: 10.3354/dao043159. [DOI] [PubMed] [Google Scholar]
- Sommer R., Tautz D. Minimal homology requirements for PCR primers. Nucl. Acids Res. 1989;17:6749. doi: 10.1093/nar/17.16.6749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soowannayan, C., Flegel, T.W., Sithigorngul, P., Slater, J., Hyatt, A., Cramerri, S., Wise, T., Crane, M.S.J., Cowley, J.A., McCulloch, R.J., Walker, P.J., 2003. Detection and differentiation of yellow head complex viruses using monoclonal antibodies. Dis. Aquat. Org. 57, 193–200. [DOI] [PubMed]
- Spann K.M., Vickers J.E., Lester R.J.G. Lymphoid organ virus of Penaeus monodon from Australia. Dis. Aquat. Org. 1995;23:127–134. [Google Scholar]
- Spann K.M., Cowley J.A., Walker P.J., Lester R.J.G. A yellow-head-like virus from Penaeus monodon cultured in Australia. Dis. Aquat. Org. 1997;31:169–179. [Google Scholar]
- Spann K.M., Donaldson R.A., Cowley J.A., Walker P.J. Differences in the susceptibility of some penaeid prawn species to gill-associated virus (GAV) infection. Dis. Aquat. Org. 2000;42:221–225. doi: 10.3354/dao042221. [DOI] [PubMed] [Google Scholar]
- Spann K.M., McCulloch R.J., Cowley J.A., East I.J., Walker P.J. Detection of gill-associated virus (GAV) by in situ hybridization during acute and chronic infections of Penaeus monodon and P. esculentus shrimp. Dis. Aquat. Org. 2003;56:1–10. doi: 10.3354/dao056001. [DOI] [PubMed] [Google Scholar]
- Steinhauer D.A., Holland J.J. Rapid evolution of RNA viruses. Ann. Rev. Microbiol. 1987;47:409–433. doi: 10.1146/annurev.mi.41.100187.002205. [DOI] [PubMed] [Google Scholar]
- Tang K.F.J., Lightner D.V. A yellow head virus gene probe: nucleotide sequence and application to in situ hybridization. Dis. Aquat. Org. 1999;35:165–173. doi: 10.3354/dao035165. [DOI] [PubMed] [Google Scholar]
- Tang K.F.J., Spann K.M., Owens L., Lightner D.V. In situ detection of Australian gill-associated virus with a yellow head virus gene probe. Aquaculture. 2002;205:1–5. doi: 10.1016/S0044-8486(01)00666-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker, P.J., Cowley, J.A., Spann, K.M., Hodgson, R.A.J., Hall, M.R., Withyachumnarnkul, B., 2001. Yellow head complex viruses: transmission cycles and topographical distribution in the Asia-Pacific region. In: Browdy, C.L., Jory, D.E. (Eds.), The New Wave, Proceedings of the Special Session on Sustainable Shrimp Farming. The World Aquaculture Society, Baton Rouge, Louisiana, pp. 227–237.
- Wang C.S., Tang K.F.J., Kou G.H., Chen S.N. Yellow head disease-like virus infection in Kuruma shrimp Penaeus japonicus cultured in Taiwan. Fish Path. 1996;31:177–182. [Google Scholar]
- Wongteerasupaya C., Sriurairatana S., Vickers J.E., Akrajamorn A., Boonsaeng V., Panyim S., Tassanakajon A., Withyachumnarnjul B., Flegel T.W. Yellow-head virus of Penaeus monodon is an RNA virus. Dis. Aquat. Org. 1995;22:45–50. [Google Scholar]
- Wongteerasupaya C., Tongcheua W., Boonsaeng V., Panyim S., Tassanakajon A., Withyachumnarnkul B., Flegel T.W. Detection of yellow-head virus of Penaeus monodon by RT-PCR amplification. Dis. Aquat. Org. 1997;31:181–186. [Google Scholar]