

Abstract
Exacerbations of asthma and COPD are major causes of morbidity, mortality, and health-care costs. Over the last decade, studies using new molecular diagnostic techniques have established that respiratory viruses are a major cause of exacerbations of both asthma and COPD. The most prevalent viruses detected during exacerbations are the rhinoviruses. Despite the burden of disease associated with exacerbations, little is known about the mechanisms of virus-induced exacerbations of airway diseases. Exacerbations are associated with increased airway inflammation in patients with both asthma and COPD, but many questions remain unanswered regarding the key inflammatory cells and mediators involved. Identifying the key inflammatory mediators involved in exacerbations holds the promise of developing diagnostic and prognostic markers of exacerbation. In addition, such studies can identify new therapeutic targets for the development of novel drugs for the prevention and treatment of exacerbations.
Key words: asthma, COPD, exacerbations, viruses
Abbreviations: ENA, epithelial-derived neutrophil attractant; IFN, interferon; IL, interleukin; LTB4, leukotriene B4; NF, nuclear factor; PCR, polymerase chain reaction; RANTES, regulated upon activation, normal T-cell expressed and secreted; Th, T helper; TLR, Toll-like receptor; TNF, tumor necrosis factor
Much of the morbidity, mortality, and excess health-care utilization associated with asthma and COPD are related to episodes of acute deterioration in health that are termed exacerbations. The precise definition of an exacerbation remains controversial, but most clinicians recognize an exacerbation as an increase in respiratory symptoms that usually causes a patient to seek medical help. Asthma exacerbations are associated with shortness of breath, cough, wheezing, and chest tightness, and are accompanied by decreases in expiratory airflow manifested by reductions in peak expiratory flow. COPD exacerbations are typically associated with shortness of breath, cough, increased sputum volume, and sputum purulence, and nonspecific symptoms such as fatigue and malaise. Changes in measures of airflow are generally smaller and more variable than in patients with asthma.1 Acute exacerbations can result in excess medication use, emergency department visits, hospitalization, and even death. They are the main drivers of asthma-related costs, accounting for almost 50% of total costs. COPD patients experience a median of 2.5 exacerbations per year, and their frequency increases with increased severity of the disease.2 Exacerbations result in a faster decline in lung function in patients with COPD and so have a direct effect on progression of the disease.3
Evidence for Viruses as a Cause of Exacerbations
Asthma
Asthma exacerbations are associated with several factors, including allergen exposure, air pollution, and stress, but the major cause of exacerbations is respiratory virus infection. An association between colds and asthma exacerbations has long been recognized, but early studies yielded low virus detection rates of approximately 10%.4 These studies used virus detection methods that have low sensitivity for rhinoviruses and coronaviruses, which between them account for the majority of colds. The optimum method for virus detection is with polymerase chain reaction (PCR)-based methods, and studies using PCR have shown that respiratory viruses are responsible for a much higher proportion of asthma exacerbations than was previously suspected. In a study,5 of children in the United Kingdom, viruses were detected in 80 to 85% of exacerbations; the most common viruses detected were rhinoviruses (50% of total exacerbations or 66% in which a virus was detected). Respiratory viruses have also been detected in a high proportion of more severe exacerbations requiring hospitalization. A total of 84% of children seen in the emergency department in a South African study6 tested positive for a respiratory virus, as did 61% of children > 3 years of age who were admitted to the hospital with wheezing illness in the United States.7 Viruses have also been implicated in the pathogenesis of asthma exacerbations in adults. The first study8 using PCR to detect respiratory viruses in adults reported a respiratory virus in 44% of exacerbations (60% rhinoviruses). Subsequent studies in patients with more severe exacerbations leading to presentation to the emergency department detected a virus in 76%9 and 78%10 of cases in two studies from Australia, and in 55% of cases in a study in the United States.11 The prominent role of rhinovirus in asthma exacerbations has been further highlighted by a Canadian study12 characterizing an annual September epidemic of asthma hospitalizations occurring first in school-aged children followed by preschool children and adults. Respiratory viruses can act synergistically with other factors that cause asthma exacerbations. Admission to the hospital with an acute asthma exacerbation is strongly associated with the combination of sensitization and exposure to an allergen, and concurrent viral infection.13 The presence of high ambient levels of nitrogen dioxide prior to a viral infection is associated with more lower respiratory tract symptoms and greater falls in peak expiratory flow during the exacerbation.14
COPD
Factors associated with COPD exacerbations include changes in air temperature and concentrations of air pollutants, but most exacerbations are associated with symptoms of respiratory infection. Historically, bacteria have been considered the main infective cause, but their exact role remains contentious as bacteria are only found in approximately half of COPD exacerbations and are also found in patients who are clinically stable. Older studies detected viruses in only 10 to 20% of exacerbations15; however, as is the case in asthma patients, more recent studies using PCR have revealed that viruses have a more prominent role in the etiology of exacerbations. In a report16 from the East London COPD cohort, a respiratory virus was identified in 39% of patients with exacerbations who were treated as outpatients, with rhinoviruses accounting for 58% of the viruses present. Two studies17, 18 in COPD patients with more severe exacerbations requiring hospital admission detected a respiratory virus in 56%17 and 64%18 of patients. In COPD patients with very severe exacerbations requiring intubation and mechanical ventilation, viral infection was identified in 47%.19 Most studies have focused exclusively on bacterial or viral infection, but a recent study20 that carried out sampling for both in sputum samples found evidence of coinfection in 25% of patients with exacerbations. Together, these studies suggest that as many as 40 to 60% of acute exacerbations of COPD are associated with respiratory virus infection.
Mechanisms of Virus-Induced Exacerbations
Despite the epidemiologic evidence linking respiratory virus infection to exacerbations of COPD and asthma, the cellular and molecular mechanisms by which viruses cause exacerbations remain undetermined. The underlying pathology of asthma and COPD differ markedly, and therefore a key question is whether the mechanisms of virus-induced exacerbations in the two diseases are similar or differ. The answer to this question is of more than academic interest as current treatments for exacerbations are inadequate, and new treatments are urgently needed. Much of our knowledge regarding the mechanisms of virus-induced asthma exacerbations is derived from studies using experimental rhinovirus infection in asthmatic volunteers as a model of exacerbation. In view of the data available from the rhinovirus model and its role as the most commonly identified virus in patients with both asthma and COPD, this review will focus mainly on the mechanisms of rhinovirus-induced exacerbations.
Rhinovirus and the Lower Airway
A key issue regarding virus-induced exacerbations of airway diseases is whether upper respiratory tract viruses such as rhinoviruses can also infect the lower airway. The prevailing view for many years was that they could not, based on evidence that the optimal temperature for the growth of rhinoviruses was 33°C. However Papadopoulos et al21 demonstrated that rhinovirus can replicate in the lower airway epithelium by using in situ hybridization to exclude sample contamination from the upper airways. Therefore, viral infection of the lower airway is likely to occur and to contribute to virus-induced exacerbations of asthma and COPD.
The extent of epithelial cell destruction observed in the airway varies according to virus type. Influenza typically causes extensive epithelial necrosis, whereas rhinovirus causes only patchy damage.22 The respiratory epithelium has important regulatory roles and contributes to the immune response following virus infection through the production of inflammatory mediators, cytokines, and chemokines. The disease syndrome following respiratory virus infection is a consequence both of the direct harmful effects of the virus itself and of immunopathology resulting from the host immune response. This has led to the concept that rhinovirus infection is at least in part an immune-mediated disease with proinflammatory cytokines, chemokines, and inflammatory cell products producing pathology, perhaps more than a direct cytotoxic effect of the virus.
Virus Infection and Asthma
Inflammatory Mediators
Viral infection in asthmatic patients induces more lower respiratory tract symptoms and falls in lung function than that in nonasthmatic patients,23 but the molecular basis of the greater sensitivity of asthmatic patients to viral infection remains obscure. In vitro infection of airway epithelial cells with rhinovirus induces the secretion of a host of inflammatory mediators. This also occurs in vivo in both experimental and naturally acquired viral infections. The neutrophil chemokine interleukin (IL)-8 and the proinflammatory cytokine IL-6 have been detected in nasal samples during virus infections in asthmatic patients.24, 25 In the lower respiratory tract, increases in Il-6, IL-8, and the chemokine regulated on activation, normal T-cell expressed and secreted (RANTES) have been documented in the sputum of asthmatic patients after experimental rhinovirus infection,10, 26 and IL-8 has been detected in the sputum of children with naturally occurring exacerbations.27 While it is well-recognized that viral infection induces proinflammatory mediators, it is unclear whether the inflammatory response to viral infection differs quantitatively or qualitatively in asthmatic patients. One experimental rhinovirus infection study25 reported increased levels of IL-8 and IL-1β in nasal lavage samples in asthmatic patients but not in control subjects; however, another study24 reported no differences in IL-6, IL-8, IL-11, and granulocyte-monocyte-colony stimulating factor levels in either nasal lavage or sputum samples. A recent study10 in patients with naturally occurring virus-associated asthma exacerbations found increased levels of IL-10 messenger RNA in the sputum of asthmatic patients compared to virus-infected healthy subjects, but no differences in the level of RANTES or IL-8 between the two groups. These conflicting results highlight the need for further studies evaluating the inflammatory profile (preferably in the lower airway) in well-characterized patients during exacerbations. Studies with control subjects of nonasthmatic patients will help to ascertain whether the inflammatory response in asthmatic patients differs from that of healthy subjects.
Cellular Response
The production of chemokines by epithelial cells in response to a viral infection leads to an influx of leukocytes into the airway. These cells are an essential part of the innate and adaptive immune responses but can also result in airway pathology. The release of inflammatory cell products such as neutrophil elastase from neutrophils, major basic protein and eosinophil cationic protein from eosinophils, and reactive oxygen species can cause tissue damage. In stable patients with asthma, the eosinophil and CD4+ T cells have been identified as key cellular components of the asthma phenotype, but the cellular response during exacerbations is more heterogeneous. Severe asthma exacerbations in children are associated with increased inflammatory cell numbers and the presence of both neutrophil and eosinophil markers in sputum samples.27 Increased levels of sputum neutrophils have been reported in virus-associated exacerbations in adults, whereas exacerbations in which no virus is detected have a higher proportion of eosinophils.9 Experimental rhinovirus infection studies have reported increased numbers of neutrophils in BAL fluid samples28 but not in sputum samples.26 Few studies have compared the inflammatory cellular response to viral infection in asthmatic patients and healthy subjects. Increased numbers of lymphocytes and eosinophils in bronchial biopsy specimens are present after experimental rhinovirus infection in both asthmatic patients and healthy subjects29; however, at 6 weeks postinfection the eosinophilia persists in the asthmatic patients only. A study30 of naturally occurring colds in asthmatic patients and healthy subjects found a greater total sputum inflammatory cell count and neutrophil count with a similar differential count in the asthmatic patients. Therefore, it would seem that virus-induced exacerbations are at least partially driven by neutrophilic inflammation, and this may account for why therapy with inhaled corticosteroids is effective at suppressing (eosinophilic) airway inflammation in stable patients with asthma but are less successful at preventing exacerbations. New treatments may need to target neutrophils and neutrophil chemokines if virus-induced exacerbations are to be prevented or ameliorated.
Antiviral Immunity and Asthma
The adaptive immune response in asthma patients is associated with a T-helper (Th) type 2 cytokine profile (ie, IL-4, IL-5, and IL-13), whereas adequate antiviral immune responses require the Th1 cytokines interferon (IFN)-γ and IL-12. Th1 and Th2 immune responses demonstrate mutual inhibition; therefore, within an airway with a preexisting Th2 microenvironment there may be inhibition of Th1 immune responses. There is some clinical evidence that imbalances in Th1/Th2 immune responses influence the outcome of viral infections. Peripheral blood mononuclear cells from asthmatic subjects exposed to rhinovirus have produced significantly lower levels of IFN-γ and IL-12 with a lower IFN-γ/IL-4 ratio than in nonasthmatic patients.31 Gern et al32 showed an inverse relationship between the IFN-γ/IL-5 ratio in sputum samples and both the peak cold symptoms and time to virus clearance from sputum samples in asthmatic patients infected with rhinovirus, suggesting that a stronger Th1 immune response is associated with less severe colds and faster viral clearance. There is also evidence that weak Th1 responses are associated with more severe disease in infections with another respiratory virus—respiratory syncytial virus.33 It has been suggested that this may be another mechanism through which virus infection can exacerbate a preexisting Th2-mediated lung disease.
Most immunologic research into asthma has focused on the role of the adaptive immune response in disease pathogenesis, but evidence is emerging suggesting that innate immunity may be impaired in asthmatic patients. Wark et al34 have shown that bronchial epithelial cells obtained from asthmatic patients support markedly increased rhinovirus replication compared to cells from nonasthmatic patients. This is accompanied by reduced apoptosis of epithelial cells in the asthmatic patients and impaired production of the antiviral cytokine IFN-β. Impaired IFN-β production and cell apoptosis result in greater virus replication, eventually leading to cytotoxic cell death with the release of inflammatory mediators and large numbers of intact viral particles. The administration of IFN-β restores the virus protection observed in epithelial cells from normal airways. If confirmed in vivo, it will be interesting to see whether these novel observations translate into new therapies aimed at augmenting or replacing deficient IFN-β production in asthma patients. The key differences in the innate and acquired antiviral responses between asthmatic patients and nonasthmatic patients are summarized in Figure 1 .
Figure 1.
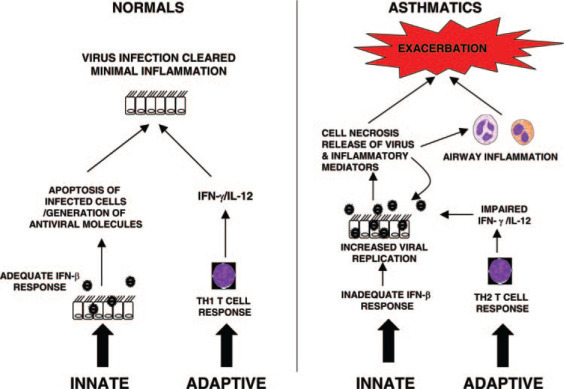
Mechanisms of virus-induced asthma exacerbations. In the normal airway epithelial cells, viral infection induces the production of IFN-β, which in turn induces apoptosis in virus-infected cells and limits virus replication. This is followed by an adaptive immune response characterized by Th1 cells that produce IFN-γ and IL-12, leading to a strong antiviral response, rapid clearance of the virus, and minimal inflammation. In asthmatic patients, both innate and adaptive antiviral immunity may be impaired, resulting in cell necrosis and the release of inflammatory mediators and virus. The increased viral load and levels of inflammatory mediators released from necrotic cells result in uncontrolled airway inflammation and exacerbation.
Other components of the innate immune response may influence the host response to viral infection such as Toll-like receptors (TLRs) and defensins. The TLRs induce antiviral responses such as type I IFNs, as well as inflammatory cytokine release and the recruitment of cells required for host defense in response to microbial products, while defensins are antimicrobial peptides with multiple effects on both the innate and acquired immune response. One TLR (TLR3) is activated by double-stranded RNA that is formed during viral replication. Rhinovirus induces the expression of both TLR3 and the defensin human β-defensin-2 in respiratory epithelial cells. In addition, TLR3 is up-regulated by allergic airway inflammation.35, 36, 37 Further studies are required to explore the possible interactions between viral infection and allergic inflammation in TLR expression and how this may influence the host response to respiratory virus infections.
Virus Infection and COPD
COPD is associated with both a pulmonary and systemic inflammatory response. Studies38 of airway inflammation in stable patients with COPD have shown that the disease is characterized by pulmonary infiltration of macrophages, neutrophils, and CD8+ T lymphocytes, together with increased expression of cytokines, chemokines, and adhesion molecules. Much less work has been carried out studying airway inflammation during exacerbations, and the results have often been conflicting. Comparing the results of different studies has often been hampered by the differing inclusion criteria (eg, chronic bronchitis vs COPD), different definitions of COPD, and different methods of airway sampling.
Inflammatory Mediators
Unlike the Th2 cytokines and eosinophil chemokines that are characteristic of asthma, COPD is associated with proinflammatory cytokines and neutrophil chemokines such as tumor necrosis factor (TNF)-α, IL-8, growth-related oncogene-α, and leukotriene B4 (LTB4). Airway inflammation is amplified during exacerbations, and the levels of inflammatory mediators are increased compared to the stable state. Increased levels of TNF-α,39, 40 IL-8,19, 39 epithelial-derived neutrophil attractant (ENA)-78,19 LTB4,41 RANTES,42 and endothelin-143 have been reported during exacerbations. IL-8 is believed to be a key neutrophil chemokine in COPD, but while some studies39, 42, 44 have reported increased IL-8 levels during exacerbations, others have not.43, 45 A study19 using bronchial biopsy specimens in patients with a severe exacerbation of COPD reported positive correlations between neutrophils and cells positive for both IL-8 and ENA-78, but the dominant CXC chemoattractant was ENA-78. Exacerbations are associated with the activation of the transcription factor nuclear factor (NF)-κB in macrophages in induced sputum samples.46 The activation of NF-κB by rhinovirus has been demonstrated in vitro and may be one mechanism whereby respiratory viruses up-regulate proinflammatory mediators in the airways, although it is not known whether rhinoviruses infect macrophages.47 Through their chemotactic effect on neutrophils (ie, IL-8, ENA-78, and LTB4), lymphocytes and monocytes (ie, RANTES) and the up-regulation of adhesion molecules (ie, TNF-α), these mediators may be central components of the increased inflammation that is characteristic of exacerbations (Fig 2 ).
Figure 2.
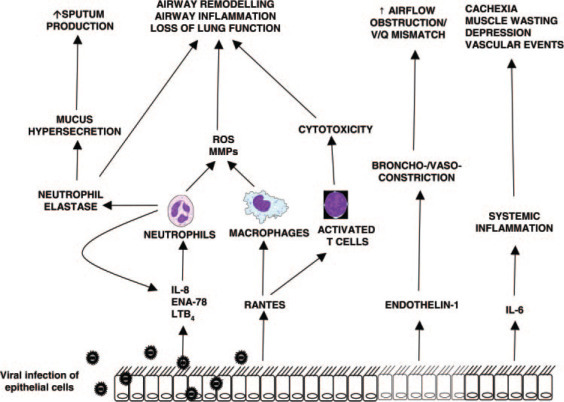
Mechanisms of virus-induced COPD exacerbations. Viral infection of epithelial cells leads to the release of proinflammatory cytokines and chemokines. Chemokines attract inflammatory cells that release toxic products, stimulating mucus production and leading to tissue damage with possible long-term loss of lung function. Some mediators such as endothelin-1 have a direct effect in causing bronchoconstriction and vasoconstriction, resulting in airflow obstruction and impaired gas exchange. MMP = matrix metalloproteinase; ROS = reactive oxygen species. V/Q = ventilation/perfusion.
Available data regarding the role of exacerbation etiology on airway inflammation has not determined whether different etiologies are associated with specific profiles of inflammatory mediators. A number of studies40, 41, 48 have reported that bacterial exacerbations are associated with higher levels of sputum IL-8, TNF-α, and LTB4 than those in which no bacterial pathogen is isolated. Virus-associated exacerbations were associated with higher levels of sputum IL-6 in one study,1 whereas in another study48 rhinovirus was not associated with significant changes in the levels of sputum inflammatory markers. Others have reported39 that increases in the levels of airway inflammatory markers occur independently of a demonstrable viral or bacterial infection.
Cellular Mechanisms
The release of cytokines by airway epithelial cells after viral infection leads to an influx of inflammatory cells. These inflammatory cells release products such as neutrophil elastase and reactive oxygen species that can cause tissue damage, stimulate mucus production, and further stimulate cytokine production. However, not all studies of the cellular inflammatory response in COPD exacerbations have had consistent results. Some studies43, 45 measuring inflammatory cell counts in induced sputum samples obtained during exacerbations have reported no differences in total cell numbers compared to the stable state, whereas others have found increases in total leukocyte counts,48 and increased neutrophil, eosinophil, and lymphocyte counts.42, 49 In a study19 of COPD patients intubated with severe exacerbations, increased numbers of neutrophils were seen in bronchial biopsy specimens compared to stable patients with COPD, but other cell types were not reported. Only one study20 has attempted to correlate the patterns of airway inflammation with exacerbation etiology. In this study,20 the levels of sputum neutrophils were elevated during exacerbation regardless of etiology, but the levels of eosinophils were increased only in patients with viral infections. The variability in the results highlights the need for further studies in carefully selected populations coupled with the determination of exacerbation etiology.
Therefore, although it is well established that COPD exacerbations are associated with increased airway inflammation, there is marked variability in the nature of the inflammatory response, and the relationship between airway inflammation and etiology has not been established. The identification of valid biomarkers of exacerbation will allow for a more objective diagnosis of exacerbations and the determination of severity. In addition, a marker that distinguishes between viral and bacterial infection has the potential for reducing the overuse of antibiotics and the targeted use of antiviral agents when these become available.
Conclusions
Despite the advances made in treatments for both asthma and COPD, acute exacerbations remain a major cause of morbidity and mortality. Recent years have seen the epidemiologic evidence linking viruses with exacerbations getting ever stronger, and the stage is now set for progress in identifying important mechanisms of virus-induced asthma exacerbations. Viral infection is associated with airways inflammation, but studies of the cellular and molecular response to infection have not shown clear-cut qualitative or quantitative differences between asthmatic patients and nonasthmatic subjects. There are no studies comparing virus-induced inflammation in COPD patients with healthy control subjects. The recent evidence of impaired innate immunity in asthmatic patients suggests that this may be a key mechanism in the pathogenesis of virus-induced asthma exacerbations. Further work is needed to identify whether similar mechanisms are involved in COPD exacerbations. Identifying the key inflammatory mediators involved in exacerbations, as well as the host defense mechanisms providing protection, holds the promise of developing new therapeutic agents and so reducing the burden of disease associated with asthma and COPD exacerbations.
Footnotes
The authors have reported to the ACCP that no significant conflicts of interest exist with any companies/organizations whose products or services may be discussed in this article.
Reproduction of this article is prohibited without written permission from the American College of Chest Physicians (www.chestjournal.org/misc/reprints.shtml).
References
- 1.Seemungal TA, Harper-Owen R, Bhowmik A. Detection of rhinovirus in induced sputum at exacerbation of chronic obstructive pulmonary disease. Eur Respir J. 2000;16:677–683. doi: 10.1034/j.1399-3003.2000.16d19.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Donaldson GC, Seemungal TA, Patel IS. Longitudinal changes in the nature, severity and frequency of COPD exacerbations. Eur Respir J. 2003;22:931–936. doi: 10.1183/09031936.03.00038303. [DOI] [PubMed] [Google Scholar]
- 3.Donaldson GC, Seemungal TA, Bhowmik A. Relationship between exacerbation frequency and lung function decline in chronic obstructive pulmonary disease. Thorax. 2002;57:847–852. doi: 10.1136/thorax.57.10.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beasley R, Coleman ED, Hermon Y. Viral respiratory tract infection and exacerbations of asthma in adult patients. Thorax. 1988;43:679–683. doi: 10.1136/thx.43.9.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Johnston SL, Pattemore PK, Sanderson G. Community study of role of viral infections in exacerbations of asthma in 9–11 year old children. BMJ. 1995;310:1225–1229. doi: 10.1136/bmj.310.6989.1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kling S, Donninger H, Williams Z. Persistence of rhinovirus RNA after asthma exacerbation in children. Clin Exp Allergy. 2005;35:672–678. doi: 10.1111/j.1365-2222.2005.02244.x. [DOI] [PubMed] [Google Scholar]
- 7.Heymann PW, Carper HT, Murphy DD. Viral infections in relation to age, atopy, and season of admission among children hospitalized for wheezing. J Allergy Clin Immunol. 2004;114:239–247. doi: 10.1016/j.jaci.2004.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nicholson KG, Kent J, Ireland DC. Respiratory viruses and exacerbations of asthma in adults. BMJ. 1993;307:982–986. doi: 10.1136/bmj.307.6910.982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wark PA, Johnston SL, Moric I. Neutrophil degranulation and cell lysis is associated with clinical severity in virus-induced asthma. Eur Respir J. 2002;19:68–75. doi: 10.1183/09031936.02.00226302. [DOI] [PubMed] [Google Scholar]
- 10.Grissell TV, Powell H, Shafren DR. Interleukin-10 gene expression in acute virus-induced asthma. Am J Respir Crit Care Med. 2005;172:433–439. doi: 10.1164/rccm.200412-1621OC. [DOI] [PubMed] [Google Scholar]
- 11.Atmar RL, Guy E, Guntupalli KK. Respiratory tract viral infections in inner-city asthmatic adults. Arch Intern Med. 1998;158:2453–2459. doi: 10.1001/archinte.158.22.2453. [DOI] [PubMed] [Google Scholar]
- 12.Johnston NW, Johnston SL, Norman GR. The September epidemic of asthma hospitalization: school children as disease vectors. J Allergy Clin Immunol. 2006;117:557–562. doi: 10.1016/j.jaci.2005.11.034. [DOI] [PubMed] [Google Scholar]
- 13.Green RM, Custovic A, Sanderson G. Synergism between allergens and viruses and risk of hospital admission with asthma: case-control study. BMJ. 2002;324:763. doi: 10.1136/bmj.324.7340.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chauhan AJ, Inskip HM, Linaker CH. Personal exposure to nitrogen dioxide (NO2) and the severity of virus-induced asthma in children. Lancet. 2003;361:1939–1944. doi: 10.1016/S0140-6736(03)13582-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smith CB, Golden CA, Kanner RE. Association of viral andMycoplasma pneumoniaeinfections with acute respiratory illness in patients with chronic obstructive pulmonary diseases. Am Rev Respir Dis. 1980;121:225–232. doi: 10.1164/arrd.1980.121.2.225. [DOI] [PubMed] [Google Scholar]
- 16.Seemungal T, Harper-Owen R, Bhowmik A. Respiratory viruses, symptoms, and inflammatory markers in acute exacerbations and stable chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2001;164:1618–1623. doi: 10.1164/ajrccm.164.9.2105011. [DOI] [PubMed] [Google Scholar]
- 17.Rohde G, Wiethege A, Borg I. Respiratory viruses in exacerbations of chronic obstructive pulmonary disease requiring hospitalisation: a case-control study. Thorax. 2003;58:37–42. doi: 10.1136/thorax.58.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tan WC, Xiang X, Qiu D. Epidemiology of respiratory viruses in patients hospitalized with near-fatal asthma, acute exacerbations of asthma, or chronic obstructive pulmonary disease. Am J Med. 2003;115:272–277. doi: 10.1016/s0002-9343(03)00353-x. [DOI] [PubMed] [Google Scholar]
- 19.Qiu Y, Zhu J, Bandi V. Biopsy neutrophilia, neutrophil chemokine and receptor gene expression in severe exacerbations of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2003;168:968–975. doi: 10.1164/rccm.200208-794OC. [DOI] [PubMed] [Google Scholar]
- 20.Papi A, Bellettato CM, Braccioni F. Infections and airway inflammation in chronic obstructive pulmonary disease severe exacerbations. Am J Respir Crit Care Med. 2006;173:1114–1121. doi: 10.1164/rccm.200506-859OC. [DOI] [PubMed] [Google Scholar]
- 21.Papadopoulos NG, Bates PJ, Bardin PG. Rhinoviruses infect the lower airways. J Infect Dis. 2000;181:1875–1884. doi: 10.1086/315513. [DOI] [PubMed] [Google Scholar]
- 22.Arruda E, Boyle TR, Winther B. Localization of human rhinovirus replication in the upper respiratory tract byin situhybridization. J Infect Dis. 1995;171:1329–1333. doi: 10.1093/infdis/171.5.1329. [DOI] [PubMed] [Google Scholar]
- 23.Corne JM, Marshall C, Smith S. Frequency, severity, and duration of rhinovirus infections in asthmatic and non-asthmatic individuals: a longitudinal cohort study. Lancet. 2002;359:831–834. doi: 10.1016/S0140-6736(02)07953-9. [DOI] [PubMed] [Google Scholar]
- 24.Fleming HE, Little FF, Schnurr D. Rhinovirus-16 colds in healthy and in asthmatic subjects: similar changes in upper and lower airways. Am J Respir Crit Care Med. 1999;160:100–108. doi: 10.1164/ajrccm.160.1.9808074. [DOI] [PubMed] [Google Scholar]
- 25.de Kluijver J, Grunberg K, Pons D. Interleukin-1β and interleukin-1ra levels in nasal lavages during experimental rhinovirus infection in asthmatic and non-asthmatic subjects. Clin Exp Allergy. 2003;33:1415–1418. doi: 10.1046/j.1365-2222.2003.01770.x. [DOI] [PubMed] [Google Scholar]
- 26.Grunberg K, Smits HH, Timmers MC. Experimental rhinovirus 16 infection: effects on cell differentials and soluble markers in sputum in asthmatic subjects. Am J Respir Crit Care Med. 1997;156:609–616. doi: 10.1164/ajrccm.156.2.9610079. [DOI] [PubMed] [Google Scholar]
- 27.Norzila MZ, Fakes K, Henry RL. Interleukin-8 secretion and neutrophil recruitment accompanies induced sputum eosinophil activation in children with acute asthma. Am J Respir Crit Care Med. 2000;161:769–774. doi: 10.1164/ajrccm.161.3.9809071. [DOI] [PubMed] [Google Scholar]
- 28.Jarjour NN, Gern JE, Kelly EA. The effect of an experimental rhinovirus 16 infection on bronchial lavage neutrophils. J Allergy Clin Immunol. 2000;105:1169–1177. doi: 10.1067/mai.2000.106376. [DOI] [PubMed] [Google Scholar]
- 29.Fraenkel DJ, Bardin PG, Sanderson G. Lower airways inflammation during rhinovirus colds in normal and in asthmatic subjects. Am J Respir Crit Care Med. 1995;151:879–886. doi: 10.1164/ajrccm/151.3_Pt_1.879. [DOI] [PubMed] [Google Scholar]
- 30.Pizzichini MM, Pizzichini E, Efthimiadis A. Asthma and natural colds: inflammatory indices in induced sputum: a feasibility study. Am J Respir Crit Care Med. 1998;158:1178–1184. doi: 10.1164/ajrccm.158.4.9712082. [DOI] [PubMed] [Google Scholar]
- 31.Papadopoulos NG, Stanciu LA, Papi A. A defective type 1 response to rhinovirus in atopic asthma. Thorax. 2002;57:328–332. doi: 10.1136/thorax.57.4.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gern JE, Vrtis R, Grindle KA. Relationship of upper and lower airway cytokines to outcome of experimental rhinovirus infection. Am J Respir Crit Care Med. 2000;162:2226–2231. doi: 10.1164/ajrccm.162.6.2003019. [DOI] [PubMed] [Google Scholar]
- 33.Roman M, Calhoun WJ, Hinton KL. Respiratory syncytial virus infection in infants is associated with predominant Th-2-like response. Am J Respir Crit Care Med. 1997;156:190–195. doi: 10.1164/ajrccm.156.1.9611050. [DOI] [PubMed] [Google Scholar]
- 34.Wark PA, Johnston SL, Bucchieri F. Asthmatic bronchial epithelial cells have a deficient innate immune response to infection with rhinovirus. J Exp Med. 2005;201:937–947. doi: 10.1084/jem.20041901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hewson CA, Jardine A, Edwards MR. Toll-like receptor 3 is induced by and mediates antiviral activity against rhinovirus infection of human bronchial epithelial cells. J Virol. 2005;79:12273–12279. doi: 10.1128/JVI.79.19.12273-12279.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fransson M, Adner M, Erjefalt J. Up-regulation of Toll-like receptors 2, 3 and 4 in allergic rhinitis. Respir Res. 2005;6:100. doi: 10.1186/1465-9921-6-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Proud D, Sanders SP, Wiehler S. Human rhinovirus infection induces airway epithelial cell production of human β-defensin 2 bothin vitroandin vivo. J Immunol. 2004;172:4637–4645. doi: 10.4049/jimmunol.172.7.4637. [DOI] [PubMed] [Google Scholar]
- 38.Saetta M, Turato G, Maestrelli P. Cellular and structural bases of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2001;163:1304–1309. doi: 10.1164/ajrccm.163.6.2009116. [DOI] [PubMed] [Google Scholar]
- 39.Aaron SD, Angel JB, Lunau M. Granulocyte inflammatory markers and airway infection during acute exacerbation of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2001;163:349–355. doi: 10.1164/ajrccm.163.2.2003122. [DOI] [PubMed] [Google Scholar]
- 40.Sethi S, Muscarella K, Evans N. Airway inflammation and etiology of acute exacerbations of chronic bronchitis. Chest. 2000;118:1557–1565. doi: 10.1378/chest.118.6.1557. [DOI] [PubMed] [Google Scholar]
- 41.Gompertz S, O’Brien C, Bayley DL. Changes in bronchial inflammation during acute exacerbations of chronic bronchitis. Eur Respir J. 2001;17:1112–1119. doi: 10.1183/09031936.01.99114901. [DOI] [PubMed] [Google Scholar]
- 42.Fujimoto K, Yasuo M, Urushibata K. Airway inflammation during stable and acutely exacerbated chronic obstructive pulmonary disease. Eur Respir J. 2005;25:640–646. doi: 10.1183/09031936.05.00047504. [DOI] [PubMed] [Google Scholar]
- 43.Roland M, Bhowmik A, Sapsford RJ. Sputum and plasma endothelin-1 levels in exacerbations of chronic obstructive pulmonary disease. Thorax. 2001;56:30–35. doi: 10.1136/thorax.56.1.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Drost EM, Skwarski KM, Sauleda J. Oxidative stress and airway inflammation in severe exacerbations of COPD. Thorax. 2005;60:293–300. doi: 10.1136/thx.2004.027946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bhowmik A, Seemungal TA, Sapsford RJ. Relation of sputum inflammatory markers to symptoms and lung function changes in COPD exacerbations. Thorax. 2000;55:114–120. doi: 10.1136/thorax.55.2.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Caramori G, Romagnoli M, Casolari P. Nuclear localisation of p65 in sputum macrophages but not in sputum neutrophils during COPD exacerbations. Thorax. 2003;58:348–351. doi: 10.1136/thorax.58.4.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Papi A, Johnston SL. Rhinovirus infection induces expression of its own receptor intercellular adhesion molecule 1 (ICAM-1) via increased NF-κB-mediated transcription. J Biol Chem. 1999;274:9707–9720. doi: 10.1074/jbc.274.14.9707. [DOI] [PubMed] [Google Scholar]
- 48.Hurst JR, Perera WR, Wilkinson TM. Systemic and upper and lower airway inflammation at exacerbation of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2006;173:71–78. doi: 10.1164/rccm.200505-704OC. [DOI] [PubMed] [Google Scholar]
- 49.Mercer PF, Shute JK, Bhowmik A. MMP-9, TIMP-1 and inflammatory cells in sputum from COPD patients during exacerbation. Respir Res. 2005;6:151. doi: 10.1186/1465-9921-6-151. [DOI] [PMC free article] [PubMed] [Google Scholar]