

Abstract
Ste24, an integral membrane protein zinc metalloprotease, is found in every kingdom of eukaryotes. It was discovered approximately 20 years ago by yeast genetic screens identifying it as a factor responsible for processing the yeast mating a-factor pheromone. In animals, Ste24 processes prelamin A, a component of the nuclear lamina; mutations in the human ortholog of Ste24 diminish its activity, giving rise to genetic diseases of accelerated aging (progerias). Additionally, lipodystrophy, acquired from the standard highly active antiretroviral therapy used to treat AIDS patients, likely results from off-target interactions of HIV (aspartyl) protease inhibitor drugs with Ste24. Ste24 possesses a novel “α-barrel” structure, consisting of a ring of seven transmembrane α-helices enclosing a large (> 12,000 Å3) interior volume that contains the active-site and substrate-binding region; this “membrane-interior reaction chamber” is unprecedented in integral membrane protein structures. Additionally, the surface of the membrane-interior reaction chamber possesses a strikingly large negative electrostatic surface potential, adding additional “functional mystery.” Recent publications implicate Ste24 as a key factor in several endoplasmic reticulum processes, including the unfolded protein response, a cellular stress response of the endoplasmic reticulum, and removal of misfolded proteins from the translocon. Ste24, with its provocative structure, enigmatic mechanism, and recently emergent new biological roles including “translocon unclogger” and (non-enyzmatic) broad-spectrum viral restriction factor, presents far differently than before 2016, when it was viewed as a “CAAX protease” responsible for cleavage of prenylated (farnesylated or geranylgeranylated) substrates. The emphasis of this review is on Ste24 of the “Post-CAAX-Protease Era.”
Abbreviations: ZMP, zinc metalloprotease; TM, transmembrane; L5D, Loop 5 Domain; CTD, C-terminal domain
Keywords: Ste24, metalloprotease, membrane protein, gluzincin, α-barrel.
Graphical abstract
Highlights
-
•
Ste24 is a eukaryotic integral membrane protein of novel structure.
-
•
Ste24 is a gluzincin ZMP whose structure/function relationships are poorly explored.
-
•
ZMP core, ZMP accessory, and “ɑ-barrel modules form the Ste24 tripartite architecture.
-
•
Emergent biology of Ste24 includes roles as a translocon unclogger and a viral restriction factor.
Introduction
Ste24 was discovered more than 20 years ago through yeast genetic screens identifying its mutation as causing a mating Sterility phenotype. Specifically, Saccharomyces cerevisiae Ste24 (ScSte24) is responsible for proteolytic processing of the yeast a-factor mating pheromone [[1], [2], [3]]. Additionally, Ste24 was shown [1,2] to contain the HExxH zinc metalloprotease (ZMP) consensus sequence [4]. In humans (Homo sapiens), HsSte24 (often referred to in the literature as ZMPSTE24) was identified to play a role in processing prelamin A, an intermediate filament protein, into mature Lamin A, a component of the nuclear lamin [5,6]. Prior to 2016, the vast preponderance of research and scientific literature on Ste24 described it as a “CAAX protease,” targeting proteins possessing a C-terminal tetrapeptide motif of (prenylated, farnesylated or geranylgeranylated) Cysteine, Aliphatic, Aliphatic, X (any residue) and cleaving the “AAX” tripeptide from the C-terminus [7]. Ste24, in yeast and humans, shares functional redundancy as a CAAX protease (cleaving a-factor or prelamin A, respectively) with Ras-converting enzyme 1 (Rce1) [1]. In both a-factor and prelamin A, Ste24 cleaves at a second non-prenylated site several residues N-terminal to the CAAX box in a reaction that is Ste24-dependent [2,8]. A significant motivation for this review is to present more recent published results, from our lab and others, which both indicate additional significant biological roles for Ste24, and very strongly suggest that Ste24 is not primarily a CAAX protease (and may likely not be one at all).
The Unique “α-Barrel” Structure of Ste24
X-ray crystal structures of yeast (Saccharomyces mikatae) and human Ste24, SmSte24, and HsSte24, respectively, were published concurrently in 2013 [9,10]. The structures are highly similar, with an RMSD (Cα) of 1.7 Å; also, HsSte24 can functionally complement yeast Ste24 in vivo [3]. Ste24 is a multi-domain ZMP constructed of seven transmembrane (TM) α-helices, two of which, TMs VI and VII, provide residues coordinating the zinc and the catalytic base, and a pair of membrane-interfacial domains, the Loop 5 Domain (L5D) and C-terminal domain (CTD) (Figure 1(a)). The membrane-spanning portion of the structure consists of seven TM α-helices, most of which are bent or kinked. The distortion of these TM α-helices results in fenestrations between neighboring helical elements likely providing putative substrate entry/exit points (see section “Ste24 is not an intramembrane enzyme”) (Figure 1(b)). By analogy to (gram-negative bacterial and mitochondrial) outer membrane β-barrels, the TM helices of Ste24 form an “α-barrel” [12], which encompasses a voluminous cavity (> 12,000 Å3) within the membrane interior (Figure 1(c)). The ZMP active site is within this “reaction cavity,” immediately indicating that the protein substrates proteolyzed by Ste24 must (somehow) enter this cavity for processing. In addition to the α-barrel, two interfacial extramembranous domains, the L5D and CTD, enclose the side of the α-barrel proximal to the enzyme's active site (Figure 1(a) and (b)). Another distinctive aspect of the α-barrel is its large negative electrostatic (interior) surface potential (Figure 1(d)), which has been previously noted [10]. Due to the amphipathic nature of the TM α-helices of Ste24, TM helix prediction programs based on primary amino acid sequence fail to identify all of them (unpublished observation).
Figure 1.

Structural overview of HsSte24. (a) Topology diagram of HsSte24. HsSte24 possesses seven TM α-helices, which span the membrane, with helices VI and VI containing the catalytic gluzincin ZMP 335HExxH339…E415 consensus motif [11]. Residues His335, His339, and Glu415 ligate the zinc (shown as a gold sphere) and Glu336 acts as the catalytic base [10]. TM α-helices are labeled I-VII. Two large domains encircle the coordinated zinc: the mixed α-helix-β-sheet L5D positioned between TM α-helices V and VI, and the α-helical CTD between TM α-helices I and VII. The Ste24 L5D of higher eukaryotes, as compared to Ste24 enzymes from fungi, plants, and protists, contains a variable length insertion of unknown function (5–37 residues; 37 residues in HsSte24) after the α3-helix; this insertion sequence is indicated by the bolded asterisk (*) [12]. (b) Van der Waals surface representation of the structure of HsSte24 (PDB 6BH8) [13]. Membrane boundaries, from the Orientations of Proteins in Membranes (OPM) database (https://opm.phar.umich.edu/) [14], are displayed as parallel black lines with the luminal and cytoplasmic (CP)/nucleoplasmic (NP) boundaries indicated. The color-coding presented in figure (a) is maintained. For clarity and viewer orientation, the L5D and CTD domains are specifically labeled. Fenestrations in the HsSte24 structure are indicated by black boxes. (c) Ribbon representation of the structure of HsSte24. HsSte24 contains a large cavity of greater than 12,000 Å3, visualized as a light gray surface, as measured with the CASTp webserver (http://sts.bioe.uic.edu/castp/index.html) [15]. (d) Electrostatics of the HsSte24 cavity. The electrostatic potential of the surface of the inner cavity of HsSte24 was calculated with the PDB2PQR server (http://nbcr-222.ucsd.edu/pdb2pqr_2.1.1/) [16] and APBS [17,18]. The orientation of HsSte24 is identical to that of (c), with HsSte24 shown as a gray ribbon with its catalytic gluzincin residues depicted as gray sticks. The electrostatic surface potential (in units of kT/e), reveals a strongly negative electrostatic surface potential of the HsSte24 “reaction cavity.”
Ste24 Enzymology
Ste24 is not an intramembrane enzyme
A hallmark of intramembrane proteases is the presence of their active sites within the membrane interior, i.e., the scissile bonds of their substrates are also located within the more hydrophobic portion of the bilayer. In contrast, the active site of Ste24 is located at the membrane interface (Figure 2(a)), consistent with the location of prenylated (or other surface-bound) substrates. The splayed TM helices V and VI, partially occluded by the L5D, were posited to serve as an “entrance portal” for interfacially-bound substrates at the cytoplasmic/nucleoplasmic leaflet (Figure 1(b)) [9,10]. The authors of the SmSte24 structure further suggested that splayed TM helices VII and I, partially occluded by the CTD, served as an “exit portal” [9]; the authors of the (original) HsSte24 structure suggest that entrance and exit portals are identical [10]. Currently, no experimental results exist to characterize how proteins enter the α-barrel reaction cavity, in which the substrate-binding groove and active site are located. Although variants of “fenestration portal” models have been hypothesized, other modes of access, such as through the cytoplasmic/nucleoplasmic membrane surface occupied by L5D and CTD, cannot currently be excluded.
Figure 2.

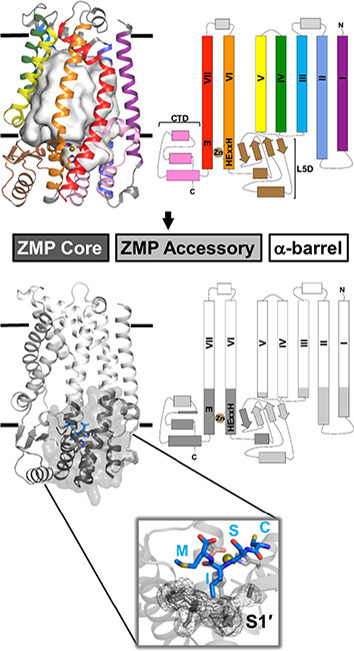
HsSte24 is an integral membrane protease possessing a tripartite architecture. (a) Location of the HsSte24 active-site as compared to the intramembrane proteases Rce1 (PDB 4CAD) [19], Site-2-Protease (S2P) (PDB 3B4R) [20], and the rhomboid protease, GlpG (PDB 2XTV) [21]. The membrane boundaries predicted by OPM [14] are displayed as parallel black lines with the luminal and cytoplasmic (CP)/nucleoplasmic (NP) boundaries indicated for all four structures. The active sites of HsSte24 (a ZMP), Rce1 (a glutamate-dependent protease), S2P (a ZMP), and GlpG (a serine protease) are indicated by the dashed circle. While Rce1, S2P, and GlpG cleave substrates within the membrane bilayer, the HsSte24 active-site is positioned outside the membrane. (b) Comparison of HsSte24 with the soluble gluzincin thermolysin (TLN). The extra-membrane lobe of HsSte24 (gray carbon) shares structural homology with thermolysin (TLN) (green carbon) (PDB 1LNF) [22] at the zinc-containing active site. The conserved α-helices, which contain the HExxE…E motif residues (shown as sticks) and a neighboring β-strand comprise a conserved structural motif in gluzincins. (c) Active-site superimpositions of HsSte24 and thermolysin. The active sites were aligned via the Pymol program SUPER [23] using the four conserved gluzincin residues of each structure, HExxH…E, to seed the alignment. Examination of the two active-sites reveals additional essential residues (i.e., outside the HExxH…E gluzincin motif) for substrate cleavage including elements of the Ste24 L5D, Asn265, and Ala266 of the β3 element, and CTD, His459 of the α6/α7 loop and Arg465 of the α7-helix, generating a “sandwich-like” motif around the zinc atom. Peptide substrate orientation in Ste24 has been determined indirectly, by comparison of the crystal structure of a yeast ortholog of Ste24 [9] to substrate and inhibitor bound structures of thermolysin [24], and directly, by structures of complexes of HsSte24 with the tetrapeptide CSIM [10] and the inhibitor phosphoramidon [13] (also see Figure 3). (d) The tripartite architecture of HsSte24. Bioinformatic and structural analysis of the Ste24 enzyme family, and identification of soluble gluzincin ZMP structural orthologs to Ste24 enabled conceptualization and development of a three-component modular tripartite architecture of Ste24 [12]. This tripartite architecture, depicted as three-dimensional structure (left-hand panel) and two-dimensional topology representation (right-hand panel), consists of (i) a chiefly cytoplasmic “ZMP core” module (shown as purple ribbon), (ii) a mixed soluble/membrane-associated “ZMP accessory” module (shown as lavender ribbon), and (iii) a heptahelical, membrane-spanning “α-barrel” module (shown as light-gray ribbon). The gluzincin HExxH…E motif, located in the ZMP core, is shown as sticks and the zinc ion as a gold sphere. The color-coding scheme is conserved between the three- and two-dimensional representations.
Ste24 is a gluzincin ZMP
The HExxH consensus sequence [4], where the two conserved histidines bind the catalytic zinc, is the motif of the Zincin family of ZMPs [11]. Identification of the third Zn-ligating amino acid provides further classification: “HExxH…E/H,” in which glutamate is most often present at the third position, denotes the gluzincin family of ZMPs [11]. The similarity of Ste24 to soluble gluzincins was recognized in the two original papers describing the yeast and human Ste24 crystal structures [9,10], and recently analyzed in much greater detail [12]. The soluble portion of the HsSte24 structure shares a conserved active-site architecture with the soluble gluzincin thermolysin (Figure 2(b)). The structural superposition of Ste24 and thermolysin [22] is shown in Figure 2(c), which also further illustrates the interfacial positioning of the Ste24 active site. The active sites of multiple soluble gluzincins, thermolysin (MEROPS database [26] family designation M4), neprilysin (M13), and a ZMP from Geobacter sulfurreducens (M48C), align well with the active site of Ste24 (subfamily M48A) [9,10,12].
The tripartite architecture of Ste24
Although the relationship between soluble gluzincin ZMPs and Ste24 is well established, much of Ste24 bears no resemblance whatsoever to them (Figure 2(b)). Moreover, as previously mentioned, the structure of the α-barrel is completely novel. What further analysis, if any, can be performed to aid in understanding, and to guide future studies, of Ste24? A very recent detailed bioinformatic and structural study of Ste24 has yielded a modular tripartite architecture representation of Ste24 [12] (Figure 2(d)). Analysis of soluble gluzincins and their comparison to Ste24 localized a “ZMP core” module to portions of TM α-helices VI and VII extending beyond the membrane interior, along with regions of the L5D and CTD. The ZMP core possesses highest structural homology to the most structure- and sequence-conserved portions of the M4, M13, and M48 soluble gluzincins. Variation in function of different soluble gluzincins is conferred by regions of low sequence conservation, often retaining structural homology, that are proximal to their respective ZMP cores. In Ste24, this “ZMP accessory” module is present in those portions of TM α-helices I–III and V extending beyond the membrane interior and located at the membrane interface, and in the portions of the L5D and CTD that are not in the ZMP core. The majority of the residues in Ste24 are in the α-barrel module. Ste24 is a ubiquitous eukaryotic integral membrane protein found in every kingdom (protist, fungus, plant, animal). Multiple sequence analysis of 58 orthologous Ste24 sequences revealed that 38 residues (8% of the HsSte24 sequence) in Ste24 are absolutely conserved, a strikingly large percentage of identical residues for an evolutionarily ancient and very highly conserved protein. Using the fungal SmSte24 sequence as a reference, pairwise ortholog identities are greater than 30% across all kingdoms; if amino acid chemical conservation (charged, amphipathic, aromatic, polar) is utilized, orthologs are seen to be even more alike. In the context of the tripartite architecture of Ste24, 18, 13, and 7 absolutely conserved residues are located in the ZMP core, ZMP accessory, and α-barrel modules, respectively. Nearly all of the conserved residues in the ZMP core module can be rationalized based on comparison to soluble gluzincins [12]. Many of the conserved residues in the ZMP accessory module are rationalized to play conserved structural roles or to be involved in the interface between the ZMP accessory module and the ZMP core module (or located at the membrane interface). The seven absolutely conserved residues of the α-barrel module are especially intriguing; as most of them are distant from the “enzymatically relevant” ZMP core and ZMP accessory modules, their role(s) are currently unknown. In light of the constantly expanding scientific and clinically relevant literature of HsSte24 (see section “Existing and Emergent Biology of Ste24”), the Ste24 tripartite architecture will facilitate better understanding of the functional data. For example, mutation of the ZMP core is best understood in the context of comparable residues in homologous, soluble gluzincins. However, mutations of some ZMP accessory and all α-barrel modules are likely “Ste24 unique,” best studied by analysis of the Ste24 family itself.
Ste24: not a CAAX protease?
Ste24 and Rce1 [27] have been denoted as “CAAX proteases,” cleaving proteins possessing a C-terminal tetrapeptide motif of (prenylated, farnesylated, or geranylgeranylated) Cysteine, Aliphatic, Aliphatic, X (any residue) [7]. However, no naturally occurring Ste24-specific CAAX boxes exist [28]. In 2016, Hildebrandt et al. [28] demonstrated that Ste24 was able to cleave both prenylated substrates (derived from K-Ras4b or yeast a-factor mating pheromone) and non-prenylated substrates (derived from amyloid Aβ or insulin, known substrates of M16A-soluble ZMPs). Thus, while Rce1 absolutely requires the presence of a farnesyl or geranylgeranyl moiety for function, Ste24 does not. Further inspection of the cleavage sites of the protein substrates cut by Ste24 in this study revealed preference for P1′ being a hydrophobic residue. However, given the limited number of substrates examined to date, Ste24 substrate specificity remains largely unknown. Nonetheless, these results strongly indicate that the substrate specificity of Ste24 is very likely broader than previously thought, and that prenylation is dispensable for substrate recognition and cleavage.
Existing and Emergent Biology of Ste24
Laminopathies and (AIDS) lypodystrophies
In animals, Ste24 processes prelamin A, the precursor to the nuclear intermediate filament protein lamin A. Lamins comprise nuclear lamina, which provide mechanical stability to the nuclear envelope and function as scaffolds for DNA repair and replication complexes [29]. Mutations in either HsSte24/ZMPSTE24 or prelamin A are associated with a spectrum of accelerated aging diseases referred to as progerias [30], and the severity of different progerias appears to be correlated with the extent of loss of HsSte24 activity and/or stability [31,32]. Improper prelamin A processing is also correlated with deleterious alterations to adipose tissue localization and accumulation, known as lipodystrophy, due to the toxic effects of prelamin A accumulation leading to altered expression of genes responsible for adipocyte proliferation and differentiation [33,34]. Additionally, the inability of some AIDS patients to produce and maintain adipose tissue, acquired from antiretroviral therapy, likely results from off-target interactions of HIV (aspartyl) protease inhibitor drugs with HsSte24 [[35], [36], [37], [38]]. Because of HsSte24's roles in these syndromes, the Zmpste24 −/− mouse has been suggested as a model for senescent wound healing [39] and lipodystrophy [40].
Translocon unclogging and diabetes
A series of publications have suggested more general roles for the Ste24 family in multiple processes occurring in the endoplasmic reticulum (ER), including proper orientation of a single-pass TM α-helical protein [41] and the unfolded protein response, a cellular stress response of the ER [42]. Utilizing APYG (the “Awesome Power of Yeast Genetics” [43]), Ste24 was identified to function as a “translocon unclogger,” removing misfolded proteins that “clog” the translocon during signal recognition particle-independent protein translocation [44]. This first report of Ste24 as translocon unclogger utilized a “clogging” chimera comprising bacterial and yeast protein domains. A 2018 publication, from a different research group, identified Ste24 as having a similar function against human islet amyloid polypeptide [45], which commonly misfolds in patients with type 2 diabetes (with this “oligomer-induced proteotoxicity” inducing β-cell failure). These results, collectively, suggest a role for Ste24 in the ER-associated degradation pathway [46]. The implication of Ste24 in multiple disparate clinical indications (e.g., progerias, AIDS drug off-target side effects, diabetes) suggests a possibility that it is a “druggable target” for one or more conditions.
Ste24 is a (non-enzymatic) broad-spectrum viral restriction factor
Most human and animal viruses are enveloped, surrounded by a viral lipid bilayer membrane, requiring fusion with one or more cellular membranes of the host in order to deliver their genomes [47]. This process very commonly occurs via fusion with endosomes, in which dramatic pH-dependent conformational changes in viral fusion proteins bring viral and endosomal membranes into close apposition, driving lipid mixing of viral and endosomal membranes (“hemifusion”) and completing the process by formation of a “fusion pore,” which enables injection of viral DNA (or RNA) into the cytosol of the host/target cell [48]. This process is mediated by direct interaction of specialized structural elements of viral fusion protein ectodomains with the endosomal membrane, perturbing the endosomal membrane to permit the complex topological transformations of viral membrane fusion. As infectivity by all endocytosed enveloped viruses requires the fusion of viral and endosomal membranes, inhibition of this process could be a target for development of broad-spectrum antiviral drugs [49]. A potential source of broad-spectrum viral restriction factor proteins is the host innate immune response; in particular, interferon signaling in response to host challenge by a viral (or bacterial) pathogen induces production of more than 2000 gene products [50]. A functional genomic screen for interferon-induced host-produced viral restriction factors for influenza yielded the interferon-inducible transmembrane protein (IFITM) family; in addition to their mediation of resistance to flu, IFITM proteins discovered in this study also mediated resistance to West Nile and Dengue viruses [51]. In multiple additional studies (reviewed in [52,53]), expression of IFITM proteins reduced infectivity of more than 20 different viruses, including Ebola [54], hepatitis C [55], HIV [56], MERS- and SARS-coronaviruses [57,58], and Zika [59]. In 2017, HsSte24 was identified, by affinity purification coupled with mass spectrometry, as interacting with IFITM3 [60,61], and was characterized as an “intrinsic broad-spectrum antiviral protein,” inhibiting infection by cowpox, Ebola, influenza, Sindbis, vaccinia, vesicular stomatitis, and Zika viruses [60]. Additionally, proteolytically inactive HsSte24/ZMPSTE24 exhibited the same broad-spectrum viral restriction factor phenotype; thus, a non-proteolytic/non-enzymatic mechanism of action exists. Moreover, deletion of IFITM proteins had no effect upon the antiviral phenotype produced by HsSte24 overexpression, strongly suggesting that HsSte24 is the causative antiviral agent. In this preliminary hypothesis, the role of IFITM3 is to function as an in vivo chaperone to convey HsSte24 to endosomes, with overexpression of HsSte24 obviating the need for IFITM3 to conduct this role. However, it is also possible that in vivo complexes of IFITM3 and HsSte24 in the endosomal membrane have enhanced antiviral effects relative to HsSte24 alone. Two 2019 publications (from different labs) present convincing (and non-mutually exclusive) evidence for two different physiological roles of IFITM3: (i) increased trafficking of virus-infected endocytic vesicles to lysosomes for degradation [62] and (ii) prevention of fusion pore formation by accumulation at the sites of viral membrane fusion [63]. Moreover, this latter publication [63] states: “Of note, the presence of IFTIM3 at the sites of IAV [influenza A virus] fusion does not rule out the possibility that the antiviral effect is due to recruitment of downstream effector proteins, such as ZMPSTE24.” Thus, elucidation of the molecular mechanism of inhibition of viral membrane fusion by HsSte24 is of significance as both a fundamental membrane biophysics question and a “translational research” question, for which consideration of Ste24 as an antiviral protein therapeutic is certainly not without intrinsic merit.
Unanswered Questions, Unresolved Challenges
Ste24 may possess an allosteric site
While Ste24 can cleave prenylated C-terminal CAAX motifs, it is likely not a CAAX protease per se. However, the presence of a prenyl (farnesyl) moiety does influence substrate cleavage by Ste24. The publication first describing the crystal structure of HsSte24 [10] included mass spectrometric product analysis of peptide substrates derived from Prelamin A. (Recall that Ste24-mediated processing of Prelamin A and yeast a-factor consists of two cleavages: one “proximal” to the prenylated CAAX box and another at a non-prenylated site N-terminal from and “distal” to the CAAX motif.) A peptide with a C-terminal farnesyl-cysteine, i.e., lacking “AAX”, was cleaved with complete specificity at its predicted “distal” (non-farnesylated) site. However, a peptide substrate lacking this farnesyl-cysteine (and four additional residues N-terminal to it) now exhibited multiple cleavage products in the vicinity of the distal site, including not only the canonical Prelamin A site but also two other adjacent positions. (A later follow-up study from the same group subsequently reported only a single cleavage of the same, truncated peptide substrate lacking the farnesyl-cysteine and four additional N-terminal residues, but did report “… slow product formation …” compared to the farnesylated version [38].) Both studies demonstrate that, for peptides derived from Prelamin A, deletion of residues at a location removed from the canonical cleavage site of Ste24 has a marked effect on HsSte24 proteolytic activity. Analysis of peptides lacking the distal site also revealed some effect of farnesylation. A farnesyl-cysteine peptide containing the Prelamin A CAAX-box was cleaved at both the canonical/predicted position (removal of the “AAX”), but also generated “AX” product. However, the corresponding non-farnesylated peptide yielded no “AAX” product whatsoever.
As described previously in this review, Ste24 is a gluzincin ZMP. In the description and analysis of the tripartite architecture representation of Ste24, the ZMP core module was shown to possess a hydrophobic S1′ specificity pocket [12], similar to that observed in M4 and M13 gluzincins [[64], [65], [66]]. Correspondingly, the P1′ positions of substrates of Ste24 (and M4 and M13 gluzincins) would be expected to be nonpolar residues that could fit into the S1′ pocket. Crystal structures of enzymatically active HsSte24 in complex with phosphoramidon (a transition-state analog and competitive inhibitor of multiple soluble ZMPs) [13], and of enzymatically-inactive point mutant (E336A, with E336 being the catalytic residue of HEXXH motif) in complex with the non-farnesylated Prelamin A CAAX motif (CSIM), provide structural confirmation of the hydrophobic P1′ specificity of the gluzincin Ste24 (Figure 3 ). The multiple cleavage sites observed with non-farnesylated substrates [10] are consistent with a rather broad gluzincin substrate specificity determined primarily (or even solely) by a hydrophobic P1′ pocket; results supportive of this were observed in earlier yeast-mating studies of a-factor CAAX box substrate specificity [7]. Therefore, the presence of the prenyl moiety, which is predicted to not interact with the P1′ pocket [9,10], may interact with a posited allosteric site within Ste24, perhaps sited at a hydrophobic cleft in the inner surface of the α-barrel. Presciently, early studies characterizing cleavage of “AAX-removed” prenyl-cysteine peptides derived from Prelamin A by the “farnesylation-dependent Prelamin A endoprotease” [6], and its noncompetitive inhibition by N-acetyl farnesyl methyl cysteine, led to the suggestion of “… a specific farnesyl binding site on the enzyme which is not at the active site.”
Figure 3.

Crystal structures of HsSte24 with the gluzincin inhibitor phosphoramidon (PDB 6BH8) [13] (left-hand panels) and tetrapeptide CSIM (PDB 2YPT) [10] (right-hand panels) reveal a hydrophobic S1′ specificity pocket. (a) Schematic images of the HsSte24-bound inhibitor phosphoramidon or tetrapeptide CSIM. The side-chain moiety occupying the S1′ specificity pocket, known as the P1′ position, is indicated for each molecule. A black arrow indicates the position of the scissile bond of the bound-peptide CSIM located between the serine and isoleucine residues. In the case of phosphoramidon binding, the two phosphoramidate oxygens ligate the zinc representing a tetrahedral intermediate state of the scissile bond [25]. (b) The S1′ specify pocket of HsSte24. The phosphoramidon and CSIM peptide are shown as sticks (marine blue carbon, phosphorous colored orange). The S1′ pocket is indicated by purple van der Waals surface. The side-chains of Ala265 of β3-strand, Leu283 of the α3-helix, Val332 of TM α-helix VII, and Leu431/Leu435/Leu438 of the α5-helix form the pocket and are shown as purple. All residues forming the S1′ specify pocket derive from the ZMP core module (see purple ZMP core module in Figure 2(d)). (c) ~ 90° rotation of the images in (b). Note: The structure of the HsSte24:phosphoramidon complex was solved with wild-type enzyme, so the position of the catalytic base Glu336 is specifically indicated. The HsSte24:CSIM complex structure was solved with the catalytically inactive E336A mutant. The deposited coordinates of HsSte24:CSIM did not model the terminal Cδ1 carbon of the isoleucine side-chain; as an aid in visualization of the S1′ pocket, the Cδ1 carbon was added to the image.
Ste24: unknown substrate specificity and incompletely determined steady-state kinetics
The observations that Ste24 does not require prenylation for substrate cleavage [28] and that Ste24 acts as a translocon unclogger [44,45] provide strong evidence of biological roles more expansive than those envisaged prior to 2016. Fundamental characterization of Ste24 can contribute to our understanding of its possible biological activities. Because prenylation is not required for substrate recognition, modern mass spectrometry-based methods utilizing standard peptide libraries, such as multiplex substrate profiling by mass spectrometry [67], could be applied to determine Ste24 substrate specificity (this method has been applied successfully to the rhomboid protease [68,69]). Additionally, while multiple researchers (for examples, see [9,13,28,37,70,71]) have performed steady-state kinetic characterization of Ste24, no absolute characterization of turnover and catalytic efficiency has yet been reported. Several technical barriers currently impede development of a standard “platform” for accurate and precise Ste24 steady-state kinetics characterization. These include the following: (i) the use of detergent-solubilized and purified Ste24, as the presence of excess (micellar and monomeric) detergent can affect the actual “Ste24-accessible” concentration of interfacial (prenylated or other hydrophobic) substrates; (ii) the use of membranes containing over-expressed Ste24, when determination of Ste24 concentration in such a “protein-noisy” background is nontrivial; and (iii) challenges in the use of standard intramolecular quenched fluorescence methods for steady-state kinetics [72,73]. We have developed generalized methods for correction of systematic errors of intramolecular quenched fluorescence induced by inner-filter and non-specific quenching effects [74], which, surprisingly, had been lacking for this very frequently utilized method of protease characterization and inhibitor library screening. Additionally, the potentially broad substrate specificity of Ste24 causes concern about use of nanodiscs [75], as the molecular scaffold protein used to form the “lipidic nanoparticle” may be cleaved by Ste24. Currently, we are pursuing the use of styrene-maleic acid copolymers for “detergent-free” solubilization and purification of Ste24 [76,77]. The novel structure of Ste24, with its “reaction cavity” and active site enclosed within an α-barrel, and the possibility of an allosteric site suggest that this enzyme could display very interesting and unusual steady-state kinetics, mechanisms of inhibition/activation, etc. One publication, on which we collaborated [28], presents tantalizing evidence of positive cooperativity in the steady-state kinetics of farnesylated substrate processing by Ste24; such behavior can indicate conformational change that is slow compared with turnover [78], certainly possible in the context of the posited substrate entrance and exit portals of Ste24. However, at the time of our writing of this review, we cannot (yet) rule out being in a regime of Ste24 concentration that is overly high (compared to K M) such that systematic error causes apparent, rather than real cooperativity.
The function(s) of the α-barrel cavity and its electrostatics
The function of the voluminous α-barrel reaction cavity remains a mystery. In addition to the proposed entrance and exit portals for interfacial substrates, further fenestrations exist within the α-barrel [9,10]; soluble substrates may enter through them and diffuse to the substrate-binding groove, where they become properly positioned in the active site. Moreover, the driving force for substrate entry into the reaction cavity and their subsequent “processive processing” [9] also remain unknown. The distinctive negative electrostatic potential of the interior surface of the α-barrel may play a role akin to that described for “substrate guidance” in acetylcholinesterase [79]. However, a more serious consideration of electrostatics within the cavity merits more careful calculation. The presence of the low-dielectric membrane affects calculations [80]; a more serious concern is the very likely possibility that water entrapped within the cavity may behave quite differently from bulk water (as has been observed with water in the translocon, which exhibits anomalous diffusion [81]). Ultimately, atomistic molecular dynamics simulations may be most productive for study of both dynamics and electrostatics (the latter via particle mesh Ewald methods [82,83]).
Summary: A Paean to Ste24
Ste24 presents multiple wonders to the structural biologist, enzymologist, cell biologist, and membrane biophysicist. The recent emergence of its biological roles, from translocon unclogger to (non-enzymatic) broad-spectrum viral restriction factor, provides substantial impetus to further study this remarkable integral membrane protein ZMP. Its presence in all eukaryotes, as well as its high level of sequence conservation and homology, strongly suggests its importance. Moreover, multiple areas of applied/translational research present themselves. Experimental and theoretical challenges abound!
Acknowledgments
This research was supported by the National Institutes of Health (Grants R01GM108612 and R56AI141627 to M.C.W.). We acknowledge useful reviewer comments, which resulted in significant improvement of this review article.
Edited by Filippo Mancia
References
- 1.Boyartchuk V.L., Ashby M.N., Rine J. Modulation of Ras and a-factor function by carboxyl-terminal proteolysis. Science. 1997;275:1796–1800. doi: 10.1126/science.275.5307.1796. [DOI] [PubMed] [Google Scholar]
- 2.Fujimura-Kamada K., Nouvet F.J., Michaelis S. A novel membrane-associated metalloprotease, Ste24p, is required for the first step of NH2-terminal processing of the yeast a-factor precursor. J. Cell Biol. 1997;136:271–285. doi: 10.1083/jcb.136.2.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tam A., Nouvet F.J., Fujimura-Kamada K., Slunt H., Sisodia S.S., Michaelis S. Dual roles for Ste24p in yeast a-factor maturation: NH2-terminal proteolysis and COOH-terminal CAAX processing. J. Cell Biol. 1998;142:635–649. doi: 10.1083/jcb.142.3.635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jongeneel C.V., Bouvier J., Bairoch A. A unique signature identifies a family of zinc-dependent metallopeptidases. FEBS Lett. 1989;242:211–214. doi: 10.1016/0014-5793(89)80471-5. [DOI] [PubMed] [Google Scholar]
- 5.Hennekes H., Nigg E.A. The role of isoprenylation in membrane attachment of nuclear lamins. A single point mutation prevents proteolytic cleavage of the lamin A precursor and confers membrane binding properties. J. Cell Sci. 1994;107(Pt 4):1019–1029. doi: 10.1242/jcs.107.4.1019. [DOI] [PubMed] [Google Scholar]
- 6.Kilic F., Dalton M.B., Burrell S.K., Mayer J.P., Patterson S.D., Sinensky M. In vitro assay and characterization of the farnesylation-dependent prelamin A endoprotease. J. Biol. Chem. 1997;272:5298–5304. doi: 10.1074/jbc.272.8.5298. [DOI] [PubMed] [Google Scholar]
- 7.Trueblood C.E., Boyartchuk V.L., Picologlou E.A., Rozema D., Poulter C.D., Rine J. The CaaX proteases, Afc1p and Rce1p, have overlapping but distinct substrate specificities. Mol. Cell. Biol. 2000;20:4381–4392. doi: 10.1128/mcb.20.12.4381-4392.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Corrigan D.P., Kuszczak D., Rusinol A.E., Thewke D.P., Hrycyna C.A., Michaelis S. Prelamin A endoproteolytic processing in vitro by recombinant Zmpste24. Biochem. J. 2005;387:129–138. doi: 10.1042/BJ20041359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pryor E.E., Jr., Horanyi P.S., Clark K.M., Fedoriw N., Connelly S.M., Koszelak-Rosenblum M. Structure of the integral membrane protein CAAX protease Ste24p. Science. 2013;339:1600–1604. doi: 10.1126/science.1232048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Quigley A., Dong Y.Y., Pike A.C., Dong L., Shrestha L., Berridge G. The structural basis of ZMPSTE24-dependent laminopathies. Science. 2013;339:1604–1607. doi: 10.1126/science.1231513. [DOI] [PubMed] [Google Scholar]
- 11.Hooper N.M. Families of zinc metalloproteases. FEBS Lett. 1994;354:1–6. doi: 10.1016/0014-5793(94)01079-x. [DOI] [PubMed] [Google Scholar]
- 12.Goblirsch B.R., Pryor E.E., Jr., Wiener M.C. The tripartite architecture of the eukaryotic integral membrane protein zinc metalloprotease Ste24. Proteins. 2019;88:604–615. doi: 10.1002/prot.25841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goblirsch B.R., Arachea B.T., Councell D.J., Wiener M.C. Phosphoramidon inhibits the integral membrane protein zinc metalloprotease ZMPSTE24. Acta Crystallogr. D Struct. Biol. 2018;74:739–747. doi: 10.1107/S2059798318003431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lomize M.A., Pogozheva I.D., Joo H., Mosberg H.I., Lomize A.L. OPM database and PPM web server: resources for positioning of proteins in membranes. Nucleic Acids Res. 2012;40:D370–D376. doi: 10.1093/nar/gkr703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tian W., Chen C., Lei X., Zhao J., Liang J. CASTp 3.0: computed atlas of surface topography of proteins. Nucleic Acids Res. 2018;46:W363–W367. doi: 10.1093/nar/gky473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dolinsky T.J., Czodrowski P., Li H., Nielsen J.E., Jensen J.H., Klebe G. PDB2PQR: expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res. 2007;35:W522–W525. doi: 10.1093/nar/gkm276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baker N.A., Sept D., Joseph S., Holst M.J., McCammon J.A. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U. S. A. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jurrus E., Engel D., Star K., Monson K., Brandi J., Felberg L.E. Improvements to the APBS biomolecular solvation software suite. Protein Sci. 2018;27:112–128. doi: 10.1002/pro.3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Manolaridis I., Kulkarni K., Dodd R.B., Ogasawara S., Zhang Z., Bineva G. Mechanism of farnesylated CAAX protein processing by the intramembrane protease Rce1. Nature. 2013;504:301–305. doi: 10.1038/nature12754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Feng L., Yan H., Wu Z., Yan N., Wang Z., Jeffrey P.D. Structure of a site-2 protease family intramembrane metalloprotease. Science. 2007;318:1608–1612. doi: 10.1126/science.1150755. [DOI] [PubMed] [Google Scholar]
- 21.Wang Y., Zhang Y., Ha Y. Crystal structure of a rhomboid family intramembrane protease. Nature. 2006;444:179–180. doi: 10.1038/nature05255. [DOI] [PubMed] [Google Scholar]
- 22.Holland D.R., Hausrath A.C., Juers D., Matthews B.W. Structural analysis of zinc substitutions in the active site of thermolysin. Protein Sci. 1995;4:1955–1965. doi: 10.1002/pro.5560041001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schrodinger L.L.C. 2015. The AxPyMOL Molecular Graphics Plugin for Microsoft PowerPoint, Version 1.8. [Google Scholar]
- 24.Matthews B.W. Structural basis of the action of thermolysin and related zinc peptidases. Acc. Chem. Res. 1988;21:333–340. [Google Scholar]
- 25.Weaver L.H., Kester W.R., Matthews B.W. Crystallographic study of complex of phosphoramidon with thermolysin—model for presumed catalytic transition-state and for binding of extended substrates. J. Mol. Biol. 1977;114:119–132. doi: 10.1016/0022-2836(77)90286-8. [DOI] [PubMed] [Google Scholar]
- 26.Rawlings N.D., Barrett A.J., Thomas P.D., Huang X., Bateman A., Finn R.D. The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database. Nucleic Acids Res. 2018;46:D624–D632. doi: 10.1093/nar/gkx1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hampton S.E., Dore T.M., Schmidt W.K. Rce1: mechanism and inhibition. Crit. Rev. Biochem. Mol. Biol. 2018;53:157–174. doi: 10.1080/10409238.2018.1431606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hildebrandt E.R., Arachea B.T., Wiener M.C., Schmidt W.K. Ste24p mediates proteolysis of both isoprenylated and non-prenylated oligopeptides. J. Biol. Chem. 2016;291:14185–14198. doi: 10.1074/jbc.M116.718197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dittmer T.A., Misteli T. The lamin protein family. Genome Biol. 2011;12:222. doi: 10.1186/gb-2011-12-5-222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Worman H.J. Nuclear lamins and laminopathies. J. Pathol. 2012;226:316–325. doi: 10.1002/path.2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barrowman J., Wiley P.A., Hudon-Miller S.E., Hrycyna C.A., Michaelis S. Human ZMPSTE24 disease mutations: residual proteolytic activity correlates with disease severity. Hum. Mol. Genet. 2012;21:4084–4093. doi: 10.1093/hmg/dds233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Spear E.D., Hsu E.T., Nie L., Carpenter E.P., Hrycyna C.A., Michaelis S. ZMPSTE24 missense mutations that cause progeroid diseases decrease prelamin A cleavage activity and/or protein stability. Dis. Model. Mech. 2018 doi: 10.1242/dmm.033670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Araujo-Vilar D., Lattanzi G., Gonzalez-Mendez B., Costa-Freitas A.T., Prieto D., Columbaro M. Site-dependent differences in both prelamin A and adipogenic genes in subcutaneous adipose tissue of patients with type 2 familial partial lipodystrophy. J. Med. Genet. 2009;46:40–48. doi: 10.1136/jmg.2008.059485. [DOI] [PubMed] [Google Scholar]
- 34.Guenantin A.C., Briand N., Bidault G., Afonso P., Bereziat V., Vatier C. Nuclear envelope-related lipodystrophies. Semin. Cell Dev. Biol. 2014;29:148–157. doi: 10.1016/j.semcdb.2013.12.015. [DOI] [PubMed] [Google Scholar]
- 35.Coffinier C., Hudon S.E., Farber E.A., Chang S.Y., Hrycyna C.A., Young S.G. HIV protease inhibitors block the zinc metalloproteinase ZMPSTE24 and lead to an accumulation of prelamin A in cells. Proc. Natl. Acad. Sci. U. S. A. 2007;104:13432–13437. doi: 10.1073/pnas.0704212104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Goulbourne C.N., Vaux D.J. HIV protease inhibitors inhibit FACE1/ZMPSTE24: a mechanism for acquired lipodystrophy in patients on highly active antiretroviral therapy? Biochem. Soc. Trans. 2010;38:292–296. doi: 10.1042/BST0380292. [DOI] [PubMed] [Google Scholar]
- 37.Clark K.M., Jenkins J.L., Fedoriw N., Dumont M.E. Human CaaX protease ZMPSTE24 expressed in yeast: structure and inhibition by HIV protease inhibitors. Protein Sci. 2017;26:242–257. doi: 10.1002/pro.3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mehmood S., Marcoux J., Gault J., Quigley A., Michaelis S., Young S.G. Mass spectrometry captures off-target drug binding and provides mechanistic insights into the human metalloprotease ZMPSTE24. Nat. Chem. 2016;8:1152–1158. doi: 10.1038/nchem.2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Butala P., Szpalski C., Soares M., Davidson E.H., Knobel D., Warren S.M. Zmpste24 −/− mouse model for senescent wound healing research. Plast. Reconstr. Surg. 2012;130:788e–798e. doi: 10.1097/PRS.0b013e31826d102b. [DOI] [PubMed] [Google Scholar]
- 40.Peinado J.R., Quiros P.M., Pulido M.R., Marino G., Martinez-Chantar M.L., Vazquez-Martinez R. Proteomic profiling of adipose tissue from Zmpste24 −/− mice, a model of lipodystrophy and premature aging, reveals major changes in mitochondrial function and vimentin processing. Mol. Cell. Proteomics. 2011;10 doi: 10.1074/mcp.M111.008094. M111 008094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tipper D.J., Harley C.A. Yeast genes controlling responses to topogenic signals in a model transmembrane protein. Mol. Biol. Cell. 2002;13:1158–1174. doi: 10.1091/mbc.01-10-0488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jonikas M.C., Collins S.R., Denic V., Oh E., Quan E.M., Schmid V. Comprehensive characterization of genes required for protein folding in the endoplasmic reticulum. Science. 2009;323:1693–1697. doi: 10.1126/science.1167983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stahl F.W., Gross C.A. Ira Herskowitz (1946–2003) Cell. 2003;114:9–10. [Google Scholar]
- 44.Ast T., Michaelis S., Schuldiner M. The protease Ste24 clears clogged translocons. Cell. 2016;164:103–114. doi: 10.1016/j.cell.2015.11.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kayatekin C., Amasino A., Gaglia G., Flannick J., Bonner J.M., Fanning S. Translocon declogger Ste24 protects against IAPP oligomer-induced proteotoxicity. Cell. 2018;173:62–73. doi: 10.1016/j.cell.2018.02.026. (e9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Avci D., Lemberg M.K. Clipping or extracting: two ways to membrane protein degradation. Trends Cell Biol. 2015;25:611–622. doi: 10.1016/j.tcb.2015.07.003. [DOI] [PubMed] [Google Scholar]
- 47.Harrison S.C. Viral membrane fusion. Nat. Struct. Mol. Biol. 2008;15:690–698. doi: 10.1038/nsmb.1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.White J.M., Whittaker G.R. Fusion of enveloped viruses in endosomes. Traffic. 2016;17:593–614. doi: 10.1111/tra.12389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vigant F., Santos N.C., Lee B. Broad-spectrum antivirals against viral fusion. Nat. Rev. Microbiol. 2015;13:426–437. doi: 10.1038/nrmicro3475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Takaoka A., Yanai H. Interferon signalling network in innate defence. Cell. Microbiol. 2006;8:907–922. doi: 10.1111/j.1462-5822.2006.00716.x. [DOI] [PubMed] [Google Scholar]
- 51.Brass A.L., Huang I.C., Benita Y., John S.P., Krishnan M.N., Feeley E.M. The IFITM proteins mediate cellular resistance to influenza a H1N1 virus, West Nile virus, and dengue virus. Cell. 2009;139:1243–1254. doi: 10.1016/j.cell.2009.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Perreira J.M., Chin C.R., Feeley E.M., Brass A.L. IFITMs restrict the replication of multiple pathogenic viruses. J. Mol. Biol. 2013;425:4937–4955. doi: 10.1016/j.jmb.2013.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Smith S., Weston S., Kellam P., Marsh M. IFITM proteins-cellular inhibitors of viral entry. Curr. Opin. Virol. 2014;4:71–77. doi: 10.1016/j.coviro.2013.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wrensch F., Karsten C.B., Gnirss K., Hoffmann M., Lu K., Takada A. Interferon-induced transmembrane protein-mediated inhibition of host cell entry of Ebolaviruses. J. Infect. Dis. 2015;212(Suppl. 2):S210–S218. doi: 10.1093/infdis/jiv255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Narayana S.K., Helbig K.J., McCartney E.M., Eyre N.S., Bull R.A., Eltahla A. The interferon-induced transmembrane proteins, IFITM1, IFITM2, and IFITM3 inhibit hepatitis C virus entry. J. Biol. Chem. 2015;290:25946–25959. doi: 10.1074/jbc.M115.657346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yu J., Li M., Wilkins J., Ding S., Swartz T.H., Esposito A.M. IFITM proteins restrict HIV-1 infection by antagonizing the envelope glycoprotein. Cell Rep. 2015;13:145–156. doi: 10.1016/j.celrep.2015.08.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Huang I.C., Bailey C.C., Weyer J.L., Radoshitzky S.R., Becker M.M., Chiang J.J. Distinct patterns of IFITM-mediated restriction of filoviruses, SARS coronavirus, and influenza a virus. PLoS Pathog. 2011;7 doi: 10.1371/journal.ppat.1001258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wrensch F., Winkler M., Pohlmann S. IFITM proteins inhibit entry driven by the MERS-coronavirus spike protein: evidence for cholesterol-independent mechanisms. Viruses. 2014;6:3683–3698. doi: 10.3390/v6093683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Savidis G., Perreira J.M., Portmann J.M., Meraner P., Guo Z., Green S. The IFITMs inhibit Zika virus replication. Cell Rep. 2016;15:2323–2330. doi: 10.1016/j.celrep.2016.05.074. [DOI] [PubMed] [Google Scholar]
- 60.Fu B., Wang L., Li S., Dorf M.E. ZMPSTE24 defends against influenza and other pathogenic viruses. J. Exp. Med. 2017;214:919–929. doi: 10.1084/jem.20161270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li S., Fu B., Wang L., Dorf M.E. ZMPSTE24 is downstream effector of interferon-induced transmembrane antiviral activity. DNA Cell Biol. 2017;36:513–517. doi: 10.1089/dna.2017.3791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Spence J.S., He R., Hoffmann H.H., Das T., Thinon E., Rice C.M. IFITM3 directly engages and shuttles incoming virus particles to lysosomes. Nat. Chem. Biol. 2019;15:259–268. doi: 10.1038/s41589-018-0213-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Suddala K.C., Lee C.C., Meraner P., Marin M., Markosyan R.M., Desai T.M. Interferon-induced transmembrane protein 3 blocks fusion of sensitive but not resistant viruses by partitioning into virus-carrying endosomes. PLoS Pathog. 2019;15 doi: 10.1371/journal.ppat.1007532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Oda K., Takahashi T., Takada K., Tsunemi M., Ng K.K., Hiraga K. Exploring the subsite-structure of vimelysin and thermolysin using FRETS-libraries. FEBS Lett. 2005;579:5013–5018. doi: 10.1016/j.febslet.2005.07.089. [DOI] [PubMed] [Google Scholar]
- 65.Rawlings N.D., Barrett A.J. Evolutionary families of metallopeptidases. Methods Enzymol. 1995;248:183–228. doi: 10.1016/0076-6879(95)48015-3. [DOI] [PubMed] [Google Scholar]
- 66.Roques B.P., Noble F., Dauge V., Fournie-Zaluski M.C., Beaumont A. Neutral endopeptidase 24.11: structure, inhibition, and experimental and clinical pharmacology. Pharmacol. Rev. 1993;45:87–146. [PubMed] [Google Scholar]
- 67.O’Donoghue A.J., Eroy-Reveles A.A., Knudsen G.M., Ingram J., Zhou M., Statnekov J.B. Global identification of peptidase specificity by multiplex substrate profiling. Nat. Methods. 2012;9:1095–1100. doi: 10.1038/nmeth.2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Arutyunova E., Jiang Z., Yang J., Kulepa A.N., Young H.S., Verhelst S. An internally quenched peptide as a new model substrate for rhomboid intramembrane proteases. Biol. Chem. 2018;399:1389–1397. doi: 10.1515/hsz-2018-0255. [DOI] [PubMed] [Google Scholar]
- 69.Lapek J.D., Jr., Jiang Z., Wozniak J.M., Arutyunova E., Wang S.C., Lemieux M.J. Quantitative multiplex substrate profiling of peptidases by mass spectrometry. Mol. Cell. Proteomics. 2019;18:968–981. doi: 10.1074/mcp.TIR118.001099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hsu E.T., Vervacke J.S., Distefano M.D., Hrycyna C.A. A quantitative FRET assay for the upstream cleavage activity of the integral membrane proteases human ZMPSTE24 and yeast Ste24. Methods Mol. Biol. 2009;2019:279–293. doi: 10.1007/978-1-4939-9532-5_21. [DOI] [PubMed] [Google Scholar]
- 71.Manandhar S.P., Hildebrandt E.R., Schmidt W.K. Small-molecule inhibitors of the Rce1p CaaX protease. J. Biomol. Screen. 2007;12:983–993. doi: 10.1177/1087057107307226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fields G.B. Using fluorogenic peptide substrates to assay matrix metalloproteinases. Methods Mol. Biol. 2010;622:393–433. doi: 10.1007/978-1-60327-299-5_24. [DOI] [PubMed] [Google Scholar]
- 73.Knight C.G. Fluorimetric assays of proteolytic enzymes. Methods Enzymol. 1995;248:18–34. doi: 10.1016/0076-6879(95)48004-8. [DOI] [PubMed] [Google Scholar]
- 74.Arachea B.T., Wiener M.C. Acquisition of accurate data from intramolecular quenched fluorescence protease assays. Anal. Biochem. 2017;522:30–36. doi: 10.1016/j.ab.2017.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Denisov I.G., Grinkova Y.V., Lazarides A.A., Sligar S.G. Directed self-assembly of monodisperse phospholipid bilayer nanodiscs with controlled size. J. Am. Chem. Soc. 2004;126:3477–3487. doi: 10.1021/ja0393574. [DOI] [PubMed] [Google Scholar]
- 76.Jamshad M., Charlton J., Lin Y.P., Routledge S.J., Bawa Z., Knowles T.J. G-protein coupled receptor solubilization and purification for biophysical analysis and functional studies, in the total absence of detergent. Biosci. Rep. 2015;35 doi: 10.1042/BSR20140171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lee S.C., Knowles T.J., Postis V.L., Jamshad M., Parslow R.A., Lin Y.P. A method for detergent-free isolation of membrane proteins in their local lipid environment. Nat. Protoc. 2016;11:1149–1162. doi: 10.1038/nprot.2016.070. [DOI] [PubMed] [Google Scholar]
- 78.Porter C.M., Miller B.G. Cooperativity in monomeric enzymes with single ligand-binding sites. Bioorg. Chem. 2012;43:44–50. doi: 10.1016/j.bioorg.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ripoll D.R., Faerman C.H., Axelsen P.H., Silman I., Sussman J.L. An electrostatic mechanism for substrate guidance down the aromatic gorge of acetylcholinesterase. Proc. Natl. Acad. Sci. U. S. A. 1993;90:5128–5132. doi: 10.1073/pnas.90.11.5128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Callenberg K.M., Choudhary O.P., de Forest G.L., Gohara D.W., Baker N.A., Grabe M. APBSmem: a graphical interface for electrostatic calculations at the membrane. PLoS One. 2010;5 doi: 10.1371/journal.pone.0012722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Capponi S., Heyden M., Bondar A.N., Tobias D.J., White S.H. Anomalous behavior of water inside the SecY translocon. Proc. Natl. Acad. Sci. U. S. A. 2015;112:9016–9021. doi: 10.1073/pnas.1424483112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Darden T., York D., Pedersen L. Particle mesh Ewald—an N.log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993;98:10089–10092. [Google Scholar]
- 83.York D.M., Darden T.A., Pedersen L.G. The effect of long-range electrostatic interactions in simulations of macromolecular crystals—a comparison of the Ewald and truncated list methods. J. Chem. Phys. 1993;99:8345–8348. [Google Scholar]