

Abstract
Hepatitis A virus (HAV) is a hepatotropic member of the family Picornaviridae. HAV has several unique biological characteristics that distinguish it from other members of this family. Recent and previous studies revealed that codon usage plays a key role in HAV replication and evolution. In this study, the patterns of synonymous codon usage in HAV have been studied through multivariate statistical methods on 30 complete open reading frames (ORFs) from the available 30 full-length HAV sequences. Effective number of codons (ENC) indicates that the overall extent of codon usage bias in HAV genomes is significant. The relative dinucleotide abundances suggest that codon usage in HAV can also be strongly influenced by underlying biases in dinucleotide frequencies. These factors strongly correlated with the first major axis of correspondence analysis (COA) on relative synonymous codon usage (RSCU). The distribution of the HAV ORFs along the plane defined by the first two major axes in COA showed that different genotypes are located at different places in the plane, suggesting that HAV codon usage is also reflecting an evolutionary process. It has been very recently described that fine-tuning translation kinetics selection also contributes to codon usage bias of HAV. The results of these studies suggest that HAV genomic biases are the result of the co-evolution of genome composition, controlled translation kinetics and probably the ability to escape the antiviral cell responses.
Keywords: Hepatitis A virus, Codon usage, Evolution
1. Introduction
Due to the degeneracy of the genetic code, most amino acids are coded by more than one codon (synonymous codon usage). These synonymous codons are not used randomly. Rather, there are some codons that are used more frequently than others. Mutational pressure and translational selection are thought to be among the main factors that account for codon usage variation among genes in different organisms (Sharp and Li, 1986a, Karlin and Mrazek, 1996, Lesnik et al., 2000).
Understanding the extent and causes of biases in codon usage is essential to the comprehension of viral evolution, particularly the interplay between viruses and the immune response (Shackelton et al., 2006).
Hepatitis A virus (HAV) is a hepatotropic member of the family Picornaviridae (Wimmer and Murdin, 1991), and its viral genome consists of a 7.5-kilobase (kb), positive-stranded RNA with a single open reading frame (ORF). The ORF, which codes 2227 amino acids is organized into three functional regions termed P1, P2 and P3. P1 encodes the capsid polypeptides VP1 to VP4, whereas P2 and P3 encode non-structural polypeptides. The ORF is preceded by a 5′ untranslated region (UTR) and is followed by a 3′ UTR with a short poly A tail (Hollinger and Emerson, 2001).
The structure of HAV, its tissue tropism, and genetic distance from other members of the family Picornaviridae indicate that HAV is unique within this family (Martin and Lemon, 2006, Cristina and Costa-Mattioli, 2007).
HAV has been shown to possess a single conserved immunogenic neutralization site, and isolates from different parts of the world belong to a single serotype (Stapleton and Lemon, 1987, Hollinger and Emerson, 2001). Nevertheless, the study of the HAV evolution in cell culture revealed the presence of some antigenic variants in the mutant spectra that were generated even in the absence of immune selection (Sanchez et al., 2003). Furthermore, several escape mutants, representing antigenic variants, have been selected for their resistance to different monoclonal antibodies (MAbs), suggesting the occurrence of severe structural constraints in the HAV capsid that prevent the more extensive substitutions necessary for the emergence of a new serotype (Nainan et al., 1992, Ping and Lemon, 1992).
Very recent in vitro studies have shown the occurrence of highly conserved clusters of rare codons in the HAV capsid-coding region and that substitutions in these clusters are negatively selected, suggesting that the need to maintain such clusters play a role in the low antigenic variability of HAV (Aragones et al., 2008). Moreover, recent studies suggest that fine-tuning translation kinetics selection is also underlying codon usage bias in this specific genome region (Aragones et al., 2010). These results reveal that codon usage plays a key role in HAV replication and evolution. However, our knowledge of other factors also contributing to shaping synonymous codon usage bias and nucleotide composition in human HAV in vivo is rather scarce.
In order to gain insight into these matters, we analyzed the codon usage and base composition of all available ORFs from 30 human HAV isolates, and investigated the possible key evolutionary determinants of codon usage bias.
2. Materials and methods
2.1. Sequences
Full-length ORFs nucleotide sequences (corresponding to 2227 amino acids) were obtained for 30 human HAV isolates by mean of the use of ARSA at DDBJ database (available at: http://arsa.ddbj.nig.ac.jp/) and EMBL database (available at: http://www.ebi.ac.uk/embl/Access/index.html). For strain names, accession numbers, geographic location of isolation and genotypes, see Supplementary Material Table 1.
2.2. Codon usage analyses
In order to investigate the extent of codon usage bias in HAV, we first aligned the complete ORF code sequences from the HAV strains, using the MUSCLE program (Edgar, 2004). Once aligned, the relative synonymous codon usage (RSCU) values of each codon were determined in order to measure the synonymous codon usage (Sharp and Li, 1986b). This was done using the CodonW program (available at: http://mobyle.pasteur.fr). The RSCU is the observed frequency of a codon divided by the frequency expected, if all synonymous codons for that amino acid were used equally. If RSCU value is close to 1.0, it indicates a lack of bias (Tsai et al., 2007). RSCU values are largely independent of amino acid composition and are particularly useful in comparing codon usage between genes that differ in size and amino acid composition. The RSCU of HAV ORFs were compared with corresponding values of human cells (International Human Genome Sequencing Consortium, 2001). The effective number of codons (ENC) and the frequency of use of GC3S (G+C at synonymous variable third position codons, excluding Met, Trp, and termination codons) were also calculated by the use of the Codon W program. ENC was used to quantify the codon usage bias of an ORF (Wrigth, 1990), which is one of the best overall estimator of absolute synonymous codon usage bias (Comeron and Aguade, 1998). The ENC values range from 20 to 61. The larger the extent of codon bias in a gene, the smaller the ENC value is. In an extremely biased gene where only one codon is used for each amino acid, this value would be 20; in an unbiased gene, it would be 61. Similarly, the fraction of the G+C nucleotides not involved in the GC3S fraction (GC12) was also calculated. All these indices were also calculated using the Codon W program. The relative frequencies of dinucleotides were also calculated using this program as implemented in the Mobile server (http://mobyle.pasteur.fr).
2.3. Correspondence analysis
COA is an ordination technique that identifies the major trends in the variation of the data and distributes genes along continuous axes in accordance with these trends. COA creates a series of orthogonal axes to identify trends that explain the data variation, with each subsequent axis explaining a decreasing amount of the variation (Greenacre, 1984). Each ORF is represented as a 59-dimensional and each dimension is related to the RSCU value of each triplet (excluding AUG, UGG and stop codons). This was done using the CodonW program.
2.4. Statistical analysis
Correlation analysis was carried out using Spearman's rank correlation analysis method (Wessa, 2010; available at: www.wessa.net).
3. Results
In order to study the extent of codon bias in HAV ORFs, the average codon usage values for all triplets were calculated. The results of these studies are shown in Table 1 .
Table 1.
Codon usage (RSCUa values) in HAV strains and human cells.
| AA | Codon | HC | HAV | AA | Codon | HC | HAV | AA | Codon | HC | HAV | AA | Codon | HC | HAV |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Phe | UUU | 0.92 | 1.57 | Ser | UCU | 1.14 | 2.27 | Tyr | UAU | 0.88 | 1.53 | Cys | UGU | 0.92 | 1.65 |
| UUC | 1.08 | 0.43 | UCC | 1.32 | 0.71 | UAC | 1.12 | 0.47 | UGC | 1.08 | 0.35 | ||||
| Leu | UUA | 0.48 | 1.18 | UCA | 0.90 | 2.04 | TER | UAA | ** | ** | TER | UGA | ** | ** | |
| UUG | 0.78 | 2.48 | UCG | 0.30 | 0.11 | UAG | ** | ** | Trp | UGG | 1.00 | 1.00 | |||
| CUU | 0.78 | 1.12 | Pro | CCU | 1.16 | 2.03 | His | CAU | 0.84 | 1.59 | Arg | CGU | 0.48 | 0.23 | |
| CUC | 1.20 | 0.20 | CCC | 1.28 | 0.49 | CAC | 1.16 | 0.41 | CGC | 1.08 | 0.09 | ||||
| CUA | 0.42 | 0.28 | CCA | 1.12 | 1.43 | Gln | CAA | 0.54 | 1.02 | CGA | 0.66 | 0.13 | |||
| CUG | 2.40 | 0.74 | CCG | 0.44 | 0.05 | CAG | 1.46 | 0.98 | CGG | 1.20 | 0.03 | ||||
| Ile | AUU | 1.08 | 2.04 | Thr | ACU | 1.00 | 1.81 | Asn | AAU | 0.94 | 1.67 | Ser | AGU | 0.90 | 0.75 |
| AUC | 1.41 | 0.29 | ACC | 1.44 | 0.41 | AAC | 1.06 | 0.33 | AGC | 1.44 | 0.13 | ||||
| AUA | 0.51 | 0.67 | ACA | 1.12 | 1.68 | Lys | AAA | 0.86 | 1.28 | Arg | AGA | 1.26 | 4.16 | ||
| Met | AUG | 1.00 | 1.00 | ACG | 0.44 | 0.10 | AAG | 1.14 | 0.72 | AGG | 1.26 | 1.37 | |||
| Val | GUU | 0.72 | 2.26 | Ala | GCU | 1.08 | 2.18 | Asp | GAU | 0.92 | 1.64 | Gly | GGU | 0.64 | 1.17 |
| GUC | 0.96 | 0.38 | GCC | 1.60 | 0.62 | GAC | 1.08 | 0.36 | GGC | 1.36 | 0.46 | ||||
| GUA | 0.48 | 0.34 | GCA | 0.92 | 1.16 | Glu | GAA | 0.84 | 1.14 | GGA | 1.00 | 1.72 | |||
| GUG | 1.84 | 1.02 | GCG | 0.44 | 0.03 | GAG | 1.16 | 0.86 | GGG | 1.00 | 0.66 | ||||
RSCU, relative synonymous codon usage; AA, amino acid; HC, human cells; HAV, Hepatitis A Virus. Highly biased codons with respect to human cells are shown in bold.
Means termination codon.
Interestingly, the frequencies of codon usage in HAV ORFs are significantly different than the ones used by human cells. Particularly, extremely highly biased codon frequencies were found for Phe, His, Asn, Asp, Cys and Arg (see Table 1). Almost all extremely high preferred codons were U-ended (see Table 1).
In order to investigate if these 30 HAV strain sequences display similar composition features, the ENC values were calculated for each ORF. The ENC values obtained vary from 38.53 to 41.04 (mean ENCs value of 39.78). For results obtained for all 30 HAV strains enrolled in these studies, see Supplementary Material Table 2. Due to the fact that almost all ENC values are less than 40, the results obtained for the HAV ORFs studied reveal that codon usage in HAV is biased.
In order to investigate the patterns of synonymous codon usage, the correlations between the positions of the ORFs along the first principal axis generated by the COA and the respective GC3S and GC12 values of each strain were analyzed. The first principal axis in COA accounts for 45.34% of the total variation, while the next three principal axes in account for 10.14%, 8.63% and 6.56% of the variability, respectively. The first axis in COA is highly correlated with the GC3S and GC12 values in HAV ORFs. This result reveals that nucleotide composition plays an important key role in the codon usage bias observed in HAV ORFs (see Table 2 ).
Table 2.
Correlation analysis between the first axis values in COA and GC3S and GC12 content for 30 HAV ORFs.
| GC3S | GC12 | |
|---|---|---|
| Axis 1 | ||
| r | 0.825 | 0.702 |
| P | <0.0001 | <0.001 |
In order to detect the possibility of codon usage variation of different HAV genomes, the HAV ORFs were divided according to their HAV genotype (IA, IB, IIA, IIB, IIIA and IIIB). COA was performed on the RSCU values of each HAV ORF and the distribution of the six genotypes along the first two principal axes of COA was determined. The results of these studies are shown in Fig. 1 .
Fig. 1.
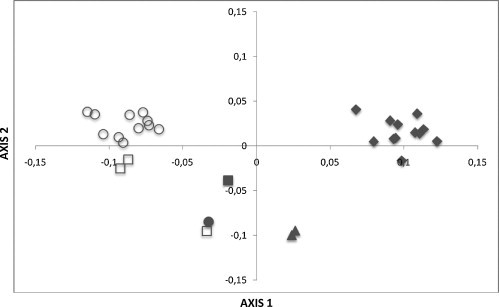
Positions of the 30 HAV ORFs in the plot of the first two major axes by correspondence analysis (COA) of relative synonymous codon usage (RSCU) values. The first and second axes account for 45.34% and 10.14% of the total variation, respectively. The HAV ORFs are divided according to their HAV genotype, genotype IA strains are indicated by a white circle (○), genotype IB by a white square (□), genotype IIA by a black circle (●), genotype IIB by a black square (■), genotype IIIA by a black diamond (♦) and genotype IIIB by a black triangle (▴).
Surprisingly, the distribution of the six genetic groups in the plane defined by the first two major axes showed that different genotypes were located at different places, suggesting that different HAV genotypes exhibit differences in their codon usage patterns (see Fig. 1).
In order to gain insight into these findings, the average codon usage values for all codons were calculated for genotype IA and IIIA strains enrolled in these studies, accounting for 24,416 and 31,108 codons, respectively. The results of these studies are shown in Supplementary Material Table 3. Interestingly, the frequencies of codon usage in the different HAV genotypes show significant different frequencies in CCA (Pro) and CGC (Arg) codons.
In order to observe if different frequencies of codon usage are found in different HAV genome regions, the same studies were repeated for the structural (P1) and non-structural regions (P2 + P3) of the HAV genome. The results of these studies are shown in Supplementary Material Table 4. Roughly similar values are obtained for both regions for most codons, although significant differences were found for Arg codons (CGC and AGG).
It has been suggested that dinucleotide biases can affect codon bias (Tao et al., 2009). To study the possible effect of dinucleotide composition on codon usage of the HAV ORFs, the relative abundances of the 16 dinucleotides in the ORFs of the 30 HAV strains were established. The results of these analyses are shown in Table 3 .
Table 3.
Relative abundance of dinucleotides in HAV ORFs.
| Relative abundance of the 16 dinucleotides |
||||||||
|---|---|---|---|---|---|---|---|---|
| UU | UC | UA | UG | CU | CC | CA | CG | |
| Mean ± SDa | 1.891 ± 0.051 | 0.901 ± 0.027 | 0.925 ± 0.025 | 1.465 ± 0.036 | 0.904 ± 0.040 | 0.506 ± 0.036 | 1.079 ± 0.024 | 0.063 ± 0.009 |
| AU | AC | AA | AG | GU | GC | GA | GG | |
|---|---|---|---|---|---|---|---|---|
| Mean ± SD | 1.493 ± 0.027 | 0.674 ± 0.026 | 1.590 ± 0.025 | 1.068 ± 0.053 | 0.893 ± 0.020 | 0.470 ± 0.016 | 1.228 ± 0.026 | 0.841 ± 0.017 |
Mean values of 30 HAV ORFs relative dinucleotide ratios ± standard deviation.
The occurrences of dinucleotides are not randomly distributed and no dinucleotides were present at the expected frequencies (Table 3). The relative abundance of CpG showed a strong deviation from the “normal range” (mean ± SD = 0.063 ± 0.009) and was markedly underrepresented. On the other had, the frequency of UpU was above the expected value (mean ± SD = 1.891 ± 0.051) (Table 3). Among the 16 dinucleotides, 14 are highly correlated with the first axis value in COA (Table 4 ). These observations indicate that the composition of dinucleotides also plays a key role in the variation found in synonymous codon usage among HAV ORFs.
Table 4.
Summary of correlation analysis between the axis in COA and sixteen dinucleotides frequencies in HAV ORFs.
| UU | UC | UA | UG | CU | CC | CA | CG | |
|---|---|---|---|---|---|---|---|---|
| Axis 1 | ||||||||
| r | −0.865 | 0.733 | −0.637 | −0.781 | 0.679 | 0.820 | −0.366 | 0.557 |
| P | <0.0001 | <0.0001 | <0.001 | <0.0001 | <0.001 | <0.0001 | 0.041 | 0.002 |
| AU | AC | AA | AG | GU | GC | GA | GG | |
|---|---|---|---|---|---|---|---|---|
| Axis 1 | ||||||||
| r | −0.625 | 0.706 | 0.431 | −0.742 | 0.798 | −0.569 | 0.699 | 0.755 |
| P | <0.001 | <0.001 | 0.019 | <0.0001 | <0.0001 | 0.002 | <0.001 | <0.0001 |
To study the possible effects of CpG under-representation on codon usage bias of HAV ORFs, the RSCU value of the eight codons that contain CpG (CCG, GCG, UCG, ACG, CGC, CGG, CGU, CGA) were analyzed. These eight codons [CCG (mean 0.05), GCG (mean 0.03), UCG (mean 0.11), ACG (mean 0.10), CGC (mean 0.09), CGG (mean 0.03) and CGU (mean 0.23), GCC (mean 0.13)] were markedly suppressed.
Besides, the position of each codon in each of the four major axes of COA was determined for the 30 HAV ORFs. Table 5 shows the codons for which the maximum and minimum values were obtained for each of the axes studied (i.e. the most divergent codons values), indicating bias in their use by HAV. As it can be seen in the table, most of the divergent codons were triplets coding for Arg.
Table 5.
Position of codons in each of the four major axes of COA for 30 HAV ORFs.
| Axis 1 |
Axis 2 |
||||
|---|---|---|---|---|---|
| Codon | Value | Aminoacid | Codon | Value | Aminoacid |
| CGC | −0.54535 | Arg | CGG | −125.916 | Arg |
| CGU | 0.46586 | Arg | CGC | 0.26321 | Arg |
| Axis 3 |
Axis 4 |
||||
|---|---|---|---|---|---|
| Codon | Value | Aminoacid | Codon | Value | Aminoacid |
| GCG | −0.53899 | Ala | CGG | −0.70233 | Arg |
| CGA | 0.1961 | Arg | CUC | 0.15466 | Leu |
In order to observe if dinucleotides frequencies may vary among different genotypes, the same studies were repeated using genotype IA and IIIA strains. The results of these studies are shown in Supplementary Material Table 5. No significant differences were observed among the two genotypes or using all 30 HAV strains representing all known HAV genotypes. A similar study conducted in order to study dinucleotide frequencies in structural and non-structural regions of the HAV genome also found no significant differences among the different regions of the HAV genome (see Supplementary Material Table 6).
4. Discussion
The results of these studies revealed that codon usage in HAV ORFs is quite different from that of human genes (see Table 1). This is in agreement with previous results found for the capsid structural region of HAV (Sanchez et al., 2003). In other members of the family Picornaviridae, like Poliovirus or foot-and-mouth disease virus (FMDV) the codon usage is very similar to that of their hosts, implying competence for tRNAs among virus and host (Sanchez et al., 2003). In these cases, competition is avoided by the induction of cellular shutoff of protein synthesis through carboxy cleavage of translation initiation factor 4G (eIF4G) by 2A and L proteases, respectively (Racaniello, 2001). HAV lacks mechanisms of inducing cellular shutoff and needs an intact eIF4G factor for the initiation of translation (Racaniello, 2001, Ali et al., 2001). Moreover, HAV has a very inefficient IRES (Whetter et al., 1994). For these reasons, HAV may be able to synthesize its proteins by adapting their codon usage to those less commonly used cellular tRNAs. This may also account for its low replicative rate (Pinto et al., 2007, Moratorio et al., 2007).
In this study, we analyzed synonymous codon usage and nucleotide compositional constraints in HAV ORFs. Interestingly, contrary to previous results found for other viruses such H5N1 Influenza A Virus (mean ENC = 50.91) (Ahn et al., 2006, Zhou et al., 2005); SARS (mean ENC = 48.99) (Zhao et al., 2008); foot-and-mouth disease virus (mean ENC = 51.42) (Zhong et al., 2007); classical swine fever virus (mean ENC = 51.7) (Tao et al., 2009) and Duck Enteritis virus (mean ENC = 52.17) (Jia et al., 2009), the ENC values found for human HAV are comparatively low (mean ENC value of 39.78), indicating that the overall extent of codon usage bias in HAV is significant. This is in agreement with recent in vitro studies on HAV capsid variability constraints (Aragones et al., 2008).
A general correlation between codon usage bias and base composition was observed in these studies. Moreover, highly significant correlations between the first axis of COA and GC3S and GC12 values were obtained for all HAV ORFs studied. These results suggest that mutational pressure significantly contributes to the codon usage bias in HAV strains. Nevertheless, as previously suggested for other viral systems, when significant distance among expected and actual ENC values are found, other factors additional to mutational bias may be also contributing to codon usage bias (Shackelton et al., 2006). This is in agreement with very recent studies on HAV populations adapted to propagate in cells with impaired protein synthesis in which fine-tuning translation kinetics selection rather than translation selection was identified as the underlying mechanism of codon usage bias in the capsid coding region (Aragones et al., 2010). Thus, both mutation pressure, as well as selection pressure for correct protein folding, play a critical role shaping HAV codon usage, indicating that HAV genomic bias is multi-factorial.
In order to detect possible codon usage variation of different genomes, the HAV ORFs were divided according to their genotype. Unexpectedly, the distribution of the six genetic groups along the first two major axes in COA showed that different genotypes are distantly located in the plane defined by the first two axes of the analysis (Fig. 1). Moreover, the frequencies of codon usage of genotype IA and IIIA showed significant differences in Pro and Arg codons (see Supplementary Material Table 3). Since species with a close genetic relationship always present a similar codon usage pattern (Sharp et al., 1988), these findings suggest that codon usage in HAV is undergoing also an evolutionary process, probably reflecting a dynamic process of mutation and selection to re-adapt its codon usage to different environments (see Fig. 1 and Supplementary Material 3).
The structural and non-structural regions of the genome roughly share the same frequencies of codon usage, except for some of the Arg codons (see Supplementary Material Table 4). This is in agreement with COA analysis, were most of the divergent codons were triplets coding for Arg (see Table 4). This reveals that the use of Arg codons plays also a role in the evolution and the variability observed among HAV strains.
The frequencies of occurrence for dinucleotides were not randomly distributed and most dinucleotides did not follow the expected frequencies in HAV ORFs (Table 3). The high correlation found between the first axis of COA and the relative dinucleotide abundances (Table 4) suggests that codon usage in HAV ORFs can also be strongly influenced by underlying biases in dinucleotide frequencies. All CpG containing codons are markedly suppressed (Table 3) in the 30 HAV strains included in the study, confirming what has been very recently noted (Bosch et al., 2010). Marked CpG deficiency has been also observed in Coronaviruses (Woo et al., 2007), vertebrate-infecting members of the family Flaviviridae (Lobo et al., 2009), Poliovirus (Rothberg and Wimmer, 1981) and other RNA viruses (Karlin et al., 1994). Moreover, polioviruses synthetically deoptimized either by codon deoptimization or codon pair deoptimization are generally marked by a higher content of CpG (and also UpA) dinucleotide (Burns et al., 2006, Burns et al., 2009, Mueller et al., 2006, Coleman et al., 2008), indicating that polioviruses have naturally evolved to eliminate these dinucleotides. CpG deficiency was proposed to be related to the immunostimulatory properties of unmethylated CpG, which were recognized by the host's innate immune system as a pathogen signature (Shackelton et al., 2006, Woo et al., 2007). Escaping from the host antiviral response may act as another selective pressure contributing to the multifactorial codon usage shaping (Vetsigian and Goldenfeld, 2009).
Thus, the results of these studies suggest that HAV genomic biases are the result from the coevolution of genome composition, the need to a controlled translation kinetics and probably the need to escape the antiviral cell responses, and thus is a model in agreement with the evolution rhetoric theory proposed by Vetsigian and Goldenfeld (2009) in which genome biases emerge by the need to increase communication with the ever changing cell environment without changing the message.
Acknowledgements
Authors acknowledge support by PEDECIBA and Agencia Nacional de Investigación e Innovación (ANII, FCE 2007_722), Uruguay. We acknowledge anonymous reviewers for important suggestions regarding this work.
Footnotes
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.virusres.2011.01.012.
Appendix A. Supplementary data
References
- Ahn I., Jeong B.J., Bae S.E., Jung J., Son H.S. Genomic analysis of Influenza A viruses, including Avian Flu (H5N1) strains. Eur. J. Epidemiol. 2006;21:511–519. doi: 10.1007/s10654-006-9031-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali I.K., McKendrich L., Morley S.J., Jackson R.J. Activity of the Hepatitis A virus IRES requires association between the Cap-binding translation initiation factor (eIF4E) and eIF4G. J. Virol. 2001;75:7854–7863. doi: 10.1128/JVI.75.17.7854-7863.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aragones L., Guix S., Ribes E., Bosch A., Pintó R.M. Fine-tuning translation kinetics selection as the driving force of codon usage bias in the Hepatitis A virus capsid. PLoS Pathog. 2010;6(3):e1000797. doi: 10.1371/journal.ppat.1000797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aragones L., Bosch A., Pinto R.M. Hepatitis A virus mutant spectra under the selective pressure of monoclonal antibodies: codon usage constraints limit capsid variability. J. Virol. 2008;82:1688–1700. doi: 10.1128/JVI.01842-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch A., Mueller S., Pinto R.M. Coding biases and viral fitness. In: Ehrenfeld E., Domingo E., Roos R.P., editors. The Picornaviruses. ASM Press; Washington, DC, USA: 2010. pp. 271–284. [Google Scholar]
- Burns C.C., Campagnoli R., Shaw J., Vincent A., Jorba J., Kew O. Genetic inactivation of poliovirus infectivity by increasing the frequencies of CpG and UpA dinucleotides within and across synonymous capsid region codons. J. Virol. 2009;83:9957–9969. doi: 10.1128/JVI.00508-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns C.C., Shaw J., Campagnoli R., Jorba J., Vincent A., Quay J., Kew O. Modulation of poliovirus replicative fitness in HeLa cells by deoptimization of synonymous codon usage in the capsid region. J. Virol. 2006;80:3259–3272. doi: 10.1128/JVI.80.7.3259-3272.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman J.R., Papamichail D., Skiena S., Futcher B., Wimmer E., Mueller S. Virus attenuation by genome-scale changes in codon pair bias. Science. 2008;320:1784–1787. doi: 10.1126/science.1155761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comeron J.M., Aguade M. An evaluation of measures of synonymous codon usage bias. J. Mol. Evol. 1998;47:268–274. doi: 10.1007/pl00006384. [DOI] [PubMed] [Google Scholar]
- Cristina J., Costa-Mattioli M. Genetic variability and molecular evolution of Hepatitis A virus. Virus Res. 2007;127:151–157. doi: 10.1016/j.virusres.2007.01.005. [DOI] [PubMed] [Google Scholar]
- Edgar R.C. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004;5:113. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenacre M. Academic Press; London: 1984. Theory and Applications of Correspondence Analysis. [Google Scholar]
- Hollinger F.B., Emerson S.U. Hepatitis A virus. In: Knipe D.M., Howley P.M., Griffin D.E., Martin M.A., Lamb R.A., Roizman B., Straus S.E., editors. 4th ed. vol. 1. Lippincott Williams & Wilkins; Philadelphia: 2001. pp. 799–840. (Fields Virology). [Google Scholar]
- International Human Genome Sequencing Consortium Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Jia R., Cheng A., Wang M., Xin H., Guo Y., Zhu D., Qi X., Zhao L., Ge H., Chen X. Analysis of synonymous codon usage in the UL24 gene of duck enteritis virus. Virus Genes. 2009;38:96–103. doi: 10.1007/s11262-008-0295-0. [DOI] [PubMed] [Google Scholar]
- Karlin S., Mrazek J. What drives codon choices in human genes? J. Mol. Biol. 1996;262:459–472. doi: 10.1006/jmbi.1996.0528. [DOI] [PubMed] [Google Scholar]
- Karlin S., Doerfler W., Cardon L.R. Why is CpG suppressed in the genomes of virtually all small eukaryotic viruses but not in those of large eukaryotic viruses? J. Virol. 1994;68:2889–2897. doi: 10.1128/jvi.68.5.2889-2897.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesnik T., Solomovici J., Deana A., Ehrlich R., Reiss C. Ribosome traffic in E. coli and regulation of gene expression. J. Theor. Biol. 2000;202:175–185. doi: 10.1006/jtbi.1999.1047. [DOI] [PubMed] [Google Scholar]
- Lobo F.P., Mota B.E.F., Pena S.D.J., Azevedo V., Macedo A.M., Tauch A., Machado C.R., Franco G.R. Virus-host coevolution: common patterns of nucleotide motif usage in Flaviviridae and their hosts. PLoS ONE. 2009;4:e6282. doi: 10.1371/journal.pone.0006282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin A., Lemon S.M. Hepatitis A virus: from discovery to vaccines. Hepatology. 2006;43:S164–S172. doi: 10.1002/hep.21052. [DOI] [PubMed] [Google Scholar]
- Moratorio G., Costa-Mattioli M., Piovani R., Romero H., Musto H., Cristina J. Bayesian coalescent inference of Hepatitis A virus populations: evolutionary rates and patterns. J. Gen. Virol. 2007;88:3039–3042. doi: 10.1099/vir.0.83038-0. [DOI] [PubMed] [Google Scholar]
- Mueller S., Papamichail D., Coleman J.R., Skiena S., Wimmer E. Reduction of the rate of poliovirus protein synthesis through large-scale codon deoptimization causes attenuation of viral virulence by lowering specific infectivity. J. Virol. 2006;80:9687–9696. doi: 10.1128/JVI.00738-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nainan O., Brinton M., Margolis H.S. Identification of amino acids located in the antibody binding sites of human hepatitis A virus. Virology. 1992;191:984–987. doi: 10.1016/0042-6822(92)90277-v. [DOI] [PubMed] [Google Scholar]
- Ping L.H., Lemon S.M. Antigenic structure of human hepatitis A virus defined by analysis of escape mutants selected against murine monoclonal antibodies. J. Virol. 1992;66:2208–2216. doi: 10.1128/jvi.66.4.2208-2216.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto R.M., Aragones L., Costafreda M.I., Ribes E., Bosch A. Codon usage and replicative strategies of hepatitis A virus. Virus Res. 2007;127:158–163. doi: 10.1016/j.virusres.2007.04.010. [DOI] [PubMed] [Google Scholar]
- Racaniello V.R. Picornaviridae: the viruses and their replication. In: Knipe D.M., Howley P.M., Griffin D.E., Lamb R.A., Martin M.A., Roizman B., Straus S.E., editors. 4th ed. vol. 1. Lippincott Williams & Wilkins; Philadelphia, PA: 2001. pp. 685–722. (Fields Virology). [Google Scholar]
- Rothberg P.G., Wimmer E. Mononucleotide and dinucleotide frequencies, and codon usage in poliovirus RNA. Nucleic Acids Res. 1981;9:6221–6229. doi: 10.1093/nar/9.23.6221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez G., Bosch A., Gomez-Mariano G., Domingo E., Pintó R.M. Evidence for quasispecies distributions in the human Hepatitis A virus genome. Virology. 2003;31:534–542. doi: 10.1016/s0042-6822(03)00483-5. [DOI] [PubMed] [Google Scholar]
- Shackelton L.A., Parrish C.R., Holmes E.C. Evolutionary basis of codon usage and nucleotide composition bias in vertebrate DNA viruses. J. Mol. Evol. 2006;62:551–563. doi: 10.1007/s00239-005-0221-1. [DOI] [PubMed] [Google Scholar]
- Sharp P.M., Cowe E., Higgins D.G., Shields D.C., Wolfe K.H., Wright F. Codon usage patterns in Escherichia coli, Bacillus subtilis, Saccharomyces cerevisiae, Schizosaccharomyces pombe, Drosophila melanogaster and Homo sapiens: a review of the considerable within-species diversity. Nucleic Acids Res. 1988;16:8207–8211. doi: 10.1093/nar/16.17.8207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp P.M., Li W.H. Codon usage in regulatory genes in Escherichia coli not reflect selection for “rare” codons. Nucleic Acids Res. 1986;14:7737–7749. doi: 10.1093/nar/14.19.7737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp P.M., Li W.H. An evolutionary perspective on synonymous codon usage in unicellular organisms. J. Mol. Evol. 1986;24:28–38. doi: 10.1007/BF02099948. [DOI] [PubMed] [Google Scholar]
- Stapleton J.T., Lemon S.M. Neutralization escape mutants define a dominant immunogenic neutralization site on Hepatitis A virus. J. Virol. 1987;61:491–498. doi: 10.1128/jvi.61.2.491-498.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao P., Dai L., Luo M., Tang F., Tien P., Pan Z. Analysis of synonymous codon usage in classical swine fever virus. Virus Genes. 2009;38:104–112. doi: 10.1007/s11262-008-0296-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai C.T., Lin C.H., Chang C.Y. Analysis of codon usage bias and base compositional constraints in iridovirus genomes. Virus Res. 2007;126:196–206. doi: 10.1016/j.virusres.2007.03.001. [DOI] [PubMed] [Google Scholar]
- Vetsigian K., Goldenfeld N. Genome rhetoric and the emergence of compositional bias. Proc. Natl. Acad. Sci. U.S.A. 2009;106:215–220. doi: 10.1073/pnas.0810122106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessa, P., 2010. Free Statistics Software, Office for Research Development and Education, version 1.1.23-r5. Available from: http://www.wessa.net.
- Whetter L.E., Day S.P., Elroystein O., Brown E.A., Lemon S.M. Low efficiency of the 5′ nontranslated region of hepatitis A virus RNA in directing cap-independent translation in permissive monkey kidney cells. J. Virol. 1994;68:5253–5263. doi: 10.1128/jvi.68.8.5253-5263.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimmer E., Murdin A. Hepatitis A and the molecular biology of picornaviruses: a case for a new genus of the family Picornaviridae. In: Hollinger L.B., Lemon S.M., Margolis H.S., editors. Viral Hepatitis and Liver Disease. Williams & Wilkins; Baltimore: 1991. pp. 1–41. [Google Scholar]
- Woo P.C.Y., Wong B.H.L., Huang Y., Lau S.K.P., Yuen K. Cytosine deamination and selection of CpG suppressed clones are the two major independent biological forces that shape codon usage bias in Coronaviruses. Virology. 2007;369:431–442. doi: 10.1016/j.virol.2007.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrigth F. The “effective number of codons” used in a gene. Gene. 1990;87:23–29. doi: 10.1016/0378-1119(90)90491-9. [DOI] [PubMed] [Google Scholar]
- Zhao S., Zhang Q., Liu X., Wang X., Zhang H., Wu Y., Jiang F. Analysis of synonymous codon usage in 11 Human Bocavirus isolates. Biosystems. 2008;92:207–214. doi: 10.1016/j.biosystems.2008.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong J., Li Y., Zhao S., Liu S., Zhang Z. Mutation pressures shapes codon usage in the GC-rich genome of foot-and-mouth disease virus. Virus Genes. 2007;35:767–776. doi: 10.1007/s11262-007-0159-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T., Gu W., Ma J., Sun X., Lu Z. Analysis of synonymous codon usage in H5N1 virus and other Influenza A viruses. Biosystems. 2005;81:77–86. doi: 10.1016/j.biosystems.2005.03.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.