

Abstract
Picornaviruses replicate their RNA genomes through a highly conserved mechanism that involves an interaction between the principal viral protease (3Cpro) and the 5′-UTR region of the viral genome. The 3Cpro catalytic site is the target of numerous replication inhibitors. This paper describes the first structural model of a complex between a picornaviral 3Cpro and a region of the 5′-UTR, stem-loop D (SLD). Using human rhinovirus as a model system, we have combined NMR contact information, small-angle X-ray scattering (SAXS) data, and previous mutagenesis results to determine the shape, position and relative orientation of the 3Cpro and SLD components. The results clearly identify a 1:1 binding stoichiometry, with pronounced loops from each molecule providing the key binding determinants for the interaction. Binding between SLD and 3Cpro induces structural changes in the proteolytic active site that is positioned on the opposite side of the protease relative to the RNA/protein interface, suggesting that subtle conformational changes affecting catalytic activity are relayed through the protein.
Keywords: NMR, SAXS, Picornavirus, Rhinovirus, Replication, 3C protease
1. Introduction
Picornaviridae is a large family of single-stranded RNA viruses (Van Regenmortel et al., 2000). Many family members cause devastating human diseases such as polio and hepatitis A while others are economically damaging, including the foot and mouth disease virus and the rhinoviruses responsible for human colds (Greenberg, 2003). Coronaviruses such as SARS share homology with picornaviruses, most notably in their mode of replication (Zhang and Yap, 2004).
The picornaviral replication cycle involves the production of a polyprotein that is subsequently cleaved into active proteins by viral proteases, principally the 3C protease, or 3Cpro (Porter, 1993). This protease is a single polypeptide of 182 amino acids composed primarily of an N-terminal α-helix followed by two anti-parallel β-barrels, each containing six strands (Matthews et al., 1994). The catalytic triad of H40, E71 and C146 lies in a cleft between the two barrels (Fig. 1 A). Numerous studies have targeted the 3Cpro proteolytic active site for drug design (Yang et al., 2006, Wang and Chen, 2007).
Fig. 1.

HRV-14 3Cpro structure, mutation data and basic residues. (A) Cartoon rendering. Two six-stranded β-barrels (peach and lavender ribbons) are joined by a long linker (yellow; residues 76–97). Basic and non-basic residues that, when mutated, inhibit RNA-binding (Andino et al., 1993; Leong et al., 1993) are shown as teal and green sticks, respectively. Side chains of other nearby basic residues are shown as dark blue sticks. The active site catalytic triad (grey spheres) is in the rear of the molecule in this view and contains the inhibitor JMC-98-3 (inhibitor not shown). (B) Surface rendering. Orientation and coloring are as in part A. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
The 3Cpro also serves a second function (Walker et al., 1995, Xiang et al., 1995). After cleavage from the remainder of the viral polyprotein, 3Cpro remains covalently linked to 3Dpol, the viral RNA-dependent RNA polymerase. This 3Cpro–3Dpol bi-protein binds via the 3Cpro segment to the untranslated 5′-end of the genomic RNA, generally stem-loop D (SLD; Fig. 2 A) from the first cloverleaf. This interaction is thought to correctly position the 3Dpol protein as a necessary step for transcribing positive sense RNA from a negative sense template. The lack of structural data featuring the 3Cpro–SLD interaction has hindered efforts targeting drug design to this second function of 3Cpro.
Fig. 2.

HRV-14 stem-loop D (SLD). (A) Sequence and schematic secondary structure with triloop bases and uracil · uracil and cytosine · uracil base pairs colored for reference in the text. (B) Solution structure (Headey et al., 2006). Coloring is similar to part A. In addition, the backbone phosphorous and singly coordinated oxygen atoms are shown in dark and light violet, respectively. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
We previously used NMR spectroscopy to determine the structure of SLD from human rhinovirus 14 (HRV-14; Fig. 2B) (Headey et al., 2006, Huang et al., 2001). Here we report an investigation into the interaction between the HRV-14 SLD and the HRV-14 3Cpro. NMR and small-angle X-ray scattering data were combined with results from previous mutational analysis (Andino et al., 1993, Leong et al., 1993) (Fig. 1) to construct a structural model of the HRV-14 3Cpro–SLD complex. The results identify the interface between 3Cpro and the RNA stem loop, showing that the RNA binds on the opposite side of the protein relative to the catalytic site, and further that there is communication between the RNA-binding and catalytic sites.
2. Materials and methods
2.1. NMR sample preparation
NMR experiments were recorded with either uniformly 15N/13C-labelled or 2D/15N/13C-labelled 3Cpro in 50 mM bis–tris-propane/MES containing 0.5 mM EDTA and 1 mM DTT at pH 6.0. Isotope-labelled 3Cpro from HRV-14 was expressed in Escherichia coli and purified using standard methods. Typically, a 50 mL overnight growth of E. coli BL21 CodonPlus cells containing the HMBP-3C plasmid was used to inoculate 1 L of LB containing chloramphenicol and ampicillin. This construct produces a histidine-tagged MBP fusion with 3Cpro that can be cleaved with thrombin (Alexandrov et al., 2001). When absorbance at 600 nm reached approximately 1.0 the cells were harvested by centrifugation at 3800 rpm (GS3 rotor) and resuspended in 500 mL of M9 minimal medium supplemented with 3.5 g of 13C-glucose and 0.5 g of 15N-ammonium chloride. After 30 min at 30 °C, induction was initiated by the addition of 0.5 mM isopropyl β-d-galactopyranoside (IPTG). For production of deuterated protein, the M9 minimal medium utilised D2O with either 2H,13C-glucose or 2H,12C-glycerol as a carbon source. The culture was grown at 30 °C until the growth curve flattened (approximately 6 h). Cells were harvested by centrifugation at 4800 rpm (GS3 rotor) at 4 °C for 20 min and resuspended in 30 mL of aqueous buffer containing 20 mM Tris and 150 mM NaCl at pH 7.5 (buffer A) with one pre-dissolved Complete™ EDTA free protease inhibitor tablet (Roche).
Cells were lysed by passing twice through a French press at a pressure of 6000 psi. The lysate was loaded onto a Ni2+–NTA (GE Healthcare) column equilibrated with buffer A. The column was washed with three column volumes of 20 mM imidazole in buffer A. H6-MBP-tagged 3C was then eluted with two column volumes of buffer A containing 250 mM imidazole. A total of 30 U of bovine thrombin (Roche) was added to the eluted fraction and this was dialyzed against 1 L of aqueous buffer containing 20 mM Tris, 1 mM DTT and 0.5 mM EDTA at pH 7.8 (buffer B) for 8 h with a change of buffer at 4 h. A QAE-Sephadex (Sigma–Aldrich) column was equilibrated with buffer B and the dialyzed protein fraction was passed through the column. The column was washed with a further three column volumes of buffer B. The flow-through and washes were combined, concentrated to approximately 250 μL and buffer exchanged to 50 mM bis–tris-propane/MES (BTM), 1 mM DTT, 0.5 mM EDTA at pH 6.0 (buffer C) via a 10 kDa molecular weight cut-off spin concentrator. The final protein concentration was determined by absorbance at 280 nm. JMC-98-3 (Webber et al., 1998) 3Cpro-specific inhibitor was a gift from S. Webber, (Agouron Pharmaceuticals, CA, USA, now part of Pfizer, CA, USA) and was added in equimolar or greater ratio to purified 3Cpro to create the 3CI sample. Partial alignment for RDC data collection was achieved via addition of a penta-ethylene glycol mono-decyl ether:hexanol mixture (Ruckert and Otting, 2000). The RNA oligomer for SLD was purchased from IDT, dissolved in the same buffer as the protein, adjusted to pH 6.0 and annealed. Aliquots of 2 mM SLD were added to a sample containing 0.15 mM 3Cpro until the RNA to protein ratio was 3:1 to create the 3CIR sample (3CI plus SLD).
2.2. X-ray scattering sample preparation
Samples used for X-ray scattering were dialyzed overnight against 50 mM MES, 0.5 mM EDTA, 1 mM DTT. The MES buffer solution contained pure water (MilliQ) treated with diethyl-pyrocarbonate (DEPC) to remove RNase activity and adjusted to pH 6.0 with 50 mM bis–tris methane. The dialysate was retained for solvent blank measurements. The post-dialysis concentration for each sample was determined by UV absorbance using E 260 nm = 22.83 mg−1 mL cm−1 and E 280 nm = 0.298 mg−1 mL cm−1 for SLD and 3C protease, respectively. The sample concentrations were: 3Cpro, 0.765 mg mL−1; SLD, 0.456 mg mL−1; 3Cpro–SLD complex, 1.07 mg mL−1. A lysozyme standard was prepared by dissolving and dialyzing the lyophilized protein (USB Corporation) in 150 mM NaCl, 40 mM sodium acetate (pH 3.8), to a final concentration of 7.7 mg mL−1.
2.3. NMR spectroscopy
NMR experiments were performed on a 700 MHz Bruker Avance spectrometer equipped with a cryoprobe, four r.f. channels and gradient pulse capabilities. In general, spectra were acquired at 22 °C using a WATERGATE (Piotto et al., 1992) solvent suppression scheme and processed in Topspin 2.0 (Bruker). The backbone resonances of 3CI were assigned via 15N-HSQC, HNCA (Kay et al., 1990, Grzesiek and Bax, 1992), HNCO (Kay et al., 1990, Grzesiek and Bax, 1992, Muhandiram and Kay, 1994), HNCACO (Clubb et al., 1992), CBCACONH (Grzesiek and Bax, 1993), and CBCANH (Grzesiek and Bax, 1992) spectra using XEASY (Bartels et al., 1995) analysis software. In order to obtain the HN chemical shift perturbation values, 1H,15N-HSQC-TROSY (Pervushin et al., 1998) spectra were acquired with the 3CI and 3CIR samples. TROSY versions of the HSQC, HNCA (Eletsky et al., 2001, Salzmann et al., 1998) and HNCO (Permi et al., 2000) experiments were used to assign the 3CIR backbone resonances with the software package Analysis (Vranken et al., 2005). The 1H–15N RDCs for the 3CI sample were obtained using an IPAP-HSQC sequence (Cordier et al., 1999).
2.4. Homology modelling, RDC-based refinement and calculation of chemical shift indexing and perturbation
A homology model of 3CI was created with Swiss Model (ExPASy website) in automatic mode using PDB accession number 2B0F (Bjorndahl et al., 2007) as a template for the 3C component. The covalently bound JMC-98-3 inhibitor was then modelled into the proteolytic active site. Coordinates for JMC-98-3 were first generated with the program Corina (Molecular Networks). The covalent bond between JMC-98-3 and 3CI C146 and a set of pseudo distance restraints based on the inhibited HRV2 crystal structure (PDB accession number 1CQQ) were used to dock JMC-98-3 into the active site of 3CI with the program CNS (Brünger et al., 1998).
1H,15N-HSQC-IPAP spectra of 3CI yielded a total of 123 well-resolved backbone 1H–15N RDCs. PALES (Zweckstetter and Bax, 2000) was used to generate an alignment tensor that was used iteratively in CNS (Brünger et al., 1998), along with the experimental RDC values, to refine the 3CI homology model. By combining 15N-HSQC, HNCO, and HNCA data, a single chemical shift perturbation (CSP) value on addition of RNA was calculated for each residue. Chemical shift indexing (CSI) (Wishart and Sykes, 1994) was performed using the Cα and C-carbonyl shifts from 3CI and 3CIR.
2.5. Small-angle X-ray scattering data acquisition, reduction, and analysis
Small-angle X-ray scattering data were acquired, reduced and analysed using an Anton Paar SAXSess with line-collimation and CCD detector as described previously (Jeffries et al., 2008). Samples were exposed for 60 min at 20 °C with 10 mm slit and integration lengths. Data reduction to I(q) vs. q (where q = (4πsinθ)/λ, 2θ is the scattering angle and λ = 1.54 Å, the wavelength of the radiation) and solvent subtractions were performed with the program SAXSquant1D (Anton Paar, Austria), while subsequent desmearing calculations were performed with the program GNOM (Semenyuk and Svergun, 1991). Radius of gyration (Rg) and zero-angle scattering (I(0)) values were initially estimated by means of Guinier analysis (Guinier and Fournet, 1955) with the program PRIMUS (Konarev et al., 2003). The programs GNOM and GIFT (Bergmann et al., 2000) were used to calculate indirect Fourier transforms of I(q) to give P(r) vs. r and associated values for I(0), Rg, and maximum dimension (Dmax). The molecular weights of the scattering particles in solution—3Cpro, SLD, and 3Cpro–SLD complex—were determined through Glatter’s method and the equation (Orthaber et al., 2000):
| (1) |
where N A is Avogadro’s number, c is the concentration of the scattering particle in g cm−3, and ΔρM 2 is the square of the scattering contrast per mass. This variable is calculated from the equation ΔρM 2 = (Δρν)2, where Δρ is the difference in scattering length density between the particle and solvent (termed the contrast and given in cm−2), and ν is the partial specific volume of the scattering particle (in cm3 g−1). The program suite MULCh (Whitten et al., 2008) and the NucProt Parameter set (Voss and Gerstein, 2005) were used to calculate values for Δρ and ν, respectively.
2.6. Shape restoration calculations from scattering data
Ab initio shape restoration calculations from experimental scattering data for the 3Cpro–SLD complex were performed with the program MONSA (Svergun and Nierhaus, 2000). This program uses multiple scattering profiles to restore the shapes of complexes and differentiate between subunits possessing different scattering densities. In the present case, a strong contrast occurs between SLD and 3Cpro due to the electron-dense phosphate backbone of the RNA. Input data for each MONSA calculation included both of the smoothed, desmeared I(q) profiles generated from GNOM for the 3Cpro–SLD complex and 3Cpro. The Rg constraints in these calculations were derived from the SAXS data for 3Cpro and the 3Cpro–SLD complex. As SLD alone showed a tendency to dimerize under these experimental conditions, we used a theoretical Rg constraint for that component calculated from the coordinates of the SLD NMR structure (Headey et al., 2006). The results of 10 independent MONSA calculations were averaged and evaluated with the program DAMAVER (Volkov and Svergun, 2003) to generate a consensus envelope for the entire complex and identify the relative positions of its protein and RNA subunits. The mean normalized spatial discrepancy between corresponding subunits in each model was 0.45 ± 0.01 and 0.51 ± 0.01 for 3Cpro and SLD, respectively, indicating that all restored shape solutions were very similar.
2.7. Rigid-body refinement of models against the scattering data
Atomic models for the 3Cpro–SLD complex that fit the scattering data were calculated through rigid-body refinement with the program SASREF6 (Petoukhov and Svergun, 2005) using the RDC-refined NMR structure of HRV-14 3Cpro and each of the nine different NMR structures within the SLD structure ensemble. These SLD structures differ primarily in the details of the triloop conformation. Ten independent calculations were performed for each combination of 3Cpro and one of the SLD structures. The SLD component was constrained to have at least one nucleotide within 10 Å of the Cα atom of the 3Cpro residues strongly implicated in RNA-binding by at least two of the following criteria: mutagenesis, NMR chemical shift perturbation, NMR signal loss, or signal doubling. This distance constraint was set to 20 Å for more moderately implicated residues to allow for the possibility that residues not directly in contact with the binding interface region may be affected by binding, but typically to a lesser extent. The best model was selected based on the fit to the scattering data as determined by the minimum χ 2 value, which for an ideal fit is 1.0. The best-fit model was energy minimized in CNS (Brünger et al., 1998) to relieve steric clashes.
3. Results
3.1. NMR data
The chemical shifts and peak intensities in NMR spectra of isotopically labelled 3Cpro in the absence and presence of SLD were compared. Spectral quality was improved by inclusion of the active site inhibitor JMC-98-3 (Webber et al., 1998). A structural model of the protein plus JMC-98-3 inhibitor, designated 3CI, was constructed based on homology modelling using the HRV-14 3Cpro solution structure (PDB: 2B0F) (Bjorndahl et al., 2007) with the JMC-98-3 inhibitor modelled into the active site. The resulting structure was refined using NH residual dipolar coupling (RDC) data. The 3Cpro consists of 182 residues and a total of 156 backbone amide peaks were assigned in the 3CI spectrum. In addition to the six prolines, twenty residues did not produce identifiable backbone amide peaks. However, the unassigned residues are sequentially isolated with the exception of the 103–112 loop that is adjacent to the proteolytic active site and is not implicated in RNA-binding (Matthews et al., 1994). Thus, the assigned peaks provide a suitable sampling of the potential interactions with RNA.
Unlabelled SLD was titrated into a sample of 2H,15N,13C-labelled 3CI with amide protons restored through the use of H2O-containing buffer. The RNA:protein ratio was increased to 3:1 to ensure full complexation of the protein component. NMR spectra obtained at 1:1 and 3:1 molar ratios are similar and clearly identify the binding stoichiometry as 1:1. Subsequent NMR analysis of 3CIR (used to designate 3CI in the presence of unlabelled SLD-RNA) was performed with the 3:1 sample. Changes induced in the 3CI 1H,15N-HSQC-TROSY spectrum upon addition of unlabelled SLD are shown in Fig. 3 . The 3CIR spectrum produced 139 assignable peaks, with peaks from 19 additional residues unassigned due to exchange broadening or shifting of peaks into crowded regions of the spectra. Conformational heterogeneity at D32, N80 and F83 (Fig. 3B) is consistent with the presence of additional local conformational exchange processes at the protein/RNA interface.
Fig. 3.

3Cpro1H,15N-HSQC-TROSY spectra. Overlay in the absence (black) and presence (red) of SLD. Significantly shifted peaks are annotated in black with peak positions indicated by black and red lines. Peaks which become unobservable upon complex formation are annotated in grey with peak positions indicated by grey lines. (A) Entire spectrum. (B) Zoom-in of boxed region from part A, showing multiple peaks for the three indicated residues in the presence of SLD. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
The secondary structure of the 3CI RDC-refined homology model was extracted with the program Procheck (Laskowski et al., 1993) (shown above Fig. 4 ). The results are consistent with previous analyses (Bjorndahl et al., 2007). 3CI consists of an N-terminal α-helix followed by two six-stranded β-barrels. Strand 5 and 5′ flank a single amino acid that lacks β-strand character in the refined model. If combined, these two strands would form the canonical strand 5. The presence of a partial helical turn from K175 to Y177 was not detected by Procheck but was displayed by Pymol using default parameters and thus appears in our figures. Loops of varying lengths separate the strands. The most extensive interstrand interval occurs between strands 6 and 7 and includes residues T76 through to V97. This lengthy loop is situated along the face opposite to the proteolytic active site, contains a short α-helix, and serves as a linker between the N-terminal and C-terminal β-barrel domains (Fig. 1, linker in yellow).
Fig. 4.

3Cpro secondary structure and chemical shift perturbation vs. amino acid number. The secondary structure calculated via Procheck using the 3CI RDC-refined structure is shown schematically above part A, with shading used to symbolize less (dark) and more (light) solvent accessible regions. The positions of catalytic triad residues H40, E71 and C146 are marked by ●. Sites of mutations shown to inhibit RNA-binding (Andino et al., 1993, Leong et al., 1993) are marked by •. Grey shaded areas extending from A to D correspond to the predominantly β-sheet regions. (A) 3CI chemical shift indexing (CSI) data. Positive and negative values indicate α-helical and β-sheet character, respectively. Gaps indicate positions of unassigned residues. (B) 3CIR CSI data. (C) Difference between B and A. Residues that are unassigned in the 3CI and/or 3CIR complex are designated as zero value. For residues that produce multiple peaks, the chemical shift of the most intense peak was used. Solid and dashed horizontal lines denote zero ± one standard deviation. (D) Chemical shift perturbation of 3CI upon titration of SLD, shown as a normalized sum of backbone chemical shifts. Grey bars represent residues for which chemical shifts could not be determined in the RNA-bound form. Dashed and dotted horizontal lines denote the mean perturbation and mean plus one standard deviation, respectively.
An alternative secondary structure analysis of 3CI on the basis of chemical shift indexing (CSI) (Wishart and Sykes, 1994) was performed (Fig. 4A). Contiguous significant positive CSI values implicate α-helix, while contiguous negative stretches indicate β-strand. The Procheck and CSI modes of secondary structure analysis, performed independently, produce largely consistent results, apart from the gap in the CSI graph due to the unassigned 103–112 stretch.
The CSI analysis of 3Cpro in the 3CI SLD complex (3CIR, Fig. 4B) differs little from the Procheck analysis (above Fig. 4A) apart from additional concentrations of unassigned residues within the N-terminal helix and in the loop between β-strands 9 and 10. Fig. 4C is a difference plot (4B minus 4A). The largest contiguously changing stretch occurs in the linker region connecting the two β-barrels and contains three residues (K82, R84, D85) implicated in RNA-binding via mutational analysis (Leong et al., 1993, Andino et al., 1993). The CSI changes alternate in sign in this region, suggesting that little regular secondary structure is formed. Nonetheless the magnitude and continuity of change suggest a significant alteration in backbone conformation. Residues 116–120 within β-strand 8 and residues 150–163 spanning β-strands 10 and 11 also show significant CSI change, in each case largely consistent with a reduction in β-strand character. Changes in strand 8 (shown along the bottom of Fig. 1A) could indicate allosteric effects as this strand does not appear to approach the RNA-binding site (see below). The loop between strands 10 and 11 contains three residues (T153, G154, K155) implicated in RNA-binding (Andino et al., 1993, Matthews et al., 1994).
A plot of chemical shift perturbations introduced by addition of SLD is presented in Fig. 4D. The height of each black bar represents a weighted average of 1H, 15N and 13C shifts for that residue while grey bars indicate residues for which 3CI shifts were assigned but 3CIR shifts were not. The largest concentration of grey bars occurs within the N-terminal α-helix and in the loop between β-strands 9 and 10. The largest measurable perturbations occur in the long central linker region connecting the two β-barrel domains of the protein, with significant shifts also noted in β-strands 1–3 and 10–11 and near the C-terminus. Note that the localised CSI changes (Fig. 4C) correlate well with regions of large chemical shift change (Fig. 4D).
The largest SLD-induced chemical shift perturbations on the 3Cpro surface cluster primarily to a patch (dark red in Fig. 5 A) that includes D32 from the loop connecting β-strands 2–3, N80, F83 and F89 from the inter-domain linker and V179 from the C-terminal region. Much smaller perturbations are observed on the opposite face of the protein where the catalytic site is located (Fig. 5A, right-hand view). Residues for which peaks become unobservable upon addition of SLD are shown in grey and include several amino acids from the N-terminal helix and E81 from the linker, as well as F25 and G144 from the catalytic face.
Fig. 5.
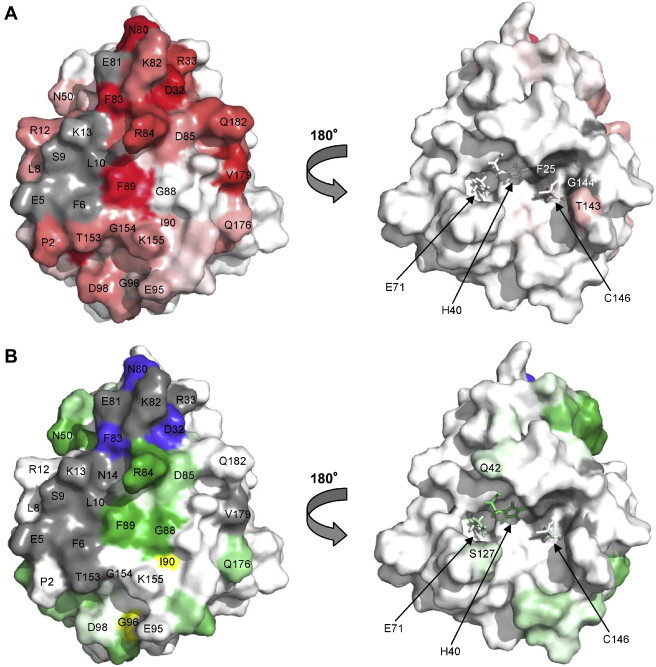
3Cpro residues affected by addition of SLD. (A) Chemical shift perturbation data (Fig. 4D), scaled from white (little change) to red (large change). Residues for which chemical shifts could not be assigned in the RNA-bound form are shown in grey. (B) RNA-induced intensity changes in the HSQC-TROSY spectra (Fig. 3) with reduction factor scaled from white (little change) to green (large reduction). Residues with significant increase in peak intensity are shown in yellow, while residues for which no peak was observed in the HSQC-TROSY spectrum are shown in grey. Dark blue indicates positions of multiple HSQC-TROSY peaks (Fig. 3B). In the left-hand view of parts A and B, the RNA-binding surface is shown. The right-hand views depict the catalytic face. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Changes in intensity of HSQC-TROSY peaks upon addition of SLD were monitored as an additional indication of changes in chemical environment. Dark green patches in Fig. 5B indicate a large reduction in intensity for resonances assigned to residues in the inter-domain linker, specifically R84, D85, G88 and F89, as well as for N50 from the loop connecting β-strands 4–5. The slight intensity increase for resonances arising from residues I90 and G96 (yellow) could indicate an increase in mobility in the C-terminal half of the linker upon SLD-binding. The three residues giving rise to multiple peaks (Fig. 3B) also produce significantly decreased intensity (dark blue in Fig. 5B). Grey patches in Fig. 5B represent residues for which the 3CIR HSQC-TROSY peak could not be assigned and include all grey patches from Fig. 5A in addition to N14, R33, K82 and T153. The resonances arising from residues at the catalytic face (Fig. 5B, right-hand view) produce far fewer significant changes in intensity.
3.2. Analysis of NMR results for use with SAXS refinement
The analyses summarized in Fig. 5A and B suggest that the top central region of the 3CI face opposite to the proteolytic active site is a key part of the SLD binding interface. This result is consistent with pre-existing mutational data (Andino et al., 1993, Leong et al., 1993) and sequence conservation analysis. The KFRDIR motif (residues 82–87) from the inter-domain linker (residues 76–97) is conserved across picornaviral 3Cpro sequences and has been implicated in RNA-binding (Leong et al., 1993, Gorbalenya et al., 1989, Shih et al., 2004). From this linker, N80, K82, F83, R84, D85 and F89 each exhibit large chemical shift changes upon complex formation while N80 and F83 produce multiple peaks, E81 and K82 peaks become unobservable and R84, D85, G88 and F89 peaks reduce significantly in intensity. Residues I86 and R87, which complete the KFRDIR motif but are partially buried along the back side of the small yellow helix in Fig. 1A, show little change on binding.
The NMR data also suggest the N-terminal α-helix (residues 2–14) is involved in RNA-binding, as many of the corresponding TROSY peaks are shifted or broadened in the 3CIR spectrum. Strands 2–3 and the intervening loop also appear to be involved. This loop includes D32, which, as noted above, produces three 3CIR HSQC-TROSY peaks (Fig. 3B) and R33. Residues from strand 10 to the midpoint of strand 11, a region that connects C146 of the catalytic triad through the protein core to the RNA-binding face, also display marked changes between the free and bound form. This region spans residues 153–155 which have been implicated in RNA-binding by mutagenesis studies in poliovirus (Andino et al., 1993). The C-terminus exhibits above average shift changes in residues 176–182. Somewhat inconsistently with the same work, we do not observe significant perturbation at K175 upon RNA-binding.
Taken together, these results show that a surface region representing several non-sequential segments of 3CI interacts with SLD. The linker region from residues 76–97 that contains the conserved KFRDIR motif is most strongly implicated, with the N-terminal helix and β-strand and loop regions from 25–38 and 145–155 also playing a role. The C-terminal residues are also perturbed. However, NMR peak intensities indicate high mobility of the C-terminus. Therefore we have limited the NMR/mutation-implicated surface used for SAXS analysis to 3Cpro residues 5–6, 9–10, 13, 32–33, 80–85, 89 and 153–155, bearing in mind that the C-terminus may fold toward the interface upon complex formation. Of these residues, D32, N80, F83, R84 and F89 were treated as strongly-implicated in RNA-binding (see Section 2.7).
3.3. SAXS data
Due to its phosphate backbone, RNA has a higher electron density and hence X-ray scattering density compared to that of a protein. The small-angle X-ray scattering from the 3Cpro–SLD complex is therefore sensitive to the positions and orientations of RNA and protein components. Scattering data were acquired for the 3Cpro, SLD and the 3Cpro–SLD complex (Fig. 6 ). These data yield basic parameters such as molecular weight, radius of gyration (Rg), and the contrast-weighted distribution of interatomic distances (P(r)) within the scattering particle (Supplementary Table 1). The experimentally determined molecular weights for 3Cpro and 3Cpro–SLD, as well as a lysozyme standard (calculated from measured I(0) values with equation (1)) agree well with the expected values based on amino acid and nucleotide sequences. The experimental Rg value for 3Cpro derived from its P(r) agrees with the theoretical value from the NMR structure (16.7 Å, calculated using the program CRYSOL (Svergun and Nierhaus, 2000)). In contrast, the SLD molecular weight determined from I(0) was approximately twice the expected value indicating that under our experimental conditions SLD is not a monomer in solution. Nonetheless, a 1:1 3Cpro–SLD complex of the expected molecular weight was formed by mixing equimolar amounts of 3Cpro to SLD (Supplementary Table 1), which is consistent with the NMR results.
Fig. 6.
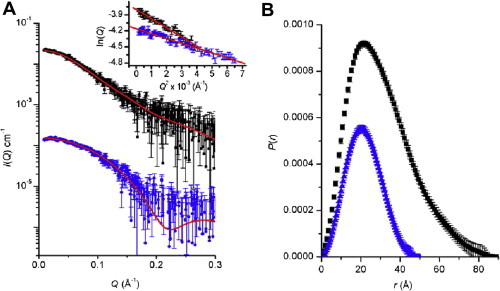
Scattering data for 3Cpro and its complex with SLD. (A) Desmeared SAXS profiles for the 3Cpro (blue) and 3Cpro–SLD complex (black) with the former profile shifted on the vertical axis for clarity. The predicted scattering profiles for the MONSA model for the 3Cpro–SLD complex and the NMR structure of 3Cpro are shown as red lines superposed with the corresponding experimental scattering data. The inset shows the expected linear Guinier plots for samples that are monodisperse, identical particles. (B) P(r) profiles calculated from the scattering data for 3Cpro (blue) with a Dmax of ∼50 Å and 3Cpro–SLD (black) with a Dmax of ∼80 Å. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Two types of structural models were generated for the 3Cpro–SLD complex. First, the molecular envelope for the 3Cpro–SLD complex was generated through ab initio shape restoration. As the protein and RNA components of the complex have different contrasts this enables the determination of the relative position of 3Cpro and SLD within the total molecular envelope. The envelope is shown as a transparent surface in Fig. 7 A and fits the scattering data with a statistically ideal χ 2 value of 1.00 (Fig. 6B). This envelope has dimensions of approximately 80 Å × 50 Å × 50 Å with the SLD component extending away from the surface of the 3Cpro at an angle of ∼30°. Independent from ab initio shape restoration, an atomic model for the complex was calculated from the NMR-derived structures for 3Cpro and SLD through rigid-body refinement. These rigid-body calculations were combined with previously derived NMR contact information to constrain the relative orientations between the subunits. The atomic model of the complex is shown as ribbons in Fig. 7A and fits the scattering data with a χ 2 value of 1.26, which is good but somewhat higher than ideal. This discrepancy may be the result of small differences between the actual conformation of SLD and/or 3Cpro in the complex compared with the corresponding structures used for the calculation, which were derived for each component in isolation. Indeed, the predicted scattering profile for the 3Cpro component of the complex fits the scattering data for uncomplexed 3Cpro relatively poorly (χ 2 = 1.44), and provides further evidence that the 3Cpro undergoes a small conformational change upon forming a complex with SLD.
Fig. 7.

Structural model for the 3Cpro–SLD complex based on combined SAXS/NMR/mutagenesis data. (A) Overlay of the molecular envelope derived by shape restoration from the scattering data with the best-fit rigid-body refined atomic model showing the triloop end of the SLD in close contact with 3CI. The stem-loop termini extend away from the protein. The RNA and protein are colored as in Fig. 2, Fig. 5A, respectively. (B) Atomic model from A colored to highlight the most strongly implicated residues in binding RNA (red). The SLD nucleotides implicated in 3Cpro-binding (by homology with coxsackie virus (Ohlenschläger et al., 2004)) are shown as spheres.
The atomic model of the complex shows the SLD triloop nestled between the end of the 3Cpro N-terminal α-helix and the inter-domain linker that contains the conserved KFRDIR motif. Residues of 3Cpro that make contact with SLD include L10 and R12–M16 from the N-terminal helix and the following strand as well as G27–R33 and C35 from β-strands 2 and 3. Other contact residues include V49 and Q52 from the loop connecting strands 4 and 5 and L77, R79–R84 and I86 from the linker that connects the two β-barrels of the protein. No residues from the C-terminal β-barrel make direct contact with SLD in this model. On the SLD molecule, nucleotides from uracil-13 to uracil-17 form contacts with the protein. This stretch includes the triloop and two bases, guanine-16 and uracil-17, from the 3′-side of stem II. Interestingly, the 31P chemical shift values of guanine-16 and uracil-17 in the uncomplexed SLD are atypical, indicating a significant departure from standard A-form geometry (Headey et al., 2006). The intermolecular interface covers an area of 653 Å2 and lies within the range of typical RNA–protein interaction interface areas (Allain et al., 2000, Johnson and Donaldson, 2006).
The SLD also closely approaches the loop that connects β-strands 2 and 3 (H31–R33). The H31–R33 loop is spatially positioned between residues R79–R84 of the inter-domain linker and the C-terminus of the protein. The N-terminal α-helix may become sufficiently mobile to approach the nearby SLD stem region, possibly explaining the loss of the corresponding peaks in the 3CIR NMR spectra if the association is transient or heterogeneous.
Both electrostatic and hydrophobic interactions play a role in complex formation. In addition to R84, other basic residues near the binding surface that may contact the SLD phosphodiester backbone include R12, K13, R33, R79 and K82. The F6, F83 and F89 residues are exposed in the uncomplexed protease and may stabilize the interaction via hydrophobic interactions such as base stacking. In the present model, the highly conserved F83 is particularly well situated for interaction with the triloop bases. The V49, L77 and I86 side chains could make additional hydrophobic contacts, as may the aliphatic regions of the many lysine and arginine residues in the area.
A conserved domain database (Marchler-Bauer et al., 2007) alignment of ten diverse 3Cpro and 3Cpro-like proteases (see Supplementary Fig. A1) was used to examine conserved residues in the contact regions. Of the residues that make contact with RNA, conserved basic positions include R12 (50% conserved), R79 (70%), K82 (70%) and R84 (100%). Conserved hydrophobic residues include L10 (60%), I15 (80%), I30 (100%), V49 (90%), L77 (100%), F83 (100%) and I86 (90%). The N14 and G29 positions are conserved and the D32 position is acidic in 70% of the 10 sequences, while the H31 position is aromatic or H in seven instances.
4. Discussion
RNA–protein interactions are by their very nature diverse due to the variety of secondary and tertiary structures that each of these biopolymers is capable of adopting. The 3Cpro tertiary structure does not appear to fit neatly into one of the previously defined classes of RNA-binding domains. However, some commonalities are apparent. For instance, we have shown that the linker connecting the two β-barrel domains of 3Cpro, which lacks regular secondary structure and is likely to possess some degree of mobility, is a key to the interaction with SLD. Proteins containing the most prolific RNA-binding domain, dubbed simply the RNA-binding domain (RBD) or RNA recognition motif (RRM), often use the flexible linker between tandem RBDs to contact a flexible region of RNA (Allain et al., 2000, Wang et al., 2001). After complex formation, each of these flexible regions changes conformation, typically becoming more highly ordered (Bouvet et al., 2001). This raises the possibility that a conformational change and an increase in rigidity may occur in the 3Cpro linker and SLD triloop. This potential rearrangement could, among other effects, help translocate the F89 aromatic ring of the connecting linker into closer contact with SLD. In addition, RBD domains possess two or three solvent-exposed aromatic bases which can form contacts with the RNA bases (Birney et al., 1993). This is consistent with the potential involvement of the exposed F83, F89 and F6 residues from 3Cpro, although the effects at F6 may simply be due to an overall shift in the position of the N-terminal α-helix. Precedence here comes from the U1A–UTR complex (Oubridge et al., 1995, Varani et al., 2000), in which the U1A C-terminal α-helix shifts position to allow the RNA UTR fragment more complete access to the RNA-binding sheet beneath.
The non-Watson–Crick base pairs (uracil-5 · uracil-23, cytosine-6 · uracil-22 and uracil-7 · uracil-21) do not appear to be directly involved in the interaction but may play an indirect role. Ideal A-form RNA contains a deep and narrow major groove which precludes the possibility of specific recognition of the base pairs within. The minor groove side provides greater access, but the bases present an essentially uniform, non-sequence-specific face in the minor groove. Specific recognition of bases must occur via the major groove. RNA stem loops containing non-canonical base pairs can present a widened major groove, as has been shown for HRV-14 SLD (Headey et al., 2006) and in other instances (Du et al., 2004, Ohlenschläger et al., 2004). Thus, contacts between the stem region closest to the triloop and the protease may benefit from the presence of the non-canonical base pairs further up the stem. The contacts between the triloop bases and 3Cpro are somewhat analogous to the binding of the sex-lethal protein to a non-paired uracil-rich tract from the drosophila transformer mRNA precursor (Handa et al., 1999). The transformer uracil-rich tract is extended but ordered, including stable sugar puckers in the atypical C2′-endo configuration. Thus it is interesting to note that each sugar of the uracil-rich stretch of implicated SLD nucleotides (uracil-13 to uracil-17) primarily adopts a C2′-endo configuration (Headey et al., 2006). In summary, while the global fold of 3Cpro may be distinct from other RNA-binding proteins investigated to date, the mechanisms of interaction may be largely conserved.
SLD stem I is highly conserved among rhinoviruses. Mutational work in which stem I adenine · uracil pairs were replaced with guanine · cytosine pairs suggests that this conservation is necessary to retain the ability to bind 3Cpro (Walker et al., 1995). This would seem at odds with the model (Fig. 7A), in which the triloop and stem II regions contact 3Cpro. On the other hand, NMR shift perturbation data for a coxsackie virus 3Cpro–SLD interaction (Ohlenschläger et al., 2004) suggest that the triloop and the nearby stem II elements contact the protein (Fig. 7B). Although 3Cpro from HRV-14 can bind SLD from other picornaviral species, the opposite is not necessarily true. For example, coxsackie virus 3Cpro is unable to bind HRV-14 SLD. Evidence suggests that this disparity is due to the presence of a triloop in HRV-14 SLD vs. a tetraloop in most other picornaviral SLD (Zell et al., 2002). These results implicate the triloop region in binding. The somewhat contradictory data regarding stem I could suggest that alterations close to the chain termini influence the global fold or stability of SLD, affecting the binding at the triloop end. That is, shape, rather than contact recognition of the specific bases in stem I, may be the key determining factor (e.g. Rieder et al., 2003, Zell et al., 2002). Sequence conservation in stem I could also reflect, to some degree, other interactions/activities that are not well characterized at present.
While the proteolytic active site face of 3Cpro experiences relatively little perturbation upon RNA binding, the largest changes on this face cluster to the vicinity of the catalytic triad (H40, E71 and C146). This includes chemical shift changes in T143, loss of peaks for F25 and G144 and intensity reduction in H40, Q42, E71 and S127. It thus appears that the signal of SLD binding to the linker side of 3Cpro is transduced into a change at the active site. This could represent a mode of regulation, fine-tuning the specificity or activity of 3Cpro as previously suggested (Peters et al., 2004, Shih et al., 2004). The mechanism of this transmission may involve the hydrophobic core of the protein. Four β-strands approach both the active site and the RNA-binding site (Fig. 8 ): strands 2 (F25–I30) and 3 (V34–I37), connected by the 31–33 loop and strands 10 (G147–L150) and 11 (F157–G163), connected by the 151–156 loop that contains T153, G154 and K155. Both of these loops show changes upon addition of SLD.
Fig. 8.
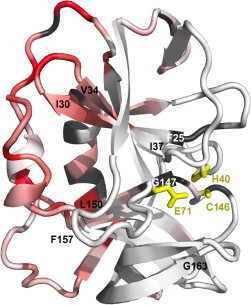
Allosteric effects at the 3Cpro proteolytic active site. Chemical shift perturbations upon addition of SLD, plotted onto a cartoon view of 3Cpro, with the catalytic triad side chains shown as yellow sticks with yellow labels. Color scheme is otherwise identical to Fig. 5A. The viewing angle is roughly intermediate between the two views of Fig. 5A. The terminal residues of four β-strands (strand 2, F25–I30; strand 3, V34–I37; strand 10, 147–150; strand 11, 157–163) are labelled for reference in the text. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
The H31 and D32 positions of the 31–33 loop that is sandwiched between the C-terminus and residues 79–84 of the linker are relatively well conserved (H or aromatic 70%; D or E 70% in conserved domain database analysis). Perturbation of this loop appears to occur via direct interactions with the RNA triloop through the 79–84 loop, or in a supporting role for this loop. The 151–156 loop is partially conserved at the G154 (50%) and K155 (70%) positions and contains 100% hydrophobic residues at the 156 and 157 positions. Changes induced in this loop may be more indirect in nature, as SLD does not closely approach these residues in the present model. In 3CI, T153 and G154 interact with F6 (90% hydrophobic) of the N-terminal helix and are within ∼4 Å of F89 of the opposing FISEDLE motif from the C-terminal half of the linker. K155 closely approaches the acidic residues in the FISEDLE loop. Thus the 151–156 loop is positioned between two important structural elements, the N-terminal α-helix and the inter-domain linker. Both of these elements directly contact the RNA. Changes induced in these elements could result in the observed (indirect) perturbation of the 151–156 loop. Note that I90 and G96 peaks increase in intensity upon SLD-binding (Fig. 5B), consistent with the C-terminal half of the linker increasing in mobility.
In turn, changes in the conformation or position of the 31–33 and 151–156 loops may transmit the observed changes to the proteolytic active site via the four associated β-strands. Strands 2 and 3 extensively hydrogen bond to each other, as do strands 10 and 11 up to the point where the longer strand 11 curves toward the protein periphery. Therefore the induced change is more likely to involve a slight reorientation of the 2–3 pair relative to the 10–11 pair than changes within each pair. The 2–3 to 10–11 contact is in fact the point of junction between the N-terminal and C-terminal β-barrel. Together with the intimate involvement of the 76–97 linker that covalently joins the two barrels, it would appear that 3Cpro–SLD complex formation may induce a subtle change in the relative positions of the two 3Cpro β-barrels that transmits RNA-binding information to the opposite, catalytic face of the protease. Identification of the 3Cpro–SLD interaction surface along with the detection of RNA-induced changes in the 3Cpro catalytic site may lead to novel avenues for drug design.
In addition to contacting the 5′-UTR (also called oriL for “Left”), picornaviral 3C proteases can also interact with a distinct internal RNA replication element (called oriI for “Internal” (Yin et al., 2003). The position of the oriI site is highly variable. For example, in poliovirus the oriI element is within the 2C coding sequence (Goodfellow et al., 2000) and in HRV-14 it is located in the VP1 (capsid protein) coding sequence (McKnight and Lemon, 1996, McKnight and Lemon, 1998). The oriI interaction leads to uridylation of the 3B protein (also known as VPg) with the resulting uridylated peptide serving as a primer for viral RNA replication. In the context of the 3C/VPg/oriI complex, a binding mechanism has been proposed that includes a 3C dimerization event (Pathak et al., 2007, Pathak et al., 2008, Shen et al., 2008), supported by cross-linking studies and crystal contacts of uncomplexed poliovirus 3Cpro (Mosimann et al., 1997) though a previous study found no evidence of dimerization (Xiang et al., 1998). NMR data from 3Cpro interactions with single-stranded 11-mer RNA sequences derived from the poliovirus oriI element (Amero et al., 2008) were combined with the above theory to propose that a poliovirus 3Cpro dimer binds to two sites on an unwound oriI element.
In the present study, we see no evidence of 3Cpro dimerization, and in fact the SAXS data is only consistent with the formation of a 1:1 3Cpro:SLD complex. Furthermore, although the bases of the SLD triloop region are unpaired, we see no evidence of significant disruption of base-pairing in the HRV-14 SLD stem regions upon complex formation. These discrepancies with previous models may reflect differences in 3Cpro interactions with oriI vs. oriL or perhaps differences between poliovirus and HRV replication mechanisms. While overlapping regions of the 3Cpro surface from poliovirus and HRV are clearly involved in interaction with single-stranded oligomers derived from the poliovirus oriI element and with the intact HRV-14 oriL, respectively, the mode of interaction may thus differ in other aspects. Along these lines, note that substitution of the poliovirus oriI element into HRV yields a chimera unable to uridylate VPg (Shen et al., 2007). Note also that the 3Cpro N-terminal α-helix amide chemical shifts were assigned in the poliovirus 3Cpro-oriI study, suggesting that the characteristics or role of the N-terminal helix may differ in poliovirus vs. HRV-14 or in binding single vs. double-stranded RNA molecules. Each of these possibilities would be consistent with the suggestion that the HRV-14 N-terminal helix may interact directly with the SLD stem region. Furthermore, a recent crystallographic study of the 3Cpro–3Dpol fusion protein from poliovirus (Marcotte et al., 2007) positions the covalently linked 3Dpol in close contact with the C–terminal region of 3Cpro. This orientation of 3Dpol, if conserved in HRV-14, would not interfere with the SLD–3Cpro contacts as determined in the present study. The 3Dpol position does, however, conflict with the poliovirus 3Cpro dimer contacts used in the previous models. It is possible that either the 3Cpro–3Cpro or 3Cpro–3Dpol contacts differ in vivo vs. in a crystal environment, or that poliovirus 3Cpro dimerizes in this fashion only after proteolytic release of 3Dpol. Further structural studies of 3Cpro–3Dpol in complex with SLD and with larger fragments of the 5′-UTR or oriI elements, along with quantitative analyses of RNA-induced effects on 3Cpro catalytic activity, will help to further delineate the molecular mechanism of genomic replication in picornaviruses and perhaps shed light on the intricate mechanisms linking replication events.
Acknowledgments
We wish to thank Wai-Ming Lee, David Libich, Trevor Loo, Giselle Soares, Joel Tyndall. Funding for this project was provided in part by grants from the Royal Society of New Zealand (Marsden Fund Award MAU301) to S.M.P. and an Australian Research Council Federation Fellowship (FF0457488) and U.S. Department of Energy Grant (DE-FG02-05ER64026) to J.T.
Footnotes
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.jsb.2009.02.010.
Appendix A. Supplementary data
References
- Alexandrov A., Dutta K., Pascal S.M. MBP fusion protein with a viral protease cleavage site: one-step cleavage/purification of insoluble proteins. Biotechniques. 2001;30:1194–1198. doi: 10.2144/01306bm01. [DOI] [PubMed] [Google Scholar]
- Allain F.H., Bouvet P., Dieckmann T., Feigon J. Molecular basis of sequence-specific recognition of pre-ribosomal RNA by nucleolin. EMBO J. 2000;19:6870–6881. doi: 10.1093/emboj/19.24.6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amero C.D., Arnold J.J., Moustafa I.M., Cameron C.E., Foster M.P. Identification of the oriI-binding site of poliovirus 3C protein by nuclear magnetic resonance spectroscopy. J. Virol. 2008;82:4363–4370. doi: 10.1128/JVI.02087-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andino R., Rieckhof G.E., Achacoso P.L., Baltimore D. Poliovirus RNA synthesis utilizes an RNP complex formed around the 5′-end of viral RNA. EMBO J. 1993;12:3587–3598. doi: 10.1002/j.1460-2075.1993.tb06032.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartels C., Xia T., Billeter M., Güntert P., Wuthrich K. The program XEASY for computer-supported NMR spectral analysis of biological macromolecules. Int. J. Biomol. NMR. 1995;6:1–10. doi: 10.1007/BF00417486. [DOI] [PubMed] [Google Scholar]
- Bergmann A., Fritz G., Glatter O. Solving the generalized indirect Fourier transformation (GIFT) by Boltzmann simplex simulated annealing (BSSA) J. Appl. Crystallogr. 2000;33:1212–1216. [Google Scholar]
- Birney E., Kumar S., Krainer A.R. Analysis of the RNA-recognition motif and RS and RGG domains: conservation in metazoan pre-mRNA splicing factors. Nucleic Acids Res. 1993;21:5803–5816. doi: 10.1093/nar/21.25.5803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorndahl T.C., Andrew L.C., Semenchenko V., Wishart D.S. NMR solution structures of the apo and peptide-inhibited human rhinovirus 3C protease (Serotype 14): structural and dynamic comparison. Biochemistry. 2007;46:12945–12958. doi: 10.1021/bi7010866. [DOI] [PubMed] [Google Scholar]
- Bouvet P., Allain F.H.T., Finger L.D., Dieckmann T., Feigon J. Recognition of pre-formed and flexible elements of an RNA stem-loop by nucleolin. J. Mol. Biol. 2001;309:763–775. doi: 10.1006/jmbi.2001.4691. [DOI] [PubMed] [Google Scholar]
- Brünger A.T., Adams P.D., Clore G.M., DeLano W.L., Gros P., Grosse-Kunstleve R.W., Jiang J.S., Kuszewski J., Nilges M., Pannu N.S., et al. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D: Biol. Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- Clubb R.T., Thanabal V., Wagner G. A constant-time 3-dimensional triple-resonance pulse scheme to correlate intraresidue H-1(N), N-15, and C-13(′) chemical-shifts in N-15–C-13-labeled proteins. J. Magn. Reson. 1992;97:213–217. [Google Scholar]
- Cordier F., Dingley A.J., Grzesiek S. A doublet-separated sensitivity-enhanced HSQC for the determination of scalar and dipolar one-bond J-couplings. J. Biomol. NMR. 1999;13:175–180. doi: 10.1023/a:1008301415843. [DOI] [PubMed] [Google Scholar]
- Du Z., Ulyanov N.B., Yu J., Andino R., James T.L. NMR structures of loop B RNAs from the stem-loop IV domain of the enterovirus internal ribosome entry site: a single C to U substitution drastically changes the shape and flexibility of RNA. Biochemistry. 2004;43:5757–5771. doi: 10.1021/bi0363228. [DOI] [PubMed] [Google Scholar]
- Eletsky A., Kienhöfer A., Pervushin K. TROSY NMR with partially deuterated proteins. J. Biomol. NMR. 2001;20:177–180. doi: 10.1023/a:1011265430149. [DOI] [PubMed] [Google Scholar]
- Goodfellow I., Chaudhry Y., Richardson A., Meredith J., Almond J.W., Barclay W., Evans D.J. Identification of a cis-acting replication element within the poliovirus coding region. J. Virol. 2000;74:4590–4600. doi: 10.1128/jvi.74.10.4590-4600.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbalenya A.E., Donchenko A.P., Blinov V.M., Koonin E.V. Cysteine proteases of positive strand RNA viruses and chymotrypsin-like serine proteases. A distinct protein superfamily with a common structural fold. FEBS Lett. 1989;243:103–114. doi: 10.1016/0014-5793(89)80109-7. [DOI] [PubMed] [Google Scholar]
- Greenberg S.B. Respiratory consequences of rhinovirus infection. Arch. Intern. Med. 2003;163:278. doi: 10.1001/archinte.163.3.278. [DOI] [PubMed] [Google Scholar]
- Grzesiek S., Bax A. An efficient experiment for sequential backbone assignment of medium-sized isotopically enriched proteins. J. Magn. Reson. 1992;99:201–207. [Google Scholar]
- Grzesiek S., Bax A. Amino acid type determination in the sequential assignment procedure of uniformly 13C/15N-enriched proteins. J. Biomol. NMR. 1993;3:185–204. doi: 10.1007/BF00178261. [DOI] [PubMed] [Google Scholar]
- Guinier A., Fournet G. Wiley; New York: 1955. Small-Angle Scattering of X-rays. [Google Scholar]
- Handa N., Nureki O., Kurimoto K., Kim I., Sakamoto H., Shimura Y., Muto Y., Yokoyama S. Structural basis for recognition of the tra mRNA precursor by the sex-lethal protein. Nature. 1999;398:579–585. doi: 10.1038/19242. [DOI] [PubMed] [Google Scholar]
- Headey S.J., Huang H., Claridge J.K., Soares G.A., Dutta K., Schwalbe M., Yang D., Pascal S.M. NMR structure of stem-loop D from human rhinovirus-14. RNA. 2006;13:351–360. doi: 10.1261/rna.313707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H., Alexandrov A., Chen X., Barnes T.W., Zhang H., Dutta K., Pascal S.M. Structure of an RNA hairpin from HRV-14. Biochemistry. 2001;40:8055–8064. doi: 10.1021/bi010572b. [DOI] [PubMed] [Google Scholar]
- Jeffries C.M., Whitten A.E., Harris S.P., Trewhella J. Small-angle X-ray scattering reveals the N-terminal domain organization of cardiac myosin binding protein C. J. Mol. Biol. 2008;377:1186–1199. doi: 10.1016/j.jmb.2008.01.080. [DOI] [PubMed] [Google Scholar]
- Johnson P., Donaldson L. RNA recognition by the Vts1p SAM domain. Nat. Struct. Mol. Biol. 2006;13:177–178. doi: 10.1038/nsmb1039. [DOI] [PubMed] [Google Scholar]
- Kay L.E., Ikura M., Tschudin R., Bax A. Three-dimensional triple-resonance NMR spectroscopy of isotopically enriched proteins. J. Magn. Reson. 1990;89:496–514. doi: 10.1016/j.jmr.2011.09.004. [DOI] [PubMed] [Google Scholar]
- Konarev P.V., Volkov V.V., Sokolova A.V., Koch M.H.J., Svergun D.I. PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 2003;36:1277–1282. [Google Scholar]
- Laskowski R.A., MacArthur M.W., Moss D.S., Thornton J.M. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993;26:283–291. [Google Scholar]
- Leong L.E., Walker P.A., Porter A.G. Human rhinovirus-14 protease 3C (3Cpro) binds specifically to the 5′-noncoding region of the viral RNA. Evidence that 3Cpro has different domains for the RNA binding and proteolytic activities. J. Biol. Chem. 1993;268:25735–25739. [PubMed] [Google Scholar]
- Marchler-Bauer A., Anderson J.B., Derbyshire M.K., DeWeese-Scott C., Gonzales N.R., Gwadz M., Hao L., He S., Hurwitz D.I., Jackson J.D., et al. CDD: a conserved domain database for interactive domain family analysis. Nucleic Acids Res. 2007;35:D237–D240. doi: 10.1093/nar/gkl951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcotte L.L., Wass A.B., Gohara D.W., Pathak H.B., Arnold J.J., Filman D.J., Cameron C.E., Hogle J.M. Crystal structure of poliovirus 3CD protein: virally encoded protease and precursor to the RNA-dependent RNA polymerase. J. Virol. 2007;81:3583–3596. doi: 10.1128/JVI.02306-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews D.A., Smith W.W., Ferre R.A., Condon B., Budahazi G., Sisson W., Villafranca J.E., Janson C.A., McElroy H.E., Gribskov C.L. Structure of human rhinovirus 3C protease reveals a trypsin-like polypeptide fold, RNA-binding site, and means for cleaving precursor polyprotein. Cell. 1994;77:761–771. doi: 10.1016/0092-8674(94)90059-0. [DOI] [PubMed] [Google Scholar]
- McKnight K.L., Lemon S.M. Capsid coding sequence is required for efficient replication of human rhinovirus 14 RNA. J. Virol. 1996;70:1941–1952. doi: 10.1128/jvi.70.3.1941-1952.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKnight K.L., Lemon S.M. The rhinovirus type 14 genome contains an internally located RNA structure that is required for viral replication. RNA. 1998;4:1569–1584. doi: 10.1017/s1355838298981006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosimann S.C., Cherney M.M., Sia S., Plotch S., James M.N. Refined X-ray crystallographic structure of the poliovirus 3C gene product. J. Mol. Biol. 1997;273:1032–1047. doi: 10.1006/jmbi.1997.1306. [DOI] [PubMed] [Google Scholar]
- Muhandiram D., Kay L.E. Gradient-enhanced triple-resonance three-dimensional NMR experiments with improved sensitivity. J. Magn. Reson. B. 1994;103:203–216. [Google Scholar]
- Ohlenschläger O., Wöhnert J., Bucci E., Seitz S., Häfner S., Ramachandran R., Zell R., Görlach M. The structure of the stemloop D subdomain of coxsackie virus B3 cloverleaf RNA and its interaction with the proteinase 3C. Structure. 2004;12:237–248. doi: 10.1016/j.str.2004.01.014. [DOI] [PubMed] [Google Scholar]
- Orthaber D., Bergmann A., Glatter O. SAXS experiments on absolute scale with Kratky systems using water as a secondary standard. J. Appl. Crystallogr. 2000;33:218–225. [Google Scholar]
- Oubridge C., Ito N., Teo C.H., Fearnley I., Nagai K. Crystallisation of RNA–protein complexes: II. The application of protein engineering for crystallisation of the U1A protein–RNA complex. J. Mol. Biol. 1995;249:409–423. doi: 10.1006/jmbi.1995.0306. [DOI] [PubMed] [Google Scholar]
- Pathak H.B., Arnold J.J., Wiegand P.N., Hargittai M.R., Cameron C.E. Picornavirus genome replication: assembly and organization of the VPg uridylylation ribonucleoprotein (initiation) complex. J. Biol. Chem. 2007;282:16202–16213. doi: 10.1074/jbc.M610608200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathak H.B., Oh H.S., Goodfellow I.G., Arnold J.J., Cameron C.E. Picornavirus genome replication: roles of precursor proteins and rate-limiting steps in oriI-dependent VPg uridylylation. J. Biol. Chem. 2008;283:30677–30688. doi: 10.1074/jbc.M806101200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Permi P., Rosevear P.R., Annila A. A set of HNCO-based experiments for measurement of residual dipolar couplings in 15N, 13C, (2H)-labeled proteins. J. Biomol. NMR. 2000;17:43–54. doi: 10.1023/a:1008372624615. [DOI] [PubMed] [Google Scholar]
- Pervushin K.V., Wider G., Wuthrich K. Single transition-to-single transition polarization transfer (ST2-PT) in [N-15,H-1]-TROSY. J. Biomol. NMR. 1998;12:345–348. doi: 10.1023/A:1008268930690. [DOI] [PubMed] [Google Scholar]
- Peters H., Kusov Y.Y., Meyer S., Benie A.J., Bäuml E., Wolff M., Rademacher C., Peters T., Gauss-Müller V. Hepatitis A virus proteinase 3C binding to viral RNA: correlation with substrate binding and enzyme dimerization. Biochem. J. 2004;385:363–370. doi: 10.1042/BJ20041153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petoukhov M.V., Svergun D.I. Global rigid body modeling of macromolecular complexes against small-angle scattering data. Biophys. J. 2005;89:1237–1250. doi: 10.1529/biophysj.105.064154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piotto M., Saudek V., Sklenár V. Gradient-tailored excitation for single-quantum NMR spectroscopy of aqueous solutions. J. Biomol. NMR. 1992;2:661–665. doi: 10.1007/BF02192855. [DOI] [PubMed] [Google Scholar]
- Porter A.G. Picornavirus nonstructural proteins: emerging roles in virus replication and inhibition of host cell functions. J. Virol. 1993;67:6917–6921. doi: 10.1128/jvi.67.12.6917-6921.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieder E., Xiang W., Paul A., Wimmer E. Analysis of the cloverleaf element in a human rhinovirus type 14/poliovirus chimera: correlation of subdomain D structure, ternary protein complex formation and virus replication. J. Gen. Virol. 2003;84:2203–2216. doi: 10.1099/vir.0.19013-0. [DOI] [PubMed] [Google Scholar]
- Ruckert M., Otting G. Alignment of biological macromolecules in novel nonionic liquid crystalline media for NMR experiments. J. Am. Chem. Soc. 2000;122:7793–7797. [Google Scholar]
- Salzmann M., Pervushin K., Wider G., Senn H., Wüthrich K. TROSY in triple-resonance experiments: new perspectives for sequential NMR assignment of large proteins. Proc. Natl. Acad. Sci. USA. 1998;95:13585–13590. doi: 10.1073/pnas.95.23.13585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenyuk A.V., Svergun D.I. GNOM—a program package for small-angle scattering data-processing. J. Appl. Crystallogr. 1991;24:537–540. [Google Scholar]
- Shen M., Wang Q., Yang Y., Pathak H.B., Arnold J.J., Castro C., Lemon S.M., Cameron C.E. Human rhinovirus type 14 gain-of-function mutants for oriI utilization define residues of 3C(D) and 3Dpol that contribute to assembly and stability of the picornavirus VPg uridylylation complex. J. Virol. 2007;81:12485–12495. doi: 10.1128/JVI.00972-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen M., Reitman Z.J., Zhao Y., Moustafa I., Wang Q., Arnold J.J., Pathak H.B., Cameron C.E. Picornavirus genome replication. Identification of the surface of the poliovirus (PV) 3C dimer that interacts with PV 3Dpol during VPg uridylylation and construction of a structural model for the PV 3C2–3Dpol complex. J. Biol. Chem. 2008;283:875–888. doi: 10.1074/jbc.M707907200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih S.R., Chiang C.Y., Chen T.C., Wu C.N., Hsu J.T.A., Lee J.C., Hwang M.J., Li M.L., Chen G.W., Hoc M.S. Mutations at KFRDI and VGK domains of enterovirus 71 3C protease affect its RNA binding and proteolytic activities. J. Biomed. Sci. 2004;11:239–248. doi: 10.1007/BF02256567. [DOI] [PubMed] [Google Scholar]
- Svergun D.I., Nierhaus K.H. A map of protein–rRNA distribution in the 70 S Escherichia coli ribosome. J. Biol. Chem. 2000;275:14432–14439. doi: 10.1074/jbc.275.19.14432. [DOI] [PubMed] [Google Scholar]
- Van Regenmortel, M.H.V., Fauquet, C.M., Bishop, D.H.L., Carstens, E., Estes, M., Lemon, S., Maniloff, J., Mayo, M.A., McGeoch, D., Pringle, C.R. et al., 2000. Virus Taxonomy. Seventh Report of the International Committee on Taxonomy of Viruses. Academic Press, New York, San Diego.
- Varani L., Gunderson S.I., Mattaj I.W., Kay L.E., Neuhaus D., Varani G. The NMR structure of the 38 kDa U1A protein–PIE RNA complex reveals the basis of cooperativity in regulation of polyadenylation by human U1A protein. Nat. Struct. Biol. 2000;7:329–335. doi: 10.1038/74101. [DOI] [PubMed] [Google Scholar]
- Volkov V.V., Svergun D.I. Uniqueness of ab initio shape determination in small-angle scattering. J. Appl. Crystallogr. 2003;36:860–864. doi: 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss N.R., Gerstein M. Calculation of standard atomic volumes for RNA and comparison with proteins: RNA is packed more tightly. J. Mol. Biol. 2005;346 doi: 10.1016/j.jmb.2004.11.072. [DOI] [PubMed] [Google Scholar]
- Vranken W.F., Boucher W., Stevens T.J., Fogh R.H., Pajon A., Llinas M., Ulrich E.L., Markley J.L., Ionides J., Laue E.D. The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins. 2005;59:687–696. doi: 10.1002/prot.20449. [DOI] [PubMed] [Google Scholar]
- Walker P.A., Leong L.E., Porter A.G. Sequence and structural determinants of the interaction between the 5′-noncoding region of picornavirus RNA and rhinovirus protease 3C. J. Biol. Chem. 1995;270:14510–14516. doi: 10.1074/jbc.270.24.14510. [DOI] [PubMed] [Google Scholar]
- Wang Q.M., Chen S.H. Human rhinovirus 3C protease as a potential target for the development of antiviral agents. Curr. Protein Pept. Sci. 2007;8:19–27. doi: 10.2174/138920307779941523. [DOI] [PubMed] [Google Scholar]
- Wang C., Grey M.J., Palmer A.G. CPMG sequences with enhanced sensitivity to chemical exchange. J. Biomol. NMR. 2001;21:361–366. doi: 10.1023/a:1013328206498. [DOI] [PubMed] [Google Scholar]
- Webber S.E., Okano K., Little T.L., Reich S.H., Xin Y., Fuhrman S.A., Matthews D.A., Love R.A., Hendrickson T.F., Patick A.K., et al. Tripeptide aldehyde inhibitors of human rhinovirus 3C protease: design, synthesis, biological evaluation, and cocrystal structure solution of P1 glutamine isosteric replacements. J. Med. Chem. 1998;41:2786–2805. doi: 10.1021/jm980071x. [DOI] [PubMed] [Google Scholar]
- Whitten A.E., Cai S., Trewhella J. MULCh: modules for the analysis of small-angle neutron contrast variation data from biomolecular complexes. J. Appl. Crystallogr. 2008;41:222–226. [Google Scholar]
- Wishart D.S., Sykes B.D. The 13C chemical-shift index: a simple method for the identification of protein secondary structure using 13C chemical-shift data. J. Biomol. NMR. 1994;4:171–180. doi: 10.1007/BF00175245. [DOI] [PubMed] [Google Scholar]
- Xiang W., Harris K.S., Alexander L., Wimmer E. Interaction between the 5′-terminal cloverleaf and 3AB/3CDpro of poliovirus is essential for RNA replication. J. Virol. 1995;69:3658–3667. doi: 10.1128/jvi.69.6.3658-3667.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang W., Cuconati A., Hope D., Kirkegaard K., Wimmer E. Complete protein linkage map of poliovirus P3 proteins: interaction of polymerase 3Dpol with VPg and with genetic variants of 3AB. J. Virol. 1998;72:6732–6741. doi: 10.1128/jvi.72.8.6732-6741.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H., Bartlam M., Rao Z. Drug design targeting the main protease, the Achilles’ heel of coronaviruses. Curr. Pharm. Des. 2006;12:4573–4590. doi: 10.2174/138161206779010369. [DOI] [PubMed] [Google Scholar]
- Yin J., Paul A.V., Wimmer E., Rieder E. Functional dissection of a poliovirus cis-acting replication element [PV-cre(2C)]: analysis of single- and dual-cre viral genomes and proteins that bind specifically to PV-cre RNA. J. Virol. 2003;77:5152–5166. doi: 10.1128/JVI.77.9.5152-5166.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zell R., Sidigi K., Bucci E., Stelzner A., Görlach M. Determinants of the recognition of enteroviral cloverleaf RNA by coxsackie virus B3 proteinase 3C. RNA. 2002;8:188–201. doi: 10.1017/s1355838202012785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.W., Yap Y.L. Exploring the binding mechanism of the main proteinase in SARS-associated coronavirus and its implication to anti-SARS drug design. Bioorg. Med. Chem. 2004;12:2219–2223. doi: 10.1016/j.bmc.2004.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zweckstetter M., Bax A. Prediction of sterically induced alignment in a dilute liquid crystalline phase: aid to protein structure determination by NMR. J. Am. Chem. Soc. 2000;122:3791–3792. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.