

Abstract
Simultaneous quantitation of two orchid viruses, cymbidium mosaic potexvirus (CymMV) and odontoglossum ringspot tobamovirus (ORSV), were carried out using the TaqMan® real-time RT-PCR, a novel detection technique that combines RT-PCR with the power of fluorescent detection. Four TaqMan® probes were synthesized, targeting at the RNA-dependent RNA polymerase (RdRp) and coat protein (CP) genes of both viruses. The reporter dye FAM (6-carboxyfluorescein) was used to label the 5′ terminus of probes specific to CymMV, while TET (tetrachloro-6-carboxyfluorescein) was used for the ORSV probes. TAMRA (6-carboxy-tetramethyl-rhodamine), which was attached at the 3′ terminus of each probe, was used as the universal quencher. With increasing amounts of standard RNA templates, the respective threshold cycle (CT) values were determined and a linear relationship was established between these CT values and the logarithm of initial template amounts. The amounts of starting templates in mixed-infected Oncidium flowers and leaves were estimated from the standard curves. As little as 104 copies or 5 fg each of CymMV and ORSV could be detected simultaneously with either the RdRp or CP gene as the target. This system offers a sensitive, high throughput and rapid method for plant virus detection.
Keywords: Cymbidium mosaic potexvirus, Odontoglossum ringspot tobamovirus, TaqMan®, Fluorogenic 5′ nuclease assay, Simultaneous quantification, One-tube RT-PCR
1. Introduction
Cymbidium mosaic potexvirus (CymMV) and odontoglossum ringspot tobamovirus (ORSV) are the two most prevalent and economically important orchid viruses (Wong et al., 1994) that have attained a worldwide distribution, infecting numerous commercially important orchid genera (Zettler et al., 1990). As such, many diagnostic techniques have been developed over the years for the detection of these two plant viruses. Reverse transcription-polymerase chain reaction (RT-PCR) has been used for the detection and identification of CymMV (Lim et al., 1993, Ryu et al., 1995) and ORSV (Ryu and Park, 1995). Detection by immunocapture-PCR of both viruses in orchids has also been reported (Barry et al., 1996). Some recent detection techniques developed at our laboratory include the use of digoxigenin (DIG)-labelled cRNA probes for the detection of both viruses (Hu and Wong, 1998), simultaneous detection of both viruses with RT-PCR using a single primer pair (Seoh et al., 1998), immuno-capillary zone electrophoresis (Eun and Wong, 1999), molecular beacons (Eun and Wong, 2000), and liquid chromatography and matrix-assisted laser desorption-ionization mass spectrometry (Tan et al., 2000).
Real-time RT-PCR is one of the latest advances in quantitative technology (Lie and Petropoulos, 1998). It is based on the use of the 5′ nuclease assay first described by Holland et al. (1991). This method exploits the 5′ nuclease activity of Taq polymerase to cleave a non-extendable, dual-labelled fluorogenic probe that is annealed to the target sequence during amplification (Lee et al., 1993, Bassler et al., 1995, Livak et al., 1995a, Livak et al., 1995b). The 5′ end of the probe is labelled with a fluorescent reporter dye (e.g. FAM (6-carboxyfluorescein)). Its emission spectrum is quenched by a second fluorescent dye (e.g. TAMRA (6-carboxy-tetramethyl-rhodamine)) at the 3′ end, through Förster Resonance Energy Transfer (FRET) (Förster, 1948).
During PCR, the fluorogenic probe, also known as the TaqMan® probe, anneals specifically between the forward and reverse primers. When the probe is cleaved by the 5′ nuclease activity of the DNA polymerase, the reporter dye becomes physically separated from the quencher dye. This releases the quenching of FAM fluorescent emission and a signal is generated. With each cycle, additional reporter dye molecules are cleaved from their respective probes, resulting in an exponential increase in fluorescence intensity that is monitored in real-time.
The fluorescence emitted at each cycle is measured by the ABI Prism® 7700 Sequence Detection System (Applied Biosystems, USA), an integrated unit capable of performing both thermal cycling and fluorescence measurement in real-time (Bassam et al., 1996), and plotted against the cycle number to generate an amplification plot that is sigmoidal in shape. The cycle number in which amplification first enters a logarithm (log) phase, denoted by C T, provides a quantitative measurement of input target sequences. By performing the RT-PCR reaction with different known amounts of starting templates and determining the respective C T values from the amplification plots, a linear relationship (standard curve) can be established between C T values and the log of starting template amounts. The amount of starting template in any unknown sample can be determined by conducting one PCR reaction to ascertain the C T value (Heid et al., 1996). In this study, two orchid viruses, namely cymbidium mosaic potexvirus (CymMV) and odontoglossum ringspot tobamovirus (ORSV) from infected orchid flower and leaf tissues were quantified simultaneously using the TaqMan® real-time RT-PCR.
2. Materials and methods
2.1. TaqMan® probe design
Four dual-labelled fluorescent probes (Table 1 ) were designed with the software program Primer Express™ (Applied Biosystems, USA) and synthesized by PE Applied Biosystems, USA. Sequence probes Cym-RdRp and Cym-CP were specific to the RNA-dependent RNA polymerase (RdRp) gene and the coat protein (CP) gene of CymMV, respectively. Cym-RdRp was designed to bind to nucleotides (nt) 1215–1238 and Cym-CP to bind to nt 5600–5625 of the CymMV genome. The 5′ and 3′ ends of these two probes were labelled with fluorescent dyes FAM (6-carboxyfluorescein, excitation wavelength=494 nm, emission wavelength=521 nm) and TAMRA (6-carboxy-tetramethyl-rhodamine), respectively. Probes ORS-RdRp and ORS-CP, respectively, specific to the RdRp gene and CP gene of ORSV, were labelled with TET (tetrachloro-6-carboxyfluorescein, excitation wavelength=519 nm, emission wavelength=537 nm) at the 5′ end and TAMRA at the 3′ end. ORS-RdRp and ORS-CP were designed to bind to nt 120–149 and 5817–5841 in the ORSV genome, respectively.
Table 1.
Nucleotide sequences of the TaqMan fluorescent probes and primers employed
| Abbreviation | Sequence | |
|---|---|---|
| TaqMan probes | ||
| CymMV | Cym-RdRp | 5′ FAM-CCAACTATTCACCGGAGCGGACCA-TAMRA 3′ |
| Detection | Cym-CP | 5′ FAM-CCCGAAGAAATCAAGGCCATAACCCA-TAMRA 3′ |
| ORSV | ORS-RdRp | 5′ TET-AACAGCCTCATTAACGATTTGGCTCAGAGA-TAMRA 3′ |
| Detection | ORS-CP | 5′ TET-TGGCAGCCGGTTCCTACTTTGACCA-TAMRA 3′ |
| Primers | ||
| CymMV | CymRdRp-F | 5′ CAAGAACAAGATACGGGAATTGG 3′ |
| CymRdRp-R | 5′ TCGAGAGCTTTGAGCAGTTGATT 3′ | |
| Detection | CymCP-F | 5′ TCACCTCCTCCATCGCCA 3′ |
| CymCP-R | 5′ AGGCCAAGGTTGTTAACCCA 3′ | |
| ORSV | ORSRdRp-F | 5′ TTGAGGCTGGTATGGGCAG 3′ |
| ORSRdRp-R | 5′ TTCGACAGCGTTATCGTAAACAC 3′ | |
| Detection | ORSCP-F | 5′ GTTCAACAGCAGTTTGCTGATGT 3′ |
| ORSCP-R | 5′ TAACCAGCGCCTGCAGG 3′ |
2.2. Primer design
For the fluorogenic 5′ nuclease assay in real-time RT-PCR, four different pairs of primers (Table 1) were designed to flank their respective TaqMan® probes. Forward primer CymRdRp-F and reverse primer CymRdRp-R were constructed to amplify the RdRp gene of CymMV from nt 1191 to 1265 such that the TaqMan® probe (CymRdRp) annealed within the PCR fragment at nt 1215 to 1238. The forward and reverse primers (CymCP-F and CymCP-R) for Cym-CP probe were designed to amplify the CP gene of CymMV from nt 5580 to 5649. Similarly, the forward and reverse primers (ORSRdRp-F and ORSRdRp-R) were designed to amplify the RdRp gene of ORSV from nt 100 to 173. The respective forward and reverse primers (ORSCP-F and ORSCP-R) for ORS-CP probe were designed to amplify the CP gene of ORSV from nt 5793 to 5866. Primers were synthesized by Genset Oligos, Singapore. A diagram showing the binding sites of the TaqMan® probes and the designed primers to the CymMV and ORSV genomes is shown in Fig. 1 .
Fig. 1.
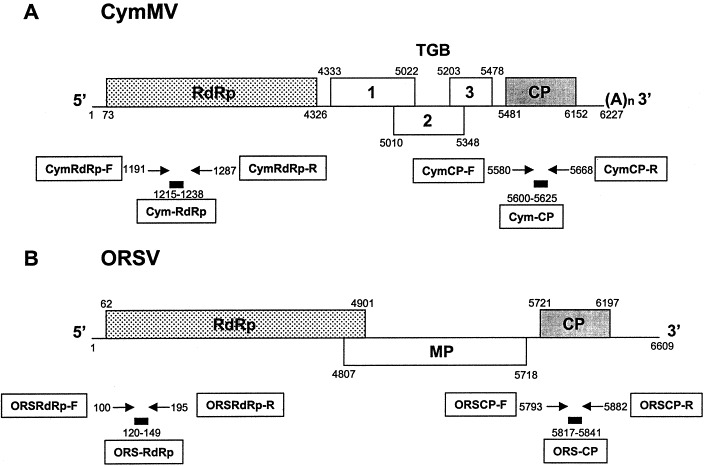
Genome organisation of (A) CymMV and (B) ORSV showing the RNA-dependent RNA polymerase (RdRp); triple gene block (TGB) 1, 2 and 3; movement protein (MP) and coat protein (CP). The positions of the four primer pairs (CymRdRp-F and CymRdRp-R, CymCP-F and CymCP-R, ORSRdRp-F and ORSRdRp-R, ORSCP-F and ORSCP-R) and the four TaqMan probes (CymRdRp, Cym-CP, ORS-RdRp, ORS-CP) are indicated.
2.3. Preparation of viral RNA standards
To quantify CymMV and ORSV in infected orchid tissues, two different sets of RNA standards were generated for each virus. The RdRp set of RNA standards was used with the RdRp TaqMan® probe and RdRp primers; while the CP set of RNA standards was used with the CP TaqMan® probe and CP primers in quantitative real-time RT-PCR. The RNA standards were obtained by in vitro transcription of viral genes.
A cDNA fragment encompassing part of the RdRp gene from nt 1 to 2341 was obtained from a BamHI (Promega, USA) digest of a full-length cDNA clone of CymMV (Yu and Wong, 1998) and purified using the QIAquick™ purification kit (Qiagen, Germany). The purified DNA was in vitro transcribed using the RiboMAX™ Large Scale RNA Production System (Promega, USA) with T3 RNA polymerase. A partial ORSV RdRp gene from nt 1 to 1371 was obtained by cutting a ORSV clone with BglII (Roche Molecular Biochemicals, Switzerland) at 37°C for 1 h.
The CymMV CP gene from nt 5481 to 6152 was amplified using the forward (CCP-F: 5′ GCTCTAGAATGGGAGAGCCCACTCCAGC 3′) and reverse (CCP-R: 5′ TCCCCCGGGTTATTCAGGTAGGGGGTGCAGG 3′) primers. The CymMV full-length cDNA clone (5 ng) was used as the template in a PCR reaction mix containing 30 pmol each of primers CCP-F and CCP-R, 0.2 mM deoxynucleoside triphosphate mix (dNTPs), 1×reaction buffer, 1.5 mM MgCl2 and 5 units of the Taq DNA Polymerase (Promega, USA). The reaction mix was subjected to 30 cycles of 96°C for 15 s, 58°C for 30 s and 72°C for 80 s with a final extension step of 72°C for 2 min after the 30 cycles. The amplified CymMV CP gene was digested with XbaI (Roche Molecular Biochemicals, Switzerland) at 37°C for 1 h followed by SmaI (Roche Molecular Biochemicals, Switzerland) at 25°C for an additional hour. The purified CymMV CP gene (100 ng) was cloned into pBluescript SK (+) (Stratagene, USA) and transformed into Escherichia coli JM109 cells. In vitro transcripts corresponding to nt 5481–6152 (672 bp) of the CymMV CP gene was transcribed from clone pCymCP (7 μg) with T3 RNA polymerase.
The CP gene of ORSV corresponding to nt 5721 to 6197 (477 bp) was amplified using forward (OCP-F: 5′ GCTCTAGAATGTCTTACACTATTACAGACC 3′) and reverse (OCP-R: 5′ TCCCCCGGGTTAGGAAGAGGTCCAAGTAAG 3′) primers. The ORSV partial clone (nt 18–4539) (10 ng) was used as template in a PCR reaction mix and thermal cycling profile as described for CymMV CP. The amplified CP gene of ORSV was similarly digested with XbaI/SmaI, cloned into pBluescript SK (+) and transformed into E. coli JM109 cells. Clone pORSCP (7 μg), which contained a 477 bp insert (nt 5721–6197) corresponding to the ORSV CP gene, was linearised, purified and transcribed in vitro with T3 RNA polymerase.
2.4. Preparation of orchid samples
Total nucleic acid (T-NA) was extracted from leaves and flowers of orchid Oncidium sp. infected with both CymMV and ORSV. Healthy orchid materials were used as negative controls. Fresh orchid tissue (0.5 g) was frozen in liquid nitrogen and ground to a fine powder. T-NA was extracted as described by Gibbs and Mackenzie (1997). The purified T-NA was dissolved in 100 μl of nuclease-free water and stored at −80°C.
2.5. Optimisation of primer concentration
A RT-PCR reaction mix was set up to provide at least four replicates for each of the nine combinations, using 50, 300 and 900 nM each of forward and reverse primers (i.e. forward:reverse primer concentration ratios of 50:50, 50:300, 50:900, 300:50, 300:300, 300:900, 900:50, 900:300, 900:900) of Cym-RdRp. The RT-PCR reactions for the nine conditions were carried out using TaqMan® Gold RT-PCR kit (Applied Biosystems, USA) with 1 fmol in vitro transcribed CymMV RdRp RNA, 200 nM of TaqMan® Cym-RdRp probe, 1×TaqMan® Buffer A, 4.0 mM of MgCl2, 300 μM of dNTP mix, 4 units (U) of RNase inhibitor, 12.5 U of Multiscribe Reverse Transcriptase and 1.25 U of AmpliTaq Gold DNA Polymerase. The ABI Prism® 7700 thermocycler was used for thermal cycling and to record changes in fluorescence intensity. The thermal cycling parameters employed were as follows: one cycle of 48°C for 30 min (reverse transcription) and 95°C for 10 min (DNA Polymerase activation); and 40 cycles of 95°C for 15 s (denaturation) and 60°C for 1 min (annealing and extension). The fluorescent intensities of both reporter dyes (FAM or TET) and quencher dye (TAMRA) were recorded throughout the amplification process and the normalised reporter signal (ΔR n) was calculated. The minimum forward and reverse primer concentrations that yield the maximum fluorescent intensity (ΔR n) were chosen as the optimal primer concentrations.
2.6. Optimisation of probe concentration
RT-PCR reactions were set up to run at least four replicates of nine different CymMV RdRp probe concentrations at 25 nM intervals from 25 to 225 nM. The TaqMan® Gold RT-PCR kit was employed for RT-PCR using the optimal primer concentrations previously determined. The minimum probe concentration that gave the minimum C T for its viral target was chosen as the optimal probe concentration. The parameter C T is defined as the cycle number at which the reporter fluorescence, generated by cleavage of the probe, crosses a fixed threshold level above the baseline fluorescence (Bassam et al., 1996). For the RT-PCR amplification plots of the different probe concentrations, the threshold was set at 10 standard deviations above the mean of baseline emission calculated from cycles 3 to 15, which is equivalent to 0.04 fluorescence (ΔR n) units.
2.7. Quantification of CymMV and ORSV
The in vitro transcribed viral RNA was diluted serially from 109 to 104 copies at one log unit intervals. To generate a standard curve, four replicates of each copy number were amplified using optimal primer and probe concentrations according to the experimental conditions described in Section 2.5. In addition to individual detection of each target gene, simultaneous detection was also performed. Either the probe combination of Cym-RdRp/ORS-RdRp or Cym-CP/ORS-CP was used to detect amplified products of the RdRp and CP genes of both viruses, respectively. Unknown amounts of viral RNA present in total nucleic acid extractions of infected orchids were amplified using the same RT-PCR reaction mix. The thermal cycling profile described in Section 2.5 was applied for all the RT-PCR reactions. Each set of experiments was repeated at least ten times.
3. Results
3.1. TaqMan® probe design
Approximately 100 sets of probes and primers for CymMV and ORSV were generated using the Primer Express™ software. Two sets of probes and primers were chosen for each virus, based on the melting temperature (T m), nucleotide (nt) length and guanidine/cysteine (GC) content (Table 2 ). The probes were designed such that the T m values was approximately 10°C higher than that of the primers, the probe length not exceeding 30 nt and its %GC content not lower than 40%.
Table 2.
Nucleotide length, melting temperature (Tm) and GC content of the probes and primers used in quantitative real-time RT-PCR
| Specificity | Start (nt) | End (nt) | Length | Tm (°C) | %GC | |
|---|---|---|---|---|---|---|
| CymMV RdRp | TaqMan probe | 1215 | 1238 | 24 | 69 | 58 |
| Forward primer | 1191 | 1213 | 23 | 59 | 43 | |
| Reverse primer | 1265 | 1287 | 23 | 59 | 43 | |
| CymMV CP | TaqMan probe | 5600 | 5625 | 26 | 69 | 50 |
| Forward primer | 5580 | 5597 | 18 | 60 | 61 | |
| Reverse primer | 5649 | 5668 | 20 | 58 | 50 | |
| ORSV RdRp | TaqMan probe | 120 | 149 | 30 | 68 | 43 |
| Forward primer | 100 | 118 | 19 | 58 | 58 | |
| Reverse primer | 173 | 195 | 23 | 58 | 43 | |
| ORSV CP | TaqMan probe | 5817 | 5841 | 25 | 70 | 56 |
| Forward primer | 5793 | 5815 | 23 | 58 | 43 | |
| Reverse primer | 5866 | 5882 | 17 | 58 | 65 |
3.2. Optimisation of primer concentration
The ΔR n value increased correspondingly when both the forward and reverse primer increased from 50 to 900 nM with forward/reverse primer concentration (nM) ratios of 50/50, 50/300, 50/900, 300/50, 900/50, 300/300, 300/900, 900/300, 900/900 yielding average ΔR n values of 0.68, 1.45, 1.67, 1.83, 2.02, 2.97, 3.13, 3.24 and 3.45, respectively. The lowest and highest ΔR n values were obtained with 50/50 and 900/900 nM forward/reverse (F/R) primer, respectively. Since the ΔR n of all the four replicates of the RT-PCR amplification plots for 300/900 nM F/R primer overlapped better (i.e. higher repeatability and stability) than those of the 900/900 and 900/300 nM F/R primers, this primer ratio of 300/900 nM F/R primer was used for subsequent experiments.
3.3. Optimisation of probe concentration
A general trend observed was that the average threshold cycle (C T) decreased with increasing probe concentration. The two lowest C T values were obtained with 200 and 225 nM of probe but all four replicate amplification plots for the 200 nM probe concentration overlapped better than that of the 225 nM probe concentration. Hence, probe concentration of 200 nM was chosen for subsequent experiments.
3.4. Quantification of CymMV and ORSV
By carrying out the RT-PCR reaction with known amounts of starting templates (104–109 copies), a linear graph was established between the C T values and the logarithm of the starting RNA copy number for both viruses (Fig. 2 A and B). Regardless of the probe or primer pair used, the C T value decreased as the starting copy number increased. Using these standard graphs, the quantity of the target genes within the extracted T-NA could be determined (Fig. 3 ). There were significantly larger copy numbers of all four gene targets in the flowers than in the leaves of the infected orchids. The T-NA of healthy plants did not yield any significant C T values, thus absence of these gene targets. In both the floral and foliar tissues, the copy number of the CymMV CP gene was approximately half that of the CymMV RdRp gene. The reverse was observed with ORSV as the copy number of the CP gene was significantly higher than that of the RdRp gene. Simultaneous quantification using either probe pair Cym-RdRp/ORS-RdRp or Cym-CP/ORS-CP with their respective F/R primer pairs yielded comparable results with those obtained when the probes were used individually. Thus, it is possible to quantitate more than one target in a single reaction tube simultaneously, provided the probes are tagged with different fluorescent reporter dyes.
Fig. 2.
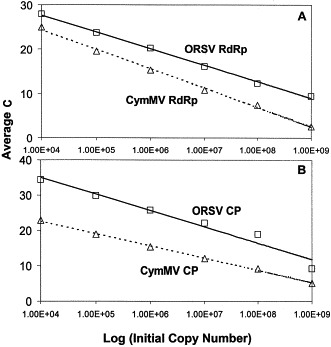
Quantification curves of (A) the RNA-dependent RNA polymerase gene and (B) coat protein gene of CymMV and ORSV. In vitro transcribed viral RNAs were serially diluted from 109 to 104 copies at one log unit intervals. The average threshold cycle (CT) was plotted against the logarithm of the initial copy numbers (1.00E+n≡1×10n).
Fig. 3.
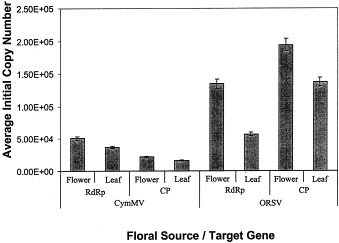
The average initial number copy number of CymMV and ORSV RNA-dependent RNA polymerase and coat protein genes per ng of total nucleic acid extracted from Oncidium flowers or leaves. Standard deviations for each vertical bar were calculated from an average of ten replicates (1.00E+n≡1×10n).
4. Discussion
The development of the TaqMan® 5′ nuclease assays represents a significant advance in nucleic acid quantification. This approach utilises the 5′ → 3′ exonuclease activity of Thermus aquaticus (Taq) DNA polymerase to cleave a dual-labelled probe annealed to a target sequence during amplification. The release of a fluorogenic tag from the 5′ end of the probe is proportional to the target sequence concentration or copy number. This increase in emission intensity is monitored at each cycle in real-time. To date, the TaqMan® fluorogenic system has been applied for the detection and quantitation of pathogens such as potato leaf roll virus (Schoen et al., 1996), feline coronaviruses (Gut et al., 1999), hepatitis C and B viruses (Mercier et al., 1999), varicella zoster virus (Hawrami and Breuer, 1999) and mycorrhizal fungi Glomus mosseae (Bohm et al., 1999). This system is also applicable for the monitoring of gene expression. Examples of such genes include the Na+/I− symporter gene in human thyroid tumours (Lazar et al., 1999) and the MYC gene in sporadic breast tumours (Bieche et al., 1999).
The TaqMan® probe design employs an energy transfer effect between matched fluorescent dyes known as the Förster resonance energy transfer through space or FRET (Förster, 1948). With FRET, light energy absorbed by a high-energy chromophore is transferred to a nearby chromophore of lower energy subject to certain geometric and spectroscopic constraints, the most important of which is the distance between the two chromophores. In the fluorogenic 5′ nuclease assay, the probes are labelled with two fluorescent dyes. One dye acts as a ‘reporter’ and the other acts as a ‘quencher’. The reporter transfers fluorescent energy to the quencher by FRET when they are in close proximity. Signal is generated when the hybridised oligo probe is cleaved by the DNA polymerase and the dyes are separated. This separation negates FRET effects and the fluorescent signal emitted by the reporter dye increases. The performance of the TaqMan® probes depends on the efficiency of reporter dye quenching, probe hybridisation; and probe cleavage by the Taq DNA polymerase (Livak et al., 1996). To facilitate efficient reporter dye quenching, the optimal probe length is between 20 and 30 nt. Probes longer than 30 nt risk forming inter- or intra-molecular structures that interfere with probe synthesis, flexibility, hybridisation, cleavage, as well as amplification process. Hence, the CymMV and ORSV probes selected for the fluorogenic assay are between 20 and 30 nt in length. For efficient hybridisation, the T m of a probe should be higher than that of the RT-PCR primers used. This is because primer-template hybrids are stabilised quickly as they are extended by the polymerase; whereas, the fluorogenic probe is not extended and therefore not stabilised. The higher T m compensates for their relative instability. The recommended T m of the probe is 70°C and the compatible RT-PCR primers should have a T m approximately 10°C lower. Thus, the T m of CymMV and ORSV probes were designed to be about 10°C higher than that of the primers. In addition, the 3′ terminus of the probe is phosphorylated to prevent extension by the polymerase during the polymerization steps.
In our experiments, the 300/900 nM F/R primer concentration was chosen although the 900/900 and 900/300 nM F/R primer concentrations yielded the highest average ΔR n values. In a RT-PCR reaction, the reverse transcriptase utilises only the reverse primer to synthesise the cDNA strand while the subsequent PCR amplification of the cDNA utilises equal amounts of forward and reverse primers. This results in a higher net loss of reverse primers and explains why the 300/900 nM F/R primer concentration exhibited higher repeatability and stability than that of the other two F/R primer concentrations. The unincorporated forward primers in the 900/900 and 900/300 nM F/R primer concentrations probably contributed to background fluorescence, widening the gap of the amplification plots.
The parameter C T is defined as the cycle number at which the reporter fluorescence, generated by the cleavage of the probe, crosses a fixed threshold value level above the baseline fluorescence emission (Bassam et al., 1996). The higher the starting copy number of nucleic acid target, the fewer the amplification cycles needed before a significant increase in fluorescence was observed and the lower the C T value. C T values indicate precise and reproducible quantification measurements because they are obtained at a stage in the reaction where reagents are not limiting. Data collected beyond the calculated C T is not considered for quantification purposes. Compared with quantification using endpoint measurements such as competitive RT-PCR, calculating C Ts greatly expands the dynamic range of quantification. It was shown above that it is possible to attain a range of five orders of magnitude in starting copy number. As little as 104 copies or 5 fg each of CymMV and ORSV can be detected simultaneously with either the RdRp or CP gene as the target. This compares favourably with other novel detection techniques such as immuno-capillary zone electrophoresis (Eun and Wong, 1999), molecular beacons (Eun and Wong, 2000) and the DARAS® system (Brown et al., 1999).
Significantly larger copy numbers of all four gene targets were observed in the flowers of infected orchid plants (Fig. 3). We postulate that the viral concentration in the flowers is higher than that of the leaves. In both floral and foliar tissues, the copy number of the CymMV CP gene was approximately half that of the CymMV RdRp gene. The opposite was observed with ORSV. The copy number of the ORSV CP gene was significantly higher than that of the RdRp gene. This could be due to the presence of higher abundant CP targets contributed by subgenomic RNAs of ORSV. The amount of CymMV subgenomic RNA may be lesser than that of ORSV.
The TaqMan® real-time RT-PCR has many advantages over conventional PCR that requires post-amplification processing such as agarose gel electrophoresis. Such steps increase the risk of inaccuracy and contamination. Accurate quantitation of the test material thus cannot be achieved. Real-time RT-PCR can be performed in a single closed tube that reduces the risk of contamination. No post-amplification steps are required and the calculation of the initial amount of starting material is performed automatically by the software programme. When coupled with a 96-well capacity, this system offers a sensitive, high throughput and rapid method for plant virus detection. The quantitative real-time RT-PCR method can be applied to the study of viral replication kinetics in protoplast systems and the determination of viral resistance levels in novel genetically engineered plants.
Acknowledgements
We wish to thank S.A. Mudzakker and Dr H. Tan of Applied Biosystems Singapore, for the use of the ABI Prism® 7700 Sequence Detection System. This work was supported by research grant RP 3972398 from the National University of Singapore (NUS), Republic of Singapore. The first two authors are research scholars from the NUS and recipients of the National Science and Technology Board Top-Up Scheme for graduate research in Biomedical Engineering.
References
- Barry K., Hu J.S., Kuehnle A.R., Sughii N. Sequence analysis and detection using immunocapture-PCR of cymbidium mosaic virus and odontoglossum ringspot virus in Hawaiian orchids. J. Phytopathol. 1996;144:179–186. [Google Scholar]
- Bassam B.J., Allen T., Flood S., Stevens J., Wyatt P., Livak K.J. Nucleic acid sequence detection systems: revolutionary automation for monitoring and reporting PCR products. Aust. Biotechnol. 1996;6:285–294. [Google Scholar]
- Bassler H.A., Flood S.J., Livak K.J., Marmaro J., Knorr R., Batt C.A. Use of fluorogenic probe in a PCR-based assay for the detection of Listeria monocytogenes. App. Environ. Microbiol. 1995;61:3724–3728. doi: 10.1128/aem.61.10.3724-3728.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieche I., Laurendeau I., Tozlu S., Olivi M., Vidaud D., Lidereau R., Vidaud M. Quantitation of MYC gene expression in sporadic breast tumors with a real-time reverse transcription-PCR assay. Cancer Res. 1999;59:2759–2765. [PubMed] [Google Scholar]
- Bohm J., Hahn A., Schubert R., Bahnweg G., Adler N., Nechwatal J., Oehlmann R., Obwald W. Real-time quantitative PCR: DNA determination in isolated spores of the mycorrhizal fungus Glomus mosseaei and monitoring of Phytophthora infestans and Phytophthora citricola in their respective host plants. J. Phytopathol. 1999;147:409–417. [Google Scholar]
- Brown A., Akinsanya A.A., Barker S.J., Brophy M., Dobb A.K., Doyle S.M., Hudson I.R., Minter S.J., Wraith M.J., Oultram J.D. Automated system for capture and detection of nucleic acids. Biotechniques. 1999;27:176–180. doi: 10.2144/99271pf01. [DOI] [PubMed] [Google Scholar]
- Eun A.J.C., Wong S.M. Detection of cymbidium mosaic potexvirus and odontoglossum ringspot tobamovirus using immuno-capillary zone electrophoresis. Phytopathology. 1999;89:522–528. doi: 10.1094/PHYTO.1999.89.6.522. [DOI] [PubMed] [Google Scholar]
- Eun A.J.C., Wong S.M. Molecular beacons: a new approach to plant virus detection. Phytopathology. 2000;90:269–275. doi: 10.1094/PHYTO.2000.90.3.269. [DOI] [PubMed] [Google Scholar]
- Förster V.Th. Zwischenmolekulare energiewanderung und fluorezenz. Ann. Phy. 1948;2:55–75. [Google Scholar]
- Gibbs A., Mackenzie A. A primer pair for amplifying part of the genome of all potyvirids by RT-PCR. J. Virol. Methods. 1997;63:9–16. doi: 10.1016/s0166-0934(96)02103-9. [DOI] [PubMed] [Google Scholar]
- Gut M., Leutenegger C.M., Huder J.B., Pedersen N.C., Lutz H. One-tube fluorogenic reverse transcription-polymerase chain reaction for the quantitation of feline coronaviruses. J. Virol. Methods. 1999;77:37–46. doi: 10.1016/S0166-0934(98)00129-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawrami K., Breuer J. Development of a fluorogenic polymerase chain reaction assay (TaqMan) for the detection and quantitation of varicella zoster virus. J. Virol. Methods. 1999;79:33–40. doi: 10.1016/s0166-0934(98)00176-1. [DOI] [PubMed] [Google Scholar]
- Heid C.A., Stevens J., Livak K.J., Williams P.M. Real time quantitative PCR. Genome Res. 1996;6:986–994. doi: 10.1101/gr.6.10.986. [DOI] [PubMed] [Google Scholar]
- Holland P.M., Abramson R.D., Watson R., Gelfand D.H. Detection of specific polymerase chain reaction product by utilising the 5′–3′ exonuclease activity of Thermus aquaticus DNA polymerase. Proc. Natl. Acad. Sci. USA. 1991;88:7276–7280. doi: 10.1073/pnas.88.16.7276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W.W., Wong S.M. The use of DIG-labelled cRNA probes for the detection of cymbidium mossaic potexvirus (CymMV) and odontogglossum ringspot tobamovirus (ORSV) in orchids. J. Virol. Methods. 1998;70:193–199. doi: 10.1016/s0166-0934(97)00187-0. [DOI] [PubMed] [Google Scholar]
- Lazar V., Bidart J.M., Caillou B., Mahe C., Lacroix L., Filetti S., Schlumberger M. Expression of the Na+/I− symporter gene in human thyroid tumors: a comparison study with other thyroid-specific genes. J. Clin. Endocrinol. Metab. 1999;84:3228–3234. doi: 10.1210/jcem.84.9.5996. [DOI] [PubMed] [Google Scholar]
- Lee L.G., Connell C.R., Bloch W. Allelic discrimination by nick-translation PCR with fluorogenic probes. Nucl. Acid Res. 1993;21:3761–3766. doi: 10.1093/nar/21.16.3761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lie Y.S., Petropoulos C.J. Advances in quantitative PCR technology: 5′ nuclease assays. Curr. Opin. Biotechnol. 1998;9:43–48. doi: 10.1016/s0958-1669(98)80082-7. [DOI] [PubMed] [Google Scholar]
- Lim S.T., Wong S.M., Yeong C.Y., Lee S.C., Goh C.J. Rapid detection of cymbidium mosaic virus by the polymerase chain reaction. J. Virol. Methods. 1993;41:37–46. doi: 10.1016/0166-0934(93)90161-j. [DOI] [PubMed] [Google Scholar]
- Livak K.J., Flood S.J., Marmaro J., Giusti W., Deets K. Oligonucleotides with fluorescent dyes at opposite ends provide a quenched probe system useful for detecting PCR product and nucleic acid hybridisation. PCR Methods Appl. 1995;4:357–362. doi: 10.1101/gr.4.6.357. [DOI] [PubMed] [Google Scholar]
- Livak K.J., Marmaro J., Todd J.A. Towards fully automated genome-wide polymorphism screening. Nat. Genet. 1995;9:341–342. doi: 10.1038/ng0495-341. [DOI] [PubMed] [Google Scholar]
- Livak K., Marmaro J., Flood S. Guidelines for designing TaqMan fluorogenic probes for 5′ nuclease assays. Perkin-Elmer Res. News. 1996;57:1–5. [Google Scholar]
- Mercier B., Burlot L., Ferec C. Simultaneous screening for HBV DNA and HCV RNA genomes in blood donations using a novel TaqMan PCR assay. J. Virol. Methods. 1999;77:1–9. doi: 10.1016/s0166-0934(98)00075-5. [DOI] [PubMed] [Google Scholar]
- Ryu K.H., Park W.M. Rapid detection and identification of odontoglossum ringspot virus by polymerase chain reaction amplification. FEMS Microbiol. Lett. 1995;133:265–269. doi: 10.1111/j.1574-6968.1995.tb07895.x. [DOI] [PubMed] [Google Scholar]
- Ryu K.H., Yoon K.E., Park W.M. Detection by RT-PCR of cymbidium mosaic virus in orchids. J. Phytopathol. 1995;143:643–646. [Google Scholar]
- Schoen C.D., Knorr D., Leone G. Detection of potato leafroll virus in dormant potato tubers by immunocapture and a fluorogenic 5′ nuclease RT-PCR assay. Phytopathology. 1996;86:993–999. [Google Scholar]
- Seoh M.L., Wong S.M., Zhang L. Simultaneous TD/RT-PCR detection of cymbidium mosaic potexvirus and odontoglossum ringspot tobamovirus with a single pair of primers. J. Virol. Methods. 1998;72:197–204. doi: 10.1016/s0166-0934(98)00018-4. [DOI] [PubMed] [Google Scholar]
- Tan S.W.L., Wong S.M., Kini R.M. Rapid simultaneous detection of two orchid viruses using LC- and/or MALDI-mass spectrometry. J. Virol. Methods. 2000;85:93–99. doi: 10.1016/s0166-0934(99)00157-3. [DOI] [PubMed] [Google Scholar]
- Wong S.M., Chng C.G., Lee Y.H., Tan K., Zettler F.W. Incidence of cymbidium mosaic and odontoglossum ringspot viruses and their significance in orchid cultivation in Singapore. Crop Protect. 1994;13:235–239. [Google Scholar]
- Yu H.H., Wong S.M. Synthesis of biologically active cDNA clones of cymbidium mosaic potexvirus using a population cloning strategy. Arch. Virol. 1998;143:1617–1620. doi: 10.1007/s007050050402. [DOI] [PubMed] [Google Scholar]
- Zettler F.W., Ko N.-J., Wisler G.C., Elliott M.S., Wong S.M. Viruses of orchids and their control. Plant Dis. 1990;74:621–626. [Google Scholar]