

Abstract
We have solved the crystal and molecular structures of hepatitis A viral (HAV) 3C proteinase, a cysteine peptidase having a chymotrypsin-like protein fold, in complex with each of three tetrapeptidyl-based methyl ketone inhibitors to resolutions beyond 1.4 Å, the highest resolution to date for a 3C or a 3C-Like (e.g. SARS viral main proteinase) peptidase. The residues of the β-hairpin motif (residues 138–158), an extension of two β-strands of the C-terminal β-barrel of HAV 3C are critical for the interactions between the enzyme and the tetrapeptide portion of these inhibitors that are analogous to the residues at the P4 to P1 positions in the natural substrates of picornaviral 3C proteinases. Unexpectedly, the Sγ of Cys172 forms two covalent bonds with each inhibitor, yielding an unusual episulfide cation (thiiranium ring) stabilized by a nearby oxyanion. This result suggests a mechanism of inactivation of 3C peptidases by methyl ketone inhibitors that is distinct from that occurring in the structurally related serine proteinases or in the papain-like cysteine peptidases. It also provides insight into the mechanisms underlying both the inactivation of HAV 3C by these inhibitors and on the proteolysis of natural substrates by this viral cysteine peptidase.
Abbreviations used: HAV, hepatitis A virus; FMDV, foot-and-mouth disease virus; TGEV, transmissible gastroenteritis coronavirus; SARS, severe acute respiratory syndrome; BBL, carboxylbenzyloxyl-L-serine-β-lactone; CMK, chloromethylketone; FMK, fluoromethylketone; Ac, acetyl; Qmm, N, N-dimethyl glutamine
Keywords: hepatitis A virus, 3C proteinase, inhibitor design, methylketone, episulfide
Introduction
Hepatitis A virus belongs to the Picornaviridae, a large family of positive single-stranded RNA viruses that also includes poliovirus (PV), foot-and-mouth disease virus (FMDV) and human rhinoviruses (HRV).1 Based on the extensive similarity in their genomic organization, it is likely that the basic features of their viral life cycles in the cell are also similar. Upon entry into the cytoplasm of susceptible cells, the viral RNA genome is translated by host ribosomes to produce a large polyprotein. Subsequent proteolytic cleavage of this polyprotein by virally encoded peptidase(s) (and in some cases by an unknown host peptidases) leads to mature viral proteins that perform key functions such as replication of the viral RNA genome, assembly of progeny virions and virus–host antagonism. The latter include inhibition of cellular mRNA translation and transcription, inhibition/induction of apoptosis, and membrane vesicularization.
In picornaviruses, most of the polyprotein is processed by the virus's 3C peptidase, although a 2A peptidase (entero- and rhinoviruses) and a small leader proteinase (aphthoviruses) participate to a minor extent.2 3C and 3C-like (3CL) peptidases also occur in caliciviruses and coronaviruses; they play major roles in polyprotein processing in these viruses, although the coronaviral polyprotein is considerably larger. These enzymes are cysteine peptidases with folds also similar to that of chymotrypsin rather than that of papain. 3CL peptidase has an additional domain that mediates homodimerization, which is thought to be critical for the enzymatic activity of 3CL.3 The catalytic mechanisms of 3C and 3CL are thought to proceed via a tetrahedral intermediate resulting from the nucleophilic attack on the carbonyl carbon of the scissile bond by the Sγ of the catalytic cysteine residue (Cys172 in HAV 3C). However, many mechanistic details remain unknown. Detailed understanding at atomic resolution of how 3C enzymes cleave their cognate substrates would greatly facilitate the rational design of anti-viral drugs. Such knowledge will help to control these dangerous pathogens in the event of a pandemic outbreak, such as the recent FMDV epidemic in England that caused significant loss of animals and the accompanying financial woes.4
The use of substrate analogue inhibitors to probe enzymatic mechanisms has a long tradition in research on proteinases. Peptidyl chloromethyl ketones react with papain and serine proteinases in dissimilar ways to yield different types of covalent adducts with the catalytic residues. In chymotrypsin, Ser195 forms a covalent linkage to the carbonyl carbon of chloromethyl ketone inhibitors, resulting in a tetrahedral geometry thought to mimic the transition state that occurs during the hydrolysis of natural substrates.5., 6., 7. In addition, the Nε2 atom of His57 attacks the halogenated α-carbon yielding a second covalent attachment between the inhibitor and the now alkylated enzyme. When the chloromethyl function is replaced with a chloroethyl group in the inhibitor, His57 is alkylated with retention of configuration at the chiral center.8 Together with kinetic studies, this indicates an internal nucleophilic displacement of chloride by the newly formed oxyanion in the tetrahedral intermediate leading to a Ser195-epoxy ether adduct that is opened by the histidine.9 , 10 In contrast, papain is inactivated by direct covalent linkage between the Sγ atom of Cys25 and the halogenated α-carbon of the same class of inhibitors.11 The imidazole ring of His159 in papain is implicated in protonating the leaving group due to its shift in orientation from a position in-plane with the Sγ atom of Cys25 to a position in-plane with the the α-carbon of the inhibitor, an atom equivalent to the N of a scissile bond.12 However, it is unclear whether reversible formation of a tetrahedral intermediate occurs prior to alkylation of Cys25.
Sequence analyses indicate that residues His44, Asp84 and Cys172 in HAV 3C should form the canonical catalytic triad.13 , 14 However, this was not confirmed structurally until the hydrogen bond between Oδ2 of Asp84 and Nδ1 of His44 was observed in the recent crystal structures of HAV 3C in complex with a β-lactone (BBL, N-benzyloxycarbonyl (Cbz) l-serine-β-lactone) inhibitor.15 Fortuitously, in these crystals, the inhibitor BBL binds covalently to His102, a surface residue near the RNA binding motif of 3C, while leaving the proteolytic active site and the catalytic Cys172 residue unobstructed. Here we report the crystal structures of HAV 3C in complex with each of three tetrapeptidyl inhibitors: N-acetyl-leucyl-alanyl-alanyl-(N,N-dimethyl)-glutamine-fluoromethylketone (Ac-LAAQmm-FMK, inhibitor Ia), N-acetyl-leucyl-phenylalanyl-phenylalanyl-glutamate-fluoromethylketone (Ac-LFFE-FMK, inhibitor II) and N-acetyl-leucyl-alanyl-alanyl-(N,N-dimethyl)-glutamine-(1,4-dioxo-3,4-dihydro-1H-phthalazin-2-yl)methylketone (Ac-LAAQmm-pMK, inhibitor Ib) (Figure 1 ). These complexes were obtained by a soaking method using the pre-grown crystals of HAV 3C-BBL.
Figure 1.
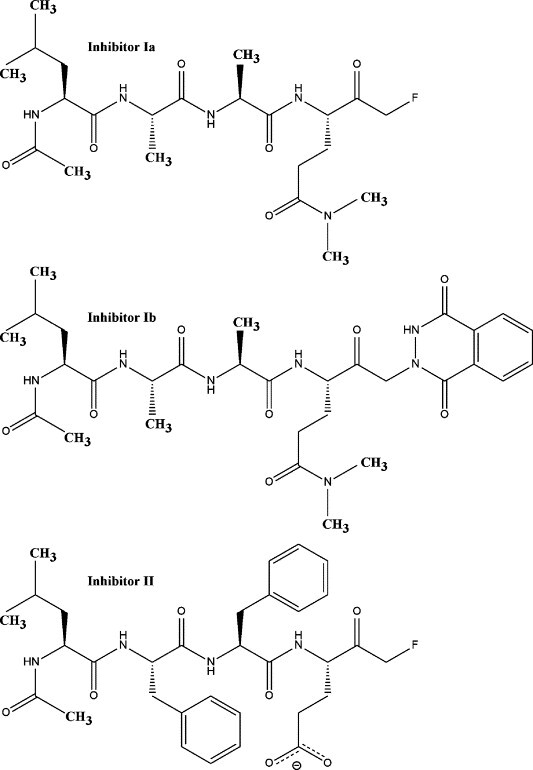
Schematics of the chemical structures of the inhibitors used in this study. Inhibitor Ia, LAAQmm-FMK; inhibitor II, LFFE-FMK; inhibitor Ib, LAAQmm-pMK.
The peptidyl portion of inhibitors Ia and Ib offers a fairly good resemblance to the most favored sequence (P4 to P1 positions) of the cleavage sites by HAV 3C in the viral polyprotein. Inhibitor II was included in this study as a mimetic of a less optimal substrate than the LAAQ sequence in inhibitors Ia and Ib, mainly due to the substitutions at the P1 (Gln->Glu) and P2 (Ala->Phe) positions of its peptidyl portion. Sequence alignment analysis of the naturally occurring cleavage sites for HAV 3C proteinase in the hepatitis A viral polyprotein indicates a clear preference for small residues such as serine and threonine at the P2 position. Available structural information strongly supports this conclusion.15 , 17 , 18 Furthermore, glutamate does occasionally occur as the P1 residue at the natural cleavage sites within picornaviral polyproteins, although at a much lower frequency than glutamine. Here, we are interested in understanding how the interactions between the HAV 3C enzyme and peptidyl inhibitors are influenced by the amino acid make-up of the inhibitors. The substrate recognition sites (S1 to S4) of HAV 3C are identified crystallographically. No covalent linkage occurred between His44 and any of the inhibitors. More interestingly, an unusual episulfide cation (thiiranium ring) is observed at the catalytic cysteine residue.
Results and Discussion
Crystallization and enzyme–inhibitor complex formation
Native and variant forms of HAV 3C protein have been crystallized in several different crystal forms.16., 17., 18. Until the recently reported crystallization of a Cys24Ser variant of HAV 3C in space group P212121, there was no structural evidence that the three catalytic residues in the proteolytic active site, His44, Asp84 and Cys172, form the canonical triad assembly as observed in chymotrypsin-like serine proteinases and other picornaviral 3C enzymes.15 Furthermore, the earlier crystal structures16., 17., 18. were of inactivated forms of HAV 3C containing either a substituted catalytic cysteine residue (Cys172Ala) or an oxidized form of Cys172, thereby complicating the study of enzyme-substrate/inhibitor interactions. Indeed, soaking of substrate-like inhibitors into pre-grown crystals of these active site variants of HAV 3C has met with little success. Here, HAV 3C crystals formed after incubating the Cys24Ser variant with BBL were used for the subsequent binding of three tetrapeptidyl-based inhibitors (Figure 1). Analysis of the soaked crystals by mass spectrometry revealed a difference in mass between 3C-BBL and 3C-BBL-inhibitor Ib, that is within experimental error of the calculated molecular mass of acetyl-LAA(N,N-dimethyl)Q-methylene (data not shown). This suggests that the phthalhydrazide group is lost during the covalent attachment of inhibitor Ib to the HAV 3C enzyme in a fashion similar to the loss of fluoride from the other two inhibitors.19
A previous study of the in vitro inhibition of HAV 3C by inhibitor Ia showed an IC50 value of 41 μM using a partial viral polyprotein (ΔP1-2A-2B) as substrate.19 Moreover, when added to HAV-infected cells at a concentration of 5 μM, inhibitor Ia reduced the viral yield by 25-fold, indicative of a much lower IC50 value in vivo. The efficacy of the inhibitor in cells is probably due to a combination of effects, including the irreversibility of inhibition and the fact that polyprotein processing is upstream of many key replicative events during HAV infection. Therefore, even partially blocking the 3C proteinase function with inhibitor Ia would have a profound impact on progeny virion production.
The inhibition of HAV 3C by inhibitor Ib had been studied initially using a short time course (15 min) and low concentrations of a fluorogenic substrate to minimize the latter's cleavage and self-quenching. Under these conditions, potent, competitive inhibition was observed with an IC50 value of 13 μM.20 In the current study, a more extensive time course study demonstrated that the initial competitive inhibition (Figure 2(a)) is followed by slow inactivation of HAV 3C by inhibitor Ib in an irreversible and time-dependent fashion (Figure 2(b)). This is in agreement with the structural data presented below that HAV 3C forms essentially the same complexes with both inhibitors Ia and Ib. The IC50 value of inhibitor Ib was estimated to be 15 μM, close to a previously reported value of 13 μM (Figure 2(a)).20 No enzymatic activity was detected after HAV 3C was incubated with inhibitor Ib for 3 h prior to the activity assay, indicating the irreversible nature of inactivation (Figure 2(b)). Control samples of HAV 3C incubated for 3 h prior to the assay did not lose activity detectably.
Figure 2.
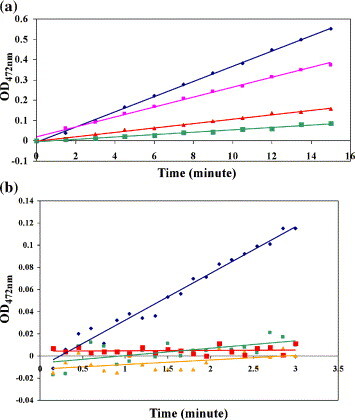
Enzymatic activity of HAV3C inhibited by inhibitor Ib. (a) The proteolytic activity of HAV 3C was assayed using a fluorogenic substrate immediately after the addition of inhibitor Ib. The data points are colored according to the concentration of inhibitor Ib present in the reaction mix: blue, 0 μM; purple, 10 μM; red, 25 μM; and green, 50 μM. (b) HAV 3C was pre-incubated with inhibitor Ib for varying periods of time before the substrate was added to assay the enzymatic activity. Dark blue, no inhibitor; green, no pre-incubation; orange, 2 h pre-incubation; red, 3 h pre-incubation. In both graphs, the y axis values are artificial fluorescence units and the x axis value times in minutes.
Structural overview
Structural analysis revealed few differences in the overall protein fold or in the 3C–BBL interactions among the three new complexes and 3C–BBL (Figure 3 ). In the three new structures, BBL was bound to the Nε2 atom of His102 in the same manner as in the 3C–BBL structure.15 The average r.m.s.d. values between the three new structures reported here and 3C-BBL are in the range of 0.23 Å to 0.30 Å over the Cα positions of all residues (Table 1 ), suggesting that little structural rearrangement is required for the binding of substrate-analogue inhibitors. This confirms our previous claim that the crystallized enzyme is in its catalytically competent conformation.15 Comparison of the r.m.s.d. values at each of the Cα positions reveals that the three new complexes differ significantly from 3C-BBL in only three regions (Figure 4 ): those comprising residues 49–51, 142–154 and 194–196. These differences suggest that enzyme–substrate complex formation involves an induced-fit mechanism, whereby subtle structural adjustments optimize enzyme–substrate interactions. Residues 142–154 form a major part of the β-hairpin motif that sits atop the catalytic residues in HAV 3C. Some of the residues in this β-hairpin help form the S sub-sites for substrate binding (vide infra). The complexes with inhibitors Ia and Ib share greater structural similarity with each other than with the inhibitor II complex (Table 1). This supports our hypothesis that inhibitors Ia and Ib react similarly in the active site of 3C despite the different substituents on the terminal α-carbon of these two inhibitors. Additionally, since both inhibitors Ia and II are fluoromethyl ketones, the differences in their tetrapeptidyl sequence must be the key cause for their structural divergences. This suggests that HAV 3C uses subtle structural adjustments to “measure up” the binding fitness of each inhibitor/substrate. The structural differences between the inhibitor Ia and II complexes are consistent with HAV 3C's preferences for (1) small residues at the P2 position of the cleavage site and (2) glutamine at the P1 position.
Figure 3.
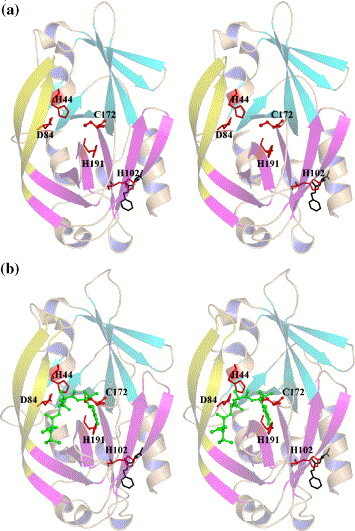
Structural overviews (in stereo) of the HAV 3C-BBL and its complexes with the three inhibitors used in this study. (a) 3C–BBL; (b) 3C–BBL–inhibitor Ia; (c) 3C–BBL–inhibitor Ib; and (d) 3C–BBL–inhibitor II. In (a) and (b), the HAV 3C polypeptide chains are shown in cartoon with the two terminal β-barrels colored in cyan (N-terminal) and magenta (C-terminal), respectively. The extended β-hairpin substructure (residues 139–158) is colored yellow. BBL molecules are show in black sticks, whereas the tetrapeptidyl inhibitors are shown in sticks and spheres and colored green. The catalytic triad, His191 and His102 are shown in red sticks. (c) and (d) Generated using the program LIGPLOT.37
Table 1.
Alignment statistics of various complexes discussed in this study
| 3C–inhibitor Ia | 3C–inhibitor II | 3C–inhibitor Ib | |
|---|---|---|---|
| 3C–BBL | 0.30a | 0.23 | 0.29 |
| 3C–inhibitor Ia | 0.20 (0.55)b | 0.05 (0.07) | |
| 3C–inhibitor II | 0.19 (0.52) |
r.m.s.d. values (Cα positions) over all protein residues and BBL (the carboxyl carbon was used in lieu of Cα) in Å.
Parentheses indicate r.m.s.d. values for inhibitors alone.
Figure 4.
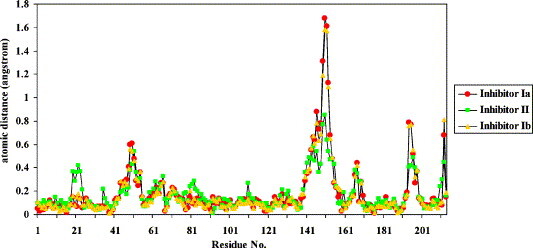
Distances between the Cα atoms obtained from the alignments of three HAV 3C–BBL–methylketone complex structures. The 3C–BBL part was aligned against previously reported 3C–BBL structure (PDB code 2CXV).
3C proteinase–inhibitor interactions and the specificity pockets of HAV 3C
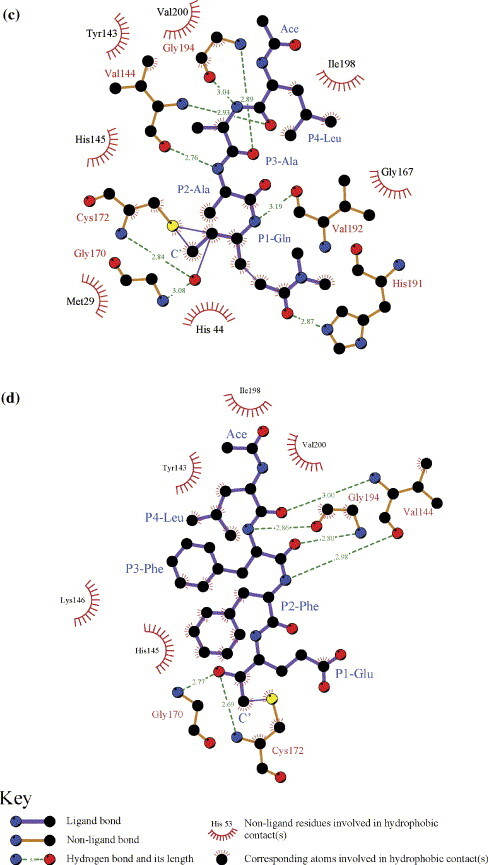
The peptidyl portions of the three ketone inhibitors form canonical β-sheet interactions with the 3C enzyme via four hydrogen bonds: those between the main-chain N of Val144 and O of P4-Leu, the main-chain O of Val144 and N of P2-Ala (or P2-Phe) on one side, and those between the main-chain O of G194 and N of P3-Ala (or P3-Phe), the main-chain N of G194 and O of P3-Ala (or P3-Phe) on the other (Figure 5 and Table 2 ). This β-ladder is slightly twisted as shown in the relative angles of the above hydrogen bonds to allow for optimal interactions between the side-chains of the inhibitor and the corresponding substrate binding sites (S sites) of the enzyme. Additionally, the main-chain N atoms of the P1 residues in inhibitors Ia and Ib (P1-Gln) form hydrogen bonds (3.2 Å) to the main-chain O atom of Val192. This interaction is not observed in the inhibitor II complex structure that has a longer corresponding distance of 3.9 Å, which likely results from the non-optimal binding of the P2-Phe residue of inhibitor II at the S2 site (see below).
Figure 5.
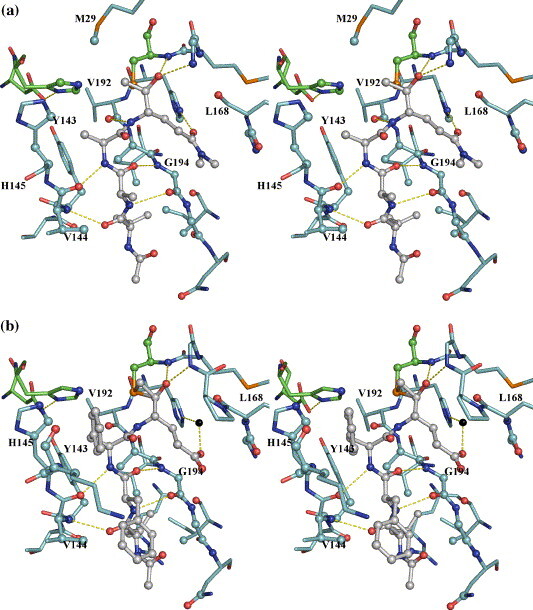
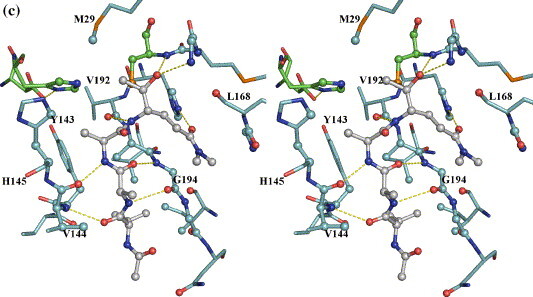
Interactions between the tetrapeptidyl inhibitors and HAV 3C in the active site. The protein residues are distinguished by cyan carbon bonds, the inhibitors by grey carbon bonds/spheres. The catalytic triad, His44, Asp84 and Cys172 are identified by green carbon bonds. Atoms of the 3C proteinase within van der Waals radius to the inhibitors are represented as spheres. The yellow broken lines mainly show the hydrogen bonds formed between the inhibitors and the 3C enzyme. For the sake of clarity, not all protein residues are labeled and solvent atoms are omitted with the exception of a water-bridged interaction between the Oε1 atom of the P1 glutamate residue of inhibitor II and the Nε2 atom of His191 in B (inhibitor II complex).
Table 2.
Interactions between the tetrapeptidyl inhibitors and HAV 3C protease (episulfide mode) and B. same
| Positiona | Inhibitor Ia | Inhibitor II | Inhibitor Ib |
|---|---|---|---|
| A. A tabulation of interactions by the components in the inhibitors | |||
| P4 | 32/9b | 34/9 | 31/8 |
| P3 | 16/2 | 28/9 | 17/2 |
| P2 | 15/4 | 36/7 | 15/4 |
| P1 | 35/3 | 30/11 | 35/3 |
| C'c | 8/2 | 7/3 | 8/2 |
| O(P1)c | 8/1 | 12 | 8/1 |
| H-bonds | P4O:Val144N (2.94)e | P4O:Val144N (3.00) | P4O:Val144N (2.93) |
| P3N:Gly194O (3.04) | P3N:Gly194O (2.86) | P3N:Gly194O (3.04) | |
| P3O:Gly194N (2.94) | P3O:Gly194N (2.80) | P3O:Gly194N (2.89) | |
| P2N:Val144O (2.72) | P2N:Val144O (2.98) | P2N:Val144O (2.76) | |
| P1N:Val192O (3.21) | Water bridged P1Oε1:HOH:His191Nε2 | P1N:Val192O (3.19) | |
| P1Oε1:His191Nε2 (2.97) | P1Oε1:His191Nε2 (2.87) | ||
| O(P1):Gly170N (3.07) | O(P1):Gly170N (2.77) | O(P1):Gly170N (3.08) | |
| O(P1):Cys172N (2.92) | O(P1):Cys172N (2.69) | O(P1):Cys172N (2.84) | |
| B. Statistics assorted by protein residues involved in the interactions (interactions with solvent omitted) | |||
| Met29 | 1 | 0 | 1 |
| His44 | 5 | 1 | 5 |
| Thr142 | 1 | 1 | 1 |
| Tyr143 | 5 | 4 | 4 |
| Val144 | 14(2)f | 14(2) | 14(2) |
| His145 | 3 | 12 | 3 |
| Lys146 | 0 | 14 | 0 |
| Arg162 | 0 | 1 | 0 |
| Gly167 | 5 | 3 | 5 |
| Leu168 | 3 | 3 | 3 |
| Pro169 | 0 | 2 | 0 |
| Gly170 | 3(1) | 3(1) | 3(1) |
| Met171 | 2 | 3 | 2 |
| Cys172 | 12(1) | 12(1) | 12(1) |
| His191 | 3(1) | 0 | 4(1) |
| Val192 | 4(1) | 2 | 4(1) |
| Ala193 | 6 | 4 | 5 |
| Gly194 | 20(2) | 22(2) | 21(2) |
| Gly195 | 5 | 3 | 5 |
| Asn196 | 2 | 3 | 2 |
| Ile198 | 3 | 3 | 3 |
| Val200 | 2 | 2 | 2 |
Residue positions with respect to scissile bond as defined by Schechter and Berger.38
Total number of van der Waals interactions/van der Waals interactions with solvent (less than or equal to 4 Å).
The terminal α-carbon atom next to the carbonyl carbon (C) of the P1 residue.
The O atom of the P1 residue.
Parentheses indicate distance in Å.
Total number of interactions (number of hydrogen bonds).
Our structural observations accord well with previous biochemical data that indicate that the P1 and P4 residues primarily determine the specificity in enzyme–substrate interactions.21 The S1, S2 and S4 sites of HAV 3C are quite well defined, whereas the S3 site is not discernible, and the side-chain of the P3 residue of each of the inhibitors simply protrudes into the solvent. The S2 site, formed by the side-chain atoms of His44, Phe48, Tyr143, His145 and Leu155 and the main-chain atoms of His44, Tyr143, Val144 and His145, is neither wide nor deep enough to accommodate a large hydrophobic residue such as Phe. Consequently, the P2-Phe residue of inhibitor II does not enter the S2 pocket. With a χ1 angle of 79°, its benzene moiety bends toward solvent at the entrance of S2 pocket, stacking against the imidazole ring of His145 (Table 2). The S4 site is a shallow depression on the surface of HAV 3C clustered with the side-chains of hydrophobic residues, namely, Tyr143, Ile198 and Val200, all forming hydrophobic interactions with the side-chain atoms of P4-Leu of the inhibitors.
Finally, the S1 site is of paramount importance in substrate recognition. It is formed by residues 192–195 as one wall and residues 167–169 as the other. His191 sits at the bottom of the S1 pocket and Leu199 at the back end opposite to Cys172. The imidazole side-chain of His191 is locked in a particular tautomeric conformation by the hydrogen bond between its Nδ1 atom and one of the two buried water molecules that are also hydrogen bonded to the carboxylate of Glu132. His191 is poised to interact with the P1 residue of the incoming peptidyl substrate: the Oε1 atom of a P1 glutamine residue would form an optimal hydrogen bond to the Nε2 atom of His191. Indeed, the Oε1 atoms of P1-Glnmm in either inhibitor Ia and Ib are at a distance of 3.0 Å and 2.9 Å to the Nε2 atom of His191. It is interesting to note that neither carboxyl oxygen atoms of P1-Glu in inhibitor II forms a direct hydrogen bond to His191. Instead, the Oε1 atom of P1-Glu is hydrogen bonded to the Nε2 atom of His191 via a bridging water molecule, which is at distances of 2.7 Å and 2.6 Å to the former and latter atoms, respectively. In the crystal structure of 3C–BBL complex, a similar water molecule forms a hydrogen bond to the Nε2 atom of His191 with a comparable distance of 2.8 Å. Similarly, a solvent molecule also forms a hydrogen bond to the equivalent P1 specificity-determining residues in the FMDV 3C (His181) and the TGEV 3CL (His162) crystal structures.22 , 23 In most serine proteinases, the water molecules interacting with the residues in the S1 specificity pockets are displaced upon binding substrate-analogue inhibitors, probably exiting the S1 site via a network of hydrogen bonding water molecules formed at the “back” end of the S1 specificity pocket.24 Similar scenes might have happened in our HAV 3C complexes with the ketone inhibitors Ia and Ib.
It is apparent that the binding of inhibitor II to HAV 3C was not sufficiently optimal to expel the solvent molecule interacting with His191 from the S1 site. Consequently, P1-Glu of inhibitor II does not bind as deeply in the S1 pocket as does the P1-Glnmm residues of inhibitors Ia and Ib. This provides strong evidence that the imidazole ring of His191 is neutral in these complexes, as otherwise a direct hydrogen bonded ion-pair interaction between P1-Glu and His191 would favor the opposite scenario. With the side-chain of His191 uncharged, the negatively charged carboxylate side-chain of P1-Glu naturally favors a more solvent-exposed location. Additionally, the non-optimal binding of P2-Phe residue of inhibitor II to the S2 site also contributes to the shallow penetration of P1-Glu in the S1 pocket. Compared to their equivalent atoms in inhibitors Ia and Ib, the Cβ and Cα atoms of P2-Phe in inhibitor II were pushed outwards solvent by 0.97 Å and 0.73 Å, respectively, to avoid clashes between the benzene ring of P2-Phe and the imidazole ring of His145. Most likely, this contributed at least partly to a movement of similar magnitude for the atoms of P1-Glu of inhibitor II (see Supplementary Data, Figure S1).
The inhibitors form an unusual episulfide linkage with the catalytic cysteine residue
In fitting the initial model of the inhibitor into the experimental electron densities near the catalytic cysteine, both the keto carbonyl carbon (CC) of the P1 residues and the adjacent α-methylene carbon (C') of the inhibitors had to be placed within 2.2 Å of the Sγ atom of Cys172. Thus, an episulfide cation fits the electron density far better than a single Sγ–C bond to either the CC or the C' atom. Although related thiiranium cations bearing only carbon substituents are well-recognized intermediates in chemical syntheses,25 , 26 including a few especially stable, structurally characterized examples,27 to the best of our knowledge there is only one report of a thiiranium (episulfide) cation having an oxyanion substituent detected as an intermediate in gas phase studies.28 In the final model of HAV 3C with inhibitor Ia, the distances between the Sγ atom of Cys172 and C' or CC (P1-Gln) are 1.83 Å and 1.82 Å, respectively (Figure 6(a)). The imidazole ring of His44 is coplanar with Sγ and C' and its Nε2 atom is at distances of 4.1 Å and 3.3 Å from Sγ and C', respectively. The distance between C' and CC is 1.56 Å, corresponding to that of a C–C single bond and indicating that the physical strain is well distributed around the entire three-membered ring. The C'-Sγ-CC angle is ∼ 51°, which is feasible due to the fact that sulfur has a larger atomic radius than either oxygen or carbon. The latter elements would not be able to form a three-membered ring structure with similar stability, which is perhaps why the epoxy ether intermediate proposed to develop during the inactivation of the serine proteinases by chloromethyl ketone inhibitors has not been observed in previous crystal structures.5 , 29 The episulfide ring structures in the inhibitor Ia and inhibitor Ib complexes are essentially identical (Table 1 and Figure 5, Figure 6).
Figure 6.
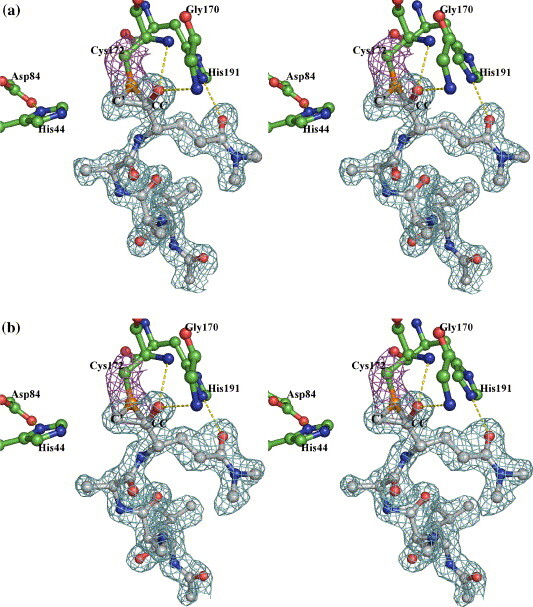
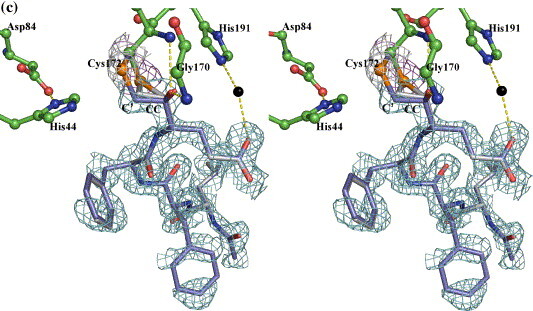
Electron densities (from a |2|Fo|−|Fc||, αcalc map contoured at 1 sigma) surrounding inhibitor Ia (a), Ib (b) and II (c). (a) and (b) Residues in 3C are distinguished by green carbon bonds/spheres, whereas the carbon atoms in the inhibitors are colored grey. The electron density contours surrounding the Sγ and Cβ atoms are colored magenta, those around the inhibitors are in cyan. Hydrogen bonds between His44 and Asp84, His191 and P1Gln, as well as those involving the oxyanion are shown as yellow broken lines. (c) Electron densities showing the two possible binding modes of inhibitor II. The color scheme is similar to that in Figure 5, with the ringless alternate conformation of the inhibitor distinguished by slate-colored carbon atoms. Hydrogen bonds similar to those in Figure 4 are shown with the exception that the His191–P1Gln direct interaction is now replaced with two hydrogen bonds bridged through a solvent molecule (black sphere).
Structural refinement of the 3C–BBL–inhibitor II complex structure using the coordinates of the episulfide ring in the 3C–BBL–inhibitor Ia complex revealed an additional positive electron density peak near the Sγ atom of Cys172 (Figure 6(c)). Further fitting indicated that this represents an alternative mode of bonding of the inhibitor characterized by a single covalent linkage between Cys172 Sγ and the C' of inhibitor II. Interestingly, this mode of bonding corresponds to that observed in the crystal structure of an inactivated papain in which Cys25 is covalently attached to a chloromethyl ketone compound.12 Furthermore, the relative orientations of the methylketone moiety of the inhibitor, the catalytic cysteine and histidine residues are also shared between these two cysteine proteinases of different folds. Nevertheless, the two enzymes made different structural adjustments in arriving at these conformations. In the case of papain, the imidazole ring of His159, in-plane with the Sγ atom of Cys25 in the apoenzyme, rotated about the Cβ–Cγ bond to become coplanar with the atom equivalent to the N atom of the scissile peptide bond. In the 3C–BBL–inhibitor II complex structure containing an episulfide ring, the Sγ atom of Cys172 is coplanar with the C' atom of the inhibitor and the imidazole ring of His44. In the alternative conformation lacking this ring, the atomic positions of His44 are unchanged, whereas the Sγ–Cβ bond of Cys172 is rotated ∼ 91° out toward the solvent, causing the Sγ atom to roll out of the common plane containing His44 imidazole ring and C'. The new position of the Sγ atom is 2.8 Å from CC atom of P1-Glu of inhibitor II, which is comparable to the average distance (2.8 Å) between Sδ and Cβ in the eight methionine residues in the HAV 3C molecules.
Possible mechanisms for inactivation of HAV 3C by substituted methyl ketone compounds
A comparison of the three structures of HAV 3C–BBL bound with tetrapeptidyl ketone inhibitors with the 3C-iodo-Val-Phe (iVF) complex structure (PDB code 1QA7) suggests that the episulfide ring structure may be an intermediate state on the iVF inactivation pathway. In the latter structure, the imidazole ring of His44 is rotated about its Cβ–Cγ bond by roughly 10° and becomes more in-plane with the carbon at the α position of the ketomethylene function of the iVF inhibitor than with the Sγ atom of Cys172. Judging from the two alternate conformations of the 3C–inhibitor II complex, it seems possible that an internal displacement reaction would resolve the episulfide ring and eventually form an alkylated Cys172 in the active site. However, it is unclear how an episulfide cation is produced in the first place. There are at least three potential routes leading to the formation of an episulfide ring (Figure 7 ). In scheme I, the Sγ atom of Cys172 attacks C' directly in a nucleophilic substitution reaction to replace the fluoride. This would then be followed by the attack of a lone electron pair of the Sγ atom on the carbonyl carbon to form the oxyanion and the episulfide cation. In schemes II and III, the Sγ atom of Cys172 first attacks the carbonyl carbon to form a tetrahedral intermediate and the oxyanion. In scheme II, a lone electron pair of Sγ then displaces the fluoride. In scheme III, the oxyanion displaces the fluoride to form a transient epoxy ether, which then rearranges to the more stable episulfide ring. As X-ray crystallography shows only snapshots of structures of interest, unless one of these proposed intermediate species can be uniformly formed and stably preserved in crystals, other experimental tools will be needed to differentiate among these mechanistic pathways.
Figure 7.
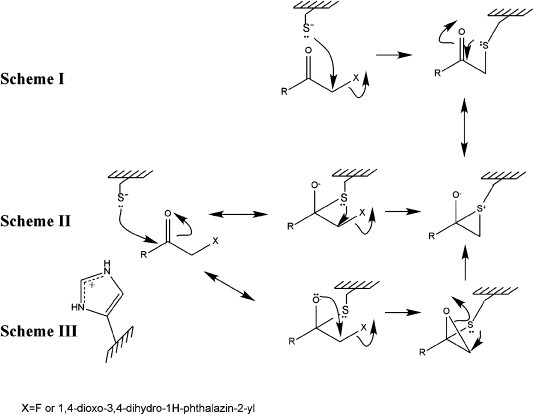
Tentative mechanisms of the inactivation of HAV 3C proteinase by the methylketone compounds used in this study. See the text for a detailed explanation.
Mechanism of hydrolysis of peptide bonds by the 3C enzyme
Although none of the reactive groups in these inhibitors represents a true peptide bond substrate for the 3C proteinase, the changes in the relative orientations between the Sγ atom of Cys172, the imidazole ring of His44 and the substituted methyl carbonyl group in the inhibitor nevertheless agree with the hypothesized roles of these catalytic residues in hydrolyzing natural peptidyl substrates. His44, with the assistance of Asp84, acts as a general base catalyst to receive a proton from Cys172, thereby producing a relatively strong nucleophilic thiolate ion. This thiolate subsequently attacks the carbonyl-carbon atom of the scissile bond, forming the tetrahedral intermediate. In the next step, His44 donates a proton to the nitrogen atom of the P1' residue making it a better leaving group. This is followed by the cleavage of the peptide bond and the formation of a thiolacyl enzyme. To fulfill its various roles in this mechanism, His44 must adjust its orientation toward Cys172 accordingly. Interestingly, the two predicted conformations of the His44-Cys172 pair correspond well to those observed in the two 3C-BBL–inhibitor II complex structures.
The only other reported structure of a 3C or 3C-like enzyme inactivated by a methyl ketone inhibitor is that the of SARS 3CL-hexapeptidyl chloromethyl ketone (CMK) complex (PDB code 1UK4).3 In that structure, the inhibitor forms a single bond with nucleophilic Cys145, corresponding to the alternate mode of bonding observed in the HAV 3C–inhibitor II complex. In the 3CL–CMK complex, the orientation of His41 with regard to Cys145 is intermediate between the two bonding modes observed in the HAV 3C–inhibitor II complex, but is more similar to that of the episulfide ring binding mode. A similar intermediate orientation is also observed in the crystal structure of HRV2 3C in complex with AG7088, a substrate analogue inhibitor30 (Table 3).
Table 3.
Crystallographic statistics of data collection and structure refinement
| PDB code | 2H9H | 2HAL | 2H6M |
|---|---|---|---|
| 3C variant | C24S | C24S | C24S |
| 3C–inhibitor complex | 3C–BBL–Ia | 3C–BBL–II | 3C–BBL–Ib |
| Space group | P212121 | P212121 | P212121 |
| a (Å) | 44.67 | 44.58 | 44.77 |
| b (Å) | 56.06 | 56.24 | 56.09 |
| c (Å) | 80.97 | 81.05 | 80.91 |
| α (°), β (°), γ (°) | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 |
| No. molecule/asymmetric unit | 1 | 1 | 1 |
| Vm (Matthews' coefficient)/% solvent content | 2.1/41.7 | 2.1/41.7 | 2.1/41.7 |
| Data collection | ALS Beamline 8.3.1 | ||
| Resolution range (Å) | 26.5–1.35 (1.40–1.35)a | 40.0–1.35 (1.40–1.35) | 46.08–1.40 (1.45–1.40) |
| Wavelength (Å) | 1.115879 | 1.115879 | 1.115869 |
| Observations | 98,591 | 10,6957 | 112,266 |
| Unique reflections | 36,919 (1013) | 38,893 (1897) | 32,357 (3192) |
| I/σ(I) | 12.7 (2.6) | 13.0 (1.50) | 13.0 (1.72) |
| Data completeness (%) | 81.2 (22.6) | 85.3 (42.4) | 79.1 (79.8) |
| Rmergeb | 0.056 (0.307) | 0.061 (0.288) | 0.068 (0.353) |
| Refinement | |||
| No. reflections used | 34,342 (1263) | 36,916 (1244) | 30,672 (2235) |
| Resolution range (Å) | 26.49–1.39 (1.43–1.39) | 20.0–1.35 (1.39–1.35) | 46.08–1.40 (1.44–1.40) |
| Free set size (%) | 5.0 | 5.0 | 5.0 |
| No. atoms | 1946 | 2073 | 1946 |
| No. waters | 263 | 391 | 263 |
| Rworkingc (%) | 18.1 (32.9) | 18.4 (23.1) | 17.7 (30.0) |
| Rfree (%) | 19.7 (29.4) | 20.4 (28.3) | 20.4 (33.0) |
| Mean B valued | 20.97/32.28/37.22 | 17.01/28.07/29.96 | 20.36/33.57/35.82 |
| r.m.s.d. from ideal geometry | |||
| Bond length (Å) | 0.009 | 0.009 | 0.008 |
| Bond angle (°) | 1.200 | 1.144 | 1.152 |
| Chirality | 0.081 | 0.076 | 0.076 |
| Ramachandran plot outliers (phi, psi) | Asp36 (51.2, –129.2) Asp84 (69.4, –69.5) | Asp36 (49.7, –127.5) Asp84 (68.1, –73.0) | Asp36 (50.0, –125.7) Asp84 (69.4, –71.0) |
Parentheses indicate values for the highest resolution shell.
Rmerge=ΣhΣj|Ihj−<Ih>|/ΣhΣjIhj, where <Ih> is the weighted mean intensity of the symmetry-related reflections Ihj.
Rworking=Σh||Fo|-|Fc||/ΣhFo, where |Fo| and |Fc| represent the observed and calculated structure factor amplitudes, respectively. Rfree is Rworking calculated with the reference set.
Average B factors of the complex/tetrapeptidyl inhibitor/solvent molecules.
Recently, crystal structures of the SARS 3CL proteinase modified by a peptidyl-based epoxide inhibitor have been obtained in two different crystal forms (PDB codes 2A5I and 2A5K).31 Remarkably, the ring-opening reaction between the inhibitor and the 3CL enzyme covalently linked the catalytic sulfur atom to the C2 atom of the inhibitor that is in a similar position as the C' atom of the halomethyl ketone inhibitors. Compared to the apoenzyme structure (PDB code 2A5A), a drastic rotation of ∼ 95° (change in the χ1 angle) of the Cβ–Sγ bond in the catalytic cysteine residue is reproducibly observed in two different crystal forms (PDB codes 2A5I and 2A5K). This indicates that 3C or 3CL proteinases, as suggested for papain, may undergo local but significant conformational changes in the active site as the hydrolysis of substrates progresses. Inhibitors aimed at structurally “trapping” these viral enzymes at one of the intermediate steps may prove extremely effective in neutralizing these essential viral components.
Materials and Methods
Production and assay of HAV 3C proteinase C24S, synthesis and testing of inhibitor Ib
Experimental procedures for enzyme production and assay, as well as the syntheses of inhibitors Ia, Ib and II (Figure 1) have been published19 , 20 and were followed in the present study. Enzyme inhibition and HMQC NMR experiments were also done according to the procedures described in the same reports.
Preparation and crystallization of the complex of HAV 3C with inhibitors
Crystals of HAV 3C proteinase variant C24S were grown and harvested as described.15 To make the 3C–BBL–ketone inhibitor triple complex, pre-grown 3C–BBL crystals were soaked in solutions containing ∼ 5 mM of the substituted methylketone inhibitor for at least 1 h before they were cryo-protected and flash-cooled for data collection. To prepare samples for mass spectrometric analysis, HAV 3C crystals before and after soaking were collected and then rinsed in large excesses of mother liquor to remove any residual or peripheral presence of the inhibitor(s).
Data collection and processing
X-ray diffraction data were collected at Beamline 8.3.1 of the Advanced Light Source (ALS) at Berkeley Lawrence National Laboratory. The programs Denzo, Scalepack and the CCP4 suite were used to process and scale the datasets32 , 33 (Table 3 ).
Structure determination and refinement
All structures were solved by molecular replacement using program Molrep in the CCP4 program suite32 and the published model of the HAV 3C-BBL complex15 (PDB code 2CXV) as a search probe. The program XFit34 was used for visualization, fine-tuned model building and adjustment. Program Refmac5 was used for the refinement of the structures.35 Crystallographic and refinement statistics are listed in Table 3.
Structure analysis and generation of Figures
The quality of the structures was assessed using the program PROCHECK.36 Intra and intermolecular contacts in various crystal structures were analyzed using the CCP4 suite of program CONTACT.32 Figures were prepared using the program Pymol† .
Protein Data Bank accession codes
The coordinates and associated structure factors have been deposited into the RCSB Protein Data Bank (PDB codes 2H9H, 2H6M and 2HAL for the HAV 3C complexes with inhibitors Ia, Ib and II complexes, respectively).
Acknowledgements
We thank Bruce Malcolm (Schering-Plough, USA) and Rajendra P. Jain (University of Alberta) for helpful discussions. X-ray diffraction data were collected at the beamline 8.3.1 of the Advanced Light Source (ALS) at the Lawrence Berkeley Lab, under an agreement with the Alberta Synchrotron Institute (ASI). The ALS is operated by the Department of Energy and supported by the National Institutes of Health. Beamline 8.3.1 was funded by the National Science Foundation, the University of California and Henry Wheeler. The ASI synchrotron access program is supported by grants from the Alberta Science and Research Authority (ASRA), the Alberta Heritage Foundation for Medical Research (AHFMR) and Western Economic Diversification (WED). This research is funded by the Canadian Institute of Health Research (CIHR), the Canada Research Chairs Program (to J.C.V. and M.N.G.J.), the Protein Engineering Centres of Excellence (PENCE), and the Natural Sciences and Engineering Research Council of Canada (NSERC). J.Y. is a recipient of the Izaak Walton Killam memorial postdoctoral fellowship at the University of Alberta. C.H. is a recipient of an NSERC postgraduate scholarship. M. N. G. J. holds the Canada Research Chair in Protein Structure and Function and J.C.V. holds the Canada Research Chair in Bioorganic and Medicinal Chemistry.
Edited by I. Wilson
Footnotes
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.jmb.2006.06.047
Appendix A. Supplementary data
Supplementary Fig. 1.
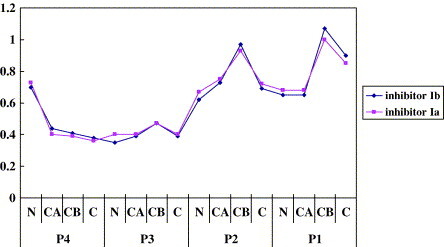
The distances between corresponding atoms in inhibitors Ia, Ib and II. The structures of HAV 3C complexed with inhibitor Ia and Ib were aligned against 3C-inhibitor II using program ALIGN. Only the main chain atoms plus Cβ are included. Y-axis values are in Å
References
- 1.Raccaniello V.R. In: Fields Virology. 4th edit. Knipe D.M., Howley P.M., Griffin D.E., Martin M.A., Lamb R.A., Roizman B., Straus S.E., editors. Lippincott Williams and Wilkins; Philadelphia, PA: 2001. Picornaviridae: the viruses and their replication; pp. 685–722. [Google Scholar]
- 2.Palmenberg A.C. Proteolytic processing of picornaviral polyprotein. Annu. Rev. Microbiol. 1990;44:603–623. doi: 10.1146/annurev.mi.44.100190.003131. [DOI] [PubMed] [Google Scholar]
- 3.Yang H., Yang M., Ding Y., Liu Y., Lou Z., Zhou Z., et al. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc. Natl Acad. Sci. USA. 2003;100:13190–13195. doi: 10.1073/pnas.1835675100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thompson D., Muriel P., Russell D., Osborne P., Bromley A., Rowland M., et al. Economic costs of the foot and mouth disease outbreak in the United Kingdom in 2001. Revue Scientifique et Technique, Office International des Épizooties. 2002;21:675–687. doi: 10.20506/rst.21.3.1353. [DOI] [PubMed] [Google Scholar]
- 5.Imperiali B., Abeles R.H. Inhibition of serine proteases by peptidyl fluoromethyl ketones. Biochemistry. 1986;25:3760–3767. doi: 10.1021/bi00361a005. [DOI] [PubMed] [Google Scholar]
- 6.McMurray J.S., Dyckes D.F. Evidence for hemiketals as intermediates in the inactivation of serine proteinases with halomethyl ketones. Biochemistry. 1986;25:2298–2301. doi: 10.1021/bi00356a067. [DOI] [PubMed] [Google Scholar]
- 7.Robertus J.D., Alden R.A., Birktoft J.J., Kraut J., Powers J.C., Wilcox P.E. An x-ray crystallographic study of the binding of peptide chloromethyl ketone inhibitors to subtilisin BPN'. Biochemistry. 1972;11:2439–2449. doi: 10.1021/bi00763a009. [DOI] [PubMed] [Google Scholar]
- 8.Kreutter K., Steinmetz A.C., Liang T.C., Prorok M., Abeles R.H., Ringe D. Three-dimensional structure of chymotrypsin inactivated with (2S)-N-acetyl-l-alanyl-l-phenylalanyl alpha-chloroethane: implications for the mechanism of inactivation of serine proteases by chloroketones. Biochemistry. 1994;33:13792–13800. doi: 10.1021/bi00250a033. [DOI] [PubMed] [Google Scholar]
- 9.Angliker H., Wikstrom P., Rauber P., Stone S., Shaw E. Synthesis and properties of peptidyl derivatives of arginylfluoromethanes. Biochem. J. 1988;256:481–486. doi: 10.1042/bj2560481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Prorok M., Albeck A., Foxman B.M., Abeles R.H. Chloroketone hydrolysis by chymotrypsin and N-methylhistidyl-57-chymotrypsin: implications for the mechanism of chymotrypsin inactivation by chloroketones. Biochemistry. 1994;33:9784–9790. doi: 10.1021/bi00198a050. [DOI] [PubMed] [Google Scholar]
- 11.Bender M.L., Brubacher L.J. The kinetics and mechanism of papain-catalysed hydrolyses. J. Am. Chem. Soc. 1966;88:5880–5889. [Google Scholar]
- 12.Drenth J., Kalk K.H., Swen H.M. Binding of chloromethyl ketone substrate analogues to crystalline papain. Biochemistry. 1976;15:3731–3738. doi: 10.1021/bi00662a014. [DOI] [PubMed] [Google Scholar]
- 13.Bazan J.F., Fletterick R.J. Viral cysteine proteases are homologous to the trypsin-like family of serine proteases: structural and functional implications. Proc. Natl Acad. Sci. USA. 1988;85:7872–7876. doi: 10.1073/pnas.85.21.7872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gorbalenya A.E., Donchenko A.P., Blinov V.M., Koonin E.V. Cysteine proteases of positive strand RNA viruses and chymotrypsin-like serine proteases. A distinct protein superfamily with a common structural fold. FEBS Letters. 1989;243:103–114. doi: 10.1016/0014-5793(89)80109-7. [DOI] [PubMed] [Google Scholar]
- 15.Yin J., Bergmann E.M., Cherney M.M., Lall M.S., Jain R.P., Vederas J.C., James M.N. Dual modes of modification of hepatitis A virus 3C protease by a serine-derived beta-lactone: selective crystallization and formation of a functional catalytic triad in the active site. J. Mol. Biol. 2005;354:854–871. doi: 10.1016/j.jmb.2005.09.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Allaire M., Chernaia M.M., Malcolm B.A., James M.N. Picornaviral 3C cysteine proteinases have a fold similar to chymotrypsin-like serine proteinases. Nature. 1994;369:72–76. doi: 10.1038/369072a0. [DOI] [PubMed] [Google Scholar]
- 17.Bergmann E.M., Cherney M.M., McKendrick J., Frormann S., Luo C., Malcolm B.A., et al. Crystal structure of an inhibitor complex of the 3C proteinase from hepatitis A virus (HAV) and implications for the polyprotein processing in HAV. Virology. 1999;265:153–163. doi: 10.1006/viro.1999.9968. [DOI] [PubMed] [Google Scholar]
- 18.Bergmann E.M., Mosimann S.C., Chernaia M.M., Malcolm B.A., James M.N. The refined crystal structure of the 3C gene product from hepatitis A virus: specific proteinase activity and RNA recognition. J. Virol. 1997;71:2436–2448. doi: 10.1128/jvi.71.3.2436-2448.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morris T.S., Frormann S., Shechosky S., Lowe C., Lall M.S., Gauss-Muller V., et al. In vitro and ex vivo inhibition of hepatitis A virus 3C proteinase by a peptidyl monofluoromethyl ketone. Bioorg. Med. Chem. 1997;5:797–807. doi: 10.1016/s0968-0896(97)88649-x. [DOI] [PubMed] [Google Scholar]
- 20.Ramtohul Y.K., James M.N., Vederas J.C. Synthesis and evaluation of keto-glutamine analogues as inhibitors of hepatitis A virus 3C proteinase. J. Org. Chem. 2002;67:3169–3178. doi: 10.1021/jo0157831. [DOI] [PubMed] [Google Scholar]
- 21.Jewell D.A., Swietnicki W., Dunn B.M., Malcolm B.A. Hepatitis A virus 3C proteinase substrate specificity. Biochemistry. 1992;31:7862–7869. doi: 10.1021/bi00149a017. [DOI] [PubMed] [Google Scholar]
- 22.Anand K., Ziebuhr J., Wadhwani P., Mesters J.R., Hilgenfeld R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science. 2003;300:1763–1767. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- 23.Birtley J.R., Knox S.R., Jaulent A.M., Brick P., Leatherbarrow R.J., Curry S. Crystal structure of foot-and-mouth disease virus 3C protease. New insights into catalytic mechanism and cleavage specificity. J. Biol. Chem. 2005;280:11520–11527. doi: 10.1074/jbc.M413254200. [DOI] [PubMed] [Google Scholar]
- 24.Bode W., Turk D., Karshikov A. The refined 1.9-Å X-ray crystal structure of d-Phe-Pro-Arg chloromethylketone-inhibited human alpha-thrombin: structure analysis, overall structure, electrostatic properties, detailed active-site geometry, and structure-function relationships. Protein Sci. 1992;1:426–471. doi: 10.1002/pro.5560010402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fox D.J., House D., Warren S. Mechanisms of sulfanyl (RS) migrations: synthesis of heterocycles. Angew Chem. Int. Ed. Engl. 2002;41:2462–2482. doi: 10.1002/1521-3773(20020715)41:14<2462::AID-ANIE2462>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 26.Fox D.J., Morley T.J., Taylor S., Warren S. Selective five- and six-membered cyclic amine syntheses via capture of episulfonium ions. Org. Biomol. Chem. 2005;3:1369–13671. doi: 10.1039/b503068b. [DOI] [PubMed] [Google Scholar]
- 27.Destro R., Lucchini V.V., Modena G., Pasquato L. X-ray structures and anionotropic rearrangements of Di-tert-butyl-substituted thiiranium and thiirenium ions. A structure-reactivity relationship. J. Org. Chem. 2000;65:3367–3370. doi: 10.1021/jo991731o. [DOI] [PubMed] [Google Scholar]
- 28.Kim J.K., Bonicamp J., Caserio M.C. Thiosulfonium ions by gas-phase methylation and thiomethylation of sulfur nucleophiles. 1. Methoxymethyl cations. J. Org. Chem. 1981;46:4230–4236. [Google Scholar]
- 29.Powers J.C. vol. 4. Marcel Dekker; New York: 1977. pp. 65–178. (Chemistry and Biochemistry of Amino Acids, Peptides and Protein). [Google Scholar]
- 30.Matthews D.A., Dragovich P.S., Webber S.E., Fuhrman S.A., Patick A.K., Zalman L.S., et al. Structure-assisted design of mechanism-based irreversible inhibitors of human rhinovirus 3C protease with potent antiviral activity against multiple rhinovirus serotypes. Proc. Natl Acad. Sci. USA. 1999;96:11000–11007. doi: 10.1073/pnas.96.20.11000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee T., Cherney M.M., Huitema C., Liu J., James K.E., Powers J.C., et al. Crystal structures of the main peptidase from the SARS coronavirus inhibited by a substrate-like aza-peptide epoxide. J. Mol. Biol. 2005;353:1137–1151. doi: 10.1016/j.jmb.2005.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Collaborative Computational Project, N The CCP4 suite: programs for protein crystallography. Acta Crystallog. sect. D. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 33.Otwinowski Z., Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 34.McRee D.E. XtalView/Xfit—A versatile program for manipulating atomic coordinates and electron density. J. Struct. Biol. 1999;125:156–165. doi: 10.1006/jsbi.1999.4094. [DOI] [PubMed] [Google Scholar]
- 35.Murshudov G.N., Vagin A.A., Lebedev A., Wilson K.S., Dodson E.J. Efficient anisotropic refinement of macromolecular structures using FFT. Acta Crystallog. sect. D. 1999;55:247–255. doi: 10.1107/S090744499801405X. [DOI] [PubMed] [Google Scholar]
- 36.Laskowski R.A., MacArthur M.W., Moss D.S., Thornton J.M. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallog. 1993;26:283–291. [Google Scholar]
- 37.Wallace A.C., Laskowski R.A., Thornton J.M. LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 1995;8:127–134. doi: 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]
- 38.Schechter I., Berger A. On the size of the active site in proteases. I. Papain. Biochem. Biophys. Res. Commun. 1967;27:157–162. doi: 10.1016/s0006-291x(67)80055-x. [DOI] [PubMed] [Google Scholar]