

Abstract
The largest extant RNA genomes are found in two diverse families of positive-strand RNA viruses, the animal Coronaviridae and the plant Closteroviridae. Comparative analysis of the viruses from the latter family reveals three levels of gene conservation. The most conserved gene module defines RNA replication and is shared with plant and animal viruses in the alphavirus-like superfamily. A module of five genes that function in particle assembly and transport is a hallmark of the family Closteroviridae and was likely present in the ancestor of all three closterovirus genera. This module includes a homologue of Hsp70 molecular chaperones and three diverged copies of the capsid protein gene. The remaining genes show dramatic variation in their numbers, functions, and origins among closteroviruses within and between the genera. Proteins encoded by these genes include suppressors of RNA silencing, RNAse III, papain-like proteases, the AlkB domain implicated in RNA repair, Zn-ribbon-containing protein, and a variety of proteins with no detectable homologues in the current databases. The evolutionary processes that have shaped the complex and fluid genomes of the large RNA viruses might be similar to those that have been involved in evolution of genomic complexity in other divisions of life.
Keywords: Virus evolution, Closteroviridae, Closterovirus, Crinivirus, Ampelovirus
1. Introduction
With their 20–30 kb genomes, the largest known RNA viruses are no match for the champions of the DNA virus world whose genomes reach 1.2 Mb (Iyer et al., 2006), let alone cellular life forms. Nevertheless, RNA viruses play a prominent role in our understanding of life's origin and evolution. Because RNA is widely believed to predate DNA as the genetic material, RNA viruses could be living fossils of the primordial RNA world (Joyce, 2002, Koonin and Martin, 2005). Therefore, at least some genes of RNA viruses are likely to encode extremely ancient, perhaps, primeval proteins involved in replication and metabolism of nucleic acids. From a somewhat different, comparative-genomic perspective, large RNA viruses provide an opportunity to investigate problems of genome complexity and its evolution on a relatively modest, tractable scale.
Closteroviruses share a conserved core of genes involved in replication with other animal and plant viruses within the alphavirus-like superfamily of positive-strand RNA viruses (Dolja et al., 1994). However, closteroviruses stand alone in regard to their genetic capacity and variability, as well as the unique morphology of their particles. It is instructive to compare the genera Closterovirus and Tobamovirus that both belong to the alphavirus-like superfamily and share the helical virion architecture (Fig. 1 ). All tobamoviruses have ∼6.5 kb genomes that code for four proteins, one of which assembles to form the rod-shaped virion (Fig. 1B and C). In contrast, the size of closterovirus genomes varies from ∼15.5 to ∼19.5 kb with a coding capacity of 10–14 proteins (Fig. 1, Fig. 2 ). Filamentous virions of closteroviruses incorporate at least five proteins that are assembled into a long body of uniform morphology and a short segmented tail (Fig. 1C). Thus, the larger amount of genetic material in closteroviruses translates into the increased structural complexity and genetic variation among individual viruses.
Fig. 1.

Comparison of the genetic and structural complexity of Beet yellows virus (BYV), genus Closterovirus, and Tobacco mosaic virus (TMV), genus Tobamovirus. (A) Genome map, functions, and evolutionary connections of BYV. The ORFs are shown as cylinders with associated protein designations. L-Pro, leader proteinase; MET, HEL, and POL, methyltransferase, RNA helicase, and RNA-dependent RNA polymerase domains of the replicase, respectively; p6, a 6-kDa protein; Hsp70h, a Hsp70-homologue; p64, a 64-kDa protein; CPm and CP, the minor and major capsid proteins, respectively; p20 and p21, the 20 and 21-kDa proteins, respectively. The sequence similarity between CP, CPm, and the C-terminal domain of p64 is illustrated with the same color. The protein functions are indicated above and below the diagram, while the evolutionary conservation patterns are shown at the bottom. (B) Genome map, functions, and evolutionary connections of TMV. P30, a 30-kDa protein. Other designations are the same as in (A). (C) Cartoons of the TMV and BYV virions. The TMV virions are rigid helical rods of ∼300 nm in length and 18 nm in diameter. The BYV virions are flexuous, helical filaments of ∼1400 nm in length and 12 nm in diameter with the ∼100 nm-long tails that are ∼8 nm in diameter. The protein composition and the length of the encapsidated viral RNA is shown.
Fig. 2.
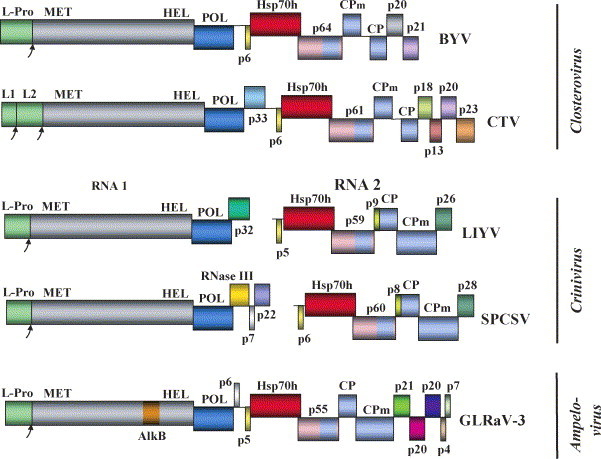
Genome maps of the selected representatives of the family Closteroviridae. The genera names are shown at the left. BYV, Beet yellows virus (Agranovsky et al., 1994); CTV, Citrus tristeza virus (Karasev et al., 1995); LIYV, Lettuce infectious yellows virus (Klaassen et al., 1995); SPCSV, Sweet potato chlorotic stunt virus (Kreuze et al., 2002); GLRaV-3, Grapevine leafroll-associated virus-3 (Ling et al., 2004). The designations and color code for the conserved proteins are the same as in Fig. 1A. L1 and L2, tandem of the leader proteinases in CTV. The unique proteins in each genome are shown in colors as dissimilar as possible with their approximate molecular weight following common ‘p’ designator, except for RNAse III and AlkB domain (see the text for further information).
Here we explore the gene repertoire of the family Closteroviridae in an attempt to reconstruct the evolutionary history of these viruses. We also use the available information on the gene functions and regulation to propose a working model of the infection cycle for a ‘generic’ closterovirus.
2. Replication-related genes
As is the case for all positive-strand RNA viruses, putative components of the closteroviral RNA replicase are expressed directly from the virion RNA (Karasev et al., 1989). The product of the 5′-terminal open reading frame (ORF) contains RNA methyltransferase (MET) and RNA helicase (HEL) domains. The RNA-dependent RNA polymerase (POL) is encoded by a downstream ORF that is presumably expressed via a +1 translational frameshift (Agranovsky et al., 1994, Karasev et al., 1995). Thus, translation of the genomic RNA results in two large polyproteins, one spanning MET–HEL, and the other one encompassing MET–HEL–POL; this second, larger polyprotein is produced in much smaller quantities due to the low frequency of frameshifting. The MET–HEL–POL module of closteroviruses is universally conserved in the entire alphavirus-like superfamily (Fig. 1A and B) (Koonin and Dolja, 1993).
A peculiar feature of the closteroviral replicases is the presence of a large variable region between the MET and HEL domains. This region is processed by an unknown mechanism to produce separate MET- and HEL-containing products (Erokhina et al., 2000). Because ectopic expression of the MET–HEL region in plants is sufficient to generate these separate products, the processing could involve either a cryptic proteolytic activity within the MET–HEL polyprotein itself, or a cellular protease (Peremyslov and Dolja, unpublished data).
In addition to their enzymatic functions, RNA replicases of alphavirus-like viruses direct assembly of the replication compartments. The principal component of these compartments is a protein shell formed by the MET–HEL protein subunits (Schwartz et al., 2002). This shell is enveloped by a membrane derived from the endoplasmic reticulum (ER) or other endomembrane. In accord with this paradigm, we found that closterovirus infection results in a drastic remodeling of the ER network that is transformed into a large vesicular factory of viral RNA. We also found that transient expression of the MET–HEL polyprotein alone is sufficient to restructure ER (Prokhnevsky et al., unpublished data). As proposed by Schwartz et al. (2002), one or a few POL-containing replication complexes are present within each virus-induced vesicle to amplify the viral genome and to produce subgenomic messenger RNA (sgRNAs).
The extreme 5′-terminal region of the closterovirus RNA encodes the papain-like leader proteinase (L-Pro) that is released from the polyprotein via autoprocessing (Agranovsky et al., 1994). Even though papain-like proteinases are common among alphavirus-like viruses, their typical role in the viral life cycle is distinct from that of L-Pro. The papain-like proteases of alphaviruses, rubiviruses, or tymoviruses are located between the replicase domains and are responsible for multiple processing events within the polyprotein (Dougherty and Semler, 1993). In contrast, the closterovirus L-Pro is located upstream of the replicase polyprotein and is not required for its processing (see above).
In addition to autocatalytic processing, L-Pro plays a prominent role in the amplification of the viral genome: elimination of L-Pro reduces the amount of viral RNA to ∼0.1% of the wild-type level (Peng and Dolja, 2000). Because L-Pro co-localizes with the replication vesicles (Zinovkin et al., 2003), its function in RNA amplification could involve either activation of the viral replicase or protection of the RNA from degradation by a host defense system.
Some closteroviruses possess a tandem of the leader proteinases that probably evolved via gene duplication. As was found using the gene swapping approach, one of these tandem proteinases (L1), but not the other (L2), can functionally substitute for a single L-Pro by enhancing virus RNA amplification (Peng et al., 2001). When expressed together, L1 and L2 act synergistically to provide for even higher levels of viral RNA. Interestingly, proliferation of the papain-like leader proteinases is also observed in some viruses of the order Nidovirales that includes the family Coronaviridae (Gorbalenya et al., 2006). This evolutionary convergence might reflect common requirements posed by the increase in the size of the viral genomes and encoded polyproteins (Peng et al., 2002).
Another feature shared by closteroviruses and nidoviruses is expression of the multiple internal genes via a nested set of the 3′-coterminal subgenomic (sg) RNAs. Usually, these sgRNAs are functionally monocistronic, each expressing only one protein from the 5′-proximal ORF. While many diverse positive-strand viruses produce from one to three subgenomic sgRNAs (Miller and Koev, 2000), nidoviruses make up to 9, and closteroviruses up to 10 sgRNAs. Although the details of transcription mechanisms may vary among the viruses of the order Nidovirales, they share certain important features (Gorbalenya et al., 2006). The short, AU-rich, transcription-regulating sequences (TRS) are present upstream from the 5′-terminal and all internal ORFs. The TRSs are thought to mediate termination of the minus-strand synthesis to produce templates for the positive-strand sgRNAs. In arteriviruses and coronaviruses, termination at the TRS of each internal ORF is followed by the template ‘jumping’ such that transcription complex lands to 5′-proximal TRS and copies the part of the 5′-leader region. This discontinuous transcription mode resulting in a mosaic structure of the sgRNAs is a unique and remarkable feature of the nidovirus genome expression mechanism (Gorbalenya et al., 2006, Pasternak et al., 2001, Sawicki et al., 2001, Zuniga et al., 2004).
The closteroviruses do not employ discontinuous transcription and have no TRS-like, common element in the regions that direct sgRNAs synthesis (Karasev et al., 1997, Kreuze et al., 2002, Peremyslov and Dolja, 2002). It is not known if, similar to smaller alpha-like viruses, closteroviral sgRNA synthesis involves internal initiation using genome-size, negative-strand template (Miller and Koev, 2000). In fact, occurrence of the minus strands of sgRNAs in closterovirus-infected cells (Dolja et al., 1990, Hilf et al., 1995) is better compatible with the nidovirus-like, minus-strand termination model. Additional support for this model comes from extensive analysis of the CTV-specific RNA species. It was found that the controller elements located upstream from each internal CTV ORF direct formation of not only the positive and negative strands of sgRNAs, but also a set of the 5′-coterminal positive-strand RNAs of subgenomic size (Gowda et al., 2001). These latter RNAs terminate at variable positions upstream of the initiation site of the corresponding sgRNAs. The simplest reconciliation of these data is that the sgRNA controller elements, which contain hairpin structures, act as terminators during transcription of both positive, and negative strands of the viral RNA. Although there were no reports of two distinct sets of subgenomic RNAs in nidoviruses, this issue seems to deserve more experimental scrutiny.
Although the functional significance, if any, of the 5′-coterminal, subgenomic-size, CTV RNAs is unknown, one may wonder if coexistence in the infected cells of the multitude of RNA species may promote formation of the recombinant RNA molecules. Indeed, plants infected with closteroviruses often contain defective RNAs (dRNAs), some of which appear to result from recombination between sgRNAs and their counterparts from the 5′-terminal region (Che et al., 2002, Rubio et al., 2000). Moreover, CTV dRNAs reminiscent of crinivirus genomic RNAs 1 and 2 were described (Che et al., 2003) suggesting that the sgRNAs may facilitate closterovirus evolution, be it genome segmentation observed in criniviruses (Klaassen et al., 1995, Liveratos et al., 2004), gene duplication (Boyko et al., 1992, Napuli et al., 2003), or recombinational accretion of the viral or cellular genes (Agranovsky et al., 1991, Kreuze et al., 2002).
Even though sgRNA controller elements of closteroviruses exhibit little or no sequence conservation within the individual virus genomes or between the related closteroviruses (Gowda et al., 2001, Kreuze et al., 2002, Peremyslov and Dolja, 2002), they likely possess common features of the higher order structures. These elements function efficiently in a heterologous genetic background, i.e., they are recognized by heterologous replicases (Peremyslov et al., 1999). The structural variability of the sgRNA controllers seems to contribute to the regulation of the level and timing of viral gene expression (Hagiwara et al., 1999, Hilf et al., 1995). On the more practical side, the promiscuity of the RNA replicases toward sgRNA controllers facilitates utilization of closteroviruses as versatile gene expression vectors. These vectors can accommodate multiple expression cassettes and efficiently produce reporters or other beneficial proteins in the infected plants (Peremyslov and Dolja, unpublished data).
It seems that the most intriguing questions pertinent to closterovirus RNA synthesis are the transcription mechanisms, the enigmatic function of the large, variable central domain of the viral replicase, and the mechanism by which L-Pro enhances RNA amplification. The answers to these questions will certainly contribute to our understanding of what does it take to replicate and express large infectious RNAs.
3. Quintuple gene block
The replication gene block of closteroviruses is rank and file of the alphavirus-like superfamily, some deviations from the prototype notwithstanding. In contrast, the five downstream genes comprise a truly unique genetic module not found outside the family Closteroviridae (Dolja et al., 1994). This quintuple gene block (QGB) codes for a ∼6-kDa hydrophobic protein (p6), an Hsp70 homologue (Hsp70h), a ∼60-kDa protein (p60), the minor capsid protein (CPm), and the major capsid protein (CP) (Fig. 1A). P6 is a single-span transmembrane protein that resides in ER and functions in virus movement from cell to cell (Alzhanova et al., 2000, Peremyslov et al., 2004b). Because p6 is not required for virus replication or assembly (Alzhanova et al., 2001, Peremyslov et al., 1998), it can be considered a conventional movement protein (MP).
CP forms a long, helical body of the flexuous, filamentous virions, and encapsidates ∼95% of the viral RNA (Fig. 1C). CPm is paralogous to CP and functions as a principal component of the short virion tail (Agranovsky et al., 1995, Tian et al., 1999). In addition to CPm, the tail assembly requires Hsp70h and p60, each of which is also an integral, although a minor tail component (Alzhanova et al., 2001, Napuli et al., 2000, Napuli et al., 2003, Peremyslov et al., 2004a, Satyanarayana et al., 2000, Satyanarayana et al., 2004). Similar to cellular molecular chaperones of Hsp70 family, closteroviral Hsp70h possess a highly conserved, N-terminal, ATPase domain and a less conserved C-terminal domain (Agranovsky et al., 1991, Bork et al., 1992). Extensive mutation analysis revealed that each of these domains is intimately involved in the virion assembly (Alzhanova et al., 2001; Prokhnevsky et al., unpublished data). It was also found that p60 possess the CP-like, virion-embedded, C-terminal domain, and the N-terminal domain, which is exposed at the virion's surface (Napuli et al., 2003). Hsp70h and p60 act cooperatively to facilitate incorporation of CPm and to define the proper tail length (Satyanarayana et al., 2004; Alzhanova et al., unpublished data). The tail encapsidates the 5′-terminal, ∼700 nt-long, RNA region (Fig. 1C) that contains a packaging signal recognized by CPm (Peremyslov et al., 2004a, Satyanarayana et al., 2004). Interestingly, virion bodies and tails can be assembled independently of each other.
Atomic force microscopy of Beet yellows virus (BYV, genus Closterovirus) virions showed that the tails are narrower than the bodies and exhibit three-segment structure (Peremyslov et al., 2004a). A pointed tip segment of the BYV tails contains a 20-kDa protein (p20) (Fig. 1C) that is dispensable for incorporation of other virion components. Inactivation of p20 results in shorter tails and apparently normal bodies. Because the orthologs of BYV p20 are not found in other closteroviruses, it is not clear if the tail segmentation pattern is a common feature of the family.
A salient functional aspect of the BYV virions is that each of the five structural proteins is also required for virus transport (Dolja, 2003). In the case of CP, this requirement could stem, simply, from the need to protect the long and degradation-prone viral RNA during its transport. Indeed, the cell-to-cell movement cycle of a closterovirus takes a day compared to two hours for TMV. However, it seems unlikely that the transport-related function of the tail is merely protective. First, CP can encapsidate the entire genome if the tail formation is prevented by mutations (Alzhanova et al., 2001). Second, the tail harbors Hsp70h, the only viral protein that was found in plasmodesmata (PD), intercellular channels through which viruses translocate (Medina et al., 1999). Third, p20 is dispensable for virion assembly and cell-to-cell movement, but is required for long-distance transport of BYV through the vascular system (Prokhnevsky et al., 2002). All this prompted interpretation of the tail as a device that evolved to facilitate cell-to-cell and systemic transport of the large closterovirus genomes (Dolja, 2003). It should be noted that this concept awaits experimental separation of assembly and transport functions for at least some tail proteins unless there is a causal relationship between the two.
Even though the QGB genes are universally conserved within the family, their order is not. In the genera Ampelovirus and Crinivirus, the order of CPm and CP is reversed, and the CPm is much larger than in the genus Closterovirus (Fig. 2) (Klaassen et al., 1995, Kreuze et al., 2002, Ling et al., 2004, Melzer et al., 2001). Other examples of QGB variation include duplication and divergence of the CPm gene in Grapevine leafroll-associated virus-1 (Fazeli and Rezaian, 2000), reshuffling of QGB in Little cherry virus (Theilmann et al., 2002), and occurrence of enigmatic additional ORFs within QGB of criniviruses and certain ampeloviruses.
4. Suppressors of RNA silencing
It has been long accepted that all non-defective plant viruses must code for at least three functions: genome replication, encapsidation, and transport within infected plants. Arguably, suppression of RNA silencing has emerged recently as a fourth universal function encoded in plant viruses (Baulcombe, 2004). Some plant viruses possess dedicated suppressors, while others delegate this function to replicational, structural, or transport proteins (Silhavy and Burgyan, 2004, Voinett, 2005). Interestingly, most of the dedicated viral suppressors represent small protein families without detectable homologous relationship to any other viral or host proteins. This tendency is often taken as evidence of relatively recent origins of the suppressors that evolved to counteract a powerful system of host defense against parasitic RNAs. The major components of RNA silencing machinery are conserved between diverse eukaryotes suggesting that this machinery emerged not much later than eukaryotes themselves (Anantharaman et al., 2002, Zamore and Haley, 2005). Search for RNA silencing suppressors in closteroviruses yielded several findings that further highlighted the trends in suppressor evolution.
Screening of the BYV genome for silencing suppressor functions showed that the 21-kDa protein (p21) is a suppressor of RNA silencing and that the orthologous proteins from other viruses of the genus Closterovirus also possess suppressor activity (Chiba et al., 2006, Reed et al., 2003). It has been found that p21 specifically binds double-stranded forms of the small interfering RNA (siRNA) or microRNA (miRNA) (Chapman et al., 2004). Thus, the molecular mechanism of p21 action is similar to that of another well-studied suppressor, p19 of Tomato bushy stunt virus (Silhavy and Burgyan, 2004): both suppressors sequester small RNA effectors of silencing and prevent their loading into the RNA-Induced Silencing Complex (RISC). However, these suppressors cannot discriminate between siRNAs that target viral genomes and endogenous siRNAs and miRNAs that are involved in regulation of plant development. As a result, accumulation of p21 or p19 in plants induces developmental abnormalities many of which are identical to symptoms of viral infection (Chapman et al., 2004). It was concluded that, at least in part, viral pathogenicity is due to interference of silencing suppressors with developmental function of plant small RNAs. Despite their mechanistic similarity, p21 and p19 appear to be structurally and evolutionarily unrelated and neither has detectable homologues outside the respective virus genera (Vargason et al., 2003, Ye and Patel, 2005).
Although Citrus tristeza virus (CTV) encodes p20, a p21-like suppressor of RNA silencing, screening of the CTV genome revealed an additional suppressor, p23, that has no homologues in other closteroviruses (Fig. 2) (Lu et al., 2004). Subsequent computer analysis of the p23 sequence showed that it has a specific form of the Zn-ribbon module that is present in a variety of cellular proteins and also in several proteins encoded by viruses of the genera Carlavirus and Vitivirus (Chiba et al., 2006). It seems likely that the latter viral proteins are distant homologues of p23. Should it be demonstrated that these proteins are suppressors of RNA silencing, this will be the first viral suppressor family represented in diverse viruses and, possibly, derived from a common host ancestor. It was also found that both CTV p20 and CP can interfere with the systemic spread of silencing, while p23 can only suppress the local silencing (Lu et al., 2004). Thus, CTV evolved a complex system of RNA silencing suppression with three components targeting distinct facets of RNA silencing response.
Another remarkable discovery in the area of closterovirus counter-defense is a two-component RNA silencing suppression system in Sweet potato chlorotic stunt virus (SPCSV) (Kreuze et al., 2005). One of the components of this system is a 22-kDa protein (p22) that possesses suppressor activity on its own (Fig. 2). This protein is unique to SPCSV and has no detectable sequence similarity to other proteins. A second component is an RNase III homologue containing both the endonuclease and the dsRNA-binding activities typically found in bona fide cellular RNases III including Dicer and Dicer-like proteins, which are involved in the generation and functioning of siRNAs and miRNAs. In addition to SPCSV, RNase III is encoded by several nucleocytoplasmic large DNA viruses (Iyer et al., 2006) where its function remains to be determined. By itself, SPCSV RNase III exhibits no suppressor activity. However, co-expression of RNase III with p22 results in a dramatic increase of RNA silencing suppression (Kreuze et al., 2005). Although the mechanism of this suppression is not known, recruitment of a protein whose homologues function in eukaryotic RNA silencing for the purpose of overcoming host defense is a remarkable example of viral resourcefulness.
It seems likely that the analysis of additional closteroviruses will yield more examples of multi-component and multilevel counter-defense systems. What could be the reason for multiplication of diverse suppressors in closteroviruses as opposed to single suppressors of smaller viruses? One possibility is that the large and slowly replicating closterovirus RNAs are more prone to degradation by the RNA silencing machinery than those of the smaller viruses. Alternatively, the increased complexity of the closterovirus genomes could be of direct relevance. Given that the antiviral siRNAs in infected plants are mostly derived from hairpin elements of the positive RNA strands (Molnar et al., 2005), controller element-rich closteroviral RNAs are likely to harbor more hairpins than the simpler genomes of other viruses.
5. Other variable genes
The closterovirus genomes are peppered with genes whose products, typically, have no identifiable orthologs in other viruses (Fig. 2). Except for BYV p20, the functions of these enigmatic proteins remain to be elucidated. Perhaps, the most illuminating example of this kind is the AlkB domain found in several plant viruses (Aravind and Koonin, 2001). Recent analysis identified AlkB domains in the polyproteins of two ampeloviruses, Grapevine leafroll-associated virus-3 (Fig. 2) and Little cherry virus, as well as in ∼20 other alpha-like plant viruses, which share filamentous particle morphology and belong to genera Allexivirus, Carlavirus, Foveavirus, Potexvirus, Trichovirus, and Vitivirus (Koonin, personal communication). In all these cases, AlkB domains are located within the multidomain viral replicases upstream of the RNA helicase domains.
Interestingly, in each of these genera, only a minority of the viruses encode AlkB domain, suggesting that it has been acquired relatively recently via horizontal gene transfer. The function of AlkB in viral infection is unknown, but it seems likely that, similarly to certain cellular AlkBs, it might be involved in RNA repair via methylation reversal (Aas et al., 2003). The fact that the majority of the AlkB-encoding viruses infect either woody or perennial plants is compatible with this hypothesis. Indeed, the extent of methylation damage is likely to be exacerbated by the prolonged virus propagation within the same infected organism and the need to be transported across long distances in the hostile phloem environment.
6. The infection cycle of a closterovirus
The available functional information provides for a relatively detailed reconstruction of the events in a closterovirus infection. In nature, most closteroviruses are inoculated into plants by insect vectors (Karasev, 2000). The initiation of the replication cycle upon virus entry into the cell requires virion disassembly followed by translation of the input viral genome. Association of the virion tail with the capped, 5′-terminal, RNA region suggests a role of the tail proteins in the RNA exposure to translation initiation machinery of the cell. As originally proposed for TMV, subsequent disassembly of the viral particle and liberation of the RNA could be mediated by the elongating ribosomes. The translated polyprotein is processed to yield L-Pro and replicase components that recruit ER and viral RNA to the membrane-bound, replication complexes. The genome-size RNA minus strand is made and used for synthesis of the genome plus strands and sgRNAs, which are exported to the cytosol. The timing and amounts of the synthesis of sgRNAs are regulated to optimize subsequent events of the virus infection cycle. For instance, sgRNAs that encode RNA silencing suppressors are produced early in infection to counteract a potent host defense system. Translation of the nascent genomes results in proliferation of replication complexes concurrent with virion assembly that is triggered by accumulation of the structural proteins.
Interestingly, the apparently normal virion tails can be formed in the absence of virion body assembly (Satyanarayana et al., 2004), while inactivation of the tail formation results in encapsidation of the entire RNA by CP (Alzhanova et al., 2001). These findings suggest that the body assembly follows the faster process of tail formation. Although CPm alone can encapsidate long stretches of RNA, assembly of the functional virion tails of the proper length requires cooperative action of CPm, Hsp70h, and p60 (Satyanarayana et al., 2004). Dispensability of BYV p20 for incorporation of other tail components suggests that this protein is added to preformed tails, possibly, via interaction with Hsp70h (Peremyslov et al., 2004a, Prokhnevsky et al., 2002).
It seems that the major reason for accumulation of large virion masses in the infected cells is to promote closterovirus acquisition and transmission by insect vectors. However, a subpopulation of the virions must be engaged in the processes of cell-to-cell movement via PD and systemic transport via the phloem. Because incorporation of each of the five virion proteins is required for one or both of these processes to occur, at least some of the transport events should involve physical translocation of the mature virions. Our understanding of the mechanistic contributions of the transport proteins remains patchy at best. It is not known how the ER-associated p6 aids the viral cell-to-cell movement. It is also not clear if CPm and p60 have specific roles in virion transport in addition to their requirement for the tail assembly. In contrast, the mounting evidence suggests that Hsp70h plays a pivotal role in virion translocation to and through PD.
It has been shown that Hsp70h accumulates in PD of the BYV-infected cells and can be targeted to PD in the absence of other viral components (Medina et al., 1999, Prokhnevsky et al., 2002). Moreover, this autonomous targeting has been shown to rely on the actomyosin motility system (Prokhnevsky et al., 2005). It is tempting to propose that one of the roles of Hsp70h present in the virion tails is to interact with myosin and chaperone the virions toward PD. This would provide an added benefit of directionality, with the virion tails put in close proximity or even inserted into PD channels. Although the form in which closteroviral genome traverses PD is not known, the apparent lack of CP within PD might suggest that virions are (partially) disassembled upon passing PD channels (Medina et al., 1999). If this is the case, tail proteins may provide for directional RNA transport, with the 5′-terminal region entering the adjacent cell and being recruited by ribosomes. The rest of the RNA could be pulled into the cytoplasm co-translationally. This speculative model is not without analogy that comes from the nuclear export of giant, ∼30 kb mRNAs of certain fly species (Daneholt, 2001). These mRNAs are co-transcriptionally assembled into the ribonucleoprotein particles that dock to the nuclear pores with their ends corresponding to the mRNA's 5′-termini. The directional transport through the pore occurs concomitantly with particle thinning and shedding some of the associated proteins. Upon entering the cytoplasm, polysomes are formed and translation begins.
The cycles of genome expression and replication, virion assembly and cell-to-cell movement, each of which takes ∼1 day, are repeated until infection reaches companion cells that have PD connection to phloem sieve elements. Loading to sieve elements allows virus to be transported long distances with the liquid flow and to unload to sink tissues of stems, leaves, and roots. In all likelihood, multiple cycles of the virus replication on the phloem route are required to ensure successful colonization of the entire plant. Two proteins that play specific roles in systemic transport of BYV are L-Pro and p20 (Peng et al., 2003, Prokhnevsky et al., 2002). Because L-Pro is a replicational enhancer, it is plausible that it aids RNA accumulation in the phloem cells. The location of p20 at the tail tip suggests a possible role in either stabilizing virions in the phloem, or guiding them through PD interconnecting sieve elements with companion cells, or both.
The onset of systemic disease takes at least 2 weeks for closteroviruses of herbaceous plants and up to a few months for those infecting woody hosts. Although the mechanisms of closteroviral pathogenicity are not entirely understood, the preferential replication in the phloem must play an important role. The massive recruitment of the ER to viral replication complexes might interfere with the normal ER function and cause phloem cytotoxicity manifested in such symptoms as vein clearing, or leaf yellowing, rolling, and wilting. The virus-coded suppressors of RNA silencing also contribute to disease phenotype by interfering with microRNA-mediated developmental processes.
The data on the last phase of the closterovirus infection cycle, the semi-persistent insect transmission, are extremely scarce (Karasev, 2000). It appears that isolated virions of at least some closteroviruses can be transmitted to plants by insect membrane feeding, meaning that virion proteins are sufficient for the process to occur. It has been proposed that the large CPm of criniviruses mediates the whitefly transmission (Tian et al., 1999), while preliminary results implicated p20 in the aphid transmission of BYV (Prokhnevsky and Dolja, unpublished data). However, slow compared to other plant viruses, the life styles of closteroviruses proved evolutionarily successful given the long and rapidly growing list of closteroviruses that infect a broad range of hosts from cucumbers to strawberries to grapevine to citrus (Aguilar et al., 2003, Martelli, 2002, Tzanetakis et al., 2005a, Tzanetakis et al., 2005b).
7. Comparative genomics and phylogenetic reconstructions
Closterovirus genome sequencing produced a genetic tapestry illustrated in Fig. 2. Among the positive-strand RNA viruses, complexity and diversity comparable to those of closteroviruses are seen only in the order Nidovirales. Because nidoviruses and closteroviruses belong to distinct evolutionary lineages, we assume that their genomes evolved independently via gene accretion by the simpler respective progenitors. An alternative to this scenario, namely, the origin of the smaller viral genomes from a very complex common ancestor, which possessed most of the genes now seen among positive-strand RNA viruses, via gradual, lineage-specific gene loss, appears to be much less plausible, if realistic at all.
To gain insight into closterovirus evolution, we conducted phylogenetic analysis of the several proteins conserved in all extant family members. The RNA-dependent RNA polymerase is the only protein universally present in all positive-strand RNA viruses (Koonin, 1991). As expected, our analysis confirmed the relationship of closteroviral polymerases with those of the other viruses from the alphavirus-like superfamily, as well as the separation of the closteroviral polymerases into three clades corresponding to the currently recognized genera (Fig. 3A). The viruses of the genera Closterovirus, Crinivirus, and Ampelovirus are predominantly transmitted by aphids, whiteflies, and mealybugs, respectively. This correlation between the polymerase phylogeny and the type of insect vectors likely reflects vector adaptation as a driving force behind divergent evolution of closteroviruses (Karasev, 2000).
Fig. 3.

Phylogenetic analysis of the four conserved closterovirus proteins and their homologues. (A) Dendrograms for the core domains of the RNA dependent RNA polymerases of closteroviruses, three animal viruses of the alphavirus-like superfamily, and SARS virus as an outgroup. Numbers indicate the number of bootstrap replicates supporting the corresponding branches; the branches with values less that 700 were collapsed. The scale bar indicates the estimated number of amino acid substitutions per site. Three genera of the family Closteroviridae are highlighted with the color-coded ellipsoids. GLRaV-2, Grapevine leafroll-associated virus-2 (Zhu et al., 1998); CYSDV, Cucurbit yellow stunting disorder virus (Aguilar et al., 2003); GLRaV-1, Grapevine leafroll-associated virus-1 (Fazeli and Rezaian, 2000); PMWaV-2, Pineapple mealybug wilt-associated virus-2 (Melzer et al., 2001); other designations are as in Fig. 2. (B) Dendrograms for the papain-like proteinase domains of the closterovirus leader proteinases. CTV and GLRaV-2 each possess two leader proteinases, L1 and L2. EAV-nsp1, the nonstructural protein-1, a papain-like leader proteinase of Equine arteritis virus used as outgroup. (C) Dendrograms for the Hsp70 homologues from the closteroviruses, the mimivirus, prokaryotes, and eukaryotes. (D) Dendrograms for the CP-like domains present in closterovirus CPs, CPms, and p60s. GLRaV-1 encodes two divergent variants of CPm, 1 and 2. PVA-CP, capsid protein of Potato virus A, an outgroup.
Sequences alignment and phylogenetic analysis were performed using the Clustal X 1.8 program (Thompson et al., 1997) and MEGA 3.1 (Kumar et al., 2004). Trees were generated using the Neighbour-Joining method (Saitou and Nei, 1987) with correction for multiple substitutions and 1000 bootstrap replicates. In all cases, similar trees and bootstrap values were obtained using the maximum parsimony method (Fitch, 1977) (not shown). The NCBI accession numbers for closteroviruses were as follows. GLRaV-3, NC004667; PMWaV-2, AF283103; GLRaV-1, AF195822; BYV, NC001598; CTV, NC001661; GLRaV-2, NC007448; LIYV, NC003617 and NC003618; SPCSV, NC004123 and NC004124; CYSDV, NC004810 and NC004809. The additional viral sequences used were NP056786 of Hepatitis E virus, NC001545 of Rubella virus RdRp, NC001547 of Alphavirus, NC004718 of SARS virus, NP705583 of Equine arteritis virus, NP734368 of Potato virus A. The Hsp70 sequences were: XP470141, XP475261, AAO17017 for O. sativa; CAA05547, NP198206, NP199802, CAB89371 for A. thaliana; NP646617 for D. discoideum; NP002146, P11021, P38646 for H. sapiens; YP 142608, AAV50662 for A. polyphaga mimivirus; NP492485 for C. elegans; Q8YW74 for Nostoc spp.; NP414555 for E. coli; NP454622 for S. enterica; NP794258 for P. syringae; XP762364 for U. maydis.
Analysis of the closteroviral L-Pro sequences also revealed clades corresponding to the three genera (Fig. 3B) indicating that RNA polymerases and proteases coevolved since the divergence of closteroviruses from the common family ancestor. The second leader proteases of CTV and GLRaV-2 (L2 in Fig. 3B) clustered with the other proteases of the same genus in accord with their proposed origin by lineage-specific duplication of the L-Pro gene (Peng et al., 2001).
The topology of the phylogenetic tree for closterovirus Hsp70h sequences also mimicked that of the RNA polymerase tree (Fig. 3C), supporting the presence of Hsp70h in the common ancestor of closteroviruses. In a broader perspective, closteroviral homologues showed no specific affinity to cellular Hsp70s from any particular group of organisms or compartments of the eukaryotic cell (Fig. 3C) (Boorstein et al., 1994). This result is indicative of the rapid evolutionary divergence due to the functional specialization of Hsp70h within the viral genetic context. The only viruses besides closteroviruses that encode Hsp70s are the mimiviruses of the protozoan Acanthamoeba (Raoult et al., 2004). Interestingly, both of the mimiviral Hsp70s cluster with their eukaryotic kin suggesting their relatively recent origin from the host and/or slower evolution (Fig. 3A). In all likelihood, closteroviruses and mimiviruses acquired Hsp70 genes independently.
The major CPs of closteroviruses belong to the protein family that includes CPs of all filamentous viruses of plants (Dolja et al., 1991). However, unlike other filamentous viruses, closteroviruses encode at least two additional proteins possessing CP-related domains, CPm and p60 (Boyko et al., 1992, Napuli et al., 2003). The phylogenetic analysis of these proteins demonstrates clear separation of the three p60 clades corresponding to the family genera (Fig. 3D). Similarly to what was found for L-Pro and Hsp70h, this supports p60 presence in the common ancestor of extant closteroviruses.
In contrast, CPms of the viruses of the genera Closterovirus and Crinivirus cluster with their respective CPs (Fig. 3D). This tree topology suggests that CPms have evolved independently in these two genera via duplication of the CP gene. This could also be the case for the genus Ampelovirus, but the affinity between ampeloviral CPms and CPs is less significant statistically, probably indicating faster evolution of CPms in this genus. This notion is also compatible with the fact that in one of the ampeloviruses, GLRaV-1, the CPm gene underwent an additional duplication (Fazeli and Rezaian, 2000).
Using the results of phylogenetic analysis, we developed the following scenario for the evolution of the family Closteroviridae (Fig. 4 ). We propose that the monophyletic closterovirus lineage descends from a plant virus of the alphavirus-like superfamily that possessed a typical MET–HEL–POL replicase, a p6-like movement protein, and a single CP forming a filamentous capsid. This progenitor evolved into the last common ancestor of closteroviruses by acquiring several new genes. Among these, acquisition of the L-Pro gene and a central domain of replicase facilitated RNA replication and conditioned expansion of the progenitor's genome. Concurrently, the Hsp70 gene was incorporated via recombination with a cellular mRNA, whereas the p60 gene originated via duplication and divergence of the CP gene. The co-evolution of Hsp70h and p60 resulted in emergence of a prototype virion tail that empowered cell-to-cell transport of the expanded genome of the ancestral closterovirus.
Fig. 4.
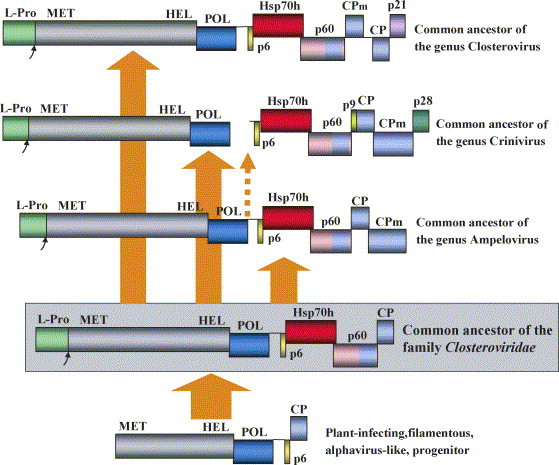
Hypothetical evolutionary scenario for the family Closteroviridae. The solid arrows show direct vertical descent. An alternative ancestry of the criniviruses by segmentation of an ancestral ampeloviral genome is shown as a dashed arrow.
Because most of the extant closteroviruses are insect-transmissible, it seems reasonable to propose that the closterovirus ancestor was also transmitted by an insect. The three current lineages of closteroviruses evolved by adaptation to the distinct types of insect vectors (Karasev, 2000). The CPms appeared at that time via duplication and divergence of the CP gene. In addition to CPm phylogenenies (Fig. 3D), this scenario is supported by the different order of CP and CPm genes in the genus Closterovirus versus the genera Crinivirus and Ampelovirus (Fig. 2). The latter two genera could arise independently from the common family ancestor (Fig. 4). Alternatively, criniviruses could evolve by the split of a single ampeloviral genomic RNA to two segments (dashed arrow in Fig. 4). The same order of the CP and CPm genes and the larger size of CPms in ampeloviruses and criniviruses compared to closteroviruses are compatible with the latter scenario.
Interestingly, evolution subsequent to emergence of CPms produced very few additional genes that would be specific to and conserved within each genus. One example is the p21-like suppressor of RNA silencing in the viruses of the genus Closterovirus. Another example is provided by the p8-like and p28-like proteins that are conserved in genus Crinivirus (Fig. 5 ). The genus Ampelovirus exhibits most genetic variation in the family. Even though the replication and assembly/movement modules are present in all known representatives of this genus, they are often perturbed by incorporation of additional unique genes or changes in the gene order (Theilmann et al., 2002).
Fig. 5.

Multiple alignments of the two Crinivirus specific-proteins. (A) The p9-like proteins. (B) The p26-like proteins. Identical residues in all four proteins are shown in red, while those in two or three proteins are shown in blue. BPYV, Beet pseudoyellows virus (Tzanetakis and Martin, 2004); PYVV, Potato yellow vein virus (Liveratos et al., 2004); SPaV, Strawberry pallidosis associated virus (Tzanetakis et al., 2005b). Nucleotide accession numbers were as follows: BPYV, NC005209; PYVV, NC006063 and NC006061; SpaV, NC005896; CYSDV, see Fig. 3. Graphic representation of the alignments was done using Vector NTI program (Invitrogen).
The numerous other novel genes acquired by closteroviruses, in addition to replication module and QGB, are virus-specific. For instance, CTV genes encoding p33, p18, p13, and p23 have no homologues in BYV or other known genus members (Fig. 2) (Pappu et al., 1994). Conspiciously, similar tendency of prolifiration of the virus- or lineage-specific genes is observed in coronaviruses and other large nidoviruses (Gorbalenya et al., 2006). Because the gene repertoirs of smaller RNA viruses tend to be more conserved in any given taxon, it seems that the unusual genetic plasticity of nidoviruses and closteroviruses is conditioned by their genome expansion. The resulting benefit of faster macroevolution via accretion of new genes may provide large RNA viruses with ability to rapidly adapt to new hosts, vectors, or changing environmental conditions.
The task of establishing the time scale for the closterovirus evolution meets the major challenges common for all viruses. These challenges include the lack of paleonthological record, the relatively fast and variable pace of molecular evolution, and the complex relations between evolution of viruses and their hosts. However, comparison of the evolutionary histories of viruses and host organisms may provide some useful insights.
As has been discussed previously, each of the three superfamilies of the positive-strand RNA viruses contains viruses that infect plants, insects, and vertebrates, suggesting that these superfamilies evolved prior to separation of the animal and plant kingdoms (Koonin and Dolja, 1993). This notion is further reinforced by finding of a plethora of picorna-like viruses in the marine environment including viruses that infect diatoms (Culley et al., 2003). Regardless of the adapted eukaryotic phylogeny, the specifics of which remain debated, plants, animals, and diatoms diverged early in the evolution of eukaryotes, and horizontal transfer of picornaviruses from land plants or animals to marine biota seems highly unlikely. Therefore, the superfamilies of positive-strand RNA viruses including alphavirus-like superfamily to which closteroviruses belong might have diverged at the dawn of eukaryotic evolution.
The lack of information on RNA viruses of red and green algae or primitive land plants limits our ability to deduce the time of origin for the last common ancestor of Closteroviridae or other extant plant virus families. However, it seems that these ancestors should have been in place, at the latest, by Jurassic (150–200 million years ago), when the great diversity of the flowering plants evolved to dominate the land (Benton and Ayala, 2003, Kenrick and Crane, 1997, Palmer et al., 2004). Because this process was accompanied closely by evolution of the potential closteroviral insect vectors, such as aphids and other Homoptera (Gaunt and Miles, 2002, Martinez-Torres et al., 2001), it seems reasonable to think that diversification of the three closteroviral genera also came about near that time.
8. Conclusion: genome complexity in RNA viruses
What is the functional significance of the increase in genome complexity seen in closteroviruses relative to the simpler plant viruses? A comparison of TMV and BYV, which both evolved from the alphavirus-like ancestors, shows that the large part of the ∼9 kb genomic surplus of BYV is dedicated to facilitating the synthesis of the virion RNA and multiple sgRNAs. The rest of the surplus was invested in the formation of the complex virion tail that empowers virus transport within and transmission between the host plants and in suppression of RNA silencing (Fig. 1). If this is the case, it almost looks like the complexity of closteroviruses arose to maintain itself.
Similar comparison between the more and less complex nidoviruses, such as SARS virus and EAV, respectively, reveals an increase in the number of RNA metabolism enzymes in the former (Gorbalenya et al., 2006). In addition to enabling replication and transcription of the large viral genomes also seen in closteroviruses, further leap in complexity is thought to confer coronaviruses with a higher fidelity of RNA synthesis and a higher degree of RNA metabolism autonomy. A greater independence of the DNA metabolism from the host cell is also one of the major tendencies in the evolution of the complex DNA viruses (Iyer et al., 2006).
Why then the largest DNA viruses with the ∼1200 kb genomes are so much larger than the largest RNA viruses with the ∼30 kb genomes? The more or less obvious answer seems to lie in the chemical properties of RNA, which is generally thought to be the original enzymatic and information-storage molecule of the ancient RNA world. It is further believed that the limitations in the RNA stability and replication fidelity resulted in the evolution of DNA as the dedicated genomic material. It seems that ∼30 kb could be close to the maximum size of RNA molecules that can function as autonomous genetic elements due to minimal stability requirements. No larger RNAs are known to be used by any viruses, transposable elements, or prokaryotic cells; although eukaryotes do have some giant mRNAs, these are likely to be short-lived and translated via special mechanisms such as coupling of translation with nucleo-cytoplasmic export (Gregorio et al., 1999).
The plant hosts impose even stricter limits on the size of viral genomes, be it RNA or DNA: at 19.3 kb, the RNA genome of CTV (Fig. 2) is the largest known plant virus genome. The single most important peculiarity in the infection cycle of plant viruses is that they must rely on direct cell-to-cell movement via PD to successfully colonize a plant (Boevink and Oparka, 2005, Lucas and Lee, 2004). The idea that the PD is a bottleneck for intercellular translocation of virions or genomes has recently gained experimental support (Gilbertson et al., 2003). The corollary of the limited size and translocation capacity of the PD channels is that viral genomes larger than ∼8 kbp for dsDNA, ∼3 kb for ssDNA, or ∼20 kb for ssRNA cannot move between the cells and therefore are eliminated during evolution.
What is the driving force behind the increase in viral genome complexity? It is certainly not the host adaptation or virus–host coevolution because both simple prokaryotic and most complex eukaryotic organisms play hosts for the simple and complex viruses alike. A simple and convincing theory of the origins of genome complexity in eukaryotes has been recently advanced by Lynch and Conery (2003). This theory states that the increase in genome complexity is a stochastic result of the relieved selection pressure that follows dramatic reductions in population size; in small populations, drift may result in fixation of genomic changes, such as duplication of numerous genes and propagation of selfish elements, which are not tolerated in larger populations with intensive purifying selection. Often, the reduction in population size would be caused by local or global environmental catastrophes that lead to abrupt drop in the population size for a few or many species at a time. Support for this non-adaptationist concept of genomic complexity comes from extensive analyses of genes, introns, and selfish DNA in a large number of complete prokaryotic and eukaryotic genomes (Koonin, 2004, Lynch and Conery, 2003).
Although a similar analysis is yet to be attempted for viruses, it is tempting to apply this concept to evolution of viral genetic complexity. What would be considered an ‘environmental catastrophe’ for a virus? The most obvious answer is the drastic reduction in a population size of the host organisms. Another possibility is the emergence of a new antiviral defense system, such as innate or acquired immunity, or RNA silencing. It is, probably, the combination of both that has driven acquisition of multiple new genes by viral genomes in certain lineages, followed by the gradual adaptation of these genes for the needs of infection. A corollary of the comparative analysis of closterovirus genomes is that their relative complexity and variability go hand in hand (Fig. 2). The abundance of unique, rare, and novel genes that appear to be picked at random by different closteroviruses is compatible with the idea of genome complexity emerging as a byproduct of increased genome entropy.
Acknowledgements
The authors are grateful to Eugene V. Koonin for stimulating discussions and critical reading of the manuscript. The work in V.V.D. lab is supported by grants from the National Institutes of Health (GM053190) and BARD (IS-3784-05), and in JPTV lab by the Academy of Finland (grants 1102003 and 1102134). J.F.K. is Junior Professional Officer funded by Sida, Sweden.
References
- Aas P.A., Otterlei M., Falnes P.O., Vagbo C.B., Skorpen F., Akbari M., Sundheim O., Bjoras M., Slupphaug G., Seeberg E., Krokan H.E. Human and bacterial oxidative demethylases repair alkylation damage in both RNA and DNA. Nature. 2003;421:859–863. doi: 10.1038/nature01363. [DOI] [PubMed] [Google Scholar]
- Agranovsky A.A., Boyko V.P., Karasev A.V., Koonin E.V., Dolja V.V. Putative 65 kDa protein of beet yellows closterovirus is a homologue of HSP70 heat shock proteins. J. Mol. Biol. 1991;217:603–610. doi: 10.1016/0022-2836(91)90517-a. [DOI] [PubMed] [Google Scholar]
- Agranovsky A.A., Koonin E.V., Boyko V.P., Maiss E., Frotschl R., Lunina N.A., Atabekov J.G. Beet yellows closterovirus: complete genome structure and identification of a leader papain-like thiol protease. Virology. 1994;198:311–324. doi: 10.1006/viro.1994.1034. [DOI] [PubMed] [Google Scholar]
- Agranovsky A.A., Lesemann D.E., Maiss E., Hull R., Atabekov J.G. “Rattlesnake” structure of a filamentous plant RNA virus built of two capsid proteins. Proc. Natl. Acad. Sci. U.S.A. 1995;92(7):2470–2473. doi: 10.1073/pnas.92.7.2470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilar J.M., Franco M., Marco C.F., Berdiales B., Rodriguez-Cerezo E., Truniger V., Aranda M.A. Further variability within the genus Crinivirus, as revealed by determination of the complete RNA sequence of Cucurbit yellow stunting disorder virus. J. Gen. Virol. 2003;84:2555–2564. doi: 10.1099/vir.0.19209-0. [DOI] [PubMed] [Google Scholar]
- Alzhanova D.V., Hagiwara Y., Peremyslov V.V., Dolja V.V. Genetic analysis of the cell-to-cell movement of beet yellows closterovirus. Virology. 2000;268(1):192–200. doi: 10.1006/viro.1999.0155. [DOI] [PubMed] [Google Scholar]
- Alzhanova D.V., Napuli A., Creamer R., Dolja V.V. Cell-to-cell movement and assembly of a plant closterovirus: roles for the capsid proteins and Hsp70 homolog. EMBO J. 2001;20:6997–7007. doi: 10.1093/emboj/20.24.6997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anantharaman V., Koonin E.V., Aravind L. Comparative genomics and evolution of proteins involved in RNA metabolism. Nucleic Acids Res. 2002;30:1427–1464. doi: 10.1093/nar/30.7.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravind, L., Koonin, E.V., 2001. The DNA repair protein AlkB, EGL-9, and leprecan define new families of 2-oxoglutarate- and iron-dependent dioxygenases. Genome Biol. 2, research/007.1. [DOI] [PMC free article] [PubMed]
- Baulcombe D. RNA silencing in plants. Nature. 2004;431:356–363. doi: 10.1038/nature02874. [DOI] [PubMed] [Google Scholar]
- Benton M.J., Ayala F.J. Dating the tree of life. Science. 2003;300:1698–1700. doi: 10.1126/science.1077795. [DOI] [PubMed] [Google Scholar]
- Boevink P., Oparka K.J. Virus–host interactions during movement process. Plant Physiol. 2005;138:1815–1821. doi: 10.1104/pp.105.066761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boorstein W.R., Ziegelhoffer T., Craig E.A. Molecular evolution of the Hsp70 multigene family. J. Mol. Evol. 1994;38:1–17. doi: 10.1007/BF00175490. [DOI] [PubMed] [Google Scholar]
- Bork P., Sander C., Valencia A. An ATPase domain common to prokaryotic cell cycle proteins, sugar kinases, actin, and Hsp70 heat shock proteins. Proc. Natl. Acad. Sci. U.S.A. 1992;89:7290–7294. doi: 10.1073/pnas.89.16.7290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyko V.P., Karasev A.V., Agranovsky A.A., Koonin E.V., Dolja V.V. Coat protein gene duplication in a filamentous RNA virus of plants. Proc. Natl. Acad. Sci. U.S.A. 1992;89:9156–9160. doi: 10.1073/pnas.89.19.9156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman E.J., Prokhnevsky A.I., Gopinath K., Dolja V.V., Carrington J.C. Viral RNA silencing suppressors inhibit the microRNA pathway at an intermediate step. Genes Dev. 2004;18:1179–1186. doi: 10.1101/gad.1201204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Che X., Dawson W.O., Bar-Joseph M. Defective RNAs of Citrus tristeza virus analogous to Crinivirus genomic RNAs. Virology. 2003;310:289–309. doi: 10.1016/s0042-6822(03)00127-2. [DOI] [PubMed] [Google Scholar]
- Che X., Mawassi M., Bar-Joseph M. A novel class of large and infectious defective RNAs of citrus tristeza virus. Virology. 2002;298:133–145. doi: 10.1006/viro.2002.1472. [DOI] [PubMed] [Google Scholar]
- Chiba, M., Reed, J.C., Prokhnevsky, A.I., Chapman, E.J., Mawassi, M., Koonin, E.V., Carrington, J.C., Dolja, V.V., 2006. Diverse suppressors of RNA silencing enhance agroinfection by a viral replicon. Virology 346, 7–14. [DOI] [PubMed]
- Culley A.I., Lang A.S., Suttle C.A. High diversity of unknown picorna-like viruses in the see. Nature. 2003;424:1054–1057. doi: 10.1038/nature01886. [DOI] [PubMed] [Google Scholar]
- Daneholt B. Packing and delivery of a genetic message. Chromosoma. 2001;110:173–185. doi: 10.1007/s004120000127. [DOI] [PubMed] [Google Scholar]
- Dolja V.V. Beet yellows virus: the importance of being different. Mol. Plant Pathol. 2003;4:91–98. doi: 10.1046/j.1364-3703.2003.00154.x. [DOI] [PubMed] [Google Scholar]
- Dolja V.V., Boyko V.P., Agranovsky A.A., Koonin E.V. Phylogeny of capsid proteins of rod-shaped and filamentous RNA plant viruses: two families with distinct patterns of sequence and probably structure conservation. Virology. 1991;184(1):79–86. doi: 10.1016/0042-6822(91)90823-t. [DOI] [PubMed] [Google Scholar]
- Dolja V.V., Karasev A.V., Agranovsky A.A. Organization of the beet yellows closterovirus genome. In: Brinton M.A., Heinz F.X., editors. New Aspects of Positive-strand RNA Viruses. ASM Press; Washington, DC: 1990. pp. 31–35. [Google Scholar]
- Dolja V.V., Karasev A.V., Koonin E.V. Molecular biology and evolution of closteroviruses: sophisticated build-up of large RNA genomes. Annu. Rev. Phytopathol. 1994;32:261–285. [Google Scholar]
- Dougherty W.G., Semler B.L. Expression of virus-encoded proteinases: functional and structural similarities with cellular enzymes. Microbiol. Rev. 1993;57(4):781–822. doi: 10.1128/mr.57.4.781-822.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erokhina T.N., Zinovkin R.A., Vitushkina M.V., Jelkmann W., Agranovsky A.A. Detection of beet yellows closterovirus methyltransferase-like and helicase-like proteins in vivo using monoclonal antibodies. J. Gen. Virol. 2000;81:597–603. doi: 10.1099/0022-1317-81-3-597. [DOI] [PubMed] [Google Scholar]
- Fazeli C.F., Rezaian M.A. Nucleotide sequence and organization of ten open reading frames in the genome of Grapevine leafroll-associated virus 1 and identification of three subgenomic RNAs. J. Gen. Virol. 2000;81:605–615. doi: 10.1099/0022-1317-81-3-605. [DOI] [PubMed] [Google Scholar]
- Fitch W.M. On the problem of discovering the most parsimonious tree. Am. Nat. 1977;111:223–257. [Google Scholar]
- Gaunt M.W., Miles M.A. An insect molecular clock dates the origin of the insects and accords with palaeontological and biogeographic landmarks. Mol. Biol. Evol. 2002;19:748–761. doi: 10.1093/oxfordjournals.molbev.a004133. [DOI] [PubMed] [Google Scholar]
- Gilbertson R.L., Sudarshana M., Jiang H., Rojas M.R., Lucas W.J. Limitations on geminivirus genome size imposed by plasmodesmata and virus-encoded movement protein: insights into DNA trafficking. Plant Cell. 2003;15:2578–2591. doi: 10.1105/tpc.015057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbalenya A.E., Enjuanes L., Ziebuhr J., Snijder E.J. Nidovirales: evolving the largest RNA virus genome. Virus Res. 2006;117:17–37. doi: 10.1016/j.virusres.2006.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowda S., Satyanarayana T., Ayllon M.A., Albiach-Marti M.R., Mawassi M., Rabindran S., Garnsey S.M., Dawson W.O. Characterization of the cis-acting elements controlling subgenomic mRNAs of citrus tristeza virus: production of positive- and negative-stranded 3′-terminal and positive-stranded 5′-terminal RNAs. Virology. 2001;286(1):134–151. doi: 10.1006/viro.2001.0987. [DOI] [PubMed] [Google Scholar]
- Gregorio C.C., Granzier H., Sorimachi H., Labeit S. Muscle assembly: a titanic achievement? Curr. Opin. Cell Biol. 1999;11:18–25. doi: 10.1016/s0955-0674(99)80003-9. [DOI] [PubMed] [Google Scholar]
- Hagiwara Y., Peremyslov V.V., Dolja V.V. Regulation of closterovirus gene expression examined by insertion of a self-processing reporter and by northern hybridization. J. Virol. 1999;73:7988–7993. doi: 10.1128/jvi.73.10.7988-7993.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilf M.E., Karasev A.V., Pappu H.R., Gumpf D.J., Niblett C.L., Garnsey S.M. Characterization of citrus tristeza virus subgenomic RNAs in infected tissue. Virology. 1995;208(2):576–582. doi: 10.1006/viro.1995.1188. [DOI] [PubMed] [Google Scholar]
- Iyer L.M., Balaji S., Koonin E.V., Aravind L. Evolutionary genomics of nucleo-cytoplasmic large DNA viruses. Virus Res. 2006;117:156–184. doi: 10.1016/j.virusres.2006.01.009. [DOI] [PubMed] [Google Scholar]
- Joyce G.F. The antiquity of RNA based evolution. Nature. 2002;418:214–221. doi: 10.1038/418214a. [DOI] [PubMed] [Google Scholar]
- Karasev A.V. Genetic diversity and evolution of closteroviruses. Annu. Rev. Phytopathol. 2000;38:293–324. doi: 10.1146/annurev.phyto.38.1.293. [DOI] [PubMed] [Google Scholar]
- Karasev A.V., Agranovsky A.A., Rogov V.V., Miroshnichenko N.A., Dolja V.V., Atabekov J.G. Virion RNA of beet yellows closterovirus: cell-free translation and some properties. J. Gen. Virol. 1989;70:241–245. [Google Scholar]
- Karasev A.V., Boyko V.P., Gowda S., Nikolaeva O.V., Hilf M.E., Koonin E.V., Niblett C.L., Cline K., Gumpf D.J., Lee R.F., Garnsey S.M., Lewandowski D.J., Dawson W.O. Complete sequence of the citrus tristeza virus RNA genome. Virology. 1995;208:511–520. doi: 10.1006/viro.1995.1182. [DOI] [PubMed] [Google Scholar]
- Karasev A.V., Hilf M.E., Garnsey S.M., Dawson W.O. Transcriptional strategy of closteroviruses: mapping the 5′ termini of the citrus tristeza virus subgenomic RNAs. J. Virol. 1997;71:6233–6236. doi: 10.1128/jvi.71.8.6233-6236.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenrick P., Crane P.R. The origin and early evolution of plants on land. Nature. 1997;389:33–39. [Google Scholar]
- Klaassen V.A., Boeshore M.L., Koonin E.V., Tian T., Falk B.W. Genome structure and phylogenetic analysis of lettuce infectious yellows virus, a whitefly-transmitted, bipartite closterovirus. Virology. 1995;208:99–110. doi: 10.1006/viro.1995.1133. [DOI] [PubMed] [Google Scholar]
- Koonin E.V. The phylogeny of RNA-dependent RNA polymerases of positive-strand RNA viruses. J. Gen. Virol. 1991;72:2197–2206. doi: 10.1099/0022-1317-72-9-2197. [DOI] [PubMed] [Google Scholar]
- Koonin E.V. A non-adaptationist perspective on evolution of genomic complexity or the continuing dethroning of man. Cell Cycle. 2004;3:280–285. [PubMed] [Google Scholar]
- Koonin E.V., Dolja V.V. Evolution and taxonomy of positive-strand RNA viruses: implications of comparative analysis of amino acid sequences. Crit. Rev. Biochem. Mol. Biol. 1993;28(5):375–430. doi: 10.3109/10409239309078440. [DOI] [PubMed] [Google Scholar]
- Koonin E.V., Martin W. On the origin of genomes and cells within inorganic compartments. Trends Genet. 2005;21:647–654. doi: 10.1016/j.tig.2005.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreuze J.F., Savenkov E.I., Cuellar W., Li X.H., Valkonen J.P. Viral class 1 RNase III involved in suppression of RNA silencing. J. Virol. 2005;79:7227–7238. doi: 10.1128/JVI.79.11.7227-7238.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreuze J.F., Savenkov E.I., Valkonen J.P.T. Complete genome sequence and analyses of the subgenomic RNAs of sweet potato chlorotic stunt virus reveal several new features for the genus Crinivirus. J. Virol. 2002;76:9260–9270. doi: 10.1128/JVI.76.18.9260-9270.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S., Tamura K., Nei M. MEGA3: integrated software for molecular evolutionary genetics analysis and sequence alignment. Brief. Bioinform. 2004;5:150–163. doi: 10.1093/bib/5.2.150. [DOI] [PubMed] [Google Scholar]
- Ling K.S., Zhu H.Y., Gonsalves D. Complete nucleotide sequence and genome organization of Grapevine leafroll-associated virus 3, type member of the genus Ampelovirus. J. Gen. Virol. 2004;85:2099–2102. doi: 10.1099/vir.0.80007-0. [DOI] [PubMed] [Google Scholar]
- Liveratos I.C., Eliasco E., Muller G., Olsthoorn R.C.L., Salazar L.F., Pleij C.W.A., Coutts R.H.A. Analysis of the RNA of Potato yellow vein virus: evidence for a tripartite genome and conserved 3′-terminal structures among members of the genus Crinivirus. J. Gen. Virol. 2004;85:2065–2075. doi: 10.1099/vir.0.79910-0. [DOI] [PubMed] [Google Scholar]
- Lu R., Folimonov A.S., Shintaku M., Li W.X., Falk B.W., Dawson W.O., Ding S.W. Three distinct suppressors of RNA silencing encoded by a 20-kb viral RNA genome. Proc. Natl. Acad. Sci. U.S.A. 2004;101:15742–15747. doi: 10.1073/pnas.0404940101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas W.J., Lee J.Y. Plasmodesmata as a supracellular control networks in plants. Nat. Rev. Mol. Cell Biol. 2004;5:712–726. doi: 10.1038/nrm1470. [DOI] [PubMed] [Google Scholar]
- Lynch M., Conery J.S. The origins of genome complexity. Science. 2003;302:1401–1404. doi: 10.1126/science.1089370. [DOI] [PubMed] [Google Scholar]
- Martelli G.P.E.A. The family Closteroviridae revised. Arch. Virol. 2002;147:2039–2044. doi: 10.1007/s007050200048. [DOI] [PubMed] [Google Scholar]
- Martinez-Torres D., Buades C., Latorre A., Moya A. Molecular systematics of aphids and their primary endosymbionts. Mol. Phylogenet. Evol. 2001;20:437–449. doi: 10.1006/mpev.2001.0983. [DOI] [PubMed] [Google Scholar]
- Medina V., Peremyslov V.V., Hagiwara Y., Dolja V.V. Subcellular localization of the HSP70-homolog encoded by beet yellows closterovirus. Virology. 1999;260(1):173–181. doi: 10.1006/viro.1999.9807. [DOI] [PubMed] [Google Scholar]
- Melzer M.J., Karasev A.V., Sether D.M., Hu J.S. Nucleotide sequence, genome organization and phylogenetic analysis of pineapple mealybug wilt-associated virus-2. J. Gen. Virol. 2001;82:1–7. doi: 10.1099/0022-1317-82-1-1. [DOI] [PubMed] [Google Scholar]
- Miller W.A., Koev G. Synthesis of subgenomic RNAs by positive-strand RNA viruses. Virology. 2000;273:1–8. doi: 10.1006/viro.2000.0421. [DOI] [PubMed] [Google Scholar]
- Molnar A., Csorba T., Lacatos L., Varallyay E., Lacomme C., Burgyan J. Plant virus-derived small interfering RNAs originate predominantly from highly structured single-stranded viral RNAs. J. Virol. 2005;79:7812–7818. doi: 10.1128/JVI.79.12.7812-7818.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napuli A.J., Alzhanova D.V., Doneanu C.E., Barofsky D.F., Koonin E.V., Dolja V.V. The 64-kDa capsid protein homolog of beet yellows virus is required for assembly of virion tails. J. Virol. 2003;77:2377–2384. doi: 10.1128/JVI.77.4.2377-2384.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napuli A.J., Falk B.W., Dolja V.V. Interaction between HSP70 homolog and filamentous virions of the Beet yellows virus. Virology. 2000;274(1):232–239. doi: 10.1006/viro.2000.0475. [DOI] [PubMed] [Google Scholar]
- Palmer J.D., Soltis D.E., Chase M.W. The plant tree of life: and overview and some points of view. Am. J. Bot. 2004;91:1437–1445. doi: 10.3732/ajb.91.10.1437. [DOI] [PubMed] [Google Scholar]
- Pappu H.R., Karasev A.V., Anderson E.J., Pappu S.S., Hilf M.E., Febres V.J., Eckloff R.M., McCaffery M., Boyko V., Gowda S. Nucleotide sequence and organization of eight 3′ open reading frames of the citrus tristeza c losterovirus genome. Virology. 1994;199(1):35–46. doi: 10.1006/viro.1994.1095. [DOI] [PubMed] [Google Scholar]
- Pasternak A.O., van den Born E., Spaan W.J., Snijder E.J. Sequence requirements for RNA strand transfer during nidovirus discontinuous subgenomic RNA synthesis. EMBO J. 2001;20:7220–7228. doi: 10.1093/emboj/20.24.7220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng C.-W., Napuli A.J., Dolja V.V. Leader proteinase of the beet yellows virus functions in long-distance transport. J. Virol. 2003;77:2843–2849. doi: 10.1128/JVI.77.5.2843-2849.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng C.W., Dolja V.V. Leader proteinase of the beet yellows closterovirus: mutation analysis of the function in genome amplification. J. Virol. 2000;74:9766–9770. doi: 10.1128/jvi.74.20.9766-9770.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng C.W., Peremyslov V.V., Mushegian A.R., Dawson W.O., Dolja V.V. Functional specialization and evolution of leader proteinases in the family Closteroviridae. J. Virol. 2001;75(24):12153–12160. doi: 10.1128/JVI.75.24.12153-12160.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng C.W., Peremyslov V.V., Snijder E.J., Dolja V. A replication-competent chimera of the plant and animal viruses. Virology. 2002;294:75–84. doi: 10.1006/viro.2001.1306. [DOI] [PubMed] [Google Scholar]
- Peremyslov V.V., Andreev I.A., Prokhnevsky A.I., Duncan G.H., Taliansky M.E., Dolja V.V. Complex molecular architecture of beet yellows virus particles. Proc. Natl. Acad. Sci. U.S.A. 2004;101:5030–5035. doi: 10.1073/pnas.0400303101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peremyslov V.V., Pan Y.-W., Dolja V.V. Movement protein of a closterovirus is a type III integral transmembrane protein localized to the endoplasmic reticulum. J. Virol. 2004;78:3704–3709. doi: 10.1128/JVI.78.7.3704-3709.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peremyslov V.V., Dolja V.V. Identification of the subgenomic mRNAs that encode 6-kDa movement protein and Hsp70 homolog of beet yellows virus. Virology. 2002;295:299–306. doi: 10.1006/viro.2002.1396. [DOI] [PubMed] [Google Scholar]
- Peremyslov V.V., Hagiwara Y., Dolja V.V. Genes required for replication of the 15.5-kilobase RNA genome of a plant closterovirus. J. Virol. 1998;72:5870–5876. doi: 10.1128/jvi.72.7.5870-5876.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peremyslov V.V., Hagiwara Y., Dolja V.V. HSP70 homolog functions in cell-to-cell movement of a plant virus. Proc. Natl. Acad. Sci. U.S.A. 1999;96:14771–14776. doi: 10.1073/pnas.96.26.14771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prokhnevsky A.I., Peremyslov V.V., Dolja V.V. Actin cytoskeleton is involved in targeting of a viral Hsp70 homolog to the cell periphery. J. Virol. 2005;79:14421–14428. doi: 10.1128/JVI.79.22.14421-14428.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prokhnevsky A.I., Peremyslov V.V., Napuli A.J., Dolja V.V. Interaction between long-distance transport factor and Hsp70-related movement protein of beet yellows virus. J. Virol. 2002;76:11003–11011. doi: 10.1128/JVI.76.21.11003-11011.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raoult D., Audic S., Robert C., Abergel C., Renesto P., Ogata H., La Scola B., Suzan M., Claverie J.-M. The 1.2-megabase genome sequence of Mimivirus. Science. 2004;306:1344–1350. doi: 10.1126/science.1101485. [DOI] [PubMed] [Google Scholar]
- Reed J.C., Kasschau K.D., Prokhnevsky A.I., Gopinath K., Pogue G.P., Carrington J.C., Dolja V.V. Suppressor of RNA silencing encoded by beet yellows virus. Virology. 2003;306:203–209. doi: 10.1016/s0042-6822(02)00051-x. [DOI] [PubMed] [Google Scholar]
- Rubio L., Yeh H.-H., Tian T., Falk B.W. A heterogeneous population of defective RNAs is associated with lettuce infectious yellows virus. Virology. 2000;271:205–212. doi: 10.1006/viro.2000.0318. [DOI] [PubMed] [Google Scholar]
- Saitou N., Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- Satyanarayana T., Gowda S., Ayllon M.A., Dawson W.O. Closterovirus bipolar virion: evidence for initiation of assembly by minor coat protein and its restriction to the genomic RNA 5′ region. Proc. Natl. Acad. Sci. U.S.A. 2004;101:799–804. doi: 10.1073/pnas.0307747100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satyanarayana T., Gowda S., Mawassi M., Albiach-Marti M.R., Ayllon M.A., Robertson C., Garnsey S.M., Dawson W.O. Closterovirus encoded HSP70 homolog and p61 in addition to both coat proteins function in efficient virion assembly. Virology. 2000;278(1):253–265. doi: 10.1006/viro.2000.0638. [DOI] [PubMed] [Google Scholar]
- Sawicki D.L., Wang T., Sawicki S.G. The RNA structures engaged in replication and transcription of the A59 strain of mouse hepatitis virus. J. Gen. Virol. 2001;82:386–396. doi: 10.1099/0022-1317-82-2-385. [DOI] [PubMed] [Google Scholar]
- Schwartz M., Chen J., Janda M., Sullivan M., den Boon J.A., Ahlquist P. A positive-strand RNA virus replication complex parallels form and function of retrovirus capsids. Mol. Cell. 2002;9:505–514. doi: 10.1016/s1097-2765(02)00474-4. [DOI] [PubMed] [Google Scholar]
- Silhavy D., Burgyan J. Effects and side effects of viral RNA silencing suppressors on short RNAs. Trends Plant Sci. 2004;9:76–83. doi: 10.1016/j.tplants.2003.12.010. [DOI] [PubMed] [Google Scholar]
- Theilmann J., Mozafari J., Reade R., Wu Z., Xie W., Jesperson G., Bernardy M., Eastwell K.C., Rochon D. Partial nucleotide sequence and genome organization of a Canadian isolate of Little cherry virus. Phytopathology. 2002;92:87–98. doi: 10.1094/PHYTO.2002.92.1.87. [DOI] [PubMed] [Google Scholar]
- Thompson J.D., Gibson T.J., Plewniak F., Jeanmougin F., Higgins D.G. The ClustalX windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;24:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian T., Rubio L., Yeh H.-H., Crawford B., Falk B.W. Lettuce infectious yellows virus: in vitro acquisition analysis using partially purified virions and the whitefly, Bemisia tabaci. J. Gen. Virol. 1999;80:1111–1117. doi: 10.1099/0022-1317-80-5-1111. [DOI] [PubMed] [Google Scholar]
- Tzanetakis I.E., Martin R.R. Complete nucleotide sequence of a strawberry isolate of Beet pseudoyellows virus. Virus Genes. 2004;28:239–246. doi: 10.1023/b:viru.0000025771.48128.f8. [DOI] [PubMed] [Google Scholar]
- Tzanetakis I.E., Postman J., Martin R.R. Characterization of a novel member of the family Closteroviridae from Mentha spp. Phytopathology. 2005;95:1043–1048. doi: 10.1094/PHYTO-95-1043. [DOI] [PubMed] [Google Scholar]
- Tzanetakis I.E., Reed J.C., Martin R.R. Nucleotide sequence, genome organization and phylogenetic analysis of Strawberry pallidosis associated virus, a new member of the genus Crinivirus. Arch. Virol. 2005;150:273–286. doi: 10.1007/s00705-004-0410-z. [DOI] [PubMed] [Google Scholar]
- Vargason J.M., Szittya G., Burgyan J., Tanaka Hall T.M. Size selective recognition of siRNA by an RNA silencing suppressor. Cell. 2003;115:799–811. doi: 10.1016/s0092-8674(03)00984-x. [DOI] [PubMed] [Google Scholar]
- Voinett O. Induction and suppression of RNA silencing: insights from viral infections. Nat. Rev. Genet. 2005;6:206–220. doi: 10.1038/nrg1555. [DOI] [PubMed] [Google Scholar]
- Ye K., Patel D.J. RNA silencing suppressor p21 of Beet yellows virus forms an RNA binding octameric ring structure. Structure. 2005;13:1375–1384. doi: 10.1016/j.str.2005.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamore P.D., Haley B. Ribo-gnome: the big world of small RNAs. Science. 2005;309:1519–1524. doi: 10.1126/science.1111444. [DOI] [PubMed] [Google Scholar]
- Zhu H.Y., Ling K.S., Goszczynski D.E., McFerson J.R., Gonsalves D. Nucleotide sequence and genome organization of grapevine leafroll-associated virus-2 are similar to beet yellows virus, the closterovirus type member. J. Gen. Virol. 1998;79:1289–1298. doi: 10.1099/0022-1317-79-5-1289. [DOI] [PubMed] [Google Scholar]
- Zinovkin, R.A., Erokhina, T.N., D.E., L., Jelkmann, W., Agranovsky, A.A., 2003. Processing and subcellular localization of the leader papain-like proteinase of Beet yellows closterovirus. J. Gen. Virol. 84, 2265–2270. [DOI] [PubMed]
- Zuniga S., Sola I., Alonso S., Enjuanes L. Sequence motifs involved in the regulation of discontinuous coronavirus subgenomic RNA synthesis. J. Virol. 2004;78:980–994. doi: 10.1128/JVI.78.2.980-994.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]