

Highlights
-
•
The whole genome sequence of feline morbillivirus (FeMV) Piuma/2015 has been obtained by the combination of SISPA and NGS starting from the infected urine sample of a cat suffering of chronic kidney disease.
-
•
The highest sequence identity was detected with early FeMVs from Hong Kong.
-
•
Sequence heterogeneity exists within European FeMVs as for the existence of FeMVs in Germany and Turkey divergent from Piuma/2015.
Keywords: Feline morbillivirus, Urine, Cat, Italy, NGS, SISPA
Abstract
Feline morbillivirus (FeMV) has been recently identified by RT-PCR in the urine sample of a nephropathic cat in Italy. In this report, we describe the whole genome sequence of strain Piuma/2015 obtained by combination of sequence independent single primer amplification method (SISPA) and next generation sequencing (NGS) starting from RNA purified from the infected urine sample. The existence in Germany and Turkey of FeMVs from cats divergent from Piuma/2015, suggests the presence of FeMV heterogeneity in Europe as it has been described previously in Japan and China.
The genus Morbillivirus of the family Paramyxoviridae comprises several viral species with a non-segmented single stranded negative RNA genome. They cause severe and often fatal infections of humans and animals including measles, distemper and the emerging pest of small ruminants (Lamb and Parks, 2013). Morbilliviruses have been very recently identified also in cats. The new viral species so far denominated feline morbillivirus (FeMV) was detected for the first time in Hong Kong (China) and apparently associated with tubule-interstitial nephritis (TIN) (Woo et al., 2012). TIN involves primary injury to renal tubules and interstitium and is the most common cause of chronic kidney disease (CKD) and one of the leading causes of deaths in housed old cats. Further studies demonstrated the presence of FeMV in Japanese domestic cats (Sakaguchi et al., 2014, Furuya et al., 2014), although without any clear clinical association with CKD. By the end of June 2015, FeMV was identified by RT-PCR in the urine sample of a stray cat suffering from CKD in Teramo, Abruzzi region, Central-Italy (Lorusso et al., 2015). Interestingly, the cat did not show other known pathogens related with kidney disease. The presence of FeMV in Europe was already evidenced in 2013 in Germany (Sieg et al., 2015) and recently also described in Turkey (acc. nos KT808322 and KT021480; Presence of feline morbillivirus-RNA in cats in Istanbul, Turkey, unpublished). FeMV has also been recently detected in USA from healthy cats and from cats suffering from CKD (Sharp et al., 2016). In this short report, it is described how the whole genome sequence of FeMV Piuma/2015 has been obtained from an infected urine sample and the genetic relation of this strain with extant FeMVs.
Urine sample was centrifuged at 3000g for 5 min to remove cell debris. Total RNA has been purified from 300 microliters (μl) of urine by using the High Pure Viral Nucleic Acid kit (Roche) following manufacturer’s guidelines. Purified total RNA was treated with RNase-free DNaseI (New England Biolabs, Ipswich, MA) at 37 °C for 10 min and quantified with Qubit® RNA HS Assay Kit (Thermo Fisher Scientific, Waltham, MA). A total quantity of 60 nanograms (ng) of RNA was used for the assessment of the sequence-independent single primer amplification (SISPA, Allander et al., 2005) with some modifications. cDNA was obtained by reverse-transcription (RT) with 200 Unit of the SuperScript® IV Reverse Transcriptase (Thermo Fisher Scientific, Waltham, MA) in presence of 1X SSIV Buffer, 50 μM of the random primer FR26RV-N, 10 mM of dNTPs mix, 100 mM of DTT, 40U of RNAse OUT RNase Inhibitor (Thermo Fisher Scientific, Waltham, MA). The reaction was incubated at 23 °C for 10 min and 50 °C for 50 min. After an inactivation step at 80 °C for 10 min, 5 units of Klenow Fragment (3′ → 5′ exo-) (New England Biolabs, Ipswich, MA) was directly added to the reaction to perform the second strand cDNA synthesis. The incubation was carried out at 37 °C for 1 h and 75 °C for 10 min. Next, 5 μl of ds cDNA was added to PCR master mix containing 1 × PfuUltra II reaction buffer, 1 μl of PfuUltra II Fusion HS DNA Polymerase (Agilent Technologies Santa Clara, CA), 10 mM of dNTPs mix and 40 μM of FR20RV primer. The incubation was performed with the following thermal conditions: 95 °C for 1 min, 40 cycles of 95 °C for 20 s, 65 °C for 20 s and 72 °C for 2 min and a final extension step of 72 °C for 3 min. The PCR product was purified by using Expin™ PCR SV (GeneAll Biotechnology CO., LTD Seoul, Korea) and then quantified by using the Qubit® DNA HS Assay Kit (Thermo Fisher Scientific, Waltham, MA). The sample was diluted to an initial concentration of 0.2 ng/μl and then 1 ng was used for library preparation by using the Nextera XT Library Prep kit (Illumina Inc., San Diego, CA) according to the manufacturer’s protocol with the only exception regarding the quantity of the transposome. We indeed added 3 μl of the tagmentation enzyme instead of the 5 μl recommended in order to avoid over fragmentation of the viral genome which would have caused quantitative and qualitative losses of the resulting library (Ullmann et al., 2015). Deep sequencing was performed on the NextSeq 500 (Illumina Inc., San Diego, CA) using the NextSeq 500/550 Mid Output Reagent Cartridge v2, 300 cycles and standard 150 bp paired-end reads.
The sequencing run delivered 1.6 GB of sequence data. The reads obtained were trimmed using an in house script that trims the first 15 nt and uses as cutoff a mean quality of 30 and a minimum length of 50 bases. The resulting 2537335 reads, ranging from 50 to 136 nt with a mean Phred score of 35, were de novo assembled using SPADES version 3.0.0 (Bankevich et al., 2012) which yielded a total number of 2472 contigs (>500 bp). A total number of 927 contigs mapped on cat genome, whereas the remaining 1545 contigs were mapped on a reference FeMV sequence (GenBank accession number JQ411016, strain M252A). The length of the final assembly (GenBank accession number KT825132) was of 16050 bp and it showed 92% nt identity to the reference sequence. As previously sequenced FeMVs, the genome was found to be 16.050 nt in length, consistent with the “rule of six” and contained 6 non overlapping genes in the order N-P/V/C-M-F-H-L, which is typical for Morbilliviruses, encoding eight structural and non-structural proteins. The amino acid (aa) lengths of the six structural proteins coded by the genome were N, 519 aa; P, 491 aa; M, 337 aa; F, 543 aa; H, 595 aa and L, 2.202 aa. Unique aa changes were identified in all putative viral structural and non-structural proteins of Piuma/2015 with respect extant FeMV strains. In particular, eight and four aa residues unique to Piuma/2015 were detected in the H and F proteins, respectively. The genome contained a 12-nt complementary 3′ leader and 5′ trailer sequence. The 3′ leader sequence was 55 nt in lenght whereas the 5′ trailer sequence was 400 nt in lenght in contrast to other morbilliviruses, which have 5′ trailer sequences of only 40 or 41 nt in lenght. The non-transcribed triplet serving as intergenic region was CTT in each gene junction except for the CTA triplet in the M-F junction, as already described in the early FeMV isolates (Woo et al., 2012).
The whole nucleotide genome sequence of the obtained Italian FeMV genome was compared to those of extant FeMVs. Nt identity between Piuma/2015 and extant FeMV sequences ranges from 88 to 94.5%. Piuma/2015 bears the highest sequence nt identity, 94.5%, with early FeMV strains 761U and 776U isolated in China in 2009 (Woo et al., 2012), the lowest (88%) with strains OtJP001 and SS1 from Japan. With the FeMV US1 strain (Sharp et al., 2016), the only complete genome available from USA, nt sequence identity is 94%. As for FeMV strains detected in Europe (from Germany and Turkey) only partial sequences of the L protein encoding gene are publicly available. Sequence identities within Piuma/2015 and the German FeMVs strains Schmusi (KR269601), Mickie (KR269600), Lisa (KR269599) and Maximilian (KP159803) are 90.6%, 89.7%, 84.1% (for both Lisa and Maximilian strains) respectively, whereas sequence identity with both FeMVs from Turkey is 81.2%. Maximum Likelihood (ML) phylogenetic analyses have been performed. In particular, ML analyses were performed by using a portion of the L protein encoding gene shared by all existing publicly available FeMVs (MLpartial) and FeMV complete genomes (MLcomplete). The more inclusive MLpartial analysis suggests the existence of at least 2 different clades within FeMVs, the first including FeMVs from Asia, USA, Italy, Turkey and strains Mickie and Schmusi from Germany, whereas the second includes German FeMV strains Lisa and Maximilian (Fig. 1 A). Thus apparently FeMVs do not seem to be geographically grouped. Within the more numerically representative clade, three different clusters are observed further evidencing FeMV heterogeneity as already described for Japanese and Chinese strains (Sakaguchi et al., 2014). Indeed the European strains cluster in the two proposed clades and within the first clade, also in different clusters. Viral heterogeneity has been also described in the US cat population. In fact, besides the presence of FeMV US1 complete genome sequence, the authors also retrieved the H protein encoding gene sequence of an additional strain, FeMV US2, showing 81% nt identity with early FeMV 776U strain from Hong Kong. On the other hand, US1 and 776U strains share 98% nt identity. Less informative is the MLcomplete analysis as for the lower number of complete genome sequences publicly available. In particular, whole genome sequences from all German strains are lacking. Anyway, the existence of different clusters within the first proposed clade observed in MLpartial is confirmed by the MLcomplete analysis showing with the same topology for the available strains (Fig. 1B). Reasonably, more sequencing efforts are warranted. Moreover, whether or not FeMVs circulate contemporarily in cats as a cloud of divergent sequences belonging to one of the two current proposed clades or more yet undetected is so far difficult to say. In this regard, previous studies showed the existence of a naturally occurring recombinant FeMV (Park et al., 2014) from Japan (strain MiJP003). In this strain the recombination event seems to have occurred within the F and H protein encoding genes involving two potential parental FeMV strains very closely related to ChJP073 from Japan and 776U from Hong Kong. According to MLcomplete these two potential parental viruses group in two different clusters of the same proposed clade. The chronic nature of the infection may be responsible for the occurrence of recombination events and also for the viral heterogeneity described so far. However, viral diversity may also be related to the existence of different viral ancestors involved in the origin of FeMVs as already suggested for the emergence of canine respiratory coronaviruses with diverse genome organization of accessory genes (Lorusso et al., 2009). Piuma/2015 does not seem to be a recombinant virus (data not shown). Overall, in this report we provide the first complete genome of FeMV from Europe. Strain Piuma/2015 has been detected in the urine sample of an old nephropathic cat (Lorusso et al., 2015). Sequence was obtained by a combination of SISPA and NGS employing total RNA purified from the infected urine sample. Isolation of FeMV on cell culture has been so far demonstrated to be difficult and time consuming (Koide et al., 2015, Sakaguchi et al., 2015). Therefore in order to have a proper genome characterization, NGS was performed from the infected urine samples to provide in a few days the genetic characteristics of the virus circumventing the need for cell culture. The SISPA method was used for the enrichment of target RNA. SISPA combines the reverse transcription step performed with random hexamers tagged with a known sequence of 20 nucleotides which is subsequently used as a primer-binding site for a second stage of amplification cycles. It is a rapid and cost effective technique for large scale de novo sequencing (Djikeng et al., 2008). This approach is indeed applied to a wide variety of sample type (blood, tissues, swabs, urine, feces, etc.) and it is useful for generating complete genome sequences straight from biological samples.
Fig. 1.
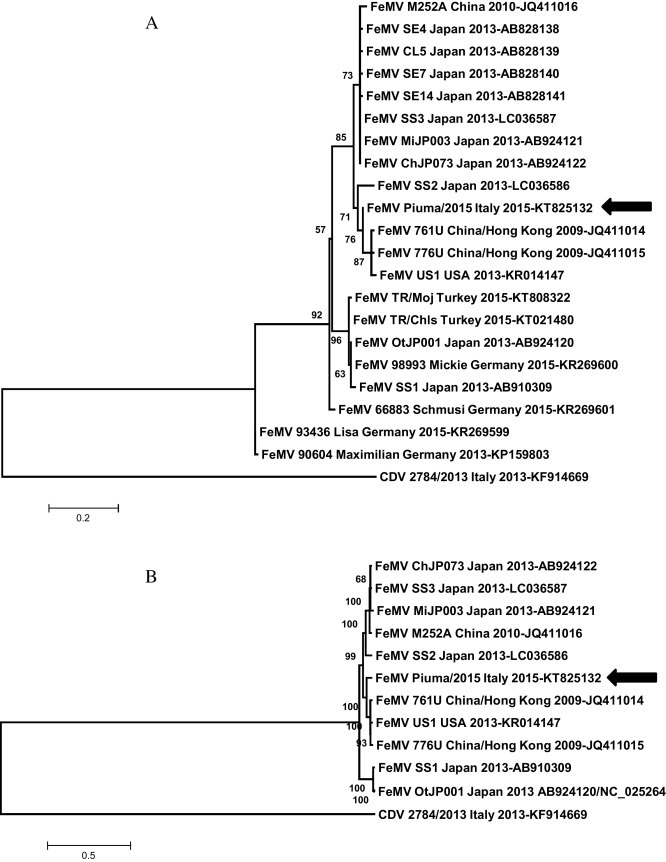
Phylogenetic analyses were inferred from partial (approximately 400 nt) L protein encoding gene of all existing FeMVs (MLpartial, 1A) and complete FeMV genomes available on line (MLcomplete, 1B). Analyses were conducted using the Maximum Likelihood (ML) method in Mega 6 (Tamura et al., 2013). The best-fit model of nucleotide substitution was identified by the Find Best DNA/protein Model available in Mega 6 as the T92 + I model for MLpartial, whereas the GTR + G + I for MLcomplete. To assess the robustness of individual nodes on the phylogenetic trees, we performed a bootstrap resampling analysis (1000 replications) using the neighbor-joining method, incorporating the ML substitution model. Canine distemper virus (CDV, Marcacci et al., 2014) served as outgroup. Bars indicate the estimated number of nt substitutions per site.
FeMV is a novel morbillivirus that apparently infects domestic cats, which have not been a natural host for previously known morbilliviruses. Moreover, cats seem so far to host a heterogenic population of novel paramyxoviruses. Indeed, in addition to the presence of FeMVs, additional feline paramyxoviruses (FPaVs) distinct from FeMVs, as they are more closely related to paramyxoviruses of rodents and bats, were also described (Sieg et al., 2015). Interestingly, also these viruses seem to be associated with CKD of cats. Therefore it seems evident that more efforts are needed to investigate the virological/serological prevalence and the antigenicity of these novel viruses in cats and, reasonably, demonstrate a clear association with clinical disease. Certainly, in a One Health approach, the discovery of novel morbilliviruses, or in general, of novel paramyxoviruses from cats highlights once more the importance of having a robust surveillance plan for viral infections of domestic animals which might potentially infect humans.
Acknowledgments
Funding was provided by the Italian Ministry of Health (Ricerca Corrente 2013 “Identificazione di standard di benessere dei cani nei canili e ruolo dei cani da affezione nella trasmissione di agenti di zoonosi e geni di resistenza agli antimicrobici all’uomo”). Mention of trade names or commercial products in this article is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the IZSAM.
References
- Allander T., Tammi M.T., Eriksson M., Bjerkner A., Tiveljung-Lindell A., Andersson B. Cloning of a human parvovirus by molecular screening of respiratory tract samples. Proc. Natl. Acad. Sci. U. S. A. 2005;102(36):12891–12896. doi: 10.1073/pnas.0504666102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankevich A., Nurk S., Antipov D., Gurevich A.A., Dvorkin M., Kulikov A.S., Lesin V.M., Nikolenko S.I., Pham S., Prjibelski A.D., Pyshkin A.V., Sirotkin A.V., Vyahhi N., Tesler G., Alekseyev M.A., Pevzner P.A. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012;19(5):455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djikeng A., Halpin R., Kuzmickas R., Depasse J., Feldblyum J., Sengamalay N., Afonso C., Zhang X., Anderson N.G., Ghedin E., Spiro D.J. Viral genome sequencing by random priming methods. BMC Genom. 2008;9:5. doi: 10.1186/1471-2164-9-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuya T., Sassa Y., Omatsu T., Nagai M., Fukushima R., Shibutani M., Yamaguchi T., Uematsu Y., Shirota K., Mizutani T. Existence of feline morbillivirus infection in Japanese cat populations. Arch. Virol. 2014;159(2):371–373. doi: 10.1007/s00705-013-1813-5. [DOI] [PubMed] [Google Scholar]
- Koide R., Sakaguchi S., Miyazawa T. Basic biological characterization of feline morbillivirus. J. Vet. Med. Sci. 2015;77(5):565–569. doi: 10.1292/jvms.14-0623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb R.A., Parks G.D. Paramyxoviridae. In: Knipe D.M., Howley P., editors. Fields Virology. 6th edn. Lippincott Williams & Wilkins; Philadelphia, PA: 2013. pp. 957–995. [Google Scholar]
- Lorusso A., Desario C., Mari V., Campolo M., Lorusso E., Elia G., Martella V., Buonavoglia C., Decaro N. Molecular characterization of a canine respiratory coronavirus strain detected in Italy. Virus Res. 2009;141:96–100. doi: 10.1016/j.virusres.2008.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorusso A., Di Tommaso M., Di Felice E., Zaccaria G., Luciani A., Marcacci M., Aste G., Boari A., Savini G. First report of feline morbillivirus in Europe. Vet. Ital. 2015;51:235–237. doi: 10.12834/VetIt.833.4136.2. [DOI] [PubMed] [Google Scholar]
- Marcacci M., Ancora M., Mangone I., Teodori L., Di Sabatino D., De Massis F., Camma' C., Savini G., Lorusso A. Whole genome sequence analysis of the arctic-lineage strain responsible for distemper in Italian wolves and dogs through a fast and robust next generation sequencing protocol. J. Virol. Methods. 2014;202:64–68. doi: 10.1016/j.jviromet.2014.02.027. [DOI] [PubMed] [Google Scholar]
- Park E.S., Suzuki M., Kimura M., Maruyama K., Mizutani H., Saito R., Kubota N., Furuya T., Mizutani T., Imaoka K., Morikawa S. Identification of a natural recombination in the F and H genes of feline morbillivirus. Virology. 2014;468–470:524–531. doi: 10.1016/j.virol.2014.09.003. [DOI] [PubMed] [Google Scholar]
- Sakaguchi S., Nakagawa S., Yoshikawa R., Kuwahara C., Hagiwara H., Asai K., Kawakami K., Yamamoto Y., Ogawa M., Miyazawa T. Genetic diversity of feline morbilliviruses isolated in Japan. J. Gen. Virol. 2014;95(Pt 7):1464–1468. doi: 10.1099/vir.0.065029-0. [DOI] [PubMed] [Google Scholar]
- Sakaguchi S., Koide R., Miyazawa T. In vitro host range of feline morbillivirus. J. Vet. Med. Sci. 2015 doi: 10.1292/jvms.15-0213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp C.R., Nambulli S., Acciardo A.S., Rennick L.J., Drexler J.F., Rima B.K., Williams T., Duprex W.P. Chronic infection of domestic cats with feline morbillivirus, United States. Emerg. Infect. Dis. 2016;22:760–762. doi: 10.3201/eid2204.151921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieg M., Heenemann K., Rückner A., Burgener I., Oechtering G., Vahlenkamp T.W. Discovery of new feline paramyxoviruses in domestic cats with chronic kidney disease. Virus Genes. 2015;51(2):294–297. doi: 10.1007/s11262-015-1232-7. [DOI] [PubMed] [Google Scholar]
- Tamura K., Stecher G., Peterson D., Filipski A., Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013;30:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullmann L.S., de Camargo Tozato C., Malossi C.D., da Cruz T.F., Cavalcante R.V., Kurissio J.K., Cagnini D.Q., Rodrigues M.V., Biondo A.W., Araujo J.P., Jr. Comparative clinical sample preparation of DNA and RNA viral nucleic acids for a commercial deep sequencing system (Illumina MiSeq) J. Virol. Methods. 2015;220:60–63. doi: 10.1016/j.jviromet.2015.04.009. [DOI] [PubMed] [Google Scholar]
- Woo P.C., Lau S.K., Wong B.H., Fan R.Y., Wong A.Y., Zhang A.J., Wu Y., Choi G.K., Li K.S., Hui J., Wang M., Zheng B.J., Chan K.H., Yuen K.Y. Feline morbillivirus, a previously undescribed paramyxovirus associated with tubulointerstitial nephritis in domestic cats. Proc. Natl. Acad. Sci. U. S. A. 2012;109(14):5435–5440. doi: 10.1073/pnas.1119972109. [DOI] [PMC free article] [PubMed] [Google Scholar]