

1. Introduction
Numerous microorganisms appear to have selected carbohydrates as the preferred attachment sites on their host cells and tissues [1]. This is not surprising, in view of the abundance of carbohydrates on the surfaces of eukaryotic cells, in the form of glycoproteins and glycolipids. Binding to the carbohydrates is mediated by microbial surface lectins, members of a large group of microbial adhesins, that play a major role in the initiation of infection and in non-opsonic phagocytosis [2], [3], [4], [5]. Host range, tissue tropism and target cell specificity demonstrated by a particular microbe are determined, at least in part, by a stereochemical fit between microbial lectins and complementary carbohydrate receptors on host cell surfaces [6]. Adhesion to insoluble carbohydrates may also play an important role in the biofouling of marine surfaces and in ecological phenomena such as biodegradation, as well as in the monitoring by bacteria of the nutrient status of the environment [7], Although a considerable number of these lectins are well defined, the existence of many more has been inferred from experiments on the effect of carbohydrates on the interaction of intact microorganisms with different target cells.
The oldest, and perhaps most thoroughly studied system of this type is the interaction between the hemagglutinin of influenza virus A and N-acetylneuraminic acid1 on cell surfaces. It is responsible not only for the adhesion of the virus to the cells, but also for fusion of the viral membrane with the host cell membrane and is also the viral component to which protective antibody is directed.
Virtually all bacterial species and genera express lectins or lectin-like activities, frequently of more than one type and with different specificities. However, it is usually not known whether individual cells coexpress multiple types of lectin or each lectin is confined to a distinct cell population. Many Gram negative bacteria (for example, Escherichia coli and Salmonellae spp.) and a few Gram positive ones (e.g. certain actinomyces), produce surface lectins that are often in the form of submicroscopic hairlike appendages known as fimbriae (pili) that protrude from the surface of the cells. The best characterized bacterial surface lectins with respect to their molecular properties, carbohydrate specificity and genetics are the type 1 fimbriae specific for mannose and the type P fimbriae specific for galabiose, [Gal(αl-4)Gal], produced by many strains of E. coli. Other examples are S fimbriae of E. coli, specific for NeuAc(α2-3)Gal, and type 2 fimbriae of oral actinomyees, specific for Gal(βl-3)GalNAc or GalNAc β– that are not so well characterized. Non-fimbrial lectins that are components of the cell surface have also been described. The case of Pseudomonas aeruginosa is unusual in that certain strains of the organism produce intracellular lectins, at least one of which appears also on the bacterial surface.
Among the fungal and protozoal lectins only a few have been studied in detail. One of these is the galactose-specific lectin of the protozoa Entamoeba histolytica. It mediates adhesion of the parasite to human colonic mucin glycoproteins and has a central role in the contact-dependent cytolysis or histolysis for which the parasite is named. A sialicacid-specific lectin has been isolated from merozoites of the human malarial parasite, Plasmodium falciparum. An unusual lectin is that of the protozoan Giardia lamblia, specific for mannose-6-phosphate, which is activated by trypsinization.
The carbohydrates to which the microbial lectins bind are in the form of glycoproteins and glycolipids (Table 1 ). Many lectins interact with both classes of glycoconjugate; this is not unexpected, since these compounds frequently contain identical oligosaccharides. Identification of the receptors for microbial lectins on animal cells (or in secretions) is based on methods such as binding of intact bacteria or of the isolated lectin either to blots of electrophoretically separated cell membrane glycoproteins [8] or to thin layer chromatograms of glycolipids extracted from the cells. Glycoproteins can also be analyzed by affinity chromatography of cell membrane extracts on Most importantly, it should be obtained from cells susceptible to infection by the organism studied. Also, antibodies to the presumed receptor should inhibit binding of the organism to the cells and cells devoid of the receptor (as seen sometimes in mutants) should lack the ability to bind the microorganism. Glycolipids that serve as native receptors for microbial lectins have been identified in a large number of cases, but information on glycoproteins that play such a role is still scarce.
Table 1.
Carbohydrates as attachment sites for infectious agentsa
| Organism | Target tissue | Carbohydrate |
Reference(s) | |
|---|---|---|---|---|
| Structure | Formb | |||
| Viruses | ||||
| Influenza type A | Respiratory tract | NeuAc(α2-6)Gal | GP | [12] |
| type B | Respiratory tract | NeuAc(α2–6)Gal | GP | [12] |
| type C | Respiratory tract | 9-O-AcNeuAc(α2–3)Gal | GP | [12] |
| Parvovirus B 19 | Erythroid cells | GalNAc(β1–3)Gal(α 1–4)Gal(β1–4) | GSL | [13] |
| Polyoma virus | Epithelial cells | NeuAc(α2–3)Gal | GP | [14] |
| Bacteria | ||||
| E. coli type 1 | Urinary tract | Man(α 1–3)[Man(α 1 –3)[Man(αl–6)] | GP | [4], [8] |
| Type P | Urinary tract | Gal(α1–4)Gal | GSL | [9] |
| Type S | Endothelial cells | NeuAc(α2–3)Gal(β1–3)GalNAc | GP | [8] |
| NeuGc(α2–3)Gal | GSL | [15] | ||
| type CFAI/II | Intestine | NeuAc(α2–3)Gal | GP | [8] |
| Type KI | Endothelial cells | GlcNAc(β1–3)GalNAc | GP | [16] |
| Type K88ac | Intestine | Gal (β1–3)GalNAc | Gp | [17] |
| Type K99 | Intestine | NeuGc(α2–3)Gal(β1–4)Glc | GSL | [18] |
| Actinomyces naeslundi | Oral | Gal(β1–4)Glc | GSL | [19] |
| GalNAc(β1–3)Galβ | GP | [20] | ||
| Neisseria gonorhoea | Genital | Gan(β1–4)Glcβ | GSL | [9] |
| NeuAc(α2–3)Gal(β1–4)GlcNAc | GP | |||
| Streptococcus sanguis | Oral | NeuAc(α2–3)Gal(β1–3)GlcNAc | GP | [21] |
| Fungi | ||||
| Candida albicans | Skin and mucosa | Gal(β1–4)Glc | GSL | [22] |
| Fuc(α1–2)Gal | GP | [23] | ||
For protozoa, see Table 3.
Predominant; GP, glycoproteins; GSL, glycolipids.
In the following we shall discuss mainly those lectins of viruses, bacteria, protozoa and fungi that are known to interact with either glycoproteins alone or with glycoproteins as well as glycolipids. We shall also deal with their role in infection and lectinophagocytosis. For bacterial lectins that appear to bind exclusively to glycolipids, see recent reviews on the subject [9], [10], [11].
2. Viruses
2.1. Sialic-acid-specific
2.1.1. Influenza virus
The ability of the virus to agglutinate erythrocytes has been first reported in 1941. It took more than a decade before it was shown that influenza virus binds to erythrocytes and other cells via N-acetylneuraminic acid residues present on the cell surface and that this binding is a prerequisite for initiation of infection [24], [25]. Other viruses, such as Sendai, Newcastle disease, polyoma and rotavirus also exhibit an affinity for sialic acid [12]. The hemagglutinin (lectin) of the influenza virus, responsible for its attachment to cells, was purified, crystallized, and studied in detail, culminating in the elucidation of the three-dimensional structure of its complex with N-acetylneuraminic-acid-containing oligosaccharides at the atomic level [26], [27], [28]. The subunit of the lectin is composed of two polypeptides, HA1 and HA2 (with molecular masses of 36 and 26 kDa, respectively), covalently linked by a single disulfide bond, and it associates non-covalently to form trimers that are located on the surface of the viral membrane [27], It is a glycoprotein, with six N-linked oligosaccharide chains attached to HA1 and one to HA2. Except for one oligomannose unit, all are complex, bi- or triantennary structures, with three of them containing sulfated galactose [29]. The carbohydrate binding site is located in a pocket of the HA 1 polypeptide chain, in a domain of the lectin protruding from the membrane, and is composed of amino acids that are largely conserved in the numerous strains of the virus [27]. Other conserved residues are found behind the pocket and seem to stabilize the architecture of the site without interacting with the carbohydrate.
Over 100 strains of influenza virus, mostly of the A- and B-types, were examined for their ability to bind to enzymatically modified erythrocytes carrying terminal N-acetylneuraminic acid attached to galactose either by an α2–3 or α2–6 linkage [12], [26]. Differences in their specificity with respect to this linkage were correlated with the species origin of the virus. Thus, human isolates preferentially agglutinated resialylated erythrocytes containing the NeuAc(α 2-6)Gal sequence, while the avian and equine isolates exhibited preference for NeuAc(α 2-3)Gal. Strains of influenza C virus (as well as coronaviruses) do not bind N-acetylneuraminic acid at all, but only recognize its derivative, 9-O-acetyl-N-acetylneuraminic acid; the 9-O-acetyl group is critical for mediating cellular attachment [30],
Comparison of the primary sequences of hemagglutinins of the human virus with those of mutants showing decreased affinity for NeuAc(α 2-6)Gal and markedly increased affinity for NeuAc(α 2-3)Gal, revealed that they differ in a single amino acid substitution, Leu226 in the parental strains being replaced by glutamine in the mutants. Similar studies with avian isolates and their variants showing the reverse change in specificity (from α 2-3 to α 2-6 linked A,-acetylncuraminic acid), again revealed a substitution only at position 226 – from glutamine to leucine. This illustrates that a single amino acid substitution can alter the sugar specificity of a lectin. Although residue 226 is located in the carbohydrate binding site of the hemagglutinin, it is not in direct contact with the bound sugar, as shown by crystallographic studies of wild-type influenza virus hemagglutinin complexed with NeuAc(α2–6)Gal(βl–4)Glc [sialyl(α2–6)lactose] and of a mutant hemagglutinin complexed with NeuAc(a2–3)Gal(βl–4)Glc [sialyl(α 2–3)lactose]. The suggestion has therefore been made that the change in specificity is due to conformational differences between the mutant and wild-type proteins.
X-ray crystallographic analysis of the complex of the hemagglutinin with bound sialyl(α 2–6)lactose, [NeuAc(α 2–6)Gal(βl–4)Glc], placed the sialic acid in the binding pocket with one side of the pyranose ring in tight contact to the protein and the other side facing the solvent [28] (Fig. 1 ). It also permitted to predict potential hydrogen bonds and van der Waals contacts of sialic acid atoms with amino acids in the binding site based on their proximity to each other. The validity of these predictions was tested by evaluating the affinity for the hemagglutinin of a series of synthetic analogs of N-acetylneuraminic acid with modified functional groups, by determining: (i) the ability of the analogs to inhibit viral attachment to cells [31] and (ii) the equilibrium dissociation constants for the binding of these analogs to the hemagglutinin [32]. It was thus confirmed that the two carboxylate oxygens of N-acetylneuraminic acid receive hydrogen bonds from the hydroxyl group of Ser-136 and from the main chain amide group at Asn-137, respectively, and that both bonds are necessary for binding. These studies also provided additional evidence on the critical importance of the hydrophobic contact between the acetamido group at C-5 of N-acetylneuraminic acid with the indole ring of Trp-153. On the other hand, the hydroxyl at C-9 does not seem to participate in ligand binding, contrary to what has been proposed from the crystal structure.
Fig. 1.
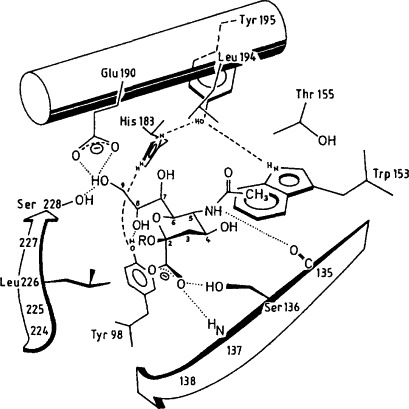
Model for the position of sialic acid in the binding pocket of influenza virus hemagglutinin. This model for the best fit has been deduced from the difference electron density maps of X-ray crystallographic studies. Some of the hydrogen bonds proposed in this model are shown by dashed lines. (Taken from ref. [31]; with permission from the authors.)
The structures of complexes of the hemagglutinin with four sialic acid analogs, with affinities 10- to 100-fold higher than that of .N-acetylneuraminie acid, were determined by high-resolution X-ray crystallography [33]. In these analogs, the sialic acid core was substituted at the 4 or 6 positions with spaced naphthyl or dansyl groups. In each of the complexes, the sialic acid moiety was equivalently positioned in the binding site of the each other interacted with hydrophobic patches and polar residues adjacent to the binding site.
In addition to the binding site discussed above, the hemagglutinin possesses a secondary site, at the interface between HA1 and HA2 polypeptide chains [34]. However, crystallographic studies have shown that sialyl(α2-6)lactose does not bind to the secondary site at all, while sialyl(α 2-3)lactose binds to this site with at least 4 times lower affinity than to the first one.
Another virus specific for sialic acid is murine polyoma virus. Two types of strains are known that differ in their specificity for sialic acid oligosaccharides: those that form large plaques bind to oligosaccharides terminating in NeuAc(α 2-3)Gal, whereas the small plaque strains also tolerate branched structures with a second, α 2-6 linked, sialic acid, e.g. NeuAc(α2-3)Galβ3[NeuAc(α 2-6)]GalNAc. These strains also differ in their ability to form tumors in mice – the small plaque strains produce few, if any, tumors, while the large plaque strains are highly tumorigenic. The critical difference in the structure of the viral protein of these strains is in residue 91, which is glycine in the small plaque strains and glutamic acid in the large plaque strains. Crystallographic studies at 3.65 A resolution of the viral protein from small plaque strains in complex with sialyl-3-lactose have shown that the combining site is in the form of a shallow groove and that both the sialic acid and the galactose form contacts with the protein [14], This is in contrast to influenza virus hemagglutinin, where only sialic acid makes such contacts.
2.2. Other specificities
Information on the existence of viral lectins with specificities other than for sialic acid is scant. Thus, parvovirus B19 binds GalNAe(βl-3)Gal(αl-4)Gal(βl-4) [13], Simplex virus binds heparan sulfate [35] and HIV recombinant envelope glycoproteins interact specifically with certain N-inked carbohydrate units of glycoproteins, e.g. of the oligomannose type, with mannose-6-phosphate and sulfated polysaccharides, such as heparin and dextran sulfate [36], [37],
3. Bacteria
The largest number of microbial lectins characterized to date are from bacterial sources (Table 1).
3.1. Mannose-specific (type 1 fimbriae)
3.1.1. Enterobacteria
Type 1 fimbriae are expressed by most strains of E. coli, as well as by other enter-obacteriaceae, such as Klebsiella pneumoniae [38], [39] (Fig. 2 ). They are heteropolymeric organelles, about 7 nm in diameter and 100 to 200 nm in length, consisting of helically arranged subunits (pilins) of several different types, assembled in a well defined order [5], [40]. The bulk of the fimbrial filament (shaft) is made up of polymers of a major subunit, Firn A, mw 17 kDa. In addition, fimbriae contain a cassette of three minor ancillary subunits, FimF, FimG and FimH. The latter is the only subunit that possesses a carbohydrate binding site and is thus responsible for the sugar specificity of the fimbriae. Although FimH is present both at the distal tip and at intervals along the length of the filament, only the subunit at the tip appears to be able to mediate mannose-sensitive adhesive interactions; the subunits at the other positions are inaccessible to the ligand. In other types of fimbriae (e.g. type P) the carbohydrate binding subunit is exclusively located to the tip.
Fig 2.
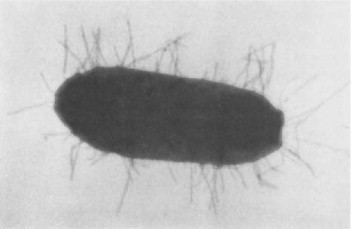
Type 1 fimbriated Eschenchia coil;
magnified 24 000 × (courtesy Dr. A. Gbarah)
Isolated FimH binds mannose-containing glycoproteins and adheres in a mannose-specific manner to human neutrophils [41], Moreover, it triggers an oxidative burst in a manner that mimics the activity of type 1 fimbriae. In addition, inert microspheres coated with FimH, but not with bovine serum albumin, are phagocytosed by neutrophils. Mutants lacking the fimH gene, but not genes encoding other fimbrial subunits, fail to bind to eukaryotic cells.
Two proteins, FimC and FimB, are involved in the biogenesis of type 1 fimbriae, without being part of the final structure. The former acts as a periplasmic chaperon that stabilizes fimbria subunits in the periplasm through the formation of distinct complexes. The subunit-chaperon complexes are targeted to FimB, an outer membrane protein, which organizes their ordered secretion into an extracellular polymer [42], Chaperons are also required for the assembly of other types of fimbriae, as well as of non-fimbrial lectins [43], [44], [45],
The expression of fimbriae is phase variable, i.e. bacteria shift periodically between a fimbriated and non-fimbriated state [38]. As a result, a given bacterial population will always contain cells of both phenotypes. The on- and off-phase variation is controlled at the transcriptional level and involves the inversion of a 314-base pair DNA segment harboring the promoter of the fim A gene [46],
Remarkable differences are found in the size and antigenic properties of the structural subunit of type 1 fimbriae (FimA) among different species of enterobacteria [47]. In contrast, a high degree of conservation is found between FimH proteins from type 1 fimbriae expressed by various species of the Enterobacteriaceae family, although they differ in their fine sugar specificity [48]. Thus, the combining sites of type 1 fimbriae of E. coli and K. pneumoniae correspond to the size of a trisaccharide and are in the form of a depression or pocket on the surface of the lectin [49]. In the case of the E. coli lectin, there are probably three adjacent subsites, each of which accommodates a monosaccharide residue. In the proximity of the combining site there is a hydrophobic binding region, as indicated by the finding that aromatic a-mannosides are significantly more powerful inhibitors than methyl a-mannoside. In contrast, several Salmonella species examined bind aromatic a-mannosides, as well as the trisaccharide Man(αl-3)Man(βl-4)GlcNAc, weaker than methyl α-mannoside, indicating that the combining site of the Salmonella lectin is probably smaller than that of E. coli and K. pneumoniae, and is devoid of a hydrophobic region.
Although very similar in their carbohydrate specificity, the lectins from E. coli and K. pneumoniae differ in their affinity for aromatic mannosides. Thus, 4-methylumbelli feryl α-mannoside is about 10-fold more effective than p-nitrophenyl α-mannoside in inhibiting yeast aggregation by type 1 fimbriated E. coli, while it is only 4 times more effective than p-nitrophenyl α-mannoside as inhibitor of K. pneumoniae [50]. Since the FimH subunits of the two organisms exhibit 88% homology, the possibility has been considered that the differences in specificity are due to differences in the presentation of the combining sites of these subunits in the fimbriae. To test this hypothesis, two types of hybrid fimbriae were genetically generated: in one of these the E. coli FimH was presented on a filament of K. pneumoniae structural subunits (EcFimH-KpFimA); in the other K. pneumoniae FimH was presented on a shaft of E. coli FimA (KpFimH-EcFimA) [50]. It was found that the specificity of the EcFimH-KpFimA hybrid with respect to aromatic a-mannosides was similar to that of native K. pneumoniae lectin, whereas that of KpFimH-EcFimA was like that of the E. coli lectin. These results indicate that the shaft on which FimH is presented plays a role in modulating the specificity of type 1 fimbriae lectins, probably by imposing conformational constraints on the carbohydrate binding subunit. The interspecies heterogeneity of the FimA subunit of enterobacteria thus ensures significant diversity in their sugar specificity and as a result, in the function of their lectins, as reflected by their ability to mediate adhesion to a particular type of animal cell. The notion that the fimbrial filament can influence the specificity of the carbohydrate binding moiety is novel and contrasts with the P fimbriae system, in which PapG appears to be the sole determinant of binding specificity.
Recently a form of type 1 fimbriae has been described which, in addition to binding carbohydrates, interacts also with non-glycosylated regions of proteins in a mannoseinhibitable manner [51]. It is not clear whether this interaction occurs via the carbohydrate binding site proper and how it is inhibited by mannose. The difference between the two functional forms of the fimbriae may be due to subtle variations in FimH or to quantitative or qualitative differences in the assembly of one or more of the subunits. In this context it should be noted that concanavalin A, a mannose/glucose-specific plant lectin, has also been shown to bind peptides in a carbohydrate-inhibitable manner [52], [53].
Type 1 fimbriated E. coli or the isolated fimbriae bind to glycoproteins from diverse sources [8]. These include a 65 kDa glycoprotein from guinea pig erythrocytes [54], the carcinoembryonic antigen, normally localized at the apical border of epithelial cells of the large intestine, secretory IgA and IgA myeloma proteins, especially those of the IgA2 subclass [55] Tamm-Horsfall glycoprotein (often referred to as uromucoid) [56], as well as several constituents of mucous layers [57] (see also section 6.1.2). The fimbriae bind to three glycoproteins derived from the cell membrane of human granulocytes (or neutrophils) [58] (Table 2 ). Two of them have been identified as components of the integrin superfamily CD11/CD18 (also known as leukocyte adhesion molecules). The fimbriated bacteria bound in a mannose-specific, dose-dependent and saturable manner to the isolated integrin in wells of microtiter plates and on SDS-PAGE gels; this binding was inhibited by monoclonal antibodies to integrin [59]. Monoclonal antibodies to CD11/CD18, but not to other granulocyte surface antigens, inhibited binding of the bacteria to the granulocytes. The same molecules serve also as receptors for type 1 fimbriae on human peritoneal macrophages [60], In addition, a glycoprotein called NCA-50, which belongs to a family of non-specific cross-reacting antigens associated with the granulocyte membrane, was reported to specifically bind type 1 fimbriae [61].
Table 2.
Glycoprotein receptors for microbial lectins
| Organism | Source of glycoprotein | Designation | Reference(s) |
|---|---|---|---|
| Mannose specific | |||
| E. coli type 1 | Human granulocytes,peritoneal macrophages | CD1 l/CD18 integrin | [59], [60] |
| Human granulocytes | NCA-50 | [61] | |
| Colonic mucosa | IgA | [55] | |
| Urine | Tamm-Horsfall glycoprotein | [56] | |
| Sialic acid specific | |||
| E. coli CFA/I | Human epithelial cells | 26 kDa gp | [62] |
| P falciparum | Human erythrocytes | Glycophorin | [63] |
| P aeruginosa, PAK | Human oral epithelium | 82 kDa, 40-50 kDa gp’s | [64] |
| Mouse corneal cells | [65] | ||
| S. sanguis | Human saliva | 400 kDa gp | [66] |
| Gal/GalNAc specific | |||
| A . naeslundii type 2 | Human oral epithelium | 160 kDa gp | [67] |
| Human saliva | 180 kDa gp | [68] | |
| B. pertussis toxin | CHO cells | 165 kDa gp | [69] |
| Human T cells, Jurkatt | 43 kDa gp; 70 kda gp | [70], [71] | |
3.2. Sialic-acid-specific
3.2.1. Escherichia coli
Certain strains of E. coli isolated from humans and farm animals express fimbrial lectins specific for glycoconjugates containing sialic acids [3], [4], [5]. This conclusion is based primarily on the observation that hemagglutination caused by these organisms is decreased or completely abolished by treatment of the erythrocytes with sialidase. Examples of such lectins are type S fimbriae of bacterial strains causing sepsis and meningitis in newborn infants, CFA (Colonization Factor Antigen) I and II of human enterotoxigenic E. coli isolates, as well as E. coli K99 fimbriae of enterotoxigenic strains isolated from piglets, calves and lambs suffering from diarrhea.
The structure of S fimbriae is very similar to that of type 1 fimbriae. They are composed of a major subunit and several minor components, of which only one binds sialic acid. In contrast, both in the CFAs and K99 fimbriae the major subunit also contains the carbohydrate binding site [72], [73]. Comparison of the amino acid sequence of the carbohydrate binding subunit of type S fimbriae with those of the major CFA I and K99 subunits revealed the presence of a common motif, rich in basic amino acids (Fig. 3 ). Site-specific mutagenesis experiments showed that a lysine and an arginine residue in this region play a part in ligand binding [74].
Fig. 3.

Sequence homology in the binding region of sialic-acid-specific bacterial lectins. Identical or functionally identical residues are underscored. Gaps have been introduced for optimal alignment.
S-fimbriated E. coli combine with α2–3 linked sialic acid residues on integral membrane glycoproteins. Limited trypsinization of human erythrocytes completely abolished binding of such bacteria, indicating that glycophorin A is their sole receptor on the erythrocytes [75]. S-fimbriated E. coli also bind to sialic acid on gangliosides, preferentially NeuGc(α2–3)Gal and NeuAc(α2–8)NeuAc [15], as well as to sulfated glycolipids [76], The latter binding apparently occurs through a different fimbrial subunit than that which interacts with sialic acid.
The specificity of CFA I is not well defined, beyond the fact that it recognizes sialic acid [77], [78]. A sialoglycoprotein with an apparent molecular weight of 26,000 is the only glycoprotein from the human erythrocyte membrane that binds CFA I fimbriated E. coli [62].
K99-bearing E. coli bind to sialylated mucus glycoproteins [79], The binding is not inhibited by sialic acid nor by other simple sugars, but by glycopeptides isolated from glycoproteins of bovine plasma, suggesting that the lectin recognizes complex carbohydrate structures. Glycopeptides bearing the terminal NeuGc(α2–3)Gal sequence strongly inhibited hemagglutination caused by E. coli K99, demonstrating the specificity of these bacteria for N-glycoloylneuraminic acid [80]. The physiological receptor for K99 on intestinal epithelial cells of pig [81] and horse [82] appears, however, to be the glycolipid N-glycoloylneuraminyllactosyl ceramide [NeuGc(α2–3)Gal(βl–4)Glcβl–Cer],
3.2.2. Streptococcus suis
The Gram positive bacterium S. suis, a common cause of sepsis, meningitis and other serious infections in young piglets and also of meningitis in humans, agglutinates human erythrocytes, but not after treatment with sialidase. Resialylation of the desialylated erythrocytes with α2–3 sialyltransferase resulted in strong agglutination of the cells by the bacteria, whereas resialylation with sialyltransferases having different specificities gave cells that were poorly agglutinated [83]. Blotting experiments revealed binding to band 3, band 4.5 and glycophorin (as well as to polyglycosyl ceramides) of human erythrocyte membranes. The involvement of glycophorin as a ligand for the bacteria on intact cells is however excluded by the finding that trypsinization of the cells does not affect their agglutination by the bacteria and by the agglutinability of En(a−) erythrocytes which are defective in glycophorin A. The ligands for S. suis are thus the sialylated poly-TV-acetyllactosamine glycans carried by band 3 and band 4.5.
3.2.3. Streptococcus sanguis
S. sanguis, an oral microorganism, adheres to saliva-coated tooth surfaces by binding to salivary glycoproteins. The binding to these glycoproteins on SDS-PAGE blots was abolished by their treatment with sialidase, as well as with hydrofluoric acid, but was not affected by peptide-N-glycanase F (PNGaseF), indicating that S. sanguis binds to sialic acid on O-linked chains of the glycoprotein(s) [84]. A 23 kDa membrane glycoprotein from human buccal epithelial cells bearing O-linked NeuAc(α2-3)Gal(βl-3)GalNAc chains was implicated as receptor for S. sanguis OMZ9 on these cells [21].
The cDNA of the sialic-acid-specific lectin of S. sanguis codes for a polypeptide of 1435 residues (calculated mw of 158.4 kDa) with three unique domains, two of which consist of repetitive amino acid sequences [66], The third, which resides near the carboxy terminus, contains 48% proline. The lectin bound to a single salivary glycoprotein of mw 400 kDa [66]. Binding was inhibited by sialic acid and was abolished by desialylation of the glycoprotein; the best inhibitor was N-acetylneuraminyllactose.
3.2.4. Helicobacter pylori
H. pylori is a pathogen which colonizes the mucus layer of human gastric tissues and is associated with gastritis and peptic ulcers (and possibly also with gastric carcinoma). It exhibits several specificities, one of which is for sialic acid. H. pylori agglutinates erythrocytes [85] and binds to mouse adrenal gland cells [86] in a sialic-acid dependent fashion. Furthermore, specific binding of H. pylori to acid glycosphingolipids extracted from human gastric mucosa, such as the ganglioside GM3, has been reported [87]. In addition, H. pylori has an affinity for fucose (see section 3.4.1) and interacts with glycolipids such as cerebroside and sulfated lactosylceramide [88], which lack both sialic acid and fucose. The inhibitory activity of lactosylceramide sulfate and GM3 ganglioside on hemagglutination induced by H. pylori was additive, consistent with the possibility that two distinct lectins are involved in the binding to sialic acid and to sulfated glycolipids, respectively. The sialic-acid-specific lectin is a fibrillar surface protein with a mw of 20 kDa. Its cDNA has been cloned, sequenced and expressed in E. coli [89]. A sequence of the lectin (residues 134-139) was found to be homologous to a region that forms part of the carbohydrate binding sites of the sialic-acid-specific lectins of S-fimbriae, K99 and CFA I (cf. Fig. 3). An antibody against a synthetic peptide containing the above sequence blocked hemagglutination of human erythrocytes by H. pylori, suggesting that in this lectin, too, it is part of the carbohydrate binding site.
3.2.5. Mycoplasma pneumoniae
M. pneumoniae, a well-established pathogen of the human respiratory tract, is another organism specific for sialic acid. It adheres to animal cells primarily through a lectin, known as PI protein (mw 170 kDa), which is densely clustered at the tip of the organism [90]. The lectin is specific for N-acetylneuraminic acid linkedα2–3 to terminal galactose residues of the poly-A1'-acetyllactosamine sequence of blood type I/i antigen, as shown by binding experiments with sialidase-treated human erythrocytes that have been resialylated by specific sialyltransferases [91]. The preference for sialic acid α2–3 linked, rather than α2–6 linked, was confirmed by the finding that the oligosaccharides and glycoproteins containing the former linkage were more inhibitory than those containing the latter one. The most potent inhibitors were glycopeptides derived from bands 3 and 4.5 of human erythrocytes, and the bovine erythrocyte glycoprotein GP-2, all rich in poly-N-acetyllactosamine chains. Further evidence for the importance of such sequences in binding the M. pneumoniae lectin was provided by experiments with human blood type i erythrocytes, whose linear poly-N-acetyllactosamine chains are susceptible to digestion with endo β-galactosidase. Following treatment with the enzyme, the binding of M. pneumoniae to the erythrocytes decreased by 85% [92],
3.3. Gal and GalNAc-specific
3.3.1. Escherichia coli
CS3, a subcomponent of CFA II of enterotoxigenic E. coli binds specifically to GalNAc(βl–4)Gal [93], This was demonstrated by inhibition studies, using well-charac terized antibodies and glycoconjugates of defined structures. Support for these findings was provided by electron microscopic experiments showing that the disaccharide, O-linked to bovine serum albumin via a spacer, localized around bacteria expressing CS3 but not around CS3-negative mutants.
Enterotoxigenic E. coli producing K88 fimbriae occur in three serological variants: ab, ac or ad, that differ in their fine carbohydrate specificity. All are galactose-specific, but whereas K88ab fimbriae recognize the sequence Gal(αl–3)Gal [94], K88ac fimbriae ap pear to bind preferentially to Gal(βl-3)GalNAc and Fuc(αl-2)Gal(βl-3/4)GlcNAc [17]. These carbohydrate structures were shown to be present in two porcine brush border glycoproteins of 210 and 240 kDa that bind K88ac fimbriae, but not K88ab and K88ad fimbriae. They were not detected in glycoproteins from brush borders of piglets that do not bind K88 fimbriated E. coli.
3.3.2. Pseudomonas aeruginosa
P. aeruginosa, an opportunistic pathogen, capable of causing infections of eye, lung, skin and other parts of the body, produces two well-characterized intracellular lectins; one is specific for galactose (PA-I), the other for fucose (PA-II, see section 3.4.2) [95]. PA-I exhibits a preference for α-galactosides, especially those with a hydrophobic aglycone [96], The cDNA of PA-I was isolated and shown to encode a chain of 121 amino acids (mw 12.7 kDa) with a predominant central hydrophilic core between two hydrophobic domains [97], PA-I agglutinates papain-treated human erythrocytes independent of blood type. However, it exhibits preferential affinity for the branched oligosaccharides bearing both A and B blood group determinants that are present in the saliva of AB secretors [98].
Although PA-I is located mainly intracellularly, evidence has been obtained that it is also exposed on the cell surface [99], This could explain the finding that injection of the purified lectin into mice protected the animal against lethal doses of the live bacteria [95].
P. aeruginosa appears to bind largely to glycolipids [1], but binding to human respiratory mucins has also been reported. The binding is sensitive to periodate oxidation of the mucins, suggesting the involvement of the carbohydrate chains of the mucins and a (putative) surface lectin(s) on the bacteria [100], Inhibition studies have shown that the organism recognizes Gal(βl–3)GleNAc and Gal(βl–4)GlcNAc, but has no affinity for sulfated glycopeptides [101]. This lectin(s) has, however, not yet been isolated and its relation to PA-I is not known.
3.3.3. Actinomyces species
Actinomyces naeslundii and Actinomyces uiscosus are prominent oral bacteria that colonize tooth and mucosal surfaces by binding to epithelial cells or other bacteria, such as S. sanguis. These interactions are mediated by-galactose/β-N-acetylgalactosamine-specific lectins, associated with type 2 fimbriae [19], [20], [102]. Although isolated type 2 fim briae alone do not agglutinate either S. sanguis or sialidase-treated erythrocytes, lactose-specific agglutination occurred when the cells were incubated with multivalent complexes, formed by crosslinking the fimbriae with small amounts of specific antibody. The actino myceslectin has not yet been purified. A fimbrial subunit gene, fimA, has been cloned, but the protein it encodes is apparently not involved in the interaction with S. sanguis.
The carbohydrate specificities of the lectins of A. uiscosus T14V and A. naeslundii WVU45 were defined using galactose-containing oligosaccharides as inhibitors of coaggregation with S. sanguis 34. The most effective disaccharide inhibitor was Gal(βl-3)Gal, which was more than 10 times as active as lactose and also more active than any galactose disaccharide tested. Receptors for the actinomyces lectins have been isolated and extensively characterized from four streptococcal strains. All are linear cell wall polysaccharides, composed of repeating hexa- (or hepta-) saccharide units linked by phosphodiester bonds to the 6-carbon of the non-reducing terminal sugar of the repeating unit and all contain A'-acetylgalactosamine. Two of them contain the sequence Gal/(βl-6)GalNAc(βl-3)Gal(al-), in the other two it is Gal/(βl-6)Gal(βl-3)GalNAc(al-) [102a]. This region is considered to be important in determining the recognition of the streptococci by the actinomyces lectins.
The binding of A. naeslundii WVU45 to sialidase-treated monolayers of epithelial cells was inhibited by pretreatment of the latter with peanut agglutinin and Bauhinia purpurea lectin [103]. Although these two lectins differ in their fine specificity, both react well with Gal(βl-3)GalNAc, and similar to the actinomyces lectins, their binding to epithelial cells is enhanced by treatment of the cells with sialidase. In contrast, Erythrina cristagalli lectin, specific for Gal(βl–4)GlcNAc, failed to inhibit bacterial binding. These and other experiments with lectins led to the conclusion that the receptor for the actinomyces lectin on epithelial cells is most likely O-linked Gal(βl-3)GalNAc. A. naeslundii binds to a glycoprotein of mw 160 kDa extracted from oral epithelial cells [67].
A. naeslundii WVU45, as well as some other strains of actinomyces expressing type 2 fimbriae, bind also to sialidase-treated polymorphonuclear leukocytes, resulting in the activation of the latter cells, phagocytosis and destruction of the bacteria. The interaction of A. naeslundii WVU45 with the leukocytes was inhibited by the same lectins that inhibited the binding of these bacteria to oral epithelial cells. The receptors for the organism on the surface of sialidase-treated polymorphonuclear cells were identified as a 130 kDa glycoprotein as well as asialoganglosides with Gal(βl-3)GalNAe termini [104],
Certain actinomyces species, such as A. naeslundii 12104 and A. uiscosus 19246 and 147, have an affinity for GalNAcβ-terminating oligosaccharides. These strains exhibit heterogeneous receptor specificities and bind to different salivary and submaxillary glycoproteins on blots [20]. A 180 kDa salivary glycoprotein that binds A. naeslundii has been isolated and characterized [68].
A special type of coaggregation of oral bacteria is that resulting from the bridging between one cell type and its partner by a third organism. This happens with Preuotella loescheii PK1295, which can serve as a bridge between Streptococcus oralis 34 and Actinomyces israelii PK14, two Gram positive oral bacteria that are otherwise unable to coaggregate. Coaggregation of P. loescheii PK1295 with S. oralis 34 is inhibited by lactose, while that with A. israelii PK14 is not, indicating that two different kinds of adhesins are involved, at least one of which is a lectin [105]. The latter has been isolated and purified to electrophoretic homogeneity, the first from oral bacteria. It is a fimbria-associated protein with a molecular weight of 450 kDa and consists of six identical subunits. In its oligomeric form it agglutinates S. oralis 34 and a variety of sialidase-treated erythrocytes in a lactose-sensitive manner, while the individual protomers blocked coaggregation between P. loescheii PK1295 and S. oralis 34 [106].
3.3.4. Rhizobia
A lectin specific for galactose has been isolated from Bradyrhizobium japonicum [107], [108]. It is a protein of 38 kDa that binds lactose about 15 times better that galactose and does not recognize N-acetylgalactosamine. The lectin is localized at one pole of the bacterial cell surface, which is coincident with the site of cell-cell contact in homotypic aggregation of the bacteria and in their adhesion to the cultured soybean cell line SB-1. Such topological distribution is consistent with a role for the lectin in the polar binding of the organism to soybean roots.
3.3.5. Myxobacteria
Myxobacteria differ from other bacteria in being social organisms. They tend to maintain close contact with each other and to aggregate into swarms. Aggregation is a developmentally regulated process that occurs as the cells differentiate from the vegetative form into mature spores. In Myxococcus xanthus, high hemagglutinating activity was found in extracts of the mature cells, but not in early vegetative cells. A lectin was purified from extracts of the aggregated stage of this organism. In solution it exists as a monomer with an apparent molecular weight of 28 kDa. The hemagglutinating activity of the lectin was not inhibited by simple sugars, only by glycoproteins such as fetuin, glycophorin and rabbit IgG, all of which contain the O-linked tetrasaccharide NeuAc(α2–3)Gal(βl–3)[NeuAc(α2–6)]GalNAc. The penultimate galactose was directly implicated in the affinity of the lectin for the saccharide, since inhibition by asialofetuin was diminished to one-fifteenth by β-galactosidase treatment. The lectin was detected on the surface of developmental, but not vegetative cells, localized in distinct patches at one or both of the cell poles. This localization suggests that the lectin may function in end-to-end cellular interactions during aggregation.
3.4. Fucose-specific
3.4.1. Helicobacter pylori
A specificity exhibited by H. pylori (in addition to that for sialic acid) is for fucose. The putative lectin discriminates between closely related fucosylated epitopes as well as carbohydrate core chains, and only binds H and Lewisb antigens expressed on lacto-series type 1 but not type 2 chains [109]. To test the hypothesis that Leb antigen functions as receptor for H. pylori and mediates its attachment to gastric pit/mucous cells [110], mice that normally do not synthesize this carbohydrate structure were genetically engineered to produce it by transfection with human αl,3/4 fucosyltransferase [111]. Expression of Leb in the transgenic mice was associated with acquisition of the ability to bind clinical isolates of H. pylori. Binding was blocked by pretreatment of the bacteria with soluble Leb–serum human albumin conjugates.
3.4.2. Pseudomonas aeruginosa
PA-II, an intracellular lectin of P. aeruginosa, is specific for fucose with an unusually high affinity (K a = 1.5 × 106 M−1) and interacts weakly also with mannose (Ka = 3.1 × 1O2 M−2) [112].
3.4.3. Vibrio cholerae
Agglutination of human group O erythrocytes by V. cholerae and adhesion of the organism to brush borders are specifically inhibited by fucose, and to a lesser extent by mannose. It has been suggested that structures on eukaryotic cell surfaces containing fucose may function as receptors for a vibrio lectin and may therefore be an important determinant of host susceptibility to these bacteria. A soluble lectin specific for fucose, produced by V. cholerae strain CA401 was purified to apparent homogeneity and was found to focus at three different pi: 6.3, 5.3 and 4.7 [113], Thus, there are apparently three distinct pi isotypes of the lectin that exist as non-covalently associated polymers of 32 kDa subunits. The lectin possessed proteolytic activity, which likewise focused at pH values 6.3, 5.3 and 4.7. It was therefore concluded that the soluble lectin is a bifunctional molecule, capable of mediating hemagglutination and proteolysis.
3.4.4. Others
Recently, a fucose-specific lectin associated with the bacterial surface of Rhizobium lupinii has been purified, but its role in the interactions between the bacteria and the lupin root has not been established [114], A fucose-specific lectin has also been isolated from the cell walls of Agrobacterium tumefaciens, a bacterium belonging to the Rhizobiaceaea family which infects dicotyledonous plants and forms crown gall [115].
3.5. Multiple specificities
3.5.1. Bordetella pertussis
Pertussis toxin, produced by virulent strains of B. pertussis, the etiological agent of whooping cough, is a classical A–B type toxin comprised of an A subunit that possesses ADP-ribosyltransferase activity and is responsible for most of the biological effects of the toxin, and a B subunit with affinity for carbohydrates. The B subunit of the pertussis toxin is a pentamer composed of four different subunits (S2–S5). The toxin acts as a hemagglutinin and exhibits dual carbohydrate specificity, due to the presence of a subunit (S2) that binds galactose-containing glycoconjugates such as lactosylceramide, and a subunit (S3) that recognizes sialylated glycoconjugates (e.g. gangliosides). Competitive inhibition studies using recombinant subunits indicated that both S2 and S3 mediate, in an additive manner, the toxin dependent attachment of B. pertussis to human macrophages [116]. It was suggested, therefore, that each subunit interacts with a different receptor(s).
The two subunits exhibit 80% homology. Their amino terminal regions contain a hexapeptide (residues 18–23) homologous to a sequence (residues 62–67) found in the carbohydrate binding pocket of wheat germ agglutinin (WGA); mutations in this hexapeptide of S2 abolished carbohydrate recognition [117]. The WGA-like region is followed by a segment (residues 30–53) which shows sequence similarity to selectins [118], a family of C-type eukaryotic lectins [119] (Fig. 4 ). The amino termini of S2 and S3 are thus a composite of a plant and an animal lectin arranged in tandem. This region appears to be important in determining the carbohydrate specificity of the subunits. Chimeric recombinant subunits S2 and S3, obtained by mutational interchange of amino acid residues 37–52, exhibited switched specificities, i.e. S2 became specific for gangliosides and S3 for lactosylceramide [120]. Concomitantly, a switch of target cell specificity occurred with respect to macrophages and ciliated cells. Mutations at Asn93 or Asn105 of S2 [121] or at Tyr82 or Lys105 of S3 [122] abolished carbohydrate binding. The sequence similarity with selectins extends to shared function, in that S2 and S3 inhibit competitively the interaction of neutrophils with surfaces coated with P-selectin and E-selectin, respectively [123],
Fig. 4.

Sequence homology between subunits S2 and S3 of Bordetellu pertussis toxin and the selectins. Identical residues are underscored. Gaps have been introduced for optimal alignment.
(Modified from ref. [123].)
The recent solution of the crystal structure of a dimeric form of pertussis toxin provided support for the composite nature of the molecule [124]. Thus, the WGA-like region of S2 superimposes on the homologous part of the three-dimensional structure of WGA (residues 62–67), while the segment homologous to the C-type lectins superimposes on the core of rat mannose binding protein, a C-type lectin.
The crystal structure of the dimeric form of the toxin in complex with a diantennary undecasaccharide isolated from human serum transferrin was determined at 3.5 Å [125]. The carbohydrate is bound to equivalent sites on S2 and S3 of the B oligomer, occupying three of the four combining sites present in the dimeric unit. Despite the relatively low resolution, NeuAc(α2-6)Gal (which forms the terminus of each branch of the undecasaccharide) was clearly identified in the binding sites with the aid of energy calculations and NMR spectroscopy. The sialic acid forms both hydrogen bonds and hydrophobic interactions with the protein, while no contacts with the galactose are seen. Surprisingly, in the crystal structure, S2 and S3 interact with sialic acid in the same way, although the two subunits appear to exhibit different sugar specificities in solution.
In vitro, the toxin binds to glycoproteins as well as glycolipids. The ability of fetuin to inhibit hemagglutination of goose erythrocytes by the toxin was abolished by treatment of the glycoprotein with sialidase and was restored by resialylation with the aid of α 2-6 sialyltransferase [126]. On blots of separated proteins from Chinese hamster ovary (CHO) cells, the intact toxin, as well as the purified B subunit, bound a single band of 165 kDa; sialidase treatment of the blots abolished binding [69], With lectin-resistant CHO mutants deficient in sialic acid, no toxin-binding component was revealed [127], The toxin did not bind to neutral glycolipids or gangliosides from CHO cells, nor to sialoparagloboside, although the latter terminates in NeuAc-N-acetyllactosamine. It appears thus that the 165 kDa glycoprotein is the receptor for pertussis toxin on CHO cells.
Toxin-binding proteins on human peripheral T lymphocytes, as well as on T-cell-derived lines (HPB-ALL and Jurkatt), all of which respond to pertussis toxin by rapid second messenger production, were detected by labeling the cells with the toxin bound to a crosslinking agent, followed by gel electrophoresis, and by binding of the toxin to blots of SDS-PAGE separated membrane proteins [70]. In all cases, a single band of 43 kDa was revealed, which was absent from non-responsive cells. The 43 kDa protein may thus be a functional receptor for the toxin. In a different report, also using peripheral T lymphocytes and Jurkatt cells and a similar labeling technique as above, a protein of 70 kDa was detected [71]. A band of the same mobility was found among several components isolated from surface-iodinated T lymphocytes by affinity chromatography on the immobilized toxin. However, it cannot be concluded that binding to this (glyco)protein was carbohydrate-mediated, since no controls with inhibitory sugars were carried out, and elution from the affinity column was with high pH and not with carbohydrate.
In addition to the toxin, virulent strains of B. pertussis express another lectin, known as filamentous hemagglutinin. It is found in a cell-associated form outside the outer membrane filamentous hemagglutinin. It is found in a cell-associated form outside the outer membrane binding with extracts of ciliated cells or standard glycolipids and has also an affinity for sulfated saccharides, such as Gal-6-sulfate, dextran sulfate and heparin. The carbohydrate binding region of the lectin was mapped to amino acids 1141— 1279 with the aid of a monoclonal antibody that blocked bacterial binding to ciliated cells and to lactosylceramide [128], Mutant strains of B. pertussis with deletions of this region did not bind to ciliated cells or macrophages and reacted poorly with the antibody. The region contains a stretch exhibiting sequence similarity with 20 amino acids (30–51) of the lectin domain of S2 of the toxin.
4. Fungi
Lectins have been found in some soilborne plant pathogenic fungi, such as Rhizoctonia solani and related species [129], [130] and in different members of the Sclerotiniaceae [131], [132], Most of these lectins are specific for galactose/N-acctylgalactosamine although some (e.g. that from Sclerotium rolfsii [132]) recognize only animal glycoproteins such as fetuin and mucin. A lectin specific for N-acetylgalactosamine has also been isolated from the nematode-trapping fungus Arthrobotrys oligospora [133]. From the same source a protein has been obtained which, although immunologically identical with that mentioned above, is not inhibited by N-acetylgalactosamine, only by glycoproteins [134].
5. Protozoa
Only a few protozoan lectins are known (Table 3 ).
Table 3.
Carbohydrate specificity of protozoal lectins
5.1. Gal and GalNAc-specific
5.1.1. Entamoeba histolytica
The pathogenic protozoan E. histolytica causes disease in humans by disruption and invasion of the colonic mucosa. It produces two lectins, specific for Gal/GalNAc [135] and for chitooligosaccharides, respectively (for the latter lectin, see section 5.3.1). The Gal/GalNAc-specific lectin has a mw of 260 kDa and is composed of two subunits (170 and 31/35 kDa) linked by disulfide bonds [136], The heavy subunit is an integral membrane protein with a large cysteine-rich extracellular domain and a short cytoplasmic tail [137], [138]. The two types of light subunit are synthesized from the same cDNA and the difference in molecular weight is probably the result of post-translational modifications, possibly of glycosylation [139]. The 31 kDa (but not the 35 kDa) isoform of the light subunit contains a phosphatidylinositol-glycan anchor; the 170/31 kDa isolectin is thus an unusual protein, in that it is composed of a transmembrane and a phosphatidylinositolglycan anchored component.
The purified lectin partially inhibits binding of amoebic trophozoites to cultured human intestinal epithelial and CHO cells. In addition, antibodies directed to the 170 kDa subunit inhibit binding of the amoeba to CHO cells, suggesting that this subunit is primarily responsible for mediating adhesion. Interestingly, certain monoclonal anti-heavy chain antibodies caused an enhancement of galactose-inhibitable adhesion of the parasite to CHO cells [140]. One of the antibodies also enhanced binding of the purified lectin to such cells, indicating that enhanced adhesion of the parasite may be due to direct activation of the galactose-binding activity of the lectin. It is thus possible that amoebae regulate their in vivo adhesion to galactose-containing substrates via changes in lectin activity. The epitopes for both the adhesion-inhibiting and adhesion-enhancing antibodies are located in the cystein-rich domain of the 170 kDa subunit, suggesting that the carbohydrate binding site may reside in this region [141].
The extent of binding of E. histolytica to wild-type CHO cells and three lectin-resistant CHO mutants with altered glycosylation patterns was directly correlated with the presence of terminal galactose residues on these cells: very poor binding was observed to CHO cell mutants deficient in galactose [142]. Moreover, galactose derivatives, in particular N-acetyllactosamine, efficiently blocked binding of the amoebae to CHO cells, while no inhibition was seen with N -acetylglucosamine or N, N'-diacetylchitobiose. The adhesion of the parasite to CHO cells was also completely inhibited by very low concentrations (1 μg/ml) of purified human colonic mucin and this inhibition was galactose-specific [143]; direct binding experiments have shown that the amoebic lectin binds the mucin with a very high affinity (Kd = 8 × 10- 11 M- 1). Therefore, the galactose-specific, rather than the N-acctylglucosamine-specific, lectin is most probably responsible for the recognition of mammalian cells by E. histolytica.
Specificity studies with a series of multivalent glycoconjugates with non-reducing terminal galactose or N-acetylgalactosamine residues confirmed the preference of the lectin for the latter monosaccharide and revealed a dramatic increase in the affinity of the lectin for its ligands when presented in multivalent form [144]. A neoglycoprotein having an average of 40 galactose residues linked to bovine serum albumin was about 17 000 times more potent as inhibitor of hemagglutination by the lectin than the monovalent sugar, and a neoglycoprotein with an average of 39 N-acetylgalactosamine residues was 140 000-fold more potent than the monosaccharide. The latter polymer bound to membranes of E. histolytica with a dissociation constant of about 10nM. These findings are in line with the above mentioned high affinity of the lectin for human colonic mucin and support the possibility that this class of glycoprotein is a potential target for E. histolytica binding in vivo.
5.2. Sialic-acid-specific
5.2.1. Plasmodium falciparum
The invasion of human erythrocytes by plasmoidal merozoites is a key event during malaria infection. Sialidase treatment of the erythrocytes renders the cells resistant to invasion by different isolates of P. falciparum, suggesting that the interaction between the parasite and the erythrocytes is mediated by a plasmodial lectin specific for sialic acid [145], Different mezoroite surface components have been proposed as the putative lectins, among them the well-defined surface antigen Pf200 [146]. Binding of the isolated antigen to erythrocytes was inhibited by sialidase treatment of the cells and by monoclonal antibodies against the glycosylated domain of glycophorin.
A protein of 175 kDa (designated EBA-175), which is released from merozoites into the culture medium, was purified by binding to, and elution from, human red blood cells [147]. Its interaction with the erythrocytes was inhibited best by NeuAc(α2-3)Gal and less by NeuAc(α2-6)Gal; free N-acetylneuraminic acid was not inhibitory. The structure of the sialic acid is critical, because removal of the 9-O-acetyl group from mouse erythrocytes, which converted the mouse sialic acid to the human form, enhanced binding of EBA-175 to the mouse cells [148].
The cDNA of the lectin was cloned; it codes for a polypeptide chain of 1435 amino acids, including a 19 residue leader sequence [149]. Antibodies against a synthetic peptide encompassing amino acid residues 1062–1103 inhibited merozoite invasion in vitro, as well as binding of the purified lectin to erythrocytes, indicating that this region may include the carbohydrate combining site of the lectin. In line with this possibility was the finding that the nucleotide sequence of this region is conserved among several plasmodium strains from various regions of the world. The inhibitory activities of the anti peptide antibodies were shown to be primarily, if not exclusively, directed against amino acids 1069–1087 [150]. Antibodies against a peptide corresponding to the above amino acid stretch inhibited the growth of the parasite similarly to antibodies against the larger peptide and, moreover, blocked the interactions of the latter antibodies with the purified lectin and with the parasite. Surprisingly however, using truncated portions of the lectin expressed in COS cells, the binding region was mapped to a cystein-rich domain closer to the N-terminal of the protein [151] The fragment (amino acids 487–673, designated as region F2) bound erythrocytes with the same pattern as native EBA-175.
The receptors for the plasmodial sialic-acid-specific lectin on human erythrocytes are the O-linked chains of glycophorin. Cells devoid of glycophorin (MKMK erythrocytes), or having glycophorin with modified O-chains (Tn and Cad) are, to a large extent, resistant to invasion [147]. Interestingly, this is also the case with En(a-) erythrocytes that contain glycophorin B, but not glycophorin A. These cells also failed to bind to EBA-175 and fragment F2 [151]. The two glycophorins contain the same 26 amino acids and eleven clustered O-linked chains at the N-terminus, but differ in the rest of the molecule. This was taken to indicate that an amino acid sequence specific for glycophorin A is necessary for binding, possibly by contributing an unique conformation to the sialic acid residues recognized by the lectin. This is supported by the finding that glycopeptide 1–64 of glycophorin A, but not a mixture of glycopeptides 1–34 and 35–64, inhibited the binding of erythrocytes to EBA-175 [151].
5.3. N-Acetylglucosamine- and chitooligosaccharide-specific
5.3.1. Entamoeba histolytica
The E. histolytica lectin specific for chitooligosaccharides is a 220 kDa plasma membrane glycoprotein that inhibits the attachment of amoebic tropozoites to layers of cultured MDCK cells [152], [153].
5.3.2. Plasmodium falciparum
Besides the sialic-acid-specific lectin (see section 5.2.1), isolates of P. falciparum from different geographical regions express a lectin specific for N-acetylglucosamine [154]. The merozoite surface of such isolates was specifically labeled with a neoglycoprotein bearing multiple residues of this monosaccharide, as evidenced by fluorescence and electron microscopy. Affinity chromatography of merozoite lysates on immobilized N-acetylglucosamine and elution with the free sugar yielded three protein bands of 45, 83 and 120 kDa. The same bands were revealed when blots of merozoite proteins were probed with the N-acetylglucosamine containing neoglycoprotein.
5.4. Mannose-6-phosphate-specific
5.4.1. Giardia lamblia
Giardia lamblia, a protozoan parasite that causes widespread diarrheal disease, expresses a surface membrane associated lectin, named taglin, which is specifically activated by limited proteolysis with trypsin, a protease present in abundance at the site of infection [155]. Trypsin-treated lysates of G. lamblia trophozoites agglutinate rabbit erythrocytes as well as enterocytes, which are the target cells of the parasite in vivo. The agglutination was best inhibited by mannose 6-phosphate; mannose 1-phosphate and other phosphorylated sugars were not inhibitory. A monoclonal antibody which inhibited hemagglutination by the activated lysate, and is thus presumably directed against the lectin, recognized a protein of 28/30 kDa on immunoblots of trophozoite lysates. The lectin activity of this protein was demonstrated by binding of erythrocytes to bands in the same molecular weight range as those recognized by the monoclonal antibody. Immunoprécipitation of G. lamblia lysates with the monoclonal antibody also yielded a protein of 28/30 kDa, which has however not been further characterized.
6. Biological roles
6.1. Infection
A major role of the microbial lectins is to mediate the adhesion of the organisms to host cells, an initial stage of infection. This has been extensively demonstrated both in vivo, in studies with isolated cells and cell cultures, and in vivo in experimental animals.
6.1.1. Viruses
The binding of the influenza virus lectin (hemagglutinin) to carbohydrates containing sialic acid on the surface of the target cells leads to the attachment of the virus to the cells. This is followed by fusion of the viral and cellular membranes, allowing release of the viral genome into the cytoplasm and subsequent replication. Removal of sialic acid from the cell membranes by sialidase abolishes binding and prevents infection, while enzymatic reattachment of sialic acid or insertion of sialic-acid-containing oligosaccharides (for example, in the form of glycolipids) into the membranes of sialidase-treated cells restores the ability of the cells to bind the virus and to be infected by it [26], [27]. Detailed knowledge of the sialic-acid-hemagglutinin interaction (cf. section 2.1.1) provides a basis for the attempts to design antiviral drugs that would block viral attachment to cells. An inhibitor, targeted to the conserved amino acids of the combining site or to the receptor carbohydrate, might be effective against influenza viruses of different subtypes. It would be independent of the antigenic changes that accompany the recurrent epidemics for which these viruses are renowned.
6.1.2. Bacteria
In the case of bacteria that possess the ability to produce fimbriae, the infectivity of the fimbriated phenotype is, as a rule, higher than that of the non-fimbriated [2], [4], Thus, when mice were infected intravesicularly with two isogenic E. coli mutants, both of which were type 1 fimbriated, but only one of which possessed mannose-binding activity, only the latter induced urinary tract infections. Also, infection by enterobacteria carrying fimbrial lectins was prevented by passive or active pre-immunization of the animals against the fimbriae. Although the anti-fimbrial antibodies inhibited bacterial adhesion in vitro, there is no compelling evidence that they are directed against the carbohydrate binding sites of the fimbrial lectin.
More significantly, in three separate systems, each employing different type 1 fimbriated enterobacterial species, mannose and methyl α-mannoside inhibited infection of the urinary tract of mice and rats (Table 4 ). In each of these glucose (or methyl α-glucoside) which is not an inhibitor of type 1 fimbriae, did not affect the infectivity of the injected bacteria. In addition, antibodies against the carbohydrate structures recognized by type 1 fimbriae on epithelial cells also prevented urinary tract infection in mice by type 1 fimbriated E. coli. Similarly, N-acetylneuraminie acid considerably reduced colonization of lung, liver and kidney by P. aeruginosa injected intravenously mice [156]. It was also demonstrated that introduction into the trachea of rabbits infected with B. pertussis of lactose or monoclonal anti-Lea antibodies resulted in the abortion of colonization of the respiratory tract by the bacteria and blocking of pulmonary edema [157] (Fig. 5 ). These findings provide some of the most convincing evidence for the central role of bacterial lectins in infection, in particular in mucosal colonization. They also illustrate the great potential of simple carbohydrates in the prevention of infections caused by bacteria that express surface lectins and raise hopes for the development of anti-adhesive drugs for human use.
Table 4.
Inhibitors of sugar-specific adhesion prevent infection in animals and humans
| Organism | Animal | Site of infection | Inhibitor | Ref. |
|---|---|---|---|---|
| Type 1 fimbriated | ||||
| Eschericha coil | Mice | Urinary tract | MeaMan | [4] |
| Mice | Gastrointestinal tract | Mannose | [4] | |
| Mice | Urinary tract | Anti-Man antibody | [4] | |
| Klebsiella Pneumoniae | Rats | Urinary tract | MeaMan | [4] |
| Shigella flexnerii | Guinea pigs | Eye | Mannose | [4] |
| K99 fimbriated | ||||
| E. coli | Calvas | Gastrointestinal tract | Glycopeptides of bovine plasma glycoproteins | [158] |
| Bordetella pertussis(toxin) | Rabbit | Lung | Lactose,anti-Lea antibody | [166] |
| Pseudomonas aeruginosa | Man | External auditory canal | Gal + Man + NeuAc | [159] |
Fig. 5.

Effect of adhesion inhibitors (A) on the colonization of the respiratory tract by Bordetella periussis and (B) on pulmonary edema in animals challenged with the virulent BP536 strain. Anti-Lea and anti-A are antibodies against blood group determinants Lea and A, respectively; anti-CR3 is the antibody against the receptor for complement fragment C3bi. VIR +, control without inhibitor.
(Modified from ref. [157].)
Other reports also support a role for bacterial lectins in natural infections. Sialylated glycoproteins, administered orally, protected colostrum-deprived, newborn calves against lethal doses of enterotoxigenic E. coli K99[158]. In a clinical trial in humans, patients with otitis externa (a painful swelling with secretion from the external auditory canal) caused by P. aeruginosa were treated at the site of infection with a solution of galactose, mannose and N-acetylneuraminic acid. The results were fully comparable to those obtained with conventional antibiotic treatment [159].
Expression of the bacterial lectins during the infection process is not always beneficial to the survival of the bacteria. Thus, type 1 fimbriated bacteria may adhere to phagocytic cells, leading to ingestion and killing of the bacteria (see section 6.2) or combine with soluble glycoproteins such as IgA. Since colonic mucosa is known to be rich in IgA2 secreting cells, it has been suggested that one important function of secretory IgA could be to bind and agglutinate intestinal bacteria and thus prevent their attachment to host cells [55]. Tamm-Horsfall glycoprotein binds mannose-specific E. coli via its single oligomannose unit [56] and may thus serve as a vehicle for the clearance of bacteria from the urinary tract.
A novel mechanism for lectin-dependent adhesion has been proposed in the binding of P. aeruginosa to animal cells, based on the observation that when lysates of this organism were mixed with the intact bacteria, the latter acquired the ability to bind to corneal epithelial culture cells and that the binding was specifically inhibited by galactose and mannose [160]. It was therefore proposed that the intracellular lectins of P. aeruginosa (PA-1 and PA-2, cf. 3.3.2, 3.4.2), released from the bacteria in vivo, bind to surviving intact cells and mediate adhesion of the organisms to galactose- or mannose-containing glycoconjugates on epithelial cells. The involvement of mannose residues as receptors for P. aeruginosa is supported by studies showing that concanavalin A, a mannose-specific lectin, inhibited binding of the bacteria to corneal epithelia [161].
The β-galactoside-specific lectins produced by oral actinomyces, such as A. naeslundii and A. viscosus, facilitate initial colonization of epithelial surfaces of the mouth and teeth by mediating the attachment of the bacteria to galactose residues either on the surface of the epithelial cells, or on the surface of other bacteria (for example, S. sanguis) which are adsorbed to the enamel of the teeth [162],
6.1.3. Fungi
The lectins of pathogenic fungi have been implicated in the specific interactions between plant pathogens and Trichoderma, a natural antagonist to other fungi and a well-known biocontrol agent, leading to the destruction of the pathogen [163]. Direct evidence for this hypothesis was obtained with the aid of a biomimetic system, in which nylon fibers coated with a fungal lectin simulate hyphae of the fungus from which the lectin is derived. When Trichoderma harzianum was allowed to grow on fibers coated with the purified lectin from Sclerotium rolfsii, it coiled around the nylon fibers and produced hooks in a pattern similar to that observed with the real host hyphae [132] (Fig. 6 ). The incidence of interaction was significantly (six times) higher with lectin-treated fibers than with untreated ones.
Fig. 6.

Scanning electron micrographs of the different stages of the interaction between Trichoderma harzianum and nylon fibers coated with purified Sclerotium rolfsii lectin. (A) A typical branching of the Trichoderma towards the fibers and contact of the branch tip with the fiber surface (bar = 10 pm); (B) subsequent elongation of the firmly attached tip along the fiber surface (bar = 1 pm); (C) Trichoderma hyphae coiled fibre, producing additional branches. The hyphal coils and branches adhere tightly to the fiber surface (bar = 10 pm).
(From ref. [132], by permission from publisher.)
6.1.4. Protozoa
The inhibitory effect of the purified Gal/GalNAc-specific lectin of E. histolytica and of anti-lectin antibodies on the binding of the parasite in vitro to animal cells and on the contact-dependent killing of the latter prompted attempts to use the lectin as a protective agent in vivo. Pre-immunization of gerbils with the purified lectin provided complete protection from liver abscesses in a majority (67%) of animals injected intrahepatically with E. histolytica trophozoites [164]. However, the sera of the immunized animals, while completely blocking adhesion at 1:10 dilution, increased adhesion when tested at 1:1000 dilution. This finding, which is analogous to the enhancement of adhesion by certain monoclonal anti-lectin antibodies mentioned above, complicates attempts to understand the role of adhesion-blocking antibodies in protection against disease. The ability to elicit protective immunity has now been mapped to a cysteine-rich fragment of the lectin, encompassing amino acids 758-1134 [165].
Facilitating the adhesion of amoebae to intestinal epithelial cells of the host is only one of the ways in which the lectins may enhance the pathogenicity of the parasite. For instance, the Gal/GalNAc-specific lectin increases the resistance of E. histolytica trophozoites to complement-dependent killing [166]. Also, once invasion has taken place and the amoebae have spread through the host, the lectins mediate binding of the parasite to other cells and tissues, in particular to hepatocytes, initiating the killing of these cells. In addition, the lectins enable the amoebae to bind bacteria carrying the appropriate sugars. The bound bacteria are subsequently ingested and serve as a source of nutrition for the parasite, increasing its virulence [167].
6.2. Non-opsonic phagocytosis
Different microorganisms in serum-free media attach readily via their surface lectins to phagocytic cells. Such attachment in the absence of opsonins is often followed by activation of the phagocytes and ingestion of the bacteria; sometimes, killing of the bacteria is observed. The process has been named lectinophagocytosis, in analogy to opsonophagocytosis, in which recognition between the microorganisms and the phago cytic cells is mediated by serum constituents termed opsonins (mainly IgG antibodies and the C3b and C3bi fragments of the C3 component of complement) [158], [169]. The best characterized system of lectinophagocytosis is that of bacteria carrying mannose-specific lectins, mainly E. coli, in the form of type 1 fimbriae. Shigella flexneri M90T, a non fimbriated organism that, unless opsonized, does not bind to or activate phagocytic cells, when transfected with the cluster of genes encoding type 1 fimbriae of E. coli, bound to human granulocytes and mouse peritoneal macrophages, and activated the latter [170]. As already mentioned (section 3.1.1), the adhesion molecules CD11/CD18 serve as the major leukocyte receptors for type 1 fimbriae [59], [60], Because CD1 lb,c/CD18 constitute the receptor CR3 for complement fragment C3bi, the findings indicate that the receptors that participate in opsonophagocytosis function in lectinophagocytosis as well.
Lectinophagocytosis mediated by bacterial surface lectins also occurs with type 2 fim briated Actinomyces species, specific for Gal(βl-3)GalNAc and Gal(βl—4)Glc [171] (cf. section 3.3.3). Phagocytosis of mutants deficient in such fimbriae is negligible. Binding and phagocytosis are markedly enhanced by pretreating the granulocytes with sialidase.
B. pertussis too adheres to, and is internalized by cultured macrophages in vitro, and alveolar macrophages in vivo, in the absence of opsonins. It is, however, not killed, but persists within the cells. Either one of the lectins (the filamentous hemagglutinin or the toxin) can mediate the adhesion of the bacteria to the macrophages. In this case, however, the hemagglutinin does not act as a lectin, since it binds through its Arg-Gly-Asp sequence rather than through the carbohydrate combining site; this binding seems to be a prerequisite for the subsequent uptake of the bacteria. The adhesion of B. pertussis to macrophages proved to be a cooperative process, involving both the toxin and the hemagglutinin. Pretreatment of the macrophages with the whole toxin, the B subunit or recombinant S2 or S3 enhanced the adhesion of hemagglutinin-producing bacterial strains, but had no effect on adhesion of hemagglutinin-deficient bacteria [116]. The enhanced binding was CDllb/CD18 dependent, as demonstrated by increased binding of erythrocytes coated with C3bi (known to bind to the above antigens) to macrophages treated as above. These results suggest a communication between the receptor(s) for the toxin and the integrin [116].
It has been argued that lectinophagocytosis may function as a defence mechanism against microbial infections in vivo at sites, such as the renal medulla and peritoneal cav ity, especially during dialysis, or in situations (e.g. in patients infected by microorganisms prior to the development of an immune response) where opsonic activity is poor [168]. Indirect evidence for such a role came from experimental infections with mixed bacterial phenotypes (isogens) of E. coli, one of which was type 1 fimbriated and the other of which was non-fimbriated. Whenever the organisms reached phagocyte-rich sites, those with the non-fimbriated phenotype survived, while at phagocyte-poor sites those with the fimbriated phenotype survived, irrespective of the bacterial species employed or the route or site of infection. The selective survival of the non-fimbriated phenotype in phagocyte-rich sites was attributed to elimination of the fimbriated phenotype by phagocytes. It was suggested that phase variation, a random on–off process that allows the bacteria to alternate between fimbriated and non-fimbriated states, and thus to choose the phenotype conducive for their survival, is an important virulence trait of type 1 fimbriated bacteria.
Recent experiments provide direct evidence for the possibility that lectinophagocytosis occurs in vivo. Injection of type 1 fimbriated, but not of non-fimbriated, E. coli cells into the peritoneal cavity of mice led to the activation of peritoneal macrophages, as measured by the release of the lysosomal enzyme, N-acetyl-β-glucosaminidase [172]. Methyl coinjected into the peritoneum with the bacteria, specifically inhibited the release of the enzyme. Furthermore, no release was observed following injection of fimbriated bacteria into a macrophage-depleted peritoneum. If indeed lectinophagocytesis will prove to be an important defense mechanism against infections, it should be taken into account in attempts to develop anti-adhesive drugs, especially those based on blocking carbohydrate mediated recognition by different infectious agents.
Footnotes
All sugars are of the D-configuration, except
References
- 1.Karlsson K.-A. Curr. Opin. Struct. Biol. 1995;5:622–635. doi: 10.1016/0959-440x(95)80054-9. [DOI] [PubMed] [Google Scholar]
- 2.Sharon N. In: The Lectins: Properties, Functions and Applications in Biology and Medicine. Liener I.E., Sharon N., Goldstein I.J., editors. Academic Press; Orlando: 1986. pp. 493–526. ch. 9. [Google Scholar]
- 3.Mirelman D., editor. Microbial Lectins and Agglutinins. John Wiley and Sons; New York: 1986. [Google Scholar]
- 4.Ofek I., Sharon N. Curr. Top. Microbiol. Immunol. 1990;152:91–113. doi: 10.1007/978-3-642-74703-8_5. [DOI] [PubMed] [Google Scholar]
- 5.Ofek I., Doyle R.J. Bacterial Adhesion to Cells and Tissues. Chapman and Hall; New York, London: 1994. [Google Scholar]
- 6.Hultgren S.J., Abraham S., Caparon M., Falk P., St. Geme J.W., III., Normark S. Cell. 1993;73:887–901. doi: 10.1016/0092-8674(93)90269-v. [DOI] [PubMed] [Google Scholar]
- 7.Yu C., Lee A.M., Bassler B.L., Roseman S. J. Biol. Chem. 1991;266:24260–24267. [PubMed] [Google Scholar]
- 8.Sharon N., Ofek I. Methods Enzymol. 1995;253:91–98. doi: 10.1016/s0076-6879(95)53010-x. [DOI] [PubMed] [Google Scholar]
- 9.Karlsson K.-A. Annu. Rev. Biochem. 1989;58:309–350. doi: 10.1146/annurev.bi.58.070189.001521. [DOI] [PubMed] [Google Scholar]
- 10.Karlsson K.-A., Ångström J., Bergström J., Lanne B. APMS (Suppl. 27) 1992;100:71–83. [PubMed] [Google Scholar]
- 11.Karlsson K.-A., Milh M.A., Ångstrom J., Bergström J., Dezfoolian H., Lanne B., Leonardsson I., Teneberg S. In: Molecular Recognition in Host-Parasite Interactions. Korhonen T.K., editor. Plenum Press; New York: 1992. pp. 115–132. [Google Scholar]
- 12.Lenz T.L. J. Genet. Virol. 1990;71:751–766. [Google Scholar]
- 13.Brown K.E., Anderson S.M., Young N.S. Science. 1993;262:114–117. doi: 10.1126/science.8211117. [DOI] [PubMed] [Google Scholar]
- 14.Stehle T., Yan Y., Benjamin T.L., Harrison S.C. Nature. 1994;369:160–163. doi: 10.1038/369160a0. [DOI] [PubMed] [Google Scholar]
- 15.Hanisch F.-G., Hacker J., Schroten H. Infect. Immun. 1993;61:2108–2115. doi: 10.1128/iai.61.5.2108-2115.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Prasadarao N.V., Wass C.A., Kim K.S. Infect. Immun. 1996;64:154–160. doi: 10.1128/iai.64.1.154-160.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Seignole D., Grange P., Duval-Iflah Y., Mouricourt M. Microbiology. 1994;140:2467–2473. doi: 10.1099/13500872-140-9-2467. [DOI] [PubMed] [Google Scholar]
- 18.Seignole D., Mouricout M., Duval-Iflah Y., Quintard B., Julien R. J. Genet. Microbiol. 1991;137:1591–1601. doi: 10.1099/00221287-137-7-1591. [DOI] [PubMed] [Google Scholar]
- 19.Strömberg N., Karlsson K.-A. J. Biol. Chem. 1990;265:11251–11258. [PubMed] [Google Scholar]
- 20.Strömberg N., Borén T., Carlen A., Olsson J. Infect. Immun. 1992;60:3278–3286. doi: 10.1128/iai.60.8.3278-3286.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Neeser J.-R., Grafström R.C., Woltz A., Brassart D., Fryder V., Guggenheim B. Glycobiology. 1995;5:97–104. doi: 10.1093/glycob/5.1.97. [DOI] [PubMed] [Google Scholar]
- 22.Jimenez-Luch V., Ginsburg V., Krivan H.C. Infect. Immun. 1990;58:2085–2090. doi: 10.1128/iai.58.7.2085-2090.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brassart D., Woltz A., Golliard M., Neeser J.-R. Infect. Immun. 1991;59:1605–1613. doi: 10.1128/iai.59.5.1605-1613.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gottschalk A. The Chemistry and Biology of Sialic Acids and Related Substances. Cambridge University Press; 1960. p. 115. [Google Scholar]
- 25.Schauer R. Glycobiology. 1991;1:449–452. doi: 10.1093/glycob/1.5.449. [DOI] [PubMed] [Google Scholar]
- 26.Paulson J.C. In: Conn P.M., editor. Vol. 2. Academic Press; New York: 1985. pp. 131–219. (The Receptors). [Google Scholar]
- 27.Wiley D.C., Skehei J.J. Annu. Rev. Biochem. 1987;56:365–394. doi: 10.1146/annurev.bi.56.070187.002053. [DOI] [PubMed] [Google Scholar]
- 28.Weis W., Brown J.H., Cusack S., Paulson J.C., Skehei J.J., Wiley D.C. Nature. 1988;333:426–431. doi: 10.1038/333426a0. [DOI] [PubMed] [Google Scholar]
- 29.Nieman H., Dabrowski J., Dabrowski U., Geyer R., Keil W., Klenk H.D., Stirm S. Eur. J. Biochem. 1985;146:523–532. doi: 10.1111/j.1432-1033.1985.tb08683.x. [DOI] [PubMed] [Google Scholar]
- 30.Vlasak R., Luytjes W., Spaan W., Palese P. Proc. Natl. Acad. Sci. USA. 1988;85:4526. doi: 10.1073/pnas.85.12.4526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kelm S., Paulson J.C., Rose U., Brossmer R., Schmid W., Bandgar B.P., Schreiner E., Hartmann M., Zbiral E. Eur. J. Biochem. 1992;205:147–153. doi: 10.1111/j.1432-1033.1992.tb16762.x. [DOI] [PubMed] [Google Scholar]
- 32.Sauter N.K., Hanson J.E., Glick G.D., Brown J.H., Crowther R.L., Park S.-J., Skehei J.J., Wiley D.C. Biochemistry. 1992;31:9609–9621. doi: 10.1021/bi00155a013. [DOI] [PubMed] [Google Scholar]
- 33.Watowich S.J., Skehei J.J., Wiley D.C. Structure. 1994;2:719–731. doi: 10.1016/s0969-2126(00)00073-3. [DOI] [PubMed] [Google Scholar]
- 34.Sauter N.K., Glick G.D., Crowther R.L., Park S.-J., Eisen M.B., Skehei J.J., Knowles J.R., Wiley D.C. Proc. Natl. Acad. Sci. 1992;89:324–327. doi: 10.1073/pnas.89.1.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shieh M.T., WuDunn D., Montgomery R.I., Esko J.D., Spear P.G. J. Cell Biol. 1992;116:1273–1281. doi: 10.1083/jcb.116.5.1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Haidar M., Gluckman J.C., Gattegno L. Glycobiology. 1992;2:429–435. doi: 10.1093/glycob/2.5.429. [DOI] [PubMed] [Google Scholar]
- 37.Mbemba E., Gluckman J.C., Gattegno L. Glycobiology. 1994;4:13–21. doi: 10.1093/glycob/4.1.13. [DOI] [PubMed] [Google Scholar]
- 38.Duguid J.P., Old D.C. In: Beachey E.H., editor. Vol. 6. Chapman and Hall; London: 1980. pp. 185–217. (Receptors and Recognition). [Google Scholar]
- 39.Klemm P. Rev. Infect. Dis. 1985;7:321–340. doi: 10.1093/clinids/7.3.321. [DOI] [PubMed] [Google Scholar]
- 40.Clegg S., Gerlach G. J. Bacterid. 1987;169:934–938. doi: 10.1128/jb.169.3.934-938.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tewari R., MacGregor J.I., Ikeda T., Little J.R., Hultgren S.J., Abraham S.N. J. Biol. Chem. 1993;268:3009–3015. [PubMed] [Google Scholar]
- 42.Jones C.H., Pinkner J.S., Nicholes A.V., Slonim L.N., Abraham S.N., Hultgren S.J. Proc. Natl. Acad. Sci. USA. 1993;90:8397–8401. doi: 10.1073/pnas.90.18.8397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hultgren S.J., Normark S., Abraham S. Annu. Rev. Microbiol. 1991;45:383–415. doi: 10.1146/annurev.mi.45.100191.002123. [DOI] [PubMed] [Google Scholar]
- 44.Jacob-Dubuisson F., Kuehn M., Hultgren S.J. Trends Microbiol. 1993;1:50–55. doi: 10.1016/0966-842x(93)90032-m. [DOI] [PubMed] [Google Scholar]
- 45.Labigne-Roussel A., Schmidt M.A., Walz W., Falkow S. J. Bacteriol. 1985;162:1285–1292. doi: 10.1128/jb.162.3.1285-1292.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Abraham S.N., Freitag C.S., Gander R.M., Clements J.R., Thomas V.L., Eisenstein B.I. Mol. Biol. Med. 1986;3:495–508. [PubMed] [Google Scholar]
- 47.Sharon N., Ofek I. In: Microbial Lectins and Agglutinins. Mirelman D., editor. Wiley; New York: 1986. pp. 55–81. [Google Scholar]
- 48.Abraham S.N., Sun D., Dale J.B., Beachey E.H. Nature. 1988;336:682–684. doi: 10.1038/336682a0. [DOI] [PubMed] [Google Scholar]
- 49.Firon N., Ofek I., Sharon N. Infect. Immun. 1984;43:1088–1090. doi: 10.1128/iai.43.3.1088-1090.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Madison B., Ofek I., Clegg S., Abraham N.S. Infect. Immun. 1994;62:843–848. doi: 10.1128/iai.62.3.843-848.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sokurenko E.V., Courtney H.S., Abraham S.N., Klemm P., Hasty D.L. Infect. Immun. 1992;60:4709–4719. doi: 10.1128/iai.60.11.4709-4719.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Oldenburg K.R., Loganathan D., Goldstein I.J., Schultz P.G., Gallop M.A. Proc. Natl. Acad. Sci. USA. 1992;89:5393–5397. doi: 10.1073/pnas.89.12.5393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Scott J.K., Loganathan D., Easley R.B., Gong X., Goldstein I.J. Proc. Natl. Acad. Sci. 1992;89:5398. doi: 10.1073/pnas.89.12.5398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Giampapa C.S., Abraham S.N., Chiang T.M., Beachey E.H. J. Biol. Chem. 1988;263:5362–5367. [PubMed] [Google Scholar]
- 55.Wold A.E., Mestecky J., Tomana M., Kobata A., Ohbayashi H., Endo T., Svanborg-Eden C. Infect. Immun. 1990;58:3073–3077. doi: 10.1128/iai.58.9.3073-3077.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kumar S., Muchmore A.V. Kidney Int. 1990;37:1395–1401. doi: 10.1038/ki.1990.128. [DOI] [PubMed] [Google Scholar]
- 57.Wadolkowski E.A., Laux D.C., Cohen P.S. Infect. Immun. 1988;56:1036–1043. doi: 10.1128/iai.56.5.1036-1043.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rodriguez-Ortega M., Ofek I., Sharon N. Infect. Immun. 1997;55:968–973. doi: 10.1128/iai.55.4.968-973.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gbarah A., Gahmberg C.G., Ofek I., Jacobi I., Sharon N. Infect. Immun. 1991;59:4524–4530. doi: 10.1128/iai.59.12.4524-4530.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gbarah A., Gahmberg C.G., Bonner G., Sharon N. J. Leukoc. Biol. 1993;54:111–113. doi: 10.1002/jlb.54.2.111. [DOI] [PubMed] [Google Scholar]
- 61.Sauter S.L., Rutherfurd S.M., Wagener C., Shively J.E., Hefta S.A. Infect. Immun. 1991;59:2485–2493. doi: 10.1128/iai.59.7.2485-2493.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pieroni P., Worobec E.A., Paranchych W., Armstrong G.D. Infect. Immun. 1988;56:1334–1340. doi: 10.1128/iai.56.5.1334-1340.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hermentin P. Parasitol. Today. 1987;3:52–55. doi: 10.1016/0169-4758(87)90214-6. [DOI] [PubMed] [Google Scholar]
- 64.Doig P., Paranchych W., Sastry P.A., Irvin R.T. Can. J. Microbiol. 1989;35:1141–1145. doi: 10.1139/m89-189. [DOI] [PubMed] [Google Scholar]
- 65.Rudner X.L., Zheng Z., Berk R.S., Irvin R.T., Hazlett L.D. Invest. Ophtalmol. Visual Sci. 1991;33:2185. [PubMed] [Google Scholar]
- 66.Demuth D.R., Golub E.E., Malamud D. J. Biol. Chem. 1990;265:7120–7126. [PubMed] [Google Scholar]
- 67.Brennan M.J., Cisar J.O., Sandberg A.L. Infect. Immun. 1986;52:840–845. doi: 10.1128/iai.52.3.840-845.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Babu J.P., Dabbous M.K., Abraham S.N. J. Periodont. Res. 1991;26:97–110. doi: 10.1111/j.1600-0765.1991.tb01632.x. [DOI] [PubMed] [Google Scholar]
- 69.Brennan M.J., David J.L., Kenimer J.G., Manclark C.R. J. Biol. Chem. 1988;263:4895–4899. [PubMed] [Google Scholar]
- 70.Rogers T.S., Corey S.J., Rosoff P.M. J. Immunol. 1990;145:678–683. [PubMed] [Google Scholar]
- 71.Clark C.G., Armstrong G.D. Infect. Immun. 1990;58:3840–3846. doi: 10.1128/iai.58.12.3840-3846.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Buhler T., Hoschutzky H., Jann K. Infect. Immun. 1991;59:3876–3882. doi: 10.1128/iai.59.11.3876-3882.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.DeGraaf F.K. Curr. Top. Microbiol. Immunol. 1990;151:29–53. doi: 10.1007/978-3-642-74703-8_2. [DOI] [PubMed] [Google Scholar]
- 74.Morschhauser J., Hoschutzky H., Jann K., Hacker J. Infect. Immun. 1990;58:2133–2138. doi: 10.1128/iai.58.7.2133-2138.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Parkkinen J., Rogers G.N., Korhonen T., Dahr W., Finne J. Infect. Immun. 1986;54:37–42. doi: 10.1128/iai.54.1.37-42.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Prasadarao N.V., Wass C.A., Hacker J., Jann K., Kim K.S. J. Biol. Chem. 1993;268:10356–10363. [PubMed] [Google Scholar]
- 77.Neeser J.-R., Chambaz A., Hoang K.Y., Link-Amster H. FEMS Microbiol. Lett. 1988;49:301–307. [Google Scholar]
- 78.Sjöberg P.O., Lindahl M., Porath J., Wadström T. Biochem. J. 1988;255:105–111. doi: 10.1042/bj2550105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mouricout M.A., Julien R.A. Infect. Immun. 1987;55:1216–1223. doi: 10.1128/iai.55.5.1216-1223.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lindahl M., Brossmer R., Wadström T. Glycoconj. J. 1987;4:51–58. [Google Scholar]
- 81.Teneberg S., Willemsen P., DeGraaf F.K., Karlsson K.A. FEBS Lett. 1990;263:10–14. doi: 10.1016/0014-5793(90)80693-d. [DOI] [PubMed] [Google Scholar]
- 82.Ono E., Abe K., Nakazawa M., Naiki M. Infect. Immun. 1989;57:907–911. doi: 10.1128/iai.57.3.907-911.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Liukkonen J., Haataja S., Tikkanen K., Kelm S., Finne J. J. Biol. Chem. 1992;267:21105–21111. [PubMed] [Google Scholar]
- 84.Murray P.A., Prakobphol A., Lee T., Hoover C.I., Fisher S.J. Infect. Immun. 1992;60:31–38. doi: 10.1128/iai.60.1.31-38.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Evans D.G., Evans D.J., Jr., Moulds J.J., Grahm D.Y. Infect. Immun. 1988;56:2896–2906. doi: 10.1128/iai.56.11.2896-2906.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Evans D.G., Evans D.J., Jr., Graham D.Y. Infect. Immun. 1989;57:2272–2278. doi: 10.1128/iai.57.8.2272-2278.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Saitoh T., Natomi H., Zhao W.L., Okuzumi K., Sugano K., Iwamori M., Nagai Y. FEBS Lett. 1991;282:385–387. doi: 10.1016/0014-5793(91)80519-9. [DOI] [PubMed] [Google Scholar]
- 88.Slomiany B.L., Piotrkowski K., Samanta A., VanHorn K., Murty V.L.N., Slomiany A. Biochem. Int. 1989;19:929–936. [PubMed] [Google Scholar]
- 89.Evans D.G., Karjalainen T.K., Evans D.J., Jr., Graham D., Lee C.-H. J. Bacteriol. 1993;175:674–683. doi: 10.1128/jb.175.3.674-683.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Razin S., Jacobs E. J. Genet. Microbiol. 1993;138:407–4022. doi: 10.1099/00221287-138-3-407. [DOI] [PubMed] [Google Scholar]
- 91.Loonies L.M., Uemura K., Childs R.A., Paulson J.C., Rogers G.N., Scudder P.R., Jr., Michalski J.-C., Hounsell E.F., Taylor-Robinson D., Feizi T. Nature. 1984;307:560–563. doi: 10.1038/307560a0. [DOI] [PubMed] [Google Scholar]
- 92.Loomes L.M., Uemura K., Feizi T. Infect. Immun. 1985;47:15–20. doi: 10.1128/iai.47.1.15-20.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wennerås C., Neeser J.-R., Svennerholm A.M. Infect. Immun. 1995;63:640–646. doi: 10.1128/iai.63.2.640-646.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Willemsen P.T., de Graaf F.K. Microbiol. Pathogenet. 1992;12:367–375. doi: 10.1016/0882-4010(92)90099-a. [DOI] [PubMed] [Google Scholar]
- 95.Gilboa-Garber N., Garber N. In: Glycoconjugates: Composition, Structure and Function. Allen H.J., Kisailus E.C., editors. Marcel Dekker; New York: 1992. pp. 541–591. [Google Scholar]
- 96.Garber N., Guempel U., Belz A., Gilboa-Garber N., Doyle R.J. Biochim. Biophys. Acta. 1992;1116:331–333. doi: 10.1016/0304-4165(92)90048-y. [DOI] [PubMed] [Google Scholar]
- 97.Avichezer D., Katcoff D.J., Garber N.C., Gilboa-Garber N. J. Biol. Chem. 1992;267:23023–23027. [PubMed] [Google Scholar]
- 98.Gilboa-Garber N., Mizrahi L. Experientia. 1985;41:681–682. doi: 10.1007/BF02007718. [DOI] [PubMed] [Google Scholar]
- 99.Glick J., Garber N. J. Genet. Microbiol. 1983;129:3085–3090. doi: 10.1099/00221287-129-10-3085. [DOI] [PubMed] [Google Scholar]
- 100.Lamblin G., Lhermitte M., Klein A., Houdre N., Scharfman A., Ramphal R., Roussel P. Am. Rev. Respirat. Dis. 1991;144:S19–S24. doi: 10.1164/ajrccm/144.3_pt_2.S19. [DOI] [PubMed] [Google Scholar]
- 101.Ramphal R., Carnoy C., Fievre S., Michalski J.-C., Houdret N., Lamblin G., Strecker G., Roussel P. Infect. Immun. 1991;59:700–704. doi: 10.1128/iai.59.2.700-704.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Cisar J.O. In: Microbial Lectins and Agglutinins. Mirelman D., editor. Wiley; New York: 1986. pp. 183–196. [Google Scholar]
- 102a.Kolenbrander P.E. In: Microbial Cell-Cell Interactions. Dworkin M., editor. Am. Soc. Microbiol.; Washington: 1991. pp. 303–329. [Google Scholar]
- 103.Brennan M.J., Cisar J.O., Vatter A.E., Sandberg A.L. Infect. Immun. 1984;46:459–464. doi: 10.1128/iai.46.2.459-464.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sandberg A.L., Ruhl S., Joralmon R.A., Brennan M.J., Sutphin M.J., Cisar J.O. Infect. Immun. 1995;63:2625–2631. doi: 10.1128/iai.63.7.2625-2631.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.London J., Hand A.R., Weiss E.I., Allen J. Infect. Immun. 1989;57:3940–3944. doi: 10.1128/iai.57.12.3940-3944.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.London J., Allen J. J. Bacteriol. 1990;172:2527–2534. doi: 10.1128/jb.172.5.2527-2534.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ho S.C., Schindler M., Wang J.L. J. Cell Biol. 1990;111:1639–1643. doi: 10.1083/jcb.111.4.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Loh J.T., Ho S.C., De Freijter A.W., Wang J.L., Schindler M. Proc. Natl. Acad. Sci. USA. 1993;90:3033–3037. doi: 10.1073/pnas.90.7.3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Borén T., Falk P., Roth K.A., Larson G., Normark S. Science. 1993;262:1892–1895. doi: 10.1126/science.8018146. [DOI] [PubMed] [Google Scholar]
- 110.Falk P., Roth K.A., Borén T., Westblom T.U., Gordon J.I., Normark S. Proc. Natl. Acad. Sci. USA. 1993;90:2035–2039. doi: 10.1073/pnas.90.5.2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Falk P.G., Bry Holgersson L., Gordon J.I. Proc. Natl. Acad. Sci. USA. 1995;92:1515–1519. doi: 10.1073/pnas.92.5.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Garber N., Guempel U., Gilboa-Garber N., Doyle R.J. FEMS Microbiol. Lett. 1987;48:331–334. [Google Scholar]
- 113.Finkelstein R.A., Hanne L.F. Infect. Immun. 1982;36:1199–1208. doi: 10.1128/iai.36.3.1199-1208.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wisniewski J.P., Monsigny M., Delmotte F.M. Biochimie. 1994;76:121–128. doi: 10.1016/0300-9084(94)90003-5. [DOI] [PubMed] [Google Scholar]
- 115.Depierreux C., Kang H.-C., Guerin B., Monsigny M., Delmotte F. Glycobiology. 1990;1:643–649. doi: 10.1093/glycob/1.6.643. [DOI] [PubMed] [Google Scholar]
- 116.van't Wout J., Burnette W.N., Mar V., Rozdzinski E., Wright S.D., Tuomanen E.I. Infect. Immun. 1992;60:3303–3308. doi: 10.1128/iai.60.8.3303-3308.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Heerze L., Chong P., Armstrong G. J. Biol. Chem. 1993;267:25810–25815. [PubMed] [Google Scholar]
- 118.Springer T.A. Nature. 1990;346:425. doi: 10.1038/346425a0. [DOI] [PubMed] [Google Scholar]
- 119.Drickamer K., Taylor M.E. Annu. Rev. Cell Biol. 1993;9:237–264. doi: 10.1146/annurev.cb.09.110193.001321. [DOI] [PubMed] [Google Scholar]
- 120.Saukkonen K., Burnette W.N., Mar V., Masure H.R., Tuomanen E. Proc. Natl. Acad. Sci. USA. 1992;89:118–122. doi: 10.1073/pnas.89.1.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Lobet Y., Feron C., Dequesne G., Simoen E., Hauser P., Locht C. J. Exp. Med. 1993;177:79–87. doi: 10.1084/jem.177.1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Loosmore S., Zealey G., Cockle S., Boux H., Chong P., Jacoob R., Klein M. Infect. Immun. 1993;61:2316–2324. doi: 10.1128/iai.61.6.2316-2324.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Rozdzinski E., Burnette W.N., Jones T., Mar V., Tuomanen E. J. Exp. Med. 1993;178:917–924. doi: 10.1084/jem.178.3.917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Stein P., Boodhoo A., Armstrong G., Cockle S., Klein M., Read R. Structure. 1994;2:45–57. doi: 10.1016/s0969-2126(00)00007-1. [DOI] [PubMed] [Google Scholar]
- 125.Stein P.E., Boodhoo A., Armstrong G.D., Heerze L.D., Cockle S.A., Klein M.H., Read R.J. Nature Struct. Biol. 1994;1:591–596. doi: 10.1038/nsb0994-591. [DOI] [PubMed] [Google Scholar]
- 126.Armstrong G.D., Howard L.A., Peppier M.S. J. Biol. Chem. 1988;263:8677–8684. [PubMed] [Google Scholar]
- 127.Witvliet M.H., Burns D.L., Brennan M.J., Poolman J.T., Manclark C.R. Infect. Immun. 1989;57:3324–3330. doi: 10.1128/iai.57.11.3324-3330.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Prasad S.M., Yin Y., Rodzinski E., Tuomanen E.I., Masure H.R. Infect. Immun. 1993;61:2780–2785. doi: 10.1128/iai.61.7.2780-2785.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Barak R., Elad Y., Chet I. Arch. Microbiol. 1986;144:346–349. [Google Scholar]
- 130.Kellens J.T.C., Peumans W.J. Mycol. Res. 1991;95:1235–1241. [Google Scholar]
- 131.Kellens J.T.C., Goldstein I.J., Peumans W.J. Mycol. Res. 1992;96:495–502. [Google Scholar]
- 132.Inbar J., Chet I. Microbiology. 1994;140:651–657. doi: 10.1099/00221287-140-3-651. [DOI] [PubMed] [Google Scholar]
- 133.Borrebaeck C.A.K., Mattiasson B., Nordbrink-Hertz B. J. Bacteriol. 1984;159:53–56. doi: 10.1128/jb.159.1.53-56.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Rosen S., Ek B., Rask L., Tunlid A. J. Genet. Microbiol. 1992;138:2663–2672. doi: 10.1099/00221287-138-12-2663. [DOI] [PubMed] [Google Scholar]
- 135.Ravdin J.I., Guerrant R.L. J. Clin. Invest. 1981;68:1305–1313. doi: 10.1172/JCI110377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Petri W.A., Jr. ASM News. 1991;57:299–306. [Google Scholar]
- 137.Tannich E., Ebert F., Horstmann R.D. Proc. Natl. Acad. Sci. USA. 1991;88:1849–1853. doi: 10.1073/pnas.88.5.1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Mann B.J., Vedvick T., Torian B., Petri W.A., Jr. Proc. Natl. Acad. Sci. USA. 1991;88:3248–3252. doi: 10.1073/pnas.88.8.3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.McCoy J.J., Mann B.J., Vedvick T.S., Pak Y., Heimark D.B., Petri W.A., Jr. J. Biol. Chem. 1993;268:24223–24231. [PubMed] [Google Scholar]
- 140.Petri W.A., Jr., Snodgrass T.L., Jackson T.F.H.G., Gathiram V., Simjee A.E., Chadee K., Chapman M.D. J. Immunol. 1990;144:4803–4809. [PubMed] [Google Scholar]
- 141.Mann B.J., Chung C.Y., Ashley J.M., Braga L.L., Snodgrass T.T. Infect. Immun. 1993;61:1772–1778. doi: 10.1128/iai.61.5.1772-1778.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Li E., Becker A., Stanley S.L., Jr. J. Exp. Med. 1998;167:1725–1730. doi: 10.1084/jem.167.5.1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Chadee K., Petri W.A., Jr., Innes D.J., Ravdin J.I. J. Clin. Invest. 1987;80:1245–1254. doi: 10.1172/JCI113199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Adler P., Wood S.J., Lee Y.C., Lee R.T., Petri W.A., Jr., Schnaar R.L. J. Biol. Chem. 1995;270:5164–5171. doi: 10.1074/jbc.270.10.5164. [DOI] [PubMed] [Google Scholar]
- 145.Breuer W.V., Ginsburg H., Cabantchik Z.I. Biochim. Biophys. Acta. 1983;755:263–271. doi: 10.1016/0304-4165(83)90213-1. [DOI] [PubMed] [Google Scholar]
- 146.Perkins M.E., Rocco L.J. J. Immunol. 1988;141:3190–3196. [PubMed] [Google Scholar]
- 147.Orlandi P.A., Klotz E.W., Haynes J.D. J. Cell Biol. 1992;116:901–909. doi: 10.1083/jcb.116.4.901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Klotz F.W., Orlandi P.A., Reuter G., Cohen S.J., Haynes J.D., Schauer R., Howard R.J., Pelese P., Miller R.H. Mol. Biochem. Parasitol. 1992;51:49–54. doi: 10.1016/0166-6851(92)90199-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Sim B.K.L., Orlandi P.A., Haynes J.D., Klotz E.W., Carter J.M., Camus D., Zegans M.E., Chulay J.D. J. Cell Biol. 1990;111:1877–1884. doi: 10.1083/jcb.111.5.1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Sim B.K.L., Carter J.M., Deal C.D., Holland C., Haynes J.D., Gross M. Exp. Parasitol. 1994;78:259–268. doi: 10.1006/expr.1994.1027. [DOI] [PubMed] [Google Scholar]
- 151.Sim B.K.L., Chitnis C.E., Wasniowska K., Hadley T.J., Miller L.H. Science. 1994;264:1941–1944. doi: 10.1126/science.8009226. [DOI] [PubMed] [Google Scholar]
- 152.Kobiler D., Mirelman D. Infect. Immun. 1980;29:221–225. doi: 10.1128/iai.29.1.221-225.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Rosales-Encina J.L., Meza I., Lopez-De-Leon A., Talamas-Rohana P., Rojkind M. J. Infect. Dis. 1987;156:790–797. doi: 10.1093/infdis/156.5.790. [DOI] [PubMed] [Google Scholar]
- 154.El Moudni B., Philippe M., Monsigny M., Schrevel J. Glycobiology. 1993;3:305–312. doi: 10.1093/glycob/3.4.305. [DOI] [PubMed] [Google Scholar]
- 155.Ward H.D., Keusch G.T., Pereira M.E.A. BioEssays. 1990;12:211–215. doi: 10.1002/bies.950120504. [DOI] [PubMed] [Google Scholar]
- 156.Ko H.L., Beuth J., Solter J., Schroten H., Uhlenbruck G., Pulverer G. Infection. 1987;15:237–240. doi: 10.1007/BF01644121. [DOI] [PubMed] [Google Scholar]
- 157.Saukkonen K., Cabellos C., Burroughs M., Prasad S., Tuomanen E. J. Exp. Med. 1991;173:1143–1149. doi: 10.1084/jem.173.5.1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Mouricout M., Petit J.M., Carias J.R., Julien R. Infect. Immun. 1990;58:98–106. doi: 10.1128/iai.58.1.98-106.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Steuer M.K., Herbst H., Beuth J., Steuer M., Pulverer G., Matthias R. Otorhinolaryngol. Nova. 1993;3:18–25. [Google Scholar]
- 160.Wentworth J.S., Austin F.E., Garber N., Gilboa-Garber N., Paterson C.A., Doyle R.J. Biofouling. 1991;4:99–104. [Google Scholar]
- 161.Blaylock W.K., Yue B.Y., Robin J.B. CLAO J. 1990;16:223–227. [PubMed] [Google Scholar]
- 162.Kolenbrander P.E., Ganeshkumar N., Cassels F.J., Hughes C.V. FASEB J. 1993;7:406–413. doi: 10.1096/fasebj.7.5.8462782. [DOI] [PubMed] [Google Scholar]
- 163.Manocha M.S., Chet Y. Can. J. Microbiol. 1990;36:69–76. doi: 10.1139/m90-133. [DOI] [PubMed] [Google Scholar]
- 164.Petri W.A., Jr., Ravdin J.I. Infect. Immun. 1991;59:97–101. doi: 10.1128/iai.59.1.97-101.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Soong C.J.G., Kain K.C., Abd-Alla M., Jackson F.H.G., Ravdin J.I. J. Infect. Dis. 1995;171:645–651. doi: 10.1093/infdis/171.3.645. [DOI] [PubMed] [Google Scholar]
- 166.Braga L.L., Ninimiya H., McCoy J.J., Eacker S., Wiedmer T., Pham C., Wood S., Sims P.J., Petri W.A., Jr. J. Clin Invest. 1992;90:1131–1137. doi: 10.1172/JCI115931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Mirelman D. Microbiol. Rev. 1987;51:272–284. doi: 10.1128/mr.51.2.272-284.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Ofek I., Sharon N. Infect. Immun. 1988;56:539–547. doi: 10.1128/iai.56.3.539-547.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Ofek I., Rest R.F., Sharon N. ASM News. 1992;58:429–435. [Google Scholar]
- 170.Gbarah A., Mirelman D., Sansonetti P.J., Verdon R., Bernhard W., Sharon N. Infect. Immun. 1993;61:1687–1693. doi: 10.1128/iai.61.5.1687-1693.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Sandberg A.L., Mudrick L.L., Cisar J.O., Metcalf J.A., Malech H.L. Infect. Immun. 1988;56:267–269. doi: 10.1128/iai.56.1.267-269.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Bernhard W., Gbarah A., Sharon N. J. Leukoc. Biol. 1992;52:343–348. doi: 10.1002/jlb.52.3.343. [DOI] [PubMed] [Google Scholar]