

Abstract
The phosphoprotein (P) of rabies virus binds the viral polymerase to the nucleoprotein (N)-RNA template for transcription and replication. By limited protease digestion we defined a monomeric C-terminal domain of P that can bind to N-RNA. The atomic structure of this domain was determined and previously described mutations that interfere with binding of P to N-RNA could now be interpreted. There appears to be two features involved in this activity situated at opposite surfaces of the molecule: a positively charged patch and a hydrophobic pocket with an exposed tryptophan side-chain. Other previously published work suggests a conformational change in P when it binds to N-RNA, which may imply the repositioning of two helices that would expose a hydrophobic groove for interaction with N. This domain of rabies virus P is structurally unrelated to the N-RNA binding domains of the phosphoproteins of Sendai and measles virus that are members of the same order of viruses, the non-segmented negative strand RNA viruses. The implications of this finding for the evolution of this virus group are discussed.
Keywords: rabies virus, replication, transcription, polymerase, evolution
Abbreviations used: P, phosphoprotein; N, nucleoprotein; L, viral polymerase; vRNA, viral RNA; PML, promyelocytic leukaemia; SAD, single anomalous dispersion; mAb, monoclonal antibody; EGS, ethylene glycolbis(succinimidyl succinate)
Introduction
Non-segmented negative strand RNA viruses are enveloped viruses with viral RNA (vRNA) in the opposite sense to that of mRNA. This order of viruses (Mononegavirales) contains the Paramyxoviridae (measles and Sendai virus), Bornaviridae, Rhabdoviridae (rabies and vesicular stomatitis virus, VSV) and Filoviridae (Ebola virus). Negative strand RNA viruses cause important human and animal disease. In 2003, measles virus killed 760,000 children, mainly in Africa and Asia (WHO report, 2003). Rabies virus kills about 50,000 people per year in these continents but is also an increasing health concern in the USA.1 The vRNA of these viruses is bound to the viral nucleoprotein (N) with a virus-family-specific nucleotide-N stoichiometry.2, 3, 4, 5 The viral RNA-dependent RNA polymerase consists of two subunits, the large protein (L) that contains the polymerase activity and a polymerase cofactor, the phosphoprotein (P) that binds L to the N-RNA.6, 7, 8, 9, 10, 11 The polymerase complex cannot transcribe or replicate the naked vRNA; vRNA has to be bound to N in order to be a functional template.12
P has a second role in the viral life-cycle as a chaperone for N that is not yet bound to vRNA (N0). When recombinant N is expressed alone in eukaryotic cells it binds non-specifically to cellular RNA to form N–RNA complexes that have the same structure as the viral N–RNA structures.5, 13, 14, 15 However, when N and P are co-expressed, a soluble complex between N0 and P is formed that does not contain RNA.16, 17, 18, 19 This N0–P complex provides the soluble N required for binding to newly replicated vRNA.
Sequence alignment of the phosphoproteins of the Lyssavirus genus (rhabdovirus genus to which the rabies virus belongs) indicates conservation and prediction of α-helical structure at the very amino terminus of the molecule (Figure 1 ; sequence conservation for residues 1–51 and predicted α-helix for residues 10–30). There is some sequence conservation and prediction of an α-helical region between residues 93 and 130, and extensive conservation from about residue 200 until the C-terminal end (Figure 1). Most published work on the structure and function of rabies virus P suggests the existence of at least two domains, an N-terminal domain (residues 1–177/184) and a C-terminal domain (residues 178/185–297).20, 21, 22 The L-binding domain is present in the first 19 amino acid residues of P.23 The domain that binds to N0 is located in the N-terminal 177 residues,20, 21, 22 whereas the C-terminal domain binds to N–RNA21, 22 (see Figure 2 ). However, deletion of the C-terminal 24 residues did not abolish binding of P to N–RNA.21
Figure 1.

Multiple sequence alignment and secondary structure (predicted or observed) for the Lyssavirus phosphoproteins. The alignment was made with ClustalW for the phosphoproteins of RABVC (rabies virus CVS strain; SWISSPROT acc. P22363), ABLV (Australian bat lyssavirus; SPTREMBL acc. Q9QSP3), EBLV1/2 (European bat lyssavirus 1 and 2; SPTREMBL acc. O56776 and SPTREMBL acc. O56780, respectively), MOKV (Mokola virus; SPTREMBL acc O56775) and LBV (Lagos bat virus; SPTREMBL acc. O56773). The secondary structures that are indicated above the alignment are predicted (indicated in green) for residues 1–185 using PHD and ESPript75 and observed (indicated in black) for the crystallised domain of rabies virus P presented here, residues 186–297.
Figure 2.

Proposed domain structure of rabies virus phosphoprotein. The domain structure of the protein is given in a bar code with the start and end of each domain indicated. The function of the domains, derived from previously published literature and from the experiments described here, are given above or below the bar.
P also has an oligomerisation domain. When P is expressed in bacteria or in insect cells, the molecule forms oligomers but it is not clear whether these are trimers or tetramers.19, 24 When Gigant et al.24 expressed an N-terminal deletion mutant of P missing the first 52 amino acid residues, the molecule was still oligomeric.
The gene for the rabies virus phosphoprotein codes for a total of five polypeptides; full-length P plus N-terminal truncated forms that arise through a leaky scanning activity of the ribosome on P mRNA.25 The shorter forms start at amino acid residues 20 (P2), 53 (P3), 69 (P4) or 83 (P5). Although the replication of rabies virus takes place in the cytoplasm, P3–5 are mainly found in the nucleus whereas P1 and P2 are cytoplasmic. The nuclear products of the P gene interact with PML (promyelocytic leukaemia) nuclear bodies that have been associated with the cellular defence mechanism against viral infections.26 The interaction of the P products with PML bodies may overcome the antiviral response of the cell. The Sendai virus P gene also codes for a number of different polypeptides that are generated by a stuttering mechanism of the polymerase that adds one or several G residues at a specific site in the mRNA for P.27, 28 Some of the viral polypeptides that result from this editing mechanism are also active in fighting the innate antiviral activity of the infected cell.29, 30
Here, we present limited protease digestion experiments on rabies virus P that show that only the C-terminal part of the protein is protease resistant. This C-terminal domain is monomeric and binds to N–RNA. In crystallisation trials with intact P, spontaneous proteolysis occurred in the crystallisation drops and only the C-terminal domain crystallised, for which the structure was determined. The previously described mutations leading to loss of binding of this domain to N22 can now be interpreted in the light of this structure. This rabies virus P domain has a structure that is different from that of the functionally analogous domains of Sendai and measles virus P31, 32 and implications for the evolution of negative strand RNA viruses are discussed.
Results
Full-length phosphoprotein produced in Escherichia coli is oligomeric24 and binds to rabies virus N–RNA15 (see also Figure 4, below). The domain structure of this recombinant protein was probed by limited protease digestion using trypsin, endoprotease Lys-C and thermolysin. These proteases digest P to protease-resistant fragments that migrate on SDS-PAGE with apparent molecular masses of 15–16 kDa (Figure 3 A–C). The fragments react with monoclonal antibody 25C2 that recognises residues 225–246 of P33 (Figure 3D). N-terminal sequencing of the fragment obtained after trypsin digestion showed that cleavage had occurred after R172. The apparent mass of the fragment from SDS-PAGE suggests that it contains residues 173–297. The N-terminal sequence of the endoprotease Lys-C digested fragment indicates digestion after K170. These experiments show that there exists a C-terminal domain of rabies virus P that is resistant to proteolysis.
Figure 4.
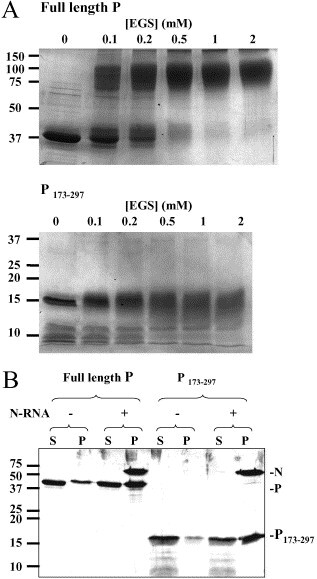
Oligomerisation states and N-RNA binding activities of full-length P and P173-297. A, Full-length P and P173-297 were incubated with different concentrations of the cross-linking agent EGS as indicated and analysed by 12% Tris–glycine SDS-PAGE for intact P and SDS-16% PAGE for P173-297. B, N-RNA templates isolated from infected BSR cells were incubated with full-length P and P173-297. After centrifugation of the N–RNA-P mixture through sucrose cushions, proteins present in the supernatants (S) and pellets (P) were analysed by SDS-16% PAGE.
Figure 3.
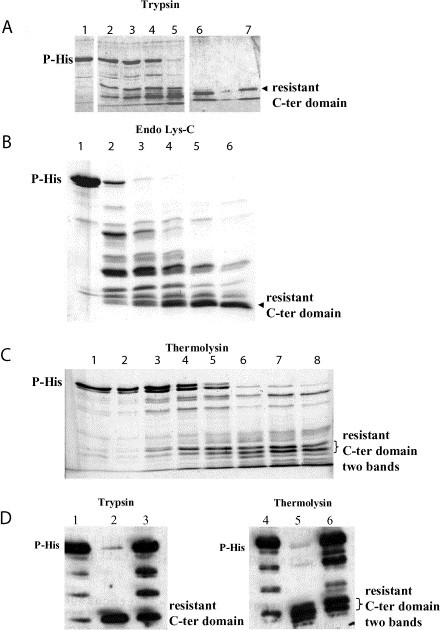
Limited proteolysis experiments on His-tagged rabies P. His-tagged P was purified from E. coli and digested for one hour at 37 °C with different proteases and analysed by SDS-16% PAGE. A, Digestion with trypsin. Lane 1, no trypsin. Lanes 2–7, trypsin/P-His ratios (w/w) of 1 : 8000, 1 : 4000, 1 : 2000, 1 : 1000, 1 : 500 and 1 : 250. The reaction was quenched by adding PMSF (10 mM final concentration). B, Digestion with endoprotease Lys-C. Lane 1, no protease. Lanes 2–6, quantities of protease used per 1 μg of P-His in units (one unit hydrolyses 1 μmol of N-p-tosyl-Gly-Pro-Lys p-nitroanilide per minute at pH 7.7 at 25 °C). Lane 2, 3.75×10−5 units; lane 3, 7.5×10−5 units; lane 4, 1.5×10−4 units; lane 5, 3×10−4 units; lane 6, 6×10−4 units. The reaction was quenched by the addition of EDTA (5 mM final concentration). C, Digestion with thermolysin. Lanes 1–8, thermolysin:P-His ratios (w/w) of: lane 1, 1 : 64; lane 2, 1 : 32; lane 3, 1 : 16; lane 4, 1 : 8; lane 5, 1 : 4; lane 6, 1 : 2; lane 7, 1 : 1; lane 8, 2 : 1. The reaction was quenched by adding EDTA (5 mM final concentration). D, Western blot of P-His after digestion with trypsin (left) and thermolysin (right) using MAb 25C2. Lane 1, no trypsin. Lanes 2 and 3, trypsin/P ratios of 1 : 2000 and 1 : 8000, respectively. Lane 4, no thermolysin. Lanes 5 and 6, thermolysin/P ratios of 1 : 1 and 1 : 4, respectively.
Cross-linking experiments showed that, under conditions that totally cross-link full-length P, the C-terminal domain derived after trypsin digestion (P173–297) remained monomeric (Figure 4 A), suggesting that this domain is a monomer in solution. Binding of P173–297 to N–RNA was compared to that of full-length P. The two proteins were incubated with N–RNA isolated from virus-infected cells. The N–RNA was then sedimented through a sucrose cushion taking with it any bound P proteins. Both full-length P and P173–297 were found to co-sediment with the N–RNA (Figure 4B), indicating that the C-terminal domain of Rabies virus P is the N–RNA-binding domain, as is the case for the C-terminal domain of Sendai virus P.
Non-tagged, full-length P was produced in insect cells and put in crystallisation drops. The molecule that crystallised was not the intact protein but a C-terminal domain corresponding to residues 186–297. Degradation of intact P to this domain by contaminating proteases was reproducible and was observed for native and for selenomethionine-substituted protein. The structure was solved using the single anomalous dispersion (SAD) method as indicated in Materials and Methods and the crystallographic details are given in Table 1 . All but the last two C-terminal residues could be modelled.
Table 1.
Data collection, phasing and refinement statistics
| Crystal | Native | Se-Met |
|---|---|---|
| Cell dimensions a, b, c (Å) | 44.3, 44.3, 106.2 | 44.05, 44.05, 105.9 |
| Wavelength (Å) | 0.9393 | 0.9795 |
| Resolution (Å) | 30–1.5 (1.6–1.5) | 30–1.9 (2.1–1.9) |
| Completeness (%) | 98.5 (93.9) | 92.7 (74.5) |
| Rmerge | 6.3 (20.7) | 4.9 (18.3) |
| Rmergeano | 4.3 (16.8) | |
| 〈I/σ(I)〉 | 16.8 (7.1) | 36.1 (12.9) |
| Redundancy | 6.4 (6.1) | 11.9 (4.0) |
| Unique reflections | 19, 783 | 16, 801 |
| Total reflections | 120, 720 | 216, 310 |
| Figure of merit | 0.31 | |
| Rcryst (%) | 17.5 | |
| Rfree (%) | 19.8 |
Values in parentheses are for the outermost resolution shell. calculated for the whole data set. with Friedel-related reflections excluded. Figure of merit=, where P(α) is the phase probability distribution and α is the phase. . Rfree was calculated as for Rcryst with 5% of the data omitted from the structural refinement.
The C-terminal domain folds as a single compact domain, with six α-helices (α1–α6), one 310 helix (η1), and a small, two-stranded antiparallel β-sheet (Figure 5 ). The overall structure resembles a pear cut along its length with a flat face and a round face (Figure 6 A and B). The top part of the half-pear is comprised of the first three helices (α1, α2, α3), with α2 and α3 packing perpendicularly to α1. The abrupt change in direction between α2 and α3, provided by P245, is necessary for their packing against α1. The lower part is comprised of the small β-sheet, with two helices (α4 and α5) packing against one side and α6 capping one edge of the β-sheet. The two parts are packed together by hydrophobic interactions between α2 and α4, with some additional hydrophobic residues contributed by the C terminus of α1. The flat face is composed mainly from α-helices, whereas the round face is made from the C terminus of α1, the short β-sheet, the 310 helix and the loops connecting these secondary structures. A positive surface potential area exists on this round face, as calculated in GRASP34 (Figure 6A–C). On the other hand, the flat face has a more symmetric charge distribution, with the main surface feature being a shallow uncharged groove, where one side is comprised of the loop between α2 and α3 and the C terminus of α4, and the other side is comprised of α6. This groove also contains a prominent hydrophobic pocket, the W-hole, composed of L244, P245, C261, W265, M287 and L291 (Figure 7 A).
Figure 5.
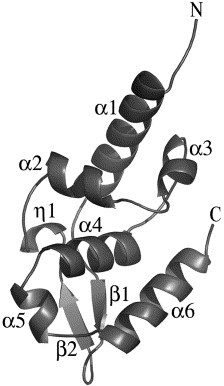
Structure of the carboxy-terminal domain of rabies virus phosphoprotein. Ribbon diagram of the secondary structure of residues 186–297 of rabies virus P, CVS strain. The amino and carboxy-terminal ends are indicated as are the secondary structure elements. The Figure was generated with PYMOL (http://www.pymol.org).
Figure 6.
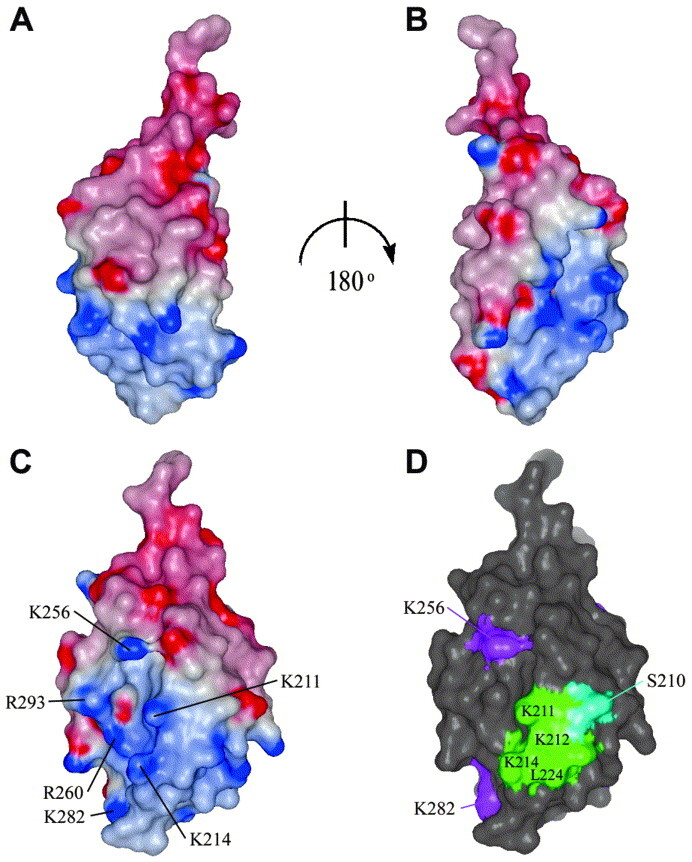
Surface representation of the C-terminal domain of rabies virus phosphoprotein. A, Side-view of the electrostatic potential map of the C-terminal domain showing its “half-pear” shape and the distribution of charges on its surface; blue indicates positively charged residues (12 kT) and red negatively charged residues (−12 kT). The amino acid residue at the top of the molecule is the well-defined W186 that is wedged in the W-hole of a neighbouring molecule in the crystal (see Figure 7). B, As A rotated by 180°. C, Front view of the round face (rotated by 90° compared to the view in A) indicating the residues that are involved in binding to N–RNA (see Table 2). D, Same view as in C indicating the residues in cluster 1 (see the text) in green. Residues that are involved in binding to N–RNA but that are not in cluster 1 are shown in magenta. S210, one of the phosphorylation sites on this domain, is in blue. The Figure was generated with Grasp34 and GLR (L. Esser, personal communication).
Figure 7.
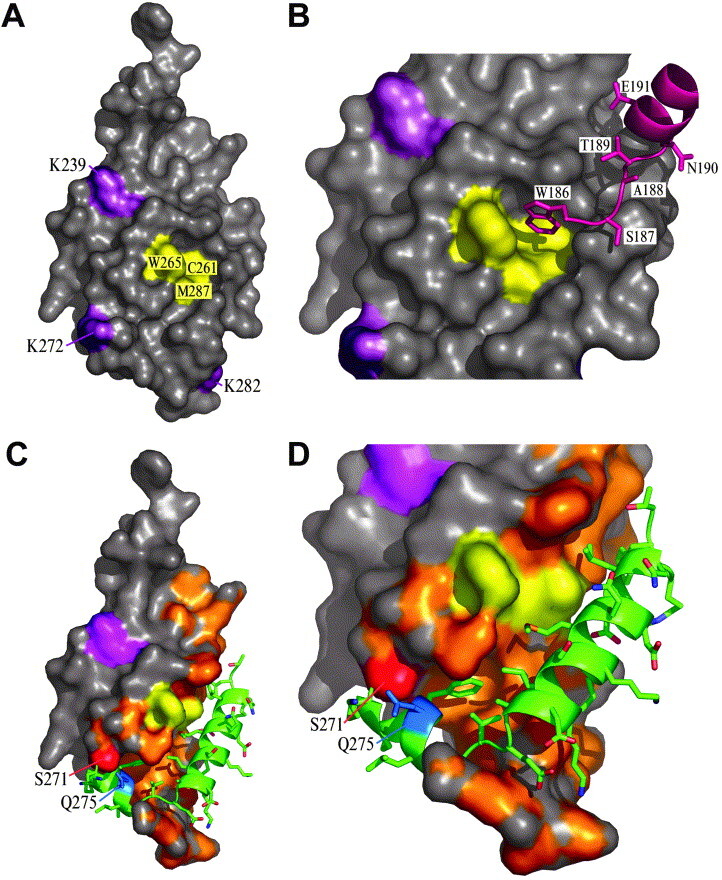
View of the flat face of the C-terminal domain of Rabies virus phosphoprotein. A, View of the flat face with residues in cluster 2 (W-hole, see the text) in yellow and other residues contributing to the binding to N-RNA in magenta. B, Detail of the W-hole and its interaction with the N-terminal W186 of a neighbouring subunit in the crystal. C, Same representation as in A with helices 5 and 6 outlined in green. The uncharged or hydrophobic surfaces on this face of the molecule and underneath helices 5 and 6 are indicated in orange. The potential phosphorylation site S271 and the centre of the epitope recognised by the mAb described by Toriumi et al.37 are indicated in red and blue, respectively. D, Higher magnification detail of C. The Figure was generated with PYMOL (http://www.pymol.org).
The C-terminal domain crystallised in trigonal space group P3121 and the asymmetric unit contains one molecule. The flat face mediates practically all of the intermolecular contacts, where two major regions of the molecule are involved in contacts with neighbours. The largest region of symmetry interaction involves residues of α1 and α2 from one molecule, which interact with residues from the last three helices (α4, α5 and α6) of a symmetry-related molecule. The smaller region is composed of the four N-terminal residues, which interact with residues in or around the W-hole from a symmetry-related neighbour. Here, W186, at the extreme N terminus of the crystallised fragment is lodged into the W-hole of its symmetry-related molecule and is therefore immobilised and well defined in the electron density map (Figure 7B).
A search of the Protein Data Bank with the full C-terminal domain using the program DALI35 did not reveal any significant homology to other proteins. Three hits with a Z-score >2.5 were found but close scrutiny showed only one with any significant structural similarity. This was from the C-terminal domain of the α-subunit from F1-ATPase with a Z-score of 2.8 and an rms value of 3.9 Å over 66 α-carbon positions, where the structural conservation is between the first four helices, (α1, α2, α3 and α4) from the C-terminal domain of P and the final four helices from the C-terminal α-helical bundle from the α-subunit of the ATPase. So the fold of the C-terminal domain of P could be described as an embellished α-helical bundle, where the small β-sheet and the 310 helix have been inserted between the first and second helix. However, the structure described here could be considered as a new fold given the importance of the β-sheet and the final two helices to the hydrophobic core packing. There is no structural homology with the N–RNA binding domains of the phosphoproteins of Sendai and measles virus.31, 32
Jacob et al.22 have produced random mutations in Mokola virus P and tested if those mutants could still bind to Mokola virus nucleoprotein in a yeast two-hybrid assay. Mokola virus is closely related to rabies virus and the sequences of the P proteins of the two viruses are very similar (Figure 2). The mutated residues in the Mokola virus P protein were compared to the corresponding residues in the rabies virus P protein. We have analysed the structural consequences of the mutations identified as having the largest effect on N-RNA binding (Table 2 ). Eleven of these mutants are expected to have a destabilising effect on the structure. Seven mutations form two major clusters on the surface. The first cluster comprises the highly conserved lysine residues (K211, K212 and K214) and L224 (Figure 6C and D). Two of the lysine residues contribute to the positive potential on the round face described above. The second cluster is comprised of three residues and is centred on the less conserved W-hole and includes C261, W265 and M287 (Figure 7). The remaining four mutations that replace lysine residues by uncharged or negatively charged amino acids are randomly spread with no discernible order (Figure 6, Figure 7). From this mutational analysis it would seem that both the positively charged patch on the round face of the domain and the W-hole on the flat face are important for binding to N-RNA.
Table 2.
Mutations in Mokola virus phosphoprotein that prevent binding to Mokola virus N are compared with corresponding residues in rabies virus P and placed in the structure of rabies virus P186-297
| RABa | MOKb | Position in structure | Effect on structure |
|---|---|---|---|
| A. Mutations that likely lead to destabilisation of the structure | |||
| S196 | D→G | α1 | Helix destabilisation |
| V197 | I→L, V | α1 | Core packing destabilisation |
| F209 | F→L | Loop | Core packing destabilisation |
| Y213 | Y→C | β1 | Core packing destabilisation |
| F215 | F→Y | β1 | Core packing destabilisation |
| M232 | M→T | Loop | Core packing destabilisation |
| D235 | D→G | α2 | Helix destabilisation |
| R260 | R→G | α4 | Loss of H-bond between α4/α6 |
| L276 | L→P | α5 | Helix destabilisation |
| N292 | N→S | α6 | Helix destabilisation |
| Y294 | Y→C | α | Core packing destabilisation |
| B. Mutations that are located on the surface of P but not in clusters 1 and 2 | |||
| K239 | K→Q | α2 | |
| K256 | K→E | loop | |
| K272 | K→A | α5 | |
| K282 | K→N, E | α6 | |
| C. Mutations that are located on the surface of P and grouped in clusters 1 and 2 | |||
| K211 | K→E | Loop | Cluster 1 |
| K212 | K→E, R | Loop | Cluster 1 |
| K214 | K→E | β1 | Cluster 1 |
| L224 | L→S | β2 | Cluster 1 |
| C261 | C→R, S | α4 | Cluster 2 (W-hole) |
| W265 | F→L | α4 | Cluster 2 (W-hole) |
| M287 | I→T | α6 | Cluster 2 (W-hole) |
Rabies virus phosphoprotein residue type and number.
Corresponding residue in Mokola virus phosphoprotein that, when mutated to the residue indicated, no longer binds to Mokola virus N in a yeast two-hybrid assay.22
Rabies virus phosphoprotein contains four potential phosphorylation sites.36 There is evidence that phosphorylation of the first two sites, S63 and S64, is mediated by a unique cellular protein kinase of 71 kDa that is packaged inside rabies virus particles, and is necessary during the infection process.36 The other two possible phosphorylation sites are S210 and S271 that can be phosphorylated in vitro by specific isomers of protein kinase C although it is not known whether this actually happens in the infected cell.36 S210 lies on a surface exposed loop between α1 and β1 and a phosphorylated serine residue can easily be created without any conformational change. Interestingly, this serine is also adjacent to the positively charged patch and the first cluster of mutations described above (Figure 6D). Thus, it is possible that phosphorylation of S210 would alter the N–RNA binding characteristics of P. S271 is located between α4 and α5 and is only partly solvent-exposed and quite constricted (Figure 7C and D). It is therefore probable that some conformational rearrangement would be required for phosphorylation to occur. S271 is located between the two clusters of mutations and close to K272 that, when mutated to A, also prevents binding of P to N (Table 2 and Figure 7A).
Discussion
Binding of rabies virus P to N-RNA
The phosphoprotein binds the viral polymerase (L) to the nucleocapsid. Here, we show that the domain of P that performs this function is a C-terminal domain from residue 186 until the C-terminal end. Residues that are implicated in the interaction with N can be found on the round and flat faces of the domain (Figure 6, Figure 7). Toriumi et al. have described a monoclonal antibody that can distinguish two conformations of rabies virus P, one when it is soluble and one when it is bound to N–RNA.37 The presumed epitope for this mAb is located around Q275 (Figure 7C and D). This suggests that binding of P to N–RNA may be associated with a conformational change resulting in the rearrangement of helices 5 and 6. These C-terminal 24 residues of P can be deleted without inhibiting the binding of P to N–RNA.21 It is, therefore, likely that α5 and α6 can occupy a conformation that is different from the one that is found in the crystal structure. Phosphorylation of S271 may have an influence on the structure of the protein in this area. Figure 7C and D show the molecule with the W-hole in yellow and helices 5 and 6 in green. These two helices lie in a hydrophobic groove (orange) and their displacement would expose this groove to the solvent. The conformation of the monomeric molecule described here is probably not stable when the two helices are displaced. Binding of P to N-RNA may involve complementary interactions from N, shielding the groove from the solvent. As mentioned above, a shallow uncharged groove is slightly exposed even with the helices in place, the W-hole making part of this surface structure.
Oligomerisation of P
Gigant et al. 24 have shown that the first 52 amino acid residues of rabies virus P are not involved in oligomerisation and here we have shown that P173-297 is monomeric as well. Because oligomerisation of Sendai virus P takes place through the formation of an inter-monomer helical coiled-coil,38 we suggest that the oligomerisation domain be situated in the predicted helical region between residues 93 and 130 (Figure 1, Figure 2). Jacob et al. have shown that a P mutant with a deletion from residue 61 to 175, so most likely monomeric, was still active in supporting transcription activity by L (Figure 2).22 This deletion mutant still has the L-binding and the N–RNA binding domains. This is in contrast to the transcription and replication activities of the Sendai virus phosphoprotein that does require the presence of the oligomerisation domain.39, 40 The difference between these two phosphoproteins could be due to a higher affinity of rabies virus phosphoprotein for its N–RNA than that of Sendai virus P for its N–RNA. The C-terminal domains of Sendai and measles virus P that bind to their N–RNAs41 are very small and flexible domains,31, 32 much smaller than the corresponding domain of rabies virus P. Another important reason why the P protein of Sendai virus has to be oligomeric in order to be active is that the binding domain for Sendai L overlaps with the oligomerisation domain38, 42 and that only oligomeric P can bind to L.
Conserved characteristics of the Mononegavirales
Viruses in this order have in common that the vRNA is in the opposite sense to that of messenger RNA, that the vRNA is bound to nucleoprotein and that the viral RNA polymerase complex is only functional on this N–RNA matrix. The N–RNA complexes of all these viruses form helical structures with different helical parameters but with N-subunits that have the same overall shape.43 It is likely that the nucleoproteins of all virus families in this order are homologous but proof for this will only come when several nucleoprotein structures have been determined. The viral polymerases (L) have also conserved sequence motifs and are, thus, homologous.44, 45 Another conserved feature of this group of viruses is the order of the genes for the structural proteins on the vRNA.
No structural similarities amongst the matrix proteins of the Mononegavirales or between the phosphoproteins of rabies virus and Sendai/measles virus
The matrix proteins of this group of viruses play an important role in the formation of the virus particles.46 They interact both with the membrane-embedded viral glycoproteins and with the nucleocapsids and their tendency to polymerise is thought to drive particle formation. These proteins also have so-called late domains that interact with host-proteins involved in cellular membrane modifications.47 However, the sequences of the matrix proteins show very little identity (sometimes less than 10% even for closely related genera like Lyssa- and Vesiculoviruses, both Rhabdoviridae) and the three structures of negative strand RNA virus matrix proteins determined so far, that of influenza virus (a segmented negative strand RNA virus),48, 49 Ebola virus50 and that of VSV,51 are all unrelated and all three present a new fold.47 The fact that the structures are not related means that these proteins do not come from the same evolutionary lineage.
As for the matrix proteins, there is very little sequence identity between the phosphoprotein sequences from the different virus families. Even within families, like the Rhabdoviridae, there is very little sequence identity. Here, we have presented the first structure of part of a rabies virus protein, the C-terminal domain of P. There is no structural similarity between the C-terminal domains of Sendai/measles virus P and that of rabies virus P described here. Again, this suggests that the C-terminal domains of these P proteins do not come from the same evolutionary lineage.
Evolution of negative strand RNA viruses
The fact that the three known structures of negative strand RNA virus matrix protein are unrelated and that the two known structures of the N–RNA-binding domains of the phosphoproteins are also different, even though they fulfil the same functions, either implies that RNA virus evolution is so extensive that homologous proteins have evolved to independent folds (although from the known matrix protein structures it is not possible to see how one structure could evolve into the other) or it means that heterologous structures have been incorporated into these viruses through heterologous recombination. Heterologous recombination has been documented for Nidovirales (Arteri- and Coronaviruses), positive strand RNA viruses, where homologous RNA recombination by the viral polymerase is part of the mechanism for the production of mRNAs.52, 53 Homologous recombination inside gene segments of segmented negative strand RNA viruses has also been shown or suggested; recombination between the polymerase genes of two Arenavirus strains has been documented54 whereas recombination in the haemagglutinin gene of the 1918 Spanish influenza virus has been suggested from sequence comparisons.55 For heterologous recombination to occur, the viral polymerase has to jump from the viral N–RNA template to a nucleoprotein-encapsidated host mRNA and then back again to the viral RNA template. Non-segmented negative strand virus nucleoprotein expressed alone in bacterial or eukaryotic cells binds in a non-specific fashion to cellular RNAs.5, 13, 56, 57, 58 Although during a virus infection most newly made N is first bound to the phosphoprotein in the N0–P complex for delivery onto newly made vRNA, it is very likely that some N will nevertheless bind to cellular RNA providing an N-cellular RNA template for the viral polymerase. It is also known that these polymerases can jump either inside the viral N-RNA template59 or from the viral template to the newly copied, N-bound complementary strand in order to form copy-back interfering particles.60 Heterologous recombination for the influenza viruses was suggested from the structure of the influenza C virus glycoprotein.61 From a comparison between the influenza A and C haemagglutinin structures it was suggested that the influenza virus haemagglutinin consists of an ancestral fusion protein combined with an independent haemagglutinin domain, for influenza C virus further complemented by an esterase domain spliced into this haemagglutinin domain. The esterase domain is homologous to bovine brain and bacterial esterases.61 Finally, when Khatchikian et al. 62 tried to select for a pathogenic turkey influenza virus to grow in chicken cells, they obtained a virulent mutant that had acquired 54 nt coming from the 28 S ribosomal RNA of the infected cells. This insertion led to a change in the presentation of the cleavage site on haemagglutinin to the cellular protease needed for proteolytic activation, allowing the virus to grow in the new host cells.
If the hypothesis on heterologous recombination will turn out to be correct, then the conserved parts of the ancestor common to all negative strand RNA viruses may be the nucleoprotein or N–RNA and the RNA-dependent RNA polymerase, i.e. the basic components of the replication machinery. The matrix proteins are not homologous nor are the phosphoproteins. The glycoproteins are probably also not conserved among all viruses. Although its structure is not yet known, the fusogenic glycoprotein of the rhabdoviruses has probably a very different structure from that of the fusion proteins of other negative strand RNA viruses because their fusion characteristics are so very different.63 Apart from these major viral proteins, many viruses of this group code for additional proteins for which no structures are known. The presence of heterologous matrix proteins in these viruses may be the reason why the shapes of the virus particles in this group are so variable, from more or less spherical (influenza, paramyxoviruses) to bullet-shaped (rhabdoviruses) and filamentous particles (filoviruses).
Materials and Methods
Limited proteolysis experiments
C-terminal His-tagged rabies virus phosphoprotein was produced in E. coli BL21 cells as described.24 Purified protein was taken up in 500 mM NaCl, 20 mM Tris–HCl (pH 8), 0.5 mM EDTA. Proteolysis experiments were performed in this buffer at 37 °C for one hour using the proteases and concentrations as indicated in the legend to Figure 3. The reactions were quenched as indicated and analysed on Tris–glycine SDS-16% (w/v) PAGE.
The Western blot shown in Figure 3D was performed with anti-P mAb 25C2 that was produced in mice immunised with recombinant P (PV strain) produced in E. coli as described.33 The epitope of this mAb was mapped to amino acid residue positions (225–246) by a peptide array assay. Briefly, peptide arrays representing the rabies virus phosphoprotein (kindly provided by M. Schwemmle, Department of Virology, Freiburg) were chemically synthesised on cellulose sheets by the SPOT synthesis technique and treated with mAb 25C2 as described.64
Functional characterisation of the C-terminal domain of P
Cross-linking analysis
About 10 μg of full-length recombinant P (see above) or P173-297 was incubated with the cross-linker ethylene glycolbis(succinimidyl succinate) (EGS) at final concentrations as indicated for Figure 4 for 30 minutes at room temperature. Reactions were quenched by the addition of 200 mM glycine and incubated for an additional 30 minutes at room temperature. The samples were then analysed by SDS-PAGE.
Binding of P and P173-297 to viral nucleocapsids
Viral nucleocapsids were isolated from virus-infected cells using a CsCl gradient as described by Schoehn et al. 15. Full-length recombinant P protein or P173-297 was incubated with nucleocapsids in 20 mM Tris–HCl (pH 7.5), 150 mM NaCl for 1.5 hours at room temperature. The mixture was then deposited on a 20% (w/w) sucrose cushion which was spun for 2.5 hours at 25,000 rpm and 4 °C in an SW55 rotor (Beckman). The pellet and supernatant fractions were analysed by SDS-16% (w/v) PAGE.
Expression and purification of native and selenomethionine-substituted P for crystallisation
Native P
High Five cells were infected with AcNPVM1 encoding full-length P65 at a multiplicity of infection of 5 p.f.u./cell. Cells were harvested 60 hours post infection, pelleted at 350g for ten minutes, resuspended in hypotonic buffer (50 mM NaCl, 20 mM Tris–HCl (pH 7.5), 1 mM EDTA in the presence of protease inhibitor cocktail “complete-EDTA free” from Roche Diagnostics), and lysed by three cycles of freezing and thawing. Cell debris was pelleted at 12,000g for 15 minutes at 4 °C and the cleared lysate was loaded onto a DEAE Sepharose Fast Flow (Pharmacia Biotech) column. The protein was eluted with a continuous 150 mM–300 mM NaCl gradient and fractions analysed by 10% Tris–glycine SDS-PAGE. Fractions containing P were pooled, concentrated by ultrafiltration (Centrifugal filter devices of Millipore) and loaded onto a Superdex 200 HR10/30 (Pharmacia Biotech) column. The protein was eluted in 20 mM Tris–HCl (pH 7.5), 150 mM NaCl, 5 mM DTT. The eluate was analysed by Tris–Tricine SDS-15% PAGE and two populations of full-length P were found in overlapping peaks (the elution volumes were far from the void volume, indicating that both populations represented soluble forms and not aggregates). Fractions were pooled separately, so as to contain either of the two populations or a mixture of both, and crystallisation trials were set up using all three fractions. Only the second population, i.e. the P eluting at the larger volume from the S200 HR10/30, crystallised.
Selenomethionine-substituted P
Sf 9 insect cells adapted to serum-free medium were infected as for the native protein. After 24 hours the medium (Sf900II plus penicillin/streptomycin; Gibco-BRL, Life Technologies) was removed and replaced with methionine-free medium (Sf900II without l-methionine and without l-cysteine; Gibco-BRL, Life Technologies). The cells were left in this medium for four hours to promote methionine depletion after which it was replaced with medium containing 50 mg/l of l-selenomethionine and 20 mg/l of l-cysteine and left for 67 hours before harvesting. Lysis and purification was as described for the native protein.
Crystallisation and data collection
Crystallization trials were all performed using full-length P in sitting drops at 20 °C, using a robot to set up drops of 2 μl (1 μl of protein solution plus 1 μl of reservoir solution). The protein concentration was 16 mg/ml in 20 mM Tris–HCl (pH 7.50), 150 mM NaCl, 5 mM DTT. Native, rod-shaped crystals grew between three and seven weeks under three reservoir conditions differing only in the salt used. Condition 1, 0.2 M MgCl2, 0.1 M Bis–Tris (pH 5.5), 25% (w/v) polyethylene glycol (PEG)3350. Condition 2, 0.2 M NaCl, 0.1 M Bis–Tris (pH 5.5), 25% PEG3350. Condition 3, 0.2 M (NH4)2SO4, 0.1 M Bis–Tris (pH 5.5), 25% PEG3350. Crystals of selenomethione-substituted P grew between three and eight weeks after setting up drops in 0.2 M MgCl2, 0.1 M Bis–Tris (pH 5.5), 25% PEG3350. The crystals were then transferred from their mother liquor to mother liquor plus 20% (v/v) glycerol prior to flash-freezing.
All X-ray data were collected at 100 K on beam line ID14-4 at the European Synchrotron Radiation Facility (ESRF). A high-resolution native data set to 1.5 Å was collected from a single crystal. A highly redundant data set from a single Se-Met crystal was collected at the peak of the Se-Met signal, as measured by X-ray fluorescence. The protein crystallised in space group P3121 with one molecule in the asymmetric unit. All the data were integrated and scaled using the XDS suite66 and data statistics are given in Table 1.
Structure determination and refinement
Two Se-Met sites were located using SOLVE67 via the single anomalous dispersion (SAD) method. The experimental phases were then improved by solvent flattening with RESOLVE.68 This gave an interpretable map and an initial poly-alanine model. Scaling of the structure factor amplitudes between the native and Se-Met data gave an R-factor of 55.5%, indicating non-isomorphism. Molecular replacement was therefore used to reposition the molecule in the unit cell. The poly-alanine model from RESOLVE68 was positioned in the cell using the AMoRe molecular replacement program,69 as implemented in CCP4.70 Input of the solution model into ARP/warp71 resulted in a nearly complete model.
Crystal structure refinement was performed using REFMAC72 and model building was performed with O.73 A randomly chosen subset of 5% of reflections for the calculation of the free R-factor was used as a monitor of refinement.74 Ordered water molecules were added at locations where there was |F o|−|Fc| density greater than 3σ above the mean and could form potential hydrogen-bonding contacts with the protein structure.
Protein Data Bank accession code
Co-ordinates have been deposited in the RCSB PDB as entry 1vyi.
Acknowledgements
We thank Drs Yves Gaudin (Gif-sur-Yvette), Dan Kolakofsky (Geneva) and Winfried Weissenhorn (EMBL-Grenoble) for numerous discussions. We gratefully acknowledge use of the crystallisation robot at the Institut de Biologie Structurale in Grenoble and thank the European Synchrotron Radiation Facility (ESRF) at Grenoble for beam-time at ID 14-4.
Edited by A. Klug
References
- 1.Messenger S.L., Smith J.S., Orciari L.A., Yager P.A., Rupprecht C.E. Emerging pattern of rabies deaths and increased viral infectivity. Emerg. Infect. Dis. 2003;9:151–154. doi: 10.3201/eid0902.020083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Egelman E.H., Wu S.S., Amrein M., Portner A., Murti G. The Sendai virus nucleocapsid exists in at least four different helical states. J. Virol. 1989;63:2233–2243. doi: 10.1128/jvi.63.5.2233-2243.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thomas D., Newcomb W.W., Brown J.C., Wall J.S., Hainfeld J.F., Trus B.L., Steven A.C. Mass and molecular composition of vesicular stomatitis virus: a scanning transmission electron microscopy analysis. J. Virol. 1985;54:598–607. doi: 10.1128/jvi.54.2.598-607.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Flamand A., Raux H., Gaudin Y., Ruigrok R.W.H. Mechanisms of rabies virus neutralisation. Virology. 1993;194:302–313. doi: 10.1006/viro.1993.1261. [DOI] [PubMed] [Google Scholar]
- 5.Mavrakis M., Kolesnikova L., Schoehn G., Becker S., Ruigrok R.W.H. Morphology of Marburg virus NP-RNA. Virology. 2002;296:300–307. doi: 10.1006/viro.2002.1433. [DOI] [PubMed] [Google Scholar]
- 6.Mellon M.G., Emerson S.U. Rebinding of transcriptase components (L and NS proteins) to the nucleocapsid template of vesicular stomatitis virus. J. Virol. 1978;27:560–567. doi: 10.1128/jvi.27.3.560-567.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hamaguchi M., Yoshida T., Nishikawa K., Naruse H., Nagai Y. Transcriptive complex of Newcastle disease virus. Both L and P proteins are required to constitute an active complex. Virology. 1983;128:105–117. doi: 10.1016/0042-6822(83)90322-7. [DOI] [PubMed] [Google Scholar]
- 8.Horikami S.M., Curran J., Kolakofsky D., Moyer S.A. Complexes of Sendai virus NP-P and P-L proteins are required for defective interfering particle genome replication in vitro. J. Virol. 1992;66:4901–8490. doi: 10.1128/jvi.66.8.4901-4908.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Portner A., Murti K.G., Morgan E.M., Kingsbury D.W. Antibodies against Sendai virus L protein: distribution of the protein in nucleocapsids revealed by immunoelectron microscopy. Virology. 1988;163:236–239. doi: 10.1016/0042-6822(88)90257-7. [DOI] [PubMed] [Google Scholar]
- 10.Curran J., Pelet T., Kolakofsky D. An acidic activation-like domain of the Sendai virus P protein is required for RNA synthesis and encapsidation. Virology. 1994;202:875–884. doi: 10.1006/viro.1994.1409. [DOI] [PubMed] [Google Scholar]
- 11.Smallwood S., Ryan K.W., Moyer S.A. Deletion analysis defines a carboxyl-proximal region of Sendai virus P protein that binds to the polymerase L protein. Virology. 1994;202:154–163. doi: 10.1006/viro.1994.1331. [DOI] [PubMed] [Google Scholar]
- 12.Emerson S.U. In: The Rhabdoviruses. Wagner R.R., editor. Plenum; New York: 1987. Transcription of vesicular stomatitis virus; pp. 245–269. [Google Scholar]
- 13.Spehner D., Kirn A., Drillien R. Assembly of nucleocapsid like structures in animal cells infected with a vaccinia virus recombinant encoding the measles virus nucleoprotein. J. Virol. 1991;65:6296–6300. doi: 10.1128/jvi.65.11.6296-6300.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Iseni F., Barge A., Baudin F., Blondel D., Ruigrok R.W.H. Characterization of rabies virus nucleocapsids and recombinant nucleocapsid-like structures. J. Gen. Virol. 1998;79:2909–2919. doi: 10.1099/0022-1317-79-12-2909. [DOI] [PubMed] [Google Scholar]
- 15.Schoehn G., Iseni F., Mavrakis M., Blondel D., Ruigrok R.W.H. Structure of recombinant rabies virus N-RNA and identification of the phosphoprotein binding site. J. Virol. 2001;75:490–498. doi: 10.1128/JVI.75.1.490-498.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Masters P.S., Banerjee A.K. Complex formation with vesicular stomatitis virus phosphoprotein NS prevents binding of nucleocapsid protein N to non-specific RNA. J. Virol. 1988;62:2658–2664. doi: 10.1128/jvi.62.8.2658-2664.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Peluso R.W. Kinetic, quantative, and functional analysis of multiple forms of the vesicular stomatitis virus nucleocapsid protein in infected cells. J. Virol. 1988;62:2799–2807. doi: 10.1128/jvi.62.8.2799-2807.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peluso R.W., Moyer S.A. Viral proteins required for the in vitro replication of vesicular stomatitis virus defective interfering particle genome RNA. Virology. 1988;162:369–376. doi: 10.1016/0042-6822(88)90477-1. [DOI] [PubMed] [Google Scholar]
- 19.Mavrakis M., Iseni F., Mazza C., Schoehn G., Ebel C., Gentzel M., et al. Isolation and characterisation of the rabies virus N°-P complex produced in insect cells. Virology. 2003;305:406–414. doi: 10.1006/viro.2002.1748. [DOI] [PubMed] [Google Scholar]
- 20.Chenik M., Chebli K., Gaudin Y., Blondel D. In vivo interaction of rabies virus phosphoprotein (P) and nucleoprotein (N): existence of two N-binding sites on P protein. J. Gen. Virol. 1994;75:2889–2896. doi: 10.1099/0022-1317-75-11-2889. [DOI] [PubMed] [Google Scholar]
- 21.Fu Z.F., Zheng Y., Wunner W.H., Koprowski H., Dietzschold B. Both the N- and the C-terminal domains of the nominal phosphoprotein of Rabies virus are involved in binding to the nucleoprotein. Virology. 1994;200:590–597. doi: 10.1006/viro.1994.1222. [DOI] [PubMed] [Google Scholar]
- 22.Jacob Y., Real E., Tordo N. Functional interaction map of lyssavirus phosphoprotein: identification of the minimal transcription domains. J. Virol. 2001;75:9613–9622. doi: 10.1128/JVI.75.20.9613-9622.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chenik M., Schnell M., Conzelmann K.K., Blondel D. Mapping the interacting domains between the rabies virus polymerase and phosphoprotein. J. Virol. 1998;72:1925–1930. doi: 10.1128/jvi.72.3.1925-1930.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gigant B., Iseni F., Gaudin Y., Knossow M., Blondel D. Neither phosphorylation nor the amino-terminal part of rabies virus phosphoprotein is required for its oligomerization. J. Gen. Virol. 2000;81:1757–1761. doi: 10.1099/0022-1317-81-7-1757. [DOI] [PubMed] [Google Scholar]
- 25.Chenik M., Chebli K., Blondel D. Translation initiation at alternate in-frame AUG codons in the rabies virus phosphoprotein mRNA is mediated by a ribosomal leaky scanning mechanism. J. Virol. 1995;69:707–712. doi: 10.1128/jvi.69.2.707-712.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blondel D., Regad T., Poisson N., Pavie B., Harper F., Pandolfi P.P., et al. Rabies virus P and small P products interact directly with PML and reorganize PML nuclear bodies. Oncogene. 2002;21:7957–7970. doi: 10.1038/sj.onc.1205931. [DOI] [PubMed] [Google Scholar]
- 27.Vidal S., Curran J., Kolakofsky D. A stuttering model for paramyxovirus P mRNA editing. EMBO J. 1990;9:2017–2022. doi: 10.1002/j.1460-2075.1990.tb08330.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hausmann S., Garcin D., Delenda C., Kolakofsky D. The versatility of paramyxovirus RNA polymerase stuttering. J. Virol. 1999;73:5568–5576. doi: 10.1128/jvi.73.7.5568-5576.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Garcin D., Latorre P., Kolakofsky D. Sendai virus C proteins counteract the interferon-mediated induction of an antiviral state. J. Virol. 1999;73:6559–6565. doi: 10.1128/jvi.73.8.6559-6565.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kato A., Kiyotani K., Sakai Y., Yoshida T., Nagai Y. The paramyxovirus, Sendai virus, V protein encodes a luxury function required for viral pathogenesis. EMBO J. 1997;16:578–587. doi: 10.1093/emboj/16.3.578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Johansson K., Bourhis J.M., Campanacci V., Cambillau C., Canard B., Longhi S. Crystal structure of the measles virus phosphoprotein domain responsible for the induced folding of the C-terminal domain of the nucleoprotein. J. Biol. Chem. 2003;278:44567–44573. doi: 10.1074/jbc.M308745200. [DOI] [PubMed] [Google Scholar]
- 32.Blanchard L., Tarbouriech N., Blackledge M., Timmins P., Burmeister W.P., Ruigrok R.W.H., Marion D. Structure and dynamics of the nucleocapsid-binding domain of the Sendai virus phosphoprotein in solution. Virology. 2004;319:201–211. doi: 10.1016/j.virol.2003.10.029. [DOI] [PubMed] [Google Scholar]
- 33.Raux H., Iseni F., Lafay F., Blondel D. Mapping of monoclonal antibody epitopes of the rabies virus P protein. J. Gen. Virol. 1997;78:119–124. doi: 10.1099/0022-1317-78-1-119. [DOI] [PubMed] [Google Scholar]
- 34.Nicholls A., Sharp K.A., Honig B. Protein folding and association: insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins Struct. Funct. Genet. 1991;11:281–296. doi: 10.1002/prot.340110407. [DOI] [PubMed] [Google Scholar]
- 35.Holm L., Sander C. Protein structure comparision by alignment of distance matricies. J. Mol. Biol. 1993;233:123–138. doi: 10.1006/jmbi.1993.1489. [DOI] [PubMed] [Google Scholar]
- 36.Gupta A.K., Blondel D., Ghoudhary S., Banerjee A.K. The phosphoprotein of rabies virus is phosphorylated by a unique cellular protein kinase and specific isomers of protein kinase C. J. Virol. 2000;74:91–98. doi: 10.1128/jvi.74.1.91-98.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Toriumi H., Honda Y., Morimoto K., Tochikura T.S., Kawai A. Structural relationship between nucleocapsid-binding activity of the rabies virus phosphoprotein (P) and exposure of epitope 402-13 located at the C terminus. J. Gen. Virol. 2002;83:3035–3043. doi: 10.1099/0022-1317-83-12-3035. [DOI] [PubMed] [Google Scholar]
- 38.Tarbouriech N., Curran J., Ruigrok R.W.H., Burmeister W.P. Tetrameric coiled coil domain of Sendai virus phosphoprotein. Nature Struct. Biol. 2000;7:777–781. doi: 10.1038/79013. [DOI] [PubMed] [Google Scholar]
- 39.Curran J., Boeck R., Lin-Marq N., Lupas A., Kolakofsky D. Paramyxovirus phosphoproteins form homotrimers as determined by an epitope dilution assay, via predicted coiled coils. Virology. 1995;214:139–149. doi: 10.1006/viro.1995.9946. [DOI] [PubMed] [Google Scholar]
- 40.Kolakofsky D., Le Mercier P., Iseni F., Garcin D. Viral RNA polymerase scanning and the gymnastics of Sendai virus RNA synthesis. Virology. 2004;318:463–473. doi: 10.1016/j.virol.2003.10.031. [DOI] [PubMed] [Google Scholar]
- 41.Curran J., Homann H., Buchholz C., Rochat S., Neubert W., Kolakofsky D. The hypervariable C-terminal tail of the Sendai paramyxovirus nucleocapsid protein is required for template function but not for RNA encapsidation. J. Virol. 1993;67:4358–4364. doi: 10.1128/jvi.67.7.4358-4364.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bowman M.C., Smallwood S., Moyer S.A. Dissection of individual functions of the Sendai virus phosphoprotein in transcription. J. Virol. 1999;73:6474–6483. doi: 10.1128/jvi.73.8.6474-6483.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schoehn G., Mavrakis M., Albertini A., Wade R., Hoenger A., Ruigrok R.W.H. 12 Å Structure of trypsin treated measles virus N-RNA. J. Mol. Biol. 2004;339:301–312. doi: 10.1016/j.jmb.2004.03.073. [DOI] [PubMed] [Google Scholar]
- 44.Poch O., Sauvaget I., Delarue M., Tordo N. Identification of four conserved motifs among the RNA-dependent polymerase encoding elements. EMBO J. 1989;8:3867–3874. doi: 10.1002/j.1460-2075.1989.tb08565.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Poch O., Blumberg B.M., Bougueleret L., Tordo N. Sequence comparison of five polymerases (L proteins) of unsegmented negative-strand RNA viruses: theoretical assignment of functional domains. J. Gen. Virol. 1990;71:1153–1162. doi: 10.1099/0022-1317-71-5-1153. [DOI] [PubMed] [Google Scholar]
- 46.Schmitt A.P., Lamb R.A. Escaping from the cell: assembly and budding of negative-strand RNA viruses. Curr. Top. Microbiol. Immunol. 2004;283:145–196. doi: 10.1007/978-3-662-06099-5_5. [DOI] [PubMed] [Google Scholar]
- 47.Timmins J., Ruigrok R.W.H., Weissenhorn W.W. Structural studies on the Ebola virus matrix protein VP40 indicate that matrix proteins of enveloped RNA viruses are analogous but not homologous. FEMS Microbiol. Letters. 2004;233:179–186. doi: 10.1111/j.1574-6968.2004.tb09480.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sha B., Luo M. Structure of a bifunctional membrane-RNA binding protein, influenza virus matrix protein M1. Nature Struct. Biol. 1997;4:239–244. doi: 10.1038/nsb0397-239. [DOI] [PubMed] [Google Scholar]
- 49.Arzt S., Baudin F., Barge A., Timmins P., Burmeister W.P., Ruigrok R.W.H. Combined results from solution studies on intact influenza virus M1 protein and from a new crystal form of its N-terminal domain show that M1 is an elongated monomer. Virology. 2001;279:439–446. doi: 10.1006/viro.2000.0727. [DOI] [PubMed] [Google Scholar]
- 50.Dessen A., Volchkov V., Dolnik O., Klenk H.D., Weissenhorn W. Crystal structure of the matrix protein VP40 from Ebola virus. EMBO J. 2000;19:4228–4236. doi: 10.1093/emboj/19.16.4228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gaudier M., Gaudin Y., Knossow M. Crystal structure of vesicular stomatitis virus matrix protein. EMBO J. 2002;21:2886–2892. doi: 10.1093/emboj/cdf284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lai M.M. RNA recombination in animal and plant viruses. Microbiol. Rev. 1992;56:61–79. doi: 10.1128/mr.56.1.61-79.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lai M.C. Recombination in large RNA viruses: Coronaviruses. Semin. Virol. 1996;7:381–388. doi: 10.1006/smvy.1996.0046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Charrel R.N., Lemasson J.-J., Garbutt M., Khelifa R., De Micc P., Feldmann H., de Lamballerie X. New insights into the evolutionary relationships between arenaviruses provided by comparative analysis of small and large segment sequences. Virology. 2003;317:191–196. doi: 10.1016/j.virol.2003.08.016. [DOI] [PubMed] [Google Scholar]
- 55.Gibbs M.J., Armstrong J.S., Gibbs A.J. Recombination in the hemagglutinin gene of the 1918 “Spanish flu”. Science. 2001;293:1842–1845. doi: 10.1126/science.1061662. [DOI] [PubMed] [Google Scholar]
- 56.Fooks A.R., Schadeck E., Liebert U.G., Dowsett A.B., Rima B.K., Steward M., et al. High-level expression of the measles virus nucleocapsid protein by using a replication-deficient adenovirus vector: induction of an MHC-1-restricted CTL response and protection in a murine model. Virology. 1995;210:456–465. doi: 10.1006/viro.1995.1362. [DOI] [PubMed] [Google Scholar]
- 57.Bhella D., Ralph A., Murphy L.B., Yeo R.P. Significant differences in nucleocapsid morphology within the Paramyxoviridae. J. Gen. Virol. 2002;83:1831–1839. doi: 10.1099/0022-1317-83-8-1831. [DOI] [PubMed] [Google Scholar]
- 58.Murphy L.B., Loney C., Murray J., Bhella D., Ashton P., Yeo R.P. Investigations into the amino-terminal domain of the respiratory syncytial virus nucleocapsid protein reveal elements important for nucleocapsid formation and interaction with the phosphoprotein. Virology. 2003;307:143–153. doi: 10.1016/s0042-6822(02)00063-6. [DOI] [PubMed] [Google Scholar]
- 59.Jennings P.A., Finch J.T., Winter G., Robertson J.S. Does the higher order structure of the influenza virus ribonucleoprotein guide sequence rearrangements in influenza viral RNA? Cell. 1983;34:619–627. doi: 10.1016/0092-8674(83)90394-x. [DOI] [PubMed] [Google Scholar]
- 60.Perrault J. Origin and replication of defective interfering particles. Curr. Topics. Microbiol. Immunol. 1981;93:151–207. doi: 10.1007/978-3-642-68123-3_7. [DOI] [PubMed] [Google Scholar]
- 61.Rosenthal P.B., Zhang X., Formanowski F., Fitz W., Wong C.H., Meier-Ewert H., et al. Structure of the haemagglutinin-esterase-fusion glycoprotein of influenza C virus. Nature. 1998;396:92–96. doi: 10.1038/23974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Khatchikian D., Orlich M., Rott R. Increased viral pathogenicity after insertion of a 28S ribosomal RNA sequence into the haemagglutinin gene of an influenza virus. Nature. 1989;340:156–157. doi: 10.1038/340156a0. [DOI] [PubMed] [Google Scholar]
- 63.Gaudin Y., Tuffereau C., Durrer P., Brunner J., Flamand A., Ruigrok R. Rabies virus-induced membrane fusion. Mol. Membr. Biol. 1999;16:21–31. doi: 10.1080/096876899294724. [DOI] [PubMed] [Google Scholar]
- 64.Billich C., Sauder C., Frank R., Herzog S., Bechter K., Takahashi K., et al. High-avidity human serum antibodies recognizing linear epitopes of Borna disease virus proteins. Biol. Psychiatry. 2002;51:979–987. doi: 10.1016/s0006-3223(02)01387-2. [DOI] [PubMed] [Google Scholar]
- 65.Préhaud C., Nel K., Bishop D.H.L. Baculovirus-expressed rabies virus M1 protein is not phosphorylated: it forms multiple complexes with expressed rabies N protein. Virology. 1992;189:766–770. doi: 10.1016/0042-6822(92)90602-l. [DOI] [PubMed] [Google Scholar]
- 66.Kabsch W. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J. Appl. Crystallog. 1993;26:795–800. [Google Scholar]
- 67.Terwilliger T.C., Berendzen J. Automated MAD and MIR structure solution. Acta Crystallog. sect. D. 1999;55:849–861. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Terwilliger T.C. Maximum-likelihood density modification. Acta Crystallog. sect. D. 2000;56:965–972. doi: 10.1107/S0907444900005072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Navaza J. AMoRe: an automated package for molecular replacement. Acta Crystallog. sect. A. 1994;50:157–163. [Google Scholar]
- 70.Collaborative Computational Project Number 4 The CCP4 suite: programs for protein crystallography. Acta Crystallog. sect. D. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 71.Perrakis A., Morris R., Lamzin V.S. Automated protein model building combined with iterative structure refinement. Nature Struct. Biol. 1999;6:458–463. doi: 10.1038/8263. [DOI] [PubMed] [Google Scholar]
- 72.Murshudov G.N., Vagin A.A., Dodson E.J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallog. sect. D. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 73.Jones T.A., Zou J.Y., Cowan S.W., Kjeldgaard M. Improved methods for building protein models in electron density maps and location of errors in these models. Acta Crystallog. sect. A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 74.Brünger A.T. The free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature. 1992;355:472–474. doi: 10.1038/355472a0. [DOI] [PubMed] [Google Scholar]
- 75.Gouet P., Courcelle E., Stuart D.I., Metoz F. ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics. 1999;15:305–308. doi: 10.1093/bioinformatics/15.4.305. [DOI] [PubMed] [Google Scholar]