

Abstract
Production of IgG in response to virus infection is central to antiviral immune effector functions and a hallmark of B cell memory. Antiviral antibodies (Abs) recognising viral glycoproteins or protein antigen displayed on the surface of virions or virus-infected cells are crucial in rendering the virus noninfectious and in eliminating viruses or infected cells, either acting alone or in conjunction with complement. In many instances, passive transfer of Abs is sufficient to protect from viral infection. Herpesviruses (HV) are equipped with a large array of immunomodulatory functions which increase the efficiency of infection by dampening the antiviral immunity. Members of the α- and β-subfamily of the Herpesviridae are distinct in encoding transmembrane glycoproteins which selectively bind IgG via its Fc domain. The Fc-binding proteins constitute viral Fcγ receptors (vFcγRs) which are expressed on the cell surface of infected cells. Moreover, vFcγRs are abundantly incorporated into the envelope of virions. Despite their molecular and structural heterogeneity, the vFcγRs generally interfere with IgG-mediated effector functions like antibody (Ab)-dependent cellular cytolysis, complement activation and neutralisation of infectivity of virions. vFcγRs may thus contribute to the limited therapeutic potency of antiherpesviral IgG in clinical settings. A detailed molecular understanding of vFcγRs opens up the possibility to design recombinant IgG molecules resisting vFcγRs. Engineering IgG with a better antiviral efficiency represents a new therapeutic option against herpesviral diseases.
Keywords: Viral FcγR, IgG, Herpesvirus, Neutralising antibodies, Immune evasion
1. Introduction: herpesvirus endurance between immune control and immune evasion
Herpesviruses (HV) which are subdivided into three subfamilies (α, β and γ) possess large double-stranded DNA genomes encoding approximately 100–200 genes. HV genomes remain latent in their natural host and can initiate a productive infection from latently infected cells resulting in virus shedding and transmission of virus progeny to a new host [1], [2]. During the long history of coevolution and cospeciation with their hosts, HV adopted multiple mechanisms to deal with immune functions that permit establishing a life-long infection with efficient replication and transmission, even in the presence of repeatedly boostered antiviral immune responses [3], [4]. Typically, the herpesviral evasion strategy prominently targets those immune responses that attack and contain the virus with the highest efficiency, i.e. CD8+ cytotoxic T cells, natural killer (NK) cells, interferons and Abs. On the other hand, fine-tuning of the host defence strategies to avoid irreversible harm has led to activation of cellular and humoral immunity that tightly control herpesviral infections in a hierarchical but also redundant manner [5]. To date, eight human herpesviruses, most of which are widespread in the population, are known (listed in Table 1 ). In the majority of cases, the viruses cause relatively mild or even asymptomatic infections in the immmunocompetent host. However, when the intricate balance between viral immune escape and the host's immunity is lost, uncontrolled virus replication leads to severe disease manifestation as is frequently observed in immunocompromised patients or congenitally infected babies [6].
Table 1.
Human herpesviruses and their FcγRs
| Subfamily | Designation | Synonyms | Genome size (kbp) | FcγRs |
|
|---|---|---|---|---|---|
| Genes | Proteins | ||||
| α | HHV-1 | Herpes simplex virus 1 | 152 | US8/US7 | gE/gl |
| HHV-2 | Herpes simplex virus 2 | 152 | US8/US7 | gE/gl | |
| HHV-3 | Varicella-zoster virus | 125 | ORF68/ORF67 | gE/gl | |
| β | HHV-5 | Cytomegalovirus | 235 | UL119–118 | gp68 |
| TRL11/IRL11 | gp34 | ||||
| HHV-6 | Human herpesvirus 6 | 162 | – | – | |
| HHV-7 | Human herpesvirus 7 | 162 | – | – | |
| γ |
HHV-4 | Epstein–Barr virus, Lymphocryptovirus | 172 | – | – |
| HHV-8 |
Kaposi's sarcoma-associated herpesvirus |
160 |
– |
– |
|
–: not reported.
The complement system and antiviral Abs form a first line of antiviral defence operating before a virion is able to attach to its cellular receptor mediating entry into the host cell to initiate the replicative cycle [7]. The complement system includes proteolytic enzymes, inflammatory proteins, cell surface receptors and proteins that cause cell death through osmotic lysis after insertion into biological membranes. Therefore, a primary requirement is that the complement cascade becomes initiated only in response to infection and cannot be initiated in a random fashion. The complement factors opsonise and lyse Ab-coated viral particles (virolysis) and infected cells (cytolysis) [8], [9]. A broad range of host cells expresses several complement-inhibiting proteins which protect tissues from complement-mediated lysis by the inhibition and dissociation of C3 convertases that are central for the amplification of the complement system [10]. Notably, the cell surface expression of CD46 and CD55, two of the complement control proteins (CCP), is strongly enhanced on the plasma membrane of HCMV-infected cells. The upregulated CD55 expression increases the capacity to regulate C3 deposition and thus protects CMV-infected cells from complement-mediated lysis [11]. The selective incorporation of CCPs, i.e. CD55, CD59 and CD46, into virions and the discovery of virus-encoded regulators of complement activation (RCA) underscore the important antiviral role of the complement system [12], [13].
Ab neutralisation of virus particles is achieved when bound Abs block the virion from productive interaction with receptors or prevent steps of entry and uncoating [14]. The combination of Abs and complement can activate the classical complement pathway to lyse enveloped viruses. The expression of the vFcγRs protects the HSV virion against this threat by interfering with the recruitment of complement [15].
The arsenal of immmuno-modulatory mechanisms of HVs is not restricted to the protection of free virus particles. HV-infected cells are protected from destruction by cytotoxic effector cells and rendered refractory to antiviral cytokines like interferons to ensure efficient virus replication. Recent reviews delineate the bewildering multitude and sophisticated mechanisms of such defence strategies in great detail [3], [4], [16], [17].
2. Antiviral effector functions of IgG
Ab neutralisation of viruses is usually demonstrated in vitro when binding of virus-specific Abs leads to a reduction in the number of infectious particles. The mechanisms leading to virus neutralisation are variable and include simple occupancy of virus surface and steric hindrance of a productive interaction with cellular receptors, causing virions to aggregate and thus reducing the concentration of infectious particles, but also complex models involving capping of critical epitopes or induction of conformational changes in viral envelope or capsid structures [18], [19]. The enhancing role of the Ab Fc part in direct neutralisation has been explained by an increased avidity of divalent Abs compared to monovalent Fab fragments [20], or its mere size in steric occupation of the virus surface [21].
Of paramount importance is the function of the Ab Fc moiety to recruit the complement system for enhancing in vivo neutralisation, by inducing Ab-dependent complement-mediated lysis (ADCL), and the Fc-receptor-mediated phagocytosis and subsequent destruction of the virus particle by professional antigen-presenting cells. The different ability of the IgG subclasses to bind complement can, therefore, explain the observation of differing neutralising capabilities of Abs sharing common variable regions but having different constant regions [22].
Only a minor fraction of the Ab repertoire that is generated during an immune response against a given virus has neutralising capacity. In addition, there are several other possible ways how Abs can act against viruses. Non-neutralising Abs may promote opsonisation and phagocytosis of a virus or contribute to antiviral defence during later stages of infection, when the virus has already entered the host cell. Recognition of viral antigens on the surface of infected cells by Abs can trigger virus-specific killing by nonspecific cytotoxic effector cells carrying Fc receptor (e.g. NK cells) in a process called Ab-dependent cellular cytotoxicity (ADCC) [23]. The Abs coating a virus-infected target cell attach to the effector cell via its FcγRs, thereby stimulating the release of perforin and granzymes from cytolytic granula which destroy the virus-infected cell.
Moreover, Abs can also block the release of progeny virus from infected cells [24] and have been shown to prevent cell-to-cell spread of viruses [25]. Multimeric IgA and IgM molecules which are transcytosed through epithelial cells have intriguing functions even beyond those of IgG: when such Igs encounter viral antigens inside of cells, they can neutralise virus infectivity even at intracellular sites [26], [27].
2.1. IgG in herpesviral infections
While Abs are powerful defence weapons against many viruses, their potency appears restricted against HVs. HVs are commonly able to reactivate productive infection from latency and to reach horizontal transmission and spread in the presence of virus-specific Abs. Likewise, preexisting CMV-specific Abs frequently fail to prevent virus transmission or exogenous superinfection with new virus strains [28], [29]. In mouse models of HV infection (mouse CMV, MCMV, murine γ-herpesvirus, MHV-68), Abs were demonstrated to be dispensable for the clearance of primary infection but only to become a limiting factor later on by preventing dissemination of recurrent virus during the post-primary phase of infection [30], [31].
The therapeutic effectiveness of hyperimmune sera against HV is limited. It is of value for preventive rather than treatment purposes in certain clinical settings. Here we will discuss the clinical use of antiherpesviral IgG, concentrating on α- and β-HVs that express FcγRs (see Section 4).
2.1.1. HCMV
In congenitally infected newborns, HCMV infection is an important cause of hearing loss as well as cognitive and motor impairments which cannot be effectively prevented or treated by current medical interventions (reviewed in Ref. [32]). Immunity to HCMV can strongly reduce the severity of disease, substantiating the need for a vaccine. In addition, HCMV infection is a major cause of disease and death in immunocompromised patients, e.g. solid organ and bone marrow transplant (BMT) recipients, cancer patients and HIV-infected individuals [33]. Pneumonia, retinitis and lesions of the gastrointestinal tract [34], [35] are the most common manifestations of HCMV disease.
The applicability of highly efficient anticytomegaloviral agents like ganciclovir and foscarnet is often limited due to the toxicity of the drugs and the development of drug-resistant HCMV. Therefore, treatment with Abs is frequently a demanding option as additional or replacement therapy. The standard formulations are intravenous infusions of either immunoglobulins pooled from normal human plasma (intravenous Ig, IVIG), or from donors screened for high anti-CMV titers (CMV-IGIV). CMV-IGIV, distributed under the trade names CytoGam® and Cytotect®, is indicated for prophylaxis of CMV disease associated with transplantation of kidney, lung, liver, pancreas and heart [36]. The latest therapeutic regimens include the combination of antivirals like gancyclovir with CMV-IGIV [37], [38], to increase the clinical benefit. The therapeutic value of CMV-IGIV has extensively been studied and reviewed [37], [39], [40], [41], [42]. Overall, most of the studies were able to demonstrate only a moderate if any reduction in CMV-associated morbidity in defined transplantation settings.
Due to the limited efficacy of polyclonal CMV-IGIV, IgG with a defined reactivity to HCMV antigens have been developed. The immunodominant CMV envelope glycoproteins gB and gH carry most of the neutralising B cell epitopes. They are involved in virus attachment, fusion with host cells and cell-to-cell spread of internal virions [43]. Therefore, gB and gH are obvious candidates for targeted Ab generation. The human monoclonal anti-gH-Ab MSL-109, or sevirumab®, has neutralising capacity but failed to exert any beneficial effect in CMV-seropositive hematopoietic stem cell transplant (HSCT) recipients [44]. Similarly, other anti-HCMV Abs including humanised mouse monoclonal IgGs capable to neutralise HCMV in vitro have not proven effective for therapeutic application in vivo [45], [46].
Several approaches using attenuated live virus or viral subunits, like gB, have been undertaken to produce HCMV vaccines, some of which were able to generate gB-specific Abs with demonstrable neutralising capacity [47]. However, no efficient HCMV vaccine is available for clinical purposes so far.
2.1.2. HSV
HSV-1 and HSV-2 share the ability to establish latent infection in sensory neurons and to accomplish recurrent cycles of reactivation, frequently leading to painful labial or genital lesions, respectively. Immunosuppression increases both the risk and the severity of HSV disease through cutaneous dissemination or infection of the central nervous system.
Several lines of circumstantial evidence suggest a protective effect of IgG against HSV-2. First, the risk of virus transmission and the extent of neonatal disease is much lower if the mother has recurrent as compared to primary genital infection [48]. Second, in mouse models of HSV-2 infection, various vaccination protocols generated Abs responses which mediated partial vaginal immunity [49], [50]. Recent efforts for vaccine generation employed glycoprotein-D (gD) subunit formulations since gD is a target for neutralising Abs due to the fact that gD directly interacts with HSV receptors on the cell surface to trigger virus entry. Two phase 3 studies of the GlaxoSmithKline HSV-2 gD-subunit vaccine demonstrated a 74% efficacy in preventing occurrence of genital herpes disease in women seronegative for both HSV-1 and HSV-2, while the vaccine was completely ineffective in decreasing HSV-2 infection in men or women seropositive for HSV-1 [51]. Similarly, the CHIRON gB2/gD2 vaccine using similar protein components, but administered with a different adjuvant, was ineffective in preventing HSV-2 infection [52].
2.1.3. VZV
VZV is the common etiological agent of two diseases, varicella (i.e. primary VZV infection or chickenpox) and zoster (i.e. shingles resulting from local reactivation of infection from endogenous VZV genomes in dorsal root ganglia cells). In immunocompetent children varicella is usually a benign, self-limited disease and no general treatment is required. In contrast, non-immune patients belonging to high-risk groups like immunocompromised individuals with exposure to VZV and newborns whose mothers undergo varicella at delivery must receive VZV immunoglobulins (VZIG). In these situations, varicella can take a life-threatening course of infection [53]. While VZIG is clearly efficient in post-exposure prophylaxis, it is not beneficial for therapy of already established VZV infection [54]. Herpes zoster occurs in the presence of VZV-specific Abs. Accordingly, VZIG has no preventive or therapeutic effect on herpes zoster, underlining the resilience of HVs to IgG.
In contrast to HCMV and HSV, active immunisation against VZV can be successfully accomplished with live attenuated virus and its use is approved in the United States since 1995. The VZV vaccine protects against severe varicella, although superinfection of vaccinees with exogenous wild-type VZV strains or reactivation of the vaccine virus may occur [55], [56], [57]. Early vaccination studies in children with leukemia suggest that the incidence of zoster might be lower in patients receiving the attenuated Oka vaccine strain than in those infected with wild-type virus [58], but at present it is not clear whether the vaccine will reduce the overall incidence of zoster in healthy vaccinated individuals.
Taken together, antiherpesviral IgG has a certain but limited prophylactic rather than therapeutic effectiveness in the control of HV spread and the progression and severity of disease. Abs alone, however, fail to provide sterile and protective immunity.
3. Cellular FcγRs
Specific surface-expressed cellular receptors are known for all Ig classes, including IgA (FcαR), IgD, (FcδR), IgE (FcεR), IgG (FcγR) and IgM (FcμR). Further Ig receptors exert essential functions in distributing Abs, like the polymeric Ig receptor which transports IgA and IgM across epithelia [59] or the neonatal FcR (FcRn) which delivers maternal IgG to the foetus across the placenta [59]. Recently, a new family consisting of several members of B-cell-expressed FcR homologs has been discovered [60], the precise function of which remains to be elucidated. All classes of cellular FcRs belong to the immunoglobulin supergene family (IgSF) of cell surface molecules. IgSF domains are characterised by a conserved structural motif termed the immunoglobulin fold (reviewed in Ref. [61]) that consists of two layers of antiparallel β-sheets, connected by variable loops. The Ig domain is an autonomous folding unit with manifold occurrence in various kinds of proteins.
The best studied and prototypic FcRs are the IgG-binding FcγRI, FcγRII, and FcγRIII which are exclusively expressed on immune cells and act at the interface of humoral and cellular immunity [62]. FcγRI (CD64) is a transmembrane protein with three extracellular Ig domains of the C2 class of Ig domains (see Fig. 1 ), which binds to human IgG-Fc with high affinity. The third, juxtamembrane domain is responsible for the high affinity binding while the first two domains alone can mediate only weak binding to IgG [63]. FcγRI is the only Fcγ receptor which strongly binds to monomeric IgG molecules, but does not signal unless IgGs are cross-linked by their specific polymeric ligands.
Fig. 1.
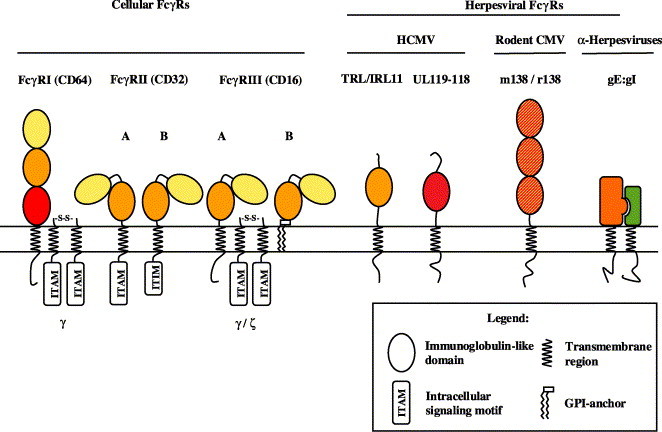
Structural composition of cellular and herpesviral FcγRs. Predicted immunoglobulin supergene family (IgSF)-like domains of cellular (left) and herpesviral (right) FcγRs are colour-coded based on their sequence relatedness. The Ig-like domains of the rodent CMV FcγRs are similarly distant to each of the three domains of cFcγRI. The α-herpesviral FcγR is composed by the gE:gI complex. gE but not gI exhibits a low sequence similarity to the second domain of cFcγRs [92]. The Ig domains of FcγRII and FcγRIII are depicted in their bent orientation that is seen in the crystal structures. The domain arrangement in the other FcγRs is unknown.
FcγRII (CD32) and FcγRIII (CD16) preferentially bind polymeric, but not monomeric IgG. They possess two extracellular Ig domains with exhibit closest similarity to the membrane distal domains of FcγRI. The FcγRII isoforms A and B are transmembrane proteins differing in their cytoplasmic portion, while FcγRIII can be expressed as a transmembrane or GPI-linked form (Fig. 1) [64]. The cellular FcγRs use distinct intracytoplasmic motifs for signal transduction into the cell. FcγRIIA and FcγRIIB contain an “immunoreceptor-tyrosine-based activation motif” (ITAM) or “immunoreceptor-tyrosine-based inhibitory motif” (ITIM), respectively, in their cytoplasmic tail. Both motifs are phosphorylated upon receptor engagement but with opposed outcome: While the ITAMs preferentially recruit src-family tyrosine kinases and induce cellular activation, ITIM phosphorylation serves to terminate signaling, mostly via the SH2-domain-containing inositol phosphatase (SHIP). FcγRI and FcγRIII do not contain intrinsic signaling properties but share the common FcR γ chain. Alternatively, FcγRIII can utilise the closely related ζ chain of the T-cell-receptor complex. The FcR γ and the ζ chain each contain an ITAM motif to induce activatory signaling events like the FcγRIIA cytoplasmic moiety [62]. The cross-linking of FcγRI-bound IgG by multivalent antigens, or the binding of preformed immune complexes with FcγRII or FcγRIII, results in clustering of the FcγR and triggering of a variety of effector mechanisms, such as ADCC, phagocytosis, the release of cytokines, enhanced antigen presentation, and the regulation of Ig production by B cells [62]. Thus, cFcγRs can be functionally considered as antigen receptors.
The Fc portion of Ig forms a well-characterised structure [65]. Its binding to cFcRs is mediated by the membrane-proximal second IgSF domain of cFcRs as revealed by recent studies resolving the crystal structures of FcγRIII in complex with IgG-Fc [66], FcγRIIA complexed with IgG-Fc [67] and FcεRI bound to IgE-Fc [68]. These studies revealed a conserved arrangement of the two Ig domains in a highly bent conformation, uncovering the Ig binding site that is located at the top of the second Ig domain concordantly in all three molecules.
Interestingly, all three FcγRs also occur as soluble forms in the serum, generated either by proteolysis [69], alternative splicing [70], or from separate genes [63]. The functional significance of these isoforms remains to be unravelled.
Studies taking advantage from gene knock-out mice have demonstrated prominent immunoregulatory properties of cFcγRs. Mice devoid of the common FcR γ chain display pleiotropic defects of immune effector cells including impaired macrophage phagocytosis and NK-cell-mediated ADCC as well as defective mast cell-dependent allergic responses [71]. The similar phenotype of FcγRIII-deficient mice illustrates the intimate functional cooperation with the γ chain [72]. The critical regulatory (inhibitory) effect of FcγRIIB on various immune cells is reflected by the hyperreactivity of the immune system in FcγRIIB-deficient animals. Due to the lack of control of B cell proliferation, FcγRIIB-deficient mice develop various forms of autoimmune diseases, either spontaneously or after experimental induction [73], [74]. Mast cells from FcγRIIB-deficient animals are hypersensitive to IgG-induced degranulation which culminates in enhanced passive cutaneous analphylaxis reactions [74].
4. Structure and functions of herpesviral FcγRs
While the cellular FcγRs play a key role in transducing the antiviral potency and specificity of IgG to cellular immunity, various pathogens exploit the same principle of binding the IgG Fc proportion for their own purpose to avoid immune destruction. Examples for Fc-binding proteins are found in gram-positive bacteria [75], in protozoa like schistosomes and trypanosomes [76], [77], [78], in coronaviruses [79], hepatitis C virus [80], and most commonly in α- and β-herpesviruses. The last group comprises cytomegaloviruses from different species [81], [82], [83], herpes simplex viruses 1 and 2 [84], varicella zoster virus (VZV) [85], the porcine herpesvirus pseudorabies virus (PRV) [86] and presumably also Marek's disease virus (MDV), an α-herpesvirus infecting chickens, where proteins homologous to the α-herpesvirus FcγRs are found [87].
4.1. The FcγR of the α-herpesviruses: the gE:gI complex
The ability of HSV-1-infected cells to bind human IgG via its Fc-part was first demonstrated in 1979 [88]. The responsible membrane proteins have subsequently been identified as a complex formed by the glycoproteins gE and gI (Fig. 1) [89], which are encoded by the HSV genes US (unique short) 7/US8 and are dispensable for HSV infection and replication in vitro. Homologous molecules to gE and gI are found in HSV-2 [84], VZV [90], PRV [91] and MDV [87]. gE and gI are exposed on the surface of infected cells and represent also the most abundant structural glycoproteins within the virus envelope. Alignment of a limited portion of the extracellular chain of gE reveals a low sequence similarity with the second domain of cFcγRs [92]. While gE alone binds only aggregated IgG with low affinity, complex formation with gI induces a high affinity receptor which is able to fix monomeric IgG. The HSV-1 gE:gI complex is a heterodimer composed of one molecule each and binds IgG with 1:1 stoichiometry. The affinity for IgG4 is highest with K D=40 nM [93]. The gE:gI complex binds IgG between the second and third Ig domain, at the Cγ2–Cγ3 interface, which is similar to the Fc-binding site of rheumatoid factors and protein A from Staphylococcus aureus. The importance of H435 of IgG1 for the interaction correlates with the failure to bind many IgG3 subclass allotypes, where the corresponding residue is an arginine [93].
The IgG-binding capability of gE:gI protects HSV-infected cells from ADCC [15]. In mice passively immunised with human anti-HSV-IgG, titers of a gE mutant with a 4-aa insert disrupting the region homologous to mammalian FcγRs were significantly reduced, whereas wild-type HSV remained unaffected. [94]. The minimal mutation introduced into the gE mutants affected the IgG-related functions of gE:gI, but did not interfere with the other important gE function, i.e. to mediate direct cell-to-cell spread of nascent virions [95].
The observed protection against antiviral IgG conferred by gE:gI was explained by “antibody bipolar bridging” (ABB) [96] (Fig. 2) . According to this model, an IgG molecule recognising an epitope on the surface of a virus-infected cell or a virion via its clonotypic antigen binding Fab domain is simultaneously sequestered by the gE:gI complex via its Fc part. Thus, the IgG Fc domain is prevented from recruiting effector mechanisms, i.e. the complement component C1q or FcγR-expressing cytotoxic effector cells. Several lines of evidence support this hypothesis. First, the competition of binding sites between gE:gI and protein A on the Fc–Cγ2–Cγ3 interface was exploited to demonstrate the masking of the Fc part of IgG through the viral FcγR when bound to HSV-infected cells via its antigen-binding sites [97]. Second, fluorescence resonance energy transfer (FRET) studies have demonstrated that the IgG molecule structure is indeed equipped with sufficient intrinsic flexibility allowing the simultaneous interaction of the Fab and Fc domains with separate ligands displayed on the same cell or virion surface [98].
Fig. 2.
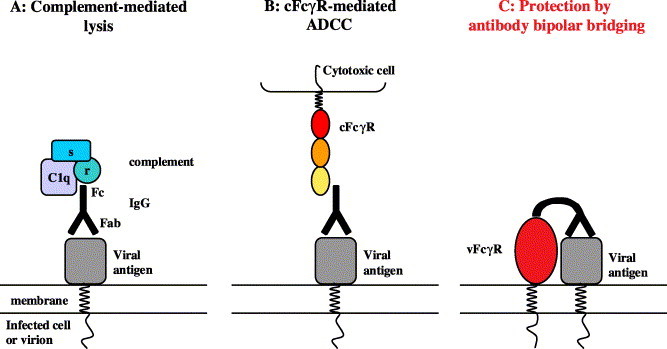
Herpesviral FcγRs protect against antiviral IgG by antibody-bipolar bridging. (A) Antiviral IgG binds to its cognate viral antigen on the surface of an infected cell or a virus particle via its Fab part. Upon binding, the Fc region recruits the C1q,r,s complex which activates the complement system via the classical pathway, resulting in antibody-dependent complement-mediated lysis (ADCL). (B) Alternatively, attachment of bound virus-specific IgG to cFcγR+-cytotoxic effector cells, e.g. NK cells, mediates cFcγR cross-linking and triggering of antibody-dependent cellular cytotoxicity (ADCC). (C) Antibody-bipolar bridging: viral FcγRs sequester the Fc part of bound antiviral IgG. This engagement blocks the recruitment of complement proteins or cFcγRs and thus mediates protection of the virus particle or infected cell from destruction.
The VZV gE vFcγR was demonstrated to undergo repeated cycles of receptor-mediated endocytosis and recycling to the plasma membrane of VZV-infected cells. This function depends on a tyrosine residue in a conserved YXXL motif that resembles endocytosis signals [99]. Most interestingly, this process is induced by Fc binding to gE. This finding suggests that the vFcγRs might be designed as a molecular device removing antiviral IgG from the surface of infected cells and thus avoids ADCC and ADCL.
4.2. HCMV FcγRs
Biochemical evidence for an HCMV-induced Fc-binding activity in infected fibroblasts has been collected over almost three decades. IgG binding was detected on the plasma membrane and at intracellular sites of infected cells, as well as in association with HCMV virions [100]. Fc binding was ascribed to various proteins with MWs between 38 and 130 kDa [101]. Only recently, three viral genes encoding two different Fc binding glycoproteins, gp68 and gp34, were identified within the HCMV AD169 genome, both of which are dispensable for viral replication in vitro [83], [102]. The equipment with two distinct FcγRs is unique for HCMV and adds another example to the bewildering array of HCMV glycoproteins by which this HV modulates immune functions. The structural composition of the HCMV-encoded vFcγRs is thoroughly different from the HSV gE:gI heterodimer in that the former represent single-chain type I transmembrane proteins showing significant sequence homology with human cFcγRs in their extraluminal part and are predicted to form IgSF-like domains [83] (Fig. 1).
gp68 is expressed during the early and late phase of HCMV infection from a hitherto unidentified spliced 4.1 kb transcript encompassing the open reading frames UL119 and UL118 from a complex transcription unit, UL115–119. The UL119–118 gene sequence encodes a 347-aa type Ia transmembrane protein with 12 potential N-glycosylation sites. The extracellular domain is predicted to contain a single V-like Immunoglobulin superfamily (IgSF) domain (aa 90–190), including a conserved disulphide bond. In comparison to cellular FcγRs, this domain displays the highest degree of homology to the unique third IgSF domain of FcγRI, the domain conferring the high affinity binding to IgG. Secondary structure algorithms concordantly predict a composition of the gp68 IgSF domain built by seven β-sheets resembling the architecture of the cFcγR IgSF domains [83]. The cytoplasmic domain of gp68 contains a modified ITIM motif (WSYKRL) which may provide a link of gp68 to cellular signaling pathways.
The glycoprotein gp34 is encoded by two identical gene copies, TRL11 and IRL11, the latter of which is present in the duplicated UL5' (unique long) segment of the laboratory HCMV strain AD169. gp34 is a type Ia transmembrane protein consisting of 234 aa. The extracellular region includes three potential N-glycosylation sites and is predicted to form one IgSF-like domain [83]. This prediction is based on homology with the second domain of the cFcγRIII/RII, which forms the essential IgG binding moiety of cFcγRs [103]. Important determinants of the three cFcγRIII binding regions for Fc fragment are conserved in the gp34 sequence. Interestingly, secondary structure calculations predict α-helical interruptions of the β-sheet structure of the IgSF domain, a feature which resembles the FcRn, where α-helices also make important contributions to the binding interface with hIgG [104]. The cytoplasmic tail of gp34 contains a conserved dileucine motif (DXXXLL) which could indicate a role in intracellular targeting of IgG–gp34 complexes to the endocytic route [105]. Thus, similar to VZV gE:gI, HCMV gp34 might remove bound IgG from the plasma membrane of infected cells to prevent ADCL and ADCC. In contrast to the HSV gE:gI FcγR, HCMV-infected cells were shown to bind to all 4 subclasses of human IgG [106] with graduated affinity (IgG1≥IgG4>IgG2>IgG3 [107]). However, the experimental setting used could not dissolve the separate Ig binding properties of gp34 and gp68. These are likely to exist given the different affinities to IgG of distinct mammalian species. Among non-human IgG, rabbit and, to a minor extent, rat IgG is recognised by gp34 while no binding was observed for gp68 [83], [106].
Ultrastructural information revealing the exact binding and contact sites of the HCMV FcγRs and the Fc ligand is not available yet. However, one conclusion with regard to the Fc molecule can be drawn form the fact that Fc-bound vFcγRs can still be precipitated with protein A [83] implying that separate binding sites must exist. This feature of HCMV FcγRs is contrary to HSV gE:gI where the S. aureus protein A competes for Fc binding.
Similar to the HSV gE:gI, both of the HCMV FcγRs are independently interfering with the neutralising function of human IgG (Reinhard, H. and Hengel H., unpublished observation). This fact is also evident from plaque reduction assays with HCMV-immune human sera performed in the presence of an excess of Fc fragments or β2-microglobulin (β2m) as a control protein. While the addition of Fc but not β2m improves the neutralising efficiency of HCMV-IgG in a dose-dependent manner, the activity of Measles virus (MV)-specific IgG is completely independent of Fc when neutralising MV virions which do not possess vFcγRs (Reinhard, H. and Hengel H., unpublished observations). This finding may have obvious implications for the in-vivo neutralisation of HCMV: In body fluids like blood, the high amount of non-immune IgG might exert a similar effect like the Fc fragment in the neutralisation assay discussed above, i.e. enhance HCMV virion neutralisation. In contrast, at sites where IgG concentrations are significantly lower than in serum, i.e. on mucosal surfaces and body fluids like saliva, breast milk and genital fluids the HCMV vFcγRs should promote virus shedding and horizontal transmission. In such an environment vFcγRs are critically required to confer resistance of HCMV particles against neutralising IgG.
Notably, the HCMV vFcγRs could also provide a molecular link for infection and replication of the human immunodeficiency virus (HIV). The HIV-induced damage of the host's immune system favours HCMV reactivation from latency and the emergence of persistent HCMV replication and recurrent disease [108]. In this context, it is interesting that HCMV infection is able to confer HIV-susceptibility to otherwise nonpermissive fibroblasts [109]. This could be explained by Ab-coated HIV particles attaching via the vFcγRs to the surface of HCMV-infected cells. By this mechanism HIV virions might be endocytosed and establish HIV infection. This could open up a cellular compartment for HIV replication where HIV may benefit from the multitude of stealth mechanisms by which HCMV avoids immune attack.
4.3. Rodent CMV FcγRs
Mouse CMV (MCMV)-infected cells can be decorated with IgG-Fc fragment both intracellularly and on the surface [82]. The genetic basis for this feature is the expression of a single gene, m138, coding for an FcγR [82]. The predicted 569-aa type I transmembrane glycoprotein has a calculated mass of 65 kDa and is synthesised during the early and late phase of MCMV replication. The MCMV FcγR is highly glycosylated reaching a MW of 105 kDa, which is reduced to a single band of 68 kDa by deglycosylation. The m138 protein can be precipitated with mouse IgG-Fc fragment coupled to protein A indicating non-overlapping binding sites as it is the case for the HCMV FcγRs. Alignment of the m138 sequence with mouse cFcγRs displays a more significant degree of homology and predicts a composition of three FcγR-related IgSF-like domains. (Fig. 1, A. Zimmermann, A. Bigl and H. Hengel, unpublished data).
Two distinct and independent m138 homologs are present in two different cytomegaloviruses of the rat (RCMV), RCMV strain Maastricht, and RCMV strain England, respectively [110], [111], [112]. Preliminary data show that both RCMV-encoded m138 homologs constitute functional vFcγRs (A. Bigl, M. Budt, S. Voigt, A. Zimmermann, unpublished observation). The MCMV model provides an unique opportunity to assess the biological role of an vFcγR for viral pathogenesis and immune control in vivo. To this end the replication of a MCMV m138 gene deletion mutant, Δm138, was analysed in mice and revealed a dramatically attenuated phenotype in vivo. Surprisingly, in μmt−/− mice lacking the Ig μ chain, defective in B cell development and devoid of Abs, the Δm138 virus was not rescued from attenuation. From this finding it was concluded that the major biological function of the MCMV FcγR must not be related to IgG binding but includes a more prominent IgG-unrelated function as it is known for HSV gE:gI [113]. However, the replacement of the m138 gene sequence by inserting the LacZ gene of Escherichia coli might have affected genes neighbouring m138, which possess common transcription units with m138 [82] and have been implicated in macrophage tropism and efficient MCMV replication in vivo [114]. Furthermore, the viral replication was investigated during primary MCMV infection, where Abs do not contribute to virus immune control [31]. Therefore, the available data do not preclude an Ab-dependent role for the MCMV FcγR at later phases of infection when Abs effectively limit virus dissemination and transmission [5], [31]. Studies introducing more subtle changes in the m138 gene without affecting adjacent genes are required to elucidate putative IgG-dependent functions of the MCMV FcγR.
5. Perspective: engineering virus-specific IgGs resisting vFcγRs?
Recent progress in Ab generation technologies allows to select Abs of perfect specificity for defined target structures which can employ distinct effector functions and can be produced in high quantities allowing therapeutic application in patients [115]. Due to the limitations of conventional antiviral drug therapy, the design of potent virus-specific Abs as a therapeutical tool is demanding. Published data support the notion that IgG has an important but certainly not exclusive immunological role against herpesviral infections, and is a potential tool of immunoprophylaxis [19], [116]. As outlined above, viral FcγRs are prime suspects for the remarkable ability of HVs to counteract IgG effector functions. Therefore, it is conceivable to assume that antiviral Abs could reach a more potent antiviral efficiency when resisting FcγR-mediated attenuation. According to this concept, an ideal IgG-Fc domain would not be bound by a vFcγR, while still retaining normal affinity to the ligands that recruit protective components of the immune system. As an example, human IgG3 Abs could represent promising candidates for an effective anti-HSV agent since this IgG subclass is a potent activator of the complement cascade [117], but is not blocked by HSV gE:gI [93]. Based on the discrimination of the HSV gE:gI FcγR between hIgG1 allotypes [118], even minimal mutations introduced into recombinant Abs could be sufficient to avoid vFcγR binding, while retaining the functional properties of the molecule with respect to cFcγRs and complement.
HCMV encodes even two FcγRs which bind all human IgG subclasses [83], although with graded affinity, making the design of a discriminating IgG-Fc domain more complex. A detailed understanding of the interaction of the CMV FcγRs with IgG-Fc including ultrastructural analysis would facilitate the modelling of effective IgGs against this virus. If a prototypic IgG-Fc domain fulfilling the requirements of selective interaction is found, this module could be fused to a large number of HCMV-reactive IgG-Fab domains. Such a cocktail of monoclonal recombinant Abs will be necessary to deal with the interstrain diversity of the major HCMV neutralising epitopes present on the glycoproteins gB and gH [119], [120], [121] in order to generate widely effective protection.
The MCMV animal model could serve as an optimal test tube to generate experimental evidence in vivo for an IgG-Fc-dependent subversive role of vFcγRs in viral pathogenesis in a natural host. Next, as a proof of principle, MCMV-specific monoclonal Abs with a therapeutic potential [116] are available which will allow to assess the therapeutic benefit of a mutated vFcγR-resistant IgG-Fc module in a variety of infection conditions in vivo. Based on this knowledge, designed IgGs could lead to a new generation of immunopharmacological tools effective against the large burden of human herpesviral diseases.
Acknowledgements
The expert technical assistance of Ingrid Deitemeier is greatly appreciated. We are grateful to Dr. K. Masihi for critical reading of the manuscript. Our work is supported by the Deutsche Forschungsgemeinschaft through SFB 421/A8 and by the European Community, grant QLRT-2001-01112.
References
- 1.Sissons J.G., Bain M., Wills M.R. Latency and reactivation of human cytomegalovirus. J. Infect. 2002;44:73–77. doi: 10.1053/jinf.2001.0948. [DOI] [PubMed] [Google Scholar]
- 2.Cohrs R.J., Gilden D.H. Human herpesvirus latency. Brain Pathol. 2001;11:465–474. doi: 10.1111/j.1750-3639.2001.tb00415.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abendroth A., Arvin A. Varicella-zoster virus immune evasion. Immunol. Rev. 1999;168:143–156. doi: 10.1111/j.1600-065x.1999.tb01289.x. [DOI] [PubMed] [Google Scholar]
- 4.Hengel H., Brune W., Koszinowski U.H. Immune evasion by cytomegalovirus-survival strategies of a highly adapted opportunist. Trends Microbiol. 1998;6:190–197. doi: 10.1016/s0966-842x(98)01255-4. [DOI] [PubMed] [Google Scholar]
- 5.Polic B., Hengel H., Krmpotic A., Trgovcich J., Pavic I., Luccaronin P. Hierarchical and redundant lymphocyte subset control precludes cytomegalovirus replication during latent infection. J. Exp. Med. 1998;188:1047–1054. doi: 10.1084/jem.188.6.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roizman B., Pellett P.E. The family herpesviridae: a brief introduction. In: Knipe D.M., Howley P.M., Griffin D.E., Lamb R.A., Martin M.A., Roizman B., Straus S.E., editors. Fields virology. 4th ed. Lippincott Williams and Wilkins; Philadelphia: 2001. pp. 2381–2397. [Google Scholar]
- 7.Zinkernagel R.M., LaMarre A., Ciurea A., Hunziker L., Ochsenbein A.F., McCoy K.D. Neutralizing antiviral antibody responses. Adv. Immunol. 2001;79:1–53. doi: 10.1016/S0065-2776(01)79001-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Favoreel H.W., Van de Walle G.R., Nauwynck H.J., Pensaert M.B. Virus complement evasion strategies. J. Gen. Virol. 2003;84:1–15. doi: 10.1099/vir.0.18709-0. [DOI] [PubMed] [Google Scholar]
- 9.Lachmann P.J., Davies A. Complement and immunity to viruses. Immunol. Rev. 1997;159:69–77. doi: 10.1111/j.1600-065x.1997.tb01007.x. [DOI] [PubMed] [Google Scholar]
- 10.Smith G.P., Smith R.A. Membrane-targeted complement inhibitors. Mol. Immunol. 2001;38:249–255. doi: 10.1016/s0161-5890(01)00047-5. [DOI] [PubMed] [Google Scholar]
- 11.Spiller O.B., Morgan B.P., Tufaro F., Devine D.V. Altered expression of host-encoded complement regulators on human cytomegalovirus-infected cells. Eur. J. Immunol. 1996;26:1532–1538. doi: 10.1002/eji.1830260719. [DOI] [PubMed] [Google Scholar]
- 12.Spear G.T., Lurain N.S., Parker C.J., Ghassemi M., Payne G.H., Saifuddin M. Host cell-derived complement control proteins CD55 and CD59 are incorporated into the virions of two unrelated enveloped viruses. Human T cell leukemia/lymphoma virus type I (HTLV-I) and human cytomegalovirus (HCMV) J. Immunol. 1995;155:4376–4381. [PubMed] [Google Scholar]
- 13.Kapadia S.B., Levine B., Speck S.H., Virgin H.W.T. Critical role of complement and viral evasion of complement in acute, persistent, and latent gamma-herpesvirus infection. Immunity. 2002;17:143–155. doi: 10.1016/s1074-7613(02)00369-2. [DOI] [PubMed] [Google Scholar]
- 14.Dimmock N.J. Neutralization of animal viruses. Curr. Top. Microbiol. Immunol. 1993;183:1–149. doi: 10.1007/978-3-642-77849-0. [DOI] [PubMed] [Google Scholar]
- 15.Dubin G., Socolof E., Frank I., Friedman H.M. Herpes simplex virus type 1 Fc receptor protects infected cells from antibody-dependent cellular cytotoxicity. J. Virol. 1991;65:7046–7050. doi: 10.1128/jvi.65.12.7046-7050.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mocarski E.S., Jr. Immunomodulation by cytomegaloviruses: manipulative strategies beyond evasion. Trends Microbiol. 2002;10:332–339. doi: 10.1016/s0966-842x(02)02393-4. [DOI] [PubMed] [Google Scholar]
- 17.Loenen W.A., Bruggeman C.A., Wiertz E.J. Immune evasion by human cytomegalovirus: lessons in immunology and cell biology. Semin. Immunol. 2001;13:41–49. doi: 10.1006/smim.2001.0294. [DOI] [PubMed] [Google Scholar]
- 18.Possee R.D., Schild G.C., Dimmock N.J. Studies on the mechanism of neutralization of influenza virus by antibody: evidence that neutralizing antibody (anti-haemagglutinin) inactivates influenza virus in vivo by inhibiting virion transcriptase activity. J. Gen. Virol. 1982;58:373–386. doi: 10.1099/0022-1317-58-2-373. [DOI] [PubMed] [Google Scholar]
- 19.Parren P.W., Burton D.R. The antiviral activity of antibodies in vitro and in vivo. Adv. Immunol. 2001;77:195–262. doi: 10.1016/S0065-2776(01)77018-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Parren P.W., Burton D.R. Antibodies against HIV-1 from phage display libraries: mapping of an immune response and progress towards antiviral immunotherapy. Chem. Immunol. 1997;65:18–56. doi: 10.1159/000319346. [DOI] [PubMed] [Google Scholar]
- 21.Lamarre A., Talbot P.J. Protection from lethal coronavirus infection by immunoglobulin fragments. J. Immunol. 1995;154:3975–3984. [PubMed] [Google Scholar]
- 22.Furebring C., Speckner A., Mach M., Sandlie I., Norderhaug L., Borrebaeck C.A. Antibody-mediated neutralization of cytomegalovirus: modulation of efficacy induced through the IgG constant region. Mol. Immunol. 2002;38:833–840. doi: 10.1016/s0161-5890(01)00119-5. [DOI] [PubMed] [Google Scholar]
- 23.Burton D.R., Woof J.M. Human antibody effector function. Adv. Immunol. 1992;51:1–84. doi: 10.1016/s0065-2776(08)60486-1. [DOI] [PubMed] [Google Scholar]
- 24.de Parseval A., Lerner D.L., Borrow P., Willett B.J., Elder J.H., Shariff D.M. Blocking of feline immunodeficiency virus infection by a monoclonal antibody to CD9 is via inhibition of virus release rather than interference with receptor binding. J. Virol. 1997;71:5742–5749. doi: 10.1128/jvi.71.8.5742-5749.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mannini-Palenzona A., Costanzo F., Fiorilli M.P., Derenzini M. Growth, spread, and extracellular localization of herpes simplex virus 1 in Vero cells in the presence of an anti-gD plaque inhibiting monoclonal antibody. New Microbiol. 1998;21:65–76. [PubMed] [Google Scholar]
- 26.Fujioka H., Emancipator S.N., Aikawa M., Huang D.S., Blatnik F., Karban T. Immunocytochemical colocalization of specific immunoglobulin A with sendai virus protein in infected polarized epithelium. J. Exp. Med. 1998;188:1223–1229. doi: 10.1084/jem.188.7.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bomsel M., Heyman M., Hocini H., Lagaye S., Belec L., Dupont C. Intracellular neutralization of HIV transcytosis across tight epithelial barriers by anti-HIV envelope protein dIgA or IgM. Immunity. 1998;9:277–287. doi: 10.1016/s1074-7613(00)80610-x. [DOI] [PubMed] [Google Scholar]
- 28.Boppana S.B., Rivera L.B., Fowler K.B., Mach M., Britt W.J. Intrauterine transmission of cytomegalovirus to infants of women with preconceptional immunity. N. Engl. J. Med. 2001;344:1366–1371. doi: 10.1056/NEJM200105033441804. [DOI] [PubMed] [Google Scholar]
- 29.Strelow L., Hansen L., Buxton K., Leid J., Edgar J., Walker J. Evidence of CMV-reinfection in a sero-positive host. 28th International Herpesvirus Workshop, Madison, WI, USA; 2003. [Google Scholar]
- 30.Doherty P.C., Christensen J.P., Belz G.T., Stevenson P.G., Sangster M.Y. Dissecting the host response to a gamma-herpesvirus. Philos. Trans. R Soc. Lond. B Biol. Sci. 2001;356:581–593. doi: 10.1098/rstb.2000.0786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jonjic S., Pavic I., Polic B., Crnkovic I., Lucin P., Koszinowski U.H. Antibodies are not essential for the resolution of primary cytomegalovirus infection but limit dissemination of recurrent virus. J. Exp. Med. 1994;179:1713–1717. doi: 10.1084/jem.179.5.1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pass R.F., Burke R.L. Development of cytomegalovirus vaccines: prospects for prevention of congenital CMV infection. Semin. Pediatr. Infect. Dis. 2002;13:196–204. doi: 10.1053/spid.2002.125863. [DOI] [PubMed] [Google Scholar]
- 33.Field A.K. Human cytomegalovirus: challenges, opportunities and new drug development. Antivir. Chem. Chemother. 1999;10:219–232. doi: 10.1177/095632029901000501. [DOI] [PubMed] [Google Scholar]
- 34.Salomon N., Perlman D.C. Cytomegalovirus pneumonia. Semin. Respir. Infect. 1999;14:353–358. [PubMed] [Google Scholar]
- 35.Hoover D.R., Peng Y., Saah A., Semba R., Detels R.R., Rinaldo C.R., Jr. Occurrence of cytomegalovirus retinitis after human immunodeficiency virus immunosuppression. Arch. Ophthalmol. 1996;114:821–827. doi: 10.1001/archopht.1996.01100140035004. [DOI] [PubMed] [Google Scholar]
- 36.Sawyer L.A. Antibodies for the prevention and treatment of viral diseases. Antiviral Res. 2000;47:57–77. doi: 10.1016/s0166-3542(00)00111-x. [DOI] [PubMed] [Google Scholar]
- 37.Zamora M.R. Use of cytomegalovirus immune globulin and ganciclovir for the prevention of cytomegalovirus disease in lung transplantation. Transpl. Infect. Dis. 2001;3(Suppl. 2):49–56. doi: 10.1034/j.1399-3062.2001.00010.x. [DOI] [PubMed] [Google Scholar]
- 38.Tzakis A.G. Cytomegalovirus prophylaxis with ganciclovir and cytomegalovirus immune globulin in liver and intestinal transplantation. Transpl. Infect. Dis. 2001;3(Suppl 2):35–39. doi: 10.1034/j.1399-3062.2001.00007.x. [DOI] [PubMed] [Google Scholar]
- 39.Wittes J.T., Kelly A., Plante K.M. Meta-analysis of CMVIG studies for the prevention and treatment of CMV infection in transplant patients. Transplant Proc. 1996;28:17–24. [PubMed] [Google Scholar]
- 40.Snydman D.R. Historical overview of the use of cytomegalovirus hyperimmune globulin in organ transplantation. Transpl. Infect. Dis. 2001;3(Suppl. 2):6–13. doi: 10.1034/j.1399-3062.2001.00002.x. [DOI] [PubMed] [Google Scholar]
- 41.Avery R.K. Prevention and treatment of cytomegalovirus infection and disease in heart transplant recipients. Curr. Opin. Cardiol. 1998;13:122–129. doi: 10.1097/00001573-199803000-00009. [DOI] [PubMed] [Google Scholar]
- 42.Ruutu T., Ljungman P., Brinch L., Lenhoff S., Lonnqvist B., Ringden O. No prevention of cytomegalovirus infection by anti-cytomegalovirus hyperimmune globulin in seronegative bone marrow transplant recipients. The Nordic BMT Group. Bone Marrow Transplant. 1997;19:233–236. doi: 10.1038/sj.bmt.1700649. [DOI] [PubMed] [Google Scholar]
- 43.Navarro D., Paz P., Tugizov S., Topp K., La Vail J., Pereira L. Glycoprotein B of human cytomegalovirus promotes virion penetration into cells, transmission of infection from cell to cell, and fusion of infected cells. Virology. 1993;197:143–158. doi: 10.1006/viro.1993.1575. [DOI] [PubMed] [Google Scholar]
- 44.Boeckh M., Bowden R.A., Storer B., Chao N.J., Spielberger R., Tierney D.K. Randomized, placebo-controlled, double-blind study of a cytomegalovirus-specific monoclonal antibody (MSL-109) for prevention of cytomegalovirus infection after allogeneic hematopoietic stem cell transplantation. Biol. Blood Marrow Transplant. 2001;7:343–351. doi: 10.1016/s1083-8791(01)80005-7. [DOI] [PubMed] [Google Scholar]
- 45.Paar D.P., Pollard R.B. Immunotherapy of CMV infections. Adv. Exp. Med. Biol. 1996;394:145–151. doi: 10.1007/978-1-4757-9209-6_15. [DOI] [PubMed] [Google Scholar]
- 46.Hamilton A.A., Manuel D.M., Grundy J.E., Turner A.J., King S.I., Adair J.R. A humanized antibody against human cytomegalovirus (CMV) gpUL75 (gH) for prophylaxis or treatment of CMV infections. J. Infect. Dis. 1997;176:59–68. doi: 10.1086/514040. [DOI] [PubMed] [Google Scholar]
- 47.Drulak M.W., Malinoski F.J., Fuller S.A., Stewart S.S., Hoskin S., Duliege A.M. Vaccination of seropositive subjects with CHIRON CMV gB subunit vaccine combined with MF59 adjuvant for production of CMV immune globulin. Viral Immunol. 2000;13:49–56. doi: 10.1089/vim.2000.13.49. [DOI] [PubMed] [Google Scholar]
- 48.Brown Z.A., Benedetti J., Ashley R., Burchett S., Selke S., Berry S. Neonatal herpes simplex virus infection in relation to asymptomatic maternal infection at the time of labor. N. Engl. J. Med. 1991;324:1247–1252. doi: 10.1056/NEJM199105023241804. [DOI] [PubMed] [Google Scholar]
- 49.Mohamedi S.A., Brewer J.M., Alexander J., Heath A.W., Jennings R. Antibody responses, cytokine levels and protection of mice immunised with HSV-2 antigens formulated into NISV or ISCOM delivery systems. Vaccine. 2000;18:2083–2094. doi: 10.1016/s0264-410x(99)00567-8. [DOI] [PubMed] [Google Scholar]
- 50.Parr M.B., Parr E.L. Vaginal immunity in the HSV-2 mouse model. Int. Rev. Immunol. 2003;22:43–63. doi: 10.1080/08830180305228. [DOI] [PubMed] [Google Scholar]
- 51.Stanberry L.R., Spruance S.L., Cunningham A.L., Bernstein D.I., Mindel A., Sacks S. Glycoprotein-D-adjuvant vaccine to prevent genital herpes. N. Engl. J. Med. 2002;347:1652–1661. doi: 10.1056/NEJMoa011915. [DOI] [PubMed] [Google Scholar]
- 52.Koelle D.M., Corey L. Recent progress in herpes simplex virus immunobiology and vaccine research. Clin. Microbiol. Rev. 2003;16:96–113. doi: 10.1128/CMR.16.1.96-113.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fleisher G., Henry W., McSorley M., Arbeter A., Plotkin S. Life-threatening complications of varicella. Am. J. Dis. Child. 1981;135:896–899. doi: 10.1001/archpedi.1981.02130340008004. [DOI] [PubMed] [Google Scholar]
- 54.Gershon A.A., Steinberg S., Brunell P.A. Zoster immune globulin. A further assessment. N. Engl. J. Med. 1974;290:243–245. doi: 10.1056/NEJM197401312900503. [DOI] [PubMed] [Google Scholar]
- 55.Hammerschlag M.R., Gershon A.A., Steinberg S.P., Clarke L., Gelb L.D. Herpes zoster in an adult recipient of live attenuated varicella vaccine. J. Infect. Dis. 1989;160:535–537. doi: 10.1093/infdis/160.3.535. [DOI] [PubMed] [Google Scholar]
- 56.Plotkin S.A., Starr S.E., Connor K., Morton D. Zoster in normal children after varicella vaccine. J. Infect. Dis. 1989;159:1000–1001. doi: 10.1093/infdis/159.5.1000. [DOI] [PubMed] [Google Scholar]
- 57.Galil K., Lee B., Strine T., Carraher C., Baughman A.L., Eaton M. Outbreak of varicella at a day-care center despite vaccination. N. Engl. J. Med. 2002;347:1909–1915. doi: 10.1056/NEJMoa021662. [DOI] [PubMed] [Google Scholar]
- 58.Hardy I., Gershon A.A., Steinberg S.P., LaRussa P. The incidence of zoster after immunization with live attenuated varicella vaccine. A study in children with leukemia. Varicella Vaccine Collaborative Study Group. N. Engl. J. Med. 1991;325:1545–1550. doi: 10.1056/NEJM199111283252204. [DOI] [PubMed] [Google Scholar]
- 59.Ghetie V., Ward E.S. Multiple roles for the major histocompatibility complex class I-related receptor FcRn. Annu. Rev. Immunol. 2000;18:739–766. doi: 10.1146/annurev.immunol.18.1.739. [DOI] [PubMed] [Google Scholar]
- 60.Davis R.S., Wang Y.H., Kubagawa H., Cooper M.D. Identification of a family of Fc receptor homologs with preferential B cell expression. Proc. Natl. Acad. Sci. U. S. A. 2001;98:9772–9777. doi: 10.1073/pnas.171308498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Williams A.F., Barclay A.N. The immunoglobulin superfamily-domains for cell surface recognition. Annu. Rev. Immunol. 1988;6:381–405. doi: 10.1146/annurev.iy.06.040188.002121. [DOI] [PubMed] [Google Scholar]
- 62.Ravetch J.V., Bolland S. IgG Fc receptors. Annu. Rev. Immunol. 2001;19:275–290. doi: 10.1146/annurev.immunol.19.1.275. [DOI] [PubMed] [Google Scholar]
- 63.Hulett M.D., Osman N., McKenzie I.F., Hogarth P.M. Chimeric Fc receptors identify functional domains of the murine high affinity receptor for IgG. J. Immunol. 1991;147:1863–1868. [PubMed] [Google Scholar]
- 64.Gessner J.E., Heiken H., Tamm A., Schmidt R.E. The IgG Fc receptor family. Ann. Hematol. 1998;76:231–248. doi: 10.1007/s002770050396. [DOI] [PubMed] [Google Scholar]
- 65.Huber R., Deisenhofer J., Colman P.M., Matsushima M., Palm W. Crystallographic structure studies of an IgG molecule and an Fc fragment. Nature. 1976;264:415–420. doi: 10.1038/264415a0. [DOI] [PubMed] [Google Scholar]
- 66.Sondermann P., Huber R., Oosthuizen V., Jacob U. The 3.2-A crystal structure of the human IgG1 Fc fragment–Fc gammaRIII complex. Nature. 2000;406:267–273. doi: 10.1038/35018508. [DOI] [PubMed] [Google Scholar]
- 67.Maxwell K.F., Powell M.S., Hulett M.D., Barton P.A., McKenzie I.F., Garrett T.P. Crystal structure of the human leukocyte Fc receptor, Fc gammaRIIa. Nat. Struct. Biol. 1999;6:437–442. doi: 10.1038/8241. [DOI] [PubMed] [Google Scholar]
- 68.Garman S.C., Wurzburg B.A., Tarchevskaya S.S., Kinet J.P., Jardetzky T.S. Structure of the Fc fragment of human IgE bound to its high-affinity receptor Fc epsilonRI alpha. Nature. 2000;406:259–266. doi: 10.1038/35018500. [DOI] [PubMed] [Google Scholar]
- 69.Bazil V., Strominger J.L. Metalloprotease and serine protease are involved in cleavage of CD43, CD44, and CD16 from stimulated human granulocytes. Induction of cleavage of L-selectin via CD16. J. Immunol. 1994;152:1314–1322. [PubMed] [Google Scholar]
- 70.Tartour E., de la Salle H., de la Salle C., Teillaud C., Camoin L., Galinha A. Identification, in mouse macrophages and in serum, of a soluble receptor for the Fc portion of IgG (Fc gamma R) encoded by an alternatively spliced transcript of the Fc gamma RII gene. Int. Immunol. 1993;5:859–868. doi: 10.1093/intimm/5.8.859. [DOI] [PubMed] [Google Scholar]
- 71.Takai T., Li M., Sylvestre D., Clynes R., Ravetch J.V. FcR gamma chain deletion results in pleiotrophic effector cell defects. Cell. 1994;76:519–529. doi: 10.1016/0092-8674(94)90115-5. [DOI] [PubMed] [Google Scholar]
- 72.Hazenbos W.L., Gessner J.E., Hofhuis F.M., Kuipers H., Meyer D., Heijnen I.A. Impaired IgG-dependent anaphylaxis and Arthus reaction in Fc gamma RIII (CD16) deficient mice. Immunity. 1996;5:181–188. doi: 10.1016/s1074-7613(00)80494-x. [DOI] [PubMed] [Google Scholar]
- 73.Yuasa T., Kubo S., Yoshino T., Ujike A., Matsumura K., Ono M. Deletion of fcgamma receptor IIB renders H-2(b) mice susceptible to collagen-induced arthritis. J. Exp. Med. 1999;189:187–194. doi: 10.1084/jem.189.1.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nakamura A., Yuasa T., Ujike A., Ono M., Nukiwa T., Ravetch J.V. Fcgamma receptor IIB-deficient mice develop Goodpasture's syndrome upon immunization with type IV collagen: a novel murine model for autoimmune glomerular basement membrane disease. J. Exp. Med. 2000;191:99–906. doi: 10.1084/jem.191.5.899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Langone J.J. Protein A of Staphylococcus aureus and related immunoglobulin receptors produced by streptococci and pneumonococci. Adv. Immunol. 1982;32:157–252. [PubMed] [Google Scholar]
- 76.Christensen P., Johansson B.G., Kronvall G. Interaction of streptococci with the Fc fragment of IgG. Acta Pathol. Microbiol. Scand., C. 1976;84:73–76. doi: 10.1111/j.1699-0463.1976.tb00001.x. [DOI] [PubMed] [Google Scholar]
- 77.De Miranda-Santos I.K., Compos-Neto A. Receptor for immunoglobulin Fc on pathogenic but not on nonpathogenic protozoa of the Trypanosomatidae. J. Exp. Med. 1981;154:1732–1742. doi: 10.1084/jem.154.6.1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Torpier G., Capron A., Ouaissi M.A. Receptor for IgG(Fc) and human beta2-microglobulin on S. mansoni schistosomula. Nature. 1979;278:447–449. doi: 10.1038/278447a0. [DOI] [PubMed] [Google Scholar]
- 79.Oleszak E.L., Leibowitz J.L. Fc receptor-like activity of mouse hepatitis virus E2 glycoprotein. Adv. Exp. Med. Biol. 1990;276:51–58. doi: 10.1007/978-1-4684-5823-7_8. [DOI] [PubMed] [Google Scholar]
- 80.Maillard P., Lavergne J.P., Siberil S., Faure G., Roohvand F., Petres S. Fcgamma receptor-like activity of hepatitis C virus core protein. J. Biol. Chem. 2004;279:2430–2437. doi: 10.1074/jbc.M311470200. [DOI] [PubMed] [Google Scholar]
- 81.Eizuru Y., Minamishima Y. Induction of Fc (IgG) receptor(s) by simian cytomegaloviruses in human embryonic lung fibroblasts. Intervirology. 1988;29:339–345. doi: 10.1159/000150065. [DOI] [PubMed] [Google Scholar]
- 82.Thale R., Lucin P., Schneider K., Eggers M., Koszinowski U.H. Identification and expression of a murine cytomegalovirus early gene coding for an Fc receptor. J. Virol. 1994;68:7757–7765. doi: 10.1128/jvi.68.12.7757-7765.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Atalay R., Zimmermann A., Wagner M., Borst E., Benz C., Messerle M. Identification and expression of human cytomegalovirus transcription units coding for two distinct Fcgamma receptor homologs. J. Virol. 2002;76:8596–8608. doi: 10.1128/JVI.76.17.8596-8608.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Para M.F., Goldstein L., Spear P.G. Similarities and differences in the Fc-binding glycoprotein (gE) of herpes simplex virus types 1 and 2 and tentative mapping of the viral gene for this glycoprotein. J. Virol. 1982;41:137–144. doi: 10.1128/jvi.41.1.137-144.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Litwin V., Sandor M., Grose C. Cell surface expression of the varicella-zoster virus glycoproteins and Fc receptor. Virology. 1990;178:263–272. doi: 10.1016/0042-6822(90)90402-d. [DOI] [PubMed] [Google Scholar]
- 86.Van de Walle G.R., Favoreel H.W., Nauwynck H.J., Pensaert M.B. Antibody-induced internalization of viral glycoproteins and gE–gI Fc receptor activity protect pseudorabies virus-infected monocytes from efficient complement-mediated lysis. J. Gen. Virol. 2003;84:939–948. doi: 10.1099/vir.0.18663-0. [DOI] [PubMed] [Google Scholar]
- 87.Jang H.K., Ono M., Kim T.J., Izumiya Y., Damiani A.M., Matsumura T. The genetic organization and transcriptional analysis of the short unique region in the genome of nononcogenic Marek's disease virus serotype 2. Virus Res. 1998;58:137–147. doi: 10.1016/s0168-1702(98)00110-5. [DOI] [PubMed] [Google Scholar]
- 88.Baucke R.B., Spear P.G. Membrane proteins specified by herpes simplex viruses: V. Identification of an Fc-binding glycoprotein. J. Virol. 1979;32:779–789. doi: 10.1128/jvi.32.3.779-789.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Johnson D.C., Frame M.C., Ligas M.W., Cross A.M., Stow N.D. Herpes simplex virus immunoglobulin G Fc receptor activity depends on a complex of two viral glycoproteins, gE and gI. J. Virol. 1988;62:1347–1354. doi: 10.1128/jvi.62.4.1347-1354.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Litwin V., Jackson W., Grose C. Receptor properties of two varicella-zoster virus glycoproteins, gpI and gpIV, homologous to herpes simplex virus gE and gI. J. Virol. 1992;66:3643–3651. doi: 10.1128/jvi.66.6.3643-3651.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Petrovskis E.A., Timmins J.G., Post L.E. Use of lambda gt11 to isolate genes for two pseudorabies virus glycoproteins with homology to herpes simplex virus and varicella-zoster virus glycoproteins. J. Virol. 1986;60:185–193. doi: 10.1128/jvi.60.1.185-193.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Dubin G., Basu S., Mallory D.L., Basu M., Tal-Singer R., Friedman H.M. Characterization of domains of herpes simplex virus type 1 glycoprotein E involved in Fc binding activity for immunoglobulin G aggregates. J. Virol. 1994;68:2478–2485. doi: 10.1128/jvi.68.4.2478-2485.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chapman T.L., You I., Joseph I.M., Bjorkman P.J., Morrison S.L., Raghavan M. Characterization of the interaction between the herpes simplex virus type I Fc receptor and immunoglobulin G. J. Biol. Chem. 1999;274:6911–6919. doi: 10.1074/jbc.274.11.6911. [DOI] [PubMed] [Google Scholar]
- 94.Nagashunmugam T., Lubinski J., Wang L., Goldstein L.T., Weeks B.S., Sundaresan P. In vivo immune evasion mediated by the herpes simplex virus type 1 immunoglobulin G Fc receptor. J. Virol. 1998;72:5351–5359. doi: 10.1128/jvi.72.7.5351-5359.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Dingwell K.S., Brunetti C.R., Hendricks R.L., Tang Q., Tang M., Rainbow A.J. Herpes simplex virus glycoproteins E and I facilitate cell-to-cell spread in vivo and across junctions of cultured cells. J. Virol. 1994;68:834–845. doi: 10.1128/jvi.68.2.834-845.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Frank I., Friedman H.M. A novel function of the herpes simplex virus type 1 Fc receptor: participation in bipolar bridging of antiviral immunoglobulin G. J. Virol. 1989;63:4479–4488. doi: 10.1128/jvi.63.11.4479-4488.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Van Vliet K.E., De Graaf-Miltenburg L.A., Verhoef J., Van Strijp J.A. Direct evidence for antibody bipolar bridging on herpes simplex virus-infected cells. Immunology. 1992;77:109–115. [PMC free article] [PubMed] [Google Scholar]
- 98.Zheng Y., Shopes B., Holowka D., Baird B. Dynamic conformations compared for IgE and IgG1 in solution and bound to receptors. Biochemistry. 1992;31:7446–7456. doi: 10.1021/bi00148a004. [DOI] [PubMed] [Google Scholar]
- 99.Olson J.K., Grose C. Endocytosis and recycling of varicella-zoster virus Fc receptor glycoprotein gE: internalization mediated by a YXXL motif in the cytoplasmic tail. J. Virol. 1997;71:4042–4054. doi: 10.1128/jvi.71.5.4042-4054.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Stannard L.M., Hardie D.R. An Fc receptor for human immunoglobulin G is located within the tegument of human cytomegalovirus. J. Virol. 1991;65:3411–3415. doi: 10.1128/jvi.65.6.3411-3415.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Xu B., Murayama T., Ishida K., Furukawa T. Characterization of IgG Fc receptors induced by human cytomegalovirus. J. Gen. Virol. 1989;70(Pt 4):893–900. doi: 10.1099/0022-1317-70-4-893. [DOI] [PubMed] [Google Scholar]
- 102.Lilley B.N., Ploegh H.L., Tirabassi R.S. Human cytomegalovirus open reading frame TRL11/IRL11 encodes an immunoglobulin G Fc-binding protein. J. Virol. 2001;75:11218–11221. doi: 10.1128/JVI.75.22.11218-11221.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sondermann P., Kaiser J., Jacob U. Molecular basis for immune complex recognition: a comparison of Fc-receptor structures. J. Mol. Biol. 2001;309:737–749. doi: 10.1006/jmbi.2001.4670. [DOI] [PubMed] [Google Scholar]
- 104.Burmeister W.P., Gastinel L.N., Simister N.E., Blum M.L., Bjorkman P.J. Crystal structure at 2.2 A resolution of the MHC-related neonatal Fc receptor. Nature. 1994;372:336–343. doi: 10.1038/372336a0. [DOI] [PubMed] [Google Scholar]
- 105.Letourneur F., Klausner R.D. A novel di-leucine motif and a tyrosine-based motif independently mediate lysosomal targeting and endocytosis of CD3 chains. Cell. 1992;69:1143–1157. doi: 10.1016/0092-8674(92)90636-q. [DOI] [PubMed] [Google Scholar]
- 106.Antonsson A., Johansson P.J. Binding of human and animal immunoglobulins to the IgG Fc receptor induced by human cytomegalovirus. J. Gen. Virol. 2001;82:1137–1145. doi: 10.1099/0022-1317-82-5-1137. [DOI] [PubMed] [Google Scholar]
- 107.Murayama T., Natsuume-Sakai S., Shimokawa K., Furukawa T. Fc receptor(s) induced by human cytomegalovirus bind differentially with human immunoglobulin G subclasses. J. Gen. Virol. 1986;67(Pt 7):1475–1478. doi: 10.1099/0022-1317-67-7-1475. [DOI] [PubMed] [Google Scholar]
- 108.Kaur A., Kassis N., Hale C.L., Simon M., Elliott M., Gomez-Yafal A. Direct relationship between suppression of virus-specific immunity and emergence of cytomegalovirus disease in simian AIDS. J. Virol. 2003;77:5749–5758. doi: 10.1128/JVI.77.10.5749-5758.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.McKeating J.A., Griffiths P.D., Weiss R.A. HIV susceptibility conferred to human fibroblasts by cytomegalovirus-induced Fc receptor. Nature. 1990;343:659–661. doi: 10.1038/343659a0. [DOI] [PubMed] [Google Scholar]
- 110.Vink C., Beuken E., Bruggeman C.A. Complete DNA sequence of the rat cytomegalovirus genome. J. Virol. 2000;74:7656–7665. doi: 10.1128/jvi.74.16.7656-7665.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Burns W.H., Barbour G.M., Sandford G.R. Molecular cloning and mapping of rat cytomegalovirus DNA. Virology. 1988;166:140–148. doi: 10.1016/0042-6822(88)90155-9. [DOI] [PubMed] [Google Scholar]
- 112.Beisser P.S., Kaptein S.J., Beuken E., Bruggeman C.A., Vink C. The Maastricht strain and England strain of rat cytomegalovirus represent different betaherpesvirus species rather than strains. Virology. 1998;246:341–351. doi: 10.1006/viro.1998.9196. [DOI] [PubMed] [Google Scholar]
- 113.Crnkovic-Mertens I., Messerle M., Milotic I., Szepan U., Kucic N., Krmpotic A. Virus attenuation after deletion of the cytomegalovirus Fc receptor gene is not due to antibody control. J. Virol. 1998;72:1377–1382. doi: 10.1128/jvi.72.2.1377-1382.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hanson L.K., Slater J.S., Karabekian Z., Ciocco-Schmitt G., Campbell A.E. Products of US22 genes M140 and M141 confer efficient replication of murine cytomegalovirus in macrophages and spleen. J. Virol. 2001;75:6292–6302. doi: 10.1128/JVI.75.14.6292-6302.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Borrebaeck C.A., Ohlin M. Antibody evolution beyond Nature. Nat. Biotechnol. 2002;20:1189–1190. doi: 10.1038/nbt1202-1189. [DOI] [PubMed] [Google Scholar]
- 116.Farrell H.E., Shellam G.R. Protection against murine cytomegalovirus infection by passive transfer of neutralizing and non-neutralizing monoclonal antibodies. J. Gen. Virol. 1991;72(Pt. 1):149–156. doi: 10.1099/0022-1317-72-1-149. [DOI] [PubMed] [Google Scholar]
- 117.Jefferis R., Pound J., Lund J., Goodall M. Effector mechanisms activated by human IgG subclass antibodies: clinical and molecular aspects. Review article. Ann. Biol. Clin. (Paris) 1994;52:57–65. [PubMed] [Google Scholar]
- 118.Armour K.L., Atherton A., Williamson L.M., Clark M.R. The contrasting IgG-binding interactions of human and herpes simplex virus Fc receptors. Biochem. Soc. Trans. 2002;30:495–500. doi: 10.1042/bst0300495. [DOI] [PubMed] [Google Scholar]
- 119.Britt W.J., Vugler L., Butfiloski E.J., Stephens E.B. Cell surface expression of human cytomegalovirus (HCMV) gp55–116 (gB): use of HCMV-recombinant vaccinia virus-infected cells in analysis of the human neutralizing antibody response. J. Virol. 1990;64:1079–1085. doi: 10.1128/jvi.64.3.1079-1085.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Urban M., Britt W., Mach M. The dominant linear neutralizing antibody-binding site of glycoprotein gp86 of human cytomegalovirus is strain specific. J. Virol. 1992;66:1303–1311. doi: 10.1128/jvi.66.3.1303-1311.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Klein M., Schoppel K., Amvrossiadis N., Mach M. Strain-specific neutralization of human cytomegalovirus isolates by human sera. J. Virol. 1999;73:878–886. doi: 10.1128/jvi.73.2.878-886.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]