

Highlights
-
•
In this review, we describe the identification of the PRRs involved in the recognition of pestiviruses, and the mechanisms of these viruses to prevent the activation of host’s innate immune response with special emphasis on viral RNases.
-
•
Most importantly, we extend these data and present our model of innate immunotolerance requiring continuous prevention of detection of immunostimulatory self nucleic acids, in contrast to the well-known long-term tolerance of the adaptive immune system targeted predominantly against proteins.
-
•
This hypothesis is very likely relevant beyond the bovine species and might answer more fundamental questions on the discrimination between “self” and “viral nonself RNA”, which are relevant also for the prevention and treatment of chronic IFN induction and autoimmunity induced by “self-RNAs”.
Keywords: Bovine viral diarrhea virus (BVDV), Persistent infection, Innate immunotolerance, Viral endoribonuclease, IFN antagonist, Self-nonself discrimination
Abstract
Pestiviruses including bovine viral diarrhea virus (BVDV), border disease virus (BDV) and classical swine fever virus (CSFV), occur worldwide and are important pathogens of livestock. A large part of their success can be attributed to the induction of central immunotolerance including B- and T-cells upon fetal infection leading to the generation of persistently infected (PI) animals. In the past few years, it became evident that evasion of innate immunity is a central element to induce and maintain persistent infection. Hence, the viral non-structural protease Npro heads the transcription factor IRF-3 for proteasomal degradation, whereas an extracellularly secreted, soluble form of the envelope glycoprotein Erns degrades immunostimulatory viral single- and double-stranded RNA, which makes this RNase unique among viral endoribonucleases. We propose that these pestiviral interferon (IFN) antagonists maintain a state of innate immunotolerance mainly pertaining its viral nucleic acids, in contrast to the well-established immunotolerance of the adaptive immune system, which is mainly targeted at proteins. In particular, the unique extension of ‘self’ to include the viral genome by degrading immunostimulatory viral RNA by Erns is reminiscent of various host nucleases that are important to prevent inappropriate IFN activation by the host’s own nucleic acids in autoimmune diseases such as Aicardi-Goutières syndrome or systemic lupus erythematosus. This mechanism of “innate tolerance” might thus provide a new facet to the role of extracellular RNases in the sustained prevention of the body’s own immunostimulatory RNA to act as a danger-associated molecular pattern that is relevant across various species.
1. Introduction
Viruses are never able to propagate and survive on their own, i.e., they are totally dependent on a host that they can infect. Consequentially, the field of virology is intimately linked to immunology as, during the long time of co-evolution, the hosts have acquired a vast array of antiviral defense mechanisms and vice versa.
Bovine viral diarrhea virus (BVDV) is probably one of the most wide-spread viruses, at least among the “terrestrial viruses”. In this mini review, we will focus on the interplay of BVDV, especially its viral RNA, with the innate immune defense of the host animals, because this is the key element to explain the long-term survival of this virus in its host population. Finally, we hypothesize that the survival strategy of BVDV to induce adaptive and, likewise, innate immunotolerance might provide a new viewpoint for the way the host handle its own, potentially immunostimulatory, self nucleic acids to prevent them from inappropriate chronic activation of the interferon (IFN) system. The pestiviral mechanisms to avoid detection and, thus, to behave identical as the body’s own RNA, might well be analogous to the mechanisms of its host not to recognize own structures, the latter being of paramount importance to prevent autoimmune reactions.
2. BVDV and the host’ interferon defence
2.1. BVD virus life cycle and persistence
Bovine viral diarrhea virus (BVDV), including the two species (also called ‘genotypes’) BVDV-I and BVDV-II, classical swine fever virus (CSFV), border disease virus (BDV) of sheep, and several tentative species belong to the genus Pestivirus in the family Flaviviridae [1]. BVDV is a cattle pathogen of major importance with a worldwide distribution. Its viral life cycle was recently described in a number of excellent reviews [2], [3], [4], [5], [6] and, thus, is only briefly summarized here.
Pestiviruses are single-stranded (ss) RNA viruses with an envelope containing three viral glycoproteins, i.e., Erns, E1, and E2. The genome with positive polarity encodes for a single large open reading frame (ORF). By virtue of a large variety of possible mutations, BVDV exists as a cytopathic (cp) and a noncytopathic (ncp) biotype, defined by their effect on cultured cells. Upon attachment of the virus particle to the cell surface, the virus enters its host cell via clathrin-mediated endocytosis. Fusion of the viral with the endosomal membrane is then initiated by acidification of the organelle. Cap-independent translation is mediated by an internal ribosomal entry site (IRES) and the polyprotein is then further processed by cellular and viral proteases into at least 12 structural and non-structural viral proteins. Virus assembly most likely occurs in intracellular vesicles and exocytosis of mature particles occurs in a non-lytic way, at least for the ncp biotype.
Infection of cattle with either biotype results in transient viremia and infected animals show no disease signs, mild diarrhea, fever, and coughing, but severe thrombocytopenia and hemorrhages have also been reported [7]. Upon resolution of infection, the animals will be protected from re-infections. By contrast, infection of pregnant cows within the first 120 days of gestation with an ncp, but not cp, biotype of BVDV may result in the birth of persistently infected (PI) calves. The clinical symptoms of such PI animals vary considerably and range from unapparent infection to severe growth retardation. Gastrointestinal and/or lung diseases are frequently described, with lung-centered pathology observed mainly in young calves and mucosal pathology predominantly in older animals ([8], and references therein). These PI calves are highly susceptible to secondary infections with other pathogens, and are at risk of developing fatal Mucosal Disease (MD). The latter can occur at any time during the life of the PI animal if mutations or recombination with viral or cellular RNA with the persisting ncp strain lead to the generation of an antigenically homologous cp biotype (for reviews, see Refs. [6], [9], [10]). However, the development of such a cp biotype in these PI animals is rather an evolutionary misfortune as the cp strain will be eliminated with the death of its host animal [10]. It is exclusively the ncp biotype of pestiviruses that is transmitted in the long term from PI animals to naïve pregnant host’s in order to produce new PI calves [5]. Hence, persistent infection at the level of the single animal is responsible for viral persistence in the host population. Consequently, it’s the PI animals that are specifically searched for and eliminated in order to eradicate BVDV in regional and national control programs [5], [11], [12], [13].
2.2. IFN induction by pestiviruses
As positive-sense RNA viruses, pestivirus replication occurs via a semi-conservative model [14] using a double-stranded (ds) RNA template to synthesize plus-strand, genomic viral RNA. Accordingly, replicative forms (RF) and replicative intermediates (RI) were detected in BVDV-infected cells, and it was estimated that the RI contain 6–7 nascent strands per template [15], [16], [17]. Thus, pestiviral replication involves the formation of dsRNA intermediates in the cytosol of infected cells as seen with a variety of RNA and DNA viruses [18]. This could be confirmed in BVDV- and CSFV-infected cultured cells by immunofluorescence or immunoelectron microscopy and flow cytometry using a dsRNA-specific monoclonal antibody [19], [20], [21]. In line with the rather unrestrained replication of the cp biotype of pestiviruses [22], the amount of plus- and minus-strand viral RNA and, thus, also of dsRNA is up to two order of magnitude higher in cells infected with cp than with ncp viruses ([19], [20], [23], and references therein).
As dsRNA is a potent pathogen-associated molecular pattern (PAMP) [24], it comes of no surprise that pestiviruses are able to induce a type-I IFN response. However, IFN induction strongly depends on the virus strain, its biotype, virulence, and the cell type being infected. Thus, infection of many cell types by pestiviruses of the cp, but not the ncp, biotype induces IFN type-I synthesis in vitro (for review, see Ref. [25]), whereas in calf testicle cells [26] and porcine plasmacytoid dendritic cells (pDC) [27], IFN type-I was induced by ncp BVDV and ncp CSFV, respectively. Notwithstanding, the amount of dsRNA present in cells infected with pestiviruses of the ncp biotype is basically able to induce IFN expression in most cell types, which become obvious as mutant ncp strains lacking the IFN antagonist Npro (see below) replicate to similar or only partially reduced levels as its wt parent strains but readily induce IFN synthesis [28], [29], [30]. Thus, it can be conceived that the threshold for IFN induction, e.g., the amount of trigger required to induce an innate immune response, and the effectiveness of the pestiviral IFN antagonists varies between different cell types.
The pattern recognition receptors (PRR) that sense the pestiviral infection are much less well characterized. Viruses are mostly recognized by virtue of their genome (for reviews, see e.g., refs. [31], [32], [33]), and thus, the cytosolic RLRs (RIG-I-like receptors, such as RIG-I, Mda-5 and LGP2) and the toll-like receptors (TLRs) in the endolysosomal compartments were obvious candidates. Transfection of total RNA extracts isolated from cp BVDV-infected cells into uninfected MDBK cells induced IFN synthesis in a 5′-triphosphate-independent manner [20], which points to Mda-5 as possible PRR. However, as RIG-I is not strictly dependent on a 5′-triphosphate moiety [34], other receptors in addition to Mda-5 could not be excluded. Accordingly, Hüsser et al., using lentivirus-mediated transduction of short hairpin RNA, nicely demonstrated that CSFV is sensed by Mda-5, RIG-I and TLR-3 in porcine PK-15 cells [35]. In addition, IFN-α secretion in pDCs induced by CSFV infection or by cell-cell contact with CSFV-infected cells was severely reduced by an oligodeoxynucleotide inhibitor of TLR7 [36]. Activation of pDCs by CSFV infection required replication of the virus, as UV inactivation or neutralization of the virus suspension with neutralizing antibody completely abrogated IFN-α release, whereas IFN expression upon cell-dependent RNA transfer was independent on infectious virus particles [36], [37].
These results show that cytosolic and endolysosomally localized PRRs are able to detect the presence of pestiviral RNA. RLRs located in the cytosol most probably detect replicative double-stranded intermediates of various length formed during viral replication in productively infected cells. However, it remains unknown how this dsRNA gets access to TLR-3 containing compartments. On the one hand, dsRNA might be liberated from infected cells that undergo spontaneous or virus-induced apoptosis and then becomes endocytosed by neighboring cells [38]. On the other hand, viral RNA present in the cytosol might be shuttled to endosomes by the formation of autophagosomes as shown for VSV and activation of TLR-7 in mouse pDCs [39]. But the fact that pestiviral replication was purported to be even enhanced by autophagy in PK-15 or MDBK cells [40], [41] argues against the latter possibility. Finally, virus replication complexes might be already formed in membranous vesicles as described, e.g., for corona- or hepatitis C viruses [42], [43], but such large membrane arrangement were not observed in pestivirus infected cells [21]. Notwithstanding, dsRNA was localized inside the lumen and outer membranes of multivesicular bodies (MVBs) in MDBK cells infected with the pestivirus strain Giraffe-1, but whether these vesicles represent autophagosomes that hide the dsRNA from detection by the innate immune system followed by disposal in lysosomes, or whether they are true sites for viral replication is currently unknown [21].
Finally, viral ssRNA represents a PAMP on its own as shown by the activation of TLR-7 in porcine pDCs [36]. This indicates that viral replication is not required to generate a danger signal, but viral RNA synthesis might be required in order to produce a sufficient amount of trigger molecules. The exact structure of the ssRNA molecule required to activate TLR-7/8 is not yet known, but AU- or GU-rich regions, or inosine-containing immunostimulatory ssRNA were reported to effectively activate TLR-7 [44], [45]. Interestingly, inosine incorporation seems to increase the RNA’s secondary structure that enables its recognition by TLR-3 [46], further demonstrating that highly structured ssRNA in addition to dsRNA is a TLR-3 agonist [47]. This is confirmed by the fact that in vitro transcribed ssRNA of the BVD viral genome possess highly structured regions resistant to serum RNases and to RNase A (preferentially ssRNases) that are able to induce activation of TLR-3 [20], [48]. The 5′- und 3′-UTRs and long-range interactions between these two regions might be especially immunostimulatory [49], [50], but most regions within the BVD viral genome seem to possess high-ordered structures able to induce TLR activation [48].
In summary, various single- and double-stranded intermediates of viral RNA replication and viral genomic ssRNA or fragments thereof that were released by premature decay of extracellular or endosomal virus particles represent immunostimulatory nucleic acids. Based on their different localizations, a variety of PRRs in different compartments, e.g., RLRs in the cytosol and TLRs in endolysosomes, might become activated by pestiviruses.
2.3. Innate immunotolerance
Infection of the fetus at an early stage of development as described above effectively bypasses the adaptive immune system by establishing self-tolerance including B- as well as T-cells. In addition, maternal neutralizing antibodies cannot cross the ruminant epitheliochorial placenta, further protecting the virus from a humoral immune response within the fetus. However, in order to succeed in establishing persistent infection, BVDV still requires to cope with the innate immune system already active from the outset. Thus, the interaction of ncp BVDV with its host bypasses the adaptive immunity by inducing central immunotolerance, as well as evades innate immunity, with the IFN system as one of the most important antiviral defense systems of the host.
The N-terminal non-structural autoprotease Npro and the envelope glycoprotein Erns are two IFN antagonists expressed by pestiviruses that are unique to this genus within the flavivirus family. In the past few years, it became more and more evident that both antagonists are required in a non-redundant way to successfully establish persistent fetal infection [51]. Npro expressed in virus infected cells is responsible for polyubiquitinylation and proteasomal degradation of the transcription factor IRF-3 in an as yet unknown manner but without requiring its proteolytic activity. By contrast, secreted Erns prevents the activation of the host’s TLRs also in non-infected cells by endonucleolytic degradation of viral RNA prior to their activation of the corresponding PRRs (for reviews, see Refs. [5], [6], [25], [52], [53]). Collectively, it appears that Npro and Erns effectively reduce or at least delay IFN induction. The fact that the highly replicating cp biotype of pestiviruses similarly express these two IFN antagonistic proteins indicate that there exists a delicate balance between the level of PAMPs and the capacity to limit their effects on the innate immune response of the host. With pestiviruses exhibiting a rather broad cell tropism, expression of Npro as the very first protein enables an efficient and fast inhibition of dsRNA-induced responses in all cells containing replicating virus. In addition, the rather unspecific cell and host tropism of soluble Erns (see below), which is even active, e.g., in canine or human cells [54], prevents TLR activation in a large variety of uninfected cells within the host animal.
Nevertheless, despite the availability of such effective inhibitory mechanisms, both biotypes of BVDV induce the expression of IFN in vivo after infection of adult animals [55], [56], [57]. By contrast, only ncp BVDV strains are immunologically silent in fetuses and PI animals [6], [58]. The latter fact might still be debated as chronic up-regulation of type-I and type-II interferon was reported in bovine fetuses infected early in utero [59]. Indeed, 48% of PI animals but only 12% of non-PI control animals expressed Mx protein in PBMCs ex vivo. However, there was no correlation with the amount of viral RNA or Erns protein and only a week positive correlation with the infectious virus titer in the plasma of the PI animals (T.T.H. Pham Blume and M. Schweizer, unpublished observation). This indicates that it is rather the increased susceptibility of PI animals to secondary infections [25] than the persisting virus itself that is the cause for the increased activation of the IFN system in these animals. Whether this increased susceptibility is related to the selective immunosuppression elicited by the expression of the IFN antagonists and whether it is more distinctive for pathogens exploiting these pathways remains to be shown. Thus, the precise adjustment of inhibitory and stimulatory triggers of the innate immune system and their spatiotemporal control in vivo are not yet sufficiently characterized. Notwithstanding, it appears that the evasion of the IFN response is the central element for ruminant pestiviruses to induce persistent infections. In the next paragraphs, we will specifically describe the mechanisms of Erns contributing to the establishment and maintenance of innate immunotolerance in PI animals, and put it into relation to other host and viral nucleases that are involved in the depletion of immunostimulatory self and nonself nucleic acids.
3. ERNS as IFN antagonist
Erns, initially termed E0, was first detected at the surface of pestiviral particles and shortly thereafter also in the supernatant of virus infected cells [60], [61]. Accordingly, Erns was detected in vivo with concentrations up to 50 ng/ml in the serum of PI animals [29]. Both, secreted and structural Erns are mostly found as disulfide-linked homodimers of around 100 kDa [62], with carbohydrates contributing approximately half of the apparent molecular weight [60]. Erns contains nine highly conserved cysteine residues that form four intramolecular disulfide bonds [63]. The C-terminal cysteine at the position 171 (C171) forms an intermolecular disulfide bond between two Erns monomers and a substitution of this residue results in a loss of the dimeric status [64]. To that effect, viruses encoding monomeric Erns are not restricted in their replication in vitro but are attenuated in vivo [64], [65].
As envelope glycoprotein, Erns plays a role in virus attachment through interactions with the cell surface glycosaminoglycans (GAGs) [66], [67]. Binding of Erns to GAGs involves a cluster of basic residues (480-KKLENKSK-487) near the C-terminus, with the lysine residues at positions 481 and 485 being critical for binding [68]. Nonetheless, pseudotyped particles containing only E1 and E2 of CSFV were still able to mediate virus entry [69]. In addition to its contribution to virus attachment, the C-terminus is folded into an amphipathic helix that anchors Erns in plane into the membrane of the viral envelope [70], [71]. Detailed analyses depicted that the amphipathic helix lies with a slight tilt within the membrane just underneath the lipid head domain [72], [73]. To ensure a sufficient number of protein molecules for the production of new virus particles, a large number of the Erns proteins remains within the infected cells in a not yet defined part of the endoplasmic reticulum (ER) by a specific retention signal located within the C-terminal amphipathic helix [74]. In summary, the C-terminus of Erns fulfills different tasks: it anchors the protein into the viral envelope, it attaches soluble Erns to the cell surface via its GAG-binding sites and it helps to maintain an appropriate ratio of cell-associated and soluble Erns.
In 1993, it was reported that, in addition to its function as envelope glycoprotein, Erns resembles ribonucleases of the RNase T2 family and indeed degrades preferentially ssRNA ([75]; for review, see Refs. [6], [25], [76]). Thereby, Erns, but not RNase-inactive mutants, potently inhibit IFN expression induced by the addition of extracellular synthetic or viral ss- or dsRNA [20], [29], [77]. According to X-ray structure analyses, however, Erns is only able to bind ss- but not dsRNA in the active site [78]. Hence, the mechanism of Erns to degrade dsRNA remains to be investigated, but we propose that it might act as a nicking endoribonuclease targeting the two strains of dsRNA individually ([79], and Lussi et al., in preparation). These in vitro results are also applicable in vivo, as BVDV and CSFV mutant viruses encoding for an Erns protein lacking its RNase activity are severely attenuated [80], [81].
Owing to the GAG-binding site within the amphipathic helix, Erns rather unspecifically binds to cell surfaces, and is thus able to act as IFN antagonist in cells of various species, e.g., caprine, ovine, canine, or human cells. Mutant proteins lacking 37 amino acid residues of the C-terminus, including the GAG-binding site, were severely limited in their inhibition of dsRNA-induced IFN expression [54]. Furthermore, binding to the cell surface was followed by an energy-dependent uptake via clathrin-mediated endocytosis, which strongly indicates that Erns cleaves its substrate in an intracellular compartment [54] (Fig. 1 B). This is corroborated by the fact that RNase-active Erns effectively inhibited activation of TLR-3 by extracellularly added dsRNA or viral ssRNA that is resistant to serum RNases of the host (Fig. 1A), and of TLR-7 by virus-infected cells in contact with pDCs [36], [54]. By contrast, activation of TLR-7 by R848 was not inhibited by Erns [54] further indicating that it does not inhibit any downstream signaling but rather degrades the viral PAMP prior to activation of TLRs (Fig. 1). As extracellular dsRNA was reported to be delivered to endosomal and – by an unknown way – to cytosolic PRRs, it cannot formally be excluded that RLRs are activated as well [82]. But Erns potently inhibits IFN induction by extracellular dsRNA, and it can thus be postulated that the dsRNA might well be degraded within an endolysosomal compartment prior to any transfer to the cytosol. Accordingly, IFN induction was unimpeded in Erns-expressing cells upon lipofectin-mediated transfection of poly(IC), whereas the effect of dsRNA added to the medium was completely blocked [29].
Fig. 1.

Pestiviral Erns inhibits ss- and dsRNA-induced type-I interferon (IFN) synthesis. Viral ss- and dsRNA, e.g., from dying infected cells, are potent pathogen-associated molecular pattern (PAMP) that are sensed by the corresponding pattern-recognition receptor (PRR) such as TLR-7 and TLR-3. Both toll-like receptors are located in endolysosomal compartments and, once bound by their substrates, they induce downstream signaling that leads to IFN expression. As many regions of the BVD viral genome are resistant to degradation by extracellular serum RNases, they effectively induce the cell’s innate immune response (A). By contrast, soluble Erns is taken up by clathrin-mediated endocytosis that enables this RNase to effectively degrade the viral PAMPs prior to TLR activation (B). The annotation of the elements in the figure is depicted in the panel on the right.
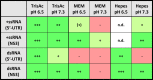
An endosomal location of Erns is also in agreement with its preference for a slightly acidic milieu. Erns is active in a broad pH range from about 4–7.5 with an optimum around a pH value of 6, measured in a 40 mM sodium- or tris-acetate buffer [83]. By contrast, Erns degraded poly(IC) but not in vitro transcribed dsRNA in cell culture medium (pH 7.2–7.4) with the latter being cut at a pH value of 4.5 [77]. However, by comparing the activity of Erns at pH 6.5 and 7.3 in tris-acetate buffer versus cell culture medium (MEM), we demonstrated that the activity of Erns is much lower in HEPES-buffered MEM irrespective of the pH tested (Table 1 ), implying that there might be an unknown inhibitory factor in the cell culture medium different from the buffer substance itself.
Table 1.
Degradation of in vitro transcribed pestiviral ss- and dsRNA [20] by Erns in various buffer systems at pH 6.5 or 7.3, with rating from very strong degradation (+++; green) to no degradation (red). TrisAc: tris-acetate buffer; MEM: Minimum Essential Medium; n.d.: not done.
 |
Based on the fact that GAG-binding of Erns is required prior to its uptake by endocytosis, we were able to effectively prevent or reverse binding of Erns to cell membranes by heparin treatment (Fig. 2 A). Furthermore, even after removal of all extracellular Erns by heparin after one hour of incubation, the viral RNase was still able to inhibit dsRNA-induced IFN expression in bovine turbinate cells up to 3–6 days later [54]. Finally, complexation of dsRNA with the human cathelicidin LL-37 completely protected the nucleic acid from degradation by Erns in vitro. Nonetheless, IFN induction by these dsRNA-LL-37 complexes was inhibited in Erns-treated cells, which suggests that the RNA dissociates from LL-37 in order to the nucleic acid being accessible to degradation by the viral RNase within the corresponding compartment (Fig. 2B). This is in complete accordance with Erns being active at pH values as low as 4.5 and with the fact that poly(IC) dissociates from LL-37 at low pH values, e.g., upon endosomal acidification [84], which is a further indication for the endolysosomal localization of Erns. The exact location of Erns inside the various cell types, and the mechanism of how it specifically encounters its targets, i.e., ss- and dsRNA, remain to be established.
Fig. 2.

Pestiviral Erns inhibit viral RNA-induced IFN synthesis in endosomal compartments. Blocking Erns from entering the cell by heparin treatment prevents this viral RNase to inhibit ss- and dsRNA-induced IFN expression (A). By contrast, despite protection from RNase degradation by complexation of nucleic acids, e.g., by LL-37, IFN expression induced by complexed viral RNA is nevertheless inhibited by Erns, as endosomal acidification leads to separation of LL-37 from the RNA that makes it immediately amenable for degradation by Erns also at low pH values (B). The annotation of the elements in the figure is depicted in the panel on the right.
4. Nucleases and innate immune activation
4.1. Viral nucleases
A number of viruses express ribonucleases, which play crucial roles in viral replication and in virus-host interactions. These viruses comprise negative-strand RNA virus of the orthomyxo-, arena-, or bunyavirus families, plus-strand RNA viruses such as nidoviruses, or DNA viruses such as herpesviruses. The role of these virus-encoded exo- and endoribonucleases, e.g., in proof-reading, mRNA cap snatching, protein synthesis shutoff, or RNA interference were described in a number of excellent reviews [42], [76], [85], [86], [87]. In the following, we therefore only describe few examples for the role of viral RNases in evasion of the host’s innate immune response.
Viruses in the order Nidovirales, including corona, arteri- and roniviruses, encode for a large number of IFN antagonists within their extraordinary large RNA genome [88]. Among them, the exonuclease ExoN within non-structural protein (nsp) 14, the endoribonuclease EndoU encoded by nsp15, and possibly the nsp1β of the arterivirus porcine reproductive and respiratory syndrome virus (PRRSV) possess RNase activity. The latter seems to be conserved only in PRRS viruses, whereas nsp1 in coronaviruses was reported to lead to host translational shut off by degrading host mRNAs through a yet unknown host endonuclease, but its precise role and conservation among the nidoviruses remain to be clarified [85], [87], [88]. The exoribonuclease ExoN within the N-terminal part of nsp14 in coronaviruses (that also harbors an N7-methyltransferase activity at the C-terminus) is responsible for ‘proof-reading’ during replication of these large nidoviruses, but additionally, a role for ExoN in degrading RNA to avoid their recognition by the host’s PRRs was suggested [42]. Finally, the endonuclease nsp15 (nsp11 in arteriviruses) cleaves ss- and dsRNA preferably at uridine residues [42], [76], [87]. The substrate of the RNase activity in nsp15 is not yet known, but it might be conceivable that nsp15 degrades viral RNA to avoid its recognition as PAMP by cellular PRRs [42]. Alternatively, nsp11 of the arterivirus PRRSV was reported to degrade MAVS mRNA [89], but this is still debatable as this conclusion was presumably drawn from experiments using single protein overexpression and the original data were not yet published.
Akin to pesti- and coronaviruses, arenavirus infections are linked to suppression of the host innate immune system. The nucleoprotein (NP) of the prototypic arenavirus lymphocytic choriomeningitis virus (LCMV) was shown to be able to inhibit an IFN type I response by blocking IRF-3 nuclear translocation [90]. This observation was later extended to other arenaviruses, including Lassa virus, and it was demonstrated that a 3′-5′ exoribonuclease activity in the C-term of NP is essential for the IFN suppression. Determination of the crystal structure of NP of Lassa and Tacaribe virus and biochemical analyses confirmed that the structure of NP contains an exonuclease domain of the DEDD family that is able to cleave 5′-triphosphate dsRNA templates in vitro. Collectively, there is strong evidence that the exonuclease activity of NP is conserved in all arenaviruses and is required to prevent activation of RIG-I by the degradation of viral PAMPs ([91], [92], and references therein).
Almost every virus encodes for at least one mechanism to prevent its nucleic acid being recognized by the host and thereby activating the host’s IFN response [31]. Overall, however, there is only few experimental evidence that viral nucleases indeed degrade their viral PAMPs to evade the host’s innate immune defense. By contrast, there are several instances of host nucleases that use a similar strategy to degrade own danger molecules that might participate in autoimmune pathologies as exemplified in the following section.
4.2. Host nucleases
Inappropriate activation of type-I interferon goes along with many autoinflammatory and autoimmune diseases, and there is strong evidence that dysregulation of various host pathways that are required to contain immunostimulatory self nucleic acids play a causative role (for recent reviews, see e.g., refs. [93], [94], [95], [96], [97], [98]). In the following, a few examples are briefly described to illustrate the role of IFNs and of nucleases regulating IFN synthesis in autoimmune diseases.
In systemic lupus erythematosus (SLE), circulating IFN-α levels correlate with disease severity. The recognition of self-DNA or self-RNA by TLRs followed by an IFN-dependent auto-amplification loop seems to be a major mechanism of disease pathogenesis. Thus, in SLE, pDCs are continuously activated by immune complexes (IC) comprising self-nucleic acids (e.g., from apoptotic or necrotic cell material) and autoantibodies to self-RNA, -DNA or nucleoproteins followed by Fc receptor mediated uptake, which ultimately leads to the secretion of large amounts of IFN type-I in a TLR-7/8 or TLR-9-dependent manner. The constant IFN production, the IFN-dependent maturation of myeloid dendritic cells followed by stimulation of autoreactive T-cells and the differentiation of B-cells into autoantibody-secreting plasma cells further aggravate the disease symptoms [95], [99], [100], [101]. Anti-inflammatory treatment of SLE by glucocorticoids, which are thought to act via inhibition of NF-κB, shows only limited success in relieving SLE disease symptoms, as the continuous activation of TLRs in pDCs by self-nucleic acids also activate NF-κB that enhances pDC survival and, thus, continued IFN secretion is not abrogated [102]. In addition to autoantibodies, the antimicrobial cathelicidin peptide LL-37 and high-mobility group box 1 protein (HMGB1, a nuclear DNA-binding protein released from necrotic or cytokine-stimulated cells) might protect the self-nucleic acids from degradation by host nucleases, and facilitate their uptake by pDCs and B-cells. Interestingly, LL-37 – which is produced by keratinocytes and neutrophils after skin injury – is similarly overexpressed in psoriatic skin lesions, and its ability to complex self-RNA and self-DNA, to enhance its retention in the endosomes of plasmacytoid and of myeloid dendritic cells, and to trigger TLR-dependent IFN synthesis might be responsible for the observed breaking of self-tolerance [100], [103], [104]. Finally, there is genetic evidence obtained from studies with various knockout mice and genome-wide association studies that support the role of innate and adaptive immune responses in the development of SLE, such as pathways involved in removal of apoptotic bodies and ICs (including DNase I and Trex-1 (DNase III)), PRR activation and IFN expression, and interference with T- and B-cell signaling [105]. Remarkably, mice with the Y-linked autoimmune accelerating (Yaa) locus show enhanced sensitivity to develop lupus depending on the background of the mice, which was attributed to a X to Y chromosomal translocation resulting in the duplication of the gene encoding for TLR-7. The latter observation might also relate to the fact that women are around 10 times more often affected by SLE than men, which might be caused by incomplete X chromosome inactivation leading to insufficient TLR-7 dosage compensation [106], [107], [108].
Aicardi-Goutières syndrome (AGS) is a genetically determined, autosomal recessive disease of progressive encephalopathy of early childhood, very similar to congenital viral infections. Some children develop early-onset SLE or a cutaneous form thereof, familial chilblain lupus. AGS is caused by mutations in Trex-1, various components of the RNase H2 complex, SAMHD1, ADAR-1 and Mda-5 (for review, see e.g., [95], [109]). Trex-1 is the main 3′-5′ DNA exonuclease in mammalian cells and might be involved in the disposal of DNA from endogenous retroelements. Recently, it was reported that Trex-1 exerts in addition RNA exonuclease activity on ssRNA and on RNA/DNA hybrids and both, DNA and RNA exonuclease activity, are lost by the mutations found in AGS patients [110], which in the end leads to spontaneous activation of the cytosolic DNA sensor cyclic GMP-AMP synthase (cGAS) followed by IFN expression [111]. RNase H2 is composed of three different subunits performing endonuclease activity cleaving RNA in RNA/DNA hybrids or it cleaves the phosphodiester bond 5′ of individual ribonucleotides in DNA duplexes. The precise role of SAMHD1 in AGS is not yet known. The wild-type enzyme is a triphosphohydrolase, which reduces the cellular dNTP pool, and is an RNase, whose endogenous RNA substrate still needs to be identified. In any case, spontaneous IFN expression in SAMHD1 knockout mice was reported to involve intracellular RNA and DNA sensors, as additional knockout of the adapter proteins MAVS or STING abolished IFN production [112]. Finally, ADAR-1 deaminates adenosine to inosine in dsRNA and was reported to suppress IFN signaling, possibly by marking endogenous dsRNA as self avoiding recognition by Mda-5 [113]. Analogously, the mutations found in Mda-5 in AGS lead to a gain-of-function phenotype with increased affinity to dsRNA and enhanced IFN expression.
Combined, mutations that cause an aberrant nucleic acid-dependent signaling and increased IFN expression were described in all of these autoimmune disorders that finally lead to disease pathology. The “IFN signature” is a hallmark of all these “type I interferonopathies” [94], and retroelements, which make up half of the human genome, seem to be an important source of endogenous ligands for the host PRRs. Finally, there is a strong overlap between the control of self-nucleic acids and the antiviral innate immune response, further highlighting the need for a tight regulation.
5. Conclusion
During viral infections, the adaptive immune system is primarily responsible for the detection of nonself proteins. As viruses lack the elements of metabolism required for independent multiplication and, therefore, depend on the enzymes of their host cells to synthesize their proteins, viruses are largely recognized by their DNA and RNA genomes. Thus, it is the innate immune system that is in charge of detecting viral nucleic acids, which makes it a formidable task for the host to separate foreign from body’s own as nucleic acids are not pathogen-specific by default. The difficulty of differentiation of self from infectious nonself nucleic acids becomes especially apparent when considering that around 10% of our own genome consists of retroviral sequences [114], [115]. To specifically detect nonself nucleic acids by the corresponding PRRs, a number of strict controls are required in order to avoid autoimmune reactions. For instance, nucleic acid-sensing TLRs are mostly confined to the endolysosomal compartment and their activation is pH dependent; modification of host nucleic acids, e.g., methylation of nucleosides or incorporation of pseudouridine, prevents their recognition by PRRs or even negatively regulates TLR activation; or sequestration might render RNA or DNA invisible to the host. Finally, if all else fails, improper activation of the innate defense is prevented by degradation of immunostimulatory nucleic acids by host RNases or DNases [96], [103]. Thus, quite some knowledge was gained on the role of host nucleases in preventing autoimmune diseases (compare Section 4.2), but most of these are localized intracellularly. By contrast, despite extracellular RNases were mentioned many times in the literature to play a role in eliminating free extracellular RNA (for instance in ref. [116]), a specific role of these RNases in the elimination of immunostimulatory self RNA has not unequivocally been demonstrated. One reason for this might be the large variety of endogenous RNases found in the serum, e.g., RNases of the RNase A and T2 family [117], [118], [119], which might prevent the establishment of single-gene knockout mice with a clear phenotype.
In order to survive in the host population, we propose the hypothesis that ruminant pestiviruses induce persistence in its host animals by completely pretending to be part of the body’s own, which is a clear advantage for the survival of the host and for successful virus transmission. As a result of the early fetal infection, BVD viral proteins already become part of the host’s own with regard to the adaptive immune system by inducing central immunotolerance. As there is no or only limited long-term innate immune memory [120], maintaining tolerance to self nucleic acids is an enduring challenge for any host. As the pestiviral genome appears to be at least partially resistant to the host’s extracellular RNases, the host’s safeguard mechanism as described above fails to prevent TLR activation by misdirected viral ss- and dsRNA. Thus, the extracellularly secreted viral endonuclease Erns might be regarded as an extension of the host’s RNase substrate specificity to avoid inappropriate activation of the innate immune system by immunostimulatory viral RNA (Fig. 1). With its capability of being endocytosed into endolysosomal compartments and with its RNase being active over a broad pH range, Erns is able to efficiently degrade viral PAMPs at all relevant compartments. Even in the case of extracellularly sequestered immunogenic RNAs that are protected from degradation by RNases, Erns effectively prevents them from stimulating TLRs as they are required to dissociate prior to activation of TLRs [84], which immediately exposes them to the pestiviral RNase (Fig. 2). Consequently, the virus in persistently infected animals is entirely tolerated by the host similarly to its own immunostimulatory nucleic acids without inducing overt disease [121]. Similarly, overexpression of TLR-7 in transgenic mice lead to a lupus-like disease phenotype, which could be partially reversed by additionally overexpressing secreted bovine RNase A [122].
The host’s IFN response is the prime antiviral defense system by inducing direct innate immune reactions and by shaping adaptive immunity. Thus, the survival strategy of BVDV consists of being non-cytopathogenic and producing less dsRNA than its cp counterpart, and expressing the IFN antagonists Npro as the first protein in order to reduce or even avoid IFN production in infected cells and Erns to degrade immunostimulatory viral RNA before they might activate the host’s PRRs. Notably, both pestiviral IFN antagonists are not only required to constantly maintain innate immunotolerance during persistent infections, but they also play an important role in acute infections [25]. Thus, RNase-inactive mutants of pestiviruses are attenuated upon acute infections [80], [81], and the evasion of the host’s IFN response by Npro and Erns upon acute infection with CSFV is also important for the virulence of the virus and the severity of immunopathology caused by the infection [123]. Thus, the pestiviral IFN antagonist, on the one hand, extend the host’s specificity to tolerate self nucleic acids to its own viral RNA during persistence and, on the other hand, participate in the evasion of the host’s IFN response during acute, transient infections, further illustrating the dichotomy of immunotolerance and the antiviral immune response.
This model might well shed new lights on fundamental questions on the innate tolerance to self nucleic acids and the specific detection of viral nonself RNA. These aspects are highly relevant also for the prevention of chronic IFN induction and autoimmunity induced by “self-RNAs” that might be fundamental beyond the mechanism of an animal disease [124]. Finally, the mechanism of the soluble pestiviral endoribonuclease Erns during persistent infection to support the virus in its strategy to pretend to be part of the body’s own might be analogous to the role of the host’s own extracellular nucleases to continuously maintain innate immunotolerance to its own immunostimulatory nucleic acids.
Acknowledgements
Many thanks to Fabienne Wyss for performing the experiments shown in Table 1, to Giuseppe Bertoni and Eveline Kindler for critically reading the manuscript, and to all the colleagues and students that were involved in our studies over the recent years. We are indebted to all of them. Our work was supported over the years by internal funds of the Institute of Veterinary Virology (now Institute of Virology and Immunology) of the Vetsuisse Faculty University of Bern, the Federal Food Safety and Veterinary Office FSVO, and the Swiss National Science Foundation.
Biographies

Matthias Schweizer received his M.Sc. in Biochemistry in 1990 from the Department of Biochemistry at the University of Zurich, Switzerland and his PhD in 1994 at the Institute of Biochemistry I at the Swiss Federal Institute of Technology (ETH) in Zurich, Switzerland. The doctoral studies were on the oxidative regulation of mitochondrial Ca2+ homeostasis and included a stay at TCOM, University of North Texas, Fort Worth (TX). After postdoctoral work at the ETH and the Institute of Veterinary Virology (IVV) at the University of Bern, he received his “habilitation” (postdoctoral lecture qualification) in Virology at the Vetsuisse Faculty of the University of Bern, Switzerland. Since 2014, he is head of the research group “virus infections of ruminants” and deputy head of the Virology section of the Institute of Virology and Immunology (IVI) of the Federal Food Safety and Veterinary Office in cooperation with the Vetsuisse Faculty of the University of Bern. His main interests are the interaction of ruminant pestiviruses with their host cells to induce persistence, the evolution of BVDV, and in applied research projects to further understand transmission and pathogenesis of pestiviral infections.

Carmela Lussi is a PhD student in the group of Dr. Matthias Schweizer at the Institute of Virology and Immunology (IVI) of the Federal Food Safety and Veterinary Office in cooperation with the Vetsuisse Faculty of the University of Bern. After receiving the Bachelor of Science in Biology, she obtained her Master of Science in Molecular Life Sciences with further specification in microbiology and immunology in 2014 at the Faculty of Science of the University of Bern. Within her PhD studies, Carmela Lussi focusses on the role of pestiviral protein Erns in innate immunotolerance.
Contributor Information
Carmela Lussi, Email: carmela.lussi@vetsuisse.unibe.ch.
Matthias Schweizer, Email: matthias.schweizer@vetsuisse.unibe.ch.
References
- 1.Pletnev A., Gould E., Heinz F.X., Meyers G., Thiel H.-J., Bukh J. Flaviviridae. In: King A.M.Q., Adams M.J., Carstens E.B., Lefkowitz E.J., editors. Virus Taxonomy. Academic Press; Oxford: 2011. pp. 1003–1020. [Google Scholar]
- 2.Neill J.D. Molecular biology of bovine viral diarrhea virus. Biologicals. 2013;41:2–7. doi: 10.1016/j.biologicals.2012.07.002. [DOI] [PubMed] [Google Scholar]
- 3.Ridpath J.F. Bovine viral diarrhea virus: global status. Vet. Clin. N. Am. Food Anim. Pract. 2010;26:105–121. doi: 10.1016/j.cvfa.2009.10.007. [DOI] [PubMed] [Google Scholar]
- 4.Rümenapf T., Thiel H.-J. Molecular biology of pestiviruses. In: Mettenleiter T.C., Sobrino F., editors. Animal Viruses: Molecular Biology. Caister Academic Press; Norwich UK: 2008. pp. 39–96. [Google Scholar]
- 5.Schweizer M., Peterhans E. Pestiviruses. Annu. Rev. Anim. Biosci. 2014;2:141–163. doi: 10.1146/annurev-animal-022513-114209. [DOI] [PubMed] [Google Scholar]
- 6.Tautz N., Tews B.A., Meyers G. The molecular biology of pestiviruses. Adv. Virus Res. 2015;93:47–160. doi: 10.1016/bs.aivir.2015.03.002. [DOI] [PubMed] [Google Scholar]
- 7.Brodersen B.W. Bovine viral diarrhea virus infections: manifestations of infection and recent advances in understanding pathogenesis and control. Vet. Pathol. 2014;51:453–464. doi: 10.1177/0300985813520250. [DOI] [PubMed] [Google Scholar]
- 8.Bachofen C., Braun U., Hilbe M., Ehrensperger F., Stalder H., Peterhans E. Clinical appearance and pathology of cattle persistently infected with bovine viral diarrhoea virus of different genetic subgroups. Vet. Microbiol. 2010;141:258–267. doi: 10.1016/j.vetmic.2009.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Becher P., Tautz N. RNA recombination in pestiviruses—cellular RNA sequences in viral genomes highlight the role of host factors for viral persistence and lethal disease. RNA Biol. 2011;8:216–224. doi: 10.4161/rna.8.2.14514. [DOI] [PubMed] [Google Scholar]
- 10.Peterhans E., Bachofen C., Stalder H.P., Schweizer M. Cytopathic bovine viral diarrhea viruses (BVDV): emerging pestiviruses doomed to extinction. Vet. Res. 2010;41:44. doi: 10.1051/vetres/2010016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bachofen C., Stalder H.P., Vogt H.R., Wegmüller M., Schweizer M., Zanoni R. Bovine Virusdiarrhöe (BVD): von der Biologie zur Bekämpfung. Berl. Munch. Tierarztl. Wochenschr. 2013;126:452–461. [PubMed] [Google Scholar]
- 12.Moennig V., Becher P. Pestivirus control programs: how far have we come and where are we going? Anim. Health Res. Rev. 2015;16:83–87. doi: 10.1017/S1466252315000092. [DOI] [PubMed] [Google Scholar]
- 13.Ståhl K., Alenius S. BVDV control and eradication in Europe—an update. Jpn. J. Vet. Res. 2012;60:S31–S39. [PubMed] [Google Scholar]
- 14.Ahlquist P. Parallels among positive-strand RNA viruses, reverse-transcribing viruses and double-stranded RNA viruses. Nat. Rev. Microbiol. 2006;4:371–382. doi: 10.1038/nrmicro1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gong Y., Shannon A., Westaway E.G., Gowans E.J. The replicative intermediate molecule of bovine viral diarrhoea virus contains multiple nascent strands. Arch. Virol. 1998;143:399–404. doi: 10.1007/s007050050296. [DOI] [PubMed] [Google Scholar]
- 16.Gong Y.H., Trowbridge R., Macnaughton T.B., Westaway E.G., Shannon A.D., Gowans E.J. Characterization of RNA synthesis during a one-step growth curve and of the replication mechanism of bovine viral diarrhoea virus. J. Gen. Virol. 1996;77:2729–2736. doi: 10.1099/0022-1317-77-11-2729. [DOI] [PubMed] [Google Scholar]
- 17.Warrilow D., Lott W.B., Greive S., Gowans E.J. Properties of the bovine viral diarrhoea virus replicase in extracts of infected MDBK cells. Arch. Virol. 2000;145:2163–2171. doi: 10.1007/s007050070046. [DOI] [PubMed] [Google Scholar]
- 18.Weber F., Wagner V., Rasmussen S.B., Hartmann R., Paludan S.R. Double-stranded RNA is produced by positive-strand RNA viruses and DNA viruses but not in detectable amounts by negative-strand RNA viruses. J. Virol. 2006;80:5059–5064. doi: 10.1128/JVI.80.10.5059-5064.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bauhofer O., Summerfield A., McCullough K.C., Ruggli N. Role of double-stranded RNA and Npro of classical swine fever virus in the activation of monocyte-derived dendritic cells. Virology. 2005;343:93–105. doi: 10.1016/j.virol.2005.08.016. [DOI] [PubMed] [Google Scholar]
- 20.Mätzener P., Magkouras I., Rümenapf T., Peterhans E., Schweizer M. The viral RNase Erns prevents IFN type-I triggering by pestiviral single- and double-stranded RNAs. Virus Res. 2009;140:15–23. doi: 10.1016/j.virusres.2008.10.015. [DOI] [PubMed] [Google Scholar]
- 21.Schmeiser S., Mast J., Thiel H.J., König M. Morphogenesis of pestiviruses: new insights from ultrastructural studies of strain giraffe-1. J.Virol. 2014;88:2717–2724. doi: 10.1128/JVI.03237-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rinck G., Birghan C., Harada T., Meyers G., Thiel H.-J., Tautz N. A cellular J-domain protein modulates polyprotein processing and cytopathogenicity of a pestivirus. J. Virol. 2001;75:9470–9482. doi: 10.1128/JVI.75.19.9470-9482.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yamane D., Kato K., Tohya Y., Akashi H. The double-stranded RNA-induced apoptosis pathway is involved in the cytopathogenicity of cytopathogenic Bovine viral diarrhea virus. J. Gen. Virol. 2006;87:2961–2970. doi: 10.1099/vir.0.81820-0. [DOI] [PubMed] [Google Scholar]
- 24.Sparrer K.M.J., Gack M.U. Intracellular detection of viral nucleic acids. Curr. Opin. Microbiol. 2015;26:1–9. doi: 10.1016/j.mib.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peterhans E., Schweizer M. BVDV: a pestivirus inducing tolerance of the innate immune response. Biologicals. 2013;41:39–51. doi: 10.1016/j.biologicals.2012.07.006. [DOI] [PubMed] [Google Scholar]
- 26.Schweizer M., Mätzener P., Pfaffen G., Stalder H.P., Peterhans E. "Self" and "nonself" manipulation of interferon defense during persistent infection: bovine viral diarrhea virus resists alpha/beta interferon without blocking antiviral activity against unrelated viruses replicating in its host cells. J. Virol. 2006;80:6926–6935. doi: 10.1128/JVI.02443-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fiebach A.R., Guzylack-Piriou L., Python S., Summerfield A., Ruggli N. Classical swine fever virus Npro limits type I interferon induction in plasmacytoid dendritic cells by interacting with interferon regulatory factor 7. J. Virol. 2011;85:8002–8011. doi: 10.1128/JVI.00330-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gil L.H.V.G., Ansari I.H., Vassilev V., Liang D.L., Lai V.C.H., Zhong W.D. The amino-terminal domain of bovine viral diarrhea virus Npro protein is necessary for alpha/beta interferon antagonism. J. Virol. 2006;80:900–911. doi: 10.1128/JVI.80.2.900-911.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Magkouras I., Mätzener P., Rümenapf T., Peterhans E., Schweizer M. RNase-dependent inhibition of extra- but not intracellular, dsRNA-induced IFN synthesis by Erns of pestiviruses. J. Gen. Virol. 2008;89:2501–2506. doi: 10.1099/vir.0.2008/003749-0. [DOI] [PubMed] [Google Scholar]
- 30.Ruggli N., Tratschin J.D., Schweizer M., McCullough K.C., Hofmann M.A., Summerfield A. Classical swine fever virus interferes with cellular antiviral defense: evidence for a novel function of Npro. J. Virol. 2003;77:7645–7654. doi: 10.1128/JVI.77.13.7645-7654.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fensterl V., Chattopadhyay S., Sen G.C. No love lost between viruses and interferons. Annu. Rev. Virol. 2015;2:549–572. doi: 10.1146/annurev-virology-100114-055249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hoffmann H.H., Schneider W.M., Rice C.M. Interferons and viruses: an evolutionary arms race of molecular interactions. Trends Immunol. 2015;36:124–138. doi: 10.1016/j.it.2015.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pandey S., Kawai T., Akira S. Microbial sensing by toll-like receptors and intracellular nucleic acid sensors. Cold Spring Harbor Perspect. Biol. 2015;7:a016246. doi: 10.1101/cshperspect.a016246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yoneyama M., Onomoto K., Jogi M., Akaboshi T., Fujita T. Viral RNA detection by RIG-I-like receptors. Curr. Opin. Immunol. 2015;32:48–53. doi: 10.1016/j.coi.2014.12.012. [DOI] [PubMed] [Google Scholar]
- 35.Hüsser L., Alves M.P., Ruggli N., Summerfield A. Identification of the role of RIG-I, MDA-5 and TLR3 in sensing RNA viruses in porcine epithelial cells using lentivirus-driven RNA interference. Virus Res. 2011;159:9–16. doi: 10.1016/j.virusres.2011.04.005. [DOI] [PubMed] [Google Scholar]
- 36.Python S., Gerber M., Suter R., Ruggli N., Summerfield A. Efficient sensing of infected cells in absence of virus particles by plasmacytoid dendritic cells is blocked by the viral ribonuclease Erns. PLoS Pathog. 2013;9:e1003412. doi: 10.1371/journal.ppat.1003412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Balmelli C., Vincent I.E., Rau H., Guzylack-Piriou L., Mccullough K., Summerfield A. Fc gamma RII-dependent sensitisation of natural interferon-producing cells for viral infection and interferon-alpha responses. Eur. J. Immunol. 2005;35:2406–2415. doi: 10.1002/eji.200525998. [DOI] [PubMed] [Google Scholar]
- 38.Nellimarla S., Mossman K.L. Extracellular dsRNA: its function and mechanism of cellular uptake. J.Interferon Cytokine Res. 2014;34:419–426. doi: 10.1089/jir.2014.0002. [DOI] [PubMed] [Google Scholar]
- 39.Lee H.K., Lund J.M., Ramanathan B., Mizushima N., Iwasaki A. Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science. 2007;315:1398–1401. doi: 10.1126/science.1136880. [DOI] [PubMed] [Google Scholar]
- 40.Fu Q., Shi H.J., Zhang H., Ren Y., Guo F., Qiao J. Autophagy during early stages contributes to bovine viral diarrhea virus replication in MDBK cells. J. Basic Microbiol. 2014;54:1044–1052. doi: 10.1002/jobm.201300750. [DOI] [PubMed] [Google Scholar]
- 41.Pei J.J., Zhao M.Q., Ye Z.D., Gou H.C., Wang J.Y., Yi L. Autophagy enhances the replication of classical swine fever virus in vitro. Autophagy. 2014;10:93–110. doi: 10.4161/auto.26843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kindler E., Thiel V. To sense or not to sense viral RNA—essentials of coronavirus innate immune evasion. Curr. Opin. Microbiol. 2014;20:69–75. doi: 10.1016/j.mib.2014.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Paul D., Bartenschlager R. Flaviviridae replication organelles: oh what a tangled web we weave. Annu. Rev. Virol. 2015;2:289–310. doi: 10.1146/annurev-virology-100114-055007. [DOI] [PubMed] [Google Scholar]
- 44.Forsbach A., Nemorin J.G., Montino C., Muller C., Samulowitz U., Vicari A.P. Identification of RNA sequence motifs stimulating sequence-specific TLR8-dependent immune responses. J. Immunol. 2008;180:3729–3738. doi: 10.4049/jimmunol.180.6.3729. [DOI] [PubMed] [Google Scholar]
- 45.Sarvestani S.T., Tate M.D., Moffat J.M., Jacobi A.M., Behlke M.A., Miller A.R. Inosine-mediated modulation of RNA sensing by toll-like receptor 7 (TLR7) and TLR8. J. Virol. 2014;88:799–810. doi: 10.1128/JVI.01571-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liao J.Y., Thakur S.A., Zalinger Z.B., Gerrish K.E., Imani F. Inosine-containing RNA is a novel innate immune recognition element and reduces RSV infection. PLoS One. 2011;6:e26463. doi: 10.1371/journal.pone.0026463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tatematsu M., Seya T., Matsumoto M. Beyond dsRNA: toll-like receptor 3 signalling in RNA-induced immune responses. Biochem. J. 2014;458:195–201. doi: 10.1042/BJ20131492. [DOI] [PubMed] [Google Scholar]
- 48.Zürcher C., Sauter K.S., Schweizer M. Pestiviral Erns blocks TLR-3-dependent IFN synthesis by LL37 complexed RNA. Vet. Microbiol. 2014;174:399–408. doi: 10.1016/j.vetmic.2014.09.028. [DOI] [PubMed] [Google Scholar]
- 49.Fricke M., Dünnes N., Zayas M., Bartenschlager R., Niepmann M., Marz M. Conserved RNA secondary structures and long-range interactions in hepatitis C viruses. RNA. 2015;21:1219–1232. doi: 10.1261/rna.049338.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hsu W.L., Chen C.L., Huang S.W., Wu C.C., Chen I.H., Nadar M. The untranslated regions of classic swine fever virus RNA trigger apoptosis. PLoS One. 2014;9:e88863. doi: 10.1371/journal.pone.0088863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Meyers G., Ege A., Fetzer C., Von Freyburg M., Elbers K., Carr V. Bovine viral diarrhea virus: prevention of persistent fetal infection by a combination of two mutations affecting Erns RNase and Npro protease. J. Virol. 2007;81:3327–3338. doi: 10.1128/JVI.02372-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Peterhans E., Schweizer M. Pestiviruses: how to outmaneuver your hosts. Vet. Microbiol. 2010;142:18–25. doi: 10.1016/j.vetmic.2009.09.038. [DOI] [PubMed] [Google Scholar]
- 53.Reid E., Charleston B. Type I and III interferon production in response to RNA viruses. J. Interferon Cytokine Res. 2014;34:649–658. doi: 10.1089/jir.2014.0066. [DOI] [PubMed] [Google Scholar]
- 54.Zürcher C., Sauter K.S., Mathys V., Wyss F., Schweizer M. Prolonged activity of the pestiviral RNase Erns as an interferon antagonist after uptake by clathrin-mediated endocytosis. J. Virol. 2014;88:7235–7243. doi: 10.1128/JVI.00672-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Charleston B., Brackenbury L.S., Carr B.V., Fray M.D., Hope J.C., Howard C.J. Alpha/beta and gamma interferons are induced by infection with noncytopathic bovine viral diarrhea virus in vivo. J. Virol. 2002;76:923–927. doi: 10.1128/JVI.76.2.923-927.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Müller-Doblies D., Ackermann M., Metzler A. In vitro and in vivo detection of Mx gene products in bovine cells following stimulation with alpha/beta interferon and viruses. Clin. Diagn. Lab. Immunol. 2002;9:1192–1199. doi: 10.1128/CDLI.9.6.1192-1199.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Smirnova N.P., Bielefeldt-Ohmann H., Van Campen H., Austin K.J., Han H., Montgomery D.L. Acute non-cytopathic bovine viral diarrhea virus infection induces pronounced type I interferon response in pregnant cows and fetuses. Virus Res. 2008;132:49–58. doi: 10.1016/j.virusres.2007.10.011. [DOI] [PubMed] [Google Scholar]
- 58.Charleston B., Fray M.D., Baigent S., Carr B.V., Morrison W.I. Establishment of persistent infection with non-cytopathic bovine viral diarrhoea virus in cattle is associated with a failure to induce type I interferon. J. Gen. Virol. 2001;82:1893–1897. doi: 10.1099/0022-1317-82-8-1893. [DOI] [PubMed] [Google Scholar]
- 59.Hansen T.R., Smirnova N.P., Webb B.T., Bielefeldt-Ohmann H., Sacco R.E., Van Campen H. Innate and adaptive immune responses to in utero infection with bovine viral diarrhea virus. Anim. Health Res. Rev. 2015;16:15–26. doi: 10.1017/S1466252315000122. [DOI] [PubMed] [Google Scholar]
- 60.Rümenapf T., Unger G., Strauss J.H., Thiel H.-J. Processing of the envelope glycoproteins of pestiviruses. J. Virol. 1993;67:3288–3294. doi: 10.1128/jvi.67.6.3288-3294.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Weiland E., Ahl R., Stark R., Weiland F., Thiel H.J. A second envelope glycoprotein mediates neutralization of a pestivirus, hog cholera virus. J. Virol. 1992;66:3677–3682. doi: 10.1128/jvi.66.6.3677-3682.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Thiel H.-J., Stark R., Weiland E., Rümenapf T., Meyers G. Hog cholera virus: molecular composition of virions from a pestivirus. J. Virol. 1991;65:4705–4712. doi: 10.1128/jvi.65.9.4705-4712.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Langedijk J.P.M., Van Veelen P.A., Schaaper W.M.M., de Ru A.H., Meloen R.H., Hulst M.M. A structural model of pestivirus Erns based on disulfide bond connectivity and homology modeling reveals an extremely rare vicinal disulfide. J. Virol. 2002;76:10383–10392. doi: 10.1128/JVI.76.20.10383-10392.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tews B.A., Schürmann E.M., Meyers G. Mutation of cysteine 171 of pestivirus Erns RNase prevents homodimer formation and leads to attenuation of classical swine fever virus. J. Virol. 2009;83:4823–4834. doi: 10.1128/JVI.01710-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.van Gennip H.G.P., Hesselink A.T., Moormann R.J.M., Hulst M.M. Dimerisation of glycoprotein Erns of classical swine fever virus is not essential for viral replication and infection. Arch. Virol. 2005;150:2271–2286. doi: 10.1007/s00705-005-0569-y. [DOI] [PubMed] [Google Scholar]
- 66.Hulst M.M., van Gennip H.G.P., Moormann R.J.M. Passage of classical swine fever virus in cultured swine kidney cells selects virus variants that bind to heparan sulfate due to a single amino acid change in envelope protein E-rns. J. Virol. 2000;74:9553–9561. doi: 10.1128/jvi.74.20.9553-9561.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Iqbal M., Flick-Smith H., McCauley J.W. Interactions of bovine viral diarrhoea virus glycoprotein Erns with cell surface glycosaminoglycans. J. Gen. Virol. 2000;81:451–459. doi: 10.1099/0022-1317-81-2-451. [DOI] [PubMed] [Google Scholar]
- 68.Iqbal M., McCauley J.W. Identification of the glycosaminoglycan-binding site on the glycoprotein Erns of bovine viral diarrhoea virus by site-directed mutagenesis. J. Gen. Virol. 2002;83:2153–2159. doi: 10.1099/0022-1317-83-9-2153. [DOI] [PubMed] [Google Scholar]
- 69.Wang Z., Nie Y.C., Wang P.G., Ding M.X., Deng H.K. Characterization of classical swine fever virus entry by using pseudotyped viruses: E1 and E2 are sufficient to mediate viral entry. Virology. 2004;330:332–341. doi: 10.1016/j.virol.2004.09.023. [DOI] [PubMed] [Google Scholar]
- 70.Fetzer C., Tews B.A., Meyers G. The carboxy-terminal sequence of the pestivirus glycoprotein Erns represents an unusual type of membrane anchor. J. Virol. 2005;79:11901–11913. doi: 10.1128/JVI.79.18.11901-11913.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tews B.A., Meyers G. The pestivirus glycoprotein Erns is anchored in plane in the membrane via an amphipathic helix. J. Biol. Chem. 2007;282:32730–32741. doi: 10.1074/jbc.M706803200. [DOI] [PubMed] [Google Scholar]
- 72.Aberle D., Muhle-Goll C., Bürck J., Wolf M., Reisser S., Luy B. Structure of the membrane anchor of pestivirus glycoprotein Erns, a long tilted amphipathic helix. PLoS Pathog. 2014;10:e1003973. doi: 10.1371/journal.ppat.1003973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Aberle D., Oetter K.M., Meyers G. Lipid binding of the amphipathic helix serving as membrane anchor of pestivirus glycoprotein Erns. PLoS One. 2015;10:e0135680. doi: 10.1371/journal.pone.0135680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Burrack S., Aberle D., Burck J., Ulrich A.S., Meyers G. A new type of intracellular retention signal identified in a pestivirus structural glycoprotein. FASEB J. 2012;26:3292–3305. doi: 10.1096/fj.12-207191. [DOI] [PubMed] [Google Scholar]
- 75.Schneider R., Unger G., Stark R., Schneider-Scherzer E., Thiel H.-J. Identification of a structural glycoprotein of an RNA virus as a ribonuclease. Science. 1993;261:1169–1171. doi: 10.1126/science.8356450. [DOI] [PubMed] [Google Scholar]
- 76.Meyers G., Rümenapf T., Ziebuhr J. Viral RNase involvement in strategies of infection. In: Nicholson A.W., editor. Ribonucleases. Springer; Berlin, Heidelberg: 2011. pp. 135–165. [Google Scholar]
- 77.Iqbal M., Poole E., Goodbourn S., McCauley J.W. Role for bovine viral diarrhea virus Erns glycoprotein in the control of activation of beta interferon by double-stranded RNA. J. Virol. 2004;78:136–145. doi: 10.1128/JVI.78.1.136-145.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Krey T., Bontems F., Vonrhein C., Vaney M.C., Bricogne G., Rümenapf T. Crystal structure of the pestivirus envelope glycoprotein Erns and mechanistic analysis of its ribonuclease activity. Structure. 2012;20:862–873. doi: 10.1016/j.str.2012.03.018. [DOI] [PubMed] [Google Scholar]
- 79.Lussi C., Schweizer M. The pestiviral IFN antagonist Erns cleaves dsRNA as nicking endoribonuclease. Cytokine. 2015;76:81–82. [Google Scholar]
- 80.Meyer C., Von Freyburg M., Elbers K., Meyers G. Recovery of virulent and RNase-negative attenuated type 2 bovine viral diarrhea viruses from infectious cDNA clones. J. Virol. 2002;76:8494–8503. doi: 10.1128/JVI.76.16.8494-8503.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Meyers G., Saalmüller A., Büttner M. Mutations abrogating the RNase activity in glycoprotein Erns of the pestivirus classical swine fever virus lead to virus attenuation. J. Virol. 1999;73:10224–10235. doi: 10.1128/jvi.73.12.10224-10235.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.DeWitte-Orr S.J., Mossman K.L. dsRNA and the innate antiviral immune response. Future Virol. 2010;5:325–341. [Google Scholar]
- 83.Windisch J.M., Schneider R., Stark R., Weiland E., Meyers G., Thiel H.-J. RNase of classical swine fever virus: biochemical characterization and inhibition by virus-neutralizing monoclonal antibodies. J. Virol. 1996;70:352–358. doi: 10.1128/jvi.70.1.352-358.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Singh D., Vaughan R., Kao C.C. LL-37 peptide enhancement of signal transduction by toll-like receptor 3 is regulated by pH. J. Biol. Chem. 2014;289:27614–27624. doi: 10.1074/jbc.M114.582973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Abernathy E., Glaunsinger B. Emerging roles for RNA degradation in viral replication and antiviral defense. Virology. 2015;479–480:600–608. doi: 10.1016/j.virol.2015.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Read G.S. Virus-encoded endonucleases: expected and novel functions. Wiley Interdiscip. Rev. RNA. 2013;4:693–708. doi: 10.1002/wrna.1188. [DOI] [PubMed] [Google Scholar]
- 87.Ulferts R., Ziebuhr J. Nidovirus ribonucleases. Structures and functions in viral replication. RNA Biol. 2011;8:295–304. doi: 10.4161/rna.8.2.15196. [DOI] [PubMed] [Google Scholar]
- 88.De Diego M.L., Nieto-Torres J.L., Jimenez-Guardeño J.M., Regla-Nava J.A., Castaño-Rodriguez C., Fernandez-Delgado R. Coronavirus virulence genes with main focus on SARS-CoV envelope gene. Virus Res. 2014;194:124–137. doi: 10.1016/j.virusres.2014.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sun Y., Han M.Y., Kim C., Calvert J.G., Yoo D. Interplay between interferon-mediated innate immunity and porcine reproductive and respiratory syndrome virus. Viruses. 2012;4:424–446. doi: 10.3390/v4040424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Martínez-Sobrido L., Zúñiga E.I., Rosario D., García-Sastre A., De La Torre J.C. Inhibition of the type I interferon response by the nucleoprotein of the prototypic arenavirus lymphocytic choriomeningitis virus. J. Virol. 2006;80:9192–9199. doi: 10.1128/JVI.00555-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jiang X., Huang Q.F., Wang W.J., Dong H.H., Ly H., Liang Y.Y. Structures of arenaviral nucleoproteins with triphosphate dsRNA reveal a unique mechanism of immune suppression. J. Biol. Chem. 2013;288:16949–16959. doi: 10.1074/jbc.M112.420521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Reynard S., Russier M., Fizet A., Carnec X., Baize S. Exonuclease domain of the Lassa virus nucleoprotein is critical to avoid RIG-I signaling and to inhibit the innate immune response. J. Virol. 2014;88:13923–13927. doi: 10.1128/JVI.01923-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Barrat F.J., Elkon K.B., Fitzgerald K.A. Importance of nucleic acid recognition in inflammation and autoimmunity. Annu. Rev. Med. 2016;67:323–336. doi: 10.1146/annurev-med-052814-023338. [DOI] [PubMed] [Google Scholar]
- 94.Crow Y.J. Type I interferonopathies: mendelian type I interferon up-regulation. Curr. Opin. Immunol. 2015;32:7–12. doi: 10.1016/j.coi.2014.10.005. [DOI] [PubMed] [Google Scholar]
- 95.Lee-Kirsch M.A., Wolf C., Kretschmer S., Roers A. Type I interferonopathies—an expanding disease spectrum of immunodysregulation. Semin. Immunopathol. 2015;37:349–357. doi: 10.1007/s00281-015-0500-x. [DOI] [PubMed] [Google Scholar]
- 96.Pelka K., Shibata T., Miyake K., Latz E. Nucleic acid-sensing TLRs and autoimmunity: novel insights from structural and cell biology. Immunol. Rev. 2016;269:60–75. doi: 10.1111/imr.12375. [DOI] [PubMed] [Google Scholar]
- 97.Rigby R.E., Rehwinkel J. RNA degradation in antiviral immunity and autoimmunity. Trends Immunol. 2015;36:179–188. doi: 10.1016/j.it.2015.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sharma S., Fitzgerald K.A., Cancro M.P., Marshak-Rothstein A. Nucleic acid-sensing receptors: rheostats of autoimmunity and autoinflammation. J. Immunol. 2015;195:3507–3512. doi: 10.4049/jimmunol.1500964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Crow M.K. Advances in understanding the role of type I interferons in systemic lupus erythematosus. Curr. Opin. Rheumatol. 2014;26:467–474. doi: 10.1097/BOR.0000000000000087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hagberg N., Rönnblom L. Systemic lupus erythematosus—a disease with a dysregulated type I interferon system. Scand. J. Immunol. 2015;82:199–207. doi: 10.1111/sji.12330. [DOI] [PubMed] [Google Scholar]
- 101.Hall J.C., Rosen A. Type I interferons: crucial participants in disease amplification in autoimmunity. Nat. Rev. Rheumatol. 2010;6:40–49. doi: 10.1038/nrrheum.2009.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Guiducci C., Gong M., Xu Z.H., Gill M., Chaussabel D., Meeker T. TLR recognition of self nucleic acids hampers glucocorticoid activity in lupus. Nature. 2010;465:937–941. doi: 10.1038/nature09102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Brencicova E., Diebold S.S. Nucleic acids and endosomal pattern recognition: how to tell friend from foe? Front. Cell. Infect. Microbiol. 2013;3:37. doi: 10.3389/fcimb.2013.00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kahlenberg J.M., Kaplan M.J. Little peptide, big effects: the role of LL-37 in inflammation and autoimmune disease. J. Immunol. 2013;191:4895–4901. doi: 10.4049/jimmunol.1302005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Dai C., Deng Y., Quinlan A., Gaskin F., Tsao B.P., Fu S.M. Genetics of systemic lupus erythematosus: immune responses and end organ resistance to damage. Curr. Opin. Immunol. 2014;31:87–96. doi: 10.1016/j.coi.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Baccala R., Hoebe K., Kono D.H., Beutler B., Theofilopoulos A.N. TLR-dependent and TLR-independent pathways of type I interferon induction in systemic autoimmunity. Nat. Med. 2007;13:543–551. doi: 10.1038/nm1590. [DOI] [PubMed] [Google Scholar]
- 107.Forsdyke D.R. X chromosome reactivation perturbs intracellular self/not-self discrimination. Immunol. Cell Biol. 2009;87:525–528. doi: 10.1038/icb.2009.39. [DOI] [PubMed] [Google Scholar]
- 108.Markle J.G., Fish E.N. SeXX matters in immunity. Trends Immunol. 2014;35:97–104. doi: 10.1016/j.it.2013.10.006. [DOI] [PubMed] [Google Scholar]
- 109.Crow Y.J., Manel N. Aicardi-Goutières syndrome and the type I interferonopathies. Nat. Rev. Immunol. 2015;15:429–440. doi: 10.1038/nri3850. [DOI] [PubMed] [Google Scholar]
- 110.Yuan F.H., Dutta T., Wang L., Song L., Gu L.Y., Qian L.Y. Human DNA exonuclease TREX1 is also an exoribonuclease that acts on single-stranded RNA. J. Biol. Chem. 2015;290:13344–13353. doi: 10.1074/jbc.M115.653915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ablasser A., Hemmerling I., Schmid-Burgk J.L., Behrendt R., Roers A., Hornung V. TREX1 deficiency triggers cell-autonomous immunity in a cGAS-dependent manner. J. Immunol. 2014;192:5993–5997. doi: 10.4049/jimmunol.1400737. [DOI] [PubMed] [Google Scholar]
- 112.Behrendt R., Schumann T., Gerbaulet A., Wittmann S., Gramberg T., Roers A. Spontaneous type I IFN response in SAMHD1-deficient mice requires both, functional intracellular RNA and DNA sensing pathways. Pediatr. Rheumatol. Online J. 2015;13(Suppl. 1):O33. [Google Scholar]
- 113.Yu Z., Chen T.Y., Cao X.T. RNA editing by ADAR1 marks dsRNA as "self". Cell Res. 2015;25:1283–1284. doi: 10.1038/cr.2015.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Volkman H.E., Stetson D.B. The enemy within: endogenous retroelements and autoimmune disease. Nat. Immunol. 2014;15:415–422. doi: 10.1038/ni.2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Yu P. The potential role of retroviruses in autoimmunity. Immunol. Rev. 2016;269:85–99. doi: 10.1111/imr.12371. [DOI] [PubMed] [Google Scholar]
- 116.Gilliet M., Cao W., Liu Y.J. Plasmacytoid dendritic cells: sensing nucleic acids in viral infection and autoimmune diseases. Nat. Rev. Immunol. 2008;8:594–606. doi: 10.1038/nri2358. [DOI] [PubMed] [Google Scholar]
- 117.Gupta S.K., Haigh B.J., Griffin F.J., Wheeler T.T. The mammalian secreted RNases: mechanisms of action in host defence. Innate Immun. 2013;19:86–97. doi: 10.1177/1753425912446955. [DOI] [PubMed] [Google Scholar]
- 118.Luhtala N., Parker R. T2 Family ribonucleases: ancient enzymes with diverse roles. Trends Biochem. Sci. 2010;35:253–259. doi: 10.1016/j.tibs.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Sorrentino S. The eight human canonical ribonucleases: molecular diversity, catalytic properties, and special biological actions of the enzyme proteins. FEBS Lett. 2010;584:2194–2200. doi: 10.1016/j.febslet.2010.04.018. [DOI] [PubMed] [Google Scholar]
- 120.Netea M.G., Latz E., Mills K.H.G., O’Neill L.A.J. Innate immune memory: a paradigm shift in understanding host defense. Nat. Immunol. 2015;16:675–679. doi: 10.1038/ni.3178. [DOI] [PubMed] [Google Scholar]
- 121.Schneider D.S., Ayres J.S. Two ways to survive infection: what resistance and tolerance can teach us about treating infectious diseases. Nat. Rev. Immunol. 2008;8:889–895. doi: 10.1038/nri2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Sun X.Z., Wiedeman A., Agrawal N., Teal T.H., Tanaka L., Hudkins K.L. Increased ribonuclease expression reduces inflammation and prolongs survival in TLR7 transgenic mice. J. Immunol. 2013;190:2536–2543. doi: 10.4049/jimmunol.1202689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Summerfield A., Ruggli N. Immune responses against classical swine fever virus: between ignorance and lunacy. Front. Vet. Sci. 2015;2:10. doi: 10.3389/fvets.2015.00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Medzhitov R. Innate immunity: quo vadis? Nat. Immunol. 2010;11:551–553. doi: 10.1038/ni0710-551. [DOI] [PubMed] [Google Scholar]