

Abstract
Extensive nonhomologous recombinations occur between the 5′ and 3′ fragments of a replicable RNA in a cell-free system composed of pure Qβ phage replicase and ribonucleoside triphosphates, providing direct evidence for the ability of RNAs to recombine without DNA intermediates and in the absence of host cell proteins. The recombination events are revealed by the molecular colony technique that allows single RNA molecules to be cloned in vitro. The observed nonhomologous recombinations are entirely dependent on the 3′ hydroxyl group of the 5′ fragment, and are due to a splicing-like reaction in which RNA secondary structure guides the attack of this 3′ hydroxyl on phosphoester bonds within the 3′ fragment.
Introduction
Intermolecular RNA recombination is a wide-spread phenomenon reported for a variety of animal, plant, and bacterial RNA-containing viruses (20, 9). It has played an important role in the evolution of RNA viruses (Lai 1992) and might have resulted in the exon–intron structure of modern genes (Gilbert 1986). Viral RNA recombination is believed to be quite different from the process of RNA trans-splicing, since it is not site-specific (Lai 1992) and is apparently associated with the RNA synthesis by viral replicases (Kirkegaard and Baltimore 1986).
Since recombinations occur rarely and randomly, they are only observed if their products can be amplified, cloned, and distinguished against a large background of nonrecombinant RNAs. So far, RNA recombination has been studied in vivo, almost exclusively on viruses or their defective interfering (DI) particles amplified in living cells under selective conditions. Thus, only those recombination events that render the particles infectious have been detected. It is therefore not surprising that most reports on RNA recombination concern homologous recombination, i.e., recombination between similar segments of related RNAs (Lai 1992). The homologous recombination can most readily be accounted for by a copy choice (template switch) reaction mechanism (Cooper et al. 1974), which implies that recombination between RNA molecules occurs via copying of the 3′ portion of the first molecule by the replicase that eventually jumps, together with the nascent strand, to a homologous region of the second molecule where it resumes RNA synthesis. This hypothesis has become commonly acknowledged and has been used to explain both homologous and nonhomologous recombinations, although it has never been proved directly, apparently because of the constraints and uncertainties inherent to the in vivo experimental models. Also, the in vivo experiments have not answered the basic questions regarding the recombination mechanism, such as whether the recombination is performed or promoted by viral replicase, by RNA molecules themselves, or by host cell proteins. Moreover, the in vivo experiments have not definitely ruled out the involvement of DNA intermediates and, therefore, the possibility that recombination occurs at the DNA, rather than at the RNA, level.
Here we report the results of our experiments on RNA recombination in a purified cell-free system. The system utilizes pure RNAs and a homogeneous preparation of Qβ replicase, the bacteriophage Qβ RNA-directed RNA polymerase, which is particularly efficient in the RNA synthesis in vitro (Chetverin and Spirin 1995). A recently developed molecular colony technique allowing individual RNA molecules to be grown as colonies (7, 10) was used in this study to detect rare recombination events. Earlier studies have provided evidence for the occurrence of both homologous (Palasingam and Shaklee 1992) and nonhomologous RNA recombination (27, 28, 25) in the Qβ phage-infected Escherichia coli cells. There have also been indications that RNA can probably recombine in the in vitro Qβ replicase reaction (Biebricher and Luce 1992).
The results show that RNA recombination did occur in such a purified system, thereby providing direct evidence for the ability of RNA molecules to recombine without DNA intermediates and in the absence of host cell components. Most of the recombinant RNAs were of a purely nonhomologous type, and the mechanism of their generation was entirely different from copy choice.
Results
Strategy of RNA Recombination Experiments in the Cell-Free Qβ System
In addition to the genomic RNA of Qβ phage, Qβ replicase can exponentially amplify in vitro a variety of small RQ RNAs that are natural phage satellites (Chetverin et al. 1991), termed so for being Replicable by Qβ replicase. These subgenomic RNAs are the most suitable for in vitro studies on RNA recombination, since they are more efficient templates for Qβ replicase than the genomic Qβ RNA. Many RQ RNAs have themselves been generated in vivo by intermolecular recombination, and their sequences can accommodate foreign inserts without losing the ability to replicate (Chetverin and Spirin 1995). In contrast to viral genomes, however, RQ RNAs lack genetic markers facilitating the selection of recombinant molecules against a large excess of parent RNAs. To overcome this problem, we devised the following experimental scheme (Figure 1A).
Figure 1.

Strategy of RNA Recombination Experiments
(A) Scheme of recombination between nonreplicable fragments of an RQ RNA leading to generation of a replicable RNA.
(B) Sequences of foreign extensions (white letters on a black background) of the 5′ and 3′ fragments of the (−)-strand of RQ135−1 RNA. A few base differences between the extensions (lowercase letters) serve as sequence markers. The fragment extensions consist of polylinker sequences providing for further manipulations of their structure at the DNA level.
Instead of the full-sized replicable molecules, the scheme employs the mutually supplementing 5′ and 3′ fragments of an RQ RNA that are prepared by transcription and that carry, at the truncated ends, artificial foreign extensions whose sequence and secondary structure may vary. Qβ replicase can start at the 3′ end of the 3′ fragment and copy it together with the foreign extension, producing a complementary strand. The reaction ceases after the first round, and RNA amplification will not occur unless a complete replicable RNA is formed by recombination between the fragments. If the recombination occurs within the extensions, the product will comprise the original RQ RNA carrying the corresponding foreign insert.
The reaction products are then analyzed by the molecular colony technique. According to this method, an RNA sample is spread over a thin Qβ replicase-containing agarose layer, which is then covered by a nylon membrane impregnated with all four ribonucleoside triphosphates. If there are replicable RNAs on the interface between the agarose and the membrane, RNA colonies appear within an hour, each colony containing up to 1012 copies of a single progenitor template (Chetverina and Chetverin 1993). While the replicable recombinant molecules will produce colonies, the nonreplicable fragments of the original RQ RNA will not. This procedure ensures positive selection of the recombination products and allows the number of recombinant molecules to be directly assessed by simply counting the RNA colonies hybridizable with the labeled 5′ fragment. The fragment is not copied by Qβ replicase and thus its complementary strand does not appear unless recombination occurs. This eliminates the background hybridization problems even when relatively high concentrations of the nonreplicating RNA fragments are used in experiments. The recombinant molecules carrying foreign inserts are detected by hybridizing the colonies with labeled oligonucleotides complementary to the foreign sequences.
In this work, we used the 5′ and 3′ fragments of the (−)-strand of RQ135−1 RNA, a 134 nt long subspecies of RQ135 RNA, which is one of the most potent Qβ replicase templates (Munishkin et al. 1991). Earlier, we found that this RNA can serve as a vector for amplification of short foreign inserts (V. I. U. and A. B. C., unpublished data). Since RNA recombination was expected to occur via the copy choice mechanism, the foreign extensions of the fragments were intentionally made almost entirely homologous to each other in order to facilitate the process (Figure 1B).
Nonreplicable RQ RNA Fragments Can Recombine to Produce Replicable RNAs
As expected, RNA colonies almost never grew when up to 2 × 1010 molecules of either the 5′ or the 3′ fragment were incubated separately (Figure 2A), confirming that these fragments cannot replicate. A few colonies that occasionally appeared in these samples (and also in the samples where no RNA was added) did not hybridize with an oligonucleotide targeted against the foreign extensions (cf. Figure 2C), and seemed to result from the airborne contamination by the wild-type RQ135 RNA (7, 10).
Figure 2.
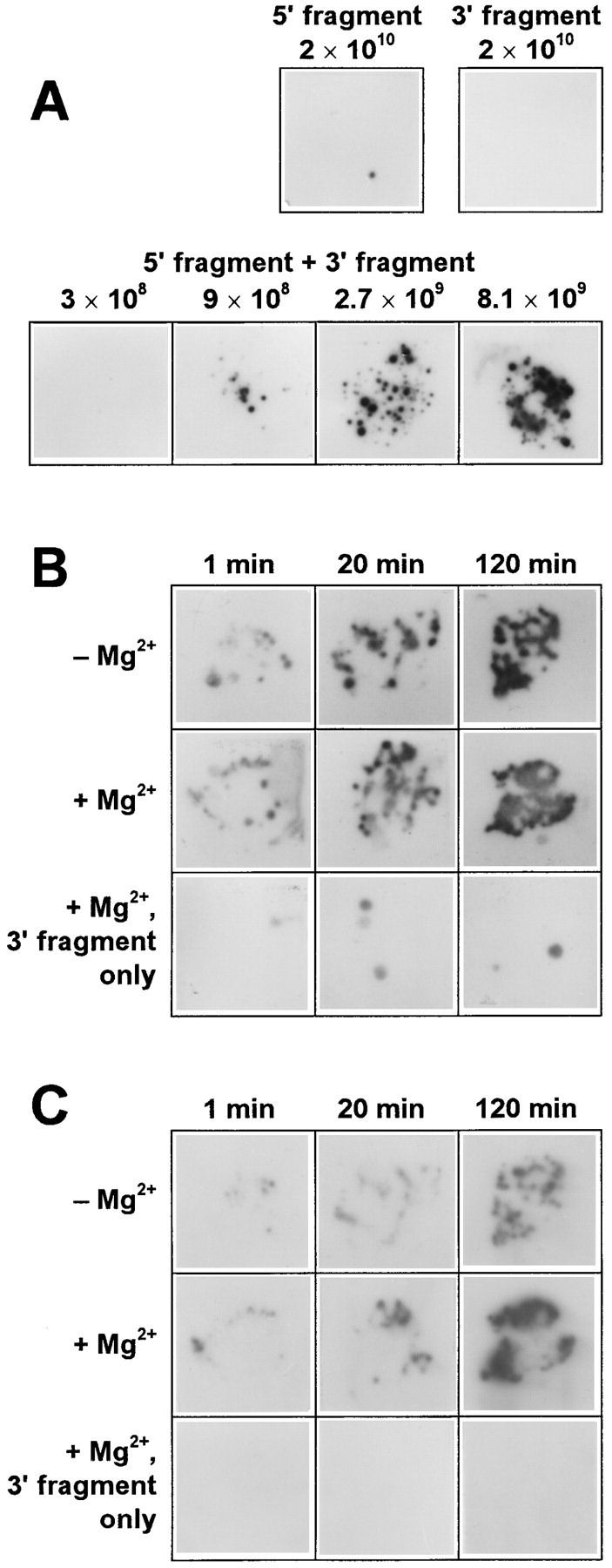
Recombination between the Extended 5′ and 3′ Fragments of RQ135−1(−) RNA
The number of replicable recombinant molecules was assessed as the number of RNA colonies hybridized with specific labeled probes.
(A) Colonies produced by the indicated number of molecules of the 5′ and 3′ fragments preincubated separately or in a mixture in a Mg2+-free buffer during 45 min. Hybridization with the labeled 5′ fragment.
(B) Colonies produced by 1010 molecules each of the mixed 5′ and 3′ fragments, or the 3′ fragment only (bottom row), preincubated in the absence or in the presence of 10 mM MgCl2 during the specified time period. Hybridization with the labeled 5′ fragment.
(C) Same as (B), but hybridization with a labeled oligonucleotide targeted against the foreign extensions.
On the contrary, up to 150 RNA colonies could be seen when 2.7 × 109 molecules (4.5 fmol) each of the 5′ and 3′ fragments were incubated together in a 10 μl aliquot (Figure 2A), indicating that replicable RNAs are formed by recombination between the fragments. Colonies did not appear in control experiments where the RNA fragments were replaced with the plasmids used for their synthesis (data not shown), which excludes the possibility that recombination is due to traces of DNA in the fragment preparations. The number of colonies corresponded to an ∼10−7 recombination frequency at the fragment concentration of 0.45 nM. The actual frequency might have been higher, since only a fraction (∼10%) of RQ135 RNA molecules are capable of producing colonies (Chetverina and Chetverin 1993) and not every recombination product can be replicable. In more dilute samples, the yield of recombinant molecules decreased roughly proportionally to the product of the fragment concentrations, as expected for a bimolecular reaction; that is to say, the recombination frequency was proportional to the RNA concentration. At a higher concentration of the fragments, the number of colonies became too large to be counted. A large proportion of the recombinant RNA colonies were hybridizable with an oligonucleotide targeted to the foreign extensions (cf. Figure 2B and Figure 2C), indicating that the extension sequences were incorporated into the recombination products. In additional experiments, we found that the complementary versions of the 5′ and 3′ fragments recombined at a similar frequency (data not shown), meaning that the ability to recombine does not depend on strand polarity.
The yield of recombinant molecules increased when the recombining fragments were preincubated up to 2 hr prior to contacting them with Qβ replicase (Figure 2B and Figure 2C), suggesting that the process of recombination includes a slow step occurring independently of the replicase action. Since this step does not depend on the presence of Mg2+ (Figure 2B), it could be a noncovalent interaction between the fragments that precedes the creation of a recombinant molecule. Indeed, the 5′ and 3′ fragments were capable of forming a noncovalent associate (heteroduplex) stable enough to survive electrophoresis under nondenaturing conditions and gel-filtration in a low-salt buffer (Figure 3A), and annealing of a mixture of the fragments efficiently promoted their recombination at a given fragment concentration (Figure 3B).
Figure 3.

Effect of Annealing of the 5′ and 3′ Fragments on Their Recombination
(A) Gel electrophoresis under nondenaturing conditions of a mixture of 1011 molecules each of the [32P]-labeled 5′ and 3′ fragments. Shown in the order of appearance: the mixture incubated for 40 min at 55°C in the presence of 0.5 M KCl; the same sample brought into a 1 mM Na-EDTA solution by passing through a Sephadex G-25 microcolumn; the desalted sample after heating for 2 min at 96°C; the mixture incubated for 40 min at 37°C in the presence of 0.5 M KCl. The last two lanes contain separate fragments.
(B) Colonies produced by the indicated number of molecules of each of the mixed 5′ and 3′ fragments that were annealed by heating to 96°C for 2 min in a Mg2+-free buffer containing 0.1 M KCl and then allowing the sample to cool down to 20°C during 1 hr (upper row), or preincubated in the same buffer at 20°C during 1 hr (lower row). Hybridization with the foreign extension-targeted oligonucleotide.
Structure of Generated Recombinant RNAs
To obtain a deeper insight into the mechanism of RNA recombination, we sequenced a total of 17 recombinant clones generated upon incubation of 1010 each of the 5′ and 3′ fragments in the absence (Figure 4A) or in the presence (Figure 4B) of Mg2+. To this end, the total RNA was extracted from an agarose lawn containing more than 100 RNA colonies, subjected to reverse transcription and polymerase chain reaction (RT-PCR) amplification, and the obtained DNA fragments were cloned in a plasmid vector. The plasmids purified from randomly picked clones were sequenced by the dideoxy chain termination method. Since each RNA colony contained at least 1011 molecules (as estimated by normalizing the autoradiography spots to a known amount of RQ135 RNA hybridized with the same probe), the extracted material contained at least 1013 recombinant RNA molecules, or 103 times as many as the number of the added 5′ and 3′ fragments. Therefore, any subsequent recombination between the fragments during reverse transcription or PCR amplification could not appreciably contribute to the cloned sequences. In control experiments, the fragment mixture was subjected to the same procedure, except the colony growth was omitted and the fragments were mixed either before (Figure 4C) or after the reverse transcription step.
Figure 4.

Sequences of Recombinant RNAs
Aligned sequences of the 5′ fragment and the 3′ fragment are shown below and above a recombinant sequence, respectively. Only partial sequences are shown; other parts of the recombinants perfectly match sequences of the respective fragments. Foreign extensions of the fragments are shown in boldface letters, and positions where the extensions differ are shown in lowercase letters. Foreign inserts in the recombinant sequences are shown in white letters on a black background. Numbers in parentheses indicate the number of clones whose sequences are identical.
(A) Sequences generated upon preincubation during 40 min prior to colony growth of a mixture of 1010 molecules each of the 5′ and 3′ fragments of RQ135−1(−) RNA in the absence of Mg2+.
(B) Sequences generated upon preincubation of the fragments in the presence of 10 mM MgCl2.
(C) Sequence of a clone isolated from products of the control RT-PCR amplification of the same mixture upon its preincubation in the presence of Mg2+.
The following observations could be made from inspection of the Qβ replicase-amplified sequences shown in Figure 4A and Figure 4B. (i) None of the sequences coincided with the primary structure of wild-type RQ135−1 RNA (Munishkin et al. 1991), and almost all included, at least partly, the foreign extensions of the 5′ and 3′ fragments. This observation strengthens the above conclusion that these molecules were formed by genuine intermolecular recombination. (ii) In most cases, crossover sites lay within the foreign extensions and were precisely identified (recombinants A1–A5, B1, and B2). All these recombinants were of a purely nonhomologous type. (iii) Surprisingly, there were no recombinants of a bona fide homologous type expected of copy choice: the nearly perfect homology between the foreign extensions was invariably ignored. Crossover sites were sometimes found within very short (1–3 nt) homologous stretches. Almost all of these crossovers involved the vector portion of either the 5′ or the 3′ fragment (recombinants B4–B6). Nevertheless, the recombinant molecules retained the UCCCU/AGGGA helix of the vector RNA (Figure 5), indicating that the relatively large proportion of this type of recombinants might have resulted from their selective amplification at the expense of those molecules in which this helix was distorted. (iv) Contribution of the 5′ and 3′ fragments to the recombinant sequences was markedly asymmetric: 12 of the 17 clones included the intact 5′ fragment (recombinants A1–A5, B1, B2), whereas the complete 3′ fragment was preserved in only one case (recombinant A5). The latter recombinant molecule was formed by end-to-end joining of the two fragments. Some of the recombinants with the intact 5′ fragment contained one to three extra nucleotides upstream from crossover sites (A2, A3, A5, B2). These nucleotides were possibly added to the 3′ terminus of the 5′ fragment during its synthesis by T7 RNA polymerase (Milligan et al. 1987) or during the subsequent incubation with Qβ replicase (Biebricher and Luce 1992). (v) All recombinants in which the 5′ fragment was abbreviated were generated in the experiment in which the fragments were preincubated in the presence of Mg2+ prior to contacting them with Qβ replicase (recombinants B4–B6 and, possibly, B3). Interestingly, these were the recombinants whose crossover sites lay within the short stretches of homology between the fragments (see item [iii]). Although the presence of Mg2+ during the preincubation step did not noticeably influence the recombination frequency (Figure 2B), the latter observation suggests that it might have affected the recombination mechanism. (vi) Crossover sites were not evenly distributed along the 3′ fragment sequence. As seen from Figure 5, they tended to concentrate at or close to the 5′ base of a helix of the putative intermolecular structure formed by the 3′ and 5′ fragments. (vii) Eight of the seventeen clones apparently were formed by adding the intact 3′ terminus of the 5′ fragment to the beginning of sequence UCUAGA in the 3′ fragment (recombinants A1–A3, B1). This crossover site was located at the 5′ base of the helix closest to the 3′ terminus of the 5′ fragment (Figure 5). Although five of these clones were identical and had been isolated from one RNA pool (recombinant A1), the probability that all of them were descendants of a single original recombinant molecule was negligibly low, less than 10−6, as expected from a random distribution of the material contained in at least 100 RNA colonies among the 9 clones isolated from that pool. Moreover, a clone with the same nucleotide sequence had been independently isolated from another RNA pool (recombinant B1), and there were three more clones in which recombination had occurred in a close proximity to this site (recombinants A4, B2, B3).
Figure 5.
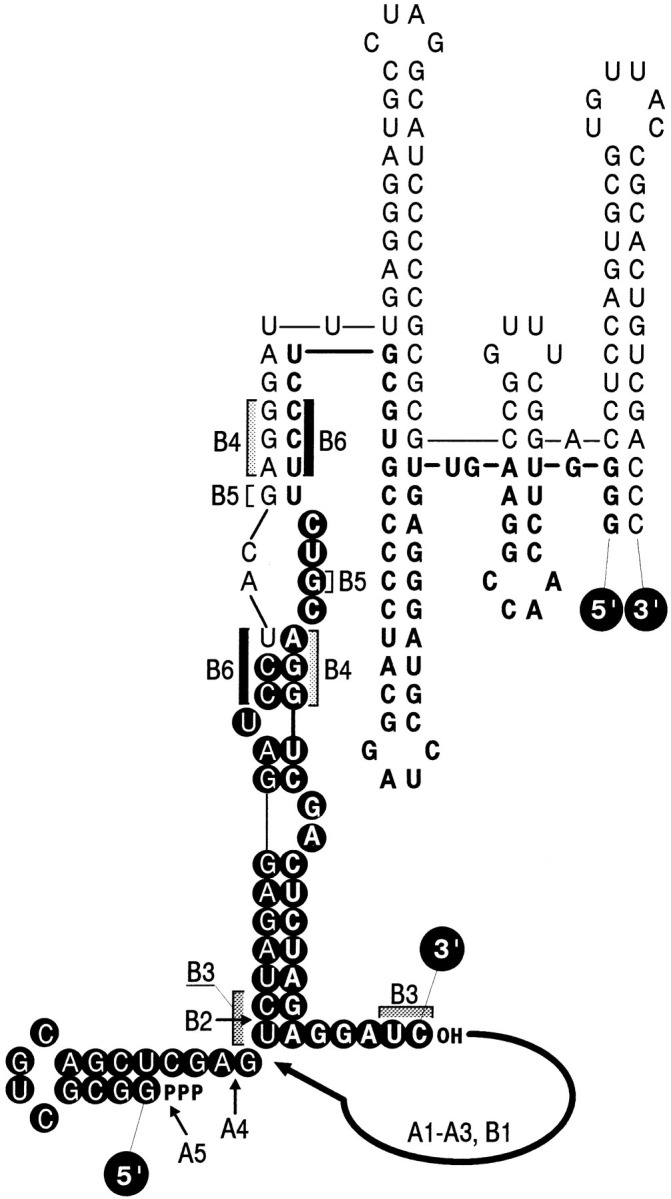
Predicted Intermolecular Secondary Structure Formed by the Extended 5′ and 3′ Fragments of RQ135−1(−) RNA
The structure is based on the secondary structure model of RQ135 RNA (Munishkin et al. 1991). The foreign extensions are folded according to the lowest free energy criterion (Zuker and Stiegler 1981). Boldface letters depict the 5′ fragment, white letters in black circles depict the foreign extensions. Arrows indicate location of the precisely identified crossover sites along the 3′ fragment; brackets with matching shading indicate homologous stretches including the imprecisely localized crossover sites. Numbered letters refer to the type of recombinant sequence (Figure 4). The big arrow pinpoints the primary site of proposed attack of the 3′ fragment by the 3′ terminal hydroxyl of the 5′ fragment (see Discussion).
To check the possibility that the relative abundance of these clones was due to selective amplification of this sort of recombinant RNAs, we compared the ability of the sequenced RNAs to replicate under conditions of colony growth. As seen from Figure 6, all of these RNAs, except recombinant A5 carrying the longest insert, were amplified by Qβ replicase with similar efficiencies, and there was no preference toward amplification of recombinants A1 or B1, which predominated among the isolated clones. It follows that their predominance reflects a high frequency of recombination at the corresponding site.
Figure 6.

Replication of Recombinant RNAs
Polyacrylamide gel electrophoresis pattern of recombinant RNAs stained with toluidine blue. Numbered letters above the gel refer to the type of recombinant sequence (Figure 4), numbers at the bottom refer to the clone number.
(A) Samples containing 1–2 μg of transcripts synthesized by T7 RNA polymerase from the cloned recombinant plasmids digested at site SmaI.
(B) Products of 10 min Qβ replicase reactions initiated with 1/400 of the amount of RNA applied to gel A. The faster migrating band is a double-stranded RNA, the slower migrating bands correspond to single strands and partial duplexes. Lane marked “0” contains material from the reaction where no RNA was added.
The control RT-PCR amplification of the fragment mixture also produced recombinant sequences. However, they migrated as a single band during gel electrophoresis, unlike the heterogeneous RT-PCR products synthesized subsequent to the amplification of the recombinant RNAs by Qβ replicase (data not shown). Furthermore, their sequencing revealed a precisely homologous recombination expected for the copy choice mechanism (Figure 4C). Since such sequences did appear only if the 5′ and 3′ RNA fragments were mixed before the cDNA synthesis step, they seemed to result from a template switch occurring during reverse transcription (Negroni et al. 1995). It follows that the RNA fragments are prone to a copy choice reaction, provided that an appropriate enzyme is used, and that the frequency of template switch by Qβ replicase, if any, is much lower than that by AMV reverse transcriptase used in these experiments.
Role of the 3′ Hydroxyl Groups in RNA Recombination
The observed features of the recombinant sequences indicate that the 3′ terminus of the 5′ fragment may be involved in the recombination process. Since the fragment contains the hydroxyl group at the terminal 3′ position as a result of transcription, we checked whether the presence of the 3′ hydroxyl is essential. To this end, the 3′ terminal ribose was oxidized with periodate, and the terminal nucleoside dialdehyde was eliminated by treatment with aniline. This resulted in a 1 nt shorter fragment that bears the phosphoryl group at the 3′ end (Steinschneider and Fraenkel-Conrat 1966b). Figure 7A (upper row) shows that such a modification of the 5′ fragment reduced the number of RNA colonies to zero, and that the removal of the 3′ terminal phosphate with calf intestine alkaline phosphatase (CIP) restored the ability of the fragment to recombine. Moreover, recombination could be switched on by adding the phosphatase directly into the sample containing a mixture of the 3′ phosphoryl 5′ fragment and the 3′ fragment (Figure 7B).
Figure 7.

Effect of 3′ Terminal Modifications
(A) Colonies produced by a mixture of 6 × 109 molecules each of the 5′ and 3′ fragments one of which was unmodified while the other was either unmodified at the 3′ terminus (… NPNOH) or modified by periodate oxidation and treatment with aniline (…NP) and subsequent dephosphorylation (…NOH). The mixtures were preincubated for 1 hr at 20°C in a Mg2+-free buffer.
(B) Colonies produced by a mixture of 6 × 109 molecules each of the 3′ phosphoryl 5′ fragment and the unmodified 3′ fragment after preincubation for 1 hr at 37°C and indicated Mg2+ concentration with or without 0.01 units of CIP. Prior to colony growth, all the samples received a sufficient amount of EDTA to chelate free Mg2+ and were then heated for 2 min at 96°C to inactivate phosphatase. RNA colonies were detected by hybridization with the foreign extension-targeted oligonucleotide.
(C) Silver-stained (Igloi 1983) products of 10 min Qβ replicase reactions initiated with 2 ng of a recombinant RNA (type A5, clone 3, cf. Figure 6), which was either unmodified at the 3′ terminus (…NPNOH) or modified by periodate oxidation (…NPNOX) and subsequent treatment with aniline (…NP) and dephosphorylation (…NOH).
In contradistinction, similar modification of the 3′ fragment had no appreciable effect on the number of RNA colonies and the number reduced, rather than increased, upon phosphatase treatment (Figure 7A, lower row). A lesser number of colonies in this case correlates with a decreased capability of a 3′ truncated recombinant RNA to be amplified by Qβ replicase (Figure 7C).
In additional experiments, we found that other 3′ terminal modifications eliminating the 3′ hydroxyl group, such as periodate oxidation and biotinylation, also annulled the recombination potential of the 5′ fragment, but not of the 3′ fragment. These observations suggest that the 3′ hydroxyl of the 5′ fragment plays a crucial role in the recombination process. Similar results were obtained for the complementary versions of the fragments (data not shown), indicating that the mechanism of recombination is the same for the strands of either polarity.
Figure 7B shows that recombination with the 3′ phosphoryl 5′ fragment could also be switched on by the Mg2+ present in the preincubation mixture at a high concentration, although to a lesser extent than by phosphatase. This effect can be explained by exposure of a reactive hydroxyl group as a result of a Mg2+-catalyzed RNA cleavage, which gives a rationale for the observed Mg2+-induced change in the sequence of some recombinant molecules (Figure 4B).
Discussion
Mechanism of RNA Recombination in the Cell-Free System
Although intermolecular RNA recombination was first reported more than 30 years ago (15, 21), its mechanism remains obscure due primarily to the absence of adequate in vitro models. Biebricher and Luce 1992 reported reproducible synthesis of a recombinant RQ RNA in the in vitro Qβ replicase reactions initiated with a different RQ RNA. However, since the synthesis was only observed under conditions selective for amplification of the recombinant RNA, and since the same RNA was studied in their laboratory earlier (Biebricher et al. 1982), it is difficult to rule out the possibility that their reactions were contaminated with the already existing recombinant RNA, given the ability of Qβ replicase to amplify even single airborne RQ RNA molecules (7, 10).
Here, we describe an efficient cell-free assay system for RNA recombination; the sensitive molecular colony technique is used to observe individual recombination events in a reaction containing only homogeneous Qβ replicase, pure ribonucleoside triphosphates, and two types of purified RNA molecules. This is a cell-free version of the Qβ phage system whose ability to perform RNA recombinations in vivo was demonstrated earlier (27, 28, 32). The recombination substrates used in these experiments were fragments of a Qβ phage satellite RNA (Chetverin et al. 1991), which itself is a product of an intermolecular recombination that had occurred in vivo (Munishkin et al. 1991). The fragments were derived from the RNA sequence by breaking it at the junction site between the in vivo duplicated segments (Chetverin et al. 1992), i.e., these RNA molecules may resemble the natural recombination substrates. The experimental conditions (temperature, pH value, concentrations of mono- and divalent cations) were close to the normal physiological conditions; this suggests that the results may be physiologically relevant.
Of course, the reported system has certain limitations. Thus, only those recombinants could be detected and studied that were amplified by Qβ replicase. The results show that in the isolated recombination products the crossover sites were exclusively located within or close to the introduced foreign sequences, and that all the recombinants retained the secondary structure elements of the original RQ RNA vector, suggesting that selection pressure rejects all molecules in which the structure of the vector is altered.
At the same time, a foreign insert as long as 52 nt (Figure 4, recombinant A5) did not eliminate the ability of the RQ135 RNA vector to replicate (Figure 6), implying that the system allows the recombination between foreign sequences of this size to be studied. In any case, the selection pressure in the in vitro system is much less pronounced than in any in vivo system, since there is no need for the ability of the recombinant RNAs to encode proteins, to be encapsidated, or to infect the cell. In addition, the in vitro system allows the recombination mechanism to be explored by changing the reaction conditions, varying composition of the reaction medium, utilizing modified RNAs or nucleotides, and by step-by-step monitoring of the recombination process. In fact, the system presents a remarkable example of quantitative biochemical assay for reactions occurring between single molecules.
This system allowed us to demonstrate directly that RNAs can recombine without the formation of DNA intermediates and without the assistance of host cell proteins, except possibly those included in the Qβ replicase heterotetramer, i.e., the translation factors EF-Tu and EF-Ts, and ribosomal protein S1 (Chetverin and Spirin 1995). Moreover, experiments in the cell-free system led us to an unexpected conclusion regarding the RNA recombination mechanism that appeared to be entirely different from copy choice.
According to the copy choice mechanism, a nascent strand synthesized by Qβ replicase on the 3′ fragment would serve as a primer for RNA synthesis on the 5′ fragment. In order to facilitate replicase switching between the recombining fragments, the fragments were provided with homologous foreign extensions (Figure 1B). Thus, sequence crossovers were expected to occur between the extensions resulting in molecules with chimeric foreign inserts in which the extension sequence of the 5′ fragment was partially replaced from the 3′ side by the homologous segment of the 3′ fragment, as it was observed for the reverse transcriptase-generated recombinants (Figure 4C). Surprisingly, none of the Qβ replicase-amplified recombinants displayed the expected structure (Figure 4A and Figure 4B). The homology between the extension sequences was invariably ignored. Moreover, in most cases, a part of the 3′ fragment extension was attached to, rather than did replace, the extension of the 5′ fragment, which was preserved intact in most of the recombinant molecules. In the extreme case, the recombinant molecule was generated by the end-to-end joining of the intact 5′ and 3′ fragments (recombinant A5).
Figure 7 demonstrates that recombination between the RNA fragments was entirely dependent on the availability of the 3′ hydroxyl group at the 5′ fragment terminus. This result not only rules out the possibility that the classical copy choice mechanism contributes to the observed recombination events, but also rejects any variation of this mechanism in which an RNA fragment serves as a primer for its own extension on another template. For example, one might argue that elimination of the terminal hydroxyl simply prevents extension of the 5′ fragment on the 3′ fragment complementary strand, which can be synthesized by Qβ replicase. This could not prevent, however, a reciprocal extension of the newly synthesized strand on the 5′ fragment, since its 3′ terminus was not modified, and only a slight (some 2-fold) reduction in the recombination frequency could be expected. The absence of recombination in this case (as well as in the reciprocal experiment with the complementary versions of the fragments) means that the newly synthesized strand lacked its recombination partner, which must be the missing complementary copy of the 5′ fragment.
It follows that the recombination occurs through a reaction between the fragments of the same polarity. This is further supported by the observation that preliminary annealing of the 5′ and 3′ fragments increased the recombination frequency by orders of magnitude (Figure 3). Together with the distribution of crossover sites within the putative secondary structure of the annealed fragments (Figure 5), the latter observation also indicates that RNA recombination is a secondary–structure guided process.
The most likely mechanism for RNA recombination, totally consistent with the above observations, can be pictured as follows. First, the recombining molecules form a noncovalent complex by means of intermolecular hydrogen bonding. Then, the 3′ terminal hydroxyl of the 5′ partner attacks a phosphate group in the sugar-phosphate backbone of the 3′ partner, resulting in the ligation of one part of the target RNA with a concomitant release of the other. Of course, the 3′ terminal hydroxyl of the 3′ fragment could also attack the 5′ fragment, but this would result in a nonreplicable molecule where the fragments are in a wrong order. Obviously, this mechanism is very similar to that operating in RNA splicing (Cech and Bass 1986). We would like to stress that our results do not rule out in principle other possible mechanisms, including copy choice. Rather, they show that those mechanisms do not appreciably contribute to the RNA recombination in our cell-free system.
This paper does not address the question of whether the observed RNA recombination required any Qβ replicase activity. At present, it seems unlikely, given the known ability of RNA to self-splice and the absence of indications that Qβ replicase can ligate RNA fragments. However, we cannot exclude the possibility that Qβ replicase acts as a protein cofactor that binds the RNA fragments and provides favorable conditions for a self-catalyzed splicing reaction.
Implications for RNA Recombination In Vivo
Although the frequency of nonhomologous recombination observed in our in vitro assay system is exceedingly low, it may nonetheless be comparable to that of RNA recombination occurring in vivo during bacteriophage and viral infection. The frequency is 10−7 per a 30 nt RNA segment at the 10−9 M fragment concentration and is roughly proportional to the RNA concentration. A straightforward extrapolation to the concentration of Qβ RNA in infected E. coli cells (up to 10−5 M; Weissmann 1974) would give the value of 10−3, i.e., similar to the frequency of the most efficient homologous recombinations in vivo (Lai 1992). Thus, the efficiency of the nonhomologous recombination reaction does not rule out, and may well argue in favor of, the physiological relevance of the in vitro reaction.
General properties of the in vitro recombination closely resemble those of the viral RNA recombination in vivo. Indeed, RNA recombination in vivo (i) can occur when one (18, 27) or even both of the recombining molecules (28, 25, 14) do not replicate; (ii) seems to be guided by the secondary structures formed between RNA molecules (19, 33, 29, 5); (iii) predominantly occurs within unpaired regions of RNA (33, 36, 5); (iv) is promoted by preannealing of the recombination partners (Dzianott et al. 1995); and (v) has an RNA concentration dependence similar to that of the in vitro recombination (Jarvis and Kirkegaard 1992). However, these properties do not distinguish between the copy choice and splicing-like mechanisms.
The possibility of a breaking and joining mechanism of a trans-splicing type in viral RNA recombination was mentioned previously, but at that time no experimental proof in support of this mechanism existed (Lai 1992). Such a proof is provided by the results reported here. At the same time, only nonhomologous recombinations were observed in this cell-free system, contrasting the in vivo observations that predominantly revealed homologous recombination (Lai 1992). This discrepancy may be partially due to selection pressure that rejects in vivo most nonhomologous recombinants. However, a majority of the poliovirus recombinants appear to be homologous even in the absence of selection (Jarvis and Kirkegaard 1992), and are presumably generated via the copy choice mechanism (Kirkegaard and Baltimore 1986).
A clue to the controversy seems to lie in the extremely different frequencies of homologous recombination in poliovirus (6 × 10−3 per 1900 nt; Jarvis and Kirkegaard 1992) and in Qβ phage from which the cell-free system was derived (only 10−8 per a similar genome length; Palasingam and Shaklee 1992). A backward extrapolation of the frequency of homologous recombination in Qβ phage to the RNA concentrations used in our in vitro experiments would give the value of 10−13 per 30 nt. Of course, this is a very rough estimate, but it helps to demonstrate that even if the copy choice mechanism operated in the in vitro system as efficiently as in Qβ phage, it would not, except fortuitously, show up on the million-fold higher background of nonhomologous recombinations.
The following concept helps to reconcile the in vivo and in vitro data. Most homologous recombinations occur by a copy choice mechanism, and their frequency is a virus-specific parameter, inasmuch as it is determined by the properties of viral replicases, such as the enzyme processivity. At the same time, nonhomologous RNA recombinations mainly occur via splicing-like reactions, probably nonenzymatic ones, and their frequency is relatively virus-nonspecific, since it is determined by the general properties of RNA, such as its secondary and/or tertiary structure. Given the fact that viruses with a high overall recombination potential (such as picorna- and coronaviruses) predominantly display homologous recombination, one could predict that the relative contribution of nonhomologous recombinations to the totality of recombination events in different viruses would increase as the overall recombination rate decreases, and this largely conforms to the available in vivo data (Lai 1992). For example, most of the nongenomic (satellite) RNAs of Qβ phage, which are less subject to selection than the genomic RNA, consist of a mosaic of fragments of both viral and cellular origin, i.e., they have formed via nonhomologous recombination (Chetverin and Spirin 1995). This hypothesis is further supported by the observation that the RNA fragments used in this work produced only nonhomologous recombinants when amplified by Qβ replicase, whereas the same fragments produced only homologous recombinants when they were copied by reverse transcriptase whose template switching ability seems to be even higher than that of poliovirus replicase (Negroni et al. 1995).
Thus, homologous and nonhomologous recombinations may be brought about by different mechanisms. Also, they seem to play different roles in biological systems. As has been pointed out (Lai 1992), the homologous recombination may be a mechanism to eliminate replication errors, and this could be particularly advantageous for viruses with large nonsegmented genomes and for those encoding large polyproteins. Therefore, the elevated capability of template switch may have been acquired by the replicases of picorna- and coronaviruses as a result of evolution. At the same time, nonhomologous recombinations provide for big and instant evolutionary jumps by enabling a virus to create new genes or to borrow them from other viruses or from the host cell, and they are also responsible for the generation of DI genomes that strongly affect viral infection. The low capability of Qβ replicase to switch between templates does not allow homologous recombination to be observed in our cell-free system. At the same time, it makes the system perfectly suited to studying nonhomologous recombinations.
RNA Secondary Structure Requirements for Nonhomologous Recombination
In agreement with the proposed splicing-like mechanism, most of the crossover sites were located within the putative intermolecular secondary structure close to the attacking 3′ terminus of the 5′ fragment, the attack occurring most frequently at the 5′ base of the nearest helix (Figure 5). In contrast to the classical splicing reaction, the sites of attack were scattered along the target sequence and the reaction was orders of magnitude less efficient. This was not unexpected, since the recombining sequences were not selected for the highest performance.
One interpretation of our data is that nearly any RNA that positions its 3′ hydroxyl in the vicinity of a stem/loop structure will lead to some level of transesterification. Indeed, this interpretation would explain why the recombination mechanism totally ignores homology between the foreign extensions, why a 3′ hydroxyl can add to a 5′-terminal triphosphate (recombinant A5), and why the splicing-like reaction even tolerates addition of CCC to the 3′ end of the attacking molecule (recombinant A3). Thus, RNAs may be intrinsically recombinogenic under physiological conditions, but the resulting low level of nonhomologous recombinants can only be appreciated when a strong selective advantage allows the recombinant molecules to outreplicate the nonrecombinants.
By using the strategy worked out in the in vitro experiments, it is now possible to perform direct experiments on the role of splicing-like reactions in nonhomologous virus recombination. At the same time, a body of indirect evidence for the existence of such a mechanism in vivo can be found in the literature. A hallmark of the splicing mechanism is that the 3′ terminus of an attacking substrate is preserved within the recombinant sequence. This is especially easy to discern if the sequence includes the 3′ terminus of an intact RNA, and such recombinants have been reported for viruses of all types of organisms. Qβ phage satellite RQ120 RNA has been generated by the addition of the 3′ terminus of E. coli tRNAAsp 1 to the last quarter of the Qβ coat protein cistron (Munishkin et al. 1988). A number of Sindbis virus DI genomes include the 3′ terminus of rat tRNAAsp attached to the beginning of the viral genome sequence (Monroe and Schlesinger 1983). Also, one half of the experimental recombinants between the genome of turnip crinkle virus and its satellite (sat-D) RNA have been generated by the addition of the 3′ terminus of sat-D to the 5′ base of a hairpin in the genomic 3′-terminal untranslated region (Carpenter et al. 1995), which is reminiscent of the secondary structure requirements for the in vitro recombination.
Thus, the splicing-like mechanism does not seem to be incompatible with the existing in vivo data, and it was observed in vitro under conditions not too different from those existing in the cell. We believe that such a mechanism does operate in vivo, bringing about nonhomologous recombination between RNA molecules, both viral and cellular. Although recombination between cellular RNAs has not yet been experimentally demonstrated, apparently because of the absence of a suitable experimental system, the sequencing of some RQ RNA molecules has shown that they consist of fragments of cellular RNAs only (28, 25). Our data support the hypothesis that the modern splicing systems, together with the exon-intron structure of genes, have evolved from the ability of cellular RNA to recombine (Gilbert 1986). The described in vitro system could provide for direct evolutionary experiments on this matter.
Experimental Procedures
RNA Fragments
The 5′ and 3′ fragments were constructed at the DNA level utilizing the selective PCR amplification of the corresponding regions of plasmid pT7RQ135−1(−) comprising a T7 promoter/RQ135−1(−) cDNA cartridge inserted into plasmid pUC18 (Morozov et al. 1993). The 5′ fragment was amplified with primer 1 (5′-CTGCAGGCATGCAAGCTTAATACGACT-3′, partially overlapping the sequence of the T7 promoter and containing site HindIII) and primer 2 (5′-AGACTCGAGCTGCAGAAGGGACGCACG-3′, complementary to positions 41–52 of the RQ135−1(−) sequence and containing site PstI). The 3′ fragment was amplified with primer 3 (5′-CGCTGCAGCTCGAGTCTAGAGGATCCTACGAGGGATTTGA-3′, matching positions 53–66 of the RQ135−1(−) sequence and introducing the foreign extension) and primer 4 (5′-CCGCGGATATCGATCCCGGGCTAACAGTG-3′, complementary to the 3′ terminus of the RQ135−1(−) sequence and containing site SmaI), and then extended at the 5′ end in two additional PCRs by employing, consecutively, primer 5 (5′-AAGCTTAATACGACTCACTATAGGCGCTGCAGCTCGAGT-3′, introducing the T7 promoter and restriction site HindIII) and primer 6 (5′-CTGCAGGCATGCAAGCTTAATACGACT-3′, extending the sequence upstream from HindIII), each in combination with primer 4. The amplified fragments were digested by restriction endonucleases HindIII and PstI (5′ fragment) or HindIII and SmaI (3′ fragment), and cloned within the pUC18 vector between the corresponding sites. The primary structures of the resulting constructs were checked by sequencing. RNA fragments used for recombination experiments were synthesized with T7 RNA polymerase (Noren et al. 1990) after digesting the plasmids at site BamHI located in the pUC18 polylinker downstream from PstI (5′ fragment) or SmaI (3′ fragment), and purified by electrophoresis through denaturing polyacrylamide gel (Milligan and Uhlenbeck 1989).
Where indicated, the fragments were modified at the 3′ end as follows. For oxidation (Steinschneider and Fraenkel-Conrat 1966a), 4 μg of RNA were incubated in the dark for 60 min at 25°C in 40 μl of a buffer (100 mM Na-acetate [pH 5.3], 10 mM EDTA) containing 35 mM NaIO4. The extent of modification was nearly 100%, judging by a streptavidin-induced gel-shift of the RNA biotinylated at the oxidized end. The oxidized RNA was used for β-elimination, essentially according to Steinschneider and Fraenkel-Conrat 1966b: 2 μg RNA were incubated for 3 hr at 25°C in 60 μl of a buffer containing 10 mM Na-acetate and 0.25 M aniline, and adjusted to pH 5.0 with acetic acid. The 3′ phosphate was removed by incubation of 1 μg of the aniline-treated RNA for 1 hr at 50°C in 20 μl of a buffer (25 mM Tris-HCl [pH 8.0], 0.1 mM EDTA) containing 2.5 units of calf intestine alkaline phosphatase (molecular biology grade, Boehringer Mannheim) with a subsequent phenol extraction and ethanol precipitation.
RNA Recombination Experiments
RNA fragments were preincubated in a buffer (80 mM Tris-HCl [pH 7.8], 1 mM EDTA, 10% glycerol, 100 mM KCl, ± 10 mM MgCl2) at 20°C during the indicated time period. A 10 μl aliquot containing the specified number of RNA molecules was spread over a 0.37 mm-thick square (18 × 18 mm) of a 2% Type IX agarose (Sigma) slab prepared in a buffer (80 mM Tris-HCl [pH 7.8], 2 mM MgCl2, 1 mM EDTA, 20% glycerol) containing, in a volume of 120 μl, 4.2 μg of a homogeneous preparation of Qβ replicase isolated as described (4, 1). The agarose was then covered with a 19 × 19 mm dried nylon membrane (Biodyne A, Pall BioSupport) containing 720 nmol MgCl2 and 120 nmol each of ATP, GTP, CTP, and UTP adjusted to pH 7.8 with Tris-OH. The final concentrations of MgCl2 and nucleotides in the medium were 8 mM and 1 mM, respectively. RNA colonies were grown during 60 min at 20°C. A replica membrane (Hybond N, Amersham) prepared by blotting the agarose for 10 min at 20°C was hybridized at 65°C in a 50% formamide buffer with the 5′ fragment of RQ135−1(−) RNA labeled by transcription of the corresponding plasmid in the presence of 50 μM [α-32P]CTP (60 Ci/mmol), as described earlier (Chetverina and Chetverin 1993). Alternatively, the original substrate membrane was hybridized at 50°C in a 20% formamide buffer with primer 3 (see above) labeled by terminal deoxynucleotidyl transferase in the presence of 5 μM [α-32P]dATP (750 Ci/mmol), according to a Promega protocol. Other details of the procedure are given elsewhere (Chetverina and Chetverin 1993).
Cloning of Recombinant Sequences
Total RNA from an agarose slab containing at least 100 recombinant RNA colonies was extracted with phenol and served as a template for cDNA synthesis by AMV reverse transcriptase, according to a Promega protocol, utilizing primer 4 (see above) complementary to the 3′ terminus of RQ135−1(−) RNA. cDNA was then amplified in a two-step PCR by employing, at the first step, primer 4 and 7 (5′-AAGCTTAATACGACTCACTATAGGGGTTCCAACCGGAAG-3′, matching the 5′ end of RQ135−1(−) RNA and including the T7 promoter sequence) and, at the second step, primer 1 and 4. PCR products were digested at sites HindIII and SmaI and inserted between the same sites in the pUC18 vector. Plasmids isolated from randomly picked clones were subjected to alkaline denaturation and sequenced by the dideoxy chain termination method using the Sequenase Version 2.0 (USB) DNA sequencing kit and protocol.
Amplification of Recombinant RNAs in Solution
Plasmids carrying recombinant sequences were digested by restriction endonuclease SmaI and used as templates for RNA synthesis by T7 RNA polymerase (Noren et al. 1990). A specified amount of phenol-extracted RNA was used to initiate RNA synthesis in 10 μl of a buffer (80 mM Tris-HCl [pH 7.8], 8 mM MgCl2, 1 mM EDTA, 20% glycerol), containing 0.25 μg of Qβ replicase and each of ATP, GTP, CTP, and UTP at a concentration of 1 mM. After a 10 min incubation at 20°C, the reaction was terminated by adding 20 μl of denaturing solution (10 mM Tris-HCl [pH 8.0], 30 mM EDTA, 0.05% SDS, 90% formamide, and 0.1% each of xylene cyanol and bromophenol blue) and heating for 2 min in a boiling bath. The samples were subjected to electrophoresis through a 5% polyacrylamide gel in the presence of 7M urea.
Acknowledgements
Correspondence should be addressed to A. B. C. We thank Prof. V. I. Agol, Prof. A. S. Spirin and Dr. E. Pilipenko for critical discussions, Dr. A. V. Finkelstein for advice in statistical estimates, V. S. Prisyazhnoy for help and advice in the experiments on terminal RNA modifications, and N. I. Androsova and I. I. Naumova for technical assistance. This work was supported by grants MTO 000 and MTO 300 from the International Science Foundation and the Russian Government, grants 93-04-06550 and 96-04-48331 from the Russian Foundation for Basic Science, grant INTAS-RFBR 95-1365, and by an International Research Scholar's award from the Howard Hughes Medical Institute to A. B. C.
References
- 1.Berestowskaya N.H., Vasiliev V.D., Volkov A.A., Chetverin A.B. Electron microscopy study of Qβ replicase. FEBS Lett. 1988;228:263–267. doi: 10.1016/0014-5793(88)80012-7. [DOI] [PubMed] [Google Scholar]
- 2.Biebricher C.K., Diekmann S., Luce R. Structural analysis of self-replicating RNA synthesized by Qβ replicase. J. Mol. Biol. 1982;154:629–648. doi: 10.1016/s0022-2836(82)80019-3. [DOI] [PubMed] [Google Scholar]
- 3.Biebricher C.K., Luce R. In vitro recombination and terminal elongation of RNA by Qβ replicase. EMBO J. 1992;11:5129–5135. doi: 10.1002/j.1460-2075.1992.tb05620.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blumenthal T. Qβ RNA replicase and protein synthesis elongation factors EF-Tu and EF-Ts. Methods Enzymol. 1979;60:628–638. doi: 10.1016/s0076-6879(79)60059-9. [DOI] [PubMed] [Google Scholar]
- 5.Carpenter C.D., Oh J.-W., Zhang C., Simon A.E. Involvement of a stem-loop structure in the location of junction sites in viral RNA recombination. J. Mol. Biol. 1995;245:608–622. doi: 10.1006/jmbi.1994.0050. [DOI] [PubMed] [Google Scholar]
- 6.Cech T.R., Bass B.L. Biological catalysis by RNA. Ann. Rev. Biochem. 1986;55:599–629. doi: 10.1146/annurev.bi.55.070186.003123. [DOI] [PubMed] [Google Scholar]
- 7.Chetverin A.B., Chetverina H.V., Munishkin A.V. On the nature of spontaneous RNA synthesis by Qβ replicase. J. Mol. Biol. 1991;222:3–9. doi: 10.1016/0022-2836(91)90729-p. [DOI] [PubMed] [Google Scholar]
- 8.Chetverin A.B., Voronin L.A., Munishkin A.V., Bondareva L.A., Chetverina H.V., Ugarov V.I. Recombination in replicating RNAs. In: Todd P., Sikdar S.K., Bier M., editors. Frontiers in Bioprocessing II. American Chemical Society; Washington, DC: 1992. pp. 44–49. [Google Scholar]
- 9.Chetverin, A.B., and Spirin, A.S. (1995). Replicable RNA vectors: prospects for cell-free gene amplification, expression and cloning. In Progress in Nucleic Acid Research and Molecular Biology, Vol. 51, W.E. Cohn and K. Moldave, eds. (San Diego, California: Academic Press), pp. 225–270. [DOI] [PubMed]
- 10.Chetverina H.V., Chetverin A.B. Cloning of RNA molecules in vitro. Nucleic Acids Res. 1993;21:2349–2353. doi: 10.1093/nar/21.10.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cooper P.D., Steiner-Pryor S., Scotti P.D., Delong D. On the nature of poliovirus genetic recombinants. J. Gen. Virol. 1974;23:41–49. doi: 10.1099/0022-1317-23-1-41. [DOI] [PubMed] [Google Scholar]
- 12.Dzianott A., Flasinski S., Bujarski J.J. Foreign complementary sequences facilitate genetic RNA recombination in brome mosaic virus. Virology. 1995;208:370–375. doi: 10.1006/viro.1995.1163. [DOI] [PubMed] [Google Scholar]
- 13.Gilbert W. The RNA world. Nature. 1986;319:618. [Google Scholar]
- 14.Hajjou M., Hill K., Subramaniam S.V., Hu J.Y., Raju R. Nonhomologous RNA-RNA recombination events at the 3′ nontranslated region of the Sindbis virus genome: hot spots and utilization of nonviral sequences. J. Virol. 1996;70:5153–5164. doi: 10.1128/jvi.70.8.5153-5164.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hirst G.K. Genetic recombination with Newcastle disease virus, poliovirus and influenza virus. Cold Spring Harbor Symp. Quant. Biol. 1962;27:303–309. doi: 10.1101/sqb.1962.027.001.028. [DOI] [PubMed] [Google Scholar]
- 16.Igloi G.L. A silver stain for the detection of nanogram amounts of tRNA following two-dimensional electrophoresis. Anal. Biochem. 1983;134:184–188. doi: 10.1016/0003-2697(83)90281-6. [DOI] [PubMed] [Google Scholar]
- 17.Jarvis T.C., Kirkegaard K. Poliovirus RNA recombination: mechanistic studies in the absence of selection. EMBO J. 1992;11:3135–3145. doi: 10.1002/j.1460-2075.1992.tb05386.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kirkegaard K., Baltimore D. The mechanism of RNA recombination in poliovirus. Cell. 1986;47:433–443. doi: 10.1016/0092-8674(86)90600-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kuge S., Saito I., Nomoto A. Primary structure of poliovirus defective-interfering particle genomes and possible generation mechanisms of the particles. J. Mol. Biol. 1986;192:473–487. doi: 10.1016/0022-2836(86)90270-6. [DOI] [PubMed] [Google Scholar]
- 20.Lai M.M.C. RNA recombination in animal and plant viruses. Microbiol. Rev. 1992;56:61–79. doi: 10.1128/mr.56.1.61-79.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ledinko N. Genetic recombination with poliovirus type 1: studies of crosses between a normal horse serum-resistant mutant and several guanidine-resistant mutants of the same strain. Virology. 1963;180:107–119. doi: 10.1016/0042-6822(63)90145-4. [DOI] [PubMed] [Google Scholar]
- 22.Milligan J.F., Groebe D.R., Witherell G.W., Uhlenbeck O.C. Oligonucleotide synthesis using T7 RNA polymerase and synthetic DNA templates. Nucleic Acids Res. 1987;15:8783–8798. doi: 10.1093/nar/15.21.8783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Milligan J.F., Uhlenbeck O.C. Determination of RNA-protein contacts using thiophosphate substitutions. Biochemistry. 1989;28:2849–2855. doi: 10.1021/bi00433a016. [DOI] [PubMed] [Google Scholar]
- 24.Monroe S.S., Schlesinger S. RNAs from two independently isolated defective interfering particles of Sindbis virus contain a cellular tRNA sequence at their 5′-ends. Proc. Natl. Acad. Sci. USA. 1983;80:3279–3283. doi: 10.1073/pnas.80.11.3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moody M.D., Burg J.L., DiFrancesco R., Lovern D., Stanik W., Lin-Goerke J., Mahdavi K., Wu Y., Farrel M.P. Evolution of host cell RNA into efficient template RNA by Qβ replicase: the origin of RNA in untemplated reactions. Biochemistry. 1994;33:13836–13847. doi: 10.1021/bi00250a038. [DOI] [PubMed] [Google Scholar]
- 26.Morozov I.Y., Ugarov V.I., Chetverin A.B., Spirin A.S. Synergism in replication and translation of messenger RNA in a cell-free system. Proc. Natl. Acad. Sci. USA. 1993;90:9325–9329. doi: 10.1073/pnas.90.20.9325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Munishkin A.V., Voronin L.A., Chetverin A.B. An in vivo recombinant RNA capable of autocatalytic synthesis by Qβ replicase. Nature. 1988;333:473–475. doi: 10.1038/333473a0. [DOI] [PubMed] [Google Scholar]
- 28.Munishkin A.V., Voronin L.A., Ugarov V.I., Bondareva L.A., Chetverina H.V., Chetverin A.B. Efficient templates for Qβ replicase are formed by recombination from heterologous sequences. J. Mol. Biol. 1991;221:463–472. doi: 10.1016/0022-2836(91)80067-5. [DOI] [PubMed] [Google Scholar]
- 29.Nagy P.D., Bujarski J.J. Targeting the site of RNA-RNA recombination in brome mosaic virus with antisense sequences. Proc. Natl. Acad. Sci. USA. 1993;90:6390–6394. doi: 10.1073/pnas.90.14.6390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Negroni M., Riccheti M., Nouvel P., Buc H. Homologous recombination by reverse transcriptase during copying of two distinct RNA templates. Proc. Natl. Acad. Sci. USA. 1995;92:6971–6975. doi: 10.1073/pnas.92.15.6971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Noren C.J., Anthony-Cahill S.J., Suich D.J., Noren K.A., Griffith M.C., Schultz P.G. In vitro suppression of an amber mutation by a chemically aminoacylated transfer RNA prepared by runoff transcription. Nucleic Acids Res. 1990;18:83–88. doi: 10.1093/nar/18.1.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Palasingam K., Shaklee P.N. Reversion of Qβ phage mutants by homologous RNA recombination. J. Virol. 1992;66:2435–2442. doi: 10.1128/jvi.66.4.2435-2442.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Romanova L.I., Blinov V.M., Tolskaya E.A., Viktorova E.G., Kolesnikova M.S., Guseva E.A., Agol V.I. The primary structure of crossover regions of intertypic poliovirus recombinants: a model of recombination between RNA genomes. Virology. 1986;155:202–213. doi: 10.1016/0042-6822(86)90180-7. [DOI] [PubMed] [Google Scholar]
- 34.Steinschneider A., Fraenkel-Conrat H. Studies of nucleotide sequences in tobacco mosaic virus ribonucleic acid. III. Periodate oxidation and semicarbazone formation. Biochemistry. 1966;5:2729–2734. doi: 10.1021/bi00872a033. [DOI] [PubMed] [Google Scholar]
- 35.Steinschneider A., Fraenkel-Conrat H. Studies of nucleotide sequences in tobacco mosaic virus ribonucleic acid. IV. Use of aniline in stepwise degradation. Biochemistry. 1966;5:2735–2743. doi: 10.1021/bi00872a034. [DOI] [PubMed] [Google Scholar]
- 36.Tolskaya E.A., Romanova L.I., Blinov V.M., Viktorova E.G., Sinyakov A.N., Kolesnikova M.S., Agol V.I. Studies on the recombination between RNA genomes of poliovirus: the primary structure and nonrandom distribution of crossover regions in the genomes of intertypic poliovirus recombinants. Virology. 1987;161:54–61. doi: 10.1016/0042-6822(87)90170-x. [DOI] [PubMed] [Google Scholar]
- 37.Weissmann C. The making of a phage. FEBS Lett. 1974;40:S10–S18. doi: 10.1016/0014-5793(74)80684-8. [DOI] [PubMed] [Google Scholar]
- 38.Zuker M., Stiegler P. Optimal computer folding of large RNA sequences using thermodynamics and auxiliary information. Nucleic Acids Res. 1981;9:133–148. doi: 10.1093/nar/9.1.133. [DOI] [PMC free article] [PubMed] [Google Scholar]