

Abstract
Reverse transcription requires two replicative template switches, called minus and plus strand strong stop transfer, and can include additional, recombinogenic switches. Donor and acceptor template homology facilitates both replicative and recombinogenic transfers, but homology-independent determinants may also contribute. Here, improved murine leukemia virus-based assays were established and the effects of varying extents of mismatches and complementarity between primer and acceptor template regions were assessed. Template switch accuracy was addressed by examining provirus structures, and efficiency was measured using a competitive titer assay. The results demonstrated that limited mismatch extension occurred readily during both minus and plus strand transfer. A strong bias for correct targeting to the U3/R junction and against use of alternate regions of homology was observed during minus strand transfer. Transfer to the U3/R junction was as accurate with five bases of complementarity as it was with an intact R, and as few as 3 nt targeted transfer to a limited extent. In contrast, 12 base recombinogenic acceptors were utilized poorly and no accurate switch was observed when recombination acceptors retained only five bases of complementarity. These findings confirm that murine leukemia virus replicative and recombinogenic template switches differ in homology requirements, and support the notion that factors other than primer–template complementarity may contribute to strong stop acceptor template recognition.
Keywords: reverse transcription, fidelity, genetic recombination
Abbreviations: pbs, primer binding site; −sssDNA, minus strand strong stop DNA
1. Introduction
Two obligate template switches, minus and plus strand transfer, occur during the synthesis of every retroviral DNA.1., 2. Minus strand synthesis initiates from the primer binding site (pbs) near the 5′ end of viral RNA and continues to the genome's 5′ end, generating a replication intermediate called minus strand strong stop DNA (−sssDNA; Figure 1(A)). Transfer of −sssDNA to the U3/R junction at the 3′ end of viral RNA allows continued minus strand synthesis. This transfer is aided by complementarity between −sssDNA and repeated regions called R, which are present at both the 5′ and 3′ ends of genomic RNA. Retroviral R repeats differ in length from 247 nt for human T-cell leukemia virus type 2 to only 16 nt in mouse mammary tumor virus.3 Human immunodeficiency virus type 1 (HIV-1) and Moloney murine leukemia virus (MLV) have 97 and 69 base R regions, respectively. Even for viruses with short Rs, minus strand transfer is apparently very efficient, since no detectable abortive −sssDNA accumulates in infected cells unless variants defective in strand transfer, such as RNaseH mutants, are studied.4., 5., 6. Short Rs suffice for some retroviruses, and incomplete Rs can support minus strand transfer.7., 8., 9., 10., 11. Thus, the whole length of complementarity present in most viral Rs is not necessary for minus strand transfer.
Figure 1.

Retroviral replication cycle. (A) Two obligate strand transfers, minus and plus strand transfer, must occur during reverse transcription. Thin lines, viral RNA; thick lines, proviral DNA; broken lines indicate RNA degraded from RNA/DNA duplexes by RNase H. (B) Recombinogenic switching during reverse transcription generates a recombinant provirus containing genetic markers from two separate templates.
Reverse transcription also requires a second replicative switch, called plus strand transfer (Figure 1(A)). This occurs when the initial product of plus strand synthesis, plus strand strong stop DNA (+sssDNA), is relocated from its site of synthesis on the primer tRNA to complementary sequences on the nascent minus strand DNA. There are 18 bases of complementarity between +sssDNA and its acceptor on minus strand DNA. Some modified templates with limited regions of homology can support plus strand transfer.12., 13., 14.
Additional template switches between genetically distinct RNAs can occur and generate recombinant viral genomes (Figure 1(B)). Although this later type of template switching has been viewed as non-essential, emerging data suggest that recombinogenic switching ordinarily occurs during most rounds of viral DNA synthesis.15., 16. It has been suggested that the reason reverse transcription is highly recombinogenic is that the need to perform replicative template switches provided a selective advantage for reverse transcriptase (RT) to become prone to recombinogenic switching as well.17., 18. Although some sequences may serve as recombination “hot” or “cold” spots, recombinogenic switching occurs at essentially random positions throughout templates.19., 20., 21., 22. Recombination frequency increases in rough proportion to the length of identical sequence on donor and acceptor templates23 and declines with decreasing extents of sequence similarity.15 Thus, sequence complementarity between the DNA synthesized on a donor template and the acceptor template region to which switching occurs is an important determinant of recombinogenic template switching.
Mounting evidence supports the notion that a more complex mechanism of acceptor recognition than mere primer–template complementarity may contribute to replicative strand transfer. One study demonstrated that although a repeated non-viral sequence of 85 nt can direct minus strand transfer, titers for non-viral repeat vectors are significantly lower than those for vectors with the native 69 nt MLV R.24 In purified reactions in vitro, both MLV and HIV-1 RT are unable to support efficient strand transfer of donor/acceptor pairs that contain heterologous R sequences (MLV RT+HIV-1 R donor/acceptor and vice versa).25., 26. Artificial R sequences are also unable to support MLV strand transfer in vitro. 27 Research from our own laboratory suggests that complementarity-independent features of the MLV U3/R junction region may contribute to −sssDNA targeting.28
The present report extends our previous work on minus strand transfer. The initial experiments established an improved assay designed to eliminate the major classes of background products observed in previous work9., 28., 29. and provided evidence that such artifacts likely affected some previous work. This new assay system was next used to study mismatch extension during minus strand transfer, and to assess the effects of limited primer-terminal complementarity on acceptor template use. Separate vectors were developed to examine plus strand transfer and recombinogenic transfer, and homology requirements for minus strand transfer were compared to those for recombinogenic switching. The data indicated that minus strand transfer and recombinogenic template switching are not equivalent events in terms of homology requirements. The findings demonstrated that targeting of minus strand transfer to the U3/R junction is aided by cis-acting determinants that allow accurate and efficient template switching in the presence of far less complementarity than that which is required during recombination.
2. Results
2.1. Improved assay to assess minus strand transfer outcomes
The study of minus strand transfer requires that two repeated sequences (designated R in Figure 2) be present on each template to act as donor and acceptor template regions. During replication in cultured cells, most MLV minus strand transfer occurs only after RT reaches the 5′ end of the donor repeat.30 However, other outcomes, such as mismatch extension, premature jump, or abortive replication (Figure 2(B)), have been reported among minus strand transfer products when acceptor and donor template regions differ.3., 11., 28., 31.
Figure 2.
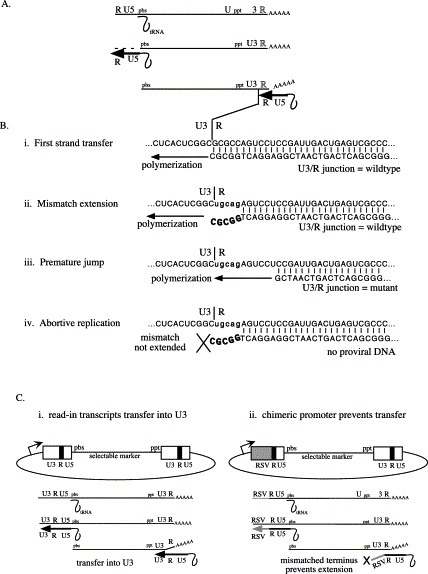
Outcomes of mismatch extension during minus strand transfer. (A) Transfer of minus strand strong stop DNA during reverse transcription. Hybridization, during minus strand transfer, of RNA sequences of the acceptor (top strand) and DNA sequences of the donor (bottom strand) at the U3/R junction. (B) Outcomes of mismatch extension. (i) Transfer from matched donor to acceptor of the parental RSVpuro vector. (ii) Transfer from parental donor to mutant acceptor containing five terminal mismatches, followed by mismatch extension. Parental sequences are regenerated following mismatch extension. (iii) Premature transfer from donor prior to completion of minus strand strong stop DNA. Mutant sequences are copied following transfer. (iv) Transfer from parental donor to mutant acceptor cannot be extended, resulting in abortive replication. (C) RSVpuro eliminates artifacts associated with vectors containing two identical LTRs. (i) Read-in transcripts from MLVpuro initiated upstream of the 5′ LTR transfer to the 3′ LTR using sequences within U3 as donor and acceptor. (ii) Read-in transcripts from RSVpuro cannot transfer to the 3′ LTR due to mismatches between U3 sequences.
In previous studies, we evaluated minus strand transfer using an MLV-based vector that was constructed from a proviral clone and contained a puromycin resistance cassette, which is referred to as pMLVpuro in this study (Figure 3(A)).28 Upon transfection into mammalian cells, pMLVpuro produces an RNA with a puromycin resistance cassette in place of virus coding sequences, and thus replication of this vector is limited to a single round. Identical R repeats at both ends of MLVpuro RNA serve as strand transfer donor and acceptor sequences. Our previous work examined reverse transcription outcomes for MLVpuro derivatives that contained mutations in the 3′ R. Integrated products of these derivatives were analyzed to determine the mutations' effects on strand transfer.28
Figure 3.

RSVpuro eliminates detectable levels of plasmid recombination. (A) Plasmid constructs used to compare plasmid recombination. Regions required for vector RNA expression are illustrated. Not to scale. (B) Comparison of RSVpuro and MLVpuro vectors. Proviral LTR sequences were amplified using primers SF223 and SF213 and subjected to restriction analysis. M, molecular weight markers; MLVpuro: undigested (lane 1), AscI (lane 2); RSVpuro: undigested (lane 3), AscI (lane 4); MLV5TMM: undigested (lane 5), AscI (lane 6), PstI (lane 7), SfoI (lane 8); RSV5TMM: undigested (lane 9), AscI (lane 10), PstI (lane 11), SfoI (lane 12). (C) Minus strand transfer with tandem acceptor sequences. Proviral LTR sequences were amplified with SF210 and SF213 and subjected to restriction analysis. RSVpuro: undigested (lane 1), AscI (lane 2); RSVtandemR: undigested (lane 3), AscI (lane 4); RSVtandem5TMM: undigested (lane 5), AscI (lane 6), PstI (lane 7).
A weakness of the MLVpuro assay system is the potential for background that masks legitimate reverse transcription products when titers of the latter are low. One cause of background is plasmid homologous recombination between LTRs during transfection, which can result in the synthesis of RNAs with wild-type sequences in place of engineered 3′ end mutations.32 Even though such recombinants arise rarely, their RNAs are reverse transcribed efficiently, and thus they generate a wild-type background that obscures low-frequency strand transfer events.28 Wild-type background can also result from the reverse transcription of rare read-in transcripts (Figure 2(C)).9., 29.
Here, a new assay vector was developed to eliminate both of these artifacts. Its key feature was a vast reduction in sequences that could support plasmid-level recombination or allow template switching on extended read-in transcripts. For this new chimeric MLV-based retroviral construct, called pRSVpuro (Figure 3(A)), the MLV U3 promoter that drives vector transcription was replaced with a Rous sarcoma virus promoter. pRSVpuro was identical to pMLVpuro except for the RSV promoter in the 5′ LTR. pMLVpuro and pRSVpuro were designed to produce identical RNAs when transfected into mammalian cells. The 5′ end of viral RNAs transcribed from these plasmids in transfected cells were determined by primer extension analysis, and the transcription start site for pRSVpuro was indistinguishable from that of pMLVpuro (data not shown).
Mismatch extension was tested in the new vector system, using a pRSVpuro derivative (pRSV5TMM) with five terminal mismatched nucleotides at the U3/R junction in the 3′ LTR (Figure 3(A)). The equivalent pMLVpuro construct (pMLV5TMM) had been tested previously, and structures judged diagnostic of mismatch extension were observed in 33% of the strand transfer products.28
In this study, vector plasmids were transiently transfected into ΦNXE packaging cells. These cells lack ecotropic receptor and thus cannot be infected by ecotropic particles. Particle-containing media were collected from transfected ΦNXE cells and used to infect D17/pJET cells. D17/pJET is a canine cell line that expresses ecotropic receptor.33., 34. Infected cells were selected with puromycin and pooled, and integrated proviruses were analyzed from cellular DNA by PCR and restriction analysis.
MLVpuro, RSVpuro, MLV5TMM, and RSV5TMM products are compared in Figure 3(B). The nature of minus strand transfer could be addressed by restriction analysis because the MLV U3/R junction contains a fortuitous AscI site (see Figure 3(A)). If strand transfer occurred precisely at the U3/R junction, an AscI site would be regenerated. If anomalous transfer occurred, the AscI site would be destroyed. For both MLVpuro and RSVpuro, >99% of the PCR products were digested with AscI (Figure 3(B), lanes 2 and 4), indicating that both constructs supported accurate minus strand transfer. However, products of the mismatched derivatives, MLV5TMM and RSV5TMM, differed significantly. The MLV5TMM construct contained a single point mutation 5′ of the U3/R junction in the upstream LTR, which converted the AscI site to an SfoI site (Figure 3(A)). Thus, digestion of integrated products with SfoI was diagnostic of MLV5TMM read-in or plasmid recombination-generated artifacts (Figure 2(C)) while digestion with PstI indicated premature jump (strand transfer occurring before minus strand strong stop DNA was completed: see Figure 2(B)), and digestion with AscI would indicate mismatch extension. For MLV5TMM, none of the product was digested by AscI while approximately 50% was digested with PstI and 50% was digested with SfoI (Figure 3(B), lanes 6–8). In contrast, products of RSV5TMM showed nearly 100% cleavage with PstI and no detectable cleavage with AscI or SfoI (Figure 3(B), lanes 10–12). This indicated that a significant portion of the MLV5TMM products were artifacts with the remainder premature jump, while premature jump occurred in nearly all the strand transfer events observed for RSV5TMM. No mismatch extension (digestion with AscI) was evident among products of either vector.
Subsequent experiments examined RSVpuro derivatives containing tandem Rs like those analyzed previously.28 Tandem R constructs contained two acceptors (R1 and R2) in the downstream LTR (Figure 3(A)), either of which could be used during minus strand transfer. If mutations were inserted in R1 at the U3/R1 junction, the use of R1 versus R2 would indicate how well the mutant acceptor competed with the wild-type.
In previous MLVpuro-based tandem R studies, when both acceptor Rs were wild-type, an analysis of products suggested both Rs were used to very similar extents, even though R1 contained only the first 46 nt of R while R2 contained all 69 nt of the native MLV R.28 When five terminal mismatches were introduced into R1, products suggestive of transfer to R1 were decreased by two-thirds, but the five base mismatched R1 did appear to be used roughly 33% as often as the fully matched acceptor.28
Here, these same tandem Rs were tested in the RSVpuro system. In modest contrast to the ∼50% observed for MLVpuro-based vectors, R2 was used during minus strand transfer 63% of the time for the RSV-based vector with wild-type sequences at both the 46 nt R1 and the 69 nt R2 (Figure 3(C), lane 3). However, a mismatched construct (RSVtandem5TMM) generated results very different from those of the mismatched MLVpuro tandem vector. No transfer to the mismatched R1 occurred and only transfer to the wild-type R2 was observed for RSVtandem5TMM (Figure 3(C), lane 5). No digestion with AscI was detected and essentially 100% digestion with PstI was observed, as expected for transfer to R2 (Figure 3(C), lanes 6 and 7).
Among controls performed for RSVpuro were tests of the frequency of the previously described minus strand transfer-associated acquisition of mutations at the U3/R junction.30 These mutations, which we have called +1G, likely result from non-templated nucleotide addition to −sssDNA followed by mismatch extension upon strand transfer.30 The +1G mutation destroys the AscI site and generates a sequence that is recognized by SfoI. As part of the current study, we digested reverse transcription products generated by wild-type infectious MLV with SfoI. The SfoI-digestible +1G product was detectable on long exposure (data not shown) albeit in lower abundance than the ∼5% frequency that we reported previously.30 However, using the single-round replication assay described in this report, the SfoI-digestible +1G product was not detected among products of either RSVpuro or MLVpuro (data not shown). Instead, AscI digestion of RSVpuro and MLVpuro products, which indicated the absence of +1G mutations, was essentially complete (see, for example, Figure 3(B), lanes 2 and 4, and Figure 3(C), lane 2). We therefore conclude that the +1G product should not significantly diminish the extent of AscI cutting, which was used to diagnose precise transfer at the U3/R junction in this report.
Taken together, results from the tandem and single R mismatch vectors confirmed that products of non-viral processes like plasmid recombination can contribute unwanted background when low-titer vectors are examined using previous assay approaches. Some outcomes that we previously interpreted as low-titer mismatch extension28 were likely due to read-in transcription or plasmid level recombination. The current findings demonstrated that the RSV constructs adopted for this study greatly reduced these artifacts to levels below detection, even for vectors with substantial primer-terminal mismatch.
2.2. Mismatch extension during minus strand transfer
The experiments above demonstrated that five bases of terminal mismatch at the U3/R junction cannot be extended detectably. Follow up work examined whether or not lesser extents of mismatch could be extended. RSVpuro variants that contained one, two, three, four or five terminal mismatches in R at the downstream U3/R junction were generated (Figure 4(B)). This series of vectors was designated “TMM”, for terminal mismatch. Each vector (1TMM, 2TMM, 3TMM, 4TMM, and 5TMM) contained a diagnostic restriction site (FauI, NheI, Fnu4HI, MseI, or PstI, respectively) introduced with the mismatch mutations. It was anticipated that one or more of the outcomes described in Figure 2(B) would result from minus strand transfer between mismatched donors and acceptors. These include mismatch extension, strand transfer prior to the completion of −sssDNA (premature jump), or no extension resulting in abortive synthesis. If mismatch extension occurred, the U3/R Asc I site would be generated from transfer of the 5′R sequences. If premature jump occurred, the restriction site that tagged the mutant sequence present in the 3′R would be present in the reverse transcription product. And finally, titer reductions and/or transfer to sites other than the U3/R junction would be seen if no extension occurred.
Figure 4.

Mismatch extension during minus strand transfer. (A) RSVpuro plasmid and vector RNA are illustrated. DNA is shown as boxes and continuous lines. RNA is shown as a broken line. (B) RNA sequences at the U3/R junction for RSVpuro and terminal mismatch derivatives containing 1, 2, 3, 4, or 5 mismatched nucleotides (1TMM, 2TMM, 3TMM, 4TMM, and 5TMM, respectively). Mismatches are shown in lower case. Diagnostic enzymes in the mutant sequences are underlined: 1TMM, FauI; 2TMM, NheI; 3TMM, Fnu4HI; 4TMM, MseI; 5TMM, PstI. (C) Proviral DNAs were PCR amplified using primers SF210 and SF212 and subjected to restriction digestion. RSVpuro: undigested (lane 1), AscI (lane 2); 1TMM: undigested (lane 3), AscI (lane 4), FauI (lane 5); 2TMM: undigested (lane 6), AscI (lane 7), NheI (lane 8); 3TMM: undigested (lane 9), AscI (lane 10), Fnu4HI (lane 11); 4TMM: undigested (lane 12), AscI (lane 13), MseI (lane 14); 5TMM: undigested (lane 15), AscI (lane 16), PstI (lane 17). Alternate target, A.T. (D) Mismatch extension (AscI digestion) was quantified by phosphorimager analysis. Percentage mismatch extension is the amount of product digested with AscI compared to the amount of product that was not digested.
Product analysis is shown in Figure 4. One, two, or three mismatches (Figure 4(C); lanes 4, 7, and 10) were extended to varying extents, as determined by digestion with AscI. Mismatch extension occurred in 48% of the strand transfers analyzed for the 1TMM vector, 33% for the 2TMM vector, and 14% for the 3TMM vector (Figure 4(D)). Premature jump was observed among products for one, two, three, four and 5 mismatches (Figure 4(C); lanes 5, 8, 11, 14, and 17), as determined by digestion with the diagnostic enzymes. The detection of alternate-length products suggests vectors with four and five mismatches exhibited alternate targeting in addition to premature jump (Figure 4(C), lanes12–17), but mismatch extension occurred in less than 2% of the analyzed proviruses (Figure 4(C), lanes 13 and 16, and Figure 4(D)).
2.3. Titer defects of mutant vectors
Puromycin resistance titers were determined using a vector competition assay (Table 1). To do this, each mutant construct was individually co-transfected into packaging cells at varying molar ratios with pRSVpuroM/B, a derivative of pRSVpuro that maintained wild-type titer but contained diagnostic restriction sites (MfeI and BamHI). Media harvested from co-transfected packaging cells was presumed to contain viral particles with mutant and RSVpuroM/B internal standard RNAs present in the same ratios as the mutant to internal standard plasmid ratios transfected. The relative success of reverse transcription and integration for mutant vectors was evaluated by PCR amplifying proviral sequences from pooled genomic DNA after selection with puromycin, followed by restriction analysis to quantify ratios of mutant versus internal standard products. This method's accuracy and reproducibility were assessed using vectors with known titers (data not shown). Table 1 shows titers determined using this approach, which are presented as order of magnitude reduction compared to the internal standard. A wild-type titer (10°) indicates that the mutant competed with the internal standard when transfected at a 1:1 ratio. One order of magnitude reduction (10−1) means that mutant titers were approximately 10% and that the mutant could compete with the standard when transfected at a ratio of 9:1. Two orders of magnitude reduction (10−2) means that vector titers were approximately 1% and that the mutant could compete when transfected at a ratio of 99:1. Three orders of magnitude reduction (10−3) represents a more than a 1000-fold decrease in titer. When the terminal mismatch vectors were tested, all displayed one order of magnitude reduction in titer (Table 1). More precise determinations indicated a titer of approximately 40% for the 1TMM vector while 4TMM and 5TMM had titers approximately 5% that of the internal standard. Restriction analysis of the proviral junctions of the 4TMM and 5TMM vectors (Figure 4(C)) revealed that no detectable mismatch extension occurred, only premature jump or mistargeting to another location, indicating that strand transfer prior to the completion of −sssDNA occurred at a rate of less than 5%. This level of premature jump is similar to a previous study in our laboratory using the MLVpuro system.30 That study determined premature strand transfer rates of 1–2%, which is likely an underestimation due to the plasmid recombination/read-in transcription artifacts we described above.
Table 1.
Viral titers determined by competition assay
| Construct | Relative titer | Construct | Relative titer |
|---|---|---|---|
| RSVpuro | 100 | 1TM | 10−2 |
| 1TMM | 10−1 | 2TM | 10−2 |
| 2TMM | 10−1 | 3TM | 10−2 |
| 3TMM | 10−1 | 4TM | 10−2 |
| 4TMM | 10−1 | 5TM | 10−2 |
| 5TMM | 10−1 | 12TM | 100 |
| RSVpuroM/B | 100 | 30Lac30 | 100–10−1 |
| 1TMMpbs | 100 | 30Lac12 | 100–10−1 |
| 3TMMpbs | 10−1 | 30Lac5 | 100–10−1 |
| 1TMMtRNA | 10−2 | 5Lac30 | 100–10−1 |
| 3TMMtRNA | 10−3 | 5Lac12 | 100–10−1 |
| 5TMintron | ≥10−3 | 5Lac5 | 10−2 |
| 12TMintron | ≥10−2 | ||
| 30TMintron | 100 |
2.4. The effects of limited terminal homology on minus strand transfer
Previous studies9., 29. indicate that 12 nt of complementarity are required to support efficient recombinogenic transfer. Here, the RSVpuro system was used to address complementarity requirements for minus strand transfer. RSVpuro derivatives (TM1, TM2, TM3, TM4, TM5, and TM12) were constructed containing only 1, 2, 3, 4, 5, or 12 nt of terminal match, respectively, to R sequences at the downstream U3/R junction (Figure 5(A)). Polyadenylation sequences from SV40 were inserted downstream of these mutant acceptors. Vectors with only 1–5 nt of terminal match exhibited titers reduced one to two orders of magnitude (Table 1). The vector with 12 nt of complementarity (TM12) displayed wild-type titer. Product restriction analysis revealed that despite its low-titer of approximately 5%, the TM5 vector accurately targeted to the U3/R junction essentially 100% of the time (Figure 5(B), lane 12). The TM4 vector targeted correctly 79% of the time and TM3 displayed 12% correct targeting (Figure 5(B), lanes 10 and 8).
Figure 5.

Limited terminal homology during minus strand transfer. (A) RNA sequences at the U3/R junction for RSVpuro and the limited terminal homology constructs. RSVpuro derivatives, TM1, TM2, TM3, TM4, TM5, and TM12, contained 1, 2, 3, 4, 5, or 12 nt of complementarity in the downstream R acceptor region. (B) Proviral DNAs from the limited terminal homology vectors were PCR amplified using primers SF223 and SF213 and subjected to restriction digestion. M, markers; RSVpuro: undigested (lane 1), AscI (lane 2); TM1: undigested (lane 3), AscI (lane 4); TM2: undigested (lane 5), AscI (lane 6); TM3: undigested (lane 7), AscI (lane 8); TM4: undigested (lane 9), AscI (lane 10); TM5: undigested (lane 11), AscI (lane 12); TM12: undigested (lane 13), AscI (lane 14).
One major alternate product was observed for TM1, TM2, TM3, and TM4 (Figure 5(B), lanes 3 through 10). Sequencing identified this target as a BssHII site in U3, upstream of the U3/R junction. This site fortuitously provided 4 nt of −sssDNA primer terminal complementarity. An interesting observation for TM4 was that despite the same amount of complementarity (4 nt) at both the U3/R junction and the BssHII site, strand transfer occurred four times more frequently to the U3/R junction. Similarly, the TM3 vector targeted to the U3/R junction 12% of the time despite the availability of more extensive complementarity at the nearby BssHII site, and did not detectably utilize either of two other fortuitous 3 nt targets in U3. Taken together, these data indicate that 5 nt of complementarity were sufficient to accurately target to the U3/R junction. The U3/R junction was used preferentially to alternate sites with equivalent amounts of complementarity for all relevant (TM3 and TM4) vectors, and as few as 3 nt could direct some accurate transfer.
Although the BssHII site was the major alternate acceptor for low-titer vectors, minor alternate products were sometimes detectable when gels were overexposed to film (not shown). Based on their abundance among detected products and the titers of the vectors that generated them, these rare products likely arose at frequencies roughly five orders of magnitude lower than matched donor/acceptor products. To determine the origins of some of these rare products, bands were excised from dried gels, eluted in TE, and re-amplified by PCR. Four alternate products were sequenced. One resulted from transfer of full length R to an alternate site, two products resulted from transfer from read-in transcripts, and one resulted from premature jump before completion of −sssDNA. The minor alternate target for full-length R transfer was located in the SV40 polyadenylation region. The point of transfer for this product contained three primer terminal complementary nucleotides, followed by one mismatch, and then three additional complementary nucleotides. One of the read-in transcripts transferred with 8 nt of fortuitous complementarity present within the SV40 polyadenylation region. The second read-in transcript transferred to a region with no sequence complementarity. Finally, the viral RNA that templated the premature jump product apparently contained plasmid sequences downstream of the SV40 polyadenylation site. These additional 3′ end sequences served as a region of extended (>20 base) complementarity between the point of premature transfer and a fortuitous acceptor in the lengthy polyadenylation read-through RNA. None of the analyzed minor products were premature transfer from within −sssDNA to any of the dozens of potential premature transfer product four base matches located in U3, thus further supporting previous work that suggests premature transfer is rare.30
2.5. Further tests of the hypothesis that minus strand transfer is preferentially targeted
To further test targeting to the U3/R junction versus other sites of −sssDNA complementarity, new vectors were constructed that were similar to the tandem R constructs above. These contained two candidate acceptors downstream of U3, separated from one another by 493 nt of lacZ (Figure 6(A)). The U3/R junction acceptor was referred to as R1 and the secondary acceptor downstream of the lacZ spacer was R2, or the decoy site. We tested constructs with either 30 nt or 5 nt of −sssDNA complementarity in R1 and 30 nt, 12 nt, or 5 nt of complementarity in R2. These vectors were referred to as 30Lac30, 30Lac12, 30Lac5, 5Lac30, 5Lac12, and 5Lac5. All vectors that retained 30 nt or 12 nt in either R displayed titers similar to the internal standard (Table 1). Only the 5Lac5 vector, which retained 5 nt of −sssDNA complementarity in both Rs, had a reduced titer (two orders of magnitude; Table 1).
Figure 6.

Minus strand transfer occurs preferentially at the U3/R junction. (A) Decoy constructs containing two acceptors (R1 and R2) separated by a lacZ spacer were tested for targeting preference during minus strand transfer. (B) Proviral DNA was PCR amplified using primers SF223 and SF213 and restriction digested. M, markers; RSVpuro: undigested (lane 1), AscI (lane 2); 30Lac30: undigested (lane 3), AscI (lane 4); 30Lac12: undigested (lane 5), AscI (lane 6); 30Lac5: undigested (lane 7), AscI (lane 8); 5Lac30: undigested (lane 9), AscI (lane 10); 5Lac12: undigested (lane 11), AscI (lane 12); 5Lac5: undigested (lane 13), AscI (lane 14).
Analysis of products' structures yielded striking results (Figure 6(B), lanes 3–14). The 30Lac30 construct targeted exclusively to the U3/R junction as did 30Lac12, 30Lac5, and 5Lac5. Some transfer to the decoy site was observed for both 5Lac30 and 5Lac12, but approximately 15–20% of strand transfer was directed to the more limited complementarity in R1 (Figure 6(B), lanes 9 and 11). Some observations provided particularly compelling support for the notion that signals in addition to complementarity are involved in directing minus strand transfer. These included use of 5 nt at the U3/R junction despite 30 nt of complementarity at a decoy site for 5Lac30, and exclusive targeting to the U3/R junction for 30Lac30, despite the presence of a decoy with equivalent complementarity.
2.6. Mismatch extension upon plus strand transfer and during tRNA-primed synthesis
RSVpuro vectors were adapted to test whether or not results with this new system would support previous reports, which used vectors with potentially artifact-prone identical LTRs12, of mismatch extension during plus strand transfer. The RSVpuro derivatives that served as plus strand transfer assay vectors here (1TMMpbs and 3TMMpbs; Figure 7(A)) contained mutations at the 3′ edge of the pbs. These mutations were designed to introduce terminal mismatches of 1 nt or 3 nt between plus strand strong stop donor and its minus strand DNA acceptor during plus strand transfer, and allowed analysis of plus strand transfer by restriction digestion. Both 1TMMpbs and 3TMMpbs vectors contained sequences that would generate a BamHI site if mismatch extension occurred upon plus strand transfer.
Figure 7.

Mismatch extension during plus stand transfer and tRNA initiation. (A) DNA sequences of WT and mutant constructs. The control construct contained point mutations flanking the pbs which generated MfeI and BamHI sites. Plus strand transfer constructs contained one or three mismatches at the 3′ edge of the pbs (1TMMpbs and 3TMMpbs, respectively). Initiation constructs contained one or three mismatches at the 5′ edge of the pbs (1TMMtRNA and 3TMMtRNA, respectively). Mismatches are shown in lower case. (B) Proviral DNA was PCR amplified with SF223 and SF213 and analyzed by restriction digestion. M, markers; RSVpuroM/B control products: undigested (lane 1), BamHI (lane 2), MfeI (lane 3); 1TMMpbs: undigested (lane 4), BamHI (lane 5), MfeI (lane 6); 3TMMpbs: undigested (lane 7), BamHI (lane 8), MfeI (lane 9); 1TMMtRNA: undigested (lane 10), BamHI (lane 11), MfeI (lane 12).
Related vectors (1TMMtRNA and 3TMMtRNA; Figure 7(A)) were used to test whether or not mismatch between the tRNA that initially primed reverse transcription and the pbs was tolerable. These vectors contained point mutations that would lead to generation of an MfeI site if a mismatched tRNA primer were extended (Figure 7(A)).
Titers for the plus strand transfer and tRNA initiation vectors were determined (Table 1) using the competition assay described above. The 1TMMpbs vector had a titer similar to the internal standard, indicating that one mismatch was well tolerated during plus strand transfer. 3TMMpbs displayed a one order of magnitude reduction. The 1TMMtRNA vector, which tested mismatch extension during tRNA-primed initiation, displayed two orders of magnitude reduction, suggesting that even a single mismatch was deleterious at this replication step. The 3TMMtRNA mismatch was even less well tolerated, and caused titers to drop three orders of magnitude.
Products of these two sets of vectors were analyzed by PCR and restriction analysis and the results are shown in Figure 7(B). RSVpuroM/B, which contained MfeI and BamHI sites flanking the pbs, was used as a control in the analysis of proviral structures. Products from RSVpuroM/B were digestible by both BamHI and MfeI, as expected (Figure 7(B), lanes 2 and 3). The 1TMMpbs products were only partially digestible with BamHI (30%; Figure 7(B), lane 5), as would result from plus strand transfer mismatch extension. The undigested portion of 1TMMpbs product was sequenced and determined to contain the mutant sequences present in the vector, likely indicating that premature jump had occurred during plus strand transfer. Note, however, that the amount of mismatch extension versus premature jump was likely not 30% and 70%, respectively, due to the nature of plus strand transfer. When mismatch extension occurs during plus strand transfer, the resulting proviral DNA initially contains heteroduplex sequences. Following integration, either strand can be repaired, or the cell may divide prior to repair and generate two daughter cells, one with wild-type and the other with mutant sequences.35., 36. None of the 3TMMpbs vector products were digestible with BamHI (Figure 7(B), lane 8) suggesting that premature jump generated all detectable strand transfer products. This was verified by product sequencing.
No evidence of mismatch extension was found among products of the vectors designed to test for such extension during tRNA-primed initiation of DNA synthesis. 1TMMtRNA DNA was not digestible with MfeI (Figure 7(B), lane 12). Sequencing of the uncut DNA revealed mutant sequences identical to the vector RNA. Possible origins of these rare products include priming by tRNA molecules that lacked at least one 3′ terminal nucleotide, pyrophosphorolysis-like removal of mismatched nucleotides in the primer prior to tRNA extension, or preferential repair of duplex DNA. Products of the 3TMMtRNA vector were not analyzed due to the severe reduction in titer exhibited by this construct.
2.7. Examining the use of limited terminal homology with a novel recombinogenic template switching assay
The complementarity observed to support accurate minus strand transfer (3 bases) was less than previously reported minimal homology values for recombinogenic switching.9., 29. However, the earlier studies were performed with vectors prone to the classes of artifacts eliminated by the vectors used here. Thus, it remained unclear whether replicative and recombinogenic switching homology requirements differed or if the above findings and previous reports' differences were due to assay sensitivity. To address this, RSVpuro derivatives that tested recombinogenic template switching were developed (Figure 8(A)). These vectors were similar to previously described forced intermolecular recombinogenic switching vectors29 but contained template switch donor and acceptor sequences on single RNA templates. This eliminated the need to determine the fraction of virions that contained both recombination donor and acceptor, since both template regions would be present in all vector-containing particles.
Figure 8.

Limited terminal homology during recombinogenic switching. (A) Plasmid structure of the puromycin-intron recombination assay vector. Intron sequences, gray boxes; recombination donor and acceptor sequences, black boxes; primers used for PCR amplification of proviral DNA, gray arrows. MLV U3 sequences labeled ΔU3 contain only the first 30 nt of U3. Drawing is not to scale. (B) Reverse transcription of the puromycin-intron recombination vector. RNA, thin lines; DNA, thick lines. Acceptor, a; donor, d. (C) Puromycin sequences in proviral DNA were PCR amplified using primers SF304 and SF308 and analyzed by restriction digestion. M, molecular weight markers; intron-containing puromycin plasmid control, pJPE657-13 undigested (lane 1); intron-less puromycin plasmid control, RSVpuro undigested (lane 2); 5TMintron: undigested (lane 3), AscI (lane 4); 12TMintron: undigested (lane 5), AscI (lane 6); 30TMintron: undigested (lane 7), AscI (lane 8); D17/pJET uninfected control (lane 9). Alternate target, A.T.; intron-less puromycin, puro.
Expression cassettes for these unusual vectors are shown in Figure 8(A) and the reverse transcription process that generated vector products is outlined in Figure 8(B). These vectors contained a single mutated LTR located toward the middle of the RNAs. This mutant LTR, which contained all of U5 but lacked most of U3 and R, provided all signals required for reverse transcription and integration but did not function as a promoter or polyadenylation signal. The vectors were designed so that their initial reverse transcription product was not the usual U5 plus R-containing −sssDNA. Instead, the initial product was an extended minus strand that resembled the DNA that would ordinarily result after minus strand transfer had occurred. Thus, these vectors were in a sense “pre-jumped”, because their structures eliminated the need to perform strand transfer of the −sssDNA intermediate, and the initial reverse transcription intermediate mimicked the postulated minus strand product involved in “forced copy choice” models for retroviral recombination.17 The only template switch required during minus strand synthesis, which occurred between the forced copy choice junction at the RNA's 5′ end and a homologous acceptor region engineered far downstream of the solo LTR, was recombinogenic rather than a mimic of strong stop transfer. The only true replicative switch these vectors required was plus strand transfer of +sssDNA.
Reverse transcription products of the one-RNA recombination vectors were RSVpuro derivatives containing a reconstituted puromycin resistance gene with an artificial intron (Figure 8(B), final line). The recombination donor on these vectors' RNAs was within a portion of the intron at the 5′ end of the vector RNA and the recombination acceptor was in a portion of the intron engineered near the vector RNA's 3′ end (Figure 8(A) and (B)). By designing these vectors so that forced recombinogenic switch occurred within an intron, some mistargeting could occur without destroying puromycin resistance. Because the splice donor was near the vector RNA's 3′ end and the splice acceptor was near the RNA's 5′ end, the vector would not generate a splicing-competent RNA until after the first round of reverse transcription.
One-RNA recombination vectors with 3′ end acceptors containing 5, 12, and 30 nt of 5′ donor identity (5TMintron, 12TMintron, and 30TMintron, respectively, for terminal match in an intron) were studied. Titers for these vectors were determined by competition assay, and indicated that the 30TMintron vector had a titer similar to the standard while both 5TMintron and 12TMintron titers were reduced two to three orders of magnitude (Table 1).
Restriction analysis of integrated products is shown in Figure 8(C). Recombinogenic transfer to the region of acceptor homology generated a diagnostic AscI site. The 30TM vector accurately transferred to the intron acceptor site, as evident from complete AscI digestion (Figure 8(C), lane 8). Approximately 41% of the 12TMintron products resulted from correct transfer, but one major alternate target was also observed (Figure 8(C), lane 5). This alternate site was used exclusively by 5TMintron (Figure 8(C), lane3), indicating that 5 nt were not sufficient for correct recombinogenic targeting. Sequencing of the major alternate target product revealed that it was an intron-less puromycin gene which could have been generated by targeting using three primer-terminal matches, followed by three mismatches, and then another four bases of primer-acceptor match (Figure 8(C), lane 2). Based on titer reductions, this product arose roughly 0.2% as frequently as recombination products directed by 30 nt of homology. When considered to serve as a surrogate internal control, the abundance of this alternate product suggested that recombinogenic acceptors with 12 nt of primer terminal complementarity were more than 100-fold less efficient than replicative acceptors of the same length.
3. Discussion
This work examined minimal acceptor template features required for efficient and accurate replicative template switching. Previous work has shown that experiments that use two-LTR vector constructs are prone to artifacts that can limit assay sensitivity.9., 12., 28., 32. Thus, our initial effort focused on developing a new assay system that removed repeated elements unnecessary to the experiments here, and demonstrating that levels of previously described read-in and plasmid recombination artifacts were reduced to below detectable levels. We used the improved vectors to re-examine some of our own results which hindsight suggested may have suffered from artifacts.28 In particular, we assessed how much mismatch extension was detectable during minus strand transfer. The new results demonstrated that while up to three bases of mismatch were extended during minus strand transfer, extension products were not detectable for four and five base mismatches. We now attribute our previous suggestion that five bases of primer-terminal mismatch could be extended during minus strand transfer28 to transfection-related and/or read-in transcript artifacts that were minimized in the new assay system.
Plus strand transfer mismatch extension was also addressed with the new assays, and the findings suggested that one but not three bases of mismatch could be extended. This result differed from a previous report, which concluded that extension of three mismatches can occur during plus strand transfer.12 However, as noted in that paper, reversion to wild-type during transfection was observed at a rate of 50–100%, likely indicative of transfection-related artifacts. Thus, the present observations are probably more accurate than the previous reports,12 and it seems likely that three mismatches cannot be extended appreciably during gammaretrovirus plus strand transfer. This plus strand result differed from those for minus strand transfer, since mismatch extension generated 12% of the minus strand transfer products for vectors with three mismatches in the present study. This may be due to inherent differences between plus and minus strand transfer, or it may reflect differences in homology lengths. MLV plus strand strong stop DNA and its acceptor contain only 18 nt of complementarity compared to 69 nt for minus strand transfer.
Data presented here suggested that initiation of reverse transcription may be more sensitive to mismatch than plus or minus strand transfer, since no mismatches between primer tRNA's 3′ end and template RNA appeared to be tolerated. This was probably not due to limiting complementarity since tRNA primed initiation and plus strand transfer both utilize 18 nt of complementarity, and one mismatch was readily tolerated during plus strand transfer. The failure to detect extension products from mismatched tRNA primers may reflect differences in RT polymerization during elongation and initiation phases, as has been described for RT in purified reactions.37., 38., 39. However, although our intention was to assess mismatch extension, we cannot rule out the possibility that the tRNA mismatch mutations exerted effects in whole or in part on other aspects of replication such as tRNA recruitment.
Other experiments tested suggestions that replicative and recombinogenic template switching differ, with the former guided in part by parameters other than donor/acceptor sequence homology. We and others have reported that context effects, such as features in U3, can contribute to minus strand transfer.28., 40. The results in the present report provide further support for this notion. Our findings confirmed previous reports that efficient recombinogenic transfer requires 12 or more bases of primer-terminal homology.29 However, efficient and accurate replicative transfer to the U3/R junction was observed with only five bases, and even 3 nt targeted transfer to a limited extent. The notion that something other than primer/template complementarity contributes to U3/R targeting was further supported by tandem R studies, which revealed a remarkable bias for the U3/R junction when two acceptors with equivalent homology competed. In addition, significant targeting to minimal U3/R junction acceptors was observed even when lengthier alternate regions of homology were available at a distal acceptor site. The preference for the U3/R junction over the distal acceptor site cannot be attributed to titer inhibition by the lacZ spacer sequences, since titers for all the decoy constructs, except the 5Lac5 construct, were nearly wild-type.
Note that although some previous reports have claimed that minus strand transfer can be directed by very short Rs,7 the finding that five bases were sufficient appears to conflict with another published report which concluded that 12 bases of complementarity are required to target minus strand transfer.9 However, the MLV-based vectors used in that study were modified to remove U3 sequences from the downstream LTR and thus lacked the sequences that our studies suggest are required to mediate U3/R targeting (Ref. 28 and see above). Therefore, we suggest the possibility that the findings based on a vector that lacks U3 sequences9 may be a better model for recombinogenic switching than for minus strand transfer. If that interpretation is adopted, the results presented here are consistent with the earlier findings. For example, our experiments demonstrated that even for native R sequences, if the sequences are placed outside their native context (as in Figure 6, Figure 8 experiments) targeting does not occur when less than 12 bases are retained. In the present work, even when 30 R nucleotides were retained at an ectopic site, there was no detectable use of these sequences when they were placed in competition with an acceptor of equal length in an authentic U3/R context. In contrast, even three R nucleotides in the U3/R context were sufficient to direct limited accurate minus strand transfer.
The initial product of retroviral reverse transcription, −sssDNA, has at times been referred to as “minus strand primer DNA”.18 This term highlights −sssDNA's role as the primer for minus strand re-initiation near the genome's 3′ end, and suggests a parallel between minus strand transfer and the initiation of genome replication for other viruses. When −sssDNA is viewed as a primer rather than as a replication intermediate, the notion that factors other than complementarity might contribute to minus strand transfer has significant precedence from other viral systems.
In contrast to the conceptually simple homology-directed mechanism postulated to direct synthesis after retroviral minus strand transfer,1., 41., 42. some other RNA viruses and retroelements rely on cis-acting elements and interaction between genome 5′ and 3′ ends. As is the case for retroviruses, hepatitis B reverse transcription involves template switching. However, although complementarity is present at hepatitis B template switch acceptor sites, there is much less complementarity present (2–18 bases) than in retroviral genomes. These limited base-pairing interactions are clearly far less important to specifying the correct site for hepatitis B DNA re-initiation than are genetically-defined distal sequence elements in that virus.31., 43. A recent study44 demonstrated that base-pairing between cis-acting sequences in duck hepatitis B virus facilitates strand transfer presumably by bringing donor and acceptor sites, present at opposite ends of the genome, into close proximity. Somewhat similar stories of cis-acting elements that specify priming specificity are emerging for RNA viruses such as coronaviruses and picornaviruses.45., 46. Although alternate mechanisms may also exist,47 3′ end recognition for these viruses appears to rely on host proteins that bind specifically to genetic elements in viral RNAs' 3′ ends. Initiation site selection mediated by cross talk between genome 5′ and 3′ ends functions in the replication of LTR-retroelements, mouse hepatitis virus, and dengue virus.48., 49., 50. Thus, in several RNA virus systems, cis-acting elements and/or proteins which bind them contribute to specifying replication product initiation sites, and it would not be surprising if similar factors functioned in the replication of retroviruses.
It remains unclear whether or not the context effects described here are mediated by trans-acting viral or host factors, or if they are guided solely by intrinsic features of the template. Context-dependent retroviral template switching “hot spots” that do not reside at strong stop transfer points have been reported.51., 52. Presumably, switching at these sites is not mediated by the putative factors that contribute to replicative switching, unless such factors are present throughout genomic RNA. Precedence for factor-independent targeting signals also comes from the observation that for at least some retroelements, partially purified RTs alone appear to recognize context-dependent initiation elements in naked RNA.53
In suggesting that homology-independent factors may contribute to strong stop acceptor template selection, we are by no means discounting the importance of donor/acceptor homology. Our results confirm that minus strand transfer efficiency to the U3/R junction is enhanced with increasing homology length until efficiency, as measured by titer, reaches parental levels at 12 bases, and that extensive regions of homology at ectopic sites are sufficient to misdirect strand transfer to those sites. However, the accurate targeting of −sssDNA with wild-type efficiency to a 12 base R in the U3/R context differs markedly from our observed >100-fold reduced efficiency and incomplete accuracy of targeting observed for 12 base acceptors during recombinogenic template switching. We hypothesize that the U3/R junction resides in a region with primer/template complementarity-independent targeting determinants that contribute to minus strand transfer, and which have been retained in MLV in addition to the R regions of primer/template complementarity.
4. Materials and Methods
4.1. Plasmids
pRSVpuro, which contains the Rous sarcoma virus (RSV) promoter was constructed as follows. Fragments containing the RSV U3 and the MLV R regions were PCR amplified from pREP8 (Invitrogen) and pMLVpuro (also called pAM86-554), respectively. These were fused by PCR using complementary overlapping primers to generate an RSV U3/MLV R fragment. This fragment replaced the upstream LTR between the SalI and SpeI sites of pMLVpuro47 to generate pRSVpuro. Variant R sequences were constructed by PCR using mutagenic primers and cloned into the downstream LTR using standard techniques. Pbs mutants were constructed by amplifying the pbs region using mutagenic primers and subcloning into pRSVpuro. pRSVpuroM/B was the parental clone for this series of mutants: as presented in Figure 7(A), this construct contains one point mutation on either side of the pbs, introducing an MfeI site upstream of the pbs and a BamHI site downstream. For constructs with 493 nt of lacZ downstream of R in the 3′ LTR, R1-lacZ-R2 sequences were PCR amplified and inserted into pRSVpuro. Sequences extend from nucleotides 21 to 514 of the lacZ coding region. PuroIntron clones were pRSVpuro derivatives subcloned from a plasmid, pJPE657-13, which contained an artificial intron that was based on intron consensus sequences55 inserted into the puromycin resistance gene of pMLVpuro. The intron was constructed by PCR amplifying pUC sequences with flanking primers containing intron consensus sequences and subcloning into pMLVpuro. The donors used in PuroIntron clones were derived from the first 30 nt of the MLV R and 5 nt, 12 nt, or 30 nt of the MLV R, located in the context of non-U3 intron sequences, served as the acceptor sequences. Note that the acceptor for the 30TMintron clone was subcloned from the 30Rlac30R construct and thus contained the 493 nt lacZ spacer downstream of the 30 nt acceptor. All PCR-generated fragments were sequenced to verify that all clones contained the intended sequences.
4.2. Cells and virus
All transfections and infections were performed in 6 cm culture dishes. Vector plasmids were transfected into ΦNXE MLV-based packaging cells56 (293T derivative) using the calcium phosphate method as previously described.57 Medium (DMEM+10% (v/v) fetal calf serum) was changed 24 hours post-transfection. Viral particle containing media were collected 48 hours post-transfection and filtered through 0.22 micron filters. Viral supernatants contained approximately 1×106 CFU/ml. Approximately 3×105 D17/pJET cells (dog osteosarcoma cells expressing murine ecotropic receptor33., 34.) were infected with 800 μl of filtered medium containing 0.8 μg/ml polybrene (hexadimethrine bromide, Sigma) for two hours. Infected cells were selected 48 hours post-infection in DMEM+10% calf serum containing 6 μg/ml puromycin. Puromycin-resistant cells were pooled, transferred to 10 cm dishes, and grown under puromycin selection until cells reached confluence.
4.3. Analysis of strand transfer products
Genomic DNA was purified using the Wizard genomic DNA purification system per the manufacturer's instructions (Promega). DNA from a 10 cm dish of cells was suspended in 100 μl TE. Proviral sequences were PCR amplified from genomic DNA (2 μl) in 100 μl reactions containing 5% (v/v) DMSO. One primer was 5′ end-labeled with [32P]ATP and T4 polynucleotide kinase. The following primers were used: SF223 (aatgaaagaccccacctgtaggtttggcaag; sense, U3), SF210 (ccagatgcggtccagccctcagcag; sense, U3), SF306 (tccgacttgtggtctcgctgtt; sense, U5), SF212 (gagctagttagctaactagtaccg; antisense, U5), SF213 (aatgaaagacccccgctgacgggta; antisense, utr), SF307 (atgctgcagcagacaagacgc; antisense, utr), SF304 (ggcctaggcttttgcaaaaagcttac; sense, upstream of puromycin), and SF308 (ccactgatatcctgtctttaac; antisense, puromycin). PCR cycles were as follows: denaturation at 94 °C for five minutes; 30 cycles of 30 second denaturation (94 °C), 30 second annealing (60 °C), and 30 second extension (72 °C).
PCR products were restriction digested and separated on 5% (w/v) polyacrylamide gels. The gels were dried and products were quantified by phosphorimager.
4.4. Titers
Viral titers were determined using a competition assay. Mutant constructs were co-transfected into ΦNXE cells with an internal standard (pRSVpuroM/B) that contained MfeI and BamHI sites flanking the pbs. Ratios of mutant to internal standard plasmid were either 1:1 (10 μg each plasmid), 9:1 (18 μg mutant+2 μg standard), or 99:1 (19.8 μg mutant+0.2 μg standard). Particle-containing media were used to infect D17/pJET cells as described above. Following puromycin selection, proviral DNA was PCR amplified using primers SF306 and SF307 (32P-end-labeled) which flank the pbs. Amplified DNA was digested with BamHI (or MfeI) and separated on 5% polyacrylamide gels. Titers were determined by quantifying undigested (mutant) and digested (internal standard) products. Titers were calculated as in the following example: for the 9:1 transfection ratio, ((% undigested mutant sequence/9)/(% digested internal standard sequence/1))×100. Controls were performed to demonstrate complete digestion of the internal standard alone in products of parallel transfections. For pRSVpuroM/B derivatives, pRSVpuro was used as the internal standard.
Acknowledgements
This work was supported by grant CA69300 from the NIH and the Midwest Chapter of the American Heart Association. We thank Dan Loeb for helpful discussion, Monica Roth for providing D17/pJET cells, and Julie Pfeiffer for the intron-containing puromycin resistance gene used in the intron-containing vectors.
Edited by J. Karn
References
- 1.Gilboa E., Mitra S.W., Goff S., Baltimore D. A detailed model of reverse transcription and tests of crucial aspects. Cell. 1979;18:93–100. doi: 10.1016/0092-8674(79)90357-x. [DOI] [PubMed] [Google Scholar]
- 2.Telesnitsky A., Goff S.P. Strong-stop strand transfer during reverse transcription. In: Skalka A.M., Goff S.P., editors. Reverse Transcriptase. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1993. pp. 49–83. [Google Scholar]
- 3.Klaver B., Berkhout B. Premature strand transfer by the HIV-1 reverse transcriptase during strong-stop DNA synthesis. Nucl. Acids Res. 1994;22:137–144. doi: 10.1093/nar/22.2.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Varmus H.E., Heasley S., Kung H.J., Oppermann H., Smith V.C., Bishop J.M., Shank P.R. Kinetics of synthesis, structure and purification of avian sarcoma virus-specific DNA made in the cytoplasm of acutely infected cells. J. Mol. Biol. 1978;120:55–82. doi: 10.1016/0022-2836(78)90295-4. [DOI] [PubMed] [Google Scholar]
- 5.Lee Y.M., Coffin J.M. Relationship of avian retrovirus DNA synthesis to integration in vitro. Mol. Cell. Biol. 1991;11:1419–1430. doi: 10.1128/mcb.11.3.1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blain S.W., Goff S.P. Effects on DNA synthesis and translocation caused by mutations in the RNase H domain of Moloney murine leukemia virus reverse transcriptase. J. Virol. 1995;69:4440–4452. doi: 10.1128/jvi.69.7.4440-4452.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berkhout B., van Wamel J., Klaver B. Requirements for DNA strand transfer during reverse transcription in mutant HIV-1 virions. J. Mol. Biol. 1995;252:59–69. doi: 10.1006/jmbi.1994.0475. [DOI] [PubMed] [Google Scholar]
- 8.Ohi Y., Clever J.L. Sequences in the 5′ and 3′ R elements of human immunodeficiency virus type 1 critical for efficient reverse transcription. J. Virol. 2000;74:8324–8334. doi: 10.1128/jvi.74.18.8324-8334.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dang Q., Hu W.S. Effects of homology length in the repeat region on minus-strand DNA transfer and retroviral replication. J. Virol. 2001;75:809–820. doi: 10.1128/JVI.75.2.809-820.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lobel L.I., Goff S.P. Reverse transcription of retroviral genomes: mutations in the terminal repeat sequences. J. Virol. 1985;53:447–455. doi: 10.1128/jvi.53.2.447-455.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ramsey C.A., Panganiban A.T. Replication of the retroviral terminal repeat sequence during in vivo reverse transcription. J. Virol. 1993;67:4114–4121. doi: 10.1128/jvi.67.7.4114-4121.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pulsinelli G.A., Temin H.M. High rate of mismatch extension during reverse transcription in a single round of retrovirus replication. Proc. Natl Acad. Sci. USA. 1994;91:9490–9494. doi: 10.1073/pnas.91.20.9490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wakefield J.K., Rhim H., Morrow C.D. Minimal sequence requirements of a functional human immunodeficiency virus type 1 primer binding site. J. Virol. 1994;68:1605–1614. doi: 10.1128/jvi.68.3.1605-1614.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wakefield J.K., Morrow C.D. Mutations within the primer binding site of the human immunodeficiency virus type 1 define sequence requirements essential for reverse transcription. Virology. 1996;220:290–298. doi: 10.1006/viro.1996.0317. [DOI] [PubMed] [Google Scholar]
- 15.An W., Telesnitsky A. Effects of varying sequence similarity on the frequency of repeat deletion during reverse transcription of a human immunodeficiency virus type 1 vector. J. Virol. 2002;76:7897–7902. doi: 10.1128/JVI.76.15.7897-7902.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu H., Jetzt A.E., Ron Y., Preston B.D., Dougherty J.P. The nature of human immunodeficiency virus type 1 strand transfers. J. Biol. Chem. 1998;273:28384–28391. doi: 10.1074/jbc.273.43.28384. [DOI] [PubMed] [Google Scholar]
- 17.Coffin J.M. Structure, replication, and recombination of retrovirus genomes: some unifying hypotheses. J. Gen. Virol. 1979;42:1–26. doi: 10.1099/0022-1317-42-1-1. [DOI] [PubMed] [Google Scholar]
- 18.Temin H.M. Retrovirus variation and reverse transcription: abnormal strand transfers result in retrovirus genetic variation. Proc. Natl Acad. Sci. USA. 1993;90:6900–6903. doi: 10.1073/pnas.90.15.6900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hu W.S., Temin H.M. Genetic consequences of packaging two RNA genomes in one retroviral particle: pseudodiploidy and high rate of genetic recombination. Proc. Natl Acad. Sci. USA. 1990;87:1556–1560. doi: 10.1073/pnas.87.4.1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hu W.S., Temin H.M. Retroviral recombination and reverse transcription. Science. 1990;250:1227–1233. doi: 10.1126/science.1700865. [DOI] [PubMed] [Google Scholar]
- 21.Jetzt A.E., Yu H., Klarmann G.J., Ron Y., Preston B.D., Dougherty J.P. High rate of recombination throughout the human immunodeficiency virus type 1 genome. J. Virol. 2000;74:1234–1240. doi: 10.1128/jvi.74.3.1234-1240.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Quinones-Mateu M.E., Gao Y., Ball S.C., Marozsan A.J., Abraha A., Arts E.J. In vitro intersubtype recombinants of human immunodeficiency virus type 1: comparison to recent and circulating in vivo recombinant forms. J. Virol. 2002;76:9600–9613. doi: 10.1128/JVI.76.19.9600-9613.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang J., Temin H.M. Retrovirus recombination depends on the length of sequence identity and is not error prone. J. Virol. 1994;68:2409–2414. doi: 10.1128/jvi.68.4.2409-2414.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cheslock S.R., Anderson J.A., Hwang C.K., Pathak V.K., Hu W.S. Utilization of nonviral sequences for minus-strand DNA transfer and gene reconstitution during retroviral replication. J. Virol. 2000;74:9571–9579. doi: 10.1128/jvi.74.20.9571-9579.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Darlix J.L., Vincent A., Gabus C., de Rocquigny H., Roques B. Trans-activation of the 5′ to 3′ viral DNA strand transfer by nucleocapsid protein during reverse transcription of HIV1 RNA. C. R. Acad. Sci. III. 1993;316:763–771. [PubMed] [Google Scholar]
- 26.Allain B., Lapadat-Tapolsky M., Berlioz C., Darlix J.L. Transactivation of the minus-strand DNA transfer by nucleocapsid protein during reverse transcription of the retroviral genome. EMBO J. 1994;13:973–981. doi: 10.1002/j.1460-2075.1994.tb06342.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Allain B., Rascle J.B., de Rocquigny H., Roques B., Darlix J.L. CIS elements and trans-acting factors required for minus strand DNA transfer during reverse transcription of the genomic RNA of murine leukemia virus. J. Mol. Biol. 1998;277:225–235. doi: 10.1006/jmbi.1997.1596. [DOI] [PubMed] [Google Scholar]
- 28.Topping R., Demoitie M.A., Shin N.H., Telesnitsky A. Cis-acting elements required for strong stop acceptor template selection during Moloney murine leukemia virus reverse transcription. J. Mol. Biol. 1998;281:1–15. doi: 10.1006/jmbi.1998.1929. [DOI] [PubMed] [Google Scholar]
- 29.Pfeiffer J.K., Telesnitsky A. Effects of limiting homology at the site of intermolecular recombinogenic template switching during Moloney murine leukemia virus replication. J. Virol. 2001;75:11263–11274. doi: 10.1128/JVI.75.23.11263-11274.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kulpa D., Topping R., Telesnitsky A. Determination of the site of first strand transfer during Moloney murine leukemia virus reverse transcription and identification of strand transfer-associated reverse transcriptase errors. EMBO J. 1997;16:856–865. doi: 10.1093/emboj/16.4.856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Loeb D.D., Gulya K.J., Tian R. Sequence identity of the terminal redundancies on the minus-strand DNA template is necessary but not sufficient for the template switch during hepadnavirus plus-strand DNA synthesis. J. Virol. 1997;71:152–160. doi: 10.1128/jvi.71.1.152-160.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Olson P., Temin H.M., Dornburg R. Unusually high frequency of reconstitution of long terminal repeats in U3-minus retrovirus vectors by DNA recombination or gene conversion. J. Virol. 1992;66:1336–1343. doi: 10.1128/jvi.66.3.1336-1343.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.O'Reilly L., Roth M.J. Second-site changes affect viability of amphotropic/ecotropic chimeric enveloped murine leukemia viruses. J. Virol. 2000;74:899–913. doi: 10.1128/jvi.74.2.899-913.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kizhatil K., Albritton L.M. Requirements for different components of the host cell cytoskeleton distinguish ecotropic murine leukemia virus entry via endocytosis from entry via surface fusion. J. Virol. 1997;71:7145–7156. doi: 10.1128/jvi.71.10.7145-7156.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Berwin B., Barklis E. Retrovirus-mediated insertion of expressed and non-expressed genes at identical chromosomal locations. Nucl. Acids Res. 1993;21:2399–2407. doi: 10.1093/nar/21.10.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang J., Tang L.Y., Li T., Ma Y., Sapp C.M. Most retroviral recombinations occur during minus-strand DNA synthesis. J. Virol. 2000;74:2313–2322. doi: 10.1128/jvi.74.5.2313-2322.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bebenek K., Abbotts J., Roberts J.D., Wilson S.H., Kunkel T.A. Specificity and mechanism of error-prone replication by human immunodeficiency virus-1 reverse transcriptase. J. Biol. Chem. 1989;264:16948–16956. [PubMed] [Google Scholar]
- 38.Lanchy J.M., Keith G., Le Grice S.F., Ehresmann B., Ehresmann C., Marquet R. Contacts between reverse transcriptase and the primer strand govern the transition from initiation to elongation of HIV-1 reverse transcription. J. Biol. Chem. 1998;273:24425–24432. doi: 10.1074/jbc.273.38.24425. [DOI] [PubMed] [Google Scholar]
- 39.Ghosh M., Williams J., Powell M.D., Levin J.G., Le Grice S.F. Mutating a conserved motif of the HIV-1 reverse transcriptase palm subdomain alters primer utilization. Biochemistry. 1997;36:5758–5768. doi: 10.1021/bi963045e. [DOI] [PubMed] [Google Scholar]
- 40.Brule F., Bec G., Keith G., Le Grice S.F., Roques B.P., Ehresmann B. In vitro evidence for the interaction of tRNA(3)(Lys) with U3 during the first strand transfer of HIV-1 reverse transcription. Nucl. Acids Res. 2000;28:634–640. doi: 10.1093/nar/28.2.634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Swanstrom R., Varmus H.E., Bishop J.M. The terminal redundancy of the retrovirus genome facilitates chain elongation by reverse transcriptase. J. Biol. Chem. 1981;256:1115–1121. [PubMed] [Google Scholar]
- 42.Haseltine W.A., Kleid D.G., Panet A., Rothenberg E., Baltimore D. Ordered transcription of RNA tumor virus genomes. J. Mol. Biol. 1976;106:109–131. doi: 10.1016/0022-2836(76)90303-x. [DOI] [PubMed] [Google Scholar]
- 43.Havert M.B., Loeb D.D. cis-acting sequences in addition to donor and acceptor sites are required for template switching during synthesis of plus-strand DNA for duck hepatitis B virus. J. Virol. 1997;71:5336–5344. doi: 10.1128/jvi.71.7.5336-5344.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu N., Tian R., Loeb D.D. Base pairing among three cis-acting sequences contributes to template switching during hepadnavirus reverse transcription. Proc. Natl Acad. Sci. USA. 2003;100:1984–1989. doi: 10.1073/pnas.0436218100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ito T., Lai M.M. Determination of the secondary structure of and cellular protein binding to the 3′-untranslated region of the hepatitis C virus RNA genome. J. Virol. 1997;71:8698–8706. doi: 10.1128/jvi.71.11.8698-8706.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Waggoner S., Sarnow P. Viral ribonucleoprotein complex formation and nucleolar-cytoplasmic relocalization of nucleolin in poliovirus-infected cells. J. Virol. 1998;72:6699–6709. doi: 10.1128/jvi.72.8.6699-6709.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Todd S., Towner J.S., Brown D.M., Semler B.L. Replication-competent picornaviruses with complete genomic RNA 3′ noncoding region deletions. J. Virol. 1997;71:8868–8874. doi: 10.1128/jvi.71.11.8868-8874.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huang P., Lai M.M. Heterogeneous nuclear ribonucleoprotein a1 binds to the 3′-untranslated region and mediates potential 5′–3′-end cross talks of mouse hepatitis virus RNA. J. Virol. 2001;75:5009–5017. doi: 10.1128/JVI.75.11.5009-5017.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cristofari G., Bampi C., Wilhelm M., Wilhelm F.X., Darlix J.L. A 5′–3′ long-range interaction in Ty1 RNA controls its reverse transcription and retrotransposition. EMBO J. 2002;21:4368–4379. doi: 10.1093/emboj/cdf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.You S., Falgout B., Markoff L., Padmanabhan R. In vitro RNA synthesis from exogenous dengue viral RNA templates requires long range interactions between 5′- and 3′-terminal regions that influence RNA structure. J. Biol. Chem. 2001;276:15581–15591. doi: 10.1074/jbc.M010923200. [DOI] [PubMed] [Google Scholar]
- 51.Li Y., Carpenter S. Cis-acting sequences may contribute to size variation in the surface glycoprotein of bovine immunodeficiency virus. J. Gen. Virol. 2001;82:2989–2998. doi: 10.1099/0022-1317-82-12-2989. [DOI] [PubMed] [Google Scholar]
- 52.Andersen E.S., Jeeninga R.E., Damgaard C.K., Berkhout B., Kjems J. Dimerization and template switching in the 5′ untranslated region between various subtypes of human immunodeficiency virus type 1. J. Virol. 2003;77:3020–3030. doi: 10.1128/JVI.77.5.3020-3030.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen B., Lambowitz A.M. De novo and primer-mediated initiation of cDNA synthesis by the mauriceville retroplasmid reverse transcriptase involve recognition of a 3′ CCA sequence. J. Mol. Biol. 1997;271:311–332. doi: 10.1006/jmbi.1997.1185. [DOI] [PubMed] [Google Scholar]
- 54.Robson N.D., Telesnitsky A. Effects of 3′ untranslated region mutations on plus-strand priming during moloney murine leukemia virus replication. J. Virol. 1999;73:948–957. doi: 10.1128/jvi.73.2.948-957.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rautmann G., Matthes H.W., Gait M.J., Breathnach R. Synthetic donor and acceptor splice sites function in an RNA polymerase B (II) transcription unit. EMBO J. 1984;3:2021–2028. doi: 10.1002/j.1460-2075.1984.tb02085.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pear W.S., Scott M.L., Nolan G.P. Generation of high titer, helper-free retroviruses by transient transfection. In: Robbins P.D., editor. Gene Therapy Protocols. Humana Press; Totowa, NJ: 1996. pp. 41–58. [Google Scholar]
- 57.Pfeiffer J.K., Topping R.S., Shin N.H., Telesnitsky A. Altering the intracellular environment increases the frequency of tandem repeat deletion during Moloney murine leukemia virus reverse transcription. J. Virol. 1999;73:8441–8447. doi: 10.1128/jvi.73.10.8441-8447.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]