

Abstract
By using biphenyl-2-ylphosphines functionalized with a remote tertiary amino group as ligand, readily available acetylenic amides are directly converted into 2-aminofurans devoid of any electron-withdrawing and hence deactivating/stabilizing substituents. These highly electron-rich furans have rarely been prepared, let alone being applied in synthesis, due to the high reactivities and low stabilities associated with their exceedingly electron-rich nature of the furan ring. In this work, these reactive furans undergo smoothly in-situ intermolecular Diels-Alder reactions to deliver highly functionalized/substituted aniline products or intramolecular ones to furnish carbazole-4-carboxylates in mostly good to excellent yields. This work offers a general and expedient access to this class of rarely studies electron-rich furans and shall open exciting opportunities for their applications.
Keywords: 1-amido-1,3diene; isomerization; gold; Diels-Alder reaction; catalysis
Graphical Abstract
What a ligand. With bifunctional ligands specifically designed for gold catalysis, acetylenic amides are transformed into 2-aminofurans in a single step efficiently. The highly electron-rich nature of these furans bestows them exceptional synthetic values but difficult to access in other means.
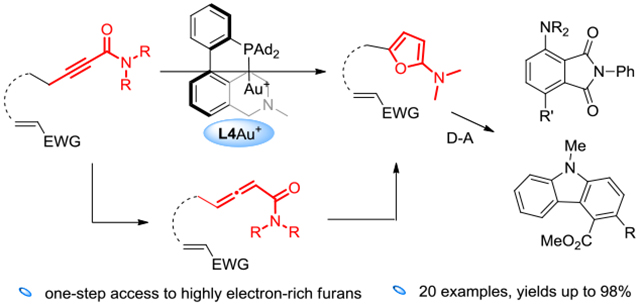
Metal-catalyzed cycloisomerization of acetylenic ketones constitutes rapid access to 2-substituted and 2,5-disubstituted furans (Scheme 1A). The general ease of substrate preparation further enhances the synthetic appeal of the approach. Mechanistically, an allenylic ketone intermediate generated in situ upon the isomerization of the substrate is proposed to further undergo metal-catalyzed cyclization to lead to the observed product. Literature precedents employ Pd[1] or Cu[2] catalysis at elevated temperature (e.g., 100 °C). There is no precedent for the related direct conversion of ynoates into valuable electron-rich 2-alkoxylfuran, let alone the more challenging synthesis of 2-aminofurans from acetylenic amide (Scheme 1B). This is likely due to the difficulty in the initial isomerization of acetylenic substrate into allenylic ester or amide in metal catalysis, where stronger basic conditions are required. Such basic environment may not be compatible with the necessity of acidic metal complexes for the subsequent cyclization step. Despite the extensive applications of electron-rich 2-siloxy/alkoxyfuran,[3] the generally more electron-rich and hence more reactive 2-aminofurans devoid of electron-withdrawing and hence stabilizing/deactivating substituents[4] are difficult to access. A reported access to a narrow subclass of these electron-rich furans[5] establishes that they are rather unstable and decompose on silica gel column, upon recrystallization or during storage. To the best of our knowledge, there is no generally applicable, let alone expedient, access to this class of highly electron-rich furan.
Scheme 1.

Synthesis of furans via isomerization of acetylenic carbonyl
In 2014, we reported a gold-catalyzedg conversion of arylalkynes and certain types of aliphatic alkynes into dienes (Scheme 2).[6] In this reaction, allenes are the reaction intermediates, and the double proton migrations are both enabled by a designed bifunctional biphenyl-2-ylphosphine ligand (i.e., L1) featuring a 3’-amino group, which acts as the proton shuttle. Notably, the gold catalysis permits the deprotonation at propargylic C-H bond (pKa in DMSO >30) by a mildly basic aniline (pKa in DMSO ~4), owing to metal ligand cooperative catalysis.[7] Our later works[8] expanded the scope of the ligand basic functionality to include differently positioned remote basic tertiary amines (see L2-L4 in Table 1 footnote) and engaged frustrated Lewis pairs in gold catalysis.[8b] Lately, we demonstrated that the initial isomerization of alkyne into allene can become asymmetric when a chiral version of L4 is employed as the gold ligand,[9] and the allenylgold intermediate can be trapped by activated aldehydes.[10]
Scheme 2.

Our prior work on isomerization of alkynes to dienes.
Table 1.
Conditions optimization.
| ||||||
|---|---|---|---|---|---|---|
| Entry | L | x(%)/y(%) | temp/time | additive | conversion | 2a/2a’b |
| 1c | L1 | 5/10 | 80 °C/15 h | - | 7% | - |
| 2 | L1 | 5/10 | 80 °C/15 h | - | 76% | <2%/23% |
| 3 | L2 | 5/10 | 80 °C/15 h | - | 21% | - |
| 4 | L3 | 5/10 | 80 °C/15 h | - | 20% | <2%/8% |
| 5 | L4 | 5/10 | 80 °C/15 h | - | 54% | 18%/35% |
| 6 | PPh3 | 5/10 | 80 °C/15 h | - | 20% | - |
| 7 | Johnphos | 5/10 | 80 °C/15 h | - | 20% | - |
| 8 | WangPhos | 5/10 | 80 °C/15 h | - | 20% | - |
| 9 | L4 | 5/10 | 80 °C/15 h | 3Å MS | 25% | - |
| 10 | L4 | 5/10 | 80 °C/15 h | Boc2Od | 100% | 72%/26% |
| 11e | L4 | 5/10 | 80 °C/15 h | Boc2Od | 100% | 73%/26% |
| 12 | L4 | 5/10 | 80 °C/30 h | Boc2Od | 100% | 91%f/0% |
All the reactions were run in DCE (0.05 M) in sealed vials.
NMR yield using triisopropylbenzene as the internal reference.
No N-phenylmaleiimide was added.
2 equiv. used.
Reaction run under Ar.
86% isolated yield.
We envisioned that the isomerization of acetylenic amide to allenylic amide can likewise be realized by a gold catalyst featuring our designed bifunctional ligands. As such, a sequence related to that in Scheme 1A should permit for the first time access to highly electron-rich 2-aminofuran in a straightforward and expedient manner.
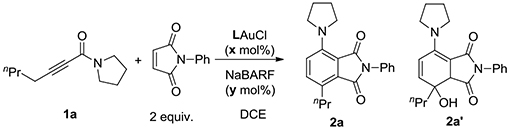
Initially, we treated the pyrrolidine-derived acetylenic amide 1a with L1AuCl (5 mol%) and NaBARF (10 mol%). To our disappointment, little reaction was observed (Table 1, entry 1). We surmised that the 2-(1-pyrrolidino)furan product might be so electron-rich that is trapped and hence deactivated the gold catalyst. To this end, we added N-phenylmaleiimide (2 equiv) to in situ trap the furan via the Diels-Alder reaction. To our delight, most of the 1a were consumed, and the bicyclic enamine 2a’ was formed in 23% NMR yield (entry 2). This product is apparently due to the ring opening of the D-A adduct 4, as outlined in Scheme 3. Its detection confirms the validity of our designed gold catalysis and the in-situ generation of the highly electron-rich 2-aminofuran 3. Dehydrative aromatization of 4 would lead to the naphthalimide product 2a, which, however, was barely detectable. Our recently developed bifunctional ligands L2-L4 featuring a remote tertiary amino group were also examined (entries 3-5). While L2 and L3 appeared inferior to L1, L4 led to better product yields, albeit a lower conversion, and moreover, the formation of 2a. These results revealed the importance of the positioning of the remote amino group. Other typical gold catalysts (entries 6-7) and our previously developed amide-functionalized phosphine WangPhos were completely ineffective, highlighting the critical importance of the basic ligand amino group. To promote dehydration in entry 5, 3 Å MS was added, but the reaction was inhibited. When 2 equivalent of Boc2O was added, the reaction was remarkably improved to afford 2a in 72% NMR along with 26% of 2a’ (entry10). Running the reaction under Ar did not noticeably improve the yields (entry 11). To convert 2a’ to 2a via dehydration and hence increase the latter’s yield, we extended the reaction time to 30 h (entry 12). Indeed, 2a was formed in 91% NMR and 86% isolated yield while no 2a’ remained.
Scheme 3.

Proposed mechanism for the formations of 2a and 2a’.
The scope of this sequential gold-catalyzed cycloisomerization and intermolecular D-A reaction was studied by using N-phenylmaleiimide as the dienophile. As shown in Table 2, entries 1-6, the ynamides can readily accommodate various N-substituents, including dimethylamino (entry 1), dibenzylamino (entry 2), piperidin-1-yl (entry 3), N-morpholinyl (entry 4), methyl(phenyl)amino (entry 5) and diphenylamino (entry 6). The reaction yields were high, ranging from 80% to 97%. The better efficiencies exhibited by the N-phenyl substrates (entries 5 and 6) are consistent with that phenyl is electron-withdrawing and therefore lowers the basicity/electron-richness of the reaction system. The N,N-dibenzylynamides possessing a 6-OTIPS (entry 7), a 6-OAc (entry 8), a 6-N(Boc)Ts (entry 9) and a 5-OTBS group (entry 10) were, however, less reactive than the non-functionalized counterpart 1c. Despite longer reaction times and/or higher catalyst loadings were used, the reactions still exhibited good efficiencies. Alkynamide 1l featuring a sterically more demanding cyclohexyl group α to the C-C triple bond underwent this tandem transformation smoothly, delivering the phthalimide 2l in 83% yield (entry 11). The substrate 1m bearing no γ-substituent also reacted successfully to form 6-amino-substituted N-phenyl-phthalimide 2m in a serviceable yield (entry 12). Finally, substrates containing either a 4- methoxyphenyl (entry 13) or a thiophen-3-yl (entry 14) substituent at the γ-position reacted smoothly to afford the biaryl products 2n and 2o, respectively, in good to excellent yields.
Table 2.
Reaction scope of sequential gold-catalyzed isomerization and D-A reaction with N-phenylmaleiimide.a
 |
 |
Reactions conditions 0.15 mmol of 1, 2 eq. of 2, L4AuCl (5 mol%), NaBARF (10 mol%), the indicated additive, 80 °C and DCE as solvent (0.05 M).
Isolated yield.
10 mol% L4AuCl and 20 mol% of NaBARF used.
30 mg 3Å MS used.
One equivalent of N-phenylphthalimide used.
The reactions of N,N-dibenzylamide 1c with less reactive dienophiles resulted in mostly little conversion, which is likely due to the inhibition of gold catalysis by the electron-rich furan intermediate 4 due to its slow D-A reaction. A comparatively lesser electron-rich 2-aminofuran 5, however, could be generated from the N-phenyl-N-methylamide 1f in the absence of any dienophile in 85% NMR yield (Scheme 4). Despite 5 is sensitive to air and acid, we were able to purify it, albeit in 35% yield, via basic alumina column chromatography under Ar atmosphere, and fully characterize it. This result lends credence to our reaction design and hence the anticipated reaction mechanism. A one-pot trapping of 5 by maleic anhydride afforded the anticipated phthalic anhydride 2f’ in 82% yield (Scheme 4). On the other hand, its one-pot reaction with naphthoquinone led to the formation of the naphtho[1,2-b]furan 6 in 85% yield. A mechanism is proposed to rationalize its formation (Scheme 4).
Scheme 4.

Reactions of the alkynamide 1f with other dienophiles.
We anticipated that facile intramolecular D-A reactions would permit the participation of a broader range of dienophiles. Moreover, the overall reaction would afford in principle increasingly complex cyclic structures of high synthetic value. As shown in Table 3, entry 1, the N,N-dibenzylynamide 7a was indeed transformed into the benzene-fused lactone 8a in a good yield. The maleate moiety apparently participated effectively in the IMDA reaction. On the other hand, the related intermolecular reaction between diethyl maleate and 1c had little conversion. With a cinnamate moiety attached to the propynamide nitrogen atom (as in 7b), the tandem gold-catalyzed cycloisomerization. The Diels-Alder reaction and aromatization occurred with excellent efficiency (entry 2). The carbazole-4-carboxylate 8b was formed in 90% isolated yield. The related reactions of 7c (entry 3) and 7d (entry 4) were equally or more efficient, delivering products with additional substitutions at the 3 position. Notably, in these reactions, no additive was needed, and the cinnamate moiety, a weak dienophile, participated in the IMDA smoothly.
Table 3.
Scope involving intramolecular D-A reactions.a
 |
Reactions were carried out with L4AuCl (5 mol%), NaBARF (10 mol%) with the indicated additive at 80 °C in DCE (0.05 M) for the indicated time.
Isolated yield.
10 mol % L4AuCl and 20 mol % of NaBARF used.
In summary, by using a remotely base-functionalized biphenyl-2-ylphosphine as ligand, we have developed general and facile access to 2-aminofurans devoid of any electron-withdrawing substituents directly from acetylenic amides. These highly electron-rich furans are of exceeding synthetic potential but have only been scantly studied due to synthetic difficulties. Combined with the ease of substrate preparation, this chemistry permits expedient studies of the synthetic utility of this important class of highly electron-rich and hence reactive furans.
Supplementary Material
Acknowledgments
We thank NSF (CHE 1800525) and NIGMS (R01GM123342) for generous financial support and NIH shared instrument grant S10OD012077 for the purchase of a 400 MHz NMR spectrometer. ZW thanks the Mellichamp Academic Initiative in Sustainability for a fellowship in support of his research.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].Sheng H, Lin S, Huang Y, Tetrahedron Lett. 1986, 27, 4893–4894. [Google Scholar]
- [2].Kel’i AV, Gevorgyan V, J. Org. Chem 2002, 67, 95–98. [DOI] [PubMed] [Google Scholar]
- [3].Boukouvalas J, Sperry J, Brimble MA, in Encyclopedia of Reagents for Organic Synthesis, John Wiley & Sons, Ltd, 2001. [Google Scholar]
- [4].(a) Padwa A, Crawford KR, Rashatasakhon P, Rose M, J. Org. Chem 2003, 68, 2609–2617; [DOI] [PubMed] [Google Scholar]; (b) Padwa A, Brodney MA, Satake K, Straub CS, J. Org. Chem 1999, 64, 4617–4626; [DOI] [PubMed] [Google Scholar]; (c) Padwa A, Dimitroff M, Waterson AG, Wu T, J. Org. Chem 1997, 62, 4088–4096; [Google Scholar]; (d) Cochran JE, Wu T, Padwa A, Tetrahedron Lett. 1996, 37, 2903–2906; [Google Scholar]; (e) Chatani N, Hanafusa T, J. Org. Chem 1987, 52, 4408–4409. [Google Scholar]
- [5].(a) Medimagh R, Marque S, Prim D, Chatti S, Heterocycles 2009, 78, 679–690; [Google Scholar]; (b) Semmelhack MF, Park J, Organometallics 1986, 5, 2550–2552. [Google Scholar]
- [6].Wang Z, Wang Y, Zhang L, J. Am. Chem. Soc 2014, 136, 8887–8890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].(a) Trincado M, Grützmacher H, in Cooperative Catalysis, Wiley-VCH Verlag GmbH & Co. KGaA, 2015, pp. 67–110; [Google Scholar]; (b) Grützmacher H, Angew. Chem. Int. Ed 2008, 47, 1814–1818; [DOI] [PubMed] [Google Scholar]; (c) Berrisford DJ, Bolm C, Sharpless KB, Angew. Chem. Int. Ed 1995, 34, 1059–1070; [Google Scholar]; (d) Askevold B, Roesky HW, Schneider S, ChemCatChem 2012, 4, 307–320; [Google Scholar]; (e) Khusnutdinova JR, Milstein D, Angew. Chem. Int. Ed 2015, 54, 12236–12273. [DOI] [PubMed] [Google Scholar]
- [8].(a) Li X, Wang Z, Ma X, Liu P.-n., Zhang L, Org. Lett 2017, 19, 5744–5747; [DOI] [PubMed] [Google Scholar]; (b) Wang Z, Ying A, Fan Z, Hervieu C, Zhang L, ACS Catal. 2017, 7, 3676–3680. [Google Scholar]
- [9].Cheng X, Wang Z, Quintanilla CD, Zhang L, J. Am. Chem. Soc 2019, 141, 3787–3791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Li T, Zhang L, J. Am. Chem. Soc 2018, 140, 17439–17443. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.