

Abstract
Exome sequencing has identified many candidate genes and mutations for human diseases, but the functional validation of these candidates is a time-consuming and costly process. Here, we describe a method which uses lentiviruses to overexpress calpain mutations that may play a role in dominant diseases such as autosomal dominant neovascular inflammatory vitreoretinopathy (ADNIV). The use of lentivirus to deliver the mutant calpain allows for a cost-effective, rapid, and efficient approach to test whether or not a candidate gene mutation from exome sequencing acts as the disease-causing allele for a human disorder. This method also provides for a comparison of different candidate mutations from a single gene identified by exome sequencing, as well as elucidating the mechanisms underlying these complex human disorders. Furthermore, this chapter focuses on two different methods to deliver mutant calpain to the cells of the eye, using either a subretinal or an intravitreal injection of the lentivirus into the mouse eye.
Keywords: Calpain, CAPN5, Lentiviral vectors, Molecular cloning, Subretinal injection, Intravitreal injection, Mouse eye
1. Introduction
Advances in next-generation exome sequencing have allowed for the identification of candidate disease alleles for complex human disorders. However, although many human genes have been identified as having potential roles in disease, the ability to functionally validate these candidate genes and mutations has progressed at a slower pace. Therefore, using lentiviruses to transiently express candidate mutations can provide a more rapid and cost-efficient method to determine whether or not a gene mutation is causative for disease, as well as allowing for the comparison of different disease alleles in transgenic mouse models.
Mutations in CAPN5, encoded by the calpain-5 gene, have been recently determined to lead to autosomal dominant neovascular inflammatory vitreoretinopathy (ADNIV, OMIM 193235, [1–6]. ADNIV is a dominant, heritable, autoimmune disorder, where the clinical presentation is found only within the eye even though the gene is widely expressed throughout the human body. ADNIV is also unusual since it shows the pathological features of a variety of eye diseases that do not normally occur together, such as retinitis pigmentosa, diabetic retinopathy, autoimmune uveitis, and proliferative vitreoretinopathy [7–9]. Thus, the study of candidate disease alleles and mutations for CAPN5 can allow for further understanding of many human eye disorders, along with elucidating the disease mechanisms underlying ADNIV. Additionally, if the expression of a single Mendelian mutation is enough to cause the ADNIV phenotype in a mouse model, it is likely that ADNIV patients with this disease allele may be good candidates for retinal gene therapy.
Here, we describe a method in which calpain mutations in the mouse eye are overexpressed using lentivirus. This method can be used to create a preclinical model for the study of ADNIV using mutant CAPN5 or to study overexpression and/or mutant forms of other calpains and their potential roles in eye disorders. This chapter describes the molecular cloning techniques needed to construct lentiviruses containing mutant calpain genes, as well as two different injection techniques, subretinal and intravitreal injections, to deliver the mutant calpain to the various cells of the eye.
2. Materials
2.1. Construction of Lentiviral Vectors
Lentiviral transfer plasmid.
Mutant calpain DNA.
High-fidelity taq PCR kit.
50× TAE: 242 g Tris-base, 57.1 mL glacial acetic acid, 100 mL of 0.5 M EDTA, add H2O to final volume of 1 L (see Note 1).
1× TAE: dilute 20 mL of 50× TAE into 980 mL of sterile H2O.
Ethidium bromide.
Gel electrophoresis equipment.
Gel chambers and gel combs.
1 kb DNA ladder.
6× DNA gel loading dye.
UV light source.
Gel DNA purification kit.
Restriction enzymes and buffers.
Calf alkaline phosphatase.
Water bath.
T4 DNA ligase and buffer.
Chemically competent E. coli.
Luria broth (LB): 10 g NaCl, 10 g tryptone, 5 g yeast extract, and 950 mL H2O. Adjust pH to 7.0 using NaOH and bring to final volume of 1 L with H2O. Autoclave on liquid cycle for 20 min at 15 psi and store at room temperature.
LB agar plates: Add 15 g agar to 1 L of LB. Autoclave on liquid cycle for 20 min at 15 psi. Add appropriate antibiotic once cooled to 60 °C and mix. Pour gently into 10 cm bacterial culture plates, and allow to cool and solidify at room temperature. Once solid, store at 4 °C.
Appropriate antibiotic: e.g., Sigma-Aldrich, ampicillin at 100 mg/mL stock and 100 μg/mL working concentration, diluted in sterile H2O and filtered before usage.
Endotoxin-free Midi-prep Plasmid Kit.
Nanodrop machine.
2.2. Lentivirus Production
HEK293T cells.
10 cm and 6-well sterile cell culture plates.
DMEM media: DMEM basal media, 15% fetal bovine serum, 2 mM l-glutamine, 1% nonessential amino acids, 1% penicillin/streptomycin, filtered through 0.22 μm filter and stored at 4 °C for up to 14 days.
Lipofectamine transfection reagent.
Lentiviral packaging plasmid.
Lentiviral VSV-G envelope expressing plasmid.
Opti-MEM media.
0.45 μm syringe filters.
10 mL disposable syringes.
Ultracentrifuge tubes and ultracentrifuge.
Sterile phosphate buffered saline (PBS).
Polybrene: 10 μg/mL.
Fluorescence stereomicroscope.
2.3. Lentivirus Delivery to the Mouse Eye
0.8–1.10 × 100 mm glass capillary tube.
Micropipette puller.
Sigmacote: silicone.
Safety-Lok 25 3/4 gauge × 12″ blood collection set.
1 mL Sub-Q 25 5/8 gauge slip-tip syringe.
Curved dressing forceps with serrations.
15-degree microsurgery knife.
Bupivacaine.
Institutionally approved anesthetics and pain medications.
3. Methods
3.1. Construction of Lentiviral Vectors
Purchase a lentiviral transfer plasmid from an appropriate source, such as Addgene. This transfer plasmid should contain a 5′ long terminal repeat (LTR), a packaging signal, and a 3′ LTR. In addition, it should include the promoter of interest for driving mutant calpain expression, whether it be ubiquitous expression of mutant calpain after lentiviral delivery, inducible mutant calpain expression such as using tetracycline-controlled transcriptional activation, or a cell-specific constitutive expression via a cell-specific promoter sequence. In addition, a fluorescence reporter is recommended (Fig. 1). If needed, a promoter sequence can be cloned into the lentiviral transfer plasmid using a similar cloning procedure as described below.
-
Amplify mutant calpain DNA using a polymerase chain reaction (PCR) with primers containing the appropriate restriction enzyme sites on the 5′ and 3′ ends for ligation into the lentiviral transfer plasmid after the promoter sequence (see Note 2). Primers should be designed to include the restriction sites and a hybridization sequence (see Note 3). The restriction site is the sequence needed for the specific restriction enzyme (based on the transfer plasmid restriction cut sites specific to the correct location to insert the calpain DNA, i.e., directly after the promoter sequence and not found in other locations in the plasmid backbone) to cut at either end of the amplified mutant calpain DNA (see Note 4). The hybridization sequence is the 18–21 bp that matches the ends of the mutant calpain DNA to be amplified by the primer (see Note 5). When designing the primers, it is ideal to have a melting temperature that is between 50 and 60 °C, and primer pairs should have melting temperatures within 5 C of each other. Additionally, primers perform best when kept at a 40–60% G/C content, beginning and ending with 1–2 G/C pairs. Remember to reverse complement the reverse primer, so that the primers bind facing each other and amplify the gene of interest. Furthermore, since it is important to amplify the correct calpain sequence with only the mutation of interest, high-fidelity taq polymerase should be used for the PCR. A standard PCR protocol is as follows, and this can be modified based on the enzyme, buffer, and melting temperature of the primers:
PCR Mix:- 2 μL template DNA (10–500 ng).
- 5 μL 10× buffer with MgCl2.
- 1 μL dNTP.
- 2.5 μL forward primer (10 μM stock).
- 2.5 μL reverse primer (10 μM stock).
- 0.2 μL Taq DNA polymerase (5 units/μL).
- 32.8 μL sterile H2O for a 50 μL total volume.
The basic PCR protocol rules are to keep the annealing temperature at approximately 5 °C below the primer melting temperatures. The DNA extension step is 1–2 min per kilobase of the expected mutant calpain gene. An example of a PCR run is as follows:- Step 1: Denaturation for 2 min at 94 °C.
- Step 2: Denaturation for 30 s at 94 °C.
- Step 3: Anneal for 30 s at 55 °C.
-
Step 4: Extension for 2 min at 74 °C.Repeat steps 2–4 for 25–30 cycles.
- Step 5: Extension for 5 min at 74 °C.
- Step 6: Cool to 4 °C until ready to use.
Dissolve 1 g of agarose into 100 mL of 1× TAE buffer using a microwave (see Note 6). When agarose is completely dissolved, let the solution cool for approximately 5 min (until the flask can be touched by hand), and add 0.5 μg/mL ethidium bromide (EtBr, see Note 7). Pour the solution into a gel tray with a wide gel comb, and allow to cool at room temperature until solid. Be cautious to avoid air bubbles and pour evenly into the gel tray. Once solid, place the solidified gel into a gel box (an electrophoresis system), and fill the gel box with 1× TAE until the top of the gel is lightly covered. Remove the gel comb, and make sure that air bubbles are removed from the loading lanes.
Add 6× loading dye to the PCR samples. Load the PCR product in one gel lane. In a nearby lane, load the appropriate DNA ladder for identifying the correctly amplified calpain cDNA based on its size in kilobases (see Note 8). Run the gel at approximately 100 V, running from the negative to positive electrode, until the dye has reached near the bottom of the gel (run through approximately 80% of the gel). Turn off the voltage, remove the gel from the gel box using nitrile gloves, and image the gel over a UV light source while wearing the appropriate face and eye protection. If a band is detectable at the correct size, cut out the band from the gel using a scalpel blade (Fig. 2). Purify the DNA from the gel using a gel purification kit, as can be purchased from companies such as Qiagen.
- The lentiviral transfer plasmid should be digested by the appropriate restriction enzymes, to linearize it for ligation of the cDNA (Fig. 3). It is recommended to digest the insert sequence purified in Subheading 3.1, step 4 at the same time. Heat a water bath to 37 °C. then, add the following in a 1.5 mL Eppendorf tube:
- 1–3 μg DNA (one tube for the lentiviral transfer plasmid and another for the mutant calpain DNA).
- 1 μL of the restriction enzyme(s).
- 3 μL of the appropriate 10× buffer for the restriction enzyme(s).
- 3 μL bovine serum albumin (BSA).
- Sterile H2O to a 30 μL final volume.
- Digest in the 37 °C water bath for at least 4 h (see Note 9), adding calf alkaline phosphatase (CIP) to the lentiviral transfer plasmid after 3 h.
Gel purify the digested transfer plasmid and insert following steps 3 and 4 (see Note 10).
-
Ligate the digested mutant calpain cDNA into the linearized transfer plasmid. It is recommended to start with a 1:3 (vector-insert) ratio; however this ratio can be modified as needed for ligation efficiency.
Vector-insert ligation:- 25 ng vector DNA.
- 75 ng insert DNA.
- 10× or 5× ligase buffer (see Note 11).
- 1 μL T4 DNA ligase.
- H2O to a 10 μL final volume.
- Ligate overnight at 16 °C.
Warm two antibiotic-resistant LB agar plates at room temperature (see Subheading 2 for making LB agar plates).
Chill 2 vials of chemically competent E. coli cells on ice for approximately 15 min. Add 5 μL of the ligated transfer plasmid containing the mutant calpain cDNA to one vial of competent cells and 5 μL of the linearized transfer plasmid alone to another vial of competent cells (vector only control). Very gently mix, but be careful not to damage competent cells. Leave on ice for 30 min. Warm a water bath to 42 °C. After 30 min, heat shock the competent cell vials by placing in the 42 °C water bath for 45 s. Place them back on ice for 2 min, then add 250 μL of LB broth to each tube, and warm while shaking at 37 °C for 1 h. After 1 h of recovery at 37 °C, spread 100 μL of each of the competent cell solutions onto an individual drug-resistant LB agar plate, and place it overnight at 37 °C (see Note 12). The remaining competent cell solutions can be placed at 4 °C overnight in case the experiment will need to be repeated the following day. Do not keep them longer than 24 h; repeat with new competent cells if a repeat experiment is needed after 24 h.
Compare the number of colonies grown overnight on the LB agar plates. Colony numbers should be higher on the plate containing the transfer plasmid with the ligated calpain cDNA, in comparison to the plate containing vector alone. A pipet tip can be used to scrape a small amount of each colony from the plate, and place into 3 mL of LB. Approximately 10 colonies should be picked with a separate, clean pipet tip and placed into individual, sterile tubes with LB broth (see Note 13). Allow the bacteria to grow while shaking for 12–18 h at 37 °C. After growth, purify the DNA using an endotoxin-free midi-prep kit, which can be purchased from suppliers such as Qiagen (see Note 14).
Nanodrop the samples to determine DNA concentration, and confirm correct orientation and presence of insert by sequencing. It is also important to ensure that no other mutations occurred during the cloning procedure. At this point, the plasmid is ready for lentivirus production.
Fig. 1.

Schematic of lentiviral transfer plasmid. A lentiviral transfer plasmid containing 5′ and 3′ long terminal repeats (LTRs), as well as the promoter sequence driving the expression of mutant calpain and the eukaryotic elongation factor 1 alpha (EF1a) promoter driving constitutive expression of a green fluorescent protein (eGFP)
Fig. 2.

Gel image with DNA plasmids. An example of a gel imaged over a UV light source showing the 1 kb DNA ladder (lane 1), an uncut linear DNA plasmid (lane 2), a linearized DNA plasmid after a single restriction enzyme cut (lane 3), and a cut DNA plasmid (lane 4, top band) after restriction enzyme digest of an insert sequence (bottom band)
Fig. 3.
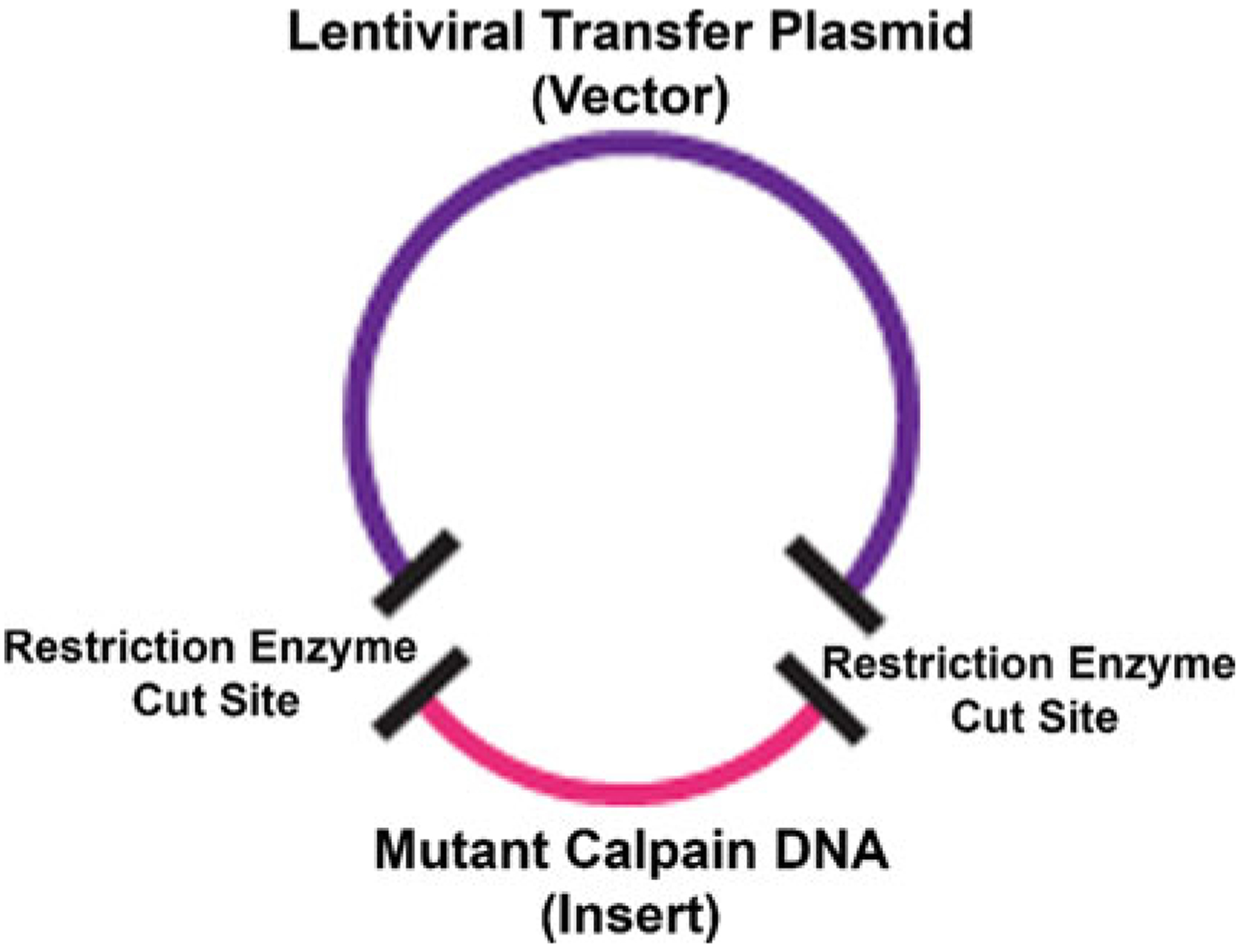
Schematic of vector and insert digestion and ligation. A vector backbone (the lentiviral transfer plasmid) will be cut by restriction enzymes (sites marked by black bands). The insert DNA sequence (mutant calpain DNA) will be digested with the same restriction enzyme cut sites (black bands). The insert DNA sequence can then be ligated with a T4 DNA ligase to the vector backbone to create a circular plasmid containing both the vector and the insert
3.2. Lentivirus Production
Each of the following steps should be performed in sterile tissue culture hoods and incubators, in a biosafety level 2+ (BL2+) room for safety when working with virus. Follow the appropriate institutional guidelines.
The day before transfection, plate 2.5 × 106 HEK293T cells in a 10 cm dish (approximately 40% confluency). After approximately 24 h, the cells should reach 70% confluency and be ready for transfection (see Note 15).
Add 5 μg of the mutant calpain lentiviral transfer plasmid DNA (from Subheading 3.1), 7.5 μg of psPAX2 packaging vector (available through Addgene), and 2.5 μg of pMD2.G VSV-G envelope expressing plasmid (available through Addgene) in a 15 mL tube.
Add 40 μL of Lipofectamine transfection reagent dropwise onto 1 mL of Opti-MEM media in another 15 mL conical tube (see Note 16). Vortex the mixture briefly, and let sit at room temperature for 5 min.
Add Opti-MEM and transfection reagent mixture to DNA, and let sit for 20 min at room temperature.
Change the media on the HEK293T cells, replacing with 9 mL of fresh DMEM media. Then add the 1 mL of Opti-MEM containing DNA to the HEK293T cells, and incubate for 24 h. From this point forward, all equipment should be disposable and bleached to kill viral particles after use.
Change to fresh DMEM media the following day, and 48 h later, begin collecting viral supernatant.
To collect viral supernatant, pipette the media from the HEK293T cells into a disposable 10 mL syringe with a 0.45 μm filter at the tip. Filter the supernatant to collect only viral particles into an ultracentrifuge tube. Spin the virus in an ultracentrifuge at approximately 20,000–25,000 rpm for 90 min at 4 °C. Remove the supernatant from the pellet, and resuspend the pellet in 100 μL sterile phosphate buffered saline (PBS). Mix via pipetting and let sit at 4 °C overnight. Aliquot virus and store at −80 °C long-term (see Note 17).
Determine viral titer. Plates 75,000 HEK293T cells into each well of a 6-well culture plate.
24 h later, thaw an aliquot of lentivirus using a 37 °C water bath. Prepare five different dilutions of the virus in DMEM media containing 10 μg/mL polybrene. Recommended dilutions are 1:500, 1:200, 1:100, 1:50, and 1:25 in 1.5 mL total media. Remove the media from 5 wells of the 6-well HEK293T cell plate, and add each viral dilution to an individual well. Count the number of HEK293T cells in the unchanged well. Incubate for 48 h.
After 48 h, aspirate media from the HEK293T cells, and replace with 1 mL sterile PBS. Take cells to a fluorescent microscope, and count the number of fluorescent cells (i.e., cells that received the virus). If a well has more than 50% of cells fluorescent, titer concentration can be miscalculated, so only count wells with less than half of the cells transfected by the virus.
Calculate the viral titer in transduction units (TU) per mL. Multiply the number of cells that were transduced (the number of HEK293T cells in the untreated well from Subheading 3.2, step 10) by the percent of fluorescent cells (Subheading 3.2, step 11) and by the dilution factor (e.g., 200, if it was the well receiving the 1:200 viral dilution). Then divide this number by 1.5 mL, i.e., the volume of media containing virus that was added to each well (see Note 18).
3.3. Delivery of Lentivirus to the Mouse Eye
Further details and video of the subretinal injection procedure into the perinatal mouse eye have been previously published [10]. Here, details for both a subretinal and intravitreal injection are described to deliver the lentivirus to the mouse eye at either perinatal or adult stages of development.
Using a Sutter P-97 pipette puller or other similar equipment, heat and pull a glass capillary tube to create a 100-μm-diameter needle with a blunt, open tip (see Note 19). This allows for injection into the intravitreal or subretinal space without damage to the eye while also providing a clear needle for visualization of the lentivirus during the injection procedure. Aspirate silicone through the needle in order to coat the inner walls, and provide passage of the virus through the needle without the virus sticking to the inner walls. After aspiration of silicone through the needle, allow for the needle to dry in a sterile container.
Prepare the surgical suite based on institutional animal welfare guidelines and protocols. All procedures should be performed within the barrier facility if animals are to be kept alive after surgery.
Obtain a 25 3/4 gauge blood collection set with Luer lock, remove the needle from the tubing by cutting the tubing, and attach the 100 μm needle to the cut end of the plastic tubing. Fill a 1 mL Sub-Q 26 5/8 gauge slip-tip syringe with sterile saline, remove the needle end of the syringe, and attach the syringe to the Luer lock. Eject some of the sterile saline through the tubing and 100 μm needle, ensuring that the setup has been prepared appropriately and there is no leakage. Keep the prepared injection apparatus on a sterile, clean surface during the surgical procedure.
Prepare the animal for surgery following the locally approved animal handling protocol, as anesthesia will vary based on the institution and age of the mouse (see Note 20). When the mouse is fully under anesthesia, with no movement or response to toe pinches, the surgical procedure can begin. The following procedure should be performed under a dissection microscope for close visualization of the eye (see Note 21).
Proptose the eye by pulling apart the eyelids and pinching them underneath the eye to hold it in the proptosed position (see Note 22). Applying slight pressure on either side underneath the eye with dressing forceps can help to hold the eye in the proptosed position.
There are two main routes for delivering lentivirus to the eye. One delivers the virus to the subretinal space, where it is localized between the retinal pigment epithelium (RPE) and the photoreceptor cells. The other delivers the virus to the intravitreal space, where it will mainly affect the bipolar and ganglion cells of the eye (Fig. 4). For a subretinal injection, using a 15-degree microsurgery blade, make a small, scleral incision at or directly posterior to the globe’s equator (see Note 23). The incision should be just large enough for the insertion of the 100 μm needle, and no larger. In pigmented eyes, the darker choroid-RPE will become visible at the incision site. If the animal is albino and the eye is not pigmented, it is recommended to mark the tip of the 15-degree surgical blade with a surgical marking pen, which will leave an ink spot to denote the incision site. For an intravitreal injection, small scrapes can be made with the 15-degree microsurgery blade until a clear fluid (the vitreous) refluxes from the incision site.
Aspirate a small, approximately 1 μL bubble of air into the end of the injection needle, in order to separate the lentivirus from the sterile saline. Place the appropriate amount of lentivirus, with a recommended viral titer of 2 × 107 TU/mL (Table 1), onto a sterile, flat surface such as the inner lid of a 10 cm culture plate. For the subretinal injection, aspirate the lentivirus into the tip of the 100 μm needle, and carefully insert the needle into the incision site and advance it parallel to the outer eye wall in order to enter the subretinal space. For intravitreal injection, insert the needle into the deeper incision site, and advance the needle just slightly parallel to the outer wall to avoid hitting the lens of the eye. It is not necessary to go too deep within the vitreous space for this injection as the virus can more easily spread throughout the vitreous.
Apply very light pressure on the syringe plunger to inject the lentivirus. In the subretinal space, moderate back pressure will be felt during injection. The virus creates a subretinal bleb between the RPE and the photoreceptor cells, causing a temporary retinal detachment at the injection site that will heal within 24 h (see Note 24). For the intravitreal injection, very little pressure will be felt, and the virus will be easily expelled into the vitreous space. Care must be taken to inject the lentivirus without causing injection of the air bubble or reflux of the virus back out from the injection site. It is also important to remove the needle following the same incision course, to avoid excess damage to the retina and eye (see Note 25).
Place the eye back under the eyelids by applying slight pressure with forceps and pushing down on the eye. Provide pain medication as approved by the institutional animal protocol and welfare guidelines. It is recommended to use a 25 gauge needle to place a single drop of 0.25% bupivacaine (diluted in sterile saline) at the injection site for the comfort of the mouse. Allow the mouse to wake from anesthesia on a heating pad, and then move it to a clean cage after the mouse is ambulatory (see Note 26). Mice should be monitored after surgery following the institutional guidelines for any signs of distress, and if present, follow the veterinary guidelines for further treatment.
Fig. 4.
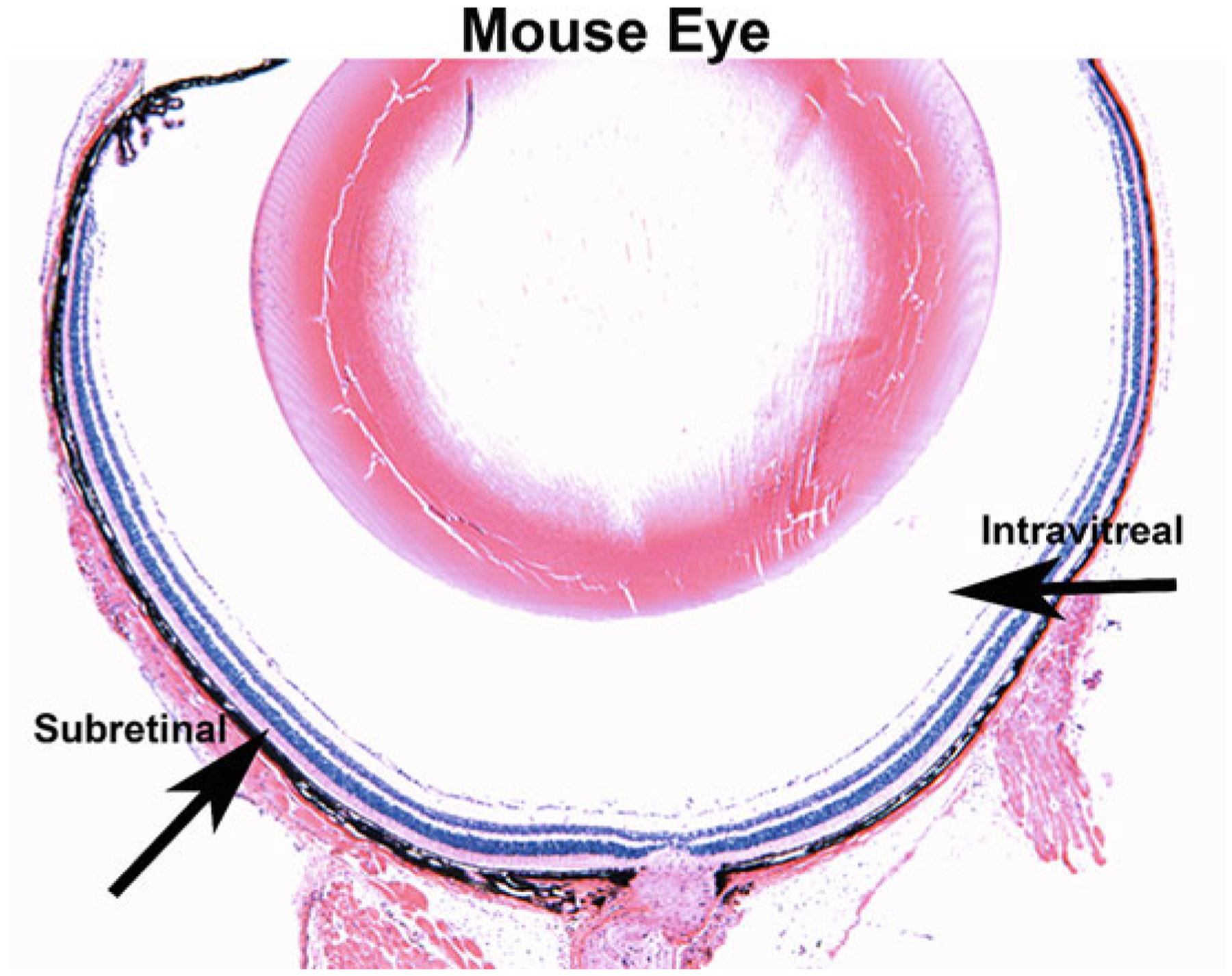
Subretinal and intravitreal injection sites in the mouse eye. A cross-section of a mouse eye stained with hematoxylin and eosin. Subretinal and intravitreal injection locations are marked by black arrows, where the subretinal injection creates a viral bleb between the photoreceptor cells and the retinal pigment epithelium (RPE) and the intravitreal injection places the virus into the vitreous fluid of the eye
Table 1.
Amount of virus based on injection location and age of the mouse. Amounts of virus in microliters that are recommended for injection into the subretinal and intravitreal space of the mouse eye, at perinatal and adult stages of development
| Age | Injection location | Amount of virus (μL) |
|---|---|---|
| Perinatal | Subretinal | 0.5–0.8 |
| Perinatal | Intravitreal | 0.8–1.0 |
| Adult | Subretinal | 0.8–1.0 |
| Adult | Intravitreal | 1.0–1.5 |
4. Notes
TBE can be used instead of TAE.
It is best to amplify the mutant calpain from plasmid DNA, provided by companies such as OriGene or Addgene.
A leader sequence can be added in the primer design to assist with the restriction enzyme digestion. The leader sequence is approximately 3–6 base pairs (bp) added to the ends of the primer. If used, the PCR product should be digested by the appropriate restriction enzymes prior to ligation.
Make sure that the restriction enzymes will not cut within the mutant calpain DNA.
Make sure to include the start site and full length of the mutant calpain DNA for correct translation in vivo.
This takes approximately 2 min, and stopping and swirling the solution while heating help dissolve the agarose without boiling over the TAE.
EtBr is a mutagen and should be handled in a fume hood with nitrile gloves.
For cutting DNA bands, it is helpful to leave an empty lane between samples when loading and running the gel.
The digestion step can be extended if the restriction enzyme does not have STAR activity, as it will not incorrectly cut outside of its designated sequence.
It is recommended to run uncut products on the gel as well as the digested sample to ensure that the correct linearized transfer plasmid and insert are purified from the gel.
Avoid too many freeze thaws of ligase buffer containing ATP by making small aliquots of the ligase buffer when first purchasing and only thawing each aliquot when needed.
Some plasmids grow more efficiently at 30 °C; check plasmid information before growing overnight.
A pipet tip can be used to scrape the remainder of each individual colony. The tip of the pipet can be inserted into a PCR tube containing PCR mix with primers designed to detect the vector backbone and insert sequence. Swirling the pipet tip in the PCR mix provides the plasmid DNA for amplification, and this PCR method can be used to screen the colonies before growing overnight for the presence of the insert and its orientation in the vector backbone.
A diagnostic restriction enzyme digest can be performed before sequencing to determine whether or not the insert is present in the plasmid. 500 ng of DNA is typically used for a diagnostic digest, and only 1 h of digestion is necessary for visualizing the insert after running the product on a gel.
HEK293T cells can lift easily off of a cell culture plate. Therefore, autoclaved 0.2% gelatin can be added to the culture plate for 20 min. Gelatin is then aspirated, and the culture plate is washed once with sterile PBS and can be used for plating the HEK293T cells on the gelatin coating.
Other transfection reagents can be used; it is important to follow the manufacturer’s protocols depending on which one is used for the lentiviral production.
Virus is good at −80 °C for at least 1 year, but viral titer may slightly decline over time and should be checked periodically after thawing aliquots.
Calculating more than one of the wells (the five different dilutions) and averaging the titer calculated from each one are recommended for better accuracy of the TU/mL.
Needles smaller than 100 μm can be used; however it is known that 100 μm needles allow for the injection of viruses without risk of damage to the eye or clogging of the needle. Also, when preparing the pulled capillary needles, if glass or debris ends up inside the needle, it can be washed by flushing with 70% ethanol. Allow for the ethanol to fully evaporate or flush afterward with sterile saline before use.
Intraperitoneal injection of anesthetic is recommended for this protocol, as the nose cone for isoflurane can block access to the eye.
The entire surgical procedure after anesthesia should take under 5 min, and therefore a heating pad during the procedure is not necessary and there is no risk for cataracts. However, when beginning to learn this procedure, more time may be needed. Uninjected adult eyes should be covered with a lubricant, and the procedure may need to be performed on a heated surface to maintain normal body temperature of the mouse.
In perinatal mice under 2 weeks of age, Vannas scissors should be used to cut at the eyelid indentation to open the eyelids. Be careful to not make too large of an incision, under 1.5 mm, as the eye will not remain proptosed and will slip back under the eyelids. If needed, a circular punch the size of a mouse eye can be made in a hardened, plastic surface, and this can be placed over the eye after proptosing the eye; the plastic can be pressed down with a small amount of pressure, being careful not to harm the animal or inhibit breathing, and this can help keep the eye accessible during the surgical procedure.
To avoid making too deep of an initial cut and damaging the retina, it is recommended to make a couple very short scrapes to open an incision into the scleral space, rather than one deeper cut. A small, acupuncture needle or similar needle can also be used to make the initial incision. Anything with a small, sharp tip that will reduce damage or size of the incision into the eye.
For subretinal injection, holding the needle in place for an additional 15 s within the subretinal space after injection of the lentivirus can help to avoid the back pressure flushing the virus back out of the incision site.
Although the incision site and/or temporary retinal detachment will heal within 24 h, the eye will remain soft for approximately 1 week, so further injections should be avoided during this timeframe.
For perinatal mice, gentle toe pinches can help them fully recover from anesthetic and regain their pink coloring before being placed back with their mother for warmth and care.
Acknowledgment
VBM is supported by NIH grants [R01EY026682, R01EY024665, R01EY025225, R01EY024698, R21AG050437, and P30EY026877], The Doris Duke Charitable Foundation Grant #2013103, and Research to Prevent Blindness (RPB), New York, NY.
References
- 1.Wert KJ, Bassuk AG, Wu WH, Gakhar L, Coglan D, Mahajan M, Wu S, Yang J, Lin CS, Tsang SH, Mahajan VB (2015) CAPN5 mutation in hereditary uveitis: the R243L mutation increases calpain catalytic activity and triggers intraocular inflammation in a mouse model. Hum Mol Genet 24:4584–4598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bassuk AG, Yeh S, Wu S, Martin DF, Tsang SH, Gakhar L, Mahajan VB (2015) Structural modeling of a novel CAPN5 mutation that causes uveitis and neovascular retinal detachment. PLoS One 10:e0122352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wert KJ, Skeie JM, Bassuk AG, Olivier AK, Tsang SH, Mahajan VB (2014) Functional validation of a human CAPN5 exome variant by lentiviral transduction into mouse retina. Hum Mol Genet 23:2665–2677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mahajan VB, Lin JH (2013) Lymphocyte infiltration in CAPN5 autosomal dominant neovascular inflammatory vitreoretinopathy. Clin Ophthalmol 7:1339–1345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rowell HA, Bassuk AG, Mahajan VB (2012) Monozygotic twins with CAPN5 autosomal dominant neovascular inflammatory vitreoretinopathy. Clin Ophthalmol 6:2037–2044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mahajan VB, Skeie JM, Bassuk AG, Fingert JH, Braun TA, Daggett HT, Folk JC, Sheffield VC, Stone EM (2012) Calpain-5 mutations cause autoimmune uveitis, retinal neovascularization, and photoreceptor degeneration. PLoS Genet 8:e1003001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pastor JC, de la Rua ER, Martin F (2002) Proliferative vitreoretinopathy: risk factors and pathobiology. Prog Retin Eye Res 21:127–144 [DOI] [PubMed] [Google Scholar]
- 8.Frank RN (2004) Diabetic retinopathy. N Engl J Med 350:48–58 [DOI] [PubMed] [Google Scholar]
- 9.Caspi RR (2010) A look at autoimmunity and inflammation in the eye. J Clin Invest 120:3073–3083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wert KJ, Skeie JM, Davis RJ, Tsang SH, Mahajan VB (2012) Subretinal injection of gene therapy vectors and stem cells in the perinatal mouse eye. J Vis Exp 69:4286. [DOI] [PMC free article] [PubMed] [Google Scholar]