

Abstract
Activating mutations in PIK3CA are frequent in human breast cancer, and phosphoinositide 3-kinase alpha (PI3Kα) inhibitors have been approved for therapy. To characterize determinants of sensitivity to these agents, we analyzed PIK3CA-mutant cancer genomes and observed the presence of multiple PIK3CA mutations in 12 to 15% of breast cancers and other tumor types, most of which (95%) are double mutations. Double PIK3CA mutations are in cis on the same allele and result in increased PI3K activity, enhanced downstream signaling, increased cell proliferation, and tumor growth. The biochemical mechanisms of dual mutations include increased disruption of p110α binding to the inhibitory subunit p85α, which relieves its catalytic inhibition, and increased p110α membrane lipid binding. Double PIK3CA mutations predict increased sensitivity to PI3Kα inhibitors compared with single-hotspot mutations.
Across all human cancers, PIK3CA is the most frequently mutated oncogene (1). It codes for p110α, the catalytic subunit of the phosphoinositide 3-kinase alpha (PI3Kα) complex, which is necessary for normal growth and proliferation (2). PI3Kα, which is composed of p110α and the regulatory subunit p85α, catalyzes the phosphorylation of the lipid phosphatidylinositol 4,5-bisphosphate (PIP2) to phosphatidylinositol 3,4,5-trisphosphate (PIP3), which in turn initiates a downstream signaling cascade involving the activation of AKT and mammalian target of rapamycin (mTOR) (3). PI3Kα is activated by binding to membrane-bound receptor tyrosine kinases (RTKs) and can be constitutively activated by oncogenic mutations. Many distinct cancer-associated PIK3CA mutations have been identified, including hotspot single–amino acid substitutions in the helical (E542K and E545K) or kinase (H1047R) domains (4). These mutations are considered oncogenic in multiple cancer histologies (5, 6), including breast cancer (7, 8), in which PIK3CA mutations are present in 40% of estrogen receptor–positive (ER+), human epidermal growth factor receptor 2− negative (HER2−) primary and metastatic tumors (9), and are a target for cancer therapy.
On the basis of this hypothesis, several PI3K inhibitors with various degrees of specificity have been studied in clinical trials in patients with PIK3CA-mutant breast cancer. Initial studies with less specific compounds showed activity, although toxicities precluded their clinical development (10–13). More recently, the selective PI3Kα inhibitor alpelisib has shown improved tolerability (14–16), and a large randomized phase 3 clinical trial has shown improved progression-free survival (PFS) in patients with ER+ PIK3CA-mutant metastatic breast cancer (17). As a result, alpelisib has been recently approved by the Food and Drug Administration for therapy in patients with advanced ER+ breast cancer harboring PIK3CA mutations.
In early clinical trials, we had observed that there was also a population of patients who displayed deep and prolonged clinical benefit from alpelisib (14, 18). In search of genomic signals of improved clinical response to PI3K inhibitors, we identified double PIK3CA mutations as a biomarker candidate. This finding prompted us to undertake a comprehensive analysis of the prevalence of these mutations and investigate their potential biological relevance and correlation with sensitivity to PI3Kα inhibitors.
Results
Durable responses to alpelisib in some patients with double–PIK3CA-mutant breast cancer
We previously reported on a patient with breast cancer who had a highly durable response to alpelisib monotherapy and eventually developed acquired resistance through convergent PTEN mutations (18). In the primary and metastatic lesions of this patient, we also detected the presence of double PIK3CA mutations, with equal variant allele frequencies (VAFs) of both mutations (fig. S1A). We analyzed data from a phase 1 clinical trial (n = 51 patients) investigating alpelisib with an aromatase inhibitor in heavily pretreated patients with ER+ metastatic breast cancer (NCT01870505), in which tumor PIK3CA mutational status was determined by the next-generation sequencing (NGS) platform MSK-IMPACT (Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets) (19). Patients with double–PIK3CA-mutant tumors had a longer median PFS than did patients with single-mutant tumors or wild-type (WT) tumors, but this was not statistically significant because of the small number of cases (fig. S1B). We hypothesized that this sensitivity was due to PI3K inhibition rather than the hormonal therapy because patients with double–PIK3CA-mutant tumors do not have improved PFS when they are treated with aromatase inhibition or the ER degrader fulvestrant alone compared with patients with single-mutant or WT tumors on retrospective analysis (fig. S1C).
Double–PIK3CA-mutant tumors are frequent in breast cancer and other tumor histologies
We analyzed a publicly available cancer-patient cohort (n = 70,754) across different histologies from the cBioPortal for Cancer Genomics (20, 21) and identified a total of 4526 PIK3CA-mutant tumors, 576 (13%) of which contained multiple PIK3CA mutations (Fig. 1A and table S1). We recapitulated these findings using a cohort from our institution enriched for metastatic tumors (n = 28,139) across different cancer types sequenced by MSK-IMPACT (19). We identified 3740 PIK3CA-mutant tumors, 451 (12%) of which contained multiple PIK3CA mutations (fig. S2A and table S2). In both the cBioPortal and MSK-IMPACT cohorts, breast, uterine, and colorectal cancers had the greatest number of multiple–PIK3CA-mutant tumors. We also analyzed individual breast cancer subsets and found similar frequencies of multiple–PIK3CA-mutant breast cancer in METABRIC (Molecular Taxonomy of Breast Cancer International Consortium) (13%) (22), TCGA (The Cancer Genome Atlas) (11%) (23), and other data sets (8%) (fig. S2B) (24–26). Most (88 to 96%) multiple–PIK3CA-mutant tumors in all of these patient cohorts carry exactly two mutations (fig. S2C).
Fig. 1. Double PIK3CA mutations are frequent across all cancers, including breast cancer, and are in cis on the same allele.
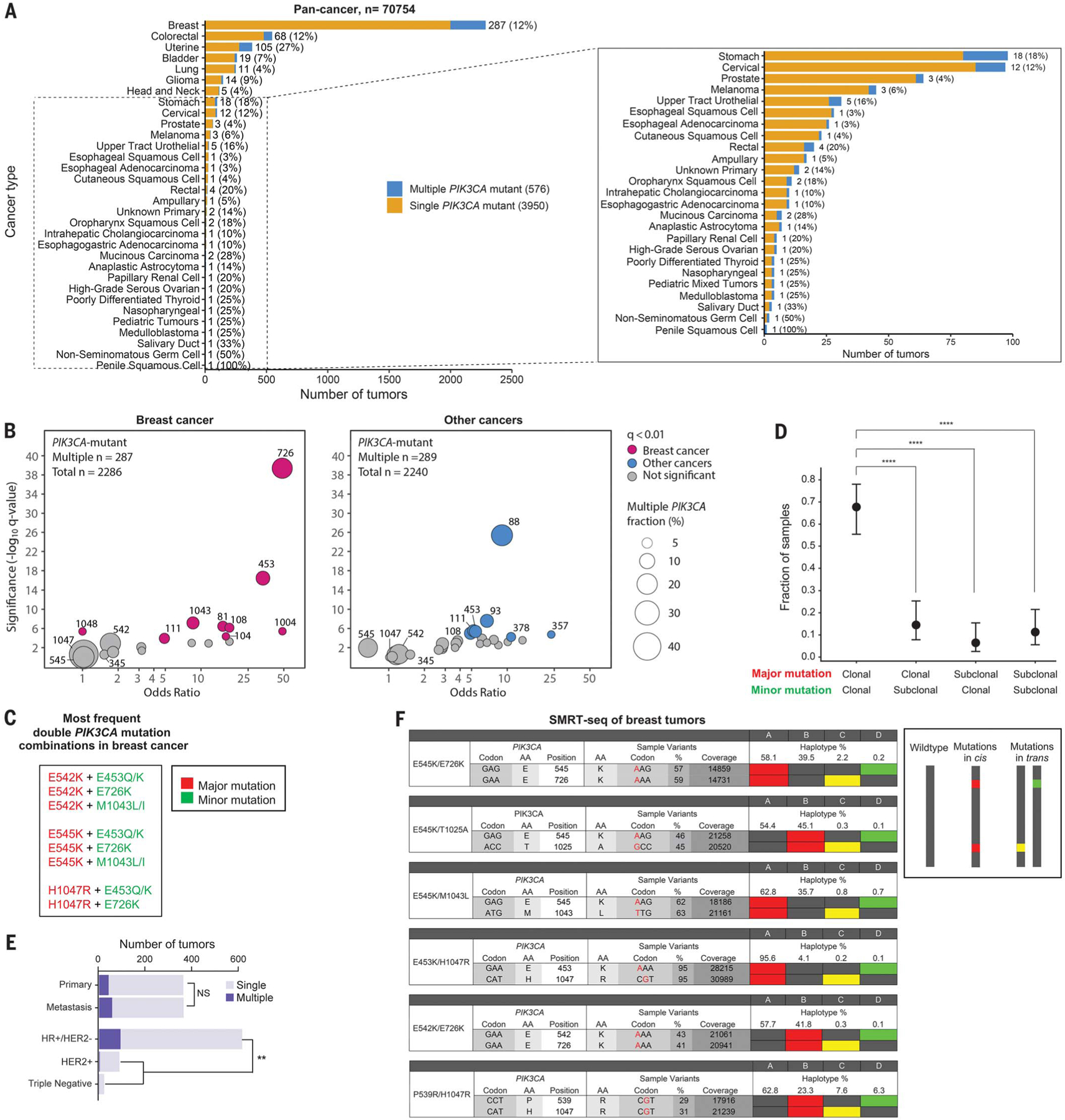
(A) Bar plot showing number and frequency of multiple–PIK3CA-mutant tumors among PIK3CA-mutant tumors across different cancer histologies (cBioPortal). Cancer types with <100 cases with PIK3CA mutations are magnified in the inset. (B) Codon enrichment analysis of significantly recurrent PIK3CA amino acid mutations in multiple–PIK3CA-mutant tumors (cBioPortal). Samples containing the same double mutant are depicted as a circle, sized according to the number of samples. Samples colored in pink (breast tumors) or blue (non-breast tumors) are those with an FDR-corrected P value (q-value) <0.01. Statistics were calculated independently by using two-sided Fisher’s exact tests. (C) List of the most frequent double–PIK3CA-mutation combinations in breast cancer (cBioPortal and MSK IMPACT). (D) Clonality analysis by FACETS (27) of multiple–PIK3CA-mutant breast tumors containing major and minor mutations (n = 62 tumors) (MSKCC dataset) (9). Data are mean ± 95% CI. ****P < 0.0001 by two-sided Fisher’s exact test. (E) Bar chart of frequency of multiple–PIK3CA-mutant breast tumors among primary versus metastatic cancers and by receptor subtype (MSKCC dataset) (9). NS, not significant; **P < 0.01 by two-sided Fisher’s exact test. (F) SMRT-seq phasing of allelic configuration of PIK3CA double-mutant breast tumors. Cis mutations are shown as red vertical squares, trans mutations are single yellow or green squares, and WT sequences are gray vertical squares, in order of amplicon frequency. Single-letter abbreviations amino acid residues: A, Ala; C, Cys; D, Asp; E, Glu; F, Phe; G, Gly; H, His; I, Ile; K, Lys; L, Leu; M, Met; N, Asn; P, Pro; Q, Gln; R, Arg; S, Ser; T, Thr; V, Val; W, Trp; Y, Tyr.
We next investigated potential patterns of comutation. In most double–PIK3CA-mutant breast tumors, one of the mutations was either a helical or kinase domain major-hotspot mutation (involving E542, E545, or H1047) (fig. S2D and tables S1 and S2), which are the most common alterations in single-mutant tumors. We performed codon enrichment analysis and determined that second-site E726, E453, and M1043 mutations were most significantly enriched in multiple-mutant tumors compared with single-mutant tumors in the cBioPortal (Fig. 1B) and MSK-IMPACT (fig. S1E) breast cancer datasets; this is compared with E542, E545, or H1047 mutations, which are equally distributed between single- and multiple-mutant tumors. Almost all tumors containing second-site E726, E453, or M1043 mutations in cBioPortal (n = 70; 88%) and MSK-IMPACT (n = 43; 100%) also contained E542, E545, or H1047 mutations (fig. S2D and tables S1 and S2). In the non–breast-cancer cohorts, E88 and E93 second-site mutations were the most statistically significantly enriched (Fig. 1B and fig. S2E). Thus, the most frequent double–PIK3CA-mutant tumor combinations in breast cancer are composed of a canonical “major-mutant” hotspot (involving either E542, E545, or H1047) combined with a second “minor-mutant” site (involving either E453, E726, or M1043) (Fig. 1C). These recurrent mutational sites appear to be specific to breast cancer compared with other cancer histologies.
To determine whether double mutants are in the same cell, we used the FACETS (Fraction and Allele-Specific Copy Number Estimates from Tumor Sequencing) tool (27) to analyze clonality of double-mutant tumors from a large clinically annotated breast cancer cohort (n = 1918) that our group has recently published (9). Of the tumors that contained the most frequent double-mutant combinations in breast cancer—E545K or H1047R major hotspots and E453, E726, or M1043 minor mutations—most (42 of 62; 68%) are clonal for both mutations (Fig. 1D). This was concordant with interpatient VAFs of multiple-mutant breast tumors from cBioPortal (fig. S2F), which follow a 1:1 linear distribution. We performed additional clinicogenomic analysis of double–PIK3CA-mutant breast tumors from METABRIC (22) and our published cohort (9) (Fig. 1E and fig. S3). Double PIK3CA mutations are enriched in hormone receptor–positive (HR+), HER2−breast cancers compared with other receptor subtypes (including HER2+ and triple-negative breast cancers) (15.4% versus 5.4%; P = 0.004) (Fig. 1E) and occur at similar frequencies in therapy-naïve primary tumors and metastatic tumors (11.6% versus 15.7%; P = 0.130) (Fig. 1E). Invasive disease–free survival and overall survival are similar between patients with multiple and single PIK3CA mutations in univariate and multivariate analyses (fig. S4).
Double PIK3CA mutations are in cis on the same allele
Any two mutations in the same gene in a cell can be on the same allele (in cis) or on separate alleles (in trans). Because double PIK3CA mutations are most often clonal (in the same cell), establishing their allelic configuration is important because cis mutations would result in a single protein with two mutations, whereas trans mutations would result in two proteins with separate individual mutations, and these could have different functional consequences.
To study the allelic configuration of double mutations, we faced several technical hurdles based on our observation that the most frequent double PIK3CA mutants are located far apart in genomic DNA (fig. S2A). An initial limitation is that tumor specimens are classically preserved as formalin-fixed, paraffin-embedded (FFPE) samples, which results in fragmented genomic DNA and RNA of ~200 nucleotides, prohibiting the phasing of recurrent double PIK3CA mutations (fig. S5A). We overcame this by obtaining fresh, frozen tumor samples from patients who were known, by MSK-IMPACT, to carry two PIK3CA mutations in their tumors. This could be done only for patients with metastatic disease (because most patients who underwent primary breast tumor resection had only FFPE samples available). In addition, even with fresh, frozen tumor samples, current NGS library construction methods limit the allelic phasing of fragments to ~300 nucleotides, again prohibiting this type of analysis for the most recurrent double PIK3CA mutations (fig. S5A). To resolve this technical limitation, we applied two alternative approaches. First, from our initial double-mutant E545K/E726K breast tumor with high VAF, we performed bacterial colony Sanger sequencing and found that 14 of the 14 (100%) mutant cDNA inserts contained double mutations in cis (fig. S5B). The same technique was applied to the double–PIK3CA-mutant BT20 breast cancer cell line P539R/H1047R, in which 13 of the 14 (92%) mutant cDNA inserts contained double mutations in cis (fig. S5C).
Although Sanger sequencing of bacterial colonies can be used to determine the allelic configuration of double mutants, it is a heterologous system, exhibits low efficiency in biopsies with low cancer-cell fraction, and is indirect for some double mutants that are far apart in the gene and that require separate priming reactions. To solve these limitations, we used single-molecule real-time sequencing (SMRT-seq) (fig. S5D) (28), which uses long-range sequencing of circular DNA templates, enabling direct phasing of the allelic configuration of all recurrent double PIK3CA mutants that are far apart in the gene.
We first analyzed BT20 cells as a control and three additional double–PIK3CA-mutant breast cancer cell lines with unknown allelic configurations: CAL148 (D350N/H1047R), MDA-MB-361 (E545K/K567R), and HCC202 (E545K/L866F) (fig. S5E). Whereas BT20 and CAL148 cell lines contain cis mutations, MDA-MB-361 cells contain trans mutations. HCC202 contains E545K and L866F mutations in trans but also E545K and I391M mutations in cis. Thus, we concluded that SMRT-seq is feasible to phase the allelic configuration of PIK3CA mutations and recurate known cell line mutations.
Six fresh tumor samples from patients were obtained, including samples with E542K-E726K, E545K-E726K, E453K-H1047R, and E545K-M1043L double PIK3CA mutations, which are representative of the most frequent double mutants in breast cancer (Fig. 1C). Samples were analyzed by SMRT-seq, and all contained double mutations in cis (Fig. 1F).
We also used MSK-IMPACT (table S3) and RNA sequencing (table S4) on breast tumors from TCGA (23) to investigate the allelic configuration of less frequent double PIK3CA mutants located close together in the gene. These findings support that double PIK3CA mutations are mainly found as cis mutations in breast cancer.
Double PIK3CA mutations in cis hyperactivate PI3K and enhance proliferation
We hypothesized that cis PIK3CA mutants demonstrate a hypermorphic function because they code for a single protein molecule with both major and minor mutations of varying activating capacities. Taken individually, the minor PIK3CA mutations E453, E726, and M1043 demonstrate mild transforming activity in vitro compared with the major mutations E542, E545, and H1047 (29). E542K and E545K single-hotspot mutants are predicted to have similar mechanisms of activation (30), and we posited that mutations at the same amino acid position also have similar mechanisms. Thus, we explored the effects of double PIK3CA mutations on the activation of the PI3K pathway and cell proliferation focusing on the E453Q/E545K, E453Q/H1047R, E545K/E726K, E726K/H1047R, and E545K/M1043L cis mutants and their constituent single mutants.
We stably overexpressed each cis mutant and constituent single mutant in MCF10A breast epithelial cells and NIH-3T3 mouse fibroblasts, both of which have been previously used to characterize PIK3CA mutations (7, 31), and in MCF7 ER+ breast cancer cells engineered to carry a PIK3CA WT background (32). Double PIK3CA mutations in cis increased downstream PI3K pathway signaling when compared with single-hotspot mutants, as evidenced by increased phosphorylation of AKT, PRAS40, and S6 under serum starvation in MCF10A cells (Fig. 2A), NIH-3T3 cells (Fig. 2B), and MCF7 cells (fig. S6A). All cis mutants are capable of additional stimulation by growth factor, as shown by platelet-derived growth factor- BB (PDGF-BB) or insulin-like growth factor 1 (IGF1) stimulation of NIH-3T3 cells (fig. S6B), although certain phosphoproteins are not further stimulated by growth factor (e.g., pS6 under IGF-1 stimulation). Cis mutants prolonged downstream signaling kinetics, as demonstrated by the E726K/H1047R MCF10A mutant, which maintains increased phosphorylated AKT up to 48 hours (Fig. 2C). Cis mutants displayed increased proliferation compared with single-hotspot mutants (Fig. 2D and fig. S6C). By contrast, mutations in trans do not increase MCF10A cell signaling (fig. S6D) and growth proliferation (fig. S6E) more than single mutations do, as demonstrated by E726K and H1047R.
Fig. 2. Double PIK3CA mutations in cis activate the PI3K pathway in vitro and in vivo more than single PIK3CA mutations.
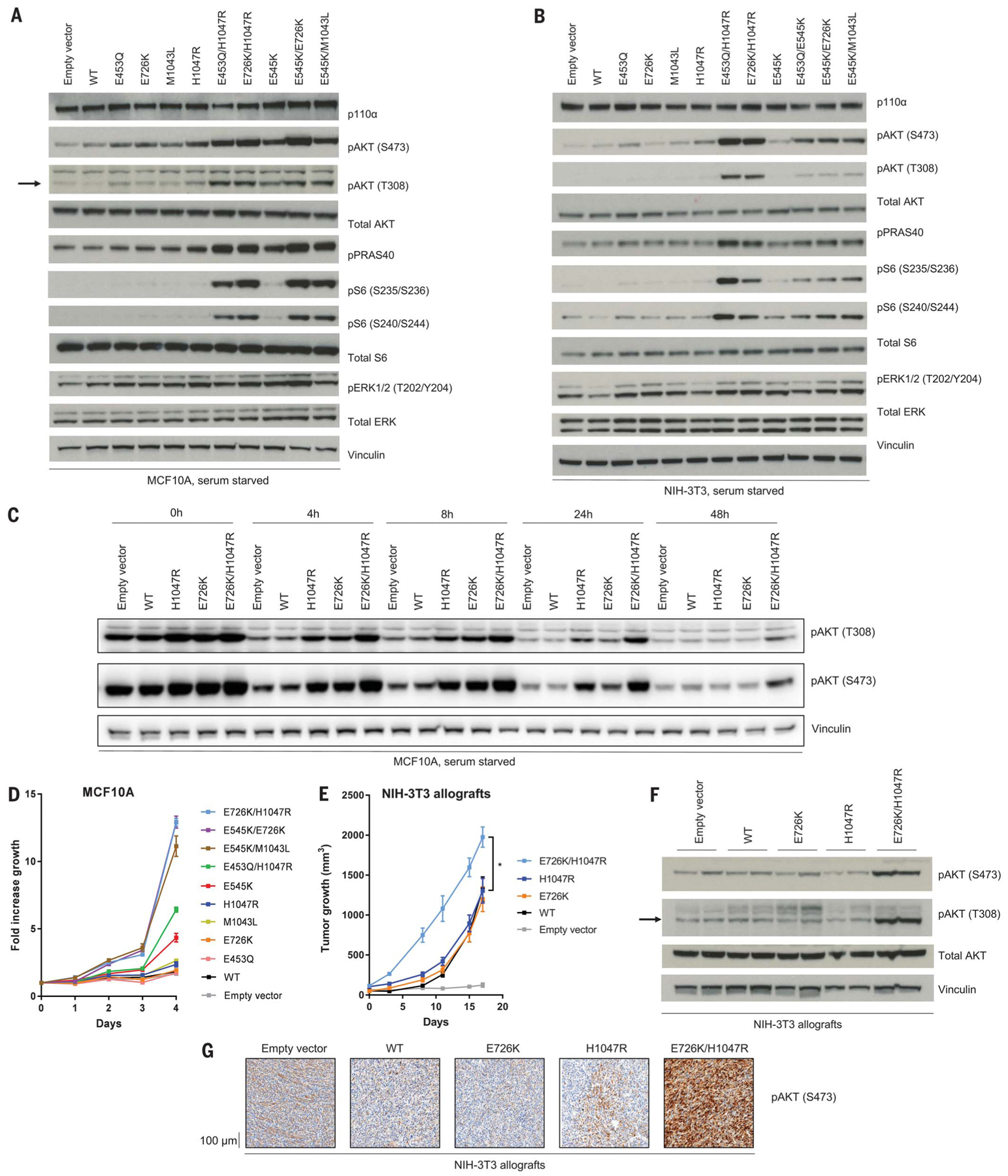
(A and B) Western blotting of PI3K effectors of PIK3CA mutant stably transduced (A) MCF10A cells and (B) NIH-3T3 cells, serum starved for 1 day. The arrow indicates pAKT (T308). (C) Western blotting of PI3K effectors of PIK3CA mutant MCF10A cells, serum starved for the indicated time points. (D) Growth proliferation time course of PIK3CA mutant MCF10A cells serum starved over 4 days. Data are mean ± SEM (n = 3 replicates). (E) NIH-3T3 murine allograft tumor growth. Data are mean ± SEM (n = 4, 4, 4, 4, 3). *P < 0.05 by two-sided Student’s t test. (F and G) Western blotting for PI3K effectors (F) and immunohistochemistry of pAKT (S473) (G) of PIK3CA mutant murine allograft tumors.
We next investigated whether cis mutant cells enhance tumor growth in vivo compared with single mutants. NIH-3T3 allografts expressing the E726K/H1047R cis mutant demonstrated increased tumor growth compared with H1047R or E726K (Fig. 2E). There was no difference in tumor growth between the single mutants and WT, supporting the notion that in some model systems, single-hotspot PIK3CA mutations are weakly oncogenic (33, 34). In parallel to the enhanced tumorigenicity, and in concordance with our observations in cell culture, E726K/H1047R cis mutant NIH-3T3 tumors exhibited higher activation of the PI3K pathway, as shown by increased phosphorylation of AKT on western blotting (Fig. 2F) and by immunohistochemistry (Fig. 2G).
Double PIK3CA mutations in cis combine biochemical effects of single mutants
We next investigated the biochemical mechanism of activation of PI3Kα by double PIK3CA mutations in cis. p110α is constitutively bound to p85α, and this interaction stabilizes its structure, inhibiting its catalytic activity (35, 36). The prevailing model of PI3Kα activation occurs through the engagement of p85α with phosphotyrosines on RTK signaling complexes. This interaction translates to a partial release of p85α from p110α, which relieves catalytic inhibition (37). Single oncogenic mutations recapitulate these events in distinctive ways in the absence of phosphotyrosine binding, by weakening the interactions between p110α and p85α (mutants that we term “disrupters”) (37) or promoting binding to the membrane (mutants that we term “binders”) (37–39). We structurally mapped the constitutive single mutants and postulated that E545K and E453Q act as disrupters, whereas E726K, H1047R, and M1043L act as binders (Fig. 3, A and B; fig. S7, A to D, and supplementary text). Notably, none of these mutants are involved directly in the PI3Kα catalytic mechanism (40). We purified recombinant full-length PI3Kα complexes containing single and double cis p110α mutations (fig. S8A and supplementary text) to dissect the biochemical mechanisms by which these double PIK3CA mutations in cis modulate p85α disruption, lipid binding, and kinase activity.
Fig. 3. Double PIK3CA mutations in cis activate PI3K through a combination protein disrupter-membrane binder mechanism.
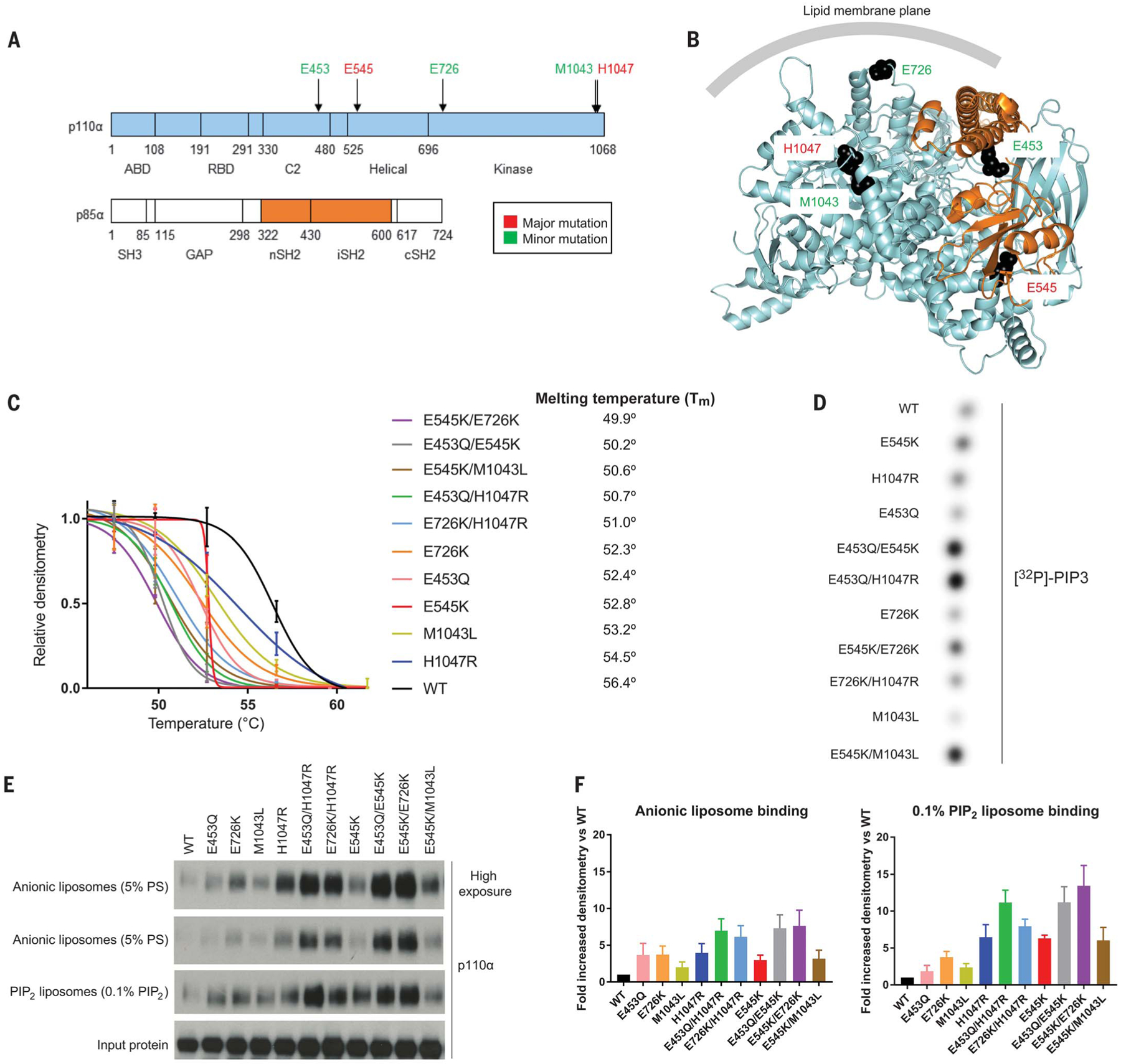
(A) Domain schematic of p110α and p85α with minor and major mutation sites indicated. ABD, adaptor binding domain; C2, protein kinase C homology–2 domain; GAP, GTPase activating protein; SH3, Src homology 3; RBD, Ras binding domain. (B) Crystal structure of PI3K complex (Protein Data Bank ID 4OVU) (39). Recurrent major and minor mutation sites are shown as spheres and colored as in (A). (C) Thermal shift assays of recombinant PI3K complexes. Western blot densitometry was performed, normalized to measurements of the lowest temperature, and data were fit to Boltzmann sigmoidal curves, from which the midpoint melting temperature (Tm) was determined. Data are mean ± SEM (n = 2 replicates). (D) In vitro radioactive lipid kinase assay of recombinant PI3K complexes (representative radiograph from one experiment, n = 3). (E) Liposome sedimentation assays of cis and single p110α mutant recombinant PI3K complexes blotted for p110α with quantifications for (F) anionic liposomes and 0.1% PIP2-containing liposomes. Data are mean ± SEM (n = 3 for each). PS, phosphatidylserine.
We modeled cis mutant disruption of p85α using thermal shift assays, which expose proteins to increasing levels of heat to determine the melting temperature. Unstable proteins will readily denature and aggregate at lower temperatures. p110α depends on its interaction with p85α to properly fold, and weakening their association renders them thermally labile (36, 41). All of the cis mutants we tested demonstrated additively increased thermal instability, as quantified by decreased melting temperatures, compared with each of their constituent minor and major mutants (Fig. 3C and fig. S8B).
We measured basal recombinant kinase activity using radioactive in vitro kinase assays, assessing for levels of radiolabeled 32P-PIP3 by thin layer chromatography. E453Q/E545K, E453Q/H1047R, E545K/E726K, and E545K/M1043L cis mutants demonstrated increased basal kinase activity compared with each of their constituent minor or major mutants (Fig. 3D).
To assess whether cis mutants increase lipid binding, we performed liposome sedimentation assays with liposomes containing anionic lipids (modeled after the inner leaflet of the plasma membrane) with and without 0.1% PIP2 (the physiologic concentration), given differential contributions to lipid binding to PI3K (42). E453Q/E545K, E453Q/H1047R, E545K/E726K, and E726K/H1047R cis mutants showed additively increased binding to anionic and PIP2 liposomes compared with their constituent single mutants (Fig. 3, E and F).
Double PIK3CA mutations in cis are hypersensitive to PI3K inhibition in cells
Our biochemical data suggest that double PIK3CA mutations in cis increase PI3Kα activity. However, median inhibitory concentration (IC50) values for the PI3Kα inhibitors alpelisib and GDC-0077 are similar among the recombinant single and cis mutant PI3Kα complexes (fig. S8C). Similar phenomena have been observed with other targetable oncogenes, where-in both WT and translocated and/or mutant proteins are inhibited at clinically attainable drug concentrations (43, 44).
Our functional data suggest that double PIK3CA mutants in cis result in a constitutive activation of PI3K signaling, implying that cells bearing these mutations are more dependent on the PI3K pathway for proliferation and survival. Therefore, we tested whether cis mutant cells exhibit differential sensitivity to PI3Kα inhibitors. Whereas in the absence of pharmacological pressure cis mutant signaling is increased compared with single mutants, treatment with the PI3Kα inhibitors alpelisib or GDC-0077 (45) results in a similar inhibition of phosphorylated AKT and S6 among all the MCF10A clones (Fig. 4, A and B). Similar results were obtained in NIH-3T3 cells (fig. S9A) and MCF7 cells (fig. S9B). We then used our MCF10A cell culture models to test cell growth on PI3Kα inhibition. E545K and H1047R major-hotspot mutants are more sensitive to alpelisib (Fig. 4C) and GDC-0077 (Fig. 4D) compared with minor mutants and WT. In turn, all cis mutants are synergistically more sensitive to alpelisib and GDC-0077 compared with single major and minor hotspots (Fig. 4, C and D) with respect to IC50, Emax, and the area under the curve (46) (fig. S9C). Cis mutants are also more sensitive to the downstream PI3K pathway mTORC1 allosteric inhibitor everolimus compared with single mutants (Fig. S9D). By contrast, mutations in trans are less sensitive to alpelisib, compared with cis mutants, and are no more sensitive than the single major mutant, as demonstrated by E726K/H1047R (fig. S9E).
Fig. 4. Double PIK3CA mutations in cis confer increased sensitivity to PI3Kα inhibition compared with single PIK3CA mutations in cells.
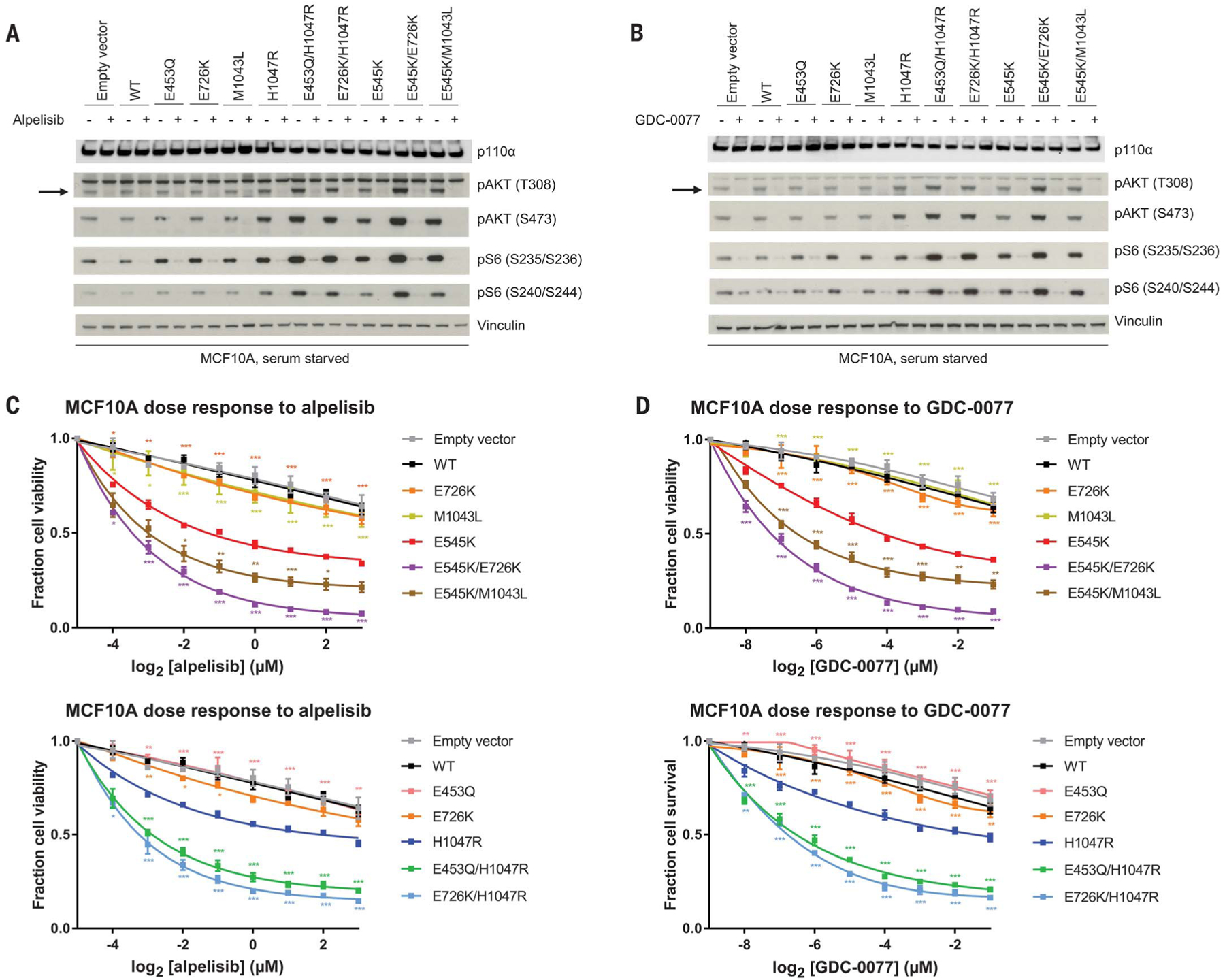
(A and B) Western blotting of PI3K effectors of PIK3CA mutant stably transduced MCF10A cells, serum starved for 1 day and then exposed to dimethyl sulfoxide (−) and (A) alpelisib (1 μM) (+) for 1 hour or (B) GDC-0077 (62.5 nM) (+) for 1 hour. The arrow indicates pAKT (T308). (C and D) Dose-response survival curves for MCF10A cell lines treated with (C) alpelisib or (D) GDC-0077 under serum starvation for 4 days. E545K-containing cis mutants (top) and H1047R-containing cis mutants (bottom) are compared with single PIK3CA mutants. Data are mean ± SEM (n = 3 replicates) and were fit to asymmetric, five-parameter sigmoidal curves. ***P < 0.001, **P < 0.01, *P < 0.05 by two-way analysis of variance corrected for multiple comparisons by Tukey’s test compared with E545K (top) or H1047R (bottom).
Multiple–PIK3CA-mutant tumors exhibit increased response to PI3K inhibition in patients
We investigated the potential correlation between multiple PIK3CA mutations and clinical response to PI3K inhibitors in metastatic breast cancer. We analyzed response data from SANDPIPER (12), a phase 3 clinical trial that tested the efficacy of the PI3Kα/γ/δ inhibitor taselisib (GDC-0032) in combination with fulvestrant, versus a placebo and fulvestrant, in a metastatic ER+ breast cancer patient population enriched with PIK3CA mutations (n = 631 patients). This is, to date, the largest randomized clinical study testing a PI3K inhibitor in a PIK3CA-mutant patient population.
Many patients with metastatic ER+ breast cancer enrolled in this trial had bone metastases, which renders DNA sequencing particularly challenging (47). Thus, we used circulating tumor DNA (ctDNA), which has been used in previous breast cancer clinical trials (10, 17, 48, 49), to detect the presence of mutations (fig. S10). Of the 631 patients in the trial, 598 had plasma samples available for analysis, of which 508 were adequate for testing (Fig. 5A). Of the 339 patients with detected PIK3CA mutations, 66 (19%) had two or more PIK3CA mutations (Fig. 5A). Notably, this is slightly higher than the frequency we observed from archival tumor testing (12%) and may reflect the ability of ctDNA to detect global tumoral heterogeneity compared with tumor biopsy of a single site.
Fig. 5. Multiple PIK3CA mutations detected by ctDNA confer increased sensitivity to taselisib compared with single PIK3CA mutations in patients.
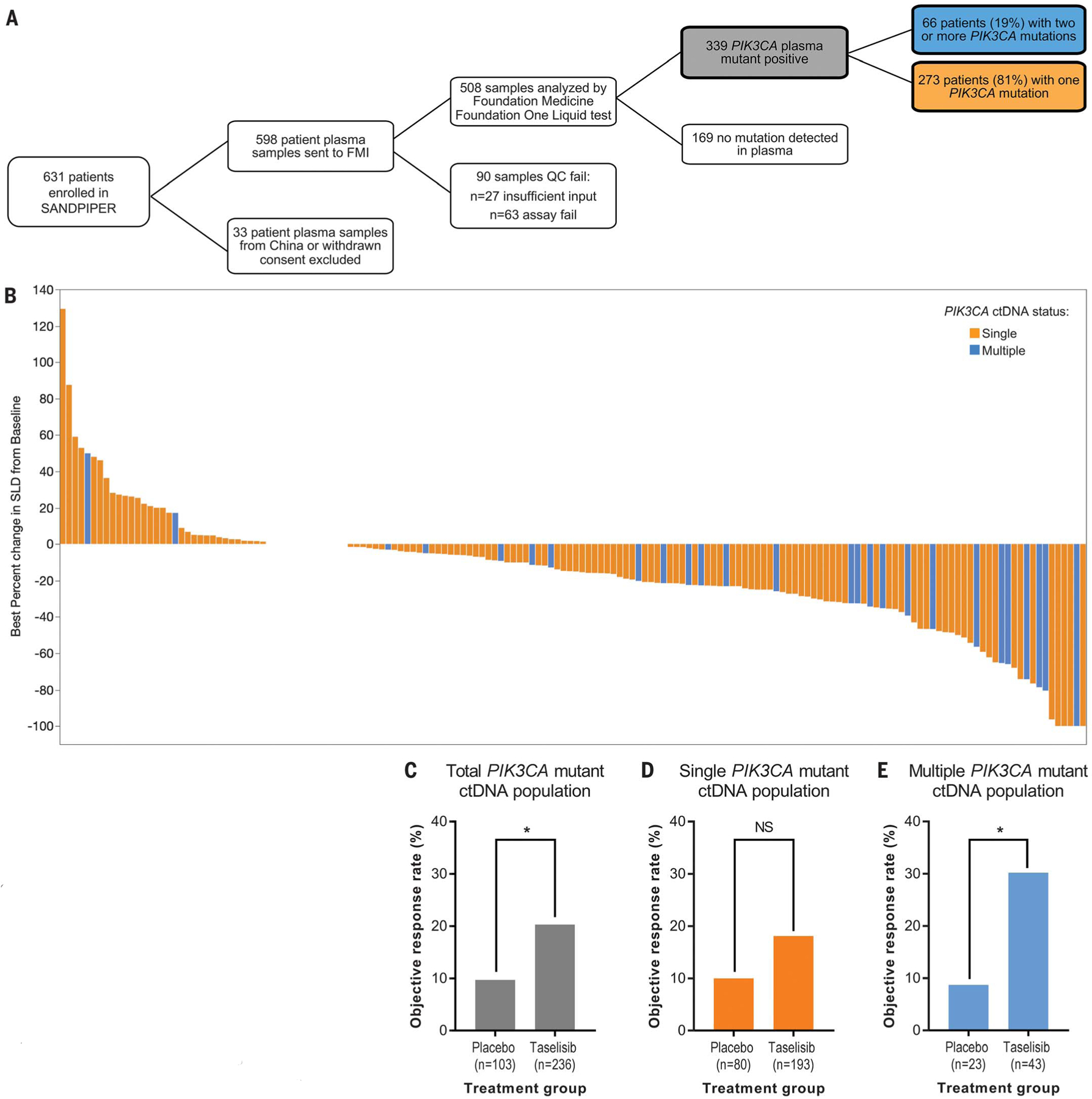
(A) Schematic showing plasma sample acquisition from patients in the SANDPIPER clinical trial (12) and analysis and sequencing of ctDNA specimens to determine PIK3CA mutational status. FMI, Foundation Medicine, Inc.; QC, quality control. (B) Waterfall plot denoting the range of tumor shrinkage [measured by percentage change of the sum of the longest dimensions (SLD) of target lesions compared with baseline] for each individual patient in SANDPIPER who received taselisib and fulvestrant by ctDNA single versus multiple PIK3CA mutation status. (C to E) ORRs (RECIST version 1.1) of placebo versus taselisib arms from the SANDPIPER clinical trial of (C) ctDNA PIK3CA-mutant total population (9.7% versus 20.3%; 95% CI 4.8 to 16.7% versus 15.5 to 25.9%; P = 0.0202), (D) single ctDNA PIK3CA-mutant subpopulation (10.0% versus 18.1%; 95% CI 4.4 to 18.1% versus 13.0 to 24.2%; P = 0.0981), and (E) multiple ctDNA PIK3CA-mutant subpopulation [8.7% versus 30.2%; 95% CI 1.6 to 26.8% versus 18.4 to 44.9%; P = 0.0493). Data are number of patients who responded divided by the total number for a particular subgroup and 95% CI (by the Blyth-Still-Casella method). The CI for the difference in ORRs between the two treatment arms was determined by using the normal approximation to the binomial distribution. Response rates in the treatment arms were compared (P value) by using the stratified Cochran-Mantel-Haenszel test. *P < 0.05. n, number of patients.
The waterfall plot (Fig. 5B) shows the individual PIK3CA-mutant patient responses to taselisib. We examined differences in the objective response rate (ORR), which is defined as tumor shrinkage of ≥30% by RECIST version 1.1 criteria (50). PIK3CA-mutant patients in the taselisib arm (n = 236) had an ORR of 20.3% versus 9.7% compared with those in the placebo arm (n = 103) [95% confidence interval (CI) 15.5 to 25.9% versus 4.8 to 16.7%; P = 0.0202] (Fig. 5C). This result confirms that the presence of PIK3CA mutations predicts response to PI3Kα inhibition (10, 11, 13, 17).
We then compared responses of patients with single versus multiple mutations. Single–PIK3CA-mutant patients in the taselisib arm (n = 193) had an ORR of 18.1% versus 10.0% compared with those in the placebo arm (n = 80), a non–statistically significant difference (95% CI 13.0 to 24.2% versus 4.4 to 18.1; P = 0.0981) (Fig. 5D). On the contrary, multiple–PIK3CA-mutant patients in the taselisib arm (n = 43) achieved an ORR of 30.2% versus 8.7% compared with those in the placebo arm (n = 23), which is a statistically significant difference (95% CI 18.4 to 44.9% versus 1.6 to 26.8%; P = 0.0493) (Fig. 5E). These findings confirm our initial observation that breast cancer patients with multiple-mutant tumors may achieve higher clinical benefit from PI3Kα inhibition compared with those with single-mutant tumors.
Discussion
In this work, we have identified double mutations in cis as a distinctive and relatively common genomic alteration in PIK3CA, the most frequently mutated oncogene in human cancer (51). Double PIK3CA mutations in cis activate PI3K pathway cellular signaling and promote growth more so than single mutants do through a combination biochemical mechanism of increased membrane binding and increased p85α disinhibition. The overall consequence of these cis mutations is a phenotype of enhanced oncogenicity and greater sensitivity to PI3Kα inhibitors. We propose that against a backdrop of moderately oncogenic single PIK3CA major mutations, the addition of a weakly oncogenic minor mutation [which comprise ~60% of PIK3CA oncogenic mutations (30)] in cis may synergize and result in a PIK3CA-hypermorphic phenotype.
Although cancers can accumulate numerous mutations in functionally relevant genes, some tumors depend on one gene for the development and maintenance of the malignant phenotype, a dependency that has been described as oncogene addiction (52, 53). Oncogene addiction has been successfully therapeutically exploited with a series of oncogene-targeted therapies that have altered the natural history of a number of previously highly lethal human cancers (54–56). Our results implicate a model of oncogene addiction to double-mutant PIK3CA in breast cancer. This would result in a greater response to PI3Kα inhibition compared with single mutations, both in preclinical models and in the clinic. Our clinical findings, however, should be interpreted with caution. Although the SANDPIPER study met its primary PFS endpoint, a high percentage of patients experienced toxicities that limited prolonged clinical exposure to this agent and resulted in a suboptimal risk/benefit ratio. One possible explanation for this toxicity is that taselisib, despite its greater sensitivity for mutant PI3Kα than for WT PI3Kα, also displays activity against PI3Kγ and PI3Kδ isoforms (57). The results from clinical studies testing other, more specific PI3Kα inhibitors will be critical to confirm the generalizability of our findings and ascertain whether an increased ORR will translate into a PFS benefit.
The common practice of sequencing only certain single-nucleotide variants or some but not all exons across a gene likely underestimates the frequency of multiple mutations in PIK3CA mutant cancers at <1% (58, 59). In fact, the true frequency is ~10 to 19%, which translates to a clinically meaningful number of patients who may derive additional benefit from this targeted therapy. PI3Kα inhibitors are now a standard of care in PIK3CA-mutant ER+ metastatic breast cancer and are being explored in other PIK3CA-mutant tumor histologies (60). Our findings provide a rationale for testing whether patients with multiple–PIK3CA-mutant tumors are markedly sensitive to PI3Kα inhibitors.
Supplementary Material
ACKNOWLEDGMENTS
We thank M. Berger and D. Solit for sharing MSK-IMPACT data; A. Santiago-Zayas and N. Mannino for help with procuring patient samples; E. de Stanchina and the MSKCC Antitumor Assessment Core Facility for assistance with allografts; M. Asher from MSKCC Pathology Core for tissue staining; Z. Yaari and D. Heller for assistance with differential light scattering; M. Kumar from Bellbrook Labs for helpful discussions regarding kinase assays; and the patients, their families, and the investigators of the SANDPIPER study for their participation.
Funding: This work was supported by grants from the NIH [awards P30 CA008748 and R01 CA190642 (M.S. and J.B.), R21 CA223789 (M.S.), and R35 CA197588 (L.C.C.)]. This work was also supported by grants from Stand Up to Cancer (Cancer Drug Combination Convergence Team), the V Foundation, the National Science Foundation (G.X., R.R., and M.S.), and the Breast Cancer Research Foundation. N.V. is supported by the Susan G. Komen Career Catalyst Research Grant, the Conquer Cancer Foundation of ASCO/Breast Cancer Research Foundation Young Investigator Award, and a grant from the Society of MSK. N.V. and E.R. are supported by the Fund for Innovation in Cancer Informatics. N.V., E.T., G.X., and M.S. are supported by a gift from B. Smith and her husband.
Footnotes
Competing interests: N.V. reports consultant and advisory board activities for Novartis and Petra Pharmaceuticals. P.R. reports consultant and advisory board activities for Novartis and has received research support from Illumina and Grail. M.T.C. is employed by Genentech. M.N.D is an Eli Lilly employee and reports consulting activities for Novartis, Pfizer, Roche, Genentech, and G1 Therapeutics. K.J. reports consultant and advisory board activities for Novartis, Spectrum Pharmaceuticals, ADC Therapeutics, Pfizer, Bristol-Myers Squibb, Jounce Therapeutics, and Taiho Oncology and research funding from Novartis, Clovis Oncology, Genentech, AstraZeneca, ADC Therapeutics, Novita Pharmaceuticals, Debio Pharmaceuticals, and Pfizer. S.C. has received research funds in the past from Novartis and Eli Lilly and ad hoc consulting honoraria from Novartis, Sermonix, Context Therapeutics, and Revolution Medicines. R.R. is a consultant for the Spanish National Cancer Research Centre and is on the scientific advisory board of Aimed Bio, a company developing new compounds for cancer therapy. F.S. and T.R.W. are employed by Genentech and have equity in Roche. L.S.F. is an employee of Oric Pharmaceuticals and is a former employee of Genentech. L.C.C. is a founder and member of the board of directors and scientific advisory board of Agios Pharmaceuticals; he is also a cofounder, member of the scientific advisory board, and shareholder of Petra Pharmaceuticals. These companies are developing novel therapies for cancer. L.C.C. laboratory receives some funding support from Petra Pharmaceuticals. M.S. is on the scientific advisory board of Menarini Ricerche and the Bioscience Institute; has received research funds from Puma Biotechnology, Daiichi-Sankio, Targimmune, Immunomedics, and Menarini Ricerche; and is a cofounder of Medendi.org. M.S. has received research funds from Menarini Ricerche, which markets MEN1611. J.B. is an employee of AstraZeneca; is on the Board of Directors of Foghorn; and is a past board member of Varian Medical Systems, Bristol-Myers Squibb, Grail, Aura Biosciences, and Infinity Pharmaceuticals. He has performed consulting and/or advisory work for Grail, PMV Pharma, ApoGen, Juno, Eli Lilly, Seragon, Novartis, and Northern Biologics. He has stock or other ownership interests in PMV Pharma, Grail, Juno, Varian, Foghorn, Aura, Infinity Pharmaceuticals, and ApoGen, as well as Tango and Venthera, of which he is a cofounder. He has previously received honoraria or travel expenses from Roche, Novartis, and Eli Lilly. J.B. is an employee of AstraZeneca, which is currently developing capivasertib, an AKT inhibitor. J.B. is a past board member of Infinity Pharmaceuticals, which markets duvelisib and IPI-549. J.B. has been a paid consultant and/or advisor for Novartis, which markets alpelisib, buparlisib, and everolimus. J.B. has stock or other ownership interests in Venthera (of which he is a cofounder), which is developing topical PI3K inhibitors for dermatological conditions. Companies that have developed or are developing PI3K inhibitors, for which coauthors on this study have a disclosure, include Novartis, Genentech, AstraZeneca, Eli Lilly, Pfizer, Roche, Infinity Pharmaceuticals, Petra, and Venthera. N.V. and J.B. are inventors on a patent application (PCT/US2019/047879) submitted by MSKCC that is related to the use of multiple PIK3CA mutations as a biomarker for clinical response to PI3K inhibitors. No potential conflicts of interests were disclosed by the other authors.
Data and materials availability: The sequencing data have been deposited in the NCBI Sequence Read Archive under accession number PRJNA566077. GDC-0077 was obtained on a material transfer agreement from Genentech. Other data that support the findings of this study are available from the corresponding authors on request.
SUPPLEMENTARY MATERIALS
science.sciencemag.org/content/366/6466/714/suppl/DC1
Materials and Methods
Supplementary Text
Figs. S1 to S10
Tables S1 to S4
References (61–71)
View/request a protocol for this paper from Bio-protocol.
REFERENCES AND NOTES
- 1.Bailey MH et al. , Cell 174, 1034–1035 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Whitman M, Downes CP, Keeler M, Keller T, Cantley L, Nature 332, 644–646 (1988). [DOI] [PubMed] [Google Scholar]
- 3.Fruman DA et al. , Cell 170, 605–635 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Samuels Y et al. , Science 304, 554(2004). [DOI] [PubMed] [Google Scholar]
- 5.Samuels Y et al. , Cancer Cell 7, 561–573 (2005). [DOI] [PubMed] [Google Scholar]
- 6.Kang S, Bader AG, Vogt PK, Proc. Natl. Acad. Sci. U.S.A 102, 802–807 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Isakoff SJ et al. , Cancer Res. 65, 10992–11000 (2005). [DOI] [PubMed] [Google Scholar]
- 8.Zhao JJ et al. , Cancer Cell 3, 483–495 (2003). [DOI] [PubMed] [Google Scholar]
- 9.Razavi P et al. , Cancer Cell 34, 427–438.e6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baselga J et al. , Lancet Oncol. 18, 904–916 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Di Leo A et al. , Lancet Oncol. 19, 87–100 (2018). [DOI] [PubMed] [Google Scholar]
- 12.Baselga J et al. , J. Clin. Oncol 36 (suppl.), LBA1006(2018). [Google Scholar]
- 13.Saura C et al. , Lancet Oncol. 20, 1226–1238 (2019). [DOI] [PubMed] [Google Scholar]
- 14.Juric D et al. , J. Clin. Oncol 36, 1291–1299 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mayer IA et al. , Clin. Cancer Res 23, 26–34 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Juric D et al. , JAMA Oncol. 5, e184475(2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.André F et al. , N. Engl. J. Med 380, 1929–1940 (2019). [DOI] [PubMed] [Google Scholar]
- 18.Juric D et al. , Nature 518, 240–244 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheng DT et al. , J. Mol. Diagn 17, 251–264 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cerami E et al. , Cancer Discov. 2, 401–404 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gao J et al. , Sci. Signal 6, pl1(2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Curtis C et al. , Nature 486, 346–352 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cancer Genome Atlas Network, Nature 490, 61–70 (2012).23000897 [Google Scholar]
- 24.Banerji S et al. , Nature 486, 405–409 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stephens PJ et al. , Nature 486, 400–404 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wagle N et al. , Cancer Res. 78, 5371(2018). [Google Scholar]
- 27.Shen R, Seshan VE, Nucleic Acids Res. 44, e131(2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eid J et al. , Science 323, 133–138 (2009). [DOI] [PubMed] [Google Scholar]
- 29.Zhang Y et al. , Cancer Cell 31, 820–832.e3 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao L, Vogt PK, Proc. Natl. Acad. Sci. U.S.A 105, 2652–2657 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ikenoue T et al. , Cancer Res. 65, 4562–4567 (2005). [DOI] [PubMed] [Google Scholar]
- 32.Beaver JA et al. , Clin. Cancer Res 19, 5413–5422 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Berenjeno IM et al. , Nat. Commun 8, 1773(2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kinross KM et al. , J. Clin. Invest 122, 553–557 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang CH et al. , Science 318, 1744–1748 (2007). [DOI] [PubMed] [Google Scholar]
- 36.Yu J et al. , Mol. Cell. Biol 18, 1379–1387 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Burke JE, Perisic O, Masson GR, Vadas O, Williams RL, Proc. Natl. Acad. Sci. U.S.A 109, 15259–15264 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mandelker D et al. , Proc. Natl. Acad. Sci. U.S.A 106, 16996–17001 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miller MS et al. , Oncotarget 5, 5198–5208 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maheshwari S et al. , J. Biol. Chem 292, 13541–13550 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Croessmann S et al. , Clin. Cancer Res 24, 1426–1435 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hon WC, Berndt A, Williams RL, Oncogene 31, 3655–3666 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Druker BJ et al. , Nat. Med 2, 561–566 (1996). [DOI] [PubMed] [Google Scholar]
- 44.Sharma SV, Settleman J, Genes Dev. 21, 3214–3231 (2007). [DOI] [PubMed] [Google Scholar]
- 45.Edgar K et al. , Cancer Res. 77, 156(2017). [Google Scholar]
- 46.Fallahi-Sichani M, Honarnejad S, Heiser LM, Gray JW, Sorger PK, Nat. Chem. Biol 9, 708–714 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Singh VM et al. , Ann. Diagn. Pathol 17, 322–326 (2013). [DOI] [PubMed] [Google Scholar]
- 48.Baselga J et al. , N. Engl. J. Med 366, 520–529 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Turner NC et al. , N. Engl. J. Med 373, 209–219 (2015). [DOI] [PubMed] [Google Scholar]
- 50.Eisenhauer EA et al. , Eur. J. Cancer 45, 228–247 (2009). [DOI] [PubMed] [Google Scholar]
- 51.Kandoth C et al. , Nature 502, 333–339 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Weinstein IB, Joe AK, Nat. Clin. Pract. Oncol 3, 448–457 (2006). [DOI] [PubMed] [Google Scholar]
- 53.Zhou P et al. , Oncogene 11, 571–580 (1995). [PubMed] [Google Scholar]
- 54.Slamon DJ et al. , N. Engl. J. Med 344, 783–792 (2001). [DOI] [PubMed] [Google Scholar]
- 55.Druker BJ et al. , N. Engl. J. Med 344, 1031–1037 (2001). [DOI] [PubMed] [Google Scholar]
- 56.Lynch TJ et al. , N. Engl. J. Med 350, 2129–2139 (2004). [DOI] [PubMed] [Google Scholar]
- 57.Ndubaku CO et al. , J. Med. Chem 56, 4597–4610 (2013). [DOI] [PubMed] [Google Scholar]
- 58.Saal LH et al. , Cancer Res. 65, 2554–2559 (2005). [DOI] [PubMed] [Google Scholar]
- 59.Yuan TL, Cantley LC, Oncogene 27, 5497–5510 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jhaveri K et al. , Cancer Res. 78, CT046(2018). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.