

Abstract
The development of genetic engineering techniques has speeded up the growth of the biotechnological industry, resulting in a significant increase in the number of recombinant protein products on the market. The deep knowledge of protein function, structure, biological interactions, and the possibility to design new polypeptides with desired biological activities have been the main factors involved in the increase of intensive research and preclinical and clinical approaches. Consequently, new biological entities with added value for innovative medicines such as increased stability, improved targeting, and reduced toxicity, among others have been obtained. Proteins are complex nanoparticles with sizes ranging from a few nanometers to a few hundred nanometers when complex supramolecular interactions occur, as for example, in viral capsids. However, even though protein production is a delicate process that imposes the use of sophisticated analytical methods and negative secondary effects have been detected in some cases as immune and inflammatory reactions, the great potential of biodegradable and tunable protein nanoparticles indicates that protein-based biotechnological products are expected to increase in the years to come.
Keywords: Protein nanoparticles, Gene therapy, Bioengineering, Nanomedicine, Innovative medicines, New biological entities, Biomaterials, Biopharmaceuticals
I. Introduction
The design of new chemical entities (NCE) for diagnosis and treatment of human diseases has relied on the discovery of active chemical drugs from a diverse library of compounds or from naturally occurring molecules.1, 2 Further chemical modifications improve pharmacokinetic properties to obtain a final product with a known mechanism of action and decreased toxicity.3 Nonetheless, using such approaches, the final products present low specificity for their target molecules, interacting with many other molecules and accumulating in some tissues, disturbing the correct homeostasis of the system. In some cases, the adverse effects of drug administration exceed pharmacological effect and despite the concise mechanism of action of the drug over the target molecule representing an improvement in the patient's state, the treatment has to be prevented or discontinued.4 In fact, although a maintained steady increase in the number of launched NCE has been observed in the last years, the question arises whether this classical approach has already exhausted the discovery of innovative molecules.5
On the other hand, macromolecular new biological entities (NBE) have been used to supplement cellular deficiencies or to inhibit cellular pathways exploiting their relatively specific mode of action. Proteins and peptides have been obtained first from their natural source or produced as recombinant versions after the development of genetic engineering techniques in the late 1970s. However, the delivery of biological entities is sometimes hampered by its low half-life in the bloodstream by unspecific degradation, resulting in an expensive and ineffective process. Nevertheless, some solutions have already been explored for biopharmaceuticals to increase solubility and stability and to reduce immunogenicity including postranslational modifications such as glycosylation and covalent conjugation of polyethylene glycol.6
Thus, one of the main objectives in the use of drugs (for either NCE or NBE) is the need to optimize the delivery system to reduce the pharmacological dose which would consequently represent a concomitant reduction in toxicity and cost. In that scenario, new delivery approaches have been implemented using biological interactions such as antigen–antibody binding (immunoliposomes)7 or more sophisticated interactions including the binding between nutrient concentrator SPARC (secreted protein acidic and rich in cysteine) and albumin in the treatment of some types of cancer (Abraxane®).8, 9
Proteins can be then used for their targeting qualities as molecular delivery vehicles both for the specific delivery of drugs or nucleic acids in gene therapy approaches and by themselves as therapeutic molecules. One of the interesting characteristics of proteins is their ability to form intermolecular driven complexes as sophisticated and structurally perfect as in the case of viral capsids. In addition, through the use of genetic engineering, recombinant proteins can be tuned to include additional properties to optimize drug delivery and nucleic acid delivery in gene therapy.
In this chapter, the main available strategies to develop protein-based nanovehicles or biopharmaceuticals will be described. In this context, several parameters will be defined such as proper formulation, stability, immunogenicity, and delivery to the correct cell type and cell compartment. Modular protein engineering, virus-like particles (VLPs), and other self-assembling entities are envisioned as modulatable novel protein nanoparticles able to include many desirable properties in the correct delivery of drugs and nucleic acids. Finally, some successful examples of protein nanoparticles on the market will be described in addition to protein products currently in clinical trials and under preclinical research in order to envision which type of protein nanoparticles will be available soon on the market.
II. Protein Nanoparticle Formulation and Biological Barriers
When a protein-only nanoparticle is meant to be used as a vector to deliver therapeutic nucleic acid, drug, or peptide, there are several steps that the nanoparticle has to perform to successfully get inside the target cell. In the first instance, it is necessary to obtain the proper formulation of the complex with the therapeutic molecule to generate a vehicle capable of being transported in the blood if a systemic administration is needed and retaining a significant stability before reaching the target cell.10, 11 In addition, the biological system poses specific barriers that have to be overcome such as membranes (cytoplasmic, endocytic, and nuclear), degradation (protease degradation induced by acid denaturalization in lysosomes, cytosolic proteosomes, and nucleases), cytosolic transport, and nuclear entry if necessary.12, 13 For central nervous system therapies, the blood–brain barrier (BBB) represents the main bottleneck, and for that, a specific strategy has to be designed.14 Furthermore, the therapeutic complex has to be flexible enough in order to release the therapeutic molecule in the specific cell compartment.
Thus, several protein motifs have been described to overcome each and every process described earlier so that a modular multifunctional protein can be generated including those modules that are necessary to achieve its goal. In order to get a rational construction of the multifunctional vector, each step has to be carefully taken into account so as to overcome every step which is needed to achieve its final goal (Table I ).
Table I.
Selection of Peptide Motifs Used in Gene Therapy and Drug Delivery to Improve Protein Nanovehicle Performance
| Peptide motif | Sequence | References |
|---|---|---|
| Nucleic acid condensation peptides | ||
| Polylysine | (KKKKKKKKKKKKKKKKKKKK)n | 16, 17, 18 |
| Polylysine containing peptides | YKAKKKKKKKKWK and derivatives | 19, 20, 21, 22 |
| Salmon protamine | PRRRRSSSRPVRRRRRPRVSRRRRRRGGRRRR | 23, 24, 25 |
| GAL4 | MKLLSSIEQACDICRLKKLKCSKEKPKCAKCLKNNWECRYSPK | 30, 31 |
| Blood–brain barrier (BBB) peptides | ||
| g7 | H2N-Gly-L-Phe-d-Thr-Gly-L-Phe-L-Leu-L-Ser(O-β-d-glucose)-CONH2 | 107 |
| RVG | YTIWMPENPRPGTPCDIFTNSRGKRASNG | 56 |
| Tat | YGRKKRRQRRR | 108 |
| R9 | RRRRRRRRR | 14 |
| Cell-penetrating peptides (CPP) | ||
| Tat | GRKKRRQRRPPQ | 36, 37, 38, 39, 40, 41 |
| R9 | RRRRRRRRR | 42 |
| Penetratin | CRQIKIWFQNRRMKWKK | 43, 44 |
| bPrPp | MVKSKIGSWILVLFVAMWSDVGLCKKRPKP | 43 |
| Transportan | CLIKKALAALAKLNIKLLYGASNLTWG | 44, 45, 46 |
| Receptor-specific ligands (ligand/receptor) | ||
| RGD/integrins (mainly αvβ3) | GRGDSP | 47, 48 |
| CXCL12/CXCR4 | KPVSLSYRCPCRFFESHVARANVKHLKILNTPNCALQIVARLKNNNRQVCIDPKLKWIQEYLEKALN | 49, 50 |
| Transferrin receptor ligand (12Aa)/transferrin receptor | THRPPMWSPVWP | 51, 52 |
| EGF/EGF receptor | NPVVGYIGERPQYRDL | 53, 54 |
| Asioaloglycoprotein/asioaloglycoprotein receptor | 55 | |
| RVG/acetil-colin receptor | YTIWMPENPRPGTPCDIFTNSRGKRASNG | 56 |
| PLAEIDGIELTY/integrin a9b1 | PLAEIDGIELTY | 57 |
| Molossin (RGD)/integrin | ICRRARGDNPDDRCT | 58 |
| Secretin/Secretin receptor | HSDGTFTSELSRLRDSARLQRLLQGLV | 59 |
| NL4 (loop 4 of nerve growth factor)/TrkA | CTTTHTFVKALTMDGKQAAWRFIRIDTAC | 60 |
| Neurotensin/Neurotensin receptor (NTRH) | ELYENKPRRPYIL | 61 |
| LSIPPKA, FQTPPQL, LTPATAI/LOX-1 | LSIPPKA, FQTPPQL, LTPATAI | 62 |
| Monoclonal Abs/antigen recognized by the antibody | – | 63, 64, 65 |
| Endosomal escape fusiogenic peptides | ||
| HA-2 | GLFGAIAGFIENGWEGMIDGWYG | 12, 69, 70 |
| GALA | WEAALAEALAEALAEHLAEALAEALEALAA | 12, 71, 72, 73, 74 |
| KALA | WEAKLAKALAKALAKHLAKALAKALKACEA | 12, 75 |
| JTS-1 | GLFEALLELLESLWELLLEA | 12, 19 |
| ppTG20 | GLFRALLRLLRSLWRLLLRA | 12, 76 |
| PPTG1 | GLFKALLKLLKSLWKLLLKA | 12, 76 |
| Melittin | GIGAVLKVLTTGLPALISWIKRKRQQ | 12, 77 |
| Tat | GRKKRRQRRRPPQ | 12, 39, 40 |
| Penetratin | RQIKIWFQNRRMKWKK | 12, 78, 79 |
| Transportant | GWTLNSAGYLLGKINLKALAALAKKIL | 12, 45, 46 |
| INF 7 | GLFEAIEGFIENGWEGMIDGWYG | 12, 80 |
| Endosomal escape histidine-rich peptides | ||
| CHK6HC | CHKKKKKKHC | 12, 22 |
| H5WYG | GLFHAIAHFIHGGWHGLIHGWYG | 12, 80, 81, 82 |
| LAH4 | KKALLALALHHLAHLALHLALALKKA | 12, 83 |
| Nuclear import peptides | ||
| SV40 large T antigen | PKKKRKV | 36 |
| Tat | VIH transcription factor | 37 |
| EBNA-1 | Epstein–Barr virus | 91 |
| Melittin | Honeybee venom (Apis mellifera) | 77 |
| M1 (c-myc transcription factor) | PAAKRVKLD | 92 |
| M2 (c-myc transcription factor) | RQRRNELKRSP | 92 |
| GAL4 amino terminal domain | Transcription factor | 93 |
| Protamines | Sperm DNA condensation protein | 23 |
| Histone H1 | Nuclear DNA condensation protein | 94, 95 |
| M9 (heterogeneous nuclear ribonucleoprotein A1 (hnRNP A1) | NQSSNFGPMKGGNFGGRSSGPYGGGGQYFAKPRNQGGY | 96 |
| Vp3 | SV40 estructural protein Vp3 | 97 |
| Adenovirus E1 protein C-terminus | KRPRP | 98 |
| Xenopus N1 protein | VRKKRKTEEESPLKDKDAKKSKQE | 99 |
| Fibroblast growth factor 3 (FDF3) | RLRRDAGGRGGVYEHLGGAPRRRK | 100 |
| Poly ADP-ribose polymerase (PARP) | KRKGDEVDGVDECAKKSKK | 101 |
| Xenopus protein nucleoplasmine | KRPAATKKAGQAKKKK | 102 |
A. Interaction with Drugs and Nucleic Acids
The DNA/RNA condensation or drug interaction with the protein vector is a critical step in the formulation of protein nanoparticles for gene therapy. They have to remain attached to the vector during the whole transport process through the body and the cell until it can be released in the desired localization within the target cell. Highly positively charged peptides containing a large number of arginines or polylysines have been used to promote electrostatic interactions since nucleic acids are highly negatively charged molecules.15, 16, 17, 18, 19, 20, 21, 22 Natural DNA-condensing proteins as nuclear histidines or protamines can also be used to bind nucleic acids.22, 23, 24, 25 Protamine, which is the protein that replaces histidines during the spermatogenesis process, is a sperm chromatin component and just as the histidines do, it has very high DNA condensation ability to protect nucleic acids form cytosolic endonucleases.23, 26 In addition, as soon as the complex reaches the cellular nucleus, protamine is degraded by chromatin-remodeling proteins, releasing the transported DNA allowing its expression.15, 23 In contrast, polycationic DNA condensation modules such as polylysines and polyarginines—even they can present higher DNA condensation ability depending on the polycationic chain length—usually present lower DNA-releasing ability, interfering negatively with the accessibility of cellular transcription factors and DNA expression capacity.15
All these DNA condensation modules described above interact with any DNA that is incubated in an unspecific way. However, there are proteins such as GAL4 that are able to recognize specific DNA sequences27, 28, 29 and that permit to bind and condensate specific DNA sequences in the final vector.30, 31
B. Protein Stability in Serum
In many cases, the multifunctional protein vector is in vivo administrated by the systemic route in order to travel in the blood and reach the target cells. That exposes the vector to all blood components, making it susceptible to be degraded. Thus, it is completely necessary that the vector remain in the blood long enough to be able to reach the target cells. It has also been described that naked DNA has an estimated half-life in blood of minutes10; so protein nanovehicles in gene therapy, among other properties, are intended to protect nucleic acids from degradation.
One important factor when the vector is exposed to the blood is that it can be recognized by the immune system components and produces an immune response against the vector. Thus, it is also very important to try to make the vector as less antigenic as possible in order to avoid being degraded or even being toxic to the organism.32
C. Defeating Biological Barriers
1. Cell Binding and Internalization
Peptide uptake or internalization involves a step before the protein binding to the cell surface. This attachment can be either specific or unspecific but in all cases the promotion of its internalization is required.33
Positively charged peptides usually bind the cellular surface by unspecific electrostatic interactions with the negatively charged cell surface proteoglicans. This kind of peptides can be used in the multifunctional protein if specific targeting is not required.33 Cell-penetrating peptides (CPPs) have been widely described as unspecific cell-binding and internalization peptides34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46 (see also the chapter “Peptide Nanoparticles for Oligonucleotide Delivery” by Lehto et al. in this volume). However, specific interactions can be obtained by incorporating cell receptor ligands if cell or tissue targeting is required for the therapeutic action. Moreover, some of those ligand–receptor interactions promote the ligand–receptor complex internalization. Many peptides have been described in the literature as receptor-specific ligands so any of them can be added to the multifunctional proteins in order to confer them cell specificity.47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62 The most natural specific ligands that can also be used for cell targeting are monoclonal antibodies.32, 63, 64, 65 In addition, if no specific peptides are available for an intended target, new specific binding peptides can be found by using phage display66 or combinatorial chemistry.67
2. Endosomal Escape
Several internalization pathways are possible depending on the vector properties,27, 33 including endocytosis (clathrin/caveolae-mediated, clathrin/caveolae-independent), macropinocytosis, and non-endocytic pathways.
It is known that more than one internalization pathway can be performed at the same time but usually the peptide-based vector uses endocytic pathways.68 Moreover, it seems that proteins that interact with a specific cellular receptor are internalized by the clathrin-mediated endocytic pathway.33 Most of the generated endosomal vesicles will converge to late endosomes that eventually will fuse with cellular lysosomes.15, 33 Remaining in the cellular endosomes, the multifunctional protein will be degraded, so it is strictly necessary that the internalized multifunctional proteins be released into the cellular cytoplasm escaping from degradation.
Several peptides have been described that are able to promote endosomal escape and can be classified into two types depending on their escape mechanism: fusiogenic peptides and histidine-rich peptides.36 The fusiogenic peptides are small peptides that have hydrophobic amino acids (Aa-s) interspersed at constant intervals with negatively charged Aa-s.12, 19, 39, 40, 45, 46, 69, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, 80 Thus, when early endosomes become late endosomes, their low pH induces a conformational change in the peptide, which adopts a alpha-helix structure, in an amphipathic structure able to fuse with the endosomal membrane, leading to pore formation and releasing all the endosomal content into the cell cytoplasm.36 The histidine-rich peptides are small peptides with a high histidine content whose endosmolytic activity is mediated by a mechanism called “proton sponge”.12, 22, 80, 81, 82, 83 When the endosomal pH becomes low in late stages, the imidazole groups of the histidines are protonated and attract endosomal Cl− ions, buffering against the proton pump. Thus, the endosomes collapse by an osmolytic swelling process and the endosomal content is released to the cell cytoplasm.36 Further details are given in the chapter “Peptide Nanoparticles for Oligonucleotide Delivery” by Lehto et al. in this volume.
3. Vector Stability in the Cytosol
Once the protein has achieved the cellular cytosol, it can be degraded by cellular proteases or by the cellular proteosome system.84 It is important to avoid this process, especially if the protein has to reach the cellular nucleus. If the final target of the nanoparticle is the cellular cytoplasm, it is necessary that it remain there at least long enough to perform its therapeutical action.
Several peptide proteosome inhibitors have been described that are able to avoid this type of protein degradation. By adding these peptides to the final protein vector it is possible to protect it and enhance cytoplasmatic stability. Epstein–Barr virus nuclear antigen 1 (EBNA1) contains a proteosome inhibitor consisting of glycine–alanine repeats able to prevent proteosomal proteolysis. It has been shown that a minimum of 4 Aa-s Gly-Ala repeats are necessary to achieve such protective activity.85, 86, 87 If the protein vector is carrying nucleic acids (DNA or RNA), degradation by the cytosolic endonucleases has to be taken into account, so it is also very important to protect this nucleic acid in order to maintain its integrity. Some DNA/RNA condensing peptides as protamines also protect the DNA against cytoplasmic endonucleases and enhance its stability as has been described above.15
4. Intracytosolic Mobility
The cellular cytoplasm is a very crowded and compartmentalized environment where cellular organelles and cytoskeleton make the free diffusion of macromolecules such as protein vectors difficult. However, cytoskeleton elements such as microtubules are used by endosomes and other cytosolic macromolecules for intracytosolic mobility.33 Dyneins have been described as being capable of carrying those macromolecules and endosomes along the microtubules in a retrograde transport toward the nucleus. Some small peptides that are able to bind dyneins have been identified. They can be added to the multifunctional protein vector in order to mediate an intracytosolic mobility toward the cellular nucleus.36 Several dynein-binding proteins have been identified in viruses that are able to use this transport system. Comparing those protein sequences, a consensus peptide sequence (KSTQT) that is able to bind to the dynein LC8 light chain has been identified.88
5. Nuclear DNA Delivery and Expression
Molecules lower than 45 kDa/10–30 nm are able to enter in the cellular nucleus by passive diffusion. However, macromolecules higher than 45 kDa/10–30 nm generally require an active transport system through the nuclear pore system. This transport mechanism generally requires a specific targeting signal peptide named nuclear localization signal (NLS). These signaling peptides are usually rich in basic Aa-s, which are recognized by the cellular importines and actively transported through the nuclear pore.15, 89 Monopartite or bipartite NLS sequences which are NLS peptides that have one or two NLS recognized sequences respectively have been described.12 Thus, these peptidic sequences can be added into the final multifunctional protein if nuclear localization is required in order to express a carried DNA. It has been reported that a single NLS sequence is sufficient to transport the vector to the nucleus and that a large number of NLS sequences can result in inhibition of its activity.90
One of the most used NLS signal peptides are fragments derived from the 111–135 Aa-s of the simian virus SV40 large tumor antigen (T-ag). Other NLS sequences can be found in GAL4, protamines, or Tat.23, 36, 37, 77, 91, 92, 93, 94, 95, 96, 97, 98, 99, 100, 101, 102
It is important that when the transported DNA reaches the cellular nucleus, it has to be released in order to be accessible to the nuclear transcription factors and achieve the desired expression level. Thus, while designing the multifunctional protein vector, this aspect has to be taken into account.
Once the DNA has been released in the cell nucleus, it will be necessary to control its expression level depending on which therapeutic action is being promoted. When the goal is to kill a cell as in cancer therapies, the uncontrolled DNA expression levels would not be a problem. However, when a specific protein expression level is required, achieving good control is very important.13 Some expression systems have been developed that can be pharmacologically regulated by oral drug formulation.103 Cell-specific promoters and enhancers can be also used in order to confer high cell specificity to the therapy.104, 105
D. Ways to Get Over the BBB
The BBB is a hermetic barrier that only allows nonlipophilic molecules smaller than 400 Da to cross it. However, some human proteins such as insulin, transferrin, insulin-like growth factor, or leptins are able to go across it by receptor-mediated transporters. Thus, the most important factor limiting central nervous system-targeting therapeutics is the presence of the BBB.106 Finding the way to cross it will be the main challenge.
Some peptides have been described that are able to reach the brain crossing the BBB. Moreover, it has been seen that they can be associated with another molecule and transported through the barrier. Thus, they could be interesting candidates to be included in the multifunctional vectors if central nervous system targeting is required.14, 56, 107, 108
Antibodies have also been described that bind transferrin and insulin receptors and that are able to cross the BBB efficiently. They can be conjugated with large molecules, allowing its translocation to the central nervous system.63, 64, 109, 110, 111
III. Multifunctional Proteins
A. Protein Engineering: Direct Evolution, De Novo Synthesis, and Rational Design
The development of genetic engineering techniques has increased the natural repertoire of proteins for the design of useful and/or valuable proteins with the aim to obtain new proteins with desired functions. There are three main strategies leading to the construction of engineered proteins: (a) direct evolution, (b) de novo protein design, and (c) rational design.
Directed evolution has developed quickly to become a method of choice for protein engineers in order to create enzymes having desired properties for all kind of processes. Over the past decade, this technique has become a daily part of the molecular toolbox of every biochemist. This is emphasized by the increasing number of publications about the subject.112
In nature, evolution and creation of new functionalities is achieved by mutagenesis, recombination, and survival of the fittest. Directed evolution mimics this and is a process of iterative cycles of producing mutants and finding the mutant with the desired properties. Mutations can be introduced at specific places using site-directed mutagenesis or throughout the gene by random mutagenesis. Several mutagenesis techniques have been developed in order to avoid codon bias.113, 114 The first technique used to mimic evolution was DNA shuffling.115 This method is based on the mixing and subsequent joining of different related small DNA fragments in order to form a complete new gene. In the process of shuffling, the recombination frequency is dependent on the degree of homology. A high level of recombination is important to get all possible combinations of mutations. Since recombination can be biased, several methods to overcome problems arising from the use of shuffling in the early years were tackled by novel strategies, all having their own advantages and disadvantages.112 The products obtained by these methods have to be screened for desired qualities and not all of them can be easily screened.
De novo protein design offers the broadest possibility for new structures. It is based on searches for amino acid sequences that are compatible with a three-dimensional protein backbone template using in silico techniques. Several research groups in the field have applied in silico methods to design the hydrophobic cores of proteins, with the novel sequences being validated with experimental data.116 In silico protein design has allowed novel functions on templates originally lacking those properties, modifying existing functions, and increasing protein stability or specificity. Beyond any doubt, intense research activities are ongoing in the field, the potential of which is simply enormous.117 So far there have been numerous examples of full sequences designed “from scratch” that were confirmed to fold into the target three-dimensional structures by experimental data.118 The zinc-finger protein designed by Dahiyat and Mayo119 was the first one to appear by this method.
Rational design of proteins is based on the modification or insertion of selected amino acids or domains in a polypeptide chain backbone to obtain proteins with new or altered biological functions. When using that strategy, a detailed knowledge of the structure and function of the backbone protein is needed to make desired changes. This generally has the advantage of being inexpensive and technically feasible. However, a major drawback of this approach is that detailed structural knowledge of a protein is often unavailable or it can be extremely difficult to predict the effects of various mutations. Modular engineering enables, by using simple DNA recombinant techniques, the construction of chimerical polypeptides in which selected domains, potentially from different origins, provide the required activities. An equilibrate combination and spatial distribution of such partner elements has generated promising prototypes, able to deliver expressible DNA or molecules to tissue culture but also to specific cell types in whole organisms.120 Modular fusion proteins that combine distinct functions required for cell type-specific uptake and intracellular delivery of DNA or drugs present an attractive approach for the development of self-assembling vectors for targeted gene or drug delivery.121 One of the first examples was described by the group of Uherek et al. They combined a cell-specific target module (antibody fragment specific for the tumor-associated ErbB2 antigen), a DNA-binding domain (Gal4), and a translocation domain for endosomal escape.121
In this context, many strategies for the construction of safer vehicles are being explored and the number of nonviral prototype vectors for gene and drug delivery is noticeably increasing. Here, the common steps that an approach like this might explore are presented (Fig. 1 ).
Fig. 1.
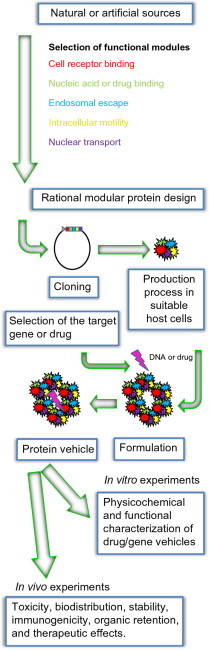
Scheme in the development of protein nanoparticles for drug delivery and gene therapy.
B. Designing a Protein Nanoparticle
When designing a new protein for drug or gene delivery there are many critical aspects, namely (a) design of the vehicle itself, required functions, stability, etc.; (b) production of the protein, suitable expression system, purification procedure, scaling up process, etc.; (c) characterization of the vehicle by physicochemical and functional tests; and finally (d) the administration route and regulatory guidance for biological products. Although all these aspects belong to different disciplines, they have to be overviewed together. Here, the major needs of a modular protein for gene and drug delivery are presented.
To enhance the physicochemical stability of the cargo molecules and their resistance to nuclease/protease-mediated degradation, protein vehicles should ideally exhibit, like their natural counterparts (viruses), nucleic-acid binding and condensing properties.27 Such abilities are, in general, conferred by cationic segments of the main scaffold molecules that interact with nucleic acids, mainly through electrostatic interactions. In addition, such complexes need to efficiently release the nucleic acid in the nucleus (if the cargo is a therapeutic gene), for which endosomal escape is required. Such functions have been found in some peptides in many natural molecules and they are suitable for functionalizing protein vehicles.
The ability to bind a particular cell type with high specificity is especially significant in a systemic delivery in which appropriate biodistribution and tissue targeting are essential.122 For nuclear targeting, only naked short nucleic acids can freely enter the nucleus of nondividing cells via free diffusion through the nuclear pore. Large molecules require active transport mediated by NLSs that are often found in viral proteins. Because the molecular mass of plasmidic DNA varies from to 2 to 10 MDA, DNA that is to be expressed, and essentially any macromolecular complex for nucleic acid delivery, requires NLSs.123 The role and types of functional modules peptides used for all these purposes will be discussed in depth in the following sections.
Finally, which protein or peptide is better for a given cargo is to be determined empirically and only few rules can be taken literally.38, 124
C. Production of Protein Nanoparticles
Some steps in the production of a protein-based vehicle after molecular cloning such as protein production and protein purification125 might be experimentally labor intense with a variable success rate. For that reason, when small proteins are needed, solid-phase peptide synthesis44 guarantees the process. However, the classical procedure of biological production allows scaling up the process in most of the cases and the production of larger polypeptides and full-length proteins.
Generally, in protein nanoparticle approaches, the protein is composed by different modules of natural sources such as the cell-penetrating peptide transactivator of transcription (TAT) derived from the TAT of the human immunodeficiency virus (HIV)126 or artificial sequences not present in any organism such as the polylysine DNA-condensing sequence.127
Once it has been defined which modules will be part of the protein, it is important to define the order they will have in the final construct. It has been demonstrated by Boekle and coworkers using melittin conjugated to polyethylenimine (PEI) that depending on the side of the linkage (C- or N-terminus), the lytic activity could be changed. Some other modules have the need to be in a determined position for its correct function.128
When producing a protein for gene or drug delivery, it is important to know the origin of its domains to choose the most suitable expression system for its production. For instance, if any module naturally carries a posttranslational modification that is essential for its biological function, the expression system chosen will have to be able to reproduce the same crucial modification.
The main biological production systems for protein drugs are described below.
Escherichia coli is the most widely used prokaryotic organism for the expression of recombinant proteins.129 The use of this host is relatively simple and inexpensive.130 Added advantages include its short duplication time, growth to high cell densities, ease of cultivation, and high yields of the recombinant product. However, since it lacks fundamental prerequisites for efficient secretion, recombinant proteins manufactured by E. coli systems are mainly produced as inclusion bodies.125, 131 Moreover, posttranscriptional modifications are not achieved with this system. There are many examples of proteins for gene delivery produced in E. coli with probed efficiency.132, 133
Like E. coli, yeasts can be grown cheaply and rapidly and are amenable to high-cell-density fermentations. Besides possessing complex posttranslational modification pathways, they offer the advantage of being neither pyrogenic nor pathogenic and are able to secrete more efficiently. Species established in industrial production procedures are Saccharomyces cerevisiae, Kluyveromyces lactis, Pichia pastoris, and Hansenulapolymorpha. S. cerevisiae is the best genetically characterized eukaryotic organism among them all and is still the prevalent yeast species in pharmaceutical production processes.131 In spite of their physiological advantageous properties and natively high expression and secretion capacity, the employability of yeasts in some cases, however, might reach a limit, particularly when the pharmacological activity of the product is impaired by the glycosylation pattern. In such cases, either a postsynthetic chemical modification has to be considered or the employment of more highly developed organisms. Most examples of nanoparticles produced in yeast are for VLPs.134
Animal cell expression systems show the highest similarity to human cells regarding the pattern and capacity of posttranslational modifications and the codon bias. However, their culture is more complicated and costlier and usually yields lower product titers. Among the known systems, insect cells infected by baculovirus vectors have reached popularity since they are considered to be more stress-resistant, easier to handle, and more productive compared with mammalian systems and are thus frequently employed for high-throughput protein expression. For commercial application, scale-up related questions have to be solved.135, 136, 137 Preferably applied in pharmaceutical production processes are mammalian systems like chinese hamster ovary (CHO) cells and baby hamster kidney (BHK) cells. These systems are genetically more stable and easier to transform and handle in scale-up processes, to grow faster in adherent and submerged cultures, and to be more similar to human cells and more consistent in their complete spectrum of modification.138 In some cases, mammalian cell systems can be the only choice for the preparation of correctly modified proteins.
Peptides, being complex and unique complex molecules with regard to its chemical and physical properties, can be produced synthetically by the solid-phase method.139, 140 This technology can be used to avoid problems related to biological production. General advantages of synthetic peptides are that they are very stable compounds, solid-phase chemistry produces highly standardized peptides, and the crucial polycation component is provided by a “natural” polycation, thus minimizing toxicity.141
However, some disadvantages related to synthetic peptides have been reported such as the difficulty to synthesize long and well-folded oligopeptides, peptides with multiple cysteine, methionine, arginine, and tryptophan residues due to technical limitations or production cost.141
D. Physicochemical Characterization
When working with protein nanoparticles, it is very important to characterize them physically and functionally in order to understand their behavior.
The size and charge of protein/cargo particles are crucial properties which influence rates of diffusion, binding to polyanionic components of connective tissues, transversal of anatomical barriers, binding of serum proteins, attachment to cells, and mechanisms of endocytosis, among other factors. Stability in physiological salt solutions is a key issue for in vivo delivery, as salt is found everywhere in the body.141 Mixing a multivalent polycation and DNA results in electrostatic binding of both molecules, with charge neutralization of DNA and a particle formation named conjugate. Charge neutralization can be easily seen by retardation gel assays and particle formation by dynamic light scattering (DLS). DLS is a good method to see particle formation but not to quantify relative number of particles of different sizes.142
To visualize particles, many groups have used transmission electron microscopy (TEM)15, 143 with good results while others have used fluid particle image analyzer (FPIA) to photograph individual particles in physiological solutions.58
The net charge of protein/cargo particles is an important variable. Generally, optimal gene delivery for cell lines requires a net positive charge but, as stated previously, it has to be determined empirically. One of the best techniques to determine the net charge is by calculating the Zeta potential that measures the electrophoretic mobility of particles.144
Despite the fact that physical characterization is a key element, understanding and testing the functionality and pharmacokinetics of a gene or drug is the most important part of its development process. Most of the initial tests are done using cell lines in in vitro experiments using reporter genes, RNA, or drugs.145, 146 Quantifying the percentage of transfected cells or drug-induced changes is a very valuable tool to evaluate nanoparticle performance in both nuclear and cytoplasmic delivery, respectively. In addition, in vitro experiments may be designed to select a candidate for the in vivo experiments from a group of possible therapy vectors.
The quantitative kinetics of particle binding, the molecular basis of particle interactions with target cell membranes, the efficiency of particle internalization, and endosomal escape are all poorly understood.141
Interaction of particles with plasma membranes prior to protein internalization can be either unspecific or specific. Untargeted delivery normally is the consequence of electrostatic interactions between anionic ligands in the cell surface and cationic components of the vehicle. On the other hand, targeted delivery to specific membrane molecules is a more sophisticated approach. It aims to improve cell specificity and efficiency, by directing to molecules, only expressed or overexpressed in a particular cell type, that initiate internalization by endocytosis. Targeting moieties include many types of molecules and is discussed afterwards.
Internalization of particles, its mechanisms, and kinetics are not well known and most studies about nanoparticle delivery do not focus on this aspect. There are several endocytic pathways each initiated by different ligands.147 Enhancing the delivery by addition of chloroquine, a synthetic molecule used primarily for the prophylaxis and treatment of malaria that disrupts endosomes,148 is an accepted parameter to demonstrate endosomal localization of particles.
Endosomal escape is the area most intensively investigated but is poorly understood. An important practical point to note is that some reagents that are used can be toxic.141 To enhance this step, anionic fusogenic peptides can be used. These peptides fuse to membranes in an acidic-dependent manner causing its disruption.149
In gene delivery approaches, translocation of DNA expression plasmids into the cell nucleus involves an active, energy-dependent process through the nuclear pore complex.150 Directly injected DNA into the cytosol is usually, but not always, poorly transferred to the nucleus150, 151 and because of that, the use of proteins carrying cationic nuclear-localizing sequences (such as that of SV40 large T antigen) has been widely used to overcome this step.143
IV. Natural Self-Assembling Protein Nanoparticles: VLPs
Ideal drug delivery and gene therapy vehicles must accomplish some desired features such as appropriate packaging size for its cargo, target cell-specificity, safe and efficient cargo delivery, and protection against immune recognition, or capability to escape immune recognition. Moreover, these vehicles must avoid inflammatory toxicity and rapid clearance.152
In this context, viral vectors have been exploited as one of the vehicles of choice. Viruses are nano-sized (15–400 nm) supramolecular nucleoprotein-based entities, covered or not with a lipid bilayer (enveloped/nonenveloped viruses) that satisfy, into relatively simple structures, outstanding properties and functions that are relevant to drug and gene delivery. Viruses are able to recognize and interact specifically with cells by receptor-mediated binding, internalize, escape from endosomes, and uncoat and release nucleic acids in different cellular compartments. They are also capable of transcribing and translating their viral proteins to self-assemble into new infectious virus particles and exit the host cell.120, 153, 154, 155
Despite all these relevant properties of viral vectors or some other rising vehicles in drug and gene delivery such as cationic liposomes, their therapeutic use presents some limitations and risks because of the complexity of production, limited packaging capacity, insertional mutagenesis and gene inactivation, low probability of integration, reduced efficacy of repeat administration or reduced expression overtime, unfavorable immunological recognition or strong immune response against vehicle and transgene, inflammatory toxicity, and rapid clearance.120, 152 In this context, virus capsids or VLPs, produced by recombinant capsid proteins but lacking the viral genome, have noticeably emerged as a safer alternative to viral vectors.
A. Structure of Protein Self-Assembled Nanovehicles
VLPs are classically described as self-assembling, nonreplicative and nonpathogenic, highly organized supramolecular multiprotein nanoparticles (coats) (ranging from 20 to 100 nm) that can be formed from the minimal spontaneous self-assembling of one or more viral structural capsid proteins. It has been described that the self-assembling process of the structural viral proteins for VLP formation involves both spontaneous assembly, under favorable experimental conditions, and the requirement of scaffold proteins as catalysts.156, 157 Therefore, VLPs are considered protein “coats”, “shells”, or “boxes” that lack the viral genome, still conserve the structure, morphology, and some properties of viruses. Some of these properties such as cellular tropism and uptake, intracellular trafficking, membrane translocation, and transfer of nucleic acids or molecules across the cytoplasmic, endosomal, and nuclear membranes are important for drug delivery and gene therapy.120, 153, 155, 158, 159, 160 Usually, the degree of similarity of VLPs and their viruses depends on the number of proteins incorporated into the constructs.161, 162
Since the first description in 1983 of the viral DNA packaging into mouse polyomavirus (MPyV) VLPs and its transduction in vitro, 163 VLPs of different viruses such as papillomaviruses,164, 165, 166 hepatitis B, C, and E viruses,167, 168, 169 polyomaviruses,163, 170, 171, 172, 173, 174, 175, 176, 177, 178, 179 lentivirus,180, 181 rotavirus,145, 182 parvovirus,183, 184 and norovirus185 have been generated.
B. Characteristic Features of VLPS and Their Limitations
VLPs offer some structure, dynamics, characteristic features, and functions that make them appealing bionanomaterials to be exploited in the biomedicine arena as drug and gene delivery vehicles and are discussed in detail afterward.
On the one hand, viral coat proteins have the ability to spontaneously self-assemble, which ensures the formation of highly organized, regular, repetitive structurally stable, and very low morphological polydisperse particles that provide useful properties to be used as scaffolds for bioimaging, synthesis of bionanomaterials, and as nanocarriers in drug and gene therapy.186 In addition, homogeneity of particle size and composition is a desired production factor when developing therapeutic molecules. The overexpression of structural viral proteins in a convenient expression system renders recombinant proteins capable of being folded and assembled in discrete organized nanoparticles with a defined size corresponding to the natural capsid geometry.187, 188, 189 Moreover, even though VLPs are structurally stable particles, some biochemical and structural studies have observed that viral capsids and bacteriophages may show some structurally dynamic properties varying in shape, size, or rearrangements of the coat proteins, in response to different factors such as pH.190, 191, 192, 193
On the other hand, VLPs are considered biologically safe nanostructures since they are not infectious (lack of viral genome) and do not replicate, representing a safer alternative to viral vectors.160, 194, 195, 196, 197 However, they can elicit immune and inflammatory responses, especially when repeated administration is needed.152 It has to be also noted that when used in vaccination, VLPs could show excellent adjuvant properties and the majority of VLPs stimulate strong cellular and humoral immune responses as direct immunogens.198 It has been suggested that recombinant VLPs derived from infection of insect cells with baculovirus or even those derived from prokaryotic systems could be contaminated with different residual components of these host cells, contributing those impurities to the adjuvant properties.153
One interesting property of VLPs is that coat viral proteins present an enormous elasticity and adaptability to be modified chemically and/or by protein genetic engineering154, 160, 199 to incorporate multiple directed functionalities, in order to be addressed in biomedical applications such as drug delivery or gene therapy. It has been recently reviewed that chemically and/or genetically modified VLPs, including CPMV, CCMV, MS2, M13 bacteriophages, and other virus-based nanoparticles,155, 186 could maintain their structural integrity and improve their physical stability154 and, moreover, these modifications could also confer desired cell-targeting properties to the nanovehicle.153, 154, 155, 186, 200, 201 VLPs can be successfully engineered with spatial precision to incorporate (attached or genetically displayed on the surface) targeting tissue-specific ligands such as epidermal growth factor (EGFR) and antibodies, or other molecules such as oligonucleotides, peptides, gold, and other metals, target proteins, carbohydrates, polymers, fluorophores, quantum dots, drugs, or small molecules.152, 154, 155 Moreover, one of the potential benefits of such modifications is that the specific geometric rearrangement confers precise recognition patterns.200, 201
Furthermore, accessibility of the materials carried within the particle and the ability of inclusion and separation of nucleic acids, small molecules, and unusual cargoes with appropriate charge is another outstanding feature and key advantage of VLPs that has also made them excellent vessels for gene and drug delivery.152, 195 As described above, VLPs can be used as empty nanocarriers to transport molecules chemically attached on their surface or can be loaded ex vivo with therapeutic small molecules such as drugs, DNAs, mRNAs, siRNAs, oligonucleotides, quantum dots, magnetic nanoparticles, or proteins.155, 157, 160 VLPs of different papillomavirus and polyomavirus have been widely characterized and used for directed delivery in biomedical applications.132, 165, 173, 174, 194, 202 Osmotic shock and in vitro self-assembling of VLP subunits in the presence of the cargo have been the two main strategies used to packaged nucleic acid or other small molecules. It has to be taken into account that some attachment of the cargo on the VLP surface can occur.195 Besides, diversity of natural tropism including liver for hepatitis B VLPs, spleen for some papillomavirus and polyomavirus VLPs, antigen-presenting cells for certain papillomavirus VLPs, and glial cells for human polyomavirus JC (JCV) VLPs, among others152 is one of the key advantages offered by VLPs providing a wide spectrum of specific targeting and distribution profiles depending on the directed application. Although each VLP has its own characteristic receptors, entry pathway, and intracellular trafficking, it has been demonstrated that tropism of VLPs could be customized, modifying the residues identified as ligands of the cellular receptor on VLPs’ surface or even varying the delivery routes.155, 189, 203
Another key advantage of VLPs is that they can be easily produced by using a wide range of hosts and expression systems, each of them with its own conditionings.162 In the past years, there has been an increasing need to improve and optimize efficient large-scale production systems, process control and monitoring, and up- and down-streaming processes.153, 157, 159, 204 Production of VLPs usually involves transfection of the cell host expression system of choice with a plasmid encoding one or more viral structural proteins, further and rigorous purification for the removal of immunogenic cellular contaminants, and quality control of the produced VLP and encapsulation of the cargo ex vivo before administration.152, 158 The most frequent and convenient expression systems, adaptable to large-scale processes are (1) yeast cells176, (2) mammalian cells, (3) insect cells infected with recombinant baculovirus205, 206, (4) bacteria204, 207, (5) green plants infected with modified viruses208, 209, and (6) cell-free systems.163, 204 The preparative and large-scale manufacture of VLPs in some of these hosts has been reviewed by Pattenden et al. and can be classified into two main methods of bioprocessing: in vivo and in vitro systems.157 In addition, the capability of in vitro dissociation and reassociation of VLPs contribute to the application of easy and more accurate purification methods than those of viral vectors.152, 157 Furthermore, depending on the expression system, the resulting VLP might be significantly different even though expressing the same viral proteins. Thus, a broad spectrum of VLPs could be customized depending on the VLP type, the number of proteins needed for VLP assembling, and the targeted final application.158, 210
As described above, VLPs have great potential as nanocarriers in drug and gene delivery. At the same time, although there is an increasing flow of developments in this area, these vehicles also present some limitations that should be addressed and taken into account, such as residual cellular components, variable yield of functional VLPs after disassembly/reassembly process, immunostimulation and unsuitability for repeated administration, tolerance to the transgene, ineffective therapeutic molecule loading, and low transfection rates.152
C. Tuning VLPS (Chemically or Genetically) for Their Uses/Applications in Gene Therapy and Drug Delivery
Due to their versatile nanoparticulate structure and morphology, and nonreplicative and noninfecting nature combined with their natural immunogenic properties and ease production, VLPs have principally emerged as an excellent alternative tool to attenuate viruses for vaccination.152, 153, 204, 210 There are currently commercialized upon the US Food and Drug Administration (FDA) approvals of some VLP-based vaccines that effectively protect humans from hepatitis B virus (HBV) (GlaxoSmithKline's Engerix® and Merck and Co., Inc.'s Recombivax HB®) and human papillomavirus (HPV) (Cervarix®, an HPV 16/18 VLP vaccine developed by GlaxoSmithKline's and Gardasil® developed by Merck against types 6, 11, 16, and 18 HPV). Other immunogenic VLP-based vaccines are already under clinical trials, preclinical test, or basic investigation including HBV,211, 212 HIV,180, 213 influenza virus,214 parvovirus,159 Norwalk virus,185 rotavirus,182 and Ebola virus.215, 216
Although VLP-based vaccines have been primarily developed for their use against the corresponding virus, in the last decades genetic engineering or chemical modifications have been applied in order to generate chimeric VLPs. Thus, on the one hand, commonly short heterologous peptide epitopes or full proteins that are unable to form VLPs or that are unsafe for vaccination have been presented on surface-exposed loops or fused to N- or C-exposed termini of structural viral capsid proteins on VLPs.154, 161, 210 Different HPV,217, 218, 219 HBV,220, 221 parvovirus,222, 223 and chimeric polyoma VLPs have been engineered170, 175 and tested for different applications including vaccination against viral or bacterial diseases, against virus-induced tumors, and more recently, for immunotherapy of nonviral cancer.161, 210 On the other hand, chemical bioconjugation for covalent coupling of protein epitopes and small molecules to lysines, cysteines, or tyrosine residues of VLP surfaces has been applied in viral or cancer vaccines.200 Chackerian et al. have demonstrated the efficient induction of protective autoantibodies using self-antigen conjugation to HPV VLPs.224
It is important to point out that VLPs can also be engineered to incorporate heterologous cell-specific ligands to cell receptors, thus altering their cellular tropism.154, 155, 186, 201 This great convertibility and flexibility of VLPs to be modified (chemically and/or genetically), their high stability, natural and diverse tropism, their nanocontainer properties, and their ability to enter in the cell and incorporate, bind, and deliver nucleic acids and small molecules have positioned VLPs as appealing entities not only for vaccination applications but also for a broad spectrum of other diverse and emerging applications in nanomedicine and nanotechnology such as immunotherapy against cancer,210, 225 gene therapy delivery of therapeutic genes into specific cells,161, 165, 171, 184, 226, 227 and targeted delivery of drugs and small molecules using VLPs as nanocarriers.174, 196
Although there is no commercial VLP as vector in gene therapy, since the initial work in 1970 of uncoating polyoma pseudovirus in mouse embryo cells as gene delivery vector228 and the establishment in 1983 of the viral DNA packaging into MPyV VLPs and its transduction in vitro, 163 different VLPs such as HBV and hepatitis E virus,229 HPV and polyomavirus nanoparticles172, 178, 229 have been modified toward the specific delivery of therapeutic genes and proteins in different target cells, organs, and tissues in vitro and in vivo by systemic injection229 or oral administration.230 For example, recombinant VP1-based polyomavirus VLPs can encapsulate in vitro exogenous DNA, and deliver it by cell surface sialic acid residues to human brain cells and fetal kidney epithelial cells.178 Furthermore, VLPs have recently emerged as novel nanocarriers or nanocontainers to store unnatural cargos, deliver modified oligonucleotides,154 synthetic small interfering RNAs, and plasmids expressing short hairpin RNAs as therapy to downregulate gene expression.171, 231 In this context, Chou et al. have recently described the use of JCV VLPs as an efficient vector for delivering RNAi in vitro using murine macrophage RAW 264.7 cells and in vivo using BALB/c mice in silencing the cytokine gene of IL-10 without significant cytotoxicity for systemic lupus erythematosus gene therapy.171
One of the key aspects in targeted gene and drug delivery is cell-specific delivery. It is important to point out that VLPs are tunable nanoparticles that can also be chemically or genetically engineered to modify their natural cellular tropism in order to diversify the range of therapeutic applications in targeted gene or drug delivery.154, 201 Some effective approaches to modify the natural cellular tropism include:
-
(1)
Genetic engineering of VLP chimeras incorporating heterologous cell-specific short peptides that contain recognition sites of target cell receptors.232 In this context, polyoma and papillomavirus, with solved atomic structures of their major structural capsid proteins, have been extensively used to obtain chimeric VLPs as delivery vector systems.165, 233 However, this approach has some bioprocessing limitations such as low production levels as a consequence of VLP modification, alterations of size and properties of the VLPs that could affect the structural interactions and conformations for VLP assembly, disassembly and packaging, and low transduction efficiencies.157
-
(2)
Chemical bioconjugation of purified VLPs with epitope-containing peptides234, 235 or a wide range of small molecules conferring cell-specific targeting such as transferrins, folic acid, or other targeting molecules. As an example, CMPV VLPs have been successfully conjugated with Tfn using “click” chemistry236 and with NHS-ester-derivatized folic acid, demonstrating both as internalized into HeLa cells and KB cells, respectively.183, 184
-
(3)
High-throughput library and directed evolution method is a rational approach that has been recently used to engineer viral vectors with the desired tropism properties.237
-
(4)
Pseudotyping, which consists of replacing the envelope protein of one virus species by the envelope protein of another virus species.238
-
(5)
Modification of the delivery route of the VLPs. It has been shown that the levels of expression of β-galactosidase in heart, lung, kidney, spleen, liver, and brain are different depending on the delivery route of polyomavirus VP1 VLPs.203
The great accessibility and reactivity showed by VLPs, as well as their ability to serve as nanocarriers, which made them suitable to be exploited in gene therapy, have also been applied to targeted drug delivery.195 Genetic modification and/or chemical functionalization of exposed amino acid residues on the capsid surface in order to attach small molecules, such as markers or bioactives molecules, is one of the most common approaches applied to target drug delivery.174, 239 As an example, canine parvovirus (CPV) VLPs produced in a baculovirus expression system and exhibiting natural tropism to transferrin receptors (TfRs) were chemically modified on accessible lysines of the capsid surface with fluorescent dye molecules and delivered to tumor cells. Derivatization of CPV-VLPs did not interfere with the binding and internalization into tumor cells.183, 184
One limitation of VLPs in gene therapy is the low efficiency of gene transduction due to inefficient DNA packaging. However, a recent study presented a novel in vivo DNA packaging of JCV VLPs in E. coli that effectively reduced human colon carcinoma volume in a nude mouse model. In this study, the exogenous plasmid DNA was transformed into the JCV VP1 expressing E. coli. The packaging of the second plasmid occurs simultaneously as the in vivo assembly of the JCV VLP. Even though it is still not clear how the plasmid DNA molecules are encapsidated in the VLP, the authors showed that gene transduction efficiency by their in vivo package system was about 80% in contrast to the 1–2% of gene transduction efficiency achieved by the in vitro osmotic shock system.226 In addition, the administration of exogenous proteins may induce the immune system response, reducing therapy effectiveness or causing undesirable secondary effects, albeit immunological response of protein nanoparticles can be modulated.240
V. Nonviral Self-Assembling Proteins
Spontaneous protein self-assembly to form ordered oligomers is a common event in biology. It can prove advantageous in terms of genome-size minimization, formation of large structures, stabilization of complexes, and inclusion of functional features.241 It has been widely documented that cellular oligomer proteins as well as viral capsids are stabilized by several weak noncovalent interactions as hydrophobic interaction, electrostatic energy, and Van der Waals forces.242, 243, 244 These interactions result in a complex quaternary structure described by three symmetry point groups named cyclic (Cn), dihedral (Dm), and cubic (T, O, I).245, 246
The development of computational techniques to predict protein–protein interactions using solved 3D protein structures makes it possible to predict and/or strengthen experimental data performing in in silico approaches.247 Furthermore, its use opens up the possibility to design proteins not only displaying specific biological functions but also interesting intermolecular interactions to obtain increased multivalency in the resulting complexes. Moreover, it should be considered that not only whole proteins can self-assemble in smart nanoparticles; oligopeptides are also capable of forming organized structures. Many applications are possible due to the enormous quantity of different combinations and features that can be exploited with peptides.248, 249
Furthermore, protein–protein interactions are not the unique parameters involved in particle formation, nucleic acid–peptide interactions, salt concentration, order of mix, and ratio between nucleic acid and protein can also strongly influence the condensation process.250, 251
Due to their natural tendency to self-assemble forming highly ordered structures, viruses provide a wide variety of scaffold proteins which are used as gene/drug carriers. Among them, VLPs have been reviewed in the previous section. However, simple bacterial proteins can be also utilized as carriers for gene delivery. For example, heat shock proteins (HSP) from hyperthermophilic archeaon Methanococcus jannaschii can assemble in a small structure of 24 subunits having an octahedral symmetry. These 12 nm structures are stable at high temperature, up to 70 °C, and wide range of pH. Residue modifications are allowed to elicit specific attachment of small molecules.186, 252
In bacteria, bacterial microcompartments (BMC) which are intracellular organelles consisting of enzymes encapsulated within polyhedral, protein-only shells, somewhat similar to viral capsids, have been described. BMCs are composed of a few thousand copies of a few repeated protein species (including one or more enzymes involved in specific metabolic pathways), and with sizes of around 100–150 nm in cross section. The general role of BMCs is to confine toxic or volatile metabolic intermediates, while allowing enzyme substrates, products, and cofactors to pass.
The first described BMC, the carboxysome, was isolated in the early 1970s253, 254 and has been found to contain both CO2-fixing ribulose bisphosphate carboxylase/oxygenase (RuBisCO)253, 254 and carbonic anhydrase255, 256, 257 enzymes. Carboxysomes’ function is to enhance autotrophic CO2 fixation at low CO2 levels.
Other BMCs were later identified in cyanobacteria and some chemoautotroph bacteria. Among them, BMC proteins have been later found to be encoded in the propanediol utilization operon (pdu) of the heterotroph Salmonella258 and by an operon for metabolizing ethanolamine (eut) in enteric bacterial species, including Salmonella and Escherichia. 259 Salmonella enterica forms a polyhedral organelle during growth on 1,2-propanediol (1,2-PD) as a sole carbon and energy source, but not during growth on other carbon sources.260, 261
The pdu organelles’ function is to minimize the harmful effects of a toxic intermediate of 1,2-PD degradation (propionaldehyde).261, 262, 263 Other studies have shown that a polyhedral organelle is involved in ethanolamine utilization (eut) by S. enterica. 259 The function of the eut microcompartment is to metabolize ethanolamine without allowing the release of acetaldehyde into the cytosol, therefore minimizing the potentially toxic effects of excess aldehyde in the bacterial cytosol264, 265, 266 and also preventing volatile acetaldehyde from diffusing across cell membrane.267
So far, about 1700 proteins containing BMC domains have been identified, covering at least 10 different bacterial phyla. The typical BMC protein consists of approximately 90 amino acids, with an alpha/beta fold pattern.268, 269 Some individual BMC proteins self-assemble to form hexamers, which further assemble side by side to form the flat facets of the shell.268, 270, 271 The formation of icosahedral, closed shells from such flat layers was elucidated in part by structural studies in carboxysomes: some BMC proteins assemble to form pentamers, which are located at and form the vertices of the icosahedral shell.270
Mechanisms directing enzyme encapsulation within protein-based BMCs have been studied during the last years. It has been described that, in some carboxysomes, protein CcmM is used as a scaffold to form interactions between both shell proteins and enzymes,272, 273 through a CcmM C-terminal region with homology to the small subunit of RuBisCO.274 Other studies revealed that pdu shells can self-assemble without needing interior enzymes275 and that carboxysomes can self-assemble in vivo when RuBisCO has been deleted.276
Regarding properties of the encapsulated enzymes, in the pdu BMC some of the internal enzymes are encapsulated by specific N-terminal targeting sequences.275, 277 In this line, Sutter and colleagues278 described a conserved C-terminal amino acid sequence that mediates the physical interaction of an iron-dependent peroxidase (DyP) or a protein closely related to ferritin (Flp) with a specific type of BMC (encapsulins).
In another example, an icosahedral enzyme complex, lumazine synthase (AaLS) from Bacillus subtilis and Aquifex aeolicus, was engineered to encapsulate target molecules by means of charge complementarity and can also be modified to give different characteristics to the assembled structure.279, 280
Moreover, enzymatic subunits, like E2 of pyruvate dehydrogenase from Bacillus stearotermophilus, can be modified to be used in gene delivery. E2 peptides naturally form a dodecahedron of 60 subunits of 24 nm in diameter allowing modification for drug-like accommodation. The assembling/disassembling of these structures can be modulated by changing the operative pH in the experimental environment. These nanoparticles can also be functionalized with antigens for vaccine development.281, 282
According to these results, specific targeting sequences could be of use in biotechnological applications to package proteins inside the stable self-assembled icosahedral shell of BMCs, offering appealing opportunities to manipulate in the laboratory such nanocages to fill them with therapeutic molecules. The simplicity of this system makes it very attractive for engineering studies to design, mimicking nature, new applications in biotechnology, providing a new, intriguing platform of microbial origin for drug delivery.
Bovine serum albumin (BSA) is able to form microspheres after sonochemical treatment in aqueous medium. Chemical effects of ultrasound radiation and coupling with an anticancer drug such as Taxol (paclitaxel) led to the assembling of a spherical carrier with an average diameter of 120 nm. BSA particles resulting from S–S bonds, due to HO2 radical formation, are able to release the encapsulated Taxol in cancer tissue with best results if compared with mere Taxol treatment. This drug for breast cancer treatment is commercially available.283, 284
Also little cationic peptides can lead to self-assembling particles. Among others, arginine-rich cationic peptides are widely known as good tools for gene delivery. For example, purified R9-tailored GFP in solution is described to form nanodisk particles 20 nm in diameter. This structure is proved to be induced by the 9 arg tails and is able to bind and condense DNA. These nanodisks are also able to deliver DNA toward the nucleus where the reporter gene is expressed.285
On the other hand, the expression of recombinant proteins over physiological rates can cause a bad functioning of cellular quality control system, leading to self-organizing, pseudo-spherical, protein aggregates known as inclusion bodies. These mechanically stable nanoparticles, ranging from 50 to 500 nm in diameter, were considered for a long time as undesired bio-products. Recently, it became clearer that they are suitable for medical approaches when utilized as scaffold surface to promote cellular proliferation.286, 287, 288
One of the most difficult goals for a foreign gene delivery is to reach the nucleus. An approach to overpass this obstacle is by fusing an NLS in a nonessential position of a DNA-binding protein. Such type of modification has been described for a tetracycline repressor protein (TetR) fused with an SV40 NLS. The TetR–NLS affinity and specificity to TetO DNA sequence is exploited to form spontaneous protein–DNA complexes which allow an enhancing of DNA transportation into the nucleus and subsequent expression of foreign genes, combining the two peculiar characteristics of each fusion component.289
VI. Medical Applications of Protein Nanoparticles
There is still a tremendous gap between progresses made in protein-based nanoparticle research for drug delivery and clinical reality. Hundreds of publications in basic research describe the combination of two or more functional elements in a single protein nanoparticle, by which the delivery of a carried drug is enhanced. These agents act by improving critical steps in the drug delivery process, such as increasing the systemic stability or tissue specificity, favoring internalization, endosomal escape, and entry into the nucleus, or transporting therapeutic material through the BBB, in in vitro and in vivo studies.
Besides the human recombinant therapeutic proteins currently on the market (or functional segments of them), there are also some fusion proteins approved for clinical use (most by incorporating an antibody fragment or a ligand to enhance cell specificity). Sadly no gene therapy trials have so far used full protein carriers in vivo, but rather peptide-functionalized vehicles.
Bottlenecking the gap between research and clinical application, the US FDA/European Medicines Agency (EMEA) only approves human proteins, to avoid the risk of an immune response that could affect not only the effectiveness of the nanoparticle but also challenge patients’ health. Another critical factor is the administration route, where the protein is degraded before arriving at the target; this problem could be solved or minimized by the use of protein d-isomers, PEGylation, or the design of protecting groups for labile sites. Despite the current situation mentioned above, there are many good examples of multifunctional modular proteins that, when carrying therapeutic material, can improve the prognosis in vivo in animal models for different diseases. These examples are reviewed below, along with those few protein nanoparticles that are currently on the market or in clinical trials.
A. Therapeutic Protein Nanoparticles Currently in the Market
Albumin is a natural protein transporter of hydrophobic molecules throughout plasma that has been approved by the FDA to reversibly bind water-insoluble anticancer agents, as is the case of albumin-bound (nab) paclitaxel, Abraxane®. This albumin-nab technology-based drug is in use in patients with metastatic breast cancer who have failed combination therapy, and it is the first protein nanoparticle approved by the FDA. Albumin potentiates paclitaxel concentration within the tumor by increasing paclytaxel endothelial transcytosis through caveolae formation. It also contributes to the fact that tumors secrete an albumin-binding protein SPARC (also called BM-40) to attract and keep albumin-bound nutrients inside the tumor cell.290 The albumin–paclitaxel complex was not formally considered a nanoparticle in the United States (due to an average size of 130 nm) but only so in Europe.
Apart from whole recombinant therapeutic proteins being currently commercialized, there are also some examples of vehicles formed by chimerical proteins with target ligands already in the market. DAB389IL-2 (denileukin diftitox or Ontak) is a fusion of Diphtheria toxin catalytic and translocation domains for lethal effect and interleukin-2 (IL-2) to gain cell specificity in the treatment of persistent or recurrent T-cell lymphoma. Belatacept (BMS-224818) is a CTLA4-Ig fusion protein formed by the cytotoxic T-lymphocyte-associated antigen 4 joined to an immunoglobulin G1 Fc fragment fusion protein, developed by Bristol–Miers–Squibb. Etanercept (Enbrel) fusion tumor necrosis factor receptor (TNFR), which binds and inhibits specifically TNF activity, to an immune globulin G1 Fc, to prevent inflammation mediated by TNF in autoimmune diseases like arthritis and psoriasis.
On the other hand, fusion proteins which include an antihuman epidermal growth factor receptor 2 (HER2) monoclonal antibody that binds tumor cell surfaces, among them the so-called “trastuzumab” (commercialized as Herceptin by Roche), associated to DM-1, an antimitotic drug, aimed at improving the treatment of breast cancer.
Finally, VLPs, that is, empty viral entities formed by the self-assembly of a viral capsid protein, are the only truly protein nanoparticles (architectonically speaking) which are currently used in clinical practice. HBsAg recombinant protein of HBV expressed in yeast and the capsid L1 recombinant protein of HPV (types 6, 11, 16, and 18) administered currently as vaccines tend to form spontaneously VLPs that elicit T and B immune response. Recently, there have been preclinical and clinical trials to test the security and efficacy of VLP vaccines against Chikungunya291 and seasonal influenza virus (http://www.medpagetoday.com/MeetingCoverage//ICAAC/22129), respectively. Influenza VLP vaccines have proven to provide complete protection against H1N1 2009 flu pandemics,292 within a record preparation time when compared to 9 months for traditional vaccines. The use of VLPs as a delivery system for drugs or nucleic acids in gene therapy is still under investigation.194
Drugs and proteins may be transformed through pegylation, a process that can assist them in overcoming some of the potential problems that delay the adoption of protein nanoparticles for clinical use. The covalent attachment of PEG can reduce immunogenicity and antigenicity by hiding the particle from the immune system, can increase the circulating time by reducing renal clearance, and can also improve the water solubility of a hydrophobic particle. The use of pegylation has been approved for commercial use by the FDA and EMEA, and some examples of pegylated protein products are Adagen® (PEG-bovine adenosine deaminase), the first pegylated protein approved by the FDA in 1990, Pegasys® (PEG-interferon alpha), and Oncaspar® (PEG-l-asparaginase).
B. Therapeutic Protein Nanoparticles Currently in Clinical Trials
The majority of protein nanoparticles studied in clinical trials (http://clinicaltrias.gov) are fusion proteins composed of a therapeutic protein/peptide and a target cell-specific ligand. An example is ALT-801, a biologic compound composed of IL-2 genetically fused to a humanized soluble T-cell receptor directed against the p53-derived antigen. The clinical trials evaluated whether directing IL-2 activity using ALT-801 to the patient's tumor sites that overexpress p53 results in clinical benefits (NCT01029873, NCT00496860). Another ligand joined to IL-2 is L19, a tumor-targeted immunocytokine constituted of a single chain fragment variable (scFv) directed against the ED-B domain of fibronectin, one of the most important markers for neoangiogenesis. L19–IL-2 is in a Phase I/II study for patients with solid tumors and renal cell carcinoma (RCC) (NCT01058538). L19 has also been fused to TNFα with the intention to target TNFα directly to tumor tissues resulting in high and sustained intralesional bioactive TNFα concentrations. The L19TNFα is under clinical trial using isolated inferior limb perfusion (ILP) with the standard treatment with melphalan 10 mg/l limb volume in subjects affected by stage III/IV limb melanoma (NCT01213732). NGR-hTNF is another bifunctional protein which combines a tumor-homing peptide (NGR) that selectively binds to amino peptidase N/CD13 highly expressed on tumor blood vessels, thus affecting tumor vascular permeability, and hTNF, with direct anticancer activity. NGR-hTNF is undergoing 14 clinical trials as a single agent to treat different cancers, as well as in combination with chemotherapy agents.
Another strategy to direct a therapeutic protein to the target cell is through fusion to a growth factor receptor ligand. An example is TP-38, a recombinant chimerical protein composed of the EGFR binding ligand (TGF-α) and a genetically engineered form of the Pseudomonas exotoxin, PE-38, to treat recurrent grade IV malignant brain tumors (NCT00071539).
Many clinical trials are based on a therapeutic protein fused to a targeting antibody, as is the case of APC8015. This drug stimulates the immune system and stops cancer cells from growing by the combination of biological therapies with Bevacizumab®, an already approved monoclonal antibody that locates tumor cells and kills them in a specific way (NCT00849290). There are also many putative protein drugs against cancer which include antibodies anti-integrins (e.g., cilengitide and IMGN388), sometimes in combination with classical therapies. A recently developed tool, the nanobodies or single domain antibodies,293 have several advantages: small size (only 12–15 KDa), which lowers the possibility of triggering immune response, safety in clinical trials (NCT01020383), and is easy to be joined to different kinds of compounds. All these features make nanobodies competent drugs against different diseases, and have been tested in vivo as bifunctional proteins associated to a prodrug, very efficient in mice cancer xenografts.294
Even though CPPs are very useful tools to deliver drugs and in gene therapy (see the chapter “Peptide Nanoparticles for Oligonucleotide Delivery” by Lehto et al. in this volume), their toxicity and endosomal entrapment slows their inclusion for systemic delivery in clinical trials. Nevertheless, there are a few examples of use to prevent undesirable cell proliferation in coronary artery bypass grafts, as is the case of a CPP (R-Ahx-R)4AhxB–PMO conjugate targeted to human c-myc to be applied ex vivo. The trial, in phase II, has been completed in 2009 (NCT00451256). Another case is PsorBan®, a product patented for the treatment of psoriasis based on a cyclosporine–polyarginine conjugate of local application, which circumvents the specificity problem of intravenous (i.v.) application. It is in clinical trial phase III, but not yet in the market. Finally, KAI-9803, a PKCδ inhibitor peptide conjugated to Tat to function as an intravenous drug for the treatment of acute myocardial infarction, is currently in phase 2b clinical trial (NCT00785954, KAI pharmaceuticals).
C. Therapeutic Protein Nanoparticles in Preclinical Models of Human Diseases
There are many proteins, often organized as nanoparticles, that when associated to a drug, therapeutic protein, peptide, or nucleic acid increase the therapeutic efficacy of a cargo alone in the treatment of various diseases. Some of them proved effective in animal models, which are discussed in more detail in this section, with relevant examples listed in Table II .
Table II.
Representative Examples of Protein Nanoparticles That, Acting as Carriers, Improve The Efficiency of Cargo Alone in the Treatment of Diseases Using In Vivo models
| Carrier | Cargo | Administration route | Disease | References |
|---|---|---|---|---|
| VP-22 | Gata4 | Transplant of transfected cells | Myocardial infartion | 295 |
| (RXR)4XB | Dystrophin exon skipping PMO | i.p.a | Duchenne muscular dystrophy | 296 |
| Tat-ErbB2 | STAT3BP | i.p. | Breast cancer xenograft | 302 |
| Penetratin | scFVs-radionuclide | i.v.b | Colon carcinoma xenograft | 305 |
| 8R | Taxol | i.p. | i.p. tumor xenografts | 297 |
| Penetratin | Caveolin-1 | i.p. | Inflammation models | 298 |
| Tat-HA | Bcl-xL | i.p. | Cerebral ischemia | 313 |
| Protamine-Erb2 Ab Fab | c-myc, MDM2, VEGF-siRNA | i.v. | Breast cancer | 301 |
| 9-d-arginine-RVG | JEV-siRNA | i.v. | JEV infection | 56 |
| Pegylated Pep-3 | Cyclin B1 -PNA | i.v. | Human prostate carcinoma xenograft | 299 |
| Chol-MPG-8 | Cyclin B1-siRNA | i.v. | Prostate and lung cancer xenografts | 300 |
| Tat | pVHL | i.p. | Mice with renal tumors dorsally implanted | 310 |
| Tat | MHC class I antigens | s.c.c | Dendritic cell vaccine for tumor regresion | 311 |
Intraperitoneal.
Intravenous.
Subcutaneous.
These nanoparticles may simply be (a) a CPP to promote nonspecific internalization,295, 296, 297, 298, 299, 300 (b) a peptide to confer cargo specificity by joining a receptor distinctive of a cell type, including scFvs or peptides obtained by phage display,301 and (c) a mixture of both,302 since as observed in several studies the CPP does not reduce ligand specificity and increases nanoparticle potency.303, 304, 305 Complex and multifunctional vehicles including endosomal escape peptides enhance the therapeutic potency of the complex, or other domains that allow their selective activation in certain contexts.306, 307
Apart from the cases listed in Table II, the spectrum of additional examples of multidomain protein nanoparticles tested in vivo is wide, and a considerable proportion of them include CPPs, mainly Tat and polyarginines. A classical Tat fusion protein is the transducible d-isomer RI-TATp53C′' fusion protein that activates p53 protein in cancer cells, but not in normal cells. RI-TATp53C′ treatment in terminal peritoneal carcinomatosis and peritoneal lymphoma preclinical models results in significant increases in life span (higher than sixfold) and full recovery from the disease.308 There are also several studies in vivo using Tat-fused therapeutic proteins which have proven effective in treating tumors309, 310, 311 and cerebral ischemia312, 313 when applied intraperitoneally (i.p.).
Regarding polyarginines, Kumar and colleagues have presented two different models in which a bifunctional peptide formed by nine arginines (9R) and a specific ligand constitute an effective siRNA vehicle. In the first model, a chimerical peptide derived from rabies virus glycoprotein (to confer neuronal specificity) fused to 9- d-arginines (RVG-9R), was able to transport si-RNA across the BBB and silence specific gene expression in the brain when applied intravenously.56 In the second model, a CD7-specific single-chain antibody was conjugated to oligo-9-arginine peptide (scFvCD7-9R) for T cell-specific antiviral siRNA delivery in humanized mice reconstituted with human lymphocytes. In HIV-infected humanized mice, this treatment controlled viral replication and prevented the disease-associated CD4 T cell loss. Moreover, it effectively suppressed viremia in infected mice.314
Some other examples of polyarginines in tumor models are 9-d-arginines fused to a tumor-suppressor peptide, which stopped tumor growth in hepatocellular carcinoma-bearing mice when applied intraperitoneally, and also colesteryl oligoarginines carrying VEGF siRNA, which inhibited tumor growth in colon adenocarcinoma after local application.315 Another BBB-crossing peptide is g7, which is able to transport nanoparticles loaded with Loperamide.107
In general, the partner fusion peptide can confer specificity instead of penetrability, as is the case of EGFR Fab fragment associated to liposomes that contain anticancer drug, which increases efficiency of anticancer effect in EGF overexpressing xenograft tumors316; in addition, RGD-4 C-doxorubicin in human breast xenografts increases efficacy and diminishes toxicity.317
In many conjugates, the therapeutic peptide of the chimerical proteins is a toxin. Anthrax lethal toxin has been modified to be activated by methaloproteases, and it has probed to be effective for human xenografted tumors such as melanoma, lung, and colorectal cancer.318 Anthrax toxin has also been associated to antibodies or growth factors for lethal effects specifically on cancer cells.319 The specific cytotoxicity desired to treat a tumor might derive from a tissue factor, which promotes clotting to restrict blood supply in tumor vessels, fused to peptides that provide specificity, like V-CAM antibodies, fibronectin, and integrin ligands.320
Eventually, drug activity may decrease when conjugated to a carrier protein, although if the entry of the drug is favored, the overall balance of activity can be much more efficient.321 On the other hand, the use of noncovalent bond drug carrier could avoid interfering with the activity of the drug.
An important issue in a preclinical study to be considered for a clinical trial is the administration route. In in vivo experiments, most of the protein nanoparticles are administered by local or intraperitoneal injection, avoiding systemic spreading and clearance in the vascular system, in a way very similar to in vitro experiments. The FDA and EMEA, on the other hand, will preferentially approve i.v. and oral administrations rather than intraperitoneal or local injections except for very accessible tissues. Another relevant issue is the number of active domains to be included in a therapeutic protein carrier, an issue that seems to be relevant for the functionality of the construct. For example, the CPP neutralization of a ligand may depend on the CPP/ligand ratio that is in the vehicle.322 It has also been observed that the integrin binding power of RGD-containing motives increases with the number of RGD domains over the monomer until a maxim of four moieties.323 Another example is Tat activity empowerment when attached to molecules that form tetramers, such as beta-galactosidase108 and p-53.324
Some multidomain protein carriers allow the drug entrance only in selected target cells by tailored smart selective mechanisms.325 For instance, CPPs neutralized by polyanions are activated and enter the cells when they are released by metalloproteases326 or by lowering the pH,327 both situations being very common in tumors.
CPP-morpholino oligomer (PMO) nanoparticles have also shown their effectiveness in treating viral infections by inhibiting viral replication, as demonstrated with the carrier (R-ahx-R) 4AhxB-PMO administered i.v. in animal models infected with picornaviruses, i.p. in mice infected with coronaviruses and flaviviruses, and the carrier R9F2C-PMO administered also i.p. in mice infected with Ebola virus. Furthermore, it has also been shown in some of these studies that the efficacy of the treatment is dependent on the incorporation of arginine-rich peptides in the nanoparticle.328
A good example of how a CPP can improve the internalization of a therapeutic protein is the case of insulin. The instability and low absorption in the digestive tract of insulin prevents its oral administration, even though it would be very convenient for a daily administrated drug. In recent studies, noncovalent conjugation of insulin to different CPPs enhances its absorption without toxic intestinal effect, l-penetratin being the most efficient as insulin carrier.329
Among the protein nanoparticles tested in vivo, it is worth making special mention of Trojan horses generated in Pardridge's laboratory to cross the BBB, through a strategy of fusing within a chimerical peptide the therapeutic protein which has to reach the CNS to a monoclonal antibody against the human insulin receptor (HIRMAb). This Trojan horse is very potent for humans and primates, and has proven effective to transport β-glucuronidase, α-l-iduronidase, GDNF, Abeta amyloid peptides, paroxonase, etc., with potential benefits in diseases like mucopolysaccharidosis type VII, Hurler syndrome, Parkinson, Alzheimer, and organophosphates toxicity, respectively.330
There are also promising results when protein nanoparticles have been tested as carriers for gene therapy in vivo, some examples being listed in Table II. In this regard, the use of modular proteins generated by insertional mutagenesis of β-galactosidase condensing the SOD gene are able to protect neurons against ischemic injury133; a bifunctional galactosylated polylysine is able to conjugate plasmid DNA and to differentially promote expression in hepatocytes that display asialoglycoprotein receptor331; a suicide multidomain protein particle formed by herpes simplex virus thymidine kinase (HSV-TK) conjugated to transferrin (Tf) by a biotin-streptavidin bridging, which, administered i.v. in K562 massively metastasized nude mice, was able to reduce tumor size and to increase mouse survival.332
VII. Conclusions
In this chapter, proteins and peptides have been envisioned as potent biotechnological tools for the development of new biocompatible biological entities that can be used as therapeutic agents by themselves or as nanovehicles for the delivery of associated drugs. Proteins are nanostructures that can form complex high-order entities such as VLPs, resulting in appropriate cages for the internalization of therapeutic molecules. In addition, the design of modular proteins displaying selected functions has been possible by using in silico approximations to the feasibility of recombinant protein production. This approach has demonstrated the versatility of such molecules in the generation of novel delivery nanovehicles opening up the possibility of new functional combinations to enhance the specific interaction with the target tissue. Such tunable specificity in the delivery of drugs, nucleic acids, or other proteins is one of the main properties that make multifunctional proteins appealing as more rational delivery vehicles.
The presence on the market of such complex entities, which started with the approval of Insulin for the treatment of diabetes, has been increasing over the past years, and this tendency is expected to continue. In fact, there are some products in clinical trials that will probably end up being approved and some more are being explored in preclinical experiments which might enter in clinical trials.
Acknowledgments
The authors appreciate the financial support received through grants BFU2010-17450 from MICINN, PS0900165 from FISS, and 2009SGR-108 from AGAUR. The authors also acknowledge the support of the CIBER de Bioingeniería, Biomateriales y Nanomedicina (CIBER-BBN), an initiative funded by the VI National R&D&i Plan 2008–2011, Iniciativa Ingenio 2010, Consolider Program, CIBER Actions and financed by the Instituto de Salud Carlos III with assistance from the European Regional Development Fund.
References
- 1.Shun T.Y., Lazo J.S., Sharlow E.R., Johnston P.A. Identifying actives from HTS data sets: practical approaches for the selection of an appropriate HTS data-processing method and quality control review. J Biomol Screen. 2011;16:1–14. doi: 10.1177/1087057110389039. [DOI] [PubMed] [Google Scholar]
- 2.Vuorela P., Leinonen M., Saikku P., Tammela P., Rauha J.P., Wennberg T. Natural products in the process of finding new drug candidates. Curr Med Chem. 2004;11:1375–1389. doi: 10.2174/0929867043365116. [DOI] [PubMed] [Google Scholar]
- 3.Zhao H.Y., Akritopoulou-Zanze I. When analoging is not enough: scaffold discovery in medicinal chemistry. Expert Opin Drug Discovery. 2010;5:123–134. doi: 10.1517/17460440903584874. [DOI] [PubMed] [Google Scholar]
- 4.Adamina M., Joerger M. Dose-toxicity models in oncology. Expert Opin Drug Metab Toxicol. 2011;7:201–211. doi: 10.1517/17425255.2011.543674. [DOI] [PubMed] [Google Scholar]
- 5.Schmid E.F., Smith D.A. Is declining innovation in the pharmaceutical industry a myth? Drug Discov Today. 2005;10:1031–1039. doi: 10.1016/S1359-6446(05)03524-5. [DOI] [PubMed] [Google Scholar]
- 6.Veronese F.M., Mero A. The impact of PEGylation on biological therapies. BioDrugs. 2008;22:315–329. doi: 10.2165/00063030-200822050-00004. [DOI] [PubMed] [Google Scholar]
- 7.Raja S.M., Clubb R.J., Ortega-Cava C., Williams S.H., Bailey T.A., Duan L. Anticancer activity of celastrol in combination with ErbB2-targeted therapeutics for treatment of ErbB2-overexpressing breast cancers. Cancer Biol Ther. 2011;11:263–276. doi: 10.4161/cbt.11.2.13959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Desai N., Trieu V., Damascelli B., Soon-Shiong P. SPARC expression correlates with tumor response to albumin-bound paclitaxel in head and neck cancer patients. Transl Oncol. 2009;2:59–64. doi: 10.1593/tlo.09109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Desai N.P., Trieu V., Hwang L.Y., Wu R., Soon-Shiong P., Gradishar W.J. Improved effectiveness of nanoparticle albumin-bound (nab) paclitaxel versus polysorbate-based docetaxel in multiple xenografts as a function of HER2 and SPARC status. Anticancer Drugs. 2008;19:899–909. doi: 10.1097/CAD.0b013e32830f9046. [DOI] [PubMed] [Google Scholar]
- 10.Houk B.E., Martin R., Hochhaus G., Hughes J.A. Pharmacokinetics of plasmid DNA in the rat. Pharm Res. 2001;18:67–74. doi: 10.1023/a:1011078711008. [DOI] [PubMed] [Google Scholar]
- 11.Wang W. Instability, stabilization, and formulation of liquid protein pharmaceuticals. Int J Pharm. 1999;185:129–188. doi: 10.1016/s0378-5173(99)00152-0. [DOI] [PubMed] [Google Scholar]
- 12.Martin M.E., Rice K.G. Peptide-guided gene delivery. AAPS J. 2007;9:E18–E29. doi: 10.1208/aapsj0901003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mastrobattista E., van der Aa M.A., Hennink W.E., Crommelin D.J. Artificial viruses: a nanotechnological approach to gene delivery. Nat Rev Drug Discov. 2006;5:115–121. doi: 10.1038/nrd1960. [DOI] [PubMed] [Google Scholar]
- 14.Gabathuler R. Approaches to transport therapeutic drugs across the blood-brain barrier to treat brain diseases. Neurobiol Dis. 2010;37:48–57. doi: 10.1016/j.nbd.2009.07.028. [DOI] [PubMed] [Google Scholar]
- 15.Glover D.J., Ng S.M., Mechler A., Martin L.L., Jans D.A. Multifunctional protein nanocarriers for targeted nuclear gene delivery in nondividing cells. FASEB J. 2009;23:2996–3006. doi: 10.1096/fj.09-131425. [DOI] [PubMed] [Google Scholar]
- 16.Tiera M.J., Winnik F.O., Fernandes J.C. Synthetic and natural polycations for gene therapy: state of the art and new perspectives. Curr Gene Ther. 2006;6:59–71. doi: 10.2174/156652306775515510. [DOI] [PubMed] [Google Scholar]
- 17.Mannisto M., Vanderkerken S., Toncheva V., Elomaa M., Ruponen M., Schacht E. Structure-activity relationships of poly(L-lysines): effects of pegylation and molecular shape on physicochemical and biological properties in gene delivery. J Control Release. 2002;83:169–182. doi: 10.1016/s0168-3659(02)00178-5. [DOI] [PubMed] [Google Scholar]
- 18.Ward C.M., Read M.L., Seymour L.W. Systemic circulation of poly(L-lysine)/DNA vectors is influenced by polycation molecular weight and type of DNA: differential circulation in mice and rats and the implications for human gene therapy. Blood. 2001;97:2221–2229. doi: 10.1182/blood.v97.8.2221. [DOI] [PubMed] [Google Scholar]
- 19.Gottschalk S., Sparrow J.T., Hauer J., Mims M.P., Leland F.E., Woo S.L. A novel DNA-peptide complex for efficient gene transfer and expression in mammalian cells. Gene Ther. 1996;3:448–457. [PubMed] [Google Scholar]
- 20.Plank C., Tang M.X., Wolfe A.R., Szoka F.C., Jr. Branched cationic peptides for gene delivery: role of type and number of cationic residues in formation and in vitro activity of DNA polyplexes. Hum Gene Ther. 1999;10:319–332. doi: 10.1089/10430349950019101. [DOI] [PubMed] [Google Scholar]
- 21.McKenzie D.L., Collard W.T., Rice K.G. Comparative gene transfer efficiency of low molecular weight polylysine DNA-condensing peptides. J Pept Res. 1999;54:311–318. doi: 10.1034/j.1399-3011.1999.00104.x. [DOI] [PubMed] [Google Scholar]
- 22.McKenzie D.L., Smiley E., Kwok K.Y., Rice K.G. Low molecular weight disulfide cross-linking peptides as nonviral gene delivery carriers. Bioconjug Chem. 2000;11:901–909. doi: 10.1021/bc000056i. [DOI] [PubMed] [Google Scholar]
- 23.Sorgi F.L., Bhattacharya S., Huang L. Protamine sulfate enhances lipid-mediated gene transfer. Gene Ther. 1997;4:961–968. doi: 10.1038/sj.gt.3300484. [DOI] [PubMed] [Google Scholar]
- 24.Brewer L.R., Corzett M., Balhorn R. Protamine-induced condensation and decondensation of the same DNA molecule. Science. 1999;286:120–123. doi: 10.1126/science.286.5437.120. [DOI] [PubMed] [Google Scholar]
- 25.Masuda T., Akita H., Harashima H. Evaluation of nuclear transfer and transcription of plasmid DNA condensed with protamine by microinjection: the use of a nuclear transfer score. FEBS Lett. 2005;579:2143–2148. doi: 10.1016/j.febslet.2005.02.071. [DOI] [PubMed] [Google Scholar]
- 26.Balhorn R. The protamine family of sperm nuclear proteins. Genome Biol. 2007;8:227. doi: 10.1186/gb-2007-8-9-227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ferrer-Miralles N., Vazquez E., Villaverde A. Membrane-active peptides for non-viral gene therapy: making the safest easier. Trends Biotechnol. 2008;26:267–275. doi: 10.1016/j.tibtech.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 28.Chan C.K., Jans D.A. Enhancement of MSH receptor- and GAL4-mediated gene transfer by switching the nuclear import pathway. Gene Ther. 2001;8:166–171. doi: 10.1038/sj.gt.3301366. [DOI] [PubMed] [Google Scholar]
- 29.Fominaya J., Wels W. Target cell-specific DNA transfer mediated by a chimeric multidomain protein: novel non-viral gene delivery system. J Biol Chem. 1996;271:10560–10568. doi: 10.1074/jbc.271.18.10560. [DOI] [PubMed] [Google Scholar]
- 30.Box M., Parks D.A., Knight A., Hale C., Fishman P.S., Fairweather N.F. A multi-domain protein system based on the HC fragment of tetanus toxin for targeting DNA to neuronal cells. J Drug Target. 2003;11:333–343. doi: 10.1080/1061186310001634667. [DOI] [PubMed] [Google Scholar]
- 31.Baleja J.D., Thanabal V., Wagner G. Refined solution structure of the DNA-binding domain of GAL4 and use of 3J(113Cd,1H) in structure determination. J Biomol NMR. 1997;10:397–401. doi: 10.1023/a:1018332327565. [DOI] [PubMed] [Google Scholar]
- 32.Bessis N., GarciaCozar F.J., Boissier M.C. Immune responses to gene therapy vectors: influence on vector function and effector mechanisms. Gene Ther. 2004;11(Suppl. 1):S10–S17. doi: 10.1038/sj.gt.3302364. [DOI] [PubMed] [Google Scholar]
- 33.Vazquez E., Ferrer-Miralles N., Villaverde A. Peptide-assisted traffic engineering for nonviral gene therapy. Drug Discov Today. 2008;13:1067–1074. doi: 10.1016/j.drudis.2008.08.008. [DOI] [PubMed] [Google Scholar]
- 34.Richard J.P., Melikov K., Vives E., Ramos C., Verbeure B., Gait M.J. Cell-penetrating peptides: a reevaluation of the mechanism of cellular uptake. J Biol Chem. 2003;278:585–590. doi: 10.1074/jbc.M209548200. [DOI] [PubMed] [Google Scholar]
- 35.Said H.F., Saleh A.F., Abes R., Gait M.J., Lebleu B. Cell penetrating peptides: overview and applications to the delivery of oligonucleotides. Cell Mol Life Sci. 2010;67:715–726. doi: 10.1007/s00018-009-0186-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vazquez E., Ferrer-Miralles N., Mangues R., Corchero J.L., Schwartz S., Villaverde A. Modular protein engineering in emerging cancer therapies. Curr Pharm Des. 2009;15:893–916. doi: 10.2174/138161209787582084. [DOI] [PubMed] [Google Scholar]
- 37.Rudolph C., Plank C., Lausier J., Schillinger U., Muller R.H., Rosenecker J. Oligomers of the arginine-rich motif of the HIV-1 TAT protein are capable of transferring plasmid DNA into cells. J Biol Chem. 2003;278:11411–11418. doi: 10.1074/jbc.M211891200. [DOI] [PubMed] [Google Scholar]
- 38.Fawell S., Seery J., Daikh Y., Moore C., Chen L.L., Pepinsky B. Tat-mediated delivery of heterologous proteins into cells. Proc Natl Acad Sci USA. 1994;91:664–668. doi: 10.1073/pnas.91.2.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brooks H., Lebleu B., Vives E. Tat peptide-mediated cellular delivery: back to basics. Adv Drug Deliv Rev. 2005;57:559–577. doi: 10.1016/j.addr.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 40.Vives E., Brodin P., Lebleu B. A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J Biol Chem. 1997;272:16010–16017. doi: 10.1074/jbc.272.25.16010. [DOI] [PubMed] [Google Scholar]
- 41.Vives E. Cellular uptake [correction of utake] of the Tat peptide: an endocytosis mechanism following ionic interactions. J Mol Recognit. 2003;16:265–271. doi: 10.1002/jmr.636. [DOI] [PubMed] [Google Scholar]
- 42.Wender P.A., Mitchell D.J., Pattabiraman K., Pelkey E.T., Steinman L., Rothbard J.B. The design, synthesis, and evaluation of molecules that enable or enhance cellular uptake: peptoid molecular transporters. Proc Natl Acad Sci USA. 2000;97:13003–13008. doi: 10.1073/pnas.97.24.13003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lundberg P., El-Andaloussi S., Sutlu T., Johansson H., Langel U. Delivery of short interfering RNA using endosomolytic cell-penetrating peptides. FASEB J. 2007;21:2664–2671. doi: 10.1096/fj.06-6502com. [DOI] [PubMed] [Google Scholar]
- 44.Muratovska A., Eccles M.R. Conjugate for efficient delivery of short interfering RNA (siRNA) into mammalian cells. FEBS Lett. 2004;558:63–68. doi: 10.1016/S0014-5793(03)01505-9. [DOI] [PubMed] [Google Scholar]
- 45.Pooga M., Hallbrink M., Zorko M., Langel U. Cell penetration by transportan. FASEB J. 1998;12:67–77. doi: 10.1096/fasebj.12.1.67. [DOI] [PubMed] [Google Scholar]
- 46.Pooga M., Kut C., Kihlmark M., Hallbrink M., Fernaeus S., Raid R. Cellular translocation of proteins by transportan. FASEB J. 2001;15:1451–1453. doi: 10.1096/fj.00-0780fje. [DOI] [PubMed] [Google Scholar]
- 47.Hart S.L. Integrin-mediated vectors for gene transfer and therapy. Curr Opin Mol Ther. 1999;1:197–203. [PubMed] [Google Scholar]
- 48.Kessler T., Bieker R., Padro T., Schwoppe C., Persigehl T., Bremer C. Inhibition of tumor growth by RGD peptide-directed delivery of truncated tissue factor to the tumor vasculature. Clin Cancer Res. 2005;11:6317–6324. doi: 10.1158/1078-0432.CCR-05-0389. [DOI] [PubMed] [Google Scholar]
- 49.Amara A., Gall S.L., Schwartz O., Salamero J., Montes M., Loetscher P. HIV coreceptor downregulation as antiviral principle: SDF-1alpha-dependent internalization of the chemokine receptor CXCR4 contributes to inhibition of HIV replication. J Exp Med. 1997;186:139–146. doi: 10.1084/jem.186.1.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liang X. CXCR4, inhibitors and mechanisms of action. Chem Biol Drug Des. 2008;72:97–110. doi: 10.1111/j.1747-0285.2008.00681.x. [DOI] [PubMed] [Google Scholar]
- 51.Daniels T.R., Delgado T., Helguera G., Penichet M.L. The transferrin receptor part II: targeted delivery of therapeutic agents into cancer cells. Clin Immunol. 2006;121:159–176. doi: 10.1016/j.clim.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 52.Joung I., Harber G., Gerecke K.M., Carroll S.L., Collawn J.F., Engler J.A. Improved gene delivery into neuroglial cells using a fiber-modified adenovirus vector. Biochem Biophys Res Commun. 2005;328:1182–1187. doi: 10.1016/j.bbrc.2005.01.080. [DOI] [PubMed] [Google Scholar]
- 53.Liu X., Tian P.K., Ju D.W., Zhang M.H., Yao M., Cao X.T. Systemic genetic transfer of p21WAF-1 and GM-CSF utilizing of a novel oligopeptide-based EGF receptor targeting polyplex. Cancer Gene Ther. 2003;10:529–539. doi: 10.1038/sj.cgt.7700596. [DOI] [PubMed] [Google Scholar]
- 54.Wolschek M.F., Thallinger C., Kursa M., Rossler V., Allen M., Lichtenberger C. Specific systemic nonviral gene delivery to human hepatocellular carcinoma xenografts in SCID mice. Hepatology. 2002;36:1106–1114. doi: 10.1053/jhep.2002.36372. [DOI] [PubMed] [Google Scholar]
- 55.Wu Y.T., Jiaang W.T., Lin K.G., Huang C.M., Chang C.H., Sun Y.L. A new N-acetylgalactosamine containing peptide as a targeting vehicle for mammalian hepatocytes via asialoglycoprotein receptor endocytosis. Curr Drug Deliv. 2004;1:119–127. doi: 10.2174/1567201043479939. [DOI] [PubMed] [Google Scholar]
- 56.Kumar P., Wu H., McBride J.L., Jung K.E., Kim M.H., Davidson B.L. Transvascular delivery of small interfering RNA to the central nervous system. Nature. 2007;448:39–43. doi: 10.1038/nature05901. [DOI] [PubMed] [Google Scholar]
- 57.Schneider H., Harbottle R.P., Yokosaki Y., Kunde J., Sheppard D., Coutelle C. A novel peptide, PLAEIDGIELTY, for the targeting of alpha9beta1-integrins. FEBS Lett. 1998;429:269–273. doi: 10.1016/s0014-5793(98)00612-7. [DOI] [PubMed] [Google Scholar]
- 58.Collins L., Fabre J.W. A synthetic peptide vector system for optimal gene delivery to corneal endothelium. J Gene Med. 2004;6:185–194. doi: 10.1002/jgm.482. [DOI] [PubMed] [Google Scholar]
- 59.McKay T., Reynolds P., Jezzard S., Curiel D., Coutelle C. Secretin-mediated gene delivery, a specific targeting mechanism with potential for treatment of biliary and pancreatic disease in cystic fibrosis. Mol Ther. 2002;5:447–454. doi: 10.1006/mthe.2002.0560. [DOI] [PubMed] [Google Scholar]
- 60.Zeng J., Too H.P., Ma Y., Luo E.S., Wang S. A synthetic peptide containing loop 4 of nerve growth factor for targeted gene delivery. J Gene Med. 2004;6:1247–1256. doi: 10.1002/jgm.610. [DOI] [PubMed] [Google Scholar]
- 61.Martinez-Fong D., Navarro-Quiroga I., Ochoa I., varez-Maya I., Meraz M.A., Luna J. Neurotensin-SPDP-poly-L-lysine conjugate: a nonviral vector for targeted gene delivery to neural cells. Brain Res Mol Brain Res. 1999;69:249–262. doi: 10.1016/s0169-328x(99)00114-x. [DOI] [PubMed] [Google Scholar]
- 62.White S.J., Nicklin S.A., Sawamura T., Baker A.H. Identification of peptides that target the endothelial cell-specific LOX-1 receptor. Hypertension. 2001;37:449–455. doi: 10.1161/01.hyp.37.2.449. [DOI] [PubMed] [Google Scholar]
- 63.Pardridge W.M., Buciak J.L., Friden P.M. Selective transport of an anti-transferrin receptor antibody through the blood-brain barrier in vivo. J Pharmacol Exp Ther. 1991;259:66–70. [PubMed] [Google Scholar]
- 64.Boado R.J., Zhang Y., Zhang Y., Pardridge W.M. Humanization of anti-human insulin receptor antibody for drug targeting across the human blood-brain barrier. Biotechnol Bioeng. 2007;96:381–391. doi: 10.1002/bit.21120. [DOI] [PubMed] [Google Scholar]
- 65.Jeong J.H., Lee M., Kim W.J., Yockman J.W., Park T.G., Kim Y.H. Anti-GAD antibody targeted non-viral gene delivery to islet beta cells. J Control Release. 2005;107:562–570. doi: 10.1016/j.jconrel.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 66.Balestrieri M.L., Napoli C. Novel challenges in exploring peptide ligands and corresponding tissue-specific endothelial receptors. Eur J Cancer. 2007;43:1242–1250. doi: 10.1016/j.ejca.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 67.Aina O.H., Liu R., Sutcliffe J.L., Marik J., Pan C.X., Lam K.S. From combinatorial chemistry to cancer-targeting peptides. Mol Pharm. 2007;4:631–651. doi: 10.1021/mp700073y. [DOI] [PubMed] [Google Scholar]
- 68.Lundberg M., Wikstrom S., Johansson M. Cell surface adherence and endocytosis of protein transduction domains. Mol Ther. 2003;8:143–150. doi: 10.1016/s1525-0016(03)00135-7. [DOI] [PubMed] [Google Scholar]
- 69.Wagner E., Plank C., Zatloukal K., Cotten M., Birnstiel M.L. Influenza virus hemagglutinin HA-2 N-terminal fusogenic peptides augment gene transfer by transferrin-polylysine-DNA complexes: toward a synthetic virus-like gene-transfer vehicle. Proc Natl Acad Sci USA. 1992;89:7934–7938. doi: 10.1073/pnas.89.17.7934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Plank C., Oberhauser B., Mechtler K., Koch C., Wagner E. The influence of endosome-disruptive peptides on gene transfer using synthetic virus-like gene transfer systems. J Biol Chem. 1994;269:12918–12924. [PubMed] [Google Scholar]
- 71.Subbarao N.K., Parente R.A., Szoka F.C., Jr., Nadasdi L., Pongracz K. pH-dependent bilayer destabilization by an amphipathic peptide. Biochemistry. 1987;26:2964–2972. doi: 10.1021/bi00385a002. [DOI] [PubMed] [Google Scholar]
- 72.Parente R.A., Nir S., Szoka F.C., Jr. Mechanism of leakage of phospholipid vesicle contents induced by the peptide GALA. Biochemistry. 1990;29:8720–8728. doi: 10.1021/bi00489a031. [DOI] [PubMed] [Google Scholar]
- 73.Parente R.A., Nadasdi L., Subbarao N.K., Szoka F.C., Jr. Association of a pH-sensitive peptide with membrane vesicles: role of amino acid sequence. Biochemistry. 1990;29:8713–8719. doi: 10.1021/bi00489a030. [DOI] [PubMed] [Google Scholar]
- 74.Li W., Nicol F., Szoka F.C., Jr. GALA: a designed synthetic pH-responsive amphipathic peptide with applications in drug and gene delivery. Adv Drug Deliv Rev. 2004;56:967–985. doi: 10.1016/j.addr.2003.10.041. [DOI] [PubMed] [Google Scholar]
- 75.Wyman T.B., Nicol F., Zelphati O., Scaria P.V., Plank C., Szoka F.C., Jr. Design, synthesis, and characterization of a cationic peptide that binds to nucleic acids and permeabilizes bilayers. Biochemistry. 1997;36:3008–3017. doi: 10.1021/bi9618474. [DOI] [PubMed] [Google Scholar]
- 76.Rittner K., Benavente A., Bompard-Sorlet A., Heitz F., Divita G., Brasseur R. New basic membrane-destabilizing peptides for plasmid-based gene delivery in vitro and in vivo. Mol Ther. 2002;5:104–114. doi: 10.1006/mthe.2002.0523. [DOI] [PubMed] [Google Scholar]
- 77.Ogris M., Carlisle R.C., Bettinger T., Seymour L.W. Melittin enables efficient vesicular escape and enhanced nuclear access of nonviral gene delivery vectors. J Biol Chem. 2001;276:47550–47555. doi: 10.1074/jbc.M108331200. [DOI] [PubMed] [Google Scholar]
- 78.Derossi D., Joliot A.H., Chassaing G., Prochiantz A. The third helix of the Antennapedia homeodomain translocates through biological membranes. J Biol Chem. 1994;269:10444–10450. [PubMed] [Google Scholar]
- 79.Derossi D., Chassaing G., Prochiantz A. Trojan peptides: the penetratin system for intracellular delivery. Trends Cell Biol. 1998;8:84–87. [PubMed] [Google Scholar]
- 80.Pichon C., Goncalves C., Midoux P. Histidine-rich peptides and polymers for nucleic acids delivery. Adv Drug Deliv Rev. 2001;53:75–94. doi: 10.1016/s0169-409x(01)00221-6. [DOI] [PubMed] [Google Scholar]
- 81.Midoux P., Kichler A., Boutin V., Maurizot J.C., Monsigny M. Membrane permeabilization and efficient gene transfer by a peptide containing several histidines. Bioconj Chem. 1998;9:260–267. doi: 10.1021/bc9701611. [DOI] [PubMed] [Google Scholar]
- 82.Midoux P., LeCam E., Coulaud D., Delain E., Pichon C. Histidine containing peptides and polypeptides as nucleic acid vectors. Somat Cell Mol Genet. 2002;27:27–47. doi: 10.1023/a:1022931923153. [DOI] [PubMed] [Google Scholar]
- 83.Kichler A., Leborgne C., Danos O., Bechinger B. Characterization of the gene transfer process mediated by histidine-rich peptides. J Mol Med. 2007;85:191–201. doi: 10.1007/s00109-006-0119-4. [DOI] [PubMed] [Google Scholar]
- 84.Kim J., Chen C.P., Rice K.G. The proteasome metabolizes peptide-mediated nonviral gene delivery systems. Gene Ther. 2005;12:1581–1590. doi: 10.1038/sj.gt.3302575. [DOI] [PubMed] [Google Scholar]
- 85.Leonchiks A., Stavropoulou V., Sharipo A., Masucci M.G. Inhibition of ubiquitin-dependent proteolysis by a synthetic glycine-alanine repeat peptide that mimics an inhibitory viral sequence. FEBS Lett. 2002;522:93–98. doi: 10.1016/s0014-5793(02)02897-1. [DOI] [PubMed] [Google Scholar]
- 86.Sharipo A., Imreh M., Leonchiks A., Imreh S., Masucci M.G. A minimal glycine-alanine repeat prevents the interaction of ubiquitinated I kappaB alpha with the proteasome: a new mechanism for selective inhibition of proteolysis. Nat Med. 1998;4:939–944. doi: 10.1038/nm0898-939. [DOI] [PubMed] [Google Scholar]
- 87.Sharipo A., Imreh M., Leonchiks A., Branden C., Masucci M.G. cis-Inhibition of proteasomal degradation by viral repeats: impact of length and amino acid composition. FEBS Lett. 2001;499:137–142. doi: 10.1016/s0014-5793(01)02542-x. [DOI] [PubMed] [Google Scholar]
- 88.Martinez-Moreno M., Navarro-Lerida I., Roncal F., Albar J.P., Alonso C., Gavilanes F. Recognition of novel viral sequences that associate with the dynein light chain LC8 identified through a pepscan technique. FEBS Lett. 2003;544:262–267. doi: 10.1016/s0014-5793(03)00516-7. [DOI] [PubMed] [Google Scholar]
- 89.Melchior F., Gerace L. Mechanisms of nuclear protein import. Curr Opin Cell Biol. 1995;7:310–318. doi: 10.1016/0955-0674(95)80084-0. [DOI] [PubMed] [Google Scholar]
- 90.Zanta M.A., Belguise-Valladier P., Behr J.P. Gene delivery: a single nuclear localization signal peptide is sufficient to carry DNA to the cell nucleus. Proc Natl Acad Sci USA. 1999;96:91–96. doi: 10.1073/pnas.96.1.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Fischer N., Kremmer E., Lautscham G., Mueller-Lantzsch N., Grasser F.A. Epstein-Barr virus nuclear antigen 1 forms a complex with the nuclear transporter karyopherin alpha2. J Biol Chem. 1997;272:3999–4005. doi: 10.1074/jbc.272.7.3999. [DOI] [PubMed] [Google Scholar]
- 92.Dang C.V., Lee W.M. Identification of the human c-myc protein nuclear translocation signal. Mol Cell Biol. 1988;8:4048–4054. doi: 10.1128/mcb.8.10.4048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Fominaya J., Uherek C., Wels W. A chimeric fusion protein containing transforming growth factor-alpha mediates gene transfer via binding to the EGF receptor. Gene Ther. 1998;5:521–530. doi: 10.1038/sj.gt.3300614. [DOI] [PubMed] [Google Scholar]
- 94.Bauerle M., Doenecke D., Albig W. The requirement of H1 histones for a heterodimeric nuclear import receptor. J Biol Chem. 2002;277:32480–32489. doi: 10.1074/jbc.M202765200. [DOI] [PubMed] [Google Scholar]
- 95.Baake M., Bauerle M., Doenecke D., Albig W. Core histones and linker histones are imported into the nucleus by different pathways. Eur J Cell Biol. 2001;80:669–677. doi: 10.1078/0171-9335-00208. [DOI] [PubMed] [Google Scholar]
- 96.Subramanian A., Ranganathan P., Diamond S.L. Nuclear targeting peptide scaffolds for lipofection of nondividing mammalian cells. Nat Biotechnol. 1999;17:873–877. doi: 10.1038/12860. [DOI] [PubMed] [Google Scholar]
- 97.Gharakhanian E., Takahashi J., Kasamatsu H. The carboxyl 35 amino acids of SV40 Vp3 are essential for its nuclear accumulation. Virology. 1987;157:440–448. doi: 10.1016/0042-6822(87)90286-8. [DOI] [PubMed] [Google Scholar]
- 98.Lyons R.H., Ferguson B.Q., Rosenberg M. Pentapeptide nuclear localization signal in adenovirus E1a. Mol Cell Biol. 1987;7:2451–2456. doi: 10.1128/mcb.7.7.2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kleinschmidt J.A., Seiter A. Identification of domains involved in nuclear uptake and histone binding of protein N1 of Xenopus laevis. EMBO J. 1988;7:1605–1614. doi: 10.1002/j.1460-2075.1988.tb02986.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kiefer P., Acland P., Pappin D., Peters G., Dickson C. Competition between nuclear localization and secretory signals determines the subcellular fate of a single CUG-initiated form of FGF3. EMBO J. 1994;13:4126–4136. doi: 10.1002/j.1460-2075.1994.tb06730.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Schreiber V., Molinete M., Boeuf H., de Murcia G., Menissier-de M.J. The human poly(ADP-ribose) polymerase nuclear localization signal is a bipartite element functionally separate from DNA binding and catalytic activity. EMBO J. 1992;11:3263–3269. doi: 10.1002/j.1460-2075.1992.tb05404.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Robbins J., Dilworth S.M., Laskey R.A., Dingwall C. Two interdependent basic domains in nucleoplasmin nuclear targeting sequence: identification of a class of bipartite nuclear targeting sequence. Cell. 1991;64:615–623. doi: 10.1016/0092-8674(91)90245-t. [DOI] [PubMed] [Google Scholar]
- 103.Rivera V.M., Gao G.P., Grant R.L., Schnell M.A., Zoltick P.W., Rozamus L.W. Long-term pharmacologically regulated expression of erythropoietin in primates following AAV-mediated gene transfer. Blood. 2005;105:1424–1430. doi: 10.1182/blood-2004-06-2501. [DOI] [PubMed] [Google Scholar]
- 104.Wang X., Qiu R., Tsark W., Lu Q. Rapid promoter analysis in developing mouse brain and genetic labeling of young neurons by doublecortin-DsRed-express. J Neurosci Res. 2007;85:3567–3573. doi: 10.1002/jnr.21440. [DOI] [PubMed] [Google Scholar]
- 105.Ishiwata A., Mimuro J., Mizukami H., Kashiwakura Y., Takano K., Ohmori T. Liver-restricted expression of the canine factor VIII gene facilitates prevention of inhibitor formation in factor VIII-deficient mice. J Gene Med. 2009;11:1020–1029. doi: 10.1002/jgm.1391. [DOI] [PubMed] [Google Scholar]
- 106.Pardridge W.M. Blood-brain barrier delivery. Drug Discov Today. 2007;12:54–61. doi: 10.1016/j.drudis.2006.10.013. [DOI] [PubMed] [Google Scholar]
- 107.Tosi G., Costantino L., Rivasi F., Ruozi B., Leo E., Vergoni A.V. Targeting the central nervous system: in vivo experiments with peptide-derivatized nanoparticles loaded with Loperamide and Rhodamine-123. J Control Release. 2007;122:1–9. doi: 10.1016/j.jconrel.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 108.Schwarze S.R., Ho A., Vocero-Akbani A., Dowdy S.F. In vivo protein transduction: delivery of a biologically active protein into the mouse. Science. 1999;285:1569–1572. doi: 10.1126/science.285.5433.1569. [DOI] [PubMed] [Google Scholar]
- 109.Boado R.J., Zhang Y., Zhang Y., Pardridge W.M. Genetic engineering, expression, and activity of a fusion protein of a human neurotrophin and a molecular Trojan horse for delivery across the human blood-brain barrier. Biotechnol Bioeng. 2007;97:1376–1386. doi: 10.1002/bit.21369. [DOI] [PubMed] [Google Scholar]
- 110.Boado R.J., Zhang Y., Zhang Y., Xia C.F., Wang Y., Pardridge W.M. Genetic engineering of a lysosomal enzyme fusion protein for targeted delivery across the human blood-brain barrier. Biotechnol Bioeng. 2008;99:475–484. doi: 10.1002/bit.21602. [DOI] [PubMed] [Google Scholar]
- 111.Boado R.J., Zhang Y., Zhang Y., Wang Y., Pardridge W.M. GDNF fusion protein for targeted-drug delivery across the human blood-brain barrier. Biotechnol Bioeng. 2008;100:387–396. doi: 10.1002/bit.21764. [DOI] [PubMed] [Google Scholar]
- 112.Otten L.G., Quax W.J. Directed evolution: selecting today's biocatalysts. Biomol Eng. 2005;22:1–9. doi: 10.1016/j.bioeng.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 113.Lutz S., Patrick W.M. Novel methods for directed evolution of enzymes: quality, not quantity. Curr Opin Biotechnol. 2004;15:291–297. doi: 10.1016/j.copbio.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 114.Neylon C. Chemical and biochemical strategies for the randomization of protein encoding DNA sequences: library construction methods for directed evolution. Nucleic Acids Res. 2004;32:1448–1459. doi: 10.1093/nar/gkh315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Stemmer W.P. Rapid evolution of a protein in vitro by DNA shuffling. Nature. 1994;370:389–391. doi: 10.1038/370389a0. [DOI] [PubMed] [Google Scholar]
- 116.Dahiyat B.I., Mayo S.L. Protein design automation. Protein Sci. 1996;5:895–903. doi: 10.1002/pro.5560050511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Floudas C.A., Fung H.K., McAllister S.R., Mönnigmann M., Rajgaria R. Advances in protein structure prediction and de novo protein design: a review. Chem Eng Sci. 2006;61:966–988. [Google Scholar]
- 118.Walsh S.T., Cheng H., Bryson J.W., Roder H., DeGrado W.F. Solution structure and dynamics of a de novo designed three-helix bundle protein. Proc Natl Acad Sci USA. 1999;96:5486–5491. doi: 10.1073/pnas.96.10.5486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Dahiyat B.I., Mayo S.L. De novo protein design: fully automated sequence selection. Science. 1997;278:82–87. doi: 10.1126/science.278.5335.82. [DOI] [PubMed] [Google Scholar]
- 120.Aris A., Villaverde A. Modular protein engineering for non-viral gene therapy. Trends Biotechnol. 2004;22:371–377. doi: 10.1016/j.tibtech.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 121.Uherek C., Fominaya J., Wels W. A modular DNA carrier protein based on the structure of diphtheria toxin mediates target cell-specific gene delivery. J Biol Chem. 1998;273:8835–8841. doi: 10.1074/jbc.273.15.8835. [DOI] [PubMed] [Google Scholar]
- 122.Li S.D., Huang L. Gene therapy progress and prospects: non-viral gene therapy by systemic delivery. Gene Ther. 2006;13:1313–1319. doi: 10.1038/sj.gt.3302838. [DOI] [PubMed] [Google Scholar]
- 123.Chan C.K., Jans D.A. Using nuclear targeting signals to enhance non-viral gene transfer. Immunol Cell Biol. 2002;80:119–130. doi: 10.1046/j.1440-1711.2002.01061.x. [DOI] [PubMed] [Google Scholar]
- 124.Dietz G.P., Bahr M. Delivery of bioactive molecules into the cell: the Trojan horse approach. Mol Cell Neurosci. 2004;27:85–131. doi: 10.1016/j.mcn.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 125.Dietz G.P., Bahr M. Synthesis of cell-penetrating peptides and their application in neurobiology. Methods Mol Biol. 2007;399:181–198. doi: 10.1007/978-1-59745-504-6_13. [DOI] [PubMed] [Google Scholar]
- 126.Ignatovich I.A., Dizhe E.B., Pavlotskaya A.V., Akifiev B.N., Burov S.V., Orlov S.V. Complexes of plasmid DNA with basic domain 47–57 of the HIV-1 Tat protein are transferred to mammalian cells by endocytosis-mediated pathways. J Biol Chem. 2003;278:42625–42636. doi: 10.1074/jbc.M301431200. [DOI] [PubMed] [Google Scholar]
- 127.Aris A., Villaverde A. Molecular organization of protein-DNA complexes for cell-targeted DNA delivery. Biochem Biophys Res Commun. 2000;278:455–461. doi: 10.1006/bbrc.2000.3824. [DOI] [PubMed] [Google Scholar]
- 128.Carrio M.M., Villaverde A. Role of molecular chaperones in inclusion body formation. FEBS Lett. 2003;537:215–221. doi: 10.1016/s0014-5793(03)00126-1. [DOI] [PubMed] [Google Scholar]
- 129.Baneyx F. Recombinant protein expression in Escherichia coli. Curr Opin Biotechnol. 1999;10:411–421. doi: 10.1016/s0958-1669(99)00003-8. [DOI] [PubMed] [Google Scholar]
- 130.Sorensen H.P., Mortensen K.K. Advanced genetic strategies for recombinant protein expression in Escherichia coli. J Biotechnol. 2005;115:113–128. doi: 10.1016/j.jbiotec.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 131.Schmidt F.R. Recombinant expression systems in the pharmaceutical industry. Appl Microbiol Biotechnol. 2004;65:363–372. doi: 10.1007/s00253-004-1656-9. [DOI] [PubMed] [Google Scholar]
- 132.Ou W.C., Wang M., Fung C.Y., Tsai R.T., Chao P.C., Hseu T.H. The major capsid protein, VP1, of human JC virus expressed in Escherichia coli is able to self-assemble into a capsid-like particle and deliver exogenous DNA into human kidney cells. J Gen Virol. 1999;80:39–46. doi: 10.1099/0022-1317-80-1-39. [DOI] [PubMed] [Google Scholar]
- 133.Peluffo H., Acarin L., Aris A., Gonzalez P., Villaverde A., Castellano B. Neuroprotection from NMDA excitotoxic lesion by Cu/Zn superoxide dismutase gene delivery to the postnatal rat brain by a modular protein vector. BMC Neurosci. 2006;7:35. doi: 10.1186/1471-2202-7-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Sasnauskas K., Buzaite O., Vogel F., Jandrig B., Razanskas R., Staniulis J. Yeast cells allow high-level expression and formation of polyomavirus-like particles. Biol Chem. 1999;380:381–386. doi: 10.1515/BC.1999.050. [DOI] [PubMed] [Google Scholar]
- 135.Birch J.R., Froud S.J. Mammalian cell culture systems for recombinant protein production. Biologicals. 1994;22:127–133. doi: 10.1006/biol.1994.1019. [DOI] [PubMed] [Google Scholar]
- 136.Ikonomou L., Schneider Y.J., Agathos S.N. Insect cell culture for industrial production of recombinant proteins. Appl Microbiol Biotechnol. 2003;62:1–20. doi: 10.1007/s00253-003-1223-9. [DOI] [PubMed] [Google Scholar]
- 137.Reiter M., Bluml G. Large-scale mammalian cell culture. Curr Opin Biotechnol. 1994;5:175–179. doi: 10.1016/s0958-1669(05)80032-1. [DOI] [PubMed] [Google Scholar]
- 138.Gervais A., Hammel Y.A., Pelloux S., Lepage P., Baer G., Carte N. Glycosylation of human recombinant gonadotrophins: characterization and batch-to-batch consistency. Glycobiology. 2003;13:179–189. doi: 10.1093/glycob/cwg020. [DOI] [PubMed] [Google Scholar]
- 139.Collins L., Asuni A.A., Anderton B.H., Fabre J.W. Efficient gene delivery to primary neuron cultures using a synthetic peptide vector system. J Neurosci Methods. 2003;125:113–120. doi: 10.1016/s0165-0270(03)00042-6. [DOI] [PubMed] [Google Scholar]
- 140.Kim E.S., Yang S.W., Hong D.K., Kim W.T., Kim H.G., Lee S.K. Cell-penetrating DNA-binding protein as a safe and efficient naked DNA delivery carrier in vitro and in vivo. Biochem Biophys Res Commun. 2010;392:9–15. doi: 10.1016/j.bbrc.2009.12.135. [DOI] [PubMed] [Google Scholar]
- 141.Fabre J.W., Collins L. Synthetic peptides as non-viral DNA vectors. Curr Gene Ther. 2006;6:459–480. doi: 10.2174/156652306777934865. [DOI] [PubMed] [Google Scholar]
- 142.Parker A.L., Collins L., Zhang X., Fabre J.W. Exploration of peptide motifs for potent non-viral gene delivery highly selective for dividing cells. J Gene Med. 2005;7:1545–1554. doi: 10.1002/jgm.809. [DOI] [PubMed] [Google Scholar]
- 143.Aris A., Villaverde A. Engineering nuclear localization signals in modular protein vehicles for gene therapy. Biochem Biophys Res Commun. 2003;304:625–631. doi: 10.1016/s0006-291x(03)00644-2. [DOI] [PubMed] [Google Scholar]
- 144.Chen L., McCrate J.M., Lee J.C., Li H. The role of surface charge on the uptake and biocompatibility of hydroxyapatite nanoparticles with osteoblast cells. Nanotechnology. 2011;22:105708. doi: 10.1088/0957-4484/22/10/105708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Cortes-Perez N.G., Sapin C., Jaffrelo L., Daou S., Grill J.P., Langella P. Rotavirus-like particles: a novel nanocarrier for the gut. J Biomed Biotechnol. 2010;2010:317545. doi: 10.1155/2010/317545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Alcala P., Feliu J.X., Aris A., Villaverde A. Efficient accommodation of recombinant, foot-and-mouth disease virus RGD peptides to cell-surface integrins. Biochem Biophys Res Commun. 2001;285:201–206. doi: 10.1006/bbrc.2001.5157. [DOI] [PubMed] [Google Scholar]
- 147.Marsh M., Helenius A. Virus entry: open sesame. Cell. 2006;124:729–740. doi: 10.1016/j.cell.2006.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Luthman H., Magnusson G. High efficiency polyoma DNA transfection of chloroquine treated cells. Nucleic Acids Res. 1983;11:1295–1308. doi: 10.1093/nar/11.5.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Vaccaro L., Cross K.J., Kleinjung J., Straus S.K., Thomas D.J., Wharton S.A. Plasticity of influenza haemagglutinin fusion peptides and their interaction with lipid bilayers. Biophys J. 2005;88:25–36. doi: 10.1529/biophysj.104.044537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Ludtke J.J., Zhang G., Sebestyen M.G., Wolff J.A. A nuclear localization signal can enhance both the nuclear transport and expression of 1 kb DNA. J Cell Sci. 1999;112:2033–2041. doi: 10.1242/jcs.112.12.2033. [DOI] [PubMed] [Google Scholar]
- 151.Wilke M., Fortunati E., van den Broek M., Hoogeveen A.T., Scholte B.J. Efficacy of a peptide-based gene delivery system depends on mitotic activity. Gene Ther. 1996;3:1133–1142. [PubMed] [Google Scholar]
- 152.Seow Y., Wood M.J. Biological gene delivery vehicles: beyond viral vectors. Mol Ther. 2009;17:767–777. doi: 10.1038/mt.2009.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Ludwig C., Wagner R. Virus-like particles-universal molecular toolboxes. Curr Opin Biotechnol. 2007;18:537–545. doi: 10.1016/j.copbio.2007.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Mateu M.G. Virus engineering: functionalization and stabilization. Protein Eng Des Sel. 2011;24:53–63. doi: 10.1093/protein/gzq069. [DOI] [PubMed] [Google Scholar]
- 155.Singh P., Gonzalez M.J., Manchester M. Viruses and their uses in nanotechnology. Drug Dev Res. 2006;67:23–41. [Google Scholar]
- 156.Dokland T. Scaffolding proteins and their role in viral assembly. Cell Mol Life Sci. 1999;56:580–603. doi: 10.1007/s000180050455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Pattenden L.K., Middelberg A.P., Niebert M., Lipin D.I. Towards the preparative and large-scale precision manufacture of virus-like particles. Trends Biotechnol. 2005;23:523–529. doi: 10.1016/j.tibtech.2005.07.011. [DOI] [PubMed] [Google Scholar]
- 158.Scheerlinck J.P.Y., Greenwood D.L.V. Virus-sized vaccine delivery systems. Drug Discov Today. 2008;13:882–887. doi: 10.1016/j.drudis.2008.06.016. [DOI] [PubMed] [Google Scholar]
- 159.Roldao A., Mellado M.C., Castilho L.R., Carrondo M.J., Alves P.M. Virus-like particles in vaccine development. Expert Rev Vaccines. 2010;9:1149–1176. doi: 10.1586/erv.10.115. [DOI] [PubMed] [Google Scholar]
- 160.Sanchez-Rodriguez S.P., Munch-Anguiano L., Bustos-Jaimes I. Advances in the development of virus-like particles as tools in medicine and nanoscience. Curr Chem Biol. 2010;4:231–243. [Google Scholar]
- 161.Ramqvist T., Andreasson K., Dalianis T. Vaccination, immune and gene therapy based on virus-like particles against viral infections and cancer. Expert Opin Biol Ther. 2007;7:997–1007. doi: 10.1517/14712598.7.7.997. [DOI] [PubMed] [Google Scholar]
- 162.Roy P., Noad R. Virus-like particles as a vaccine delivery system: myths and facts. Hum Vaccin. 2008;4:5–12. doi: 10.4161/hv.4.1.5559. [DOI] [PubMed] [Google Scholar]
- 163.Slilaty S.N., Aposhian H.V. Gene transfer by polyoma-like particles assembled in a cell-free system. Science. 1983;220:725–727. doi: 10.1126/science.6301016. [DOI] [PubMed] [Google Scholar]
- 164.Lenz P., Day P.M., Pang Y.Y.S., Frye S.A., Jensen P.N., Lowy D.R. Papillomavirus-like particles induce acute activation of dendritic cells. J Immunol. 2001;166:5346–5355. doi: 10.4049/jimmunol.166.9.5346. [DOI] [PubMed] [Google Scholar]
- 165.Xu Y.F., Zhang Y.Q., Xu X.M., Song G.X. Papillomavirus virus-like particles as vehicles for the delivery of epitopes or genes. Arch Virol. 2006;151:2133–2148. doi: 10.1007/s00705-006-0798-8. [DOI] [PubMed] [Google Scholar]
- 166.Buonaguro F.M., Tornesello M.L., Buonaguro L. Virus-like particle vaccines and adjuvants: the HPV paradigm. Expert Rev Vaccines. 2009;8:1379–1398. doi: 10.1586/erv.09.81. [DOI] [PubMed] [Google Scholar]
- 167.Brandenburg B., Stockl L., Gutzeit C., Roos M., Lupberger J., Schwartlander R. A novel system for efficient gene transfer into primary human hepatocytes via cell-permeable hepatitis B virus-like particle. Hepatology. 2005;42:1300–1309. doi: 10.1002/hep.20950. [DOI] [PubMed] [Google Scholar]
- 168.Li T.C., Takeda N., Miyamura T., Matsuura Y., Wang J.C., Engvall H. Essential elements of the capsid protein for self-assembly into empty virus-like particles of hepatitis E virus. J Virol. 2005;79:12999–13006. doi: 10.1128/JVI.79.20.12999-13006.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Xiang J., Wunschmann S., George S.L., Klinzman D., Schmidt W.N., LaBrecque D.R. Recombinant hepatitis C virus-like particles expressed by baculovirus: utility in cell-binding and antibody detection assays. J Med Virol. 2002;68:537–543. doi: 10.1002/jmv.10237. [DOI] [PubMed] [Google Scholar]
- 170.Andreasson K., Tegerstedt K., Eriksson M., Curcio C., Cavallo F., Forni G. Murine pneumotropic virus chimeric Her2/neu virus-like particles as prophylactic and therapeutic vaccines against Her2/neu expressing tumors. Int J Cancer. 2009;124:150–156. doi: 10.1002/ijc.23920. [DOI] [PubMed] [Google Scholar]
- 171.Chou M.I., Hsieh Y.F., Wang M., Chang J.T., Chang D., Zouali M. In vitro and in vivo targeted delivery of IL-10 interfering RNA by JC virus-like particles. J Biomed Sci. 2010;17:51. doi: 10.1186/1423-0127-17-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Gleiter S., Lilie H. Cell-type specific targeting and gene expression using a variant of polyoma VP1 virus-like particles. Biol Chem. 2003;384:247–255. doi: 10.1515/BC.2003.028. [DOI] [PubMed] [Google Scholar]
- 173.Goldmann C., Petry H., Frye S., Ast O., Ebitsch S., Jentsch K.D. Molecular cloning and expression of major structural protein VP1 of the human polyomavirus JC virus: formation of virus-like particles useful for immunological and therapeutic studies. J Virol. 1999;73:4465–4469. doi: 10.1128/jvi.73.5.4465-4469.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Goldmann C., Stolte N., Nisslein T., Hunsmann G., Luke W., Petry H. Packaging of small molecules into VP1-virus-like particles of the human polyomavirus JC virus. J Virol Methods. 2000;90:85–90. doi: 10.1016/s0166-0934(00)00226-3. [DOI] [PubMed] [Google Scholar]
- 175.Lawatscheck R., Aleksaite E., Schenk J.A., Micheel B., Jandrig B., Holland G. Chimeric polyomavirus-derived virus-like particles: the immunogenicity of an inserted peptide applied without adjuvant to mice depends on its insertion site and its flanking linker sequence. Viral Immunol. 2007;20:453–460. doi: 10.1089/vim.2007.0023. [DOI] [PubMed] [Google Scholar]
- 176.Sasnauskas K., Bulavaite A., Hale A., Jin L., Knowles W.A., Gedvilaite A. Generation of recombinant virus-like particles of human and non-human polyomaviruses in yeast Saccharomyces cerevisiae. Intervirology. 2002;45:308–317. doi: 10.1159/000067922. [DOI] [PubMed] [Google Scholar]
- 177.Voronkova T., Kazaks A., Ose V., Ozel M., Scherneck S., Pumpens P. Hamster polyomavirus-derived virus-like particles are able to transfer in vitro encapsidated plasmid DNA to mammalian cells. Virus Genes. 2007;34:303–314. doi: 10.1007/s11262-006-0028-1. [DOI] [PubMed] [Google Scholar]
- 178.Krauzewicz N., Stokrova J., Jenkins C., Elliott M., Higgins C.F., Griffin B.E. Virus-like gene transfer into cells mediated by polyoma virus pseudocapsids. Gene Ther. 2000;7:2122–2131. doi: 10.1038/sj.gt.3301322. [DOI] [PubMed] [Google Scholar]
- 179.Clark B., Caparros-Wanderley W., Musselwhite G., Kotecha M., Griffin B.E. Immunity against both polyomavirus VP1 and a transgene product induced following intranasal delivery of VP1 pseudocapsid-DNA complexes. J Gen Virol. 2001;82:2791–2797. doi: 10.1099/0022-1317-82-11-2791. [DOI] [PubMed] [Google Scholar]
- 180.Young K.R., McBurney S.P., Karkhanis L.U., Ross T.M. Virus-like particles: designing an effective AIDS vaccine. Methods. 2006;40:98–117. doi: 10.1016/j.ymeth.2006.05.024. [DOI] [PubMed] [Google Scholar]
- 181.Muratori C., Bona R., Federico M. Lentivirus-based virus-like particles as a new protein delivery tool. Methods Mol Biol. 2010;614:111–124. doi: 10.1007/978-1-60761-533-0_7. [DOI] [PubMed] [Google Scholar]
- 182.Istrate C., Hinkula J., Charpilienne A., Poncet D., Cohen J., Svensson L. Parenteral administration of RF 8-2/6/7 rotavirus-like particles in a one-dose regimen induce protective immunity in mice. Vaccine. 2008;26:4594–4601. doi: 10.1016/j.vaccine.2008.05.089. [DOI] [PubMed] [Google Scholar]
- 183.Singh P., Destito G., Schneemann A., Manchester M. Canine parvovirus-like particles, a novel nanomaterial for tumor targeting. J Nanobiotechnology. 2006;4:2. doi: 10.1186/1477-3155-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Singh P. Tumor targeting using canine parvovirus nanoparticles. Springer-Verlag; Berlin: 2009. pp. 123–141. [DOI] [PubMed] [Google Scholar]
- 185.Herbst-Kralovetz M., Mason H.S., Chen Q. Norwalk virus-like particles as vaccines. Expert Rev Vaccines. 2010;9:299–307. doi: 10.1586/erv.09.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Manchester M., Singh P. Virus-based nanoparticles (VNPs): platform technologies for diagnostic imaging. Adv Drug Deliv Rev. 2006;58:1505–1522. doi: 10.1016/j.addr.2006.09.014. [DOI] [PubMed] [Google Scholar]
- 187.Pease L.F., III, Lipin D.I., Tsai D.H., Zachariah M.R., Lua L.H., Tarlov M.J. Quantitative characterization of virus-like particles by asymmetrical flow field flow fractionation, electrospray differential mobility analysis, and transmission electron microscopy. Biotechnol Bioeng. 2009;102:845–855. doi: 10.1002/bit.22085. [DOI] [PubMed] [Google Scholar]
- 188.Stehle T., Harrison S.C. High-resolution structure of a polyomavirus VP1-oligosaccharide complex: implications for assembly and receptor binding. EMBO J. 1997;16:5139–5148. doi: 10.1093/emboj/16.16.5139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Neu U., Stehle T., Atwood W.J. The Polyomaviridae: contributions of virus structure to our understanding of virus receptors and infectious entry. Virology. 2009;384:389–399. doi: 10.1016/j.virol.2008.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Johnson J.E., Chiu W. Structures of virus and virus-like particles. Curr Opin Struct Biol. 2000;10:229–235. doi: 10.1016/s0959-440x(00)00073-7. [DOI] [PubMed] [Google Scholar]
- 191.Zhang S. Fabrication of novel biomaterials through molecular self-assembly. Nat Biotechnol. 2003;21:1171–1178. doi: 10.1038/nbt874. [DOI] [PubMed] [Google Scholar]
- 192.Ivanovska I.L., de Pablo P.J., Ibarra B., Sgalari G., MacKintosh F.C., Carrascosa J.L. Bacteriophage capsids: tough nanoshells with complex elastic properties. Proc Natl Acad Sci USA. 2004;101:7600–7605. doi: 10.1073/pnas.0308198101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Bothner B., Taylor D., Jun B., Lee K.K., Siuzdak G., Schultz C.P. Maturation of a tetravirus capsid alters the dynamic properties and creates a metastable complex. Virology. 2005;334:17–27. doi: 10.1016/j.virol.2005.01.017. [DOI] [PubMed] [Google Scholar]
- 194.Petry H., Goldmann C., Ast O., Luke W. The use of virus-like particles for gene transfer. Curr Opin Mol Ther. 2003;5:524–528. [PubMed] [Google Scholar]
- 195.Garcea R.L., Gissmann L. Virus-like particles as vaccines and vessels for the delivery of small molecules. Curr Opin Biotechnol. 2004;15:513–517. doi: 10.1016/j.copbio.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 196.Georgens C., Weyermann J., Zimmer A. Recombinant virus like particles as drug delivery system. Curr Pharm Biotechnol. 2005;6:49–55. doi: 10.2174/1389201053167202. [DOI] [PubMed] [Google Scholar]
- 197.Noad R., Roy P. Virus-like particles as immunogens. Trends Microbiol. 2003;11:438–444. doi: 10.1016/s0966-842x(03)00208-7. [DOI] [PubMed] [Google Scholar]
- 198.Grgacic E.V., Anderson D.A. Virus-like particles: passport to immune recognition. Methods. 2006;40:60–65. doi: 10.1016/j.ymeth.2006.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Carrasco C., Castellanos M., de Pablo P.J., Mateu M.G. Manipulation of the mechanical properties of a virus by protein engineering. Proc Natl Acad Sci USA. 2008;105:4150–4155. doi: 10.1073/pnas.0708017105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Fischlechner M., Donath E. Viruses as building blocks for materials and devices. Angew Chem Int Ed Engl. 2007;46:3184–3193. doi: 10.1002/anie.200603445. [DOI] [PubMed] [Google Scholar]
- 201.Lee L.A., Wang Q. Adaptations of nanoscale viruses and other protein cages for medical applications. Nanomedicine. 2006;2:137–149. doi: 10.1016/j.nano.2006.07.009. [DOI] [PubMed] [Google Scholar]
- 202.Adjei A.A. Blocking oncogenic Ras signaling for cancer therapy. J Natl Cancer Inst. 2001;93:1062–1074. doi: 10.1093/jnci/93.14.1062. [DOI] [PubMed] [Google Scholar]
- 203.May T., Gleiter S., Lilie H. Assessment of cell type specific gene transfer of polyoma virus like particles presenting a tumor specific antibody Fv fragment. J Virol Methods. 2002;105:147–157. doi: 10.1016/s0166-0934(02)00099-x. [DOI] [PubMed] [Google Scholar]
- 204.Liew M.W., Rajendran A., Middelberg A.P. Microbial production of virus-like particle vaccine protein at gram-per-litre levels. J Biotechnol. 2010;150:224–231. doi: 10.1016/j.jbiotec.2010.08.010. [DOI] [PubMed] [Google Scholar]
- 205.Kost T.A., Condreay J.P. Recombinant baculoviruses as mammalian cell gene-delivery vectors. Trends Biotechnol. 2002;20:173–180. doi: 10.1016/s0167-7799(01)01911-4. [DOI] [PubMed] [Google Scholar]
- 206.Maranga L., Cruz P.E., Aunins J.G., Carrondo M.J. Production of core and virus-like particles with baculovirus infected insect cells. Adv Biochem Eng Biotechnol. 2002;74:183–206. doi: 10.1007/3-540-45736-4_9. [DOI] [PubMed] [Google Scholar]
- 207.Lee C.D., Yan Y.P., Liang S.M., Wang T.F. Production of FMDV virus-like particles by a SUMO fusion protein approach in Escherichia coli. J Biomed Sci. 2009;16:69. doi: 10.1186/1423-0127-16-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Santi L., Huang Z., Mason H. Virus-like particles production in green plants. Methods. 2006;40:66–76. doi: 10.1016/j.ymeth.2006.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Santi L., Batchelor L., Huang Z., Hjelm B., Kilbourne J., Arntzen C.J. An efficient plant viral expression system generating orally immunogenic Norwalk virus-like particles. Vaccine. 2008;26:1846–1854. doi: 10.1016/j.vaccine.2008.01.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Ramqvist T., Dalianis T. Immunotherapeutic polyoma and human papilloma virus-like particles. Immunotherapy. 2009;1:303–312. doi: 10.2217/1750743X.1.2.303. [DOI] [PubMed] [Google Scholar]
- 211.McAleer W.J., Buynak E.B., Maigetter R.Z., Wampler D.E., Miller W.J., Hilleman M.R. Human hepatitis B vaccine from recombinant yeast. Nature. 1984;307:178–180. doi: 10.1038/307178a0. [DOI] [PubMed] [Google Scholar]
- 212.Pumpens P., Razanskas R., Pushko P., Renhof R., Gusars I., Skrastina D. Evaluation of HBs, HBc, and frCP virus-like particles for expression of human papillomavirus 16 E7 oncoprotein epitopes. Intervirology. 2002;45:24–32. doi: 10.1159/000050084. [DOI] [PubMed] [Google Scholar]
- 213.Hammonds J., Chen X., Zhang X., Lee F., Spearman P. Advances in methods for the production, purification, and characterization of HIV-1 Gag-Env pseudovirion vaccines. Vaccine. 2007;25:8036–8048. doi: 10.1016/j.vaccine.2007.09.016. [DOI] [PubMed] [Google Scholar]
- 214.Peek L.J., Middaugh C.R., Berkland C. Nanotechnology in vaccine delivery. Adv Drug Deliv Rev. 2008;60:915–928. doi: 10.1016/j.addr.2007.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Sun Y., Carrion R., Jr., Ye L., Wen Z., Ro Y.T., Brasky K. Protection against lethal challenge by Ebola virus-like particles produced in insect cells. Virology. 2009;383:12–21. doi: 10.1016/j.virol.2008.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Warfield K.L., Bosio C.M., Welcher B.C., Deal E.M., Mohamadzadeh M., Schmaljohn A. Ebola virus-like particles protect from lethal Ebola virus infection. Proc Natl Acad Sci USA. 2003;100:15889–15894. doi: 10.1073/pnas.2237038100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Freyschmidt E.J., Alonso A., Hartmann G., Gissmann L. Activation of dendritic cells and induction of T cell responses by HPV 16 L1/E7 chimeric virus-like particles are enhanced by CpG ODN or sorbitol. Antivir Ther. 2004;9:479–489. [PubMed] [Google Scholar]
- 218.Kaufmann A.M., Nieland J.D., Jochmus I., Baur S., Friese K., Gabelsberger J. Vaccination trial with HPV16 L1E7 chimeric virus-like particles in women suffering from high grade cervical intraepithelial neoplasia (CIN 2/3) Int J Cancer. 2007;121:2794–2800. doi: 10.1002/ijc.23022. [DOI] [PubMed] [Google Scholar]
- 219.Kaufmann A.M., Nieland J., Schinz M., Nonn M., Gabelsberger J., Meissner H. HPV16 L1E7 chimeric virus-like particles induce specific HLA-restricted T cells in humans after in vitro vaccination. Int J Cancer. 2001;92:285–293. doi: 10.1002/1097-0215(200102)9999:9999<::aid-ijc1181>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 220.Cheong W.S., Reiseger J., Turner S.J., Boyd R., Netter H.J. Chimeric virus-like particles for the delivery of an inserted conserved influenza A-specific CTL epitope. Antiviral Res. 2009;81:113–122. doi: 10.1016/j.antiviral.2008.10.003. [DOI] [PubMed] [Google Scholar]
- 221.Yin Y., Zhang J., Dong D., Liu S., Guo Q., Song X. Chimeric hepatitis B virus core particles carrying an epitope of anthrax protective antigen induce protective immunity against Bacillus anthracis. Vaccine. 2008;26:5814–5821. doi: 10.1016/j.vaccine.2008.08.031. [DOI] [PubMed] [Google Scholar]
- 222.Amexis G., Young N.S. Parvovirus B19 empty capsids as antigen carriers for presentation of antigenic determinants of dengue 2 virus. J Infect Dis. 2006;194:790–794. doi: 10.1086/506361. [DOI] [PubMed] [Google Scholar]
- 223.Ogasawara Y., Amexis G., Yamaguchi H., Kajigaya S., Leppla S.H., Young N.S. Recombinant viral-like particles of parvovirus B19 as antigen carriers of anthrax protective antigen. In Vivo. 2006;20:319–324. [PubMed] [Google Scholar]
- 224.Chackerian B., Lowy D.R., Schiller J.T. Conjugation of a self-antigen to papillomavirus-like particles allows for efficient induction of protective autoantibodies. J Clin Invest. 2001;108:415–423. doi: 10.1172/JCI11849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Cornuz J., Zwahlen S., Jungi W.F., Osterwalder J., Klingler K., Van M.G. A vaccine against nicotine for smoking cessation: a randomized controlled trial. PLoS One. 2008;3:e2547. doi: 10.1371/journal.pone.0002547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Chen L.S., Wang M., Ou W.C., Fung C.Y., Chen P.L., Chang C.F. Efficient gene transfer using the human JC virus-like particle that inhibits human colon adenocarcinoma growth in a nude mouse model. Gene Ther. 2010;17:1033–1041. doi: 10.1038/gt.2010.50. [DOI] [PubMed] [Google Scholar]
- 227.Ramsey J.D., Vu H.N., Pack D.W. A top-down approach for construction of hybrid polymer-virus gene delivery vectors. J Control Release. 2010;144:39–45. doi: 10.1016/j.jconrel.2010.01.031. [DOI] [PubMed] [Google Scholar]
- 228.Osterman J.V., Waddell A., Aposhian H.V. DNA and gene therapy: uncoating of polyoma pseudovirus in mouse embryo cells. Proc Natl Acad Sci USA. 1970;67:37–40. doi: 10.1073/pnas.67.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Yamada T., Iwasaki Y., Tada H., Iwabuki H., Chuah M.K., VandenDriessche T. Nanoparticles for the delivery of genes and drugs to human hepatocytes. Nat Biotechnol. 2003;21:885–890. doi: 10.1038/nbt843. [DOI] [PubMed] [Google Scholar]
- 230.Takamura S., Niikura M., Li T.C., Takeda N., Kusagawa S., Takebe Y. DNA vaccine-encapsulated virus-like particles derived from an orally transmissible virus stimulate mucosal and systemic immune responses by oral administration. Gene Ther. 2004;11:628–635. doi: 10.1038/sj.gt.3302193. [DOI] [PubMed] [Google Scholar]
- 231.Kimchi-Sarfaty C., Brittain S., Garfield S., Caplen N.J., Tang Q., Gottesman M.M. Efficient delivery of RNA interference effectors via in vitro-packaged SV40 pseudovirions. Hum Gene Ther. 2005;16:1110–1115. doi: 10.1089/hum.2005.16.1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Asokan A., Conway J.C., Phillips J.L., Li C., Hegge J., Sinnott R. Reengineering a receptor footprint of adeno-associated virus enables selective and systemic gene transfer to muscle. Nat Biotechnol. 2010;28:79–82. doi: 10.1038/nbt.1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Windram O.P., Weber B., Jaffer M.A., Rybicki E.P., Shepherd D.N., Varsani A. An investigation into the use of human papillomavirus type 16 virus-like particles as a delivery vector system for foreign proteins: N- and C-terminal fusion of GFP to the L1 and L2 capsid proteins. Arch Virol. 2008;153:585–589. doi: 10.1007/s00705-007-0025-2. [DOI] [PubMed] [Google Scholar]
- 234.Gleiter S., Lilie H. Coupling of antibodies via protein Z on modified polyoma virus-like particles. Protein Sci. 2001;10:434–444. doi: 10.1110/ps.31101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Stubenrauch K., Gleiter S., Brinkmann U., Rudolph R., Lilie H. Conjugation of an antibody Fv fragment to a virus coat protein: cell-specific targeting of recombinant polyoma-virus-like particles. Biochem J. 2001;356:867–873. doi: 10.1042/0264-6021:3560867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Sen G.S., Kuzelka J., Singh P., Lewis W.G., Manchester M., Finn M.G. Accelerated bioorthogonal conjugation: a practical method for the ligation of diverse functional molecules to a polyvalent virus scaffold. Bioconj Chem. 2005;16:1572–1579. doi: 10.1021/bc050147l. [DOI] [PubMed] [Google Scholar]
- 237.Schaffer D.V., Koerber J.T., Lim K.I. Molecular engineering of viral gene delivery vehicles. Annu Rev Biomed Eng. 2008;10:169–194. doi: 10.1146/annurev.bioeng.10.061807.160514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Kobinger G.P., Weiner D.J., Yu Q.C., Wilson J.M. Filovirus-pseudotyped lentiviral vector can efficiently and stably transduce airway epithelia in vivo. Nat Biotechnol. 2001;19:225–230. doi: 10.1038/85664. [DOI] [PubMed] [Google Scholar]
- 239.Qu Q., Sawa H., Suzuki T., Semba S., Henmi C., Okada Y. Nuclear entry mechanism of the human polyomavirus JC virus-like particle: role of importins and the nuclear pore complex. J Biol Chem. 2004;279:27735–27742. doi: 10.1074/jbc.M310827200. [DOI] [PubMed] [Google Scholar]
- 240.Raja K.S., Wang Q., Gonzalez M.J., Manchester M., Johnson J.E., Finn M.G. Hybrid virus-polymer materials. 1. Synthesis and properties of PEG-decorated cowpea mosaic virus. Biomacromolecules. 2003;4:472–476. doi: 10.1021/bm025740+. [DOI] [PubMed] [Google Scholar]
- 241.Marianayagam N.J., Sunde M., Matthews J.M. The power of two: protein dimerization in biology. Trends Biochem Sci. 2004;29:618–625. doi: 10.1016/j.tibs.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 242.Tu R.S., Tirrell M. Bottom-up design of biomimetic assemblies. Adv Drug Deliv Rev. 2004;56:1537–1563. doi: 10.1016/j.addr.2003.10.047. [DOI] [PubMed] [Google Scholar]
- 243.Leckband D. Measuring the forces that control protein interactions. Annu Rev Biophys Biomol Struct. 2000;29:1–26. doi: 10.1146/annurev.biophys.29.1.1. [DOI] [PubMed] [Google Scholar]
- 244.Meyer E.E., Rosenberg K.J., Israelachvili J. Recent progress in understanding hydrophobic interactions. Proc Natl Acad Sci USA. 2006;103:15739–15746. doi: 10.1073/pnas.0606422103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Schneider H.J. Binding mechanisms in supramolecular complexes. Angew Chem Int Ed Engl. 2009;48:3924–3977. doi: 10.1002/anie.200802947. [DOI] [PubMed] [Google Scholar]
- 246.Goodsell D.S., Olson A.J. Structural symmetry and protein function. Annu Rev Biophys Biomol Struct. 2000;29:105–153. doi: 10.1146/annurev.biophys.29.1.105. [DOI] [PubMed] [Google Scholar]
- 247.Eriksson J., Fenyo D. Modeling experimental design for proteomics. Methods Mol Biol. 2010;673:223–230. doi: 10.1007/978-1-60761-842-3_14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Kopecek J. Smart and genetically engineered biomaterials and drug delivery systems. Eur J Pharm Sci. 2003;20:1–16. doi: 10.1016/s0928-0987(03)00164-7. [DOI] [PubMed] [Google Scholar]
- 249.Zhao X., Zhang S. Molecular designer self-assembling peptides. Chem Soc Rev. 2006;35:1105–1110. doi: 10.1039/b511336a. [DOI] [PubMed] [Google Scholar]
- 250.Domingo-Espin J., Vazquez E., Ganz J., Conchillo O., Garcia-Fruitos E., Cedano J. Nanoparticulate architecture of protein-based artificial viruses is supported by protein-DNA interactions. Nanomedicine. 2011;6:1047–1061. doi: 10.2217/nnm.11.28. [DOI] [PubMed] [Google Scholar]
- 251.Tsumoto K., Luckel F., Yoshikawa K. Giant DNA molecules exhibit on/off switching of transcriptional activity through conformational transition. Biophys Chem. 2003;106:23–29. doi: 10.1016/s0301-4622(03)00138-8. [DOI] [PubMed] [Google Scholar]
- 252.Flenniken M., Willits D., Brumfield S., Young M.J., Douglas T. The small heat shock protein cage from Methanococcus jannaschii is a versatile nanoscale platform for genetic and chemical modification. Nano Lett. 2003;3:1573–1576. [Google Scholar]
- 253.Shively J.M., Decker G.L., Greenawalt J.W. Comparative ultrastructure of the thiobacilli. J Bacteriol. 1970;101:618–627. doi: 10.1128/jb.101.2.618-627.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Shively J.M., Ball F., Brown D.H., Saunders R.E. Functional organelles in prokaryotes: polyhedral inclusions (carboxysomes) of Thiobacillus neapolitanus. Science. 1973;182:584–586. doi: 10.1126/science.182.4112.584. [DOI] [PubMed] [Google Scholar]
- 255.Price G.D., Coleman J.R., Badger M.R. Association of carbonic anhydrase activity with carboxysomes isolated from the Cyanobacterium Synechococcus PCC7942. Plant Physiol. 1992;100:784–793. doi: 10.1104/pp.100.2.784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.So A.K., Espie G.S., Williams E.B., Shively J.M., Heinhorst S., Cannon G.C. A novel evolutionary lineage of carbonic anhydrase (epsilon class) is a component of the carboxysome shell. J Bacteriol. 2004;186:623–630. doi: 10.1128/JB.186.3.623-630.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Yu J.W., Price G.D., Song L., Badger M.R. Isolation of a putative carboxysomal carbonic anhydrase gene from the Cyanobacterium Synechococcus PCC7942. Plant Physiol. 1992;100:794–800. doi: 10.1104/pp.100.2.794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Chen P., Andersson D.I., Roth J.R. The control region of the pdu/cob regulon in Salmonella typhimurium. J Bacteriol. 1994;176:5474–5482. doi: 10.1128/jb.176.17.5474-5482.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Kofoid E., Rappleye C., Stojiljkovic I., Roth J. The 17-gene ethanolamine (eut) operon of Salmonella typhimurium encodes five homologues of carboxysome shell proteins. J Bacteriol. 1999;181:5317–5329. doi: 10.1128/jb.181.17.5317-5329.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Bobik T.A., Havemann G.D., Busch R.J., Williams D.S., Aldrich H.C. The propanediol utilization (pdu) operon of Salmonella enterica serovar Typhimurium LT2 includes genes necessary for formation of polyhedral organelles involved in coenzyme B(12)-dependent 1, 2-propanediol degradation. J Bacteriol. 1999;181:5967–5975. doi: 10.1128/jb.181.19.5967-5975.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Havemann G.D., Sampson E.M., Bobik T.A. PduA is a shell protein of polyhedral organelles involved in coenzyme B(12)-dependent degradation of 1,2-propanediol in Salmonella enterica serovar typhimurium LT2. J Bacteriol. 2002;184:1253–1261. doi: 10.1128/JB.184.5.1253-1261.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Havemann G.D., Bobik T.A. Protein content of polyhedral organelles involved in coenzyme B12-dependent degradation of 1,2-propanediol in Salmonella enterica serovar Typhimurium LT2. J Bacteriol. 2003;185:5086–5095. doi: 10.1128/JB.185.17.5086-5095.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Leal N.A., Havemann G.D., Bobik T.A. PduP is a coenzyme-a-acylating propionaldehyde dehydrogenase associated with the polyhedral bodies involved in B12-dependent 1,2-propanediol degradation by Salmonella enterica serovar Typhimurium LT2. Arch Microbiol. 2003;180:353–361. doi: 10.1007/s00203-003-0601-0. [DOI] [PubMed] [Google Scholar]
- 264.Rondon M.R., Horswill A.R., Escalante-Semerena J.C. DNA polymerase I function is required for the utilization of ethanolamine, 1,2-propanediol, and propionate by Salmonella typhimurium LT2. J Bacteriol. 1995;177:7119–7124. doi: 10.1128/jb.177.24.7119-7124.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Rondon M.R., Kazmierczak R., Escalante-Semerena J.C. Glutathione is required for maximal transcription of the cobalamin biosynthetic and 1,2-propanediol utilization (cob/pdu) regulon and for the catabolism of ethanolamine, 1,2-propanediol, and propionate in Salmonella typhimurium LT2. J Bacteriol. 1995;177:5434–5439. doi: 10.1128/jb.177.19.5434-5439.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Sampson E.M., Bobik T.A. Microcompartments for B12-dependent 1,2-propanediol degradation provide protection from DNA and cellular damage by a reactive metabolic intermediate. J Bacteriol. 2008;190:2966–2971. doi: 10.1128/JB.01925-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Penrod J.T., Roth J.R. Conserving a volatile metabolite: a role for carboxysome-like organelles in Salmonella enterica. J Bacteriol. 2006;188:2865–2874. doi: 10.1128/JB.188.8.2865-2874.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Kerfeld C.A., Sawaya M.R., Tanaka S., Nguyen C.V., Phillips M., Beeby M. Protein structures forming the shell of primitive bacterial organelles. Science. 2005;309:936–938. doi: 10.1126/science.1113397. [DOI] [PubMed] [Google Scholar]
- 269.Yeates T.O., Crowley C.S., Tanaka S. Bacterial microcompartment organelles: protein shell structure and evolution. Annu Rev Biophys. 2010;39:185–205. doi: 10.1146/annurev.biophys.093008.131418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Tanaka S., Kerfeld C.A., Sawaya M.R., Cai F., Heinhorst S., Cannon G.C. Atomic-level models of the bacterial carboxysome shell. Science. 2008;319:1083–1086. doi: 10.1126/science.1151458. [DOI] [PubMed] [Google Scholar]
- 271.Tanaka S., Sawaya M.R., Yeates T.O. Structure and mechanisms of a protein-based organelle in Escherichia coli. Science. 2010;327:81–84. doi: 10.1126/science.1179513. [DOI] [PubMed] [Google Scholar]
- 272.Cot S.S., So A.K., Espie G.S. A multiprotein bicarbonate dehydration complex essential to carboxysome function in cyanobacteria. J Bacteriol. 2008;190:936–945. doi: 10.1128/JB.01283-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Long B.M., Badger M.R., Whitney S.M., Price G.D. Analysis of carboxysomes from Synechococcus PCC7942 reveals multiple Rubisco complexes with carboxysomal proteins CcmM and CcaA. J Biol Chem. 2007;282:29323–29335. doi: 10.1074/jbc.M703896200. [DOI] [PubMed] [Google Scholar]
- 274.Price G.D., Howitt S.M., Harrison K., Badger M.R. Analysis of a genomic DNA region from the cyanobacterium Synechococcus sp. strain PCC7942 involved in carboxysome assembly and function. J Bacteriol. 1993;175:2871–2879. doi: 10.1128/jb.175.10.2871-2879.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Parsons J.B., Frank S., Bhella D., Liang M., Prentice M.B., Mulvihill D.P. Synthesis of empty bacterial microcompartments, directed organelle protein incorporation, and evidence of filament-associated organelle movement. Mol Cell. 2010;38:305–315. doi: 10.1016/j.molcel.2010.04.008. [DOI] [PubMed] [Google Scholar]
- 276.Menon B.B., Dou Z., Heinhorst S., Shively J.M., Cannon G.C. Halothiobacillus neapolitanus carboxysomes sequester heterologous and chimeric RubisCO species. PLoS One. 2008;3:e3570. doi: 10.1371/journal.pone.0003570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Fan C., Cheng S., Liu Y., Escobar C.M., Crowley C.S., Jefferson R.E. Short N-terminal sequences package proteins into bacterial microcompartments. Proc Natl Acad Sci USA. 2010;107:7509–7514. doi: 10.1073/pnas.0913199107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Sutter M., Boehringer D., Gutmann S., Gunther S., Prangishvili D., Loessner M.J. Structural basis of enzyme encapsulation into a bacterial nanocompartment. Nat Struct Mol Biol. 2008;15:939–947. doi: 10.1038/nsmb.1473. [DOI] [PubMed] [Google Scholar]
- 279.Seebeck F.P., Woycechowsky K.J., Zhuang W., Rabe J.P., Hilvert D. A simple tagging system for protein encapsulation. J Am Chem Soc. 2006;128:4516–4517. doi: 10.1021/ja058363s. [DOI] [PubMed] [Google Scholar]
- 280.Zhang X., Konarev P.V., Petoukhov M.V., Svergun D.I., Xing L., Cheng R.H. Multiple assembly states of lumazine synthase: a model relating catalytic function and molecular assembly. J Mol Biol. 2006;362:753–770. doi: 10.1016/j.jmb.2006.07.037. [DOI] [PubMed] [Google Scholar]
- 281.Dalmau M., Lim S., Chen H.C., Ruiz C., Wang S.W. Thermostability and molecular encapsulation within an engineered caged protein scaffold. Biotechnol Bioeng. 2008;101:654–664. doi: 10.1002/bit.21988. [DOI] [PubMed] [Google Scholar]
- 282.Dalmau M., Lim S., Wang S.W. pH-triggered disassembly in a caged protein complex. Biomacromolecules. 2009;10:3199–3206. doi: 10.1021/bm900674v. [DOI] [PubMed] [Google Scholar]
- 283.Grinberg O., Hayun M., Sredni B., Gedanken A. Characterization and activity of sonochemically-prepared BSA microspheres containing Taxol–an anticancer drug. Ultrason Sonochem. 2007;14:661–666. doi: 10.1016/j.ultsonch.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 284.Rivkin I., Cohen K., Koffler J., Melikhov D., Peer D., Margalit R. Paclitaxel-clusters coated with hyaluronan as selective tumor-targeted nanovectors. Biomaterials. 2010;31:7106–7114. doi: 10.1016/j.biomaterials.2010.05.067. [DOI] [PubMed] [Google Scholar]
- 285.Vazquez E., Roldan M., Diez-Gil C., Unzueta U., Domingo-Espin J., Cedano J. Protein nanodisk assembling and intracellular trafficking powered by an arginine-rich (R9) peptide. Nanomedicine. 2010;5:259–268. doi: 10.2217/nnm.09.98. [DOI] [PubMed] [Google Scholar]
- 286.Diez-Gil C., Krabbenborg S., Garcia-Fruitos E., Vazquez E., Rodriguez-Carmona E., Ratera I. The nanoscale properties of bacterial inclusion bodies and their effect on mammalian cell proliferation. Biomaterials. 2010;31:5805–5812. doi: 10.1016/j.biomaterials.2010.04.008. [DOI] [PubMed] [Google Scholar]
- 287.Garcia-Fruitos E., Rodriguez-Carmona E., Diez-Gil C., Ferraz R.M., Vazquez E., Corchero J.L. Surface cell growth engineering assisted by a novel bacterial nanomaterial. Adv Mater. 2009;21:4249. [Google Scholar]
- 288.Rodriguez-Carmona E., Villaverde A. Nanostructured bacterial materials for innovative medicines. Trends Microbiol. 2010;18:423–430. doi: 10.1016/j.tim.2010.06.007. [DOI] [PubMed] [Google Scholar]
- 289.Vaysse L., Harbottle R., Bigger B., Bergau A., Tolmachov O., Coutelle C. Development of a self-assembling nuclear targeting vector system based on the tetracycline repressor protein. J Biol Chem. 2004;279:5555–5564. doi: 10.1074/jbc.M311894200. [DOI] [PubMed] [Google Scholar]
- 290.Fukunaga-Kalabis M., Herlyn M. Unraveling mysteries of the multifunctional protein SPARC. J Invest Dermatol. 2007;127:2497–2498. doi: 10.1038/sj.jid.5701050. [DOI] [PubMed] [Google Scholar]
- 291.Akahata W., Yang Z.Y., Andersen H., Sun S., Holdaway H.A., Kong W.P. A virus-like particle vaccine for epidemic Chikungunya virus protects nonhuman primates against infection. Nat Med. 2010;16:334–338. doi: 10.1038/nm.2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Pushko P., Kort T., Nathan M., Pearce M.B., Smith G., Tumpey T.M. Recombinant H1N1 virus-like particle vaccine elicits protective immunity in ferrets against the 2009 pandemic H1N1 influenza virus. Vaccine. 2010;28:4771–4776. doi: 10.1016/j.vaccine.2010.04.093. [DOI] [PubMed] [Google Scholar]
- 293.Harmsen M.M., De Haard H.J. Properties, production, and applications of camelid single-domain antibody fragments. Appl Microbiol Biotechnol. 2007;77:13–22. doi: 10.1007/s00253-007-1142-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Cortez-Retamozo V., Backmann N., Senter P.D., Wernery U., De B.P., Muyldermans S. Efficient cancer therapy with a nanobody-based conjugate. Cancer Res. 2004;64:2853–2857. doi: 10.1158/0008-5472.can-03-3935. [DOI] [PubMed] [Google Scholar]
- 295.Bian J., Popovic Z.B., Benejam C., Kiedrowski M., Rodriguez L.L., Penn M.S. Effect of cell-based intercellular delivery of transcription factor GATA4 on ischemic cardiomyopathy. Circ Res. 2007;100:1626–1633. doi: 10.1161/01.RES.0000269778.75877.68. [DOI] [PubMed] [Google Scholar]
- 296.Fletcher S., Honeyman K., Fall A.M., Harding P.L., Johnsen R.D., Steinhaus J.P. Morpholino oligomer-mediated exon skipping averts the onset of dystrophic pathology in the mdx mouse. Mol Ther. 2007;15:1587–1592. doi: 10.1038/sj.mt.6300245. [DOI] [PubMed] [Google Scholar]
- 297.Dubikovskaya E.A., Thorne S.H., Pillow T.H., Contag C.H., Wender P.A. Overcoming multidrug resistance of small-molecule therapeutics through conjugation with releasable octaarginine transporters. Proc Natl Acad Sci USA. 2008;105:12128–12133. doi: 10.1073/pnas.0805374105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Bucci M., Gratton J.P., Rudic R.D., Acevedo L., Roviezzo F., Cirino G. In vivo delivery of the caveolin-1 scaffolding domain inhibits nitric oxide synthesis and reduces inflammation. Nat Med. 2000;6:1362–1367. doi: 10.1038/82176. [DOI] [PubMed] [Google Scholar]
- 299.Morris M.C., Gros E., drian-Herrada G., Choob M., Archdeacon J., Heitz F. A non-covalent peptide-based carrier for in vivo delivery of DNA mimics. Nucleic Acids Res. 2007;35:e49. doi: 10.1093/nar/gkm053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Crombez L., Morris M.C., Dufort S., drian-Herrada G., Nguyen Q., Mc M.G. Targeting cyclin B1 through peptide-based delivery of siRNA prevents tumour growth. Nucleic Acids Res. 2009;37:4559–4569. doi: 10.1093/nar/gkp451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Song E., Zhu P., Lee S.K., Chowdhury D., Kussman S., Dykxhoorn D.M. Antibody mediated in vivo delivery of small interfering RNAs via cell-surface receptors. Nat Biotechnol. 2005;23:709–717. doi: 10.1038/nbt1101. [DOI] [PubMed] [Google Scholar]
- 302.Tan M., Lan K.H., Yao J., Lu C.H., Sun M., Neal C.L. Selective inhibition of ErbB2-overexpressing breast cancer in vivo by a novel TAT-based ErbB2-targeting signal transducers and activators of transcription 3-blocking peptide. Cancer Res. 2006;66:3764–3772. doi: 10.1158/0008-5472.CAN-05-2747. [DOI] [PubMed] [Google Scholar]
- 303.Myrberg H., Zhang L., Mae M., Langel U. Design of a tumor-homing cell-penetrating peptide. Bioconj Chem. 2008;19:70–75. doi: 10.1021/bc0701139. [DOI] [PubMed] [Google Scholar]
- 304.Laakkonen P., Porkka K., Hoffman J.A., Ruoslahti E. A tumor-homing peptide with a targeting specificity related to lymphatic vessels. Nat Med. 2002;8:751–755. doi: 10.1038/nm720. [DOI] [PubMed] [Google Scholar]
- 305.Jain M., Chauhan S.C., Singh A.P., Venkatraman G., Colcher D., Batra S.K. Penetratin improves tumor retention of single-chain antibodies: a novel step toward optimization of radioimmunotherapy of solid tumors. Cancer Res. 2005;65:7840–7846. doi: 10.1158/0008-5472.CAN-05-0662. [DOI] [PubMed] [Google Scholar]
- 306.Vocero-Akbani A.M., Heyden N.V., Lissy N.A., Ratner L., Dowdy S.F. Killing HIV-infected cells by transduction with an HIV protease-activated caspase-3 protein. Nat Med. 1999;5:29–33. doi: 10.1038/4710. [DOI] [PubMed] [Google Scholar]
- 307.Bidwell G.L., III, Fokt I., Priebe W., Raucher D. Development of elastin-like polypeptide for thermally targeted delivery of doxorubicin. Biochem Pharmacol. 2007;73:620–631. doi: 10.1016/j.bcp.2006.10.028. [DOI] [PubMed] [Google Scholar]
- 308.Snyder E.L., Meade B.R., Saenz C.C., Dowdy S.F. Treatment of terminal peritoneal carcinomatosis by a transducible p53-activating peptide. PLoS Biol. 2004;2:E36. doi: 10.1371/journal.pbio.0020036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Harada H., Hiraoka M., Kizaka-Kondoh S. Antitumor effect of TAT-oxygen-dependent degradation-caspase-3 fusion protein specifically stabilized and activated in hypoxic tumor cells. Cancer Res. 2002;62:2013–2018. [PubMed] [Google Scholar]
- 310.Datta K., Sundberg C., Karumanchi S.A., Mukhopadhyay D. The 104–123 amino acid sequence of the beta-domain of von Hippel-Lindau gene product is sufficient to inhibit renal tumor growth and invasion. Cancer Res. 2001;61:1768–1775. [PubMed] [Google Scholar]
- 311.Shibagaki N., Udey M.C. Dendritic cells transduced with protein antigens induce cytotoxic lymphocytes and elicit antitumor immunity. J Immunol. 2002;168:2393–2401. doi: 10.4049/jimmunol.168.5.2393. [DOI] [PubMed] [Google Scholar]
- 312.Bright R., Raval A.P., Dembner J.M., Perez-Pinzon M.A., Steinberg G.K., Yenari M.A. Protein kinase C delta mediates cerebral reperfusion injury in vivo. J Neurosci. 2004;24:6880–6888. doi: 10.1523/JNEUROSCI.4474-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Cao G., Pei W., Ge H., Liang Q., Luo Y., Sharp F.R. In vivo delivery of a Bcl-xL fusion protein containing the TAT protein transduction domain protects against ischemic brain injury and neuronal apoptosis. J Neurosci. 2002;22:5423–5431. doi: 10.1523/JNEUROSCI.22-13-05423.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Kumar P., Ban H.S., Kim S.S., Wu H., Pearson T., Greiner D.L. T cell-specific siRNA delivery suppresses HIV-1 infection in humanized mice. Cell. 2008;134:577–586. doi: 10.1016/j.cell.2008.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Kim W.J., Christensen L.V., Jo S., Yockman J.W., Jeong J.H., Kim Y.H. Cholesteryl oligoarginine delivering vascular endothelial growth factor siRNA effectively inhibits tumor growth in colon adenocarcinoma. Mol Ther. 2006;14:343–350. doi: 10.1016/j.ymthe.2006.03.022. [DOI] [PubMed] [Google Scholar]
- 316.Mamot C., Drummond D.C., Noble C.O., Kallab V., Guo Z., Hong K. Epidermal growth factor receptor-targeted immunoliposomes significantly enhance the efficacy of multiple anticancer drugs in vivo. Cancer Res. 2005;65:11631–11638. doi: 10.1158/0008-5472.CAN-05-1093. [DOI] [PubMed] [Google Scholar]
- 317.Arap W., Pasqualini R., Ruoslahti E. Cancer treatment by targeted drug delivery to tumor vasculature in a mouse model. Science. 1998;279:377–380. doi: 10.1126/science.279.5349.377. [DOI] [PubMed] [Google Scholar]
- 318.Liu S., Wang H., Currie B.M., Molinolo A., Leung H.J., Moayeri M. Matrix metalloproteinase-activated anthrax lethal toxin demonstrates high potency in targeting tumor vasculature. J Biol Chem. 2008;283:529–540. doi: 10.1074/jbc.M707419200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Frankel A.E., Powell B.L., Duesbery N.S., Vande Woude G.F., Leppla S.H. Anthrax fusion protein therapy of cancer. Curr Protein Pept Sci. 2002;3:399–407. doi: 10.2174/1389203023380567. [DOI] [PubMed] [Google Scholar]
- 320.Hu P., Yan J., Sharifi J., Bai T., Khawli L.A., Epstein A.L. Comparison of three different targeted tissue factor fusion proteins for inducing tumor vessel thrombosis. Cancer Res. 2003;63:5046–5053. [PubMed] [Google Scholar]
- 321.Lindgren M., Rosenthal-Aizman K., Saar K., Eiriksdottir E., Jiang Y., Sassian M. Overcoming methotrexate resistance in breast cancer tumour cells by the use of a new cell-penetrating peptide. Biochem Pharmacol. 2006;71:416–425. doi: 10.1016/j.bcp.2005.10.048. [DOI] [PubMed] [Google Scholar]
- 322.Anderson D.C., Nichols E., Manger R., Woodle D., Barry M., Fritzberg A.R. Tumor cell retention of antibody Fab fragments is enhanced by an attached HIV TAT protein-derived peptide. Biochem Biophys Res Commun. 1993;194:876–884. doi: 10.1006/bbrc.1993.1903. [DOI] [PubMed] [Google Scholar]
- 323.Dijkgraaf I., Kruijtzer J.A., Liu S., Soede A.C., Oyen W.J., Corstens F.H. Improved targeting of the alpha(v)beta (3) integrin by multimerisation of RGD peptides. Eur J Nucl Med Mol Imaging. 2007;34:267–273. doi: 10.1007/s00259-006-0180-9. [DOI] [PubMed] [Google Scholar]
- 324.Kawamura K.S., Sung M., Bolewska-Pedyczak E., Gariepy J. Probing the impact of valency on the routing of arginine-rich peptides into eukaryotic cells. Biochemistry. 2006;45:1116–1127. doi: 10.1021/bi051338e. [DOI] [PubMed] [Google Scholar]
- 325.Vives E., Schmidt J., Pelegrin A. Cell-penetrating and cell-targeting peptides in drug delivery. Biochim Biophys Acta. 2008;1786:126–138. doi: 10.1016/j.bbcan.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 326.Jiang T., Olson E.S., Nguyen Q.T., Roy M., Jennings P.A., Tsien R.Y. Tumor imaging by means of proteolytic activation of cell-penetrating peptides. Proc Natl Acad Sci USA. 2004;101:17867–17872. doi: 10.1073/pnas.0408191101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Sethuraman V.A., Bae Y.H. TAT peptide-based micelle system for potential active targeting of anti-cancer agents to acidic solid tumors. J Control Release. 2007;118:216–224. doi: 10.1016/j.jconrel.2006.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Lebleu B., Moulton H.M., Abes R., Ivanova G.D., Abes S., Stein D.A. Cell penetrating peptide conjugates of steric block oligonucleotides. Adv Drug Deliv Rev. 2008;60:517–529. doi: 10.1016/j.addr.2007.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Kamei N., Morishita M., Eda Y., Ida N., Nishio R., Takayama K. Usefulness of cell-penetrating peptides to improve intestinal insulin absorption. J Control Release. 2008;132:21–25. doi: 10.1016/j.jconrel.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 330.Pardridge W.M. Biopharmaceutical drug targeting to the brain. J Drug Target. 2010;18:157–167. doi: 10.3109/10611860903548354. [DOI] [PubMed] [Google Scholar]
- 331.Perales J.C., Ferkol T., Beegen H., Ratnoff O.D., Hanson R.W. Gene transfer in vivo: sustained expression and regulation of genes introduced into the liver by receptor-targeted uptake. Proc Natl Acad Sci USA. 1994;91:4086–4090. doi: 10.1073/pnas.91.9.4086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Sato Y., Yamauchi N., Takahashi M., Sasaki K., Fukaura J., Neda H. In vivo gene delivery to tumor cells by transferrin-streptavidin-DNA conjugate. FASEB J. 2000;14:2108–2118. doi: 10.1096/fj.99-1052com. [DOI] [PubMed] [Google Scholar]