

AIRWAY HYPERRESPONSIVENESS IN ASTHMA
Asthma is described as the presence of wheezing, cough, dyspnea, and chest tightness and by variable airway narrowing and airway hyperresponsiveness to inhaled bronchoconstrictor stimuli. Reversible airway narrowing is the sine qua non of asthma. Airway hyperresponsiveness is an increased sensitivity of the airways to constrictor agonists, as indicated by a smaller concentration of the agonist needed to initiate the bronchoconstrictor response; a greater reactivity of the airways, as indicated by an increased slope of the concentration-response curve; and a greater maximal response to the agonist (Woolcock et al., 1984) ( Fig. 81.1 ). Airway hyperresponsiveness is present in virtually all asthmatics with current symptoms (Crockcroft et al., 1977); however, it can be found in patients with other airway diseases (Ramsdale et al., 1985).
Fig. 81.1.
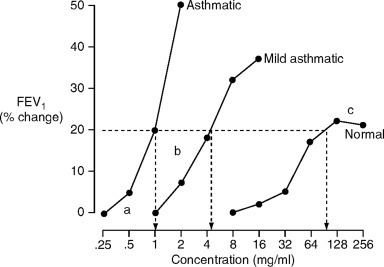
Dose-response curves to inhaled constrictor agonists in a normal subject, and asthmatic subjects with mild and severe airway hyperresponsiveness. The responses are measured as the fall in FEV1 from baseline values, and in these examples, the responses are expressed as the provocative concentrations of the agonist causing a 20% fall in FEV1. Asthmatics differ from normal subjects, with a lower threshold dose (a), a steeper slope of the dose-response (b), and both the mild asthmatic and normal subjects demonstrate a plateau response (c), which is lost in the more severe asthmatic subject.
Airway hyperresponsiveness in asthma has been the focus of extensive research over the past 30 years. This research has examined a variety of methods of measuring airway responsiveness, the clinical significance and the effects of antiasthma medications on these measurements, and the pathophysiology and pathogenesis of airway hyperresponsiveness in asthmatic patients. As a result of this research, the methods of measuring airway responsiveness have been standardized and become widely accepted. Also, there is some agreement about the clinical significance and effects of treatment on airway hyperresponsiveness.
Airway hyperresponsiveness is measured by subjects inhaling increasing concentrations of a stimulus ranging from bronchoconstrictor mediators, such as histamine (Crockcroft et al., 1977) and methacholine (Juniper et al., 1992), to physical stimuli, such as cold air (O'Byrne et al., 1982), or hypertonic or hypotonic solutions (Anderson et al., 1983). Airway hyperresponsiveness is nonspecific, in that asthmatics who are hyperresponsive to one bronchoconstrictor stimulus will be hyperresponsive to others. The bronchoconstrictor mediators most often used in clinical studies are histamine or the cholinergic agonist, methacholine. The degree of airway responsiveness correlates with the severity of asthma (Crockcroft et al., 1977) and the treatment required to control symptoms (Juniper et al., 1982).
TRANSIENT AIRWAY HYPERRESPONSIVENESS AND INFLAMMATION
The identification of stimuli that can cause asthma has proven to be important in studies of the pathogenesis of airway hyperresponsiveness in asthma. These stimuli include environmental allergens (O'Byrne, 1988), occupational sensitizing agents (Chan-Yeung and Malo, 1995), certain viruses (Lemanske et al., 1989), and the atmospheric pollutant ozone (Holtzman et al., 1979). Each of these stimuli is known to induce airway hyperresponsiveness in experimental models. Studies of the mechanisms of airway hyperresponsiveness in animal models have identified temporal associations between the presence of inflammatory cells in the airways and airway hyperresponsiveness (Holtzman et al., 1983a; Abraham et al., 1988). These studies have led to the hypothesis that activated inflammatory cells, and mediators released from these cells, are responsible for the development of airway hyperresponsiveness after inhalation of various stimuli in human subjects. Subsequently, this hypothesis was extended to suggest that persisting airway hyperresponsiveness in asthmatics, not obviously related to exposure to a specific stimulus such as allergen, is a result of structural changes in the airways caused by long-standing airway inflammation.
ALLERGEN-INDUCED AIRWAY RESPONSES
Environmental allergens are among the most important stimuli known to cause airway inflammation and airway hyperresponsiveness. The pathogenesis of allergic asthma has been greatly clarified by studying the effects of inhaled allergens in mild, allergic asthmatic subjects. Inhalation of allergen by a sensitized subject in the laboratory results in an early asthmatic response; a late asthmatic response occurs in 50% to 60% of adults and 70% to 80% of children who develop early responses. A further consequence of inhaling allergen is the development of airway hyperresponsiveness, the magnitude and duration of which appear to be related to the occurrence of late asthmatic responses (Bhalla et al., 1992).
In human subjects, allergen-induced airway responses are associated with airway inflammation, and with increases in activated airway eosinophils (Gauvreau et al., 1999) and mast cells and basophils (Gauvreau et al., 2000). Some of the mediators that cause allergen-induced bronchoconstrictor responses also have been identified. The cysteinyl leukotrienes (Taylor et al., 1991), thromboxane (Manning et al., 1991), and histamine (Roquet et al., 1997) are involved in early responses, whereas the leukotrienes (Hamilton et al., 1997) and histamine (Roquet et al., 1997) are mainly responsible for bronchoconstriction during the late response.
The identification of allergen as an important cause of asthma has resulted in the development of a variety of animal models of allergen-induced early and late responses, airway hyperresponsiveness, and airway inflammation. The species used include dogs, sheep, guinea pigs, rabbits, rats, mice, and primates.
Dogs have been used since the early 1980s to examine the mechanisms of allergen-induced airway inflammation. They are often naturally sensitized to the parasite, Ascaris suum (Chung et al., 1984), possibly by cross-reactivity to epitopes present on the parasite Toxacara canis. In addition, dogs have been sensitized at birth to ragweed (Becker et al., 1989). Sensitized dogs develop acute bronchoconstriction within 15 minutes after inhaling allergen, and have airway inflammation and airway hyperresponsiveness 12–24 hours after allergen (Chung et al., 1984). The cellular infiltrate consists of eosinophils (Woolley et al., 1995) and/or neutrophils (Chung et al., 1985). Interestingly, despite the development of allergen-induced airway inflammation, dogs do not develop allergen-induced late responses unless the endogeneous production of glucocorticosteroids is blocked by metyrapone (Sasaki et al., 1987).
The origin of the inflammatory cells recruited during allergen-induced airway inflammation and the factors that regulate this have been evaluated in dogs. A. suum-induced neutrophilic airway inflammation is associated with an increase in the progenitors for neutrophils (granuloyte-macrophage progenitors) in the bone marrow (Woolley et al., 1994), in response to the release of a hematopoeitic factor into the bloodstream (Inman et al., 1996). The presence of the serum factor, rather than the bone marrow's response to the factor, is what determines the subsequent increase in progenitors. Newly formed cells from the bone marrow are subsequently recruited into the airways after allergen inhalation (Wood et al., 1998). Therefore, the development of allergen inflammation is presumably determined by both the airway's ability to produce a factor to stimulate the bone marrow and the marrow's increased production of progenitors.
Advantages of a canine model of airway hyperresponsiveness include the size of the animal, which allows physiologic measurements to be made easily and reproducibly, and repeated access to the airways and other sites, such as the bone marrow, to evaluate inflammatory events following allergen inhalation. A major limitation of a canine model is the lack of specific immunologic reagents, although occasionally cross-reactivity with monoclonal antibodies can occur, which allows for studies in dogs with antibodies developed against human epitopes (Li et al., 1992).
Sheep are also naturally sensitized to A. suum and develop early and late responses (Delehunt et al., 1984) and airway hyperresponsiveness (Lanes et al., 1986) after inhalation of that allergen. The time course of these physiologic changes is similar to that of responses to allergen inhalation in humans. The changes are also associated with increases in airway eosinophils (Abraham et al., 1988). This model has the same advantages and suffers from many of the same limitations of dog models. It has, however, often been used to evaluate the antiallergic activity of drugs thought to be useful in asthma (Abraham et al., 1986; Soler et al., 1990; Tomioka et al., 1989).
Primates are the third large-animal species used to evaluate the pathophysiology of allergen-induced airway responses. However, because of the high cost of acquisition and maintenance, their use has largely become restricted to the pharmaceutical industry. Monkeys also are sensitized to A. suum antigen and develop early and late responses (Gundel et al., 1992), airway inflammation (Gundel et al., 1991), and airway hyperresponsiveness (Wegner et al., 1990). The airway inflammatory response consists of increases in both eosinophils and neutrophils.
Rabbits can be sensitized to ragweed (Murphy et al., 1986) or alternaria (Shampain et al., 1982) antigens shortly after birth, and when challenged develop allergen-induced early and late responses (Shampain et al., 1982), airway inflammation, and airway hyperresponsiveness (Murphy et al., 1986). Sensitization during the neonatal period is essential for the development of increases in specific IgE to the allergens (Shampain et al., 1982). The inflammatory infiltrate consists of eosinophils and neutrophils, and ablation of the airway inflammatory response prevents the development of the physiologic changes (Murphy et al., 1986).
Guinea pigs are widely used for studying allergen-induced airway responses because of the ease of sensitization, mainly to ovalbumin (Hutson et al., 1988), and the relative ease of making physiologic measurements. The Hartley strain is the most widely used. Allergen inhalation causes early and late responses, airway inflammation mainly consisting of increases in eosinophils and neutrophils (Hutson et al., 1990), and airway hyperresponsiveness. However, efforts to measure the physiologic responses in awake guinea pigs have been confounded by the large component of upper airway responses in the measurements (Johns et al., 1990). In addition, guinea pigs have an airway eosinophilia, even in a baseline state before allergen inhalation, which can make the evaluation of changes after allergen inhalation difficult to interpret, in the absence of appropriate controls.
Rats can be sensitized to ovalbumin and, when challenged, develop early and late airway responses (Eidelman et al., 1988), airway inflammation (Richards et al., 1996), and airway hyperresponsiveness (Elwood et al., 1991). Several strains of rats have been used, but Norway rats are the best characterized and develop the most consistent responses (Wang et al., 1996).
Mice have become a widely used species to investigate the immunologic mechanisms of allergen-induced airway inflammation. The success of this model is a reflection of the large number of reagents for use in mice and the ability to selectively manipulate immunologic responses by using knockouts, genetic variants, or mice that overexpress a molecule of interest. Although a variety of strains have been used to evaluate allergen-induced airway inflammation, BALB/c are the most often used because these mice develop sensitization to allergens, such as ovalbumin; increases in allergen-specific IgE; and allergen-induced airway eosinophilia (Oshiba et al., 1996).
Development of airway hyperresponsiveness associated with airway eosinophilia and increases in TH2-type cytokines is a consistent finding following allergen challenge in sensitized mice (Inman et al., 1999; Foster et al., 1996). Typically, both the inflammation and functional changes are transient, returning to baseline within 2 weeks of the brief exposure to allergen. There is a report of early and late bronchoconstrictor responses to allergen (Cieslewicz et al., 1999); however, direct measurements of airway or pulmonary resistance were not made. Several investigators have used brief allergen exposure to investigate the mechanisms of the ensuing airway hyperresponsiveness. Clearly, the development of both inflammatory responses and airway hyperresponsiveness is dependent on T cells (Hogan et al., 1998a) and, more specifically, CD40 ligand-mediated responses (Mehlhop et al., 2000). Sorting out which of the T-cell cytokines are involved in the response has proven difficult. Evidence has been presented that many of the TH2-type cytokines, including IL-4 (Corry et al., 1996), IL-5 (Foster et al., 1996), and IL-13 (Grunig et al., 1998), are involved to some extent in establishing allergen-induced airway hyperresponsiveness. However, there is also strong evidence that airway hyperresponsiveness can develop in the absence of IL-4, IL-5, and airway eosinophilia (Hogan et al., 1998b). These models have also been used to demonstrate that several mediators, including interferon gamma (Yoshida et al., 2002), IL-6 (Wang et al., 2000), IL-10 (On et al., 2002), IL-12 (Kips et al., 1996), and IL-18 (Walter et al., 2001), act to prevent or reverse allergen-induced airway hyperresponsiveness in mice. Further studies have demonstrated that conventional and experimental antiasthma drugs including corticosteroids (Leign et al., 2002), antileukotriene agents (Blain and Sirois, 2000), and phosphodiesterase-4 inhibitors (Kung et al., 2000) can prevent allergic inflammation and airway hyperresponsiveness in these models.
More recently, investigators have begun to develop models of chronic allergen challenge in mice. These studies have to some extent been successful in their aim to include aspects of airway wall remodeling, including subepithelial fibrosis, goblet cell hyperplasia, and increased smooth muscle mass, in the pathologic changes induced as a result of allergen exposure (Temelkovski et al., 1998; Tanaka et al., 2001), which are similar to the changes considered to indicate airway remodeling in asthma. It is believed that inclusion of these aspects of asthmatic-type airway pathology will make these models more relevant for studying the mechanisms of airway dysfunction. These models all have demonstrated airway hyperresponsiveness in association with both acute cellular inflammation and some features of airway wall remodeling. More recently, it has further been demonstrated in a similar model of chronic allergen exposure that airway hyperresponsiveness can persist well beyond the resolution of cellular inflammation, suggesting a fundamentally different mechanism than found in models of brief allergen exposure. Such chronic models have been used to begin to elucidate the mechanisms underlying the development of airway wall remodeling, demonstrating a role for IL-13 (Blease et al., 2001), but not IL-4 or IL-5 (Foster et al., 2000) ( Fig. 81.2 ). Further studies will be required to explore the role of these cytokines in the development of airway hyperresponsiveness, which appears to continue beyond the resolution of airway inflammation. Interestingly, it has been demonstrated that treatment with an antileukotriene agent throughout the period of chronic allergen exposure can prevent several aspects of airway wall remodeling but not the associated airway hyperresponsiveness. Whether any intervention can prevent, or more importantly reverse, the sustained airway hyperresponsiveness should be a primary aim of ongoing research with these models.
Fig. 81.2.

Mice exposed to chronic allergen challenge and studied 24 hours after the final challenge. Only mice deficient for IL-5 were protected from allergen-induced eosinophilic inflammation (top panel). However, neither IL-5 nor IL-4-deficient mice were protected from allergen-induced airway remodeling (bottom panel).
(Modified from Foster et al., 2000)
© 2005
VIRAL INFECTIONS AND AIRWAY HYPERRESPONSIVENESS
Viral airway infections can cause airway hyperresponsiveness in normal individuals and can exacerbate asthma (Busse et al., 1997; Johnston et al., 1995). It is estimated that viral infections are associated with as many as 50% of wheezing illnesses and asthma exacerbations in children and as many as 20% of those in adults (Johnston et al., 1995; Pattemore et al., 1992). Mild viral respiratory tract infections seldom cause wheezing in normal individuals, but they frequently exacerbate symptoms in asthmatics, and severe infections can cause life-threatening asthma attacks (Ferreira et al., 2002).
Several different viruses, including rhinoviruses, coronaviruses, adenovirus, influenza B, respiratory syncytial virus, and parainfluenza virus, can evoke asthmatic symptoms (Pattemore et al., 1992; Casale et al., 1997; Gern and Busse, 1995). Furthermore, exposure to influenza virus or rhinovirus can cause airway hyperresponsiveness (Empey et al., 1976; Grunberg and Sterk, 1999). Rhinovirus infections can also increase the likelihood of late allergic reactions to antigen (Lemanske et al., 1989). Respiratory viruses damage the airway epithelium and evoke the release of cytokines and inflammatory mediators that can stimulate mast cells, increase vascular permeability, attract inflammatory cells, and initiate an immune response that may have lasting consequences (Busse, 1995). The released mediators and cellular changes can lead to airway hyperresponsiveness, edema, allergic responses, and airflow obstruction (Busse, 1995; Hegele, 1997).
Some viral respiratory infections have long-lasting consequences. After respiratory syncytial virus (RSV) infection, a common cause of bronchiolitis in children, lymphocytes can acquire sensitivity to specific food or mite antigens, thereby predisposing to the onset of allergic disease (Noma et al., 1996). Furthermore, latent or persistent viral infections may be associated with long-lasting airway hyperresponsiveness and chronic inflammation. Portions of the genome of adenovirus, RSV, or Epstein-Barr virus have been detected in the lungs of some patients with chronic obstructive lung disease (Hegele, 1997; Hogg, 2001).
Airway hyperresponsiveness is a well-documented consequence of viral respiratory infection in rats and guinea pigs. After inoculation with type 1 parainfluenza virus (Sendai virus), adult rats become abnormally sensitive to intravenous methacholine (Sorkness and Lemanske, 1996).
This abnormality lasts about 4 weeks, and thus outlasts the 2-week duration of the acute inflammatory response to the infection. The hyperresponsiveness is dependent upon intact vagus nerves. When neonatal rats are infected, the methacholine hyperresponsiveness can last as long as 16 weeks (Kumar et al., 1995). Pathologic changes in the airways of young rats include thickening, fibrosis, and recruitment of macrophages, mast cells, lymphocytes, and eosinophils (Uhl et al., 1996). These changes are more severe in Brown-Norway rats than in F344 rats and are associated with increased expression of transforming growth factor-beta (TGF-β) in mucosal macrophages (Uhl et al., 1996). Rat-adapted influenza virus infection in Brown-Norway rats can increase serologic titers of allergen-specific IgE and inhibit tolerance to repeated exposure to aerosolized allergen (Lebrec et al., 1996).
One mechanism by which viral infections may lead to airway hyperresponsiveness is through their effect on M2 muscarinic receptors on airway nerves. For example, type 3 parainfluenza virus infection in guinea pigs decreases the function of M2 receptors (Jacoby and Fryer, 1999). However, M3 muscarinic receptors on airway smooth muscle are unchanged by the infection. Viral infection may decrease M2 receptor function by damaging the receptors as a result of viral neuraminidase-induced cleavage of sialic acid residues or by inducing inflammation, as suggested by the leukocyte dependency of the effect (Fryer et al., 1997).
Respiratory syncytial virus (RSV) infection in mice results in eosinophilic inflammation and increased airway responsiveness to inhaled methacholine (Schwarze et al., 1997). Both the influx of eosinophils and the hyperresponsiveness can be blocked by an antibody to IL-5 (Schwarze et al., 1999).
Another mechanism by which viral infections may lead to airway hyperresponsiveness is through an effect on lung mast cells. Neonatal rats inoculated with Sendai virus have more than 100 times as many mast cells in their lungs as their age-matched controls (Castleman et al., 1989). Mast cells become particularly concentrated in bronchiolar walls. The increased number of mast cells is first detectable at 30 days after infection and is still present at 90 days (Castleman et al., 1990). During this period the rats have methacholine hyperresponsiveness (Castleman et al., 1990). After Sendai virus infection, Brown-Norway rats have higher viral titers, less efficient viral clearance, larger increases in bronchiolar mast cells, more persistent inflammatory responses, and greater airway responsiveness than do F344 rats (Sorden and Castleman, 1995a). The increased number of mast cells results from the proliferation of mast cells, as shown by bromodeoxyuridine labeling, and from the recruitment of mast cell precursors from blood (Sorden and Castleman, 1995b).
Respiratory infections can exaggerate neurogenic inflammation in the airway mucosa (McDonald et al., 1991). For example, Sendai virus infection in rats increases the amount of plasma leakage that occurs in the airway mucosa after an injection of capsaicin (Piedimonte et al., 1990a). This augmented response coincides with the epithelial damage and influx of inflammatory cells that peak 4–6 days after inoculation (Piedimonte et al., 1990a) and can be prevented by pretreatment with dexamethasone (Piedimonte et al., 1990b).
Sendai virus infection in guinea pigs increases the bronchoconstrictor response to substance P and capsaicin (Dusser et al., 1989). The mechanism of this change may involve a decrease in substance P-degrading neutral endopeptidase (NEP) in the airway epithelium, because after infection the NEP inhibitor phosphoramidon no longer potentiates the response to substance P or capsaicin. Bronchomotor responses to acetylcholine are unaffected by the infection (Dusser et al., 1989). Similar accentuated smooth muscle contractile responses to substance P have been reported in ferret tracheas infected in vitro with human influenza virus A (Jacoby et al., 1988). Here, the activity of NEP is decreased by 50% (Jacoby et al., 1988).
The question of how viral airway infections can produce long-lasting changes, resembling the persistent nature of asthma and other chronic airway diseases, is beginning to be addressed in animal models. One approach is through experiments on virally mediated lymphocyte activation (Gern et al., 1996). Another approach focuses on latent viral infections. After intranasal inoculation with adenovirus 5, guinea pigs develop bronchiolitis that can persist for more than 6 weeks (Vitalis et al., 1996). Viral DNA can be detected by polymerase chain reaction at this time in most of the animals, and viral protein can be detected by immunohistochemistry in some (Vitalis et al., 1996). Additional research will be needed to determine the mechanism underlying the prolonged inflammatory response.
MYCOPLASMAL INFECTION AND AIRWAY HYPERRESPONSIVENESS
Respiratory infection caused by Mycoplasma pneumoniae is considered an etiologic or precipitating factor in some cases of asthma (Berkovich et al., 1970; Mok et al., 1979). Mycoplasma pulmonis infections cause a chronic respiratory disease in rats and mice (Lindsey et al., 1971; Lindsey and Cassell, 1973). The severity of M. pulmonis disease ranges from subclinical to lethal, with genetic and environmental factors playing important roles in the outcome. The organisms are prokaryotic extracellular parasites with no cell wall that attach to the luminal plasma membrane of airway epithelial cells. The initial acute inflammatory response, in which neutrophils predominate, evolves into chronic inflammation characterized by the accumulation of mucosal lymphoid tissue, peribronchial lymphoid follicles, and enlarged hilar, mediastinal, and cervical lymph nodes. Dendritic cells, T and B lymphocytes, and macrophages accumulate in the airway mucosa (Davis et al., 1982). Lymphoid tissue, which is rare or absent in the tracheal mucosa of pathogen-free rats and mice, can occupy 75% of the mucosa after M. pulmonis infection (McDonald et al., 1991). Other pathologic changes include hyperplasia of epithelial ciliated cells and goblet cells, mucous gland hypertrophy, angiogenesis, and fibrosis (McDonald et al., 1991; Schoeb et al., 1985; McIntosh et al., 1992; Cartner et al., 1995; Huang et al., 1989). These changes can result in a several-fold increase in the thickness of the airway mucosa ( Fig. 81.3 ) (McDonald et al., 1991). Lifelong inflammatory airway disease, with chronic tracheobronchitis, bronchiectasis, airway wall thickening and fibrosis, and lung consolidation, can eventually develop (McIntosh et al., 1992).
Fig. 81.3.
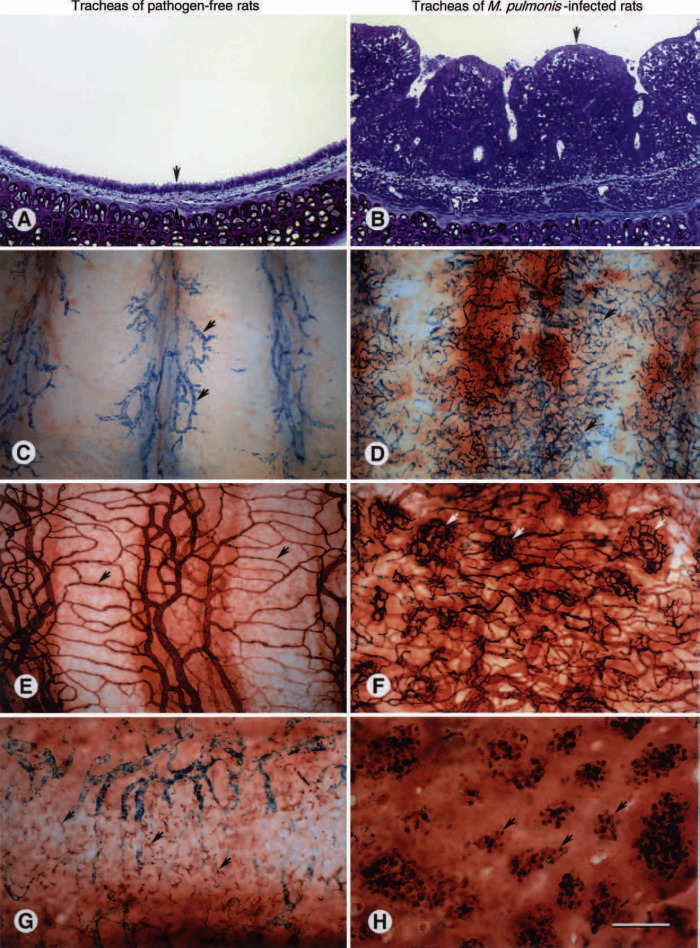
Remodeling of airway mucosa after Mycoplasma pulmonis infection. A, B. Histologic sections of rat tracheas stained with toluidine blue showing the thin mucosa of a pathogen-free rat (A) and the much thicker mucosa of a rat infected with M. pulmonis for 4 weeks (B). Arrows mark the outer and inner limits of the mucosa. C, D. Whole mounts of rat tracheas showing leaky mucosal blood vessels (arrows, blue) after an intravenous injection of substance P to mimic neurogenic inflammation. The amount of neurogenic inflammation, as reflected by the number of leaky blood vessels, is much smaller in the pathogen-free rat (C) than in the rat infected with M. pulmonis for 4 weeks (D). E, F. Whole mounts of rat tracheas showing the amount and architecture of the mucosal vasculature after staining by perfusion of biotinylated Lycopersicon esculentum lectin. Relatively sparse, straight capillaries (arrows) in a pathogen-free rat (E) contrast with the abundant, tortuous angiogenic blood vessels in a rat infected with M. pulmonis for 4 weeks (F). Some of the angiogenic blood vessels form focal networks (arrows). G, H. Whole mounts of rat tracheas stained immunohistochemically with antibody ED2 to show tissue macrophages. Tissue macrophages are irregularly shaped and scattered in the mucosa of the pathogen-free rat (G), but in the rat infected with M. pulmonis for 4 weeks they are rounded and concentrated in focal clusters around tortuous networks of angiogenic blood vessels (H). Scale bar 150 μm in A, B, G, H; 300 μm in C–F.
Although it has not yet been determined whether the airways of M. pulmonis-infected animals are hyperresponsiveness to methacholine, other forms of hyperresponsiveness do occur. For example, the newly formed microvasculature is abnormal, one manifestation of which is hyperresponsiveness to certain irritants and predisposition to plasma leakage (Fig. 81.3) (Kwan et al., 2001; McDonald, 2001). Also, airway mucous secretion is exaggerated (Huang et al., 1989).
OZONE-INDUCED AIRWAY RESPONSES
Ozone is a powerful oxidizing agent that is classified as a “secondary air pollutant.” Secondary air pollutants are not emitted into the atmosphere but formed from subsequent atmospheric chemical reactions of primary pollutants (nitrogen dioxide, sulfur dioxide, particles, carbon monoxide, and lead). Ozone is used in a variety of animal models to induce airway hyperresponsiveness and airway inflammation.
Dogs: Airway hyperresponsiveness develops in dogs after ozone inhalation (Lee et al., 1977). The airway hyperresponsiveness is most marked at 1 hour after ozone inhalation, is still present 1 day later, and is back to baseline levels by 1 week (Holtzman et al., 1983b). Ozone-induced airway hyperresponsiveness in dogs is associated with a marked reversible neutrophil influx in the epithelium and bronchoalveolar lavage (Holtzman et al., 1983a) ( Fig. 81.4 ). The dogs responsive to ozone have an increased number of epithelial cells in the lavage 1 hour after ozone inhalation (Fabbri et al., 1984). There is a close correlation between the degree of airway hyperresponsiveness and the number of neutrophils in the epithelium (Holtzman et al., 1983a). Ozone-induced airway hyperresponsiveness is prevented by depleting circulating neutrophils (O'Byrne et al., 1984). These studies suggest a causal relationship between the onset of ozone-induced hyperresponsiveness and neutrophil influx into the airways. However, other investigators have been unable to attenuate the ozone-induced airway hyperresponsiveness in dogs with cyclophosphamide-induced neutrophil depletion (Imai et al., 1990). Further evidence that the ozone-induced neutrophilia is not necessarily related to the ozone-induced airway hyperresponsiveness comes from the study of Li et al. (1992), who blocked the ozone-induced leukocyte migration with antileukocyte adhesion molecule (anti-Mo1) without inhibiting ozone-induced airway hyper-responsiveness.
Fig. 81.4.
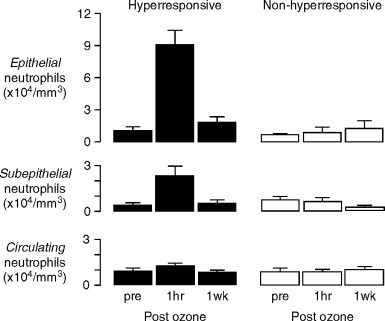
Increases in neutrophils in the epithelium and subepithelium of bronchial mucosal biopsies from dogs with ozone-induced airway hyperresponsiveness. No increases were demonstrated in dogs that did not develop ozone-induced airway hyperresponsiveness, or in circulating neutrophil numbers in either group.
(Reprinted with permission from Holtzman et al., 1983b).
© 2005
Sheep: Airway hyperresponsiveness to inhaled carbacol develops in sheep 24 hours after ozone inhalation, with no change in tracheal mucous velocity (Phipps et al., 1986). Ozone also increases in airway responsiveness to inhaled radioactively labeled histamine. The airway hyperresponsiveness parallels ozone-induced increases in epithelial permeability, which was estimated from the rate of appearance of the labeled histamine in the blood plasma (Abraham et al., 1984). Tracheal mucous velocity significantly decreases after ozone inhalation (Allegra et al., 1991). Ozone inhalation also causes a dose-dependent increase in the bronchial artery blood flow in sheep, as a reflection of vasodilatation of the bronchial vasculature (Schelegle et al., 1990).
Primates: Airway responsiveness to methacholine increases 2.5-fold in rhesus monkeys exposed weekly to ozone for 19 weeks (Menzel, 1996). After a single ozone inhalation, reinfused labeled neutrophils appear in lung tissue and bronchoalveolar lavage fluid, and this effect is maximal at 12 hours and returns to baseline by 24 hours after ozone inhalation (Hyde et al., 1992). Epithelial necrosis is seen in the trachea and bronchioles of rhesus monkeys at 1 hour and 12 hours after inhaled ozone. The epithelial necrosis and repair is associated with the presence of granulocytes in the epithelium and interstitium (Hyde et al., 1992). This is seen as early as 4 hours after inhaled ozone, and is maximal between 12 and 24 hours (Castleman et al., 1980). Eosinophils are maximally increased in the bronchial mucosa at 24 hours when epithelial necrosis and lavageable protein are also maximally increased (Hyde et al., 1992).
Rabbits: Exposure of rabbits to 2ppm ozone, 6 hours daily for 3 days, results in increased pulmonary resistance, epithelial damage, increased submucosal edema in the large airways and in terminal and respiratory bronchioles, and thickened alveolar walls in the proximal alveolar ducts (Yokoyama et al., 1989). Ozone inhalation also results in a large increase in PGE2 and PGF2α, and small increases in TxB2 and 6-keto-prostaglandin F1α. This effect decreases and eventually disappears as the animals grow toward adulthood. Acute ozone inhalation experiments have not been carried out in rabbits. In vitro ozone exposure of lung macrophages results in elaboration of PGE2 and PGF2α.
Guinea pigs: In guinea pigs, ozone inhalation causes airway hyperresponsiveness without neutrophil influx into the tracheal mucosa (Murlas and Roum, 1985). Guinea pigs exposed to ozone for 2 hours also develop airway hyperresponsiveness to subcutaneous histamine (Gordon and Amdur, 1980). Methacholine airway hyperresponsiveness occurs after ozone inhalation of 1ppm for 1.5 hours, but not after inhaling 1ppm for 0.5 hours, suggesting that the ozone-induced airway hyperresponsiveness in guinea pigs is significantly correlated with the ozone dose (the product of time and concentration). Ozone inhalation (3ppm for 1 hour) increases neutrophils in bronchoalveolar lavage in ovalbumin-sensitized and nonsensitized male guinea pigs (Campus et al., 1992). Furthermore, these investigators found that ozone inhalation increases the in vivo bronchoconstrictor response to both histamine and allergen. Neutrophils rapidly accumulated in the guinea pig lung interstitium after ozone inhalation and returned to near control values within 24 hours (Schultheis and Bassett, 1991). By contrast, neutrophils recovered in bronchoalveolar lavage peak at 3–6 hours and remain elevated for 3 days. Leukocyte depletion with cyclophosphamide in steroid-treated guinea pigs does not prevent acetylcholine airway hyperresponsiveness at 2 or 6 hours after ozone inhalation (Murlas and Ram, 1985), suggesting that airway neutrophilic infiltration is a consequence of ozone-induced mucosal injury, but is not the cause of airway hyperresponsiveness.
Rats: Exposure of rats to high ozone concentrations causes airway hyperresponsiveness, epithelial damage, and in most studies, airway neutrophil influx, but not increased vascular permeability. Rats developed acetylcholine airway hyperresponsiveness immediately after ozone inhalation, and the airway responsiveness returned to control levels by 24 hours (Evans et al., 1988). The ozone-induced acetylcholine airway hyperresponsiveness has been suggested to result from a marked inhibition of acetylcholinesterase (Gordon et al., 1981). Inhalation of lower concentration of ozone also causes airway hyperresponsiveness, but without neutrophil influx into the tracheal mucosa or increases in vascular permeability (Mustafa et al., 1983). However, a similar study, from the same laboratory, reports that the number of neutrophils in bronchoalveolar lavage is significantly increased after the same concentration of ozone (Hotchkiss et al., 1989). To investigate the importance of neutrophils in ozone-induced airway hyperresponsiveness, Joad et al. (1993) perfused rat lungs with buffer with or without neutrophils, and with or without exposure to ozone. They concluded that perfusion of neutrophils in the absence of ozone decreased pulmonary compliance and increased pulmonary resistance, total bronchoalveolar lavage protein, lung weight, and pulmonary arterial pressure, probably as a result of increased vascular leakage. However, neutrophil perfusion alone neither changed airway responsiveness to methacholine nor damaged the airway epithelium. Yet, neutrophil perfusion did have an additive effect on the ozone-induced impairment of pulmonary function and a synergistic effect on ozone-induced airway epithelial injury.
These studies suggest that neutrophils play a role in modulating the repair processes after ozone-induced injury and that plasma extravasation from alveolar capillaries does not occur following ozone inhalation.
Mice: Some investigators have suggested that the magnitude of the inflammatory response of the lungs to inhaled ozone in animals is, at least in part, under genetic control. In support of this is an 11-fold difference in inflammatory responses demonstrated in an acute ozone-induced pulmonary inflammation-susceptible strain of mice to inhaled ozone as compared with ozone-exposed control mice (Kleeberger et al., 1990). Ozone-induced airway hyperresponsiveness and an associated neutrophilic airway inflammation has been described in mice (Shore et al., 2001). The airway hyperresponsiveness, but not the neutrophilic inflammation, is mediated by tumor necrosis factor (TNF) (Shore et al., 2001).
INTERACTIONS OF OZONE- AND ALLERGEN-INDUCED AIRWAY RESPONSES
The additive or synergistic effects of ozone and allergen inhalation are potentially important clinically because high concentrations of atmospheric ozone often coincide with peak levels of airborne allergens. It also has been suggested that epithelial damage induced by ozone could exaggerate allergic reactions to inhaled foreign proteins (Osebold et al., 1980). In atopic subjects, inhaling ozone at concentrations selected to produce no detectable effects on baseline lung function increases airway responsiveness to inhaled allergen in atopic asthmatics (Molfino et al., 1991). Guinea pigs challenged with ozone also have been shown to be more sensitive to inhaled allergens (Matsumura, 1970).
Ozone inhalation increases acetylcholine airway responsiveness in dogs nonallergic and allergic to A. suum (Yanai et al., 1990). Immediately after ozone inhalation, the Ascaris-sensitive dogs were 4.7-fold more responsive to the inhaled Ascaris allergen and to acetylcholine. Two weeks later, these dogs were still significantly hyperresponsive to A. suum inhalation (1.6-fold increase), but no longer to acetylcholine.
CONCLUSIONS
Inhaled allergens, ozone, and some viruses can cause airway inflammation and airway hyperresponsiveness in humans and in a variety of laboratory animals. Studies using these stimuli have contributed significantly to the understanding of the pathophysiology of airway inflammation and airway hyperresponsiveness. Indeed, a causal link between airway inflammation and airway hyperresponsiveness was initially demonstrated in dogs exposed to ozone. The importance of inflammatory cells and various mediators in airway hyperresponsiveness remains controversial. Differences among species may stem from differences in the experimental models and types of measurements, as well as biologic differences. Overall, much more needs to be done to fully characterize the pathophysiology of allergen-, pathogen-, and ozone-induced airway hyperresponsiveness.
REFERENCES
- Abraham W.M., Delehunt J.C., Yerger L., Marchette B., Oliver W., Jr. Changes in airway permeability and responsiveness after exposure to ozone. Environ. Res. 1984;34:110–119. doi: 10.1016/0013-9351(84)90080-x. [DOI] [PubMed] [Google Scholar]
- Abraham W.M., Lanes S., Stevenson J.S., Yerger L.D. Effect of an inhaled glucocorticosteroid (budesonide) on postantigen induced increases in airway responsiveness. Bull. Eur. Physiopathol. Respir. 1986;22:387–392. [PubMed] [Google Scholar]
- Abraham W.M., Sielczak M.W., Wanner A., Perruchoud A.P., Blinder L., Ahmed A., Yerger L.D. Cellular markers of inflammation in the airways of allergic sheep with and without allergen-induced late responses. Am. Rev. Respir. Dis. 1988;138:1565–1571. doi: 10.1164/ajrccm/138.6.1565. [DOI] [PubMed] [Google Scholar]
- Allegra L., Moavero N.E., Rampoldi C. Ozone-induced impairment of mucociliary transport and its prevention with N-acetylcysteine. Am. J. Med. 1991;91:67S–71S. doi: 10.1016/0002-9343(91)90286-7. [DOI] [PubMed] [Google Scholar]
- Anderson S.D., Schoeffel R.E., Finney M. Evaluation of ultrasonically nebulised solutions for provocation testing in patients with asthma. Thorax. 1983;38:284–291. doi: 10.1136/thx.38.4.284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker A.B., Hershkovich J., Simons F.E., Simons K.J., Lilley M.K., Kepron M.W. Development of chronic airway hyperresponsiveness in ragweed-sensitized dogs. J. Appl. Physiol. 1989;66:2691–2697. doi: 10.1152/jappl.1989.66.6.2691. [DOI] [PubMed] [Google Scholar]
- Berkovich S., Millian S.J., Snyder R.D. The association of viral and mycoplasma infections with recurrence of wheezing in the asthmatic child. Ann. Allergy. 1970;28:43–49. [PubMed] [Google Scholar]
- Bhalla D.K., Daniels D.S., Luu N.T. Attenuation of ozone-induced airway permeability in rats by pretreatment with cyclophosphamide, FPL 55712, and indomethacin. Am. J. Respir. Cell Mol. Biol. 1992;7:73–80. doi: 10.1165/ajrcmb/7.1.73. [DOI] [PubMed] [Google Scholar]
- Blain J.F., Sirois P. Involvement of LTD4 in allergic pulmonary inflammation in mice: Modulation by cysLT(1) antagonist MK-571. Prostaglandins Leukot. Essent. Fatty Acids. 2000;62:361–368. doi: 10.1054/plef.2000.0167. [DOI] [PubMed] [Google Scholar]
- Blease K., Jakubzick C., Westwick J., Lukacs N., Kunkel S.L., Hogaboam C.M. Therapeutic effect of IL-13 immunoneutralization during chronic experimental fungal asthma. J. Immunol. 2001;166:5219–5224. doi: 10.4049/jimmunol.166.8.5219. [DOI] [PubMed] [Google Scholar]
- Busse W.W. Viral-infections in humans. Am. J. Respir. Crit. Care Med. 1995;151:1675–1677. doi: 10.1164/ajrccm.151.5.7735632. [DOI] [PubMed] [Google Scholar]
- Busse W.W., Gern J.E. Viruses in asthma. J. Allergy Clin. Immunol. 1997;100:147–150. doi: 10.1016/s0091-6749(97)70216-1. [DOI] [PubMed] [Google Scholar]
- Campos M.G., Segura P., Vargas M.H., Vanda B., Ponce-Monter H., Selman M. O3-induced airway hyperresponsiveness to noncholinergic system and other stimuli. J. Appl. Physiol. 1992;73:354–361. doi: 10.1152/jappl.1992.73.1.354. [DOI] [PubMed] [Google Scholar]
- Cartner S.C., Simecka J.W., Lindsey J.R., Cassel G.H., Davis J.K. Chronic respiratory mycoplasmosis in C3H/HeN and C57BL/6N mice: Lesion severity and antibody response. Infect. Immun. 1995;63:4138–4142. doi: 10.1128/iai.63.10.4138-4142.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casale T.B., Bernstein I.L., Busse W.W., LaForce C.F., Tinkelman D.G., Stoltz R.R., Dockhorn R.J., Reimann J., Su J.Q., Fick R.B., Adelman D.C. Use of an anti-IgE humanized monoclonal antibody in ragweed-induced allergic rhinitis. J. Allergy Clin. Immunol. 1997;100:110–121. doi: 10.1016/s0091-6749(97)70202-1. [DOI] [PubMed] [Google Scholar]
- Castleman W.L., Dungworth D.L., Schwartz L.W., Tyler W.S. Acute respiratory bronchiolitis: An ultrastructural and autoradiographic study of epithelial cell injury and renewal in rhesus monkeys exposed to ozone. Am. J. Pathol. 1980;98:811–840. [PMC free article] [PubMed] [Google Scholar]
- Castleman W., Owens S.B., Brundage-Anguish L.J. Acute and persistent alterations in pulmonary inflammatory cells and airway mast cells induced by Sendai virus infection in neonatal rats. Vet. Pathol. 1989;26:11–25. doi: 10.1177/030098588902600104. [DOI] [PubMed] [Google Scholar]
- Castleman W., Sorkness R.L., Lemanske R.F., McAllister P.K. Viral bronchiolitis during early life induces increased numbers of bronchiolar mast cells and airway hyperresponsiveness. Am. J. Pathol. 1990;137:821–831. [PMC free article] [PubMed] [Google Scholar]
- Chan-Yeung M., Malo J.L. Occupational asthma. N. Engl. J. Med. 1995;333:107–112. doi: 10.1056/NEJM199507133330207. [DOI] [PubMed] [Google Scholar]
- Chung K.F., Becker A.B., Frick O.L., Nadel J.A., Gold W.M. Airway inflammation and hyperresponsiveness during the late-phase response to antigen challenge in ragweed-sensitized dogs. Clin. Res. 1984;32:527a. [Google Scholar]
- Chung K.F., Becker A.B., Lazarus S.C., Frick O.L., Nadel J.A., Gold W.M. Antigen-induced airway hyperresponsiveness and pulmonary inflammation in allergic dogs. J. Appl. Physiol. 1985;58:1347–1353. doi: 10.1152/jappl.1985.58.4.1347. [DOI] [PubMed] [Google Scholar]
- Cieslewicz G., Tomkinson A., Adler A., Duez C., Schwarze J., Takeda K., Larson K.A., Lee J.J., Irvin C.G., Gelfand E.W. The late, but not early, asthmatic response is dependent on IL-5 and correlates with eosinophil infiltration. J. Clin. Invest. 1999;104:301–308. doi: 10.1172/JCI7010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cockcroft D.W., Killian D.N., Mellon J.J., Hargreave F.E. Bronchial reactivity to inhaled histamine: A method and clinical survey. Clin. Allergy. 1977;7:235–243. doi: 10.1111/j.1365-2222.1977.tb01448.x. [DOI] [PubMed] [Google Scholar]
- Corry D.B., Folkesson H.G., Warnock M.L., Erle D.J., Matthay M.A., Wiener-Kronish J.P., Locksley R.M. Interleukin 4, but not interleukin 5 or eosinophils, is required in a murine model of acute airway hyperreactivity. J. Exp. Med. 1996;183:109–117. doi: 10.1084/jem.183.1.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis J.K., Thorpe R.B., Maddox P.A., Brown M.B., Cassel G.H. Murine respiratory mycoplasmosis in F344 and LEW rats: evolution of lesions and lung lymphoid cell populations. Infect. Immunity. 1982;36:720–729. doi: 10.1128/iai.36.2.720-729.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delehunt J.C., Perruchoud A.P., Yerger L., Marchette B., Stevenson J.S., Abraham W.M. The role of slow-reacting substance of anaphylaxis in the late bronchial response after antigen challenge in allergic sheep. Am. Rev. Respir. Dis. 1984;130:748–754. doi: 10.1164/arrd.1984.130.5.748. [DOI] [PubMed] [Google Scholar]
- Dusser D.J., Jacoby D.B., Djokic T.D., Rubinstein I., Borson D.B., Nadel J.A. Virus induces airway hyperresponsiveness to tachykinins: role of neutral endopeptidase. J. Appl. Physiol. 1989;67:1504–1511. doi: 10.1152/jappl.1989.67.4.1504. [DOI] [PubMed] [Google Scholar]
- Eidelman D.H., Bellofiore S., Martin J.G. Late airway responses to antigen challenge in sensitized inbred rats. Am. Rev. Respir. Dis. 1988;137:1033–1037. doi: 10.1164/ajrccm/137.5.1033. [DOI] [PubMed] [Google Scholar]
- Elwood W., Lotvall J.O., Barnes P.J., Chung K.F. Characterization of allergen-induced bronchial hyperresponsiveness and airway inflammation in actively sensitized Brown-Norway rats. J. Allergy Clin. Immunol. 1991;88:951–960. doi: 10.1016/0091-6749(91)90253-k. [DOI] [PubMed] [Google Scholar]
- Empey D.W., Laitinen L.A., Jacobs L., Gold W.M., Nadel J.A. Mechanisms of bronchial hyperreactivity in normal subjects after upper respiratory tract infection. Am. Rev. Respir. Dis. 1976;113:131–139. doi: 10.1164/arrd.1976.113.2.131. [DOI] [PubMed] [Google Scholar]
- Evans T.W., Brokaw J.J., Chung K.F., Nadel J.A., McDonald D.M. Ozone-induced bronchial hyperresponsiveness in the rat is not accompanied by neutrophil influx or increased vascular permeability in the trachea. Am. Rev. Respir. Dis. 1988;138:140–144. doi: 10.1164/ajrccm/138.1.140. [DOI] [PubMed] [Google Scholar]
- Fabbri L.M., Aizawa H., Alpert S.E., Walters E.H., O'Byrne P.M., Gold B.D., Nadel J.A., Holtzman M.J. Airway hyper-responsiveness and changes in cell counts in bronchoalveolar lavage after ozone exposure in dogs. Am. Rev. Respir. Dis. 1984;129:288–291. [PubMed] [Google Scholar]
- Ferreira A., Williams Z., Donninger H., van Schalkwyk E.M., Bardin P.G. Rhinovirus is associated with severe asthma exacerbations and raised nasal interleukin-12. Respiration. 2002;69:136–142. doi: 10.1159/000056316. [DOI] [PubMed] [Google Scholar]
- Foster P.S., Hogan S.P., Ramsay A.J., Matthaei K.I., Young I.G. Interleukin 5 deficiency abolishes eosinophilia, airways hyperreactivity, and lung damage in a mouse asthma model. J. Exp. Med. 1996;183:195–201. doi: 10.1084/jem.183.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster P.S., Ming Y., Matthei K.I., Young I.G., Temelkovski J., Kumar R.K. Dissociation of inflammatory and epithelial responses in a murine model of chronic asthma. Lab. Invest. 2000;80:655–662. doi: 10.1038/labinvest.3780068. [DOI] [PubMed] [Google Scholar]
- Fryer A.D., Costello R.W., Yost B.L., Lobb R.R., Tedder T.F., Steeber D.A., Bochner B.S. Antibody to VLA-4, but not to L-selectin, protects neuronal M-2 muscarinic receptors in antigen-challenged guinea pig airways. J. Clin. Invest. 1997;99:2036–2044. doi: 10.1172/JCI119372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauvreau G.M., Lee J.M., Watson R.M., Irani A.M., Schwartz L.B., O'Byrne P.M. Increased numbers of both airway basophils mast cells in sputum after allergen inhalation challenge of atopic asthmatics. Am. J. Respir. Crit. Care Med. 2000;161:1473–1478. doi: 10.1164/ajrccm.161.5.9908090. [DOI] [PubMed] [Google Scholar]
- Gauvreau G.M., Watson R.M., O'Byrne P.M. Kinetics of allergen-induced airway eosinophilic cytokine production and airway inflammation. Am. J. Respir. Crit. Care Med. 1999;160:640–647. doi: 10.1164/ajrccm.160.2.9809130. [DOI] [PubMed] [Google Scholar]
- Gern J.E., Busse W.W. The effects of rhinovirus infections on allergic airway responses. Am. J. Respir. Crit. Care Med. 1995;152:S40–S45. doi: 10.1164/ajrccm/152.4_Pt_2.S40. [DOI] [PubMed] [Google Scholar]
- Gern J.E., Vrtis J., Kelly E.A., Dick C.R., Busse W.W. Rhinovirus produces nonspecific activation of lymphocytes through a monocyte-dependent mechanism. J. Immunol. 1996;157:1605–1612. [PubMed] [Google Scholar]
- Gordon T., Amdur M.O. Effect of ozone on respiratory response of guinea pigs to histamine. J. Toxicol. Environ. Health. 1980;6:185–195. doi: 10.1080/15287398009529841. [DOI] [PubMed] [Google Scholar]
- Gordon T., Taylor B.F., Amdur M.O. Ozone inhibition of tissue cholinesterase in guinea pigs. Arch. Environ. Health. 1981;36:284–288. doi: 10.1080/00039896.1981.10667639. [DOI] [PubMed] [Google Scholar]
- Grunberg K., Sterk P.J. Rhinovirus infections: Induction and modulation of airways inflammation in asthma. Clin. Exp. Allergy. 1999;29:65–73. doi: 10.1046/j.1365-2222.1999.00011.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunig G., Warnock M., Wakil A.E., Venkayya R., Brombacher F., Rennick D.M., Sheppard D., Mohrs M., Donaldson D.D., Locksley R.M., Corry D.B. Requirement for IL-13 independently of IL-4 in experimental asthma. Science. 1998;282:2261–2263. doi: 10.1126/science.282.5397.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gundel R.H., Wegner C.D., Letts L.G. Antigen-induced acute and late phase responses in primates. Am. Rev. Respir. Dis. 1992;146:369–373. doi: 10.1164/ajrccm/146.2.369. [DOI] [PubMed] [Google Scholar]
- Gundel R.H., Wegner C.D., Torcellini C.A., Clark C.C., Haynes N., Rothlein R., Smith C.W., Letts L.G. Endothelial leukocyte adhesion molecule-1 mediates antigen-induced airway inflammation and late phase airway obstruction in monkeys. J. Clin. Invest. 1991;88:1407–1411. doi: 10.1172/JCI115447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton A.L., Watson R.M., Wyile G., O'Byrne P.M. A 5-lipoxygenase activating protein antagonist, Bay x 1005, attenuates both early and late phase allergen-induced bronchoconstriction in asthmatic subjects. Thorax. 1997;52:348–354. doi: 10.1136/thx.52.4.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegele R.G. The role of viruses in chronic respiratory disease. Infectious Dis. Clin. Pract. 1997;6:167–173. [Google Scholar]
- Hogan S.P., Koskinen A., Matthaei K.I., Young I.G., Foster P.S. Interleukin-5-producing CD4+ T cells play a pivotal role in aeroallergen-induced eosinophilia, bronchial hyperreactivity, and lung damage in mice. Am. J. Respir. Crit. Care Med. 1998;157:210–218. doi: 10.1164/ajrccm.157.6.mar-1. [DOI] [PubMed] [Google Scholar]
- Hogan S.P., Matthaei K.I., Young J.M., Koskinen A., Young I.G., Foster P.S. A novel T cell-regulated mechanism modulating allergen-induced airways hyperreactivity in BALB/c mice independently of IL-4 and IL-5. J. Immunol. 1998;161:1501–1509. [PubMed] [Google Scholar]
- Hogg J.C. Role of latent viral infections in chronic obstructive pulmonary disease and asthma. Am. J. Respir. Crit. Care Med. 2001;164:S71–S75. doi: 10.1164/ajrccm.164.supplement_2.2106063. [DOI] [PubMed] [Google Scholar]
- Holtzman M.J., Cunningham J.H., Sheller J.R., Irsigler G.B., Nadel J.A., Boushey H.A. Effects of ozone on bronchial reactivity in atopic and nonatopic subjects. Am. Rev. Respir. Dis. 1979;120:1059–1067. doi: 10.1164/arrd.1979.120.5.1059. [DOI] [PubMed] [Google Scholar]
- Holtzman M.J., Fabbri L.M., O'Byrne P.M., Gold B.D., Aizawa H., Walters E.H., Alpert S.E., Nadel J.A. Importance of airway inflammation for hyperresponsiveness induced by ozone. Am. Rev. Respir. Dis. 1983;127:686–690. doi: 10.1164/arrd.1983.127.6.686. [DOI] [PubMed] [Google Scholar]
- Holtzman M.J., Fabbri L.M., Skoogh B.E., O'Byrne P.M., Walters E.H., Aizawa H., Nadel J.A. Time course of airway hyperresponsiveness induced by ozone in dogs. J. Appl. Physiol. Resp. Env. Exc. Physiol. 1983;55:1232–1236. doi: 10.1152/jappl.1983.55.4.1232. [DOI] [PubMed] [Google Scholar]
- Hotchkiss J.A., Harkema J.R., Kirkpatrick D.T., Henderson R.F. Response of rat alveolar macrophages to ozone: Quantitative assessment of population size, morphology, and proliferation following acute exposure. Exp. Lung Res. 1989;15:1–16. doi: 10.3109/01902148909069605. [DOI] [PubMed] [Google Scholar]
- Huang H.T., Haskell A., McDonald D.M. Changes in epithelial secretory cells and potentiation of neurogenic inflammation in the trachea of rats with respiratory tract infections. Anat. Embryol. (Berl). 1989;180:325–341. doi: 10.1007/BF00311165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutson P.A., Church M.K., Clay T.P., Miller P., Holgate S.T. Early and late-phase bronchoconstriction after allergen challenge of nonanesthetized guinea pigs. I. The association of disordered airway physiology to leukocyte infiltration. Am. Rev. Respir. Dis. 1988;137:548–557. doi: 10.1164/ajrccm/137.3.548. [DOI] [PubMed] [Google Scholar]
- Hutson P.A., Varley J.G., Sanjar S., Kings M., Holgate S.T., Church M.K. Evidence that neutrophils do not participate in the late-phase airway response provoked by ovalbumin inhalation in conscious, sensitized guinea pigs. Am. Rev. Respir. Dis. 1990;141:535–539. doi: 10.1164/ajrccm/141.3.535. [DOI] [PubMed] [Google Scholar]
- Hyde D.M., Hubbard W.C., Wong V., Wu R., Pinkerton K., Plopper C.G. Ozone-induced acute tracheobronchial epithelial injury: relationship to granulocyte emigration in the lung. Am. J. Respir. Cell Mol. Biol. 1992;6:481–497. doi: 10.1165/ajrcmb/6.5.481. [DOI] [PubMed] [Google Scholar]
- Imai T., Adachi M., Idaira K., Hiyama T., Suganuma T., Takahashi T., Yamaguchi H., Saito C., Maeda M., Tuzi A. The role of neutrophils in airway hyperresponsiveness in dogs after ozone exposure. Arerugi. 1990;39:90–98. [PubMed] [Google Scholar]
- Inman M.D., Denburg J.A., Ellis R., Dahlbäck M., O'Byrne P.M. Allergen-induced increases in bone marrow progenitors in airway hyperresponsive dogs: Regulation by a serum haematopoeitic factor. Am. J. Respir. Cell Mol. Biol. 1996;15:305–311. doi: 10.1165/ajrcmb.15.3.8924277. [DOI] [PubMed] [Google Scholar]
- Inman M.D., Wattie J., Denburg J.A., O'Byrne P.M. Allergen-induced increases in airway responsiveness, airway eosinophilia and bone marrow eosinophil progenitors in mice. Am. J. Respir. Cell Mol. Biol. 1999;21:473–479. doi: 10.1165/ajrcmb.21.4.3622. [DOI] [PubMed] [Google Scholar]
- Jacoby D.B., Fryer A.D. Interaction of viral infections with muscarinic receptors. Clin. Exp. Allergy. 1999;29:59–64. doi: 10.1046/j.1365-2222.1999.00010.x. [DOI] [PubMed] [Google Scholar]
- Jacoby D.B., Tamoki J., Borson D.B., Nadel J.A. Influenza infection causes airway hyperresponsiveness by decreasing enkephalinase. J. Appl. Physiol. 1988;64:2653–2658. doi: 10.1152/jappl.1988.64.6.2653. [DOI] [PubMed] [Google Scholar]
- Joad J.P., Bric J.M., Pino M.V., Hyde D.M., McDonald R.J. Effects of ozone and neutrophils on function and morphology of the isolated rat lung. Am. Rev. Respir. Dis. 1993;147:1578–1584. doi: 10.1164/ajrccm/147.6_Pt_1.1578. [DOI] [PubMed] [Google Scholar]
- Johns K., Sorkness R., Graziano F., Castleman W., Lemanske R.F., Jr. Contribution of upper airways to antigen-induced late airway obstructive response in guinea pigs. Am. Rev. Respir. Dis. 1990;142:138–142. doi: 10.1164/ajrccm/142.1.138. [DOI] [PubMed] [Google Scholar]
- Johnston S.L., Pattemore P.K., Sanderson G., Smith S., Lampe F., Josephs L., Symington P., Otoole S., Myint S.H., Tyrrell D.A.J., Holgate S.T. Community study of role of viral-infections in exacerbations of asthma in 9–11 year-old children. BMJ. 1995;310:1225–1229. doi: 10.1136/bmj.310.6989.1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juniper E.F., Frith P.A., Hargreave F.E. Airway responsiveness to histamine and methacholine: Relationship to minimum treatment to control symptoms of asthma. Thorax. 1982;37:288–291. doi: 10.1136/thx.36.8.575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juniper E.F., Frith P.A., Dunnett C., Cockcroft D.W., Hargreave F.E. Reproducibility and comparison of responses to inhaled histamine and methacholine. Thorax. 1992;89:1111–1119. doi: 10.1136/thx.33.6.705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kips J.C., Brusselle G.J., Joos G.F., Peleman R.A., Tavernier J.H., Devos R.R. Interleukin-12 inhibits antigen-induced airway hyperresponsiveness in mice. Am. J. Respir. Crit. Care Med. 1996;153:535–539. doi: 10.1164/ajrccm.153.2.8564093. [DOI] [PubMed] [Google Scholar]
- Kleeberger S.R., Bassett D.J., Jakab G.J., Levitt R.C. A genetic model for evaluation of susceptibility to ozone-induced inflammation. Am. J. Physiol. 1990;258:L313–L320. doi: 10.1152/ajplung.1990.258.6.L313. [DOI] [PubMed] [Google Scholar]
- Kumar A., Sorkness R., Kaplan M.R., Lemanske R.F. Longitudinal-study of persistent airway abnormalities post viral bronchiolitis in rats. J. Allergy Clin. Immunol. 1995;95:204. [Google Scholar]
- Kung T.T., Crawley Y., Luo B., Young S., Kreutner W., Chapman R.W. Inhibition of pulmonary eosinophilia and airway hyperresponsiveness in allergic mice by rolipram: Involvement of endogenously released corticosterone and catecholamines. Br. J. Pharmacol. 2000;130:457–463. doi: 10.1038/sj.bjp.0703308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan M.L., Gomez A.D., Baluk P., Hashizume H., McDonald D.M. Airway vasculature after mycoplasma infection: Chronic leakiness and selective hypersensitivity to substance P. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001;280:L286–L297. doi: 10.1152/ajplung.2001.280.2.L286. [DOI] [PubMed] [Google Scholar]
- Lanes S., Stevenson J.S., Codias E., Hernandez A., Sielczak M.W., Wanner A., Abraham W.M. Indomethacin and FPL-57231 inhibit antigen-induced airway hyperresponsiveness in sheep. J. Appl. Physiol. 1986;61:864–872. doi: 10.1152/jappl.1986.61.3.864. [DOI] [PubMed] [Google Scholar]
- Lebrec H., Sarlo K., Burleson G.R. Effect of influenza virus infection on ovalbumin-specific IgE responses to inhaled antigen in the rat. J. Toxicol. Environ. Health. 1996;27:619–630. doi: 10.1080/009841096160664. [DOI] [PubMed] [Google Scholar]
- Lee L.Y., Bleecker E.R., Nadel J.A. Effect of ozone on bronchomotor response to inhaled histamine aerosol in dogs. J. Appl. Physiol. Resp. Env. Exc. Physiol. 1977;43:626–631. doi: 10.1152/jappl.1977.43.4.626. [DOI] [PubMed] [Google Scholar]
- Leigh R., Vethanayagam D., Yoshida M., Watson R.M., Rerecich T., Inman M.D., O'Byrne P.M. Effects of montelukast and budesonide on airway responses and airway inflammation in asthma. Am. J. Respir. Crit. Care Med. 2002;166:1212–1217. doi: 10.1164/rccm.200206-509OC. [DOI] [PubMed] [Google Scholar]
- Lemanske R.F., Jr., Dick E.C., Swenson C.A., Vrits R.F., Busse W.W. Rhinovirus upper respiratory infection increases airway hyperreactivity and late asthmatic reactions. J. Clin. Invest. 1989;83:1–10. doi: 10.1172/JCI113843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z., Daniel E.E., Lane C.G., Arnaout M.A., O'Byrne P.M. Effect of an anti-Mol MAb on ozone-induced airway inflammation and airway hyperresponsiveness in dogs. Am. J. Physiol. 1992;263:L723–L726. doi: 10.1152/ajplung.1992.263.6.L723. [DOI] [PubMed] [Google Scholar]
- Lindsey J.R., Cassell H. Experimental Mycoplasma pulmonis infection in pathogen-free mice. Models for studying mycoplasmosis of the respiratory tract. Am. J. Pathol. 1973;72:63–90. [PMC free article] [PubMed] [Google Scholar]
- Lindsey J.R., Baker H.J., Overcash R.G., Cassell H., Hunt C.E. Murine chronic respiratory disease. Significance as a research complication and experimental production with Mycoplasma pulmonis. Am. J. Pathol. 1971;64:675–708. [PMC free article] [PubMed] [Google Scholar]
- Manning P.J., Stevens W.H., Cockcroft D.W., O'Byrne P.M. The role of thromboxane in allergen-induced asthmatic responses. Eur. Respir. J. 1991;4:667–672. [PubMed] [Google Scholar]
- Matsumura Y. The effects of ozone, nitrogen dioxide, and sulfur dioxide on the experimentally induced allergic respiratory disorder in guinea pigs. Am. Rev. Respir. Dis. 1970;102:438–443. doi: 10.1164/arrd.1970.102.3.438. [DOI] [PubMed] [Google Scholar]
- McDonald D.M. Angiogenesis and remodeling of airway vasculature in chronic inflammation. Am. J. Respir. Crit. Care Med. 2001;164:S39–S45. doi: 10.1164/ajrccm.164.supplement_2.2106065. [DOI] [PubMed] [Google Scholar]
- McDonald D.M., Schoeb T.R., Lindsey J.R. Mycoplasma pulmonis infections cause long-lasting potentiation of neurogenic inflammation in the respiratory tract of the rat. J. Clin. Invest. 1991;87:787–799. doi: 10.1172/JCI115082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh J.C., Simecka J.W., Ross S.E., Davis J.K., Miller E.J., Cassel G.H. Infection-induced airway fibrosis in two rat strains with differential susceptibility. Infect. Immun. 1992;60:2936–2942. doi: 10.1128/iai.60.7.2936-2942.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehlhop P.D., van de Rijn M., Brewer J.P., Kisselgof A.B., Geha R.S., Oettgen H.C., Martin T.R. CD40L, but not CD40, is required for allergen-induced bronchial hyperresponsiveness in mice. Am. J. Respir. Cell Mol. Biol. 2000;23:646–651. doi: 10.1165/ajrcmb.23.5.3954. [DOI] [PubMed] [Google Scholar]
- Menzel D.B. The role of free radicals in the toxicity of air pollutants (nitrogen oxides and ozone) In: Pryor W.A., editor. Free Radicals in Biology. Academic Press; New York: 1976. pp. 181–202. [Google Scholar]
- Mok J.Y., Waugh P.R., Simpson H. Mycoplasma pneumonia infection. A follow-up study of 50 children with respiratory illness. Arch. Dis. Child. 1979;54:506–511. doi: 10.1136/adc.54.7.506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molfino N.A., Wright S.C., Katz I., Tarlo S., Silverman F., McClean P.A., Szalai J.P., Raizenne M., Slutsky A.S., Zamel N. Effect of low concentrations of ozone on inhaled allergen responses in asthmatic subjects. Med. J. Aust. 1991;338:199–203. doi: 10.1016/0140-6736(91)90346-q. [DOI] [PubMed] [Google Scholar]
- Murlas C.G., Roum J.H. Bronchial hyperactivity occurs in steroid-treated guinea-pigs depleted of leukocytes by cyclophosphamide. J. Appl. Physiol. 1985;58:1630–1637. doi: 10.1152/jappl.1985.58.5.1630. [DOI] [PubMed] [Google Scholar]
- Murlas C.G., Roum J.H. Sequence of pathologic changes in the airway mucosa of guinea pigs during ozone-induced bronchial hyperreactivity. Am. Rev. Respir. Dis. 1985;131:314–320. doi: 10.1164/arrd.1985.131.3.314. [DOI] [PubMed] [Google Scholar]
- Murphy K.R., Wilson M.C., Irvin C.G., Glezen L.S., Marsh W.R., Haslett C., Henson P.M., Larsen G.L. The requirement for polymorphonuclear leukocytes in the late asthmatic response and heightened airways reactivity in an animal model. Am. Rev. Respir. Dis. 1986;134:62–68. doi: 10.1164/arrd.1986.134.1.62. [DOI] [PubMed] [Google Scholar]
- Mustafa M.G., Elsayed N.M., Graham J.A., Gardner D.E. Effects of ozone exposure on lung metabolism, influence of animal age, species, and exposure conditions. In: Lee S.D., Mustafa M.G., Mehlman M.A., editors. The Biomedical Effects of Ozone and Related Photochemical Oxidants. Princeton Scientific Publishers; 1983. pp. 57–73. [Google Scholar]
- Noma T., Mori A., Yoshizawa I. Induction of allergen-specific IL-2 responsiveness of lymphocytes after respiratory syncytial virus infection and prediction of onset of recurrent wheezing and bronchial asthma. J. Allergy Clin. Immunol. 1996;98:816–826. doi: 10.1016/s0091-6749(96)70131-8. [DOI] [PubMed] [Google Scholar]
- O'Byrne P.M. Allergen-induced airway hyperresponsiveness. J. Allergy Clin. Immunol. 1988;81:119–127. doi: 10.1016/0091-6749(88)90230-8. [DOI] [PubMed] [Google Scholar]
- O'Byrne P.M., Ryan G., Morris M., McCormack D., Jones N.L., Morse J.L., Hargreave F.E. Asthma induced by cold air and its relation to nonspecific bronchial responsiveness to methacholine. Am. Rev. Respir. Dis. 1982;125:281–285. doi: 10.1164/arrd.1982.125.3.281. [DOI] [PubMed] [Google Scholar]
- O'Byrne P.M., Walters E.H., Gold B.D., Aizawa H.A., Fabbri L.M., Alpert S.E., Nadel J.A., Holtzman M.J. Neutrophil depletion inhibits airway hyperresponsiveness induced by ozone exposure. Am. Rev. Respir. Dis. 1984;130:214–219. doi: 10.1164/arrd.1984.130.2.214. [DOI] [PubMed] [Google Scholar]
- Oh J.W., Seroogy C.M., Meyer E.H., Akbari O., Berry G., Fathman C.G., DeKruyff R.H., Umetsu D.T. CD4 T-helper cells engineered to produce IL-10 prevent allergen-induced airway hyperreactivity and inflammation. J. Allergy Clin. Immunol. 2002;110:460–468. doi: 10.1067/mai.2002.127512. [DOI] [PubMed] [Google Scholar]
- Osebold J.W., Gershwin L.J., Zee Y.C. Studies on the enhancement of allergic lung sensitization by inhalation of ozone and sulfuric acid aerosol. J. Environ. Pathol. Toxicol. Oncol. 1980;3:221–234. [PubMed] [Google Scholar]
- Oshiba A., Hamelmann E., Takeda K., Bradley K.L., Loader J.E., Larsen G.L., Gelfand E.W. Passive transfer of immediate hypersensitivity and airway hyperresponsiveness by allergen-specific immunoglobulin (Ig)E and IgG1 in mice. J. Clin. Invest. 1996;97:1398–1408. doi: 10.1172/JCI118560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattemore P.K., Johnston S.L., Bardin P.G. Viruses as precipitants of asthma symptoms. I. Epidemiology. Clin. Exp. Allergy. 1992;22:325–336. doi: 10.1111/j.1365-2222.1992.tb03094.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phipps R.J., Denas S.M., Sielczak M.W., Wanner A. Effects of 0.5 ppm ozone on glycoprotein secretion, ion and water fluxes in sheep trachea. J. Appl. Physiol. 1986;60:918–927. doi: 10.1152/jappl.1986.60.3.918. [DOI] [PubMed] [Google Scholar]
- Piedimonte G., McDonald D.M., Nadel J.A. Glucocorticoids inhibit neurogenic plasma extravasation and prevent virus-potentiated extravasation in the rat trachea. J. Clin. Invest. 1990;86:1409–1415. doi: 10.1172/JCI114855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piedimonte G., Nadel J.A., Umeno E., McDonald D.M. Sendai virus infection potentiates neurogenic inflammation in the rat trachea. J. Appl. Physiol. 1990;68:754–760. doi: 10.1152/jappl.1990.68.2.754. [DOI] [PubMed] [Google Scholar]
- Ramsdale E.H., Morris M.M., Roberts R.S., Hargreave F.E. Asymptomatic bronchial hyperresponsiveness in rhinitis. J. Allergy Clin. Immunol. 1985;75:573–577. doi: 10.1016/0091-6749(85)90032-6. [DOI] [PubMed] [Google Scholar]
- Richards I.M., Kolbasa K.P., Winterrowd G.E., Hatfield C.A., Vonderfecht S.L., Fidler S.F., Griffin R.L., Brashler J.R., Krzesicki R.F., Lane C.L., Anderson D.C., Sly L.M., Staite N.D., Chin J.E. Role of intercellular adhesion molecule-1 in antigen-induced lung inflammation in brown Norway rats. Am. J. Physiol. 1996;271:L267–L276. doi: 10.1152/ajplung.1996.271.2.L267. [DOI] [PubMed] [Google Scholar]
- Roquet A., Dahlen B., Kumlin M., Ihre E., Aanstren G., Binks S., Dahlen S.E. Combined antagonist of leukotrienes and histamine produces predominent inhibition of allergen-induced early and late phase airway obstruction in asthmatics. Am. J. Respir. Crit. Care Med. 1997;155:1856–1863. doi: 10.1164/ajrccm.155.6.9196086. [DOI] [PubMed] [Google Scholar]
- Sasaki H., Yanai M., Shimura S., Okayama H., Aikawa T., Sasaki T., Takishima T. Late asthmatic response to ascaris antigen challenge in dogs treated with metyrapone. Am. Rev. Respir. Dis. 1987;136:1459–1465. doi: 10.1164/ajrccm/136.6.1459. [DOI] [PubMed] [Google Scholar]
- Schelegle E.S., Gunther R.A., Parsons G.H., Colbert S.R., Yousef M.A., Cross C.E. Acute ozone exposure increases bronchial blood flow in conscious sheep. Respir. Physiol. 1990;82:325–335. doi: 10.1016/0034-5687(90)90102-5. [DOI] [PubMed] [Google Scholar]
- Schoeb T.R., Kervin K.C., Lindsey J.R. Exacerbation of murine respiratory mycoplasmosis in gnotobiotic F344/N rats by Sendai virus infection. Vet. Pathol. 1985;22:272–282. doi: 10.1177/030098588502200310. [DOI] [PubMed] [Google Scholar]
- Schultheis A.H., Bassett D.J. Inflammatory cell influx into ozone-exposed guinea pig lung interstitial and airways spaces. Agents Actions. 1991;34:270–273. doi: 10.1007/BF01993300. [DOI] [PubMed] [Google Scholar]
- Schwarze J., Cieslewicz G., Hamelmann E., Joetham A., Shultz L.D., Lamers M.C., Gelfand E.W. IL-5 and eosinophils are essential for the development of airway hyperresponsiveness following acute respiratory syncytial virus infection. J. Immunol. 1999;162:2997–3004. [PubMed] [Google Scholar]
- Schwarze J., Hamelmann E., Bradley K.L., Takeda K., Gelfand E.W. Respiratory syncytial virus infection results in airway hyperresponsiveness and enhanced airway sensitization to allergen. J. Clin. Invest. 1997;100:226–233. doi: 10.1172/JCI119516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shampain M.P., Behrens B.L., Larsen G.L., Henson P.M. An animal model of late pulmonary responses to Alternaria challenge. Am. Rev. Respir. Dis. 1982;126:493–498. doi: 10.1164/arrd.1982.126.3.493. [DOI] [PubMed] [Google Scholar]
- Shore S.A., Schwartzman I.N., Le Blanc B., Murthy G.G.K., Doerschuk C.M. Tumor necrosis factor receptor 2 contributes to ozone-induced airway hyperresponsiveness in mice. Am. J. Respir. Crit. Care Med. 2001;164:602–607. doi: 10.1164/ajrccm.164.4.2001016. [DOI] [PubMed] [Google Scholar]
- Soler M., Sielczak M., Abraham W.M. A bradykininantagonist blocks antigen-induced airway hyperresponsiveness and inflammation in sheep. Pulm. Pharmacol. 1990;3:9–15. doi: 10.1016/0952-0600(90)90003-2. [DOI] [PubMed] [Google Scholar]
- Sorden S.D., Castleman W.L. Virus-induced increases in airway mast cells in brown Norway rats are associated with enhanced pulmonary viral replication and persisting lymphocytic infiltration. Exp. Lung Res. 1995;21:197–213. doi: 10.3109/01902149509068827. [DOI] [PubMed] [Google Scholar]
- Sorden S.D., Castleman W.L. Virus-induced increases in bronchiolar mast cells in brown Norway rats are associated with both local mast cell proliferation and increases in blood mast cell precursors. Lab. Invest. 1995;73:197–204. [PubMed] [Google Scholar]
- Sorkness R., Lemanske R.F. Attenuation of airway hyperresponsiveness during acute viral infection using the 21-aminosteroid U-83836E in rats. Pulm. Pharmacol. Ther. 1996;9:219–222. doi: 10.1006/pulp.1996.0027. [DOI] [PubMed] [Google Scholar]
- Tanaka H., Masuda T., Tokuoka S., Komai M., Nagao K., Takahashi Y., Nagai H. The effect of allergen-induced airway inflammation on airway remodeling in a murine model of allergic asthma. Inflamm. Res. 2001;50:616–624. doi: 10.1007/PL00000243. [DOI] [PubMed] [Google Scholar]
- Taylor I.K., O'Shaughnessy K.M., Fuller R.W., Dollery C.T. Effect of a cysteinylleukotriene receptor antagonist, ICI 204–219 on allergen-induced bronchoconstriction and airway hyperactivity in atopic subjects. Lancet. 1991;337:690–694. doi: 10.1016/0140-6736(91)90277-v. [DOI] [PubMed] [Google Scholar]
- Temelkovski J., Hogan S.P., Shepherd D.P., Foster P.S., Kumar R.K. An improved murine model of asthma: Selective airway inflammation, epithelial lesions and increased methacholine responsiveness following chronic exposure to aerosolised allergen. Thorax. 1998;53:849–856. doi: 10.1136/thx.53.10.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomioka K., Garrido R., Ahmed A., Stevenson J.S., Abraham W.M. YM461, a PAF antagonist, blocks antigen-induced late airway responses and airway hyperresponsiveness in allergic sheep. Eur. J. Pharmacol. 1989;170:209–215. doi: 10.1016/0014-2999(89)90541-4. [DOI] [PubMed] [Google Scholar]
- Uhl E.W., Castleman W.L., Sorkness R.L., Busse W.W., Lemanske R.F., McAllister P.K. Parainfluenza virus-induced persistence of airway inflammation, fibrosis, and dysfunction associated with TGF-beta(1), expression in brown Norway rats. Am. J. Respir. Crit. Care Med. 1996;154:1834–1842. doi: 10.1164/ajrccm.154.6.8970378. [DOI] [PubMed] [Google Scholar]
- Vitalis T.Z., Keicho N., Itabashi S., Hayashi S., Hogg J.C. A model of latent adenovirus 5 infection in the guinea pig (Cavia porcellus) Am. J. Respir. Cell. Mol. Biol. 1996;14:225–231. doi: 10.1165/ajrcmb.14.3.8845172. [DOI] [PubMed] [Google Scholar]
- Walter D.M., Wong C.P., DeKruyff R.H., Berry G.J., Levy S., Umetsu D.T. IL-18 gene transfer by adenovirus prevents the development of and reverses established allergen-induced airway hyperreactivity. J. Immunol. 2001;166:6392–6398. doi: 10.4049/jimmunol.166.10.6392. [DOI] [PubMed] [Google Scholar]
- Wang C.G., Dimaria G., Bates J.H.T., Guttmann R.D., Martin J.G. Methacholine-induced airway reactivity of inbred rats. J. Appl. Physiol. 1986;61:2180–2186. doi: 10.1152/jappl.1986.61.6.2180. [DOI] [PubMed] [Google Scholar]
- Wang J.M., Homer R.J., Chen Q.S., Elias J.A. Endogenous and exogenous IL-6 inhibit aeroallergen-induced Th2 inflammation. J. Immunol. 2000;165:4051–4061. doi: 10.4049/jimmunol.165.7.4051. [DOI] [PubMed] [Google Scholar]
- Wegner C.D., Gundel R.H., Reilly P., Haynes N., Letts L.G., Rothlein R. Intercellular adhesion molecule-1 (ICAM-1) in the pathogenesis of asthma. Science. 1990;247:456–459. doi: 10.1126/science.1967851. [DOI] [PubMed] [Google Scholar]
- Wood L.J., Inman M.D., Denburg J.A., O'Byrne P.M. Allergen-induced increases in cell trafficking between bone marrow and lung. Am. J. Respir. Cell Mol. Biol. 1998;18:759–767. doi: 10.1165/ajrcmb.18.6.3006. [DOI] [PubMed] [Google Scholar]
- Woolcock A.J., Salome C.M., Yan K. The shape of the dose-response curve to histamine in asthmatic and normal subjects. Am. Rev. Respir. Dis. 1984;130:71–75. doi: 10.1164/arrd.1984.130.1.71. [DOI] [PubMed] [Google Scholar]
- Woolley M.J., Denburg J.A., Ellis R., Dahlback M., O'Byrne P.M. Allergen-induced changes in bone marrow progenitors and airway responsiveness in dogs and the effect of inhaled budesonide on these parameters. Am. J. Respir. Cell Mol. Biol. 1994;11:600–606. doi: 10.1165/ajrcmb.11.5.7946389. [DOI] [PubMed] [Google Scholar]
- Woolley M.J., Lane C.G., Ellis R., Stevens W.H., Woolley K.L., O'Byrne P.M. Role of eosinophils in the development of allergen-induced airway hyperresponsiveness in dogs. Am. J. Respir. Crit. Care Med. 1995;152:1508–1512. doi: 10.1164/ajrccm.152.5.7582285. [DOI] [PubMed] [Google Scholar]
- Yanai M., Ohrui T., Aikawa T., Okayama H., Sekizawa K., Maeyama K., Sasaki H., Takishima T. Ozone increases susceptibility to antigen inhalation in allergic dogs. J. Appl. Physiol. 1990;68:2267–2273. doi: 10.1152/jappl.1990.68.6.2267. [DOI] [PubMed] [Google Scholar]
- Yokoyama E., Goto H., Kawai K., Kyono H. Mechanical properties of rabbit lung with edema caused by exposure to ozone. J. Environ. Pathol. Toxicol. Oncol. 1989;9:95–108. [PubMed] [Google Scholar]
- Yoshida M., Leigh R., Matsumoto K., Wattie J., Ellis R., O'Byrne P.M., Inman M.D. Effect of interferon-gamma on allergic airway responses in interferon-gamma-deficient mice. Am. J. Respir. Crit. Care Med. 2002;166:451–456. doi: 10.1164/rccm.200202-095OC. [DOI] [PubMed] [Google Scholar]