

Abstract
The orthomyxoviruses include influenza viruses, thogotoviruses, and isaviruses. Influenza A viruses circulate in reservoir of wild aquatic birds, occasionally cause human pandemics, undergo antigenic drift and antigentic shift, and have been used as model viruses to understand many fundamental processes including receptor binding, membrane fusion, protein structure, and adaptive immunity. Influenza A and B viruses contain a hemagglutinin (HA) surface glycoprotein, the dominant surface antigen that promotes receptor binding and membrane fusion during viral entry, and a neuraminidase (NA) glycoprotein, a secondary antigen that has receptor-destroying activity necessary for virus release from infected cells. Influenza C viruses contain a single antigen, the hemagglutinin-esterase-fusion (HEF) protein, which possesses receptor binding, membrane fusion, and virus release functions. The HA and HEF proteins are prototypic, trimeric, structural class I viral fusion proteins with a membrane-distal, immunodominant (and highly variable) receptor-binding head domain and a more highly conserved membrane-proximal, metastable, stalk domain that catalyzes membrane fusion. The tetrameric NA protein has a membrane-distal globular head domain with an antiparallel β-propeller topology and a membrane-proximal coiled-coil stalk. For the influenza A and B viruses, the HA protein is the predominant target for naturally acquired and vaccine-induced adaptive immunity. Recent structural studies of broadly reactive monoclonal antibodies bound to the HA protein, along with complementary immunological experiments, suggest ways to generate “universal” influenza vaccines that are effective after antigenic drift and shift.
Keywords: Immune response, Influenza virus, Membrane fusion, Membrane protein, Protein structure, Receptor binding, Virus entry
Introduction
The family Orthomyxoviridae includes enveloped viruses that have a negative-sense, single-stranded, and segmented RNA genome. There are five different genera in the family: Influenzavirus A, B, and C, Thogotovirus, and Isavirus. Influenza viruses are classified into three types: A, B, and C. Type A influenza viruses are further classified into subtypes based on the antigenic properties of their surface antigens, hemagglutinin (HA) and neuraminidase (NA). There are 18 known HA subtypes (H1–H18) and 11 known NA subtypes (N1–N11) of influenza A. Viruses containing HA proteins from each of the first 16 HA subtypes and NAs of the first 9 NA subtypes, in different combinations, are maintained in a reservoir of wild aquatic birds. Bat-derived H17N10 and H18N11 subtypes have also been identified.
Avian influenza A viruses cause sporadic outbreaks in domestic poultry, swine, and other species. Rarely, an influenza A virus acquires the molecular properties necessary to spread efficiently in a new host species, causing a pandemic.
Human viruses that caused pandemics in the last 100 years were of the combination H1N1 in 1918, H2N2 in 1957, H3N2 in 1968, and H1N1 in 2009. Viruses that are currently circulating are of the pandemic 2009 (p2009) H1N1 and H3N2 influenza A virus combinations and influenza B virus. Recent sporadic infections in humans that have not yet caused a pandemic include influenza A viruses of H5, H7, and H9 subtypes. H1 and H3 subtype viruses are endemic in domestic pigs, which have also suffered limited outbreaks by H2, H4, H5, H7, and H9 subtypes. H7 and H3 viruses have caused outbreaks in horses, and related H3 viruses have caused outbreaks in dogs. Influenza A virus infections have also been reported in cats and marine mammals.
Different influenza virus strains are named according to their type, the species from which the virus was isolated (omitted if human), place of virus isolation, the number of the isolate, the year of isolation, and HA and NA subtypes for influenza A viruses. For example, the 282nd isolate of an H5N1 subtype virus from chickens in Hong Kong in 2006 is designated A/chicken/Hong Kong/282/2006 (H5N1). The fourth isolate of a p2009 H1N1 influenza A virus isolated from a human in California at the start of the 2009 pandemic is designated A/California/04/2009 (H1N1).
The genus Thogotovirus contains two different species, Dhori virus and Thogoto virus. Viruses from both these species were isolated from ticks, and therefore are different from influenza viruses with respect to their host range. The genus Isavirus, with the type species Infectious salmon anemia virus, is also distinct from influenza viruses A, B, and C, although numerous studies identify these isolates as members of the family Orthomyxoviridae.
The majority of the research into the family Orthomyxoviridae of viruses has been concerned with influenza viruses and, therefore, this article will focus exclusively on the structures of the influenza viral antigens.
Influenza Virus Surface Glycoproteins
The surface of type A and B influenza viruses has two major surface glycoproteins, HA and NA, whereas influenza C viruses have a single glycoprotein, hemagglutinin–esterase-fusion (HEF). Antibodies are raised to both HA and NA, although HA elicits the dominant antigenic response. Natural variation in HA amino acid sequence due to selective pressure over time results in antigenic drift, limiting the long-term effectiveness of anti-influenza vaccines. Consequently, annual updates to the vaccine composition are required to maintain a match to the viruses that are currently circulating. Recent vaccine engineering studies seek to reduce or eliminate the need to update influenza vaccines by stimulating “universal” antibody responses to conserved structural elements of the HA protein that are far less susceptible to sequence drift than the immunodominant receptor-binding domain head.
Influenza A and B Hemagglutinin Proteins
In type A and B influenza viruses, the HA protein mediates attachment of the virus to the host cell via binding to sialic acid residues on the termini of cell surface glycoproteins. After receptor binding, the virions are internalized by endocytosis. After exposure to low pH, HA mediates the fusion of viral and endosomal membranes so that the genome can be uncoated and delivered into the cytoplasm, and later the nucleus.
HA is a type I integral membrane protein that is synthesized as a single-chain polypeptide (HA0) with ectodomain, transmembrane, and cytoplasmic tail domains of approximately 513, 27, and 11 residues, respectively. In order to be capable of mediating membrane fusion, HA0 is subsequently “primed” by proteolytic cleavage of the ectodomain into two chains, HA1 and HA2. An interchain disulfide bond exists between cys-14 (HA1) and cys-137 (HA2). The monomers then associate non-covalently to form the functional trimer. The highly conserved and hydrophobic N-terminal residues of HA2, known as the fusion peptide (see below), are buried from solvent upon cleavage. It is known that these residues insert into the host membrane to facilitate virus-cell membrane fusion. The C-terminal end of HA2 has a transmembrane anchor that tethers the molecule to the viral membrane and a short cytoplasmic tail. These C-terminal domains have roles in HA protein localization on the cell surface and, subsequently, influenza virus assembly and budding.
The number of glycosylation sites in the HA molecule varies between virus species and has a marked impact on antigenicity (see below). HA proteins from avian hosts tend to have a limited number of glycosylation sites. As human pandemic viruses circulate in humans, antigenic drift tends to result in increased N-linked glycosylation, which assists antibody escape. For example, the H1 HA protein from the virus that caused the pandemic of 1918 has five glycosylation sites in HA1 and one in HA2. H1 HA proteins isolated from viruses circulating in 2002 had accumulated an additional six potential glycosylation sites with respect to the 1918 H1 HA protein. In addition to their influence on the antigenicity, glycosylation sites that are conserved across subtypes play a role in the co-translational folding of the HA protein via binding to host cell chaperones.
X-ray crystallographic studies have elucidated high-resolution structures of the HA ectodomains of numerous subtypes in uncleaved, prefusion, intermediate, and postfusion forms. Such constructs have lacked the transmembrane and cytoplasmic tail domains to promote solubility and crystal packing. Traditionally, HA ectodomain constructs were purified after cleavage from the surface of the virus. More recently, recombinant HA ectodomain constructs have been expressed by baculovirus vectors in insect cells. Additionally, x-ray crystal structures have been determined for E. coli-expressed constructs of the head domain in its neutral-pH form and the stalk domain in its postfusion form.
The first crystal structure of an HA ectodomain to be determined was from the A/Aichi/2/68 (H3N2) virus. Because of this, it is customary to refer to amino acids of all HA subtypes using “H3 numbering”. The structure of the H3N2 HA trimer in its uncleaved, prefusion state showed it to be composed of a membrane-proximal stalk and a globular multi-domain head. The stalk has at its core a triple-stranded α-helical coiled coil composed by HA2 residues and buttressing segments composed of HA1 and HA2 residues (Figure 1(A) ). The membrane-distal heads of each monomer can be subdivided into a vestigial esterase domain and a receptor-binding domain, the latter located at the very tip of each monomer. The receptor-binding domain consists of three secondary structure elements – the 190 helix (residues 190–198), the 130 loop (residues 135–138), and the 220 loop (residues 221–228) – that form the sides of each receptor binding site, with the base made up of the conserved residues Tyr-98, Trp-153, His-183, and Tyr-195.
Figure 1.

(A) Cartoon representation of influenza A virus HA protein. The receptor-binding domain of HA1 is colored blue and the vestigial esterase domain green. HA2, which forms the central α-helical stem, is colored red. (B) Cartoon representation of influenza C virus HEF protein. The receptor-binding domain of HEF1 is colored blue and the esterase domain green. HEF2, which forms the central a-helical stem, is colored red.
The HA protein mediates the first stage of virus infection via binding to terminal sialic acid sugars on host glycoproteins and glycolipids. Sialic acids are usually found in either α-2,3- or α-2,6-linkages to galactose, the predominant penultimate sugar of N-linked carbohydrate side chains. The binding preference of an HA protein for one of these linkage types correlates with the species that the virus infects. The avian enteric tract has predominantly sialic acid in the α-2,3- linkage, and all HA proteins found in the first 16 antigenic subtypes found in avian influenza viruses bind preferentially to this linkage. In cells of the human upper respiratory tract, however, sialic acid in the α-2,6-linkage predominates. Indeed, the HA proteins from human influenza viruses display a binding preference for sialic acid in the α-2,6-linkage. An avian origin has been proposed for human influenza viruses and, therefore, a change in binding specificity is required for cross-species transfer. While the precise details of the amino acid changes underlying this switch in specificity seem to be HA subtype dependent, it has been shown for both H1 and H3 HA proteins that only a small number of mutations are necessary. For example, a single mutation in H3 HA of Gln-226 to Leu-226 causes a switch in specificity from α-2,3- to α-2,6-binding preference. Insights into the amino acids involved in avian and human receptor binding have also been gained from elucidating the structures of HA proteins in complex with receptor analogs. The HA proteins from bat H17 and H18 subtypes have a distorted receptor binding pocket, do not bind the canonical glycan receptors for either humans or avians, and presumably have a novel receptor that has not yet been defined.
The structure of HA0 has also been elucidated via crystallization of a protein in which Arg-329 had been mutated to a glutamine to prevent proteolytic cleavage of the polypeptide. The structure revealed that the cleavage site is located in a prominent surface loop, which lies adjacent to a cavity not present in the cleaved-HA structure. There are three ionizable residues in this cavity that are buried from solvent after cleavage due to the burial of the nearly formed N-terminus of HA2 (fusion peptide). Thus, it has been proposed that cleavage of HA0 results in a metastable form of the protein in which a low-pH trigger has been set due to the burial of ionizable residues. A characteristic of highly pathogenic influenza viruses such as the currently circulating H5N1 viruses is an insertion of a series of basic residues adjacent to the cleavage site. This polybasic insertion would create a much more extended surface loop that is recognized by intracellular proteases. Facile intracellular cleavage allows expanded tropism, and often systemic influenza virus spread, thereby promoting enhanced pathogenicity.
In addition to receptor binding, the HA protein mediates the fusion of the viral envelope and the host cell membrane. After receptor binding, the influenza virus is internalized via endocytosis, exposing the virus to a low-pH environment that triggers a dramatic and irreversible conformational change in the HA protein. Exposure of the HA protein to low pH causes an alteration in the protonation state of residues that are buried by the fusion peptide, resulting in its removal from its buried site. Subsequently, the middle of the original long α-helix of HA2 unfolds to form a reverse turn, jack-knifing the C-terminal half backward toward the N-terminus. This results in HA2 adopting a rod-like coiled-coil structure. HA1, which was missing from the crystal structure, de-trimerizes and swings away from the HA2 but the interchain disulfide bond is maintained. As a consequence of these molecular rearrangements, the N-terminal fusion peptide, which inserts into the host membrane, and the C-terminal transmembrane anchor of HA2, which is tethered to the viral envelope, are placed at the same end of the HA molecule, thereby facilitating membrane fusion (Figure 2 ).
Figure 2.
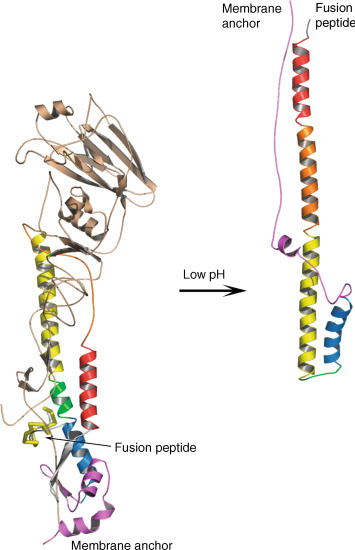
Schematic representation of the molecular rearrangements of the HA protein upon exposure to low pH. The location of the fusion peptide and membrane anchor is shown in both structures. HA1 is colored in gold and HA2 is colored to highlight equivalent regions of the polypeptide. Note that only the yellow part of HA2 adopts the same position and conformation in both forms.
The structure adopted by HA2 at the pH of membrane fusion shares a number of features with equivalent parts of other Class I membrane fusion proteins including those of human immunodeficiency virus (HIV), paramyxoviruses, coronaviruses, and Ebola virus. In each case, the molecules contain central triple-stranded coiled coils that are surrounded by three chains antiparallel to the core helices. As a consequence, the fusion peptide and membrane anchor region of each of the proteins from different virus families are at the same end of a rod-shaped molecule. Evidence suggests that part of the energy required to do the work of membrane fusion is provided by the formation of this hairpin structure.
X-ray crystal structures of the prefusion form have been determined for the influenza B virus HA ectodomain bound to α-2,3- and α-2,6-linked sialic acids. Despite HA1 and HA2 sequence identities of approximately 20% and 30%, respectively, between the HA proteins of influenza A and B, the overall folds of the proteins are similar. Nevertheless, the A and B virus HA proteins have subtle structural differences in their receptor binding pockets.
Influenza C HEF Protein
In contrast to the HA protein of influenza A and B viruses, the major glycoprotein, HEF, of the C viruses has a receptor-destroying activity, as well as receptor-binding and fusion activities. As a consequence, influenza C viruses lack the NA glycoprotein. Despite sharing only 12% sequence identity with influenza A HA proteins, both HEF and HA are structurally similar (Figure 1). HEF has a membrane-proximal, α-helical stem-like structure and a membrane-distal globular multidomain structure. The receptor-binding domains of both HEF and HA are structurally similar despite HEF binding 9-O-acetyl sialic acid rather than sialic acid. The receptor-destroying domain shows structural homology to bacterial esterases, in keeping with its activity as a 9-O-acetylesterase. The stem region of the HEF protein is similar to that observed in the HA protein except that the triple-stranded, α-helical bundle diverges at both its ends and the fusion peptide is partially exposed to solvent. The receptor-binding domain is inserted into a surface loop of the esterase domain, which itself is inserted into a surface loop of the stem. Thus, all three functions of the HEF are segregated into structurally distinct domains.
Influenza A and B Neuraminidase Proteins
After virus replication, the NA protein removes sialic acid from viral surface and cellular glycoproteins to facilitate virus release and the spread of infection to new cells or a new host. Without the ability to remove receptors displayed on virions and infected cells, viral aggregation would occur. Thus, NA is commonly known as a receptor-destroying enzyme.
The NA protein is synthesized as a single polypeptide chain that does not get cleaved after translation. NA is a Type II integral membrane protein with a short, highly conserved cytoplasmic tail and a hydrophobic transmembrane region that provides the anchor for the stalk and the head domains. Like HA, NA is a highly N-glycosylated protein and serves to influence antigenicity. As with HA, NA ectodomain constructs suitable for structural studies are made via proteolytic cleavage from the virus surface or by baculovirus expression in insect cells.
The first crystal structure of the head domain of the NA protein was of an N2 subtype, thus an N2 numbering scheme is often used even for other subtypes. The N2 NA structure showed that the molecule was homotetrameric with circular fourfold symmetry. Each monomer is composed of six topologically identical four-stranded antiparallel β-sheets that are themselves arranged like the blades of a propeller (Figure 3 ).
Figure 3.

Cartoon representation of the influenza A virus NA protein. Each monomer of the tetramer is colored differently, with the monomer at the top-left colored to emphasize the six-bladed β-propeller structure. Oseltamivir is bound to the active site and shown in stick representation and colored blue.
Sialic acid, the product of catalysis, binds in a deep pocket on the surface of the molecule, roughly in the middle of each monomer. No conformational changes occur upon binding of sialic acid. The amino acids in the active site that interact with sialic acid are highly conserved across N1–N9 NA subtypes. The active sites of the N1–N9 NA proteins contain three arginine residues, Arg 118, Arg 292, and Arg 371, that bind the carboxylate of the substrate sialic acid; Arg 152 that interacts with the acetamido substituent of the substrate; and Glu 276 that forms hydrogen bonds with the 8- and 9-hydroxyl groups of the substrate.
These properties of the active site made the NA protein an attractive target for structure-based drug design programs, and resulted in the synthesis of oseltamivir (Tamiflu) and zanamivr (Relenza), two clinically licensed anti-influenza drugs.
The crystal structure of the influenza B virus NA protein has also been elucidated and is essentially identical to the influenza A virus NA protein. The NA structures from both A and B types have the same homotetrameric arrangement and a high degree of conservation of the active site.
Influenza A Subtype Variation in Antigen Structure
Influenza A virus HA proteins belong to two major groups and several smaller clades based on structural and antigenic similarities. Group 1 HA proteins include H1, H2, H5, H6, H8, H9, H11, H12, H13, H16, H17, and H18 subtypes. Group 2 HA proteins include H3, H4, H7, H10, H14, and H15 subtypes. To date, a wide variety of x-ray crystal structures have been determined for ectodomain constructs of the HA proteins from both group 1 (including H1, H2, H5, H6, H9, H13, H16, and H17) and group 2 (including H3, H7, and H10). All of the HA proteins have the same molecular fold but HA proteins representative of each clade are distinguished by differences in the orientation of their membrane distal globular domains relative to the central trimeric coiled coil. For example, the receptor-binding domain of the H3 HA protein is rotated clockwise by 24 degree relative to the H1 HA protein. Also, the conformation of the interhelical loop of HA2 is different in the HA proteins from each clade. The clade-specific differences between the HA proteins are clustered in regions that undergo conformational changes in membrane fusion, for example near to the fusion peptide. Evidence suggests that differences in the stability of HA proteins to pH and temperature represent selection pressures in the evolution of influenza A HA proteins. In general, it has been observed that avian-adapted HA proteins are relatively unstable while mammalian-adapted HA proteins are more resistant to extracellular acid exposure, which occurs in the mammalian upper respiratory tract.
Influenza A NA proteins form three phylogenetic groups. Group 1 NA proteins include the avian-origin N1, N4, N5, and N8 subtypes. Group 2 NA proteins include the avian-origin N2, N3, N6, N7, and N9 subtypes. Group 3 NA proteins include the bat-derived N10 and N11 subtypes. High-resolution structures have been determined for NA proteins from each of the 11 known subtypes. All of the avian-origin N1–N9 NA proteins have the same homotetrameric conformation, but group-specific differences occur in the active site. Relative to group 2 NA proteins, a large cavity exists in group 1 NA proteins adjacent to the 4-amino group of oseltamivir. Upon binding of inhibitors to group 1 NA proteins, a conformational change of the 150-loop occurs that results in the active site of the two groups of NA being essentially identical. Chemical exploitation of this cavity is currently being undertaken in the development of a new generation of anti-influenza drugs to address the problem of mutations in the NA protein that cause resistance to oseltamivir and zanamivir. Just as the bat-derived H17 and H18 HA proteins do not bind sialic acid, their respective N10 and N11 NA proteins are not sialidases. The N10 and N11 NA proteins have an overall tetrameric structure similar to the N1–N9 NA proteins. Reflective of their lack of sialidase activity, the N10 and N11 NA proteins have active sites divergent, and more open, compared to the active sites of the sialidases comprising N1–N9 subtypes.
Antigenic Variation and Antibody Binding
Influenza viruses undergo a process known as “antigenic drift” whereby natural variants of the HA and NA proteins occur, due to the error-prone nature of the viral RNA polymerase. The HA protein is the major molecule recognized by the adaptive immune system of the host, although the NA protein does elicit an immune response. Upon infection of a cell, the virus causes an immune response that commonly results in the production of antibodies. In the case of the HA protein, neutralizing antibodies prevent the virus from binding to cells. While not preventing viral entry, antibodies against the NA protein prevent spread of the virus and afford some protection against challenge with the same or similar virus.
In the last 100 years, four viruses caused pandemics that contributed to millions of deaths. Following a pandemic, immune pressure causes antigenic variation that has the potential to continue until the fitness of the virus is compromised via deleterious mutations in the HA protein that affect its receptor-binding or membrane fusion properties.
Examination of the positions of amino acid substitutions of natural variants of HA proteins show that they are scattered throughout the molecule. Of the changes that are retained by circulating viruses, so-called “fixed” substitutions, the majority are located on the surface of the molecule. Whereas, in the case of H3 HA subtype viruses isolated between 1968 and 2005, two-thirds of the residues that are not retained are buried. This suggests that the “fixed” substitutions have been selected because they alter the local structure of the HA protein and prevent antibody binding.
This is supported by the fact that the location of amino acid substitutions in antigenic variants of HA proteins that have been selected by growing virus in the presence of anti-HA monoclonal antibodies (mAbs) map to the same location as the “fixed” substitutions. Thus sites of mAb-selected mutations indicate the sites at which selecting antibodies bind. These mutants escape neutralization by the selecting antibody. The antibodies nevertheless still bind, but with a much reduced affinity. There appears to be no preference in the type of amino acid change that occurs in selected variants.
Prevention of antibody recognition frequently occurs through the introduction of new glycosylation sites in the HA protein. Oligosaccharide attachment at these sites prevents recognition by masking the protein surface with oligosaccharides made by cellular enzymes. This serves to remove antibody binding sites and cause a portion of the molecule to be deemed by the immune system to be antigenically “self”.
Sites of amino acid substitutions that occur in natural and antibody-selected variants are predominantly on the surface of the membrane distal head of the HA protein, and commonly surround the receptor-binding site. This suggests that there is a link between the neutralization of viral infectivity and the prevention of virus binding to cells. Amino acid residues in the membrane-proximal stalk domain are more highly conserved, both within and across HA structural groups. Therefore, the stalk domain is the target antigen for “universal” influenza vaccines currently in development.
Crystal structures have been elucidated for HA and NA proteins in complex with antibodies (Figure 4 ). These structures have shown that fragment antigen binding (Fab) antibody fragments do indeed bind to different parts of the molecules. This supports the observation that natural variants are scattered throughout the molecules. Neutralization occurs by preventing sialic acid binding or by preventing conformational changes needed to cause membrane fusion.
Figure 4.
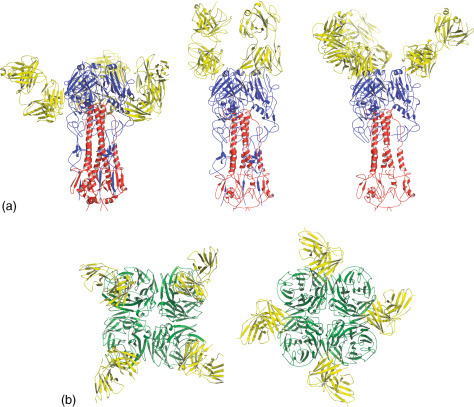
Antibody binding to influenza antigens. (A) Three different crystal structures of influenza HA proteins bound to Fab fragments. (B) Two different crystal structures of influenza NA proteins bound to Fab fragments.
Conclusion
Knowledge of the three-dimensional structures of the surface antigens of influenza viruses have had a major impact on the understanding of their role in the life cycle of influenza viruses. Insights into receptor binding, fusion mechanisms, and drug discovery have been gained. The study of these antigens from a range of influenza viruses has given insights into their structural evolution in relation to immune recognition.
Acknowledgment
Rupert Russell, the original author of a version of this reference module published in 2008, sadly died in 2012 while a Lecturer in Structural Virology at the University of St. Andrews. Minimal edits were made to this updated manuscript to honor the author of the original contribution and to honor its completeness.
Footnotes
Change History: November 2015. CJ Russell updated the “Introduction” section to include an explanation of influenza virus ecology, the 2009 H1N1 pandemic, and the emergence of H17 and H18 bat influenza viruses. Descriptions of a large number of high-resolution structures of HA and NA proteins from a wide variety of antigenic subtypes have been added. Recent advances on broadly reactive monoclonal antibodies and the pursuit of a universal influenza vaccine have been added. The references have been updated.
Further Reading
- Gamblin S.J., Skehel J.J. Influenza hemagglutinin and neuraminidase membrane glycoproteins. Journal of Biological Chemistry. 2010;285:28403–28409. doi: 10.1074/jbc.R110.129809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee P.S., Wilson I.A. Structural characterization of viral epitopes recognized by broadly cross-reactive antibodies. Current Topics in Microbiology and Immunology. 2014;386:323–341. doi: 10.1007/82_2014_413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palese P., Shaw M.L. Orthomyxoviridae. In: Knipe D.M., Howley P.M., Cohen J.I., editors. Fields Virology. 6th edn. Lippincott Williams and Wilkins; Philadelphia: 2013. pp. 1151–1185. [Google Scholar]
- Xiong X., McCauley J.W., Steinhauer D.A. Receptor binding properties of the influenza virus hemagglutinin as a determinant of host range. Current Topics in Microbiology and Immunology. 2014;385:63–91. doi: 10.1007/82_2014_423. [DOI] [PubMed] [Google Scholar]
Glossary
- Hemagglutinin
Trimeric surface glycoprotein responsible for receptor binding and membrane fusion during influenza virus entry.
- Influenza viruses
Prototypical members of the virus family Orthomyxoviridae that includes influenza A, B, and C types.
- Neuraminidase
Tetrameric surface glycoprotein necessary for viral release.