

Having described the structures of viruses and their genomes, I now move on to how these genomes function. As an introduction to the subject, I shall consider a brief outline of the probable main stages in the replication of a virus. There are many variations in detail in these stages.
-
1.
The virus particle enters the cell. At the time of entry or shortly afterwards the genome is released from the protein coat or the structure of the particle relaxes to enable the next stages to take place.
-
2.
The infecting genome is either translated directly if it is (+)-sense ssRNA, or mRNAs are formed and translated, to give early products such as the viral replicase, and perhaps other virus-specific proteins. This is described in this chapter.
-
3.
The viral replicase or replication-associated protein(s) are used to synthesize subgenomic mRNAs if required by the genome strategy. This is also described in this chapter.
-
4.
The viral replicase or replication-associated proteins are used to synthesize new viral genomes, as described in the next chapter.
-
5.
Proteins required relatively late in the viral replication cycle, such as coat protein and cell-to-cell movement protein, are synthesized.
-
6.
Coat protein subunits and viral genomes are assembled to give new virus particles, which accumulate within the cell, usually in the cytoplasm. This was described in Chapter 5.
-
7.
Infectious units of the virus move from the initially infected cell to adjacent cells and possibly through the plant to initiate a systemic infection, as described in Chapter 9.
I. INTRODUCTION
Viral genomes are expressed from mRNAs that are either the nucleic acid of positive-sense [(+)-sense] ssRNA viruses or transcripts from negative-sense [(–)-sense] or dsRNA or from ds or ss DNA viruses. Baltimore (1971) pointed out that the expression of all viral genomes, be they RNA or DNA, ss or ds, (+)- or (–)-sense, converge on the mRNA stage (Fig. 7.1 ).
Fig. 7.1.

Routing of viral genome expression through mRNA. Route I is transcription of dsDNA usually by host DNA-dependent RNA polymerase. Route II is the transcription of ssDNA to give the dsDNA template for I (e.g. geminiviruses). Route III is transcription of dsRNA, usually by virus-coded RdRp (e.g. reoviruses). Route IV is replication of (+)-strand RNA via a (–)-strand template by virus-coded RdRp–the viral (+) strand is often the template for early translation (the (+)-strand RNA viruses). Route V is transcription of (–)-strand virus genome by virus-coded RdRp (e.g. tospoviruses). Route VI is reverse transcription of RNA stage of retro- and pararetro-viruses leading to the dsDNA template for mRNA transcription.
From Baltimore (1971), with permission.
© 2002
When the encapsidated virus particle enters a susceptible plant cell, the genome must be released from the relatively stable capsid required for movement from host to host. Once the genome becomes available, it can be translated directly if (+)-sense ss RNA is present, or else the formation of mRNA can commence.
As will be described in Section V.A, expression of the viral mRNA faces various constraints imposed by the eukaryotic translation system.
In this chapter, I describe how viruses release their encapsidated genome on entry into the host cell, how they express their genetic information overcoming the various constraints imposed by the host translation system, and how the expression is regulated.
II. VIRUS ENTRY AND UNCOATING
A. Virus entry
As described in Chapters 11 and 12, plant viruses require damage of the cuticle and cell wall to be able to enter a plant cell. There have been various suggestions as to the mechanism of entry into the cell in which infection is initially established (reviewed by Shaw, 1999) (Fig. 7.2 ).
Fig. 7.2.

Proposed routes for entry of TMV particles during manual inoculation of leaves. None of these routes has been demonstrated directly and all remain unproven. Left to right: direct entry of virus particles through wound; attachment of virus particle to cell membrane and passage of virus particle or viral RNA into cell; passage of virus particle through cell wall via ectodesmata or ‘bleb’; attachment of virus particle to cell membrane and entry after invagination of membrane and formation of endocytotic vesicle; attachment of virus particle to outer cell wall and passage of viral RNA through wall into cell.
From Shaw (1999), with kind permission of the copyright holder, © The Royal Society.
© 2002
There is no evidence for a specific entry mechanism such as plasma membrane receptor sites or endocytotic uptake, and it is generally considered that ‘entry is accomplished by brute force’ (Shaw, 1999).
B. Uncoating of TMV (reviewed by Shaw, 1999)
1. Early events in intact leaves
The nature of the leaf surface, the requirement for wounding, the efficiency of the process, and other aspects of infection in intact leaves are discussed in Chapter 12. The uncoating process has been examined directly by applying TMV radioactively labelled in the protein or the RNA or in both components (e.g. Shaw, 1973; Hayashi, 1974). The following conclusions were drawn from such experiments:
-
1.
Within a few minutes of inoculation, about 10% of the RNA may be released from the virus retained on the leaf.
-
2.
Much of the RNA is in a degraded state but some full-length RNAs have been detected.
-
3.
In vivo stripping of the protein from the rod begins at a minimum of two and probably many more sites along the rod (Shaw, 1973) (but see Section II.B.2).
-
4.
The early stages of the process do not appear to depend on pre-existing or induced enzymes (Shaw, 1969).
-
5.
The process is not host-specific, at least in the early stages. However, there is a fundamental difficulty with all such experiments. Concentrated inocula must be used to provide sufficient virus for analysis, but this means that large numbers of virus particles enter cells rapidly (Fig. 7.3 ). It is impossible to know which among these particles actually establish an infection.
Fig. 7.3.
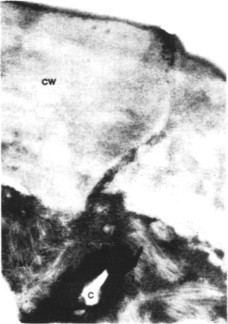
TMV particles that have entered a tobacco leaf lower epidermal cell through a wound caused by abrasive (celite). Tissue was excised and fixed immediately after inoculation. Large numbers of virus rods (TMV) are visible in the cytoplasm. CW, cell wall; C, celite.
From Plaskitt et al. (1987), with kind permission of the copyright holder, © The American Phytopathological Society.
© 2002
2. Disassembly of the virus in vitro
To initiate infection, TMV RNA must be uncoated, at least to the extent of allowing the first ORF to be translated. Most initial in vitro experiments on the disassembly of TMV were carried out under non-physiological conditions. For example, alkali or detergent (1% sodium dodecyl sulfate, SDS) cause the protein subunits to be stripped from TMV RNA beginning at the 5’-end of the RNA (the concave end of the rod) (e.g. Perham and Wilson, 1976). Controlled disassembly by such reagents yields a series of subviral rods of discrete length (e.g. Hogue and Asselin, 1984). Various cations slow down or prevent the stripping process at pH 9.0 (Powell, 1975). Durham et al. (1977) suggested that Ca2+ binding sites might act as a switch controlling disassembly of TMV in the cell. Removal of Ca2+ would result in a change in the conformation of protein subunits, leading to their disaggregation. Durham (1978) proposed that TMV (and other small viruses) might be disassembled at or within a cell membrane. The virus might be in a medium roughly 10−3 M with respect to Ca2+ outside the cell, while inside the cell Ca2+ is about 10−7 M. The ion dilution would provide free energy to help break inter-subunit bonds. These ideas remain speculative.
Wilson (1984a) found that treatment of TMV rods briefly at pH 8.0 allowed some polypeptide synthesis to occur when the treated virus was incubated in an mRNA-dependent rabbit reticulocyte lysate (Fig. 7.4 ).
Fig. 7.4.

In vitro co-translational disassembly of TMV. Electrophoretic resolution of the products of cell-free translation reactions programed with TMV RNA (lane 2) or purified TMV particles that had been pretreated at pH 8.0–8.2 (lanes 5–10). Numbers at left show positions of markers; those to the right are positions of TMV proteins. The appearance of the 126 kDa product provided evidence of a co-translational disassembly mechanism.
From Wilson (1984a), with permission.
© 2002
Wilson suggested that the alkali treatment destabilizes the 5’-end of the rod sufficiently to allow a ribosome to attach to the 5’ leader sequence and then to move down the RNA, displacing coat protein subunits as it moves by a process termed co-translational disassembly. He called the ribosome-partially-stripped-rod complexes ‘striposomes’ (Fig. 7.5 ) and suggested that a similar uncoating mechanism may occur in vivo.
Fig. 7.5.

‘Striposome’ complexes. Electron microscopic examination of the products of in vitro translation reactions programed with TMV particles. One end of some of the particles is associated with structures thought to be ribosomes. The complexes are thought to be intermediates in the co-translational disassembly process.
From Wilson (1984a), with permission.
© 2002
In contrast to the reticulocyte lysate system, in the wheatgerm system virus treated at pH 8.0 gave rise to three times as much polypeptide synthesis as isolated TMV RNA, presumably owing to protection of the RNA in the rod from nucleases before it was uncoated (Wilson, 1984b). In an in vitro protein-synthesizing system from E. coli, virus treated at pH 8.0 gave rise to significant amounts of the 126-kDa protein, whereas TMV RNA gave polypeptides of 50 kDa or less, with a substantial amount of coat protein size (Wilson 1986). Xenopus oocytes micro-injected with TMV produced at least as much immunoreactive 126-kDa protein as did oocytes injected with TMV RNA (Ph. C. Turner et al., 1987). This experiment appears to rule out a specific role for the cellulose cell wall in the uncoating of TMV in leaves. Whether it also rules out a role for the plasma membrane depends on whether intact virus particles contaminating the outside of the needle used for injection could have entered the oocytes via the cell membrane, being uncoated on the way.
Treatment of TMV in vitro with SDS for 15 seconds exposed a sequence of nucleotides from the 5’ terminus to beyond the first AUG codon. No more 5’ nucleotides were exposed during a further 15 minutes in SDS. Incubation of SDS-treated rods with wheatgerm extract, or rabbit reticulocyte lysate, led to the binding of one or two ribosomes in ≈20% of the particles (Mundry et al., 1991). Structure predictions suggest that the exposed sequence up to the first AUG exists in an extended single-stranded configuration (see Section V.C.5), which would assist in the recruitment of ribosomes.
3. Experiments with protoplasts
To obtain infection of a reasonable proportion of protoplasts it is necessary to treat the virus and/or the protoplasts in one of several ways (see Chapter 8, Section III.A.5). Electron microscopy has been used to study the entry process, and it has been suggested that poly-L-ornithine stimulates entry of TMV either by damaging the plasmalemma (Burgess et al., 1973) or by stimulating endocytotic activity (Takebe et al., 1975). Estimates of the extent of uncoating of the adsorbed TMV inoculum vary from 5% (Wyatt and Shaw, 1975) to about 30% one hour after inoculation (Zhuravlev et al., 1975), but the proportion of fully stripped RNA has not been determined. In view of the abnormal state of the cells–and particularly the nature of the suspension medium–the relevance of studies in protoplasts to the infection process in leaves following mechanical inoculation is open to question.
4. Co-translational disassembly
The initial experiments of Wilson and colleagues demonstrating co-translational disassembly in vitro suggested an attractive mechanism for a key step in the infection process. To investigate whether such a mechanism operates in vivo, Shaw et al. (1986) extracted samples from epidermal cells of tobacco leaves inoculated with TMV and identified molecules indicative an 80S ribosome moving along the RNA from the 5’-end in the manner of co-translational disassembly suggested from in vitro studies (see above, Section II.B.2). Translation complexes with the expected properties of striposomes have been isolated from the epidermis of tobacco leaves shortly after inoculation (Plaskitt et al., 1987). Subsequent experiments with protoplasts (Wu et al., 1994; Wu and Shaw, 1996, 1997) have built up a more complete picture.
The first event in co-translational disassembly is that the structure of the virion has to relax so that the 5’ terminus of the RNA is accessible to a ribosome. In vitro treatments, such as SDS or weak alkali, showed that the 68 nucleotide 5’ leader sequence, which lacks G residues, interacts more weakly with coat protein subunits than do other regions of the genome (Mundry et al., 1991). As discussed in Chapter 5 (Section III.B.5), TMV particles are stabilized by carboxylate interactions, there being two carboxyl–carboxylate bonds between adjacent subunits and one carboxylate-RNA interaction. At slightly alkaline pHs these carboxylates become protonated, leading to electrostatic repulsion. Mutagenic studies on these bonds have shown that the situation is probably more complex than initially thought but that these groups provide the key controlling mechanisms for virus disassembly (Culver et al., 1995; Lu et al., 1996, 1998a; Wang et al., 1998a).
Having initiated translation, ribosomes proceed along TMV RNA translating the 5’ ORF, the 126/183-kDa replicase protein, and displacing coat protein subunits. When the ribosomes reach the stop codon of the 126/183-kDa ORF they disengage. This raises the question of how the 3’ quarter of the particle is disassembled. Wilson (1985) suggested that the replicase might perform this task in a 3’ → 5’ direction in synthesizing the (–)-strand replication intermediate. This suggestion was supported by the experiments of Wu and Shaw (1997) who obtained evidence for co-replicational disassembly from the 3’-end. They showed that particles containing mutations in the 126- or 183-kDa ORFs were unable to undergo 3’ → 5’ disassembly in electroporated protoplasts, but that this disassembly could be complemented in trans by wild-type TMV.
Thus, TMV is uncoated in a bi-directional manner, using the co-translational mechanism for the 5’ → 3’ direction yielding the replicase which disassembles the rest of the particle in the 3’ → 5’ direction, showing that disassembly and replication are coupled processes. The process happens rapidly with the whole capsid uncoated within 20 minutes (Wu et al., 1994; Wu and Shaw, 1996) (Fig. 7.6 ).
Fig. 7.6.

Bidirectional disassembly of TMV particles in vivo. Coat protein subunits are removed in a 5’ → 3’ direction from c. 75% of the viral RNA in the first 2–3 minutes after inoculation of protoplasts. Uncoating the 3’-end of the RNA begins shortly thereafter and is completed by removal of subunits in the 3’ → 5’ direction.
From Wu et al. (1994), with kind permission of the copyright holder, © The National Academy of Sciences, USA.
© 2002
C. Uncoating of bromoviruses
The isometric particles of bromoviruses swell at pHs above 7 (see Chapter 5, Section VI.B.4.b) and it has been suggested that, under these conditions, co-translational disassembly can take place (Brisco et al., 1986a). In these in vitro experiments, swollen BMV and CCMV particles were added to a wheatgerm extract and translation products were obtained. However, it was not possible to perform translations on unswollen virus particles as the wheatgerm extract did not translate mRNAs at pHs below 7. By analogy with similar experiments on SBMV (Section II.D), in which the swelling could be controlled by both pH and Ca2+, it was concluded that swelling of the bromovirus particle was required for uncoating. Analysis by sucrose and CsCl density gradients showed that the virus–ribosome complexes contain up to four ribosomes per virus particle (Roenhorst et al., 1989).
Using mutants of CCMV that did not swell under alkaline conditions, Albert et al. (1997) found that swelling was not necessarily required for co-translational disassembly. They suggested that there is a pH-dependent structural transition in the virion, other than swelling, which enables the RNA to be accessible to the translation system. The proposed model, which is similar to ones from some vertebrate and insect viruses, postulates that the N termini of the five subunits in the pentameric capsomere undergo a major structural transition from the interior to the exterior of the virion. This provides a channel through which the RNA passes to be accessible for translation (Albert et al., 1997). However, the 5’-end of the RNA must be released, which suggests that it is located in association with a pentameric capsomere.
D. Uncoating of SBMV
In similar experiments to those described above for bromoviruses, Brisco et al. (1986a) showed that co-translational disassembly takes place on swollen SBMV particles. As the stabilization of particles of SBMV is controlled by both pH-dependent and Ca2+-mediated interactions (see Chapter 5, Section VI.B.4.e), they were able to control the swelling at the alkaline pHs required for the translation system. When swollen SBMV particles were incubated with a wheatgerm extract containing [35S]methionine, sucrose density gradient analysis showed that 80S ribosomes were associated with intact or almost intact virus particles (Shields et al., 1989). The data suggested that translation of the viral RNA begins before it is fully released from the virus particle. It is not known whether the disassembly involved release of the RNA through ‘holes’ in the pentameric capsomeres as suggested above for CCMV.
E. Uncoating of TYMV
TYMV did not co-translationally disassemble in the in vitro translation system described above for bromoviruses and SBMV (Brisco et al., 1986a).
In vitro studies show that, under various non-physiological conditions, the RNA can escape from TYMV particles without disintegration of the protein shell. Thus, at pH 11.5 in 1 M KCl the RNA is rapidly released together with a cluster of 5–8 protein subunits from the shell (Keeling and Matthews, 1982). A hole corresponding to 5–7 subunits is left in the protein shell following release of RNA by freezing and thawing (Katouzian-Safadi and Berthet-Colominas, 1983). Treatment of TYMV with 3–7% butanol at pH 7.4 leads to the rapid release of RNA and five or six protein subunits in monomer form (Matthews, 1991).
In Chinese cabbage leaves, Kurtz-Fritsch and Hirth (1972) found that about 2% of the retained TYMV inoculum was uncoated after 20 minutes. They showed that empty shells and low-molecular-weight protein were formed following RNA release.
Matthews and Witz (1985) confirmed these findings and demonstrated that a significant proportion of the retained inoculum was uncoated within 45 seconds, and that the process was complete within 2 minutes. At least 80–90% of this uncoating takes place in the epidermis. Approximately 106 particles per epidermal cell can be uncoated (see also Chapter 12, Sections II.D and II.E). The process gives rise to empty shells that have lost about 5–6 protein subunits and to low-molecular-weight protein. At the high inoculum concentrations used, most of the released RNA must be inactivated, presumably in the epidermal cells. Celite was used as an abrasive, so that the mechanism of entry proposed for TMV (Fig. 7.3) could account for the large numbers of particles entering each cell. The uncoating process just described was not confined to known hosts of TYMV.
F. Discussion
There is a dichotomy in the structural stabilization requirements of viruses in that the particles have to be stable enough to protect the viral genome when being transported outside the host yet they have to be able to present the genome to the cellular milieu for the first stages in replication. For at least some of the viruses with (+)-sense RNA genomes, the process of co-translational disassembly answers this problem. The coupled co-translational and co-replicational uncoating of TMV is an elegant process applicable to a rod- shaped virus. However, for other longer rod-shaped viruses, this may not be the process by which they are disassembled. The origin of assembly is at or near the 5’-end of the RNA (see Chapter 5, Section IV.B) and, as suggested for TMV, would be likely to present an obstacle to the translocation of the ribosomes.
It is likely that some form of co-translational disassembly takes place in vivo for the isometric viruses that swell or are permanently swollen (e.g. AMV). Whether the proposed mechanism for release of the RNA through a destabilized pentamer structure is applicable to more stable isometric viruses is still an open question as is the possible involvement of membranes in the uncoating process.
As for other viral genomes, the requirements in the first stages of infection are different to those of the (+)-sense ssRNA viruses. Viruses with dsRNA or (–)-sense ssRNA have to transcribe their genome to give mRNA. These viruses carry the viral RNA-dependent RNA polymerase in the virus particle and, presumably, transcription is an early event. It is not known whether this occurs within the virus particle, possibly in a relaxed structure, or whether the viral genome is released into the cell. However, it is most likely that this process takes place in an environment protected from cellular nucleases and that it is coupled to translation of the mRNA.
The dsDNA genomes of members of the Caulimoviridae have to be transported to the nucleus where they are transcribed to mRNA by the host RNA-dependent RNA polymerase (see Section IV.C.1; Chapter 8, Section VII.B). The coat protein of CaMV has a nuclear localization signal (Leclerc et al., 1999) that will presumably target the particle into the nucleus. Particles of some caulimoviruses and badnaviruses are particularly stable, being able to resist phenol (Hull and Covey, 1983a; Bao and Hull, 1994) and nothing is known about how they disassemble.
The ssDNA genomes of members of the Geminiviridae also have to be transported to the nucleus so that they can be replicated before being transcribed to give mRNAs. As described in Chapter 9 (Section II.C), nuclear localization signals have been recognized in some geminiviral proteins. However, nothing is known about how the particles uncoat.
III. VIRAL GENOME EXPRESSION
Genome strategy is a useful but rather vague term (see Wolstenholme and O'Connor, 1971; Matthews, 1991), which could be extended to include almost every aspect of virus structure, replication and ecology. The term has been taken to include: (1) the structure of the genome (DNA or RNA; ds or ss; if ss whether it is (+)or (–)-sense); (2) the question as to whether the nucleic acid alone is infectious; (3) general aspects of the enzymology by which the genome is replicated (e.g. the presence of nucleic acid polymerases in the virus particle, and any other enzymes concerned in nucleic acid metabolism that are coded for by the virus); and (4) the overall pattern whereby the information in the genome is transcribed and translated into viral proteins (not the detailed molecular biology of this process). However, in this chapter I will use this term to describe the kinds of strategy that have evolved among the groups of plant viruses to translate the genomic information from the mRNA stage of the infection cycle. Several selection pressures have probably been involved in this evolution. After describing the methods for studying genome strategies, I will discuss ways in which mRNAs are synthesized, the selection pressures and then the ways that viruses use to overcome these limitations.
The actual sequence of events that has led to the understanding of viral genome strategies has varied widely for different viruses. This is because of the rather haphazard manner in which a particular branch of science tends to develop. To take two examples:
-
•
All four of the definite TMV gene products, three of which are non-structural, were identified before the nucleotide sequence of the genome of that virus had been established.
-
•
At the other extreme, the full nucleotide sequences of several viruses are known, while only one or two of seven potential gene products had been characterized, and these are usually proteins found in the virus.
I shall attempt to present a summary of the various methods involved in a more logical sequence than that in which they have actually been applied to many viruses. First, we must understand the structure of the genome, and in particular the number of genome pieces, the arrangement of the ORFs in the genome, the deduced amino sequences for those ORFs, and the positions of any likely regulatory and recognition nucleotide sequences. As a second stage, we must define the ORFs that are actually functional by both in vitro and in vivo studies. We need to recognize the gene functions of the virus, either by direct studies on any viral proteins that can be isolated from infected cells, or by classic genetic studies that may reveal various biological activities. Finally, we need to match these viral gene activities with the functional ORFs. It is here that the techniques of reverse genetics can play a very important role. In reverse genetics, an alteration (base change, insertion or deletion of bases) is made at any preselected position in the genome. The consequences of the change are then studied with respect to its effect on the gene product and the product's biological function, a function that may not have been previously recognized by traditional methods. However, it must be recognized that the changes could induce secondary effects on other gene products.
A. Structure of the genome
There are several steps in determining the structure of a viral genome. The starting material is almost always nucleic acid isolated from purified virus preparations.
1. Kind of nucleic acid
Whether the nucleic acid is ss or ds, DNA or RNA, or linear or circular can be established by the various chemical, physical and enzymatic procedures outlined in Chapter 4.
2. Number of genome pieces
When virus particles housing separate pieces of a multi-partite genome differ sufficiently in size or density, they may be fractionated on density gradients and the nucleic acids isolated from the fractions. Alternatively, nucleic acids of differing size may be separated on density gradients or by gel electrophoresis. When the two pieces of a bipartite genome are of very similar size, as with some geminiviruses, the existence of two distinct parts of the genome may be inferred from hybridization experiments estimating sequence complexity. However, formal proof that the genome is in two pieces of nucleic acid can best be obtained by cloning the full length of each piece separately and demonstrating that both are required for infectivity (e.g. the geminiviruses: Hamilton et al., 1983).
3. Terminal structures
Chemical and enzymatic procedures can be used to establish the nature of any structures at the 5’ and 3’ termini of a linear nucleic acid (see Chapter 4, Section III.A.3).
4. Nucleotide sequence
Knowledge of the full nucleotide sequence of a viral genome is essential for understanding genome structure and strategy. The methods used are detailed in many publications and laboratory manuals.
5. Open reading frames (ORFs)
With the help of an appropriate computer program, the nucleotide sequence is searched for ORFs in each of the three reading frames of both (+)- and (–)-sense strands. All ORFs are tabulated, as is illustrated in Fig. 7.7 for a tymovirus.
Fig. 7.7.
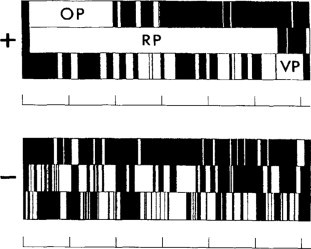
Diagram of the three triplet codon phases of the plus and minus strand RNAs of OYMV genomic RNA. White boxes indicate all ORFs that begin with an AUG and terminate with UGA, UAG or UAA. There are three ORFs considered to be significant labeled OP (overlapping protein), RP (replicase protein) and VP (coat protein).
From Ding et al. (1989), with permission.
© 2002
As shown in Fig. 7.7, a large number of ORFs may be revealed. Those ORFs that could code for polypeptides of MW less than 7–10 kDa, or that would give rise to proteins of highly improbable amino acid composition, are usually not given further consideration. However, sequence similarity between small ORFs in several viruses may indicate that they are functional (e.g. the 7–9 kDa ORFs of potexviruses and carlaviruses).
ORFs of significant size representing possible proteins of 100 amino acids or more occur in the (–)-sense strands of several viruses that are normally regarded as being (+)-stranded (e.g. CPMV RNA2: Lomonossoff and Shanks, 1983; TMV: Goelet et al., 1982; AMV RNA1 and RNA2: Cornelissen et al., 1983a,b; PapMV: AbouHaidar, 1988). There is no evidence that any of these have functional significance. However, there is no reason, in principle, why functional ORFs should not occur in the (–)-sense strand. Such ORFs are found in the geminiviruses, tospoviruses and tenuiviruses (see Chapter 6, Sections V and VII).
ORFs do not necessarily start with the conventional AUG start codon. An AUU start codon has been recognized for ORF I of RTBV (Fütterer et al., 1996) (see Section V.B.6) and a CUG start codon for the capsid protein of SBWMV (Shirako, 1998). This phenomenon raises the question of the definition of an ORF. Conventionally, it starts with an AUG codon and stops with one of the three stop codons. If non-AUG start codons are more widely used than at present thought, an ORF should be a largish in frame region without a stop codon.
6. Amino acid sequence
The amino acid sequence and MW of the potential polypeptide for each ORF of interest can be determined from the nucleotide sequence and the genetic code.
7. Regulatory signals
Various parts of the genome and particularly the 5’ and 3’ non-coding sequences are searched for relevant regulatory and recognition signals, as will be discussed in Section V.C. Regulatory sequences may also be found in coding regions.
8. mRNAs
The genomes of DNA viruses must be transcribed into one or more mRNAs. These must be identified in nucleic acids isolated from infected tissue and matched for sequence with the genomic DNA. Many plant viruses with ss (+)-sense RNA genomes have some ORFs that are translated only from a subgenomic RNA (discussed in Section V.B.2). These too must be identified to establish the strategy of the genome.
In RNA preparations isolated from virus-infected tissue or from purified virus preparations, subgenomic RNAs may be present that can be translated in vitro to give polypeptides with a range of sizes that do not correspond to ORFs in the genomic RNA. For example, Higgins et al. (1978) detected RNAs of eight discrete lengths in nucleic acid isolated from preparations of TYMV. Mellema et al. (1979) were able to associate five of these with particular polypeptides synthesized in the reticulocyte system. The full-length translation products of these RNAs and the genomic RNA overlapped with one another and shared a common amino terminus. Mellema et al. concluded that these RNAs share a common translation initiation site near their 5’ termini.
In vitro translation of TMV RNAs isolated from TMV-infected tissue gave rise to a series of products with molecular weights of 45, 55, 80 and 95 kDa (Goelet and Karn, 1982). These formed a nested set of proteins sharing C-terminal sequences and having staggered N-terminal amino acids. Goelet and Karn suggested that viral RNA may be transcribed into a set of incomplete negative-sense strands that are in turn transcribed into a set of incomplete mRNAs. Since no function in viral replication has yet been ascribed to such N-terminal or C-terminal families of proteins, they will not be further discussed.
Thus, it may be a difficult task to establish whether a viral RNA of subgenomic size is a functional mRNA or merely a partly degraded or partly synthesized piece of genomic RNA. One criterion is to first isolate an active polyribosome fraction and then isolate the presumed mRNAs. The RNAs may then be fractionated by gel electrophoresis, and those with virus-specific sequences identified by the use of appropriate hybridization probes or by PCR.
Not infrequently, genuine viral subgenomic mRNAs are encapsidated along with the genomic RNAs. These can then be isolated from purified virus preparations and characterized. When the sequence of the genomic nucleic acid is known there are two techniques available to locate precisely the 5’ terminus of a presumed subgenomic RNA. In the S1 nuclease protection procedure, the mRNA is hybridized with a complementary DNA sequence that covers the 5’ region of the subgenomic RNA. The ss regions of the hybridized molecule are removed with S1 nuclease. The DNA that has been protected by the mRNA is then sequenced. In the second method, primer extension, a suitable ss primer molecule is annealed to the mRNA. Reverse transcriptase is then used to extend the primer as far as the 5’ terminus of the mRNA and the DNA produced sequenced. Carrington and Morris (1986) used both these procedures to locate the 5’ termini of the two subgenomic RNAs of CarMV. A sequence determination that reveals a single termination nucleotide rather than several is a good indication that the subgenomic RNA under study is a single distinct species and not a set of heterogeneous molecules (e.g. Sulzinski et al., 1985).
B. Defining functional ORFs
Some of the ORFs revealed by the nucleotide sequence will code for proteins in vivo, whereas others may not. The functional ORFs can be unequivocally identified only by in vitro translation studies using viral mRNAs and by finding the relevant protein in infected cells.
1. In vitro translation of mRNAs
The monocistronic RNA of STNV was translated with fidelity in the prokaryotic in vitro system derived from Escherichia coli (Lundquist et al., 1972). However, results with other plant viral RNAs were difficult to interpret. Three systems derived from eukaryotic sources have proven useful with plant viral RNAs. The general outline of the procedure for these three systems follows.
-
1.
The RNA or RNAs of interest are purified to high degree, using density gradient centrifugation and/or Polyacrylamide gel electrophoresis. For viruses whose particles become swollen under the conditions of the in vitro protein-synthesizing system, the RNA associated with the virus may act effectively as mRNA (e.g. Brisco et al., 1986a). Alternatively, RNA may be transcribed from cloned viral cDNA or DNA.
-
2.
The RNAs are then added to the protein-synthesizing system in the presence of amino acids, one or more of which is radioactively labelled.
-
3.
After the reaction is terminated, the polypeptide products are fractionated by electrophoresis on SDS-PAGE, together with markers of known size.
-
4.
The products are located on the gels by means of the incorporated radioactivity.
The three systems are:
-
•
The rabbit reticulocyte lysate system. The cells from anaemic rabbit blood are lysed in water and centrifuged at 12 000g for 10 minutes. The supernatant fluid is then used. This is a useful system because of the virtual absence of RNase activity. Fig. 7.8 illustrates the use of this system.
-
•
The wheat embryo system. In this system, the viral RNA is added in the presence of an appropriate label to a supernatant fraction from extracted wheat embryos from which the mitochondria have been removed. This system may contain plant factors not present in animal systems.
-
•
Toad oocytes. These strictly do not constitute an in vitro system. Intact oocytes of Xenopus or Bufo are injected with the viral mRNA and incubated in a labelled medium.
Fig. 7.8.

Translation of plant viral RNAs in the rabbit reticulocyte system. The polypeptide products were fractionated by electrophoresis in a Polyacrylamide gel and located by autoradiography of the gel. (A) Products using TMV RNA as message. (B) Control with no added RNA. (C) Products using TYMV RNA. The unmarked smaller polypeptides may be incomplete transcripts of the viral message or due to endogenous mRNA. Note that no protein the size of the viral coat protein (17.5 or 20 kDa) is produced by either viral RNA.
From Briand (1978), with permission.
© 2002
Jagus (1987a,b) gives technical details of these methods and the first two systems are available in kit form from several companies. Not infrequently, when purified viral genomic RNAs are translated in cell-free systems, several viral-specific polypeptides may be produced in minor amounts in addition to those expected from the ORFs in the genomic RNA. It is unlikely that such polypeptides have any functional role in vivo. They are probably formed in vitro by one or more of the following mechanisms: (1) endonuclease cleavage of genomic RNA at specific sites; (2) proteolytic cleavage of longer products during the incubation; (3) misreading of sense codons as termination signals; and (4) secondary structure of the genomic RNA, formed under the in vitro conditions, which prevents translocation of a proportion of ribosomes along the RNA. For example, multiple polypeptides of MW below 11 kDa are synthesized in the rabbit reticulocyte system with TMV RNA (Wilson and Glover, 1983). Similarly, when the I2 subgenomic RNA of TMV is translated in vitro, a family of polypeptides besides the 30-kDa protein is produced, but only the 30-kDa protein could be detected in vivo (Ooshika et al., 1984). Such polypeptides will not be discussed further in relation to virus replication.
What criteria can be used to ‘optimize’ conditions for in vitro translation? Measurement of total radioactivity incorporated is not particularly informative. Measurements of radioactivity in individual polypeptides separated on Polyacrylamide gels are much more useful. One might aim for conditions producing (1) the greatest number of polypeptides, (2) the fewest, (3) the longest, or (4) the most of a particular known gene product. It thus becomes apparent that to obtain definitive mapping of the genome from studies on the polypeptides produced in vitro, we must also know what polypeptides are actually synthesized in vivo by the virus.
2. Methods for identifying ORFs that are functional in vivo
Virus-coded proteins, other than those found in virus particles, may be difficult to detect in vivo especially if they occur in very low amounts and are only transiently during a particular phase of the virus replication cycle. However, a battery of methods is now available for detecting virus-coded proteins in vivo and matching these with the ORFs in a sequenced viral genome. In particular, the nucleotide sequence information gives a precise estimate of the size and amino acid composition of the expected protein. Knowledge of the expected amino acid sequence can be used to identify the in vivo product either from a partial amino acid sequence of that product or by reaction with antibodies raised against either a synthetic polypeptide that matches part of the expected amino acid sequence or against the ORF or part thereof expressed in, say, E. coli.
a. Proteins found in the virus
Coat proteins are readily allocated to a particular ORF based on several criteria: (1) amino acid composition compared with that calculated for the ORF; (2) amino acid sequence of part or all of the coat protein; (3) serological reaction of an in vitro translation product with an antiserum raised against the virus; and (4) for a few viruses such as TMV, assembly of an in vitro translation product into virus particles when mixed with authentic coat protein. Viruses such as rhabdoviruses and reoviruses may be exceptional in that several of the gene products, corresponding to various ORFs in the genome, are found in purified virus preparations (see Chapter 4, Section III.B.6.d, and Chapter 6, Section VII.A).
b. Direct isolation from infected tissue or protoplasts
Healthy and virus-infected leaves or protoplasts are labelled with one or more radioactive amino acids. Cell extracts are fractionated by appropriate procedures and proteins separated by gel electrophoresis. Protein bands appearing in the samples from infected cells and not from healthy cells may be identified with the expected product of a particular ORF by comparing its mobility and its pattern of tryptic peptides with that of an in vitro translation product of the ORF (e.g. Bujarski et al., 1982). With appropriate in vivo labelling, partial amino acid sequencing of the isolated protein may allow the precise location of its coding sequence in the genome to be established (e.g. Wellink et al., 1986). In infections with some viruses such as potyviruses, large amounts of several virus-coded non-structural proteins accumulate in infected cells, facilitating the allocation of each protein to its position in the genome.
c. Serological reactions
Antisera provide a powerful set of methods for recognizing viral-coded proteins produced in vivo and identifying them with the appropriate ORF in the genome.
i. Antisera against synthetic peptides
A synthetic peptide can be prepared corresponding to part of the amino acid sequence predicted from an ORF. An antiserum is raised against the synthetic peptide and used to search for the expected protein in extracts of healthy or infected tissue or protoplasts. For example, Kibertis et al. (1983) synthesized a peptide corresponding to the C-terminal 11 amino acids of a 30-kDa ORF in the TMV genome. They were able to show that a polypeptide corresponding to this ORF was synthesized in infected protoplasts.
ii. Antisera against in vitro translation products
If an mRNA is available that is translated in vitro to give a polypeptide product clearly identified with a particular ORF or genome segment, antisera raised against the in vitro product can be used to search for the same protein in extracts of infected cells or tissue. For example, such antisera have been used to identify non-structural proteins coded for by AMV RNAs in tobacco leaves. The antisera were used in conjunction with a very sensitive immunoblotting procedure.
iii. Antisera against recombinant proteins
Recombinant proteins can be derived either in vitro from translation of RNA transcripts of a cloned gene or by expression in various E. coli systems. In the latter it is convenient to attach a ‘tag’ to the protein to enable it to be purified. Antibodies are then raised against the recombinant protein and used to search for the corresponding protein in extracts of infected plant. An example of the in vitro transcript procedure was used to establish the position of the nucleocapsid protein gene in the rhabdovirus SYNV (Zuidema et al., 1987). The E. coli procedure is exemplified by the antisera raised against the polymerase and protease gene regions in the polyprotein of RTSV (Thole and Hull, 1998). The RTSV cDNAs were placed in an E. coli vector that expressed them as fusion proteins with glutathione S-transferase that enabled them to be purified by absorption on to glutathione-agarose beads.
iv. Immunogold labeling
Antibodies produced against a synthetic peptide corresponding to part of a particular ORF in the genomic nucleic acid and labeled with gold can be used to probe infected cells for the presence of the putative gene product. This was done for the 30-kDa gene product of TMV (Tomenius et al., 1987) and examples include antibodies produced against CaMV ORF I product expressed in E. coli combined with immunogold labeling demonstrated that this protein is expressed in infected leaves (Linstead et al., 1988).
d. Comparison with genes known to be functional in other viruses
Size, location in the genome and nucleotide sequence similarities with known functional genes may give a strong indication that a particular ORF codes for a functional protein in vivo. These are frequently identified by searches of computer databases. For example, ORFs coding potential polymerases (RNA-dependent RNA polymerases and reverse transcriptases) are often recognized by the presence of characteristic motifs (see Chapter 8, Sections IV.B.1 and VII.A).
e. Presence of a well-characterised subgenomic RNA
Occasionally, a viral subgenomic RNA (sgRNA) has been well characterized but no in vivo protein product has been detected. Thus, the I1 sgRNA of TMV was recognized as a functional mRNA because (1) it is located in the polyribosome fraction from infected cells, and (2) it has a precisely defined 5’ terminus (Sulzinski et al., 1985). Thus, it is reasonable to suppose that the 5’ ORF of this subgenomic RNA is functional in vivo.
f. Presence of appropriate regulatory signals in the RNA
AUG triplets that are used to initiate protein synthesis may have a characteristic sequence of nucleotides nearby (Section V.A). Upstream of the AUG triplet there may be identifiable ribosome recognition signals. Presence of these sequences would indicate that the ORF is functional.
g. Codon usage
Frequency of codon usage has sometimes been used to indicate whether an ORF revealed in a genomic nucleotide sequence is likely to produce a functional protein (e.g. Morch et al., 1988). However, an analysis of the codon usage by RTBV showed that it used many rare codons (R. Hull, unpublished), and thus this character should not be used as a firm criterion.
h. Reoviruses
The reoviruses are a special case with respect to establishing functional ORFs. Each ds genome segment is transcribed in vitro to give an mRNA that gives a single protein product (Nuss and Peterson, 1981). On this basis, it was considered reasonable to assume that each genome piece has a single functional ORF (but see Chapter 6, Section VI.A).
C. Recognizing activities of viral genes
Before information on the sequence of nucleotides in viral genomes became available and before the advent of in vitro translation systems, there were two ways of recognizing the activities of viral genes–identification of proteins in the virus particle and classic virus genetics. These approaches are still relevant.
1. Gene products in the virus
Fraenkel-Conrat and Singer (1957) reconstituted the RNA of one strain of TMV in the protein of another strain that was recognizably different. The progeny virus produced in vivo by this in vitro ‘hybrid’ had the coat protein corresponding to the strain that provided the RNA. Since this classic experiment, it has been universally assumed that coat proteins are encoded by viral genomes. Likewise, it has usually been assumed that other proteins found as part of the virus particle are also virus coded; for example, those found in reoviruses and rhabdoviruses.
2. Classic viral genetics
Two kinds of classic genetic study have identified many biological activities of viral genomes, and both of these procedures are still useful in appropriate circumstances.
a. Allocation of functions in multi-particle viruses
The discovery of viruses with the genome divided between two or three particles opened up the possibility of locating specific functions on particular RNA species. The requirements for, and stages in, this kind of analysis are as follows.
-
1.
Purification of the virus.
-
2.
Fractionation of the genome components, either as nucleoprotein particles (on density gradients of sucrose or cesium salts) or as isolated biologically active RNA species (usually by electrophoresis on Polyacrylamide gels).
-
3.
Definition of the set of RNA molecules that constitute the minimum viral genome.
-
4.
Identification and isolation of natural strains or artificial mutants differing in some defined biological or physical properties, which will provide suitable experimental markers. For example, Dawson (1978a) isolated a set of ts mutants of CMV. One group of mutations mapped on RNA3 and the rest on RNA1.
-
5.
In vitro substitution of components from different strains or mutants in various combinations. These are inoculated to appropriate host plant species. The relevant biological or physical properties of the various combinations are determined. A particular property may then be allocated to a particular genome segment or segments.
-
6.
Back-mixing experiments. In such experiments, the parental genome pieces are isolated from the artificial hybrids, mixed in the original combinations, and tested for appropriate physical or biological properties. Such tests are necessary to show that the RNAs of the hybrids retain their identity during replication.
-
7.
Supplementation tests. These provide an alternative procedure to in vitro reassortment. Individual wild-type genome segments are added to a defective (mutant) inoculum. Restoration of the wild-type character in a particular mixture will indicate which segment controls the character (e.g. Dawson, 1978a). Transgenic plants expressing a single genome segment can also be used in supplementation tests.
-
8.
Mixing of mutants. Unfractionated preparations of two different mutants may be mixed and tested. If the wild-type property is restored it can be assumed that the two mutations are on different pieces of RNA.
Supplementation tests and mixing of mutants do not require purification and fractionation of the mutant viruses. They can provide independent confirmation of results obtained by in vitro substitution experiments (de Jager, 1976).
Various factors may complicate the analysis of reassortment experiments:
-
1.
A particular property may be determined by more than one gene, located on the same or on separate pieces of RNA.
-
2.
Some genes are pleiotropic (i.e. have more than one effect). An example is the coat protein of AMV, which is involved in encapsidation of the virus, its aphid transmission, RNA replication and the spread of the virus through the plant (Tenllado and Bol, 2000).
-
3.
If certain parts of the RNA are used to produce two proteins with different functions (e.g. by the read-through mechanism), then a single base change might induce changes in the two different functions.
-
4.
Some amino acid replacements might be ‘silent’ with respect to one property of the protein but not another.
However, by using these procedures, many activities of viral genes have been attributed to one or more of the genome segments in a multipartite virus. Local or systemic symptoms in particular hosts, host range, and proteins found in the virus particle are activities that have commonly been studied. It must be recognized that these reassortment experiments have two limitations:
-
1.
Where more than one gene is present on the RNA or DNA segment an activity cannot be allocated to a particular gene.
-
2.
Except for structural proteins, they do not prove that the gene product is responsible for the activity. In principle, the activity could be due to some direct effect of the nucleic acid itself.
b. Natural or artificially induced virus mutants
The study of naturally occurring or artificially induced mutants of a virus has allowed various virus activities to be identified. Again, many of the activities involve biological properties of the virus.
Mutants that grow at a normal (permissive) temperature but that replicate abnormally or not at all at the non-permissive (usually higher) temperature are particularly useful. Such temperature-sensitive (is) mutants are easy to score and manipulate, and most genes seem to be potentially susceptible to such mutations. They arise when a base change (or changes) in the viral nucleic acid gives rise to an amino acid substitution (or substitutions) in a protein, which results in defective function at the non-permissive temperature. Alternatively, the base change might affect the function of a non-translated part of the genome–a control element, for example. The experimental objective is to collect and study a series of ts mutants of a particular virus. To be useful for studies on replication, ts mutants must possess certain characteristics: (1) they must not be significantly ‘leaky’ at the non-permissive temperature; and (2) the rate of reversion to wild type must be low enough to allow extended culture of the mutant at both the permissive and non-permissive temperatures.
If the mutation studied occurred in the gene for the coat protein and the amino acid sequence of the coat protein is known, it is possible to locate the mutation within that protein. The location of mutations in other viral genes had to await information of nucleotide sequence and genome structure. The ts strain of TMV known as Ls1 is a good illustration. This strain replicates normally at 22°C, but at the non-permissive temperature (32°C) there is very little replication compared with the parent virus (TMV-L). Nishiguchi et al. (1980) studied the replication and movement of the two strains at the two temperatures, using fluorescent antibody staining to identify cells where virus had replicated. The results, illustrated in Fig. 7.9 , showed clearly that Ls1 was defective in a cell-to-cell movement function. However, these results did not locate the cell-to-cell function involved. This had to await a knowledge of the genome structure of TMV and the site of the mutation within that structure (see Chapter 9, Section II.D.2.a). Viral mutants and other variants are discussed further in Chapter 17.
Fig. 7.9.

Example of a ts mutant of TMV defective in cell-to-cell transport function. Fluorescent antibody staining of epidermal cells indicates the distribution of coat protein antigen 24 hours after inoculation. Tomato leaflets were infected at the permissive and non-permissive temperature with a ts strain (Ls1) or a wild-type strain (L) of TMV. Inoculated leaflets were cultured for 24 hours (a) Ls1 at 32°C; (b) Ls1 at 22°C; (c) L at 32°C; (d) L at 22°C.
From Nishiguchi et al. (1980), with permission.
© 2002
D. Matching gene activities with functional ORFs
A variety of methods is now available for attempting to match the in vivo function of particular viral gene products with a particular ORF. A few of these give unequivocal proof of function, whereas others are more or less strongly indicative of a particular function.
There are two kinds of method. In the first, which may not be generally applicable, the natural gene product produced in infected tissue is isolated, and its activity is established by direct methods. The second group of methods involves, directly or indirectly, the use of recombinant DNA technology.
1. Direct testing of protein function
Some virus-coded proteins besides coat proteins have functions that can be identified in in vitro tests.
a. In vitro translation products
Carrington and Dougherty (1987a) prepared an in vitro plasmid expression vector that allowed cell-free synthesis of particular segments of the TEV genome. The RNAs obtained were translated in rabbit reticulocyte lysates to give polypeptides that could be assessed for protease activity and the ability to act as protease substrates. In this way, they showed that the 40-kDa protein was a viral protease. Using an in vitro transcription and translation system on mutagenized cDNAs, Thole and Hull (1998) showed that a 35-kDa protein from the RTSV polyprotein has proteolytic activity, and they identified the potential cleavage sites in the polyprotein.
b. A protein isolated from infected cells
Thornbury et al. (1985) purified the protein helper component for aphid transmission from leaves infected with PVY, showing that it has a molecular weight of 58 kDa. They isolated the corresponding protein from another potyvirus and produced antisera against the two proteins. Using an in vitro aphid-feeding test, they showed that the antisera specifically inhibited aphid transmission of the virus that induced formation of the corresponding protein in infected plants. These tests demonstrated that the polypeptides were essential for helper component activity. Active CaMV aphid transmission factor was recovered after expression of a recombinant of the ORF II product and a baculovirus in insect cells (Blanc et al., 1993b); however, this protein was not active when expressed in E. coli.
2. Approaches depending on recombinant DNA technology
a. Location of spontaneous point mutations
Knowledge of nucleotide sequences in natural virus variants allows a point mutation to be located in a particular gene, even if the protein product has not been isolated. In this way, the changed or defective function can be allocated to a particular gene. The ts mutant of TMV known as Ls1 can serve again as an example. At the non-permissive temperature, it replicates and forms virus particles normally in protoplasts and infected leaf cells, but is unable to move from cell to cell in leaves. A nucleotide comparison of the Ls1 mutant and the parent virus showed that the Ls1 mutant had a single base change in the 30-kDa protein gene that substituted a serine for a proline (Ohno et al., 1983). This was a good indication, but not definitive proof, that the 30-kDa protein is involved in cell-to-cell movement.
b. Introduction of point mutations, deletions or insertions
The genomes of many DNA viruses and cDNAs to many RNA viruses have been cloned and the DNA or transcripts thereof shown to be infectious. There are numerous examples of experiments in which point mutations, deletions or insertions have been used to elucidate the function(s) of the gene produced by the modified ORF. The introduction of defined changes in particular RNA viral genes to study their biological effects, and thus define gene functions, is commonly known as reverse genetics. This approach has been of major importance in gaining understanding of the gene functions described in this book.
c. Recombinant viruses
Recombinant DNA technology can be used to construct viable viruses from segments of related virus strains that have differing properties, and thus to associate that property with a particular viral gene. For example, Saito et al. (1987a) constructed various viable recombinants containing parts of the genome of two strains of TMV, only one of which caused necrotic local lesions on plants such as Nicotiana sylvestris, which contain the N’ gene. Their results indicated that the viral factor responsible for the necrotic response in N’ plants is coded for in the coat protein gene. This response is discussed further in Chapter 10 (Section III.E.1).
Another example of the use of recombinants constructed in vitro is given by the work of Woolston et al. (1983) with CaMV. They infected plants with hybrids constructed from the genomes of an aphid-transmissible and an aphid non-transmissible strain of the virus. The results showed that aphid transmission and the synthesis of an 18-kDa protein were located in either ORF I or ORF II. Tests with a deletion indicated that ORF II was the gene involved.
It is also possible to construct viable recombinant hybrids between different viruses. Sacher et al. (1988) used biologically active cDNA clones to replace the natural coat protein gene of BMV RNA3 with the coat protein gene of SHMV. In SHMV the origin of assembly lies within the coat protein gene. In barley protoplasts co-inoculated with BMV RNAs 1 and 2, the hybrid RNA3 was replicated by trans-acting BMV factors, but was coated in TMV coat protein to give rod-shaped particles instead of the normal BMV icosahedra. However, since functions are highly integrated in a viral genome it is sure to be able to produce viable recombinants between distantly-related viruses.
One further application of recombinants is the tagging of gene products with fluorescent or other probes that report where in the plant or protoplast that gene product is being expressed or accumulates. This is described more fully in Chapter 9 (Section II.B).
d. Expression of the gene in a transgenic plant
As with mutagenesis of infectious cloned genomes of viruses, the technique of transforming plants with viral (and other) sequences has had a major impact on understanding viral genes and control functions and there are numerous examples of their expression in transgenic plants. The technique is described in detail in numerous texts, including Old and Primrose (1989) and Draper and Scott (1991). The basic features of the technique are that a construct comprising the gene of interest, a promoter, often the 35S promoter of CaMV (see Section IV.C.1) and a transcriptional terminator sequence, are introduced into suitable plant material. The plant material was originally protoplasts but now usually embryonic cell suspensions or similar meristematic tissue from which plants can be regenerated.
There are two commonly used ways of introducing the construct into the plant material. In the biolistic approach, the construct to be introduced is coated on to small microparticles which are propelled into the plant tissues by an explosive or blast of high pressure. The other approach is to use the integrating properties of the Ti plasmid of Agrobacterium tumefaciens. The construct of interest is placed in a T-DNA plasmid that retains the integrating properties (see Old and Primrose, 1989) but has tumor-inducing genes deleted. This is then co-cultivated with the plant tissue, allowing the integration of the construct. In both approaches, a selection marker, usually an antibiotic resistance gene or a herbicide tolerance gene, is included so that successful transformation events can be identified and isolated.
e. Bacterial, yeast and insect cell systems
There are numerous bacterial systems that are used for the expression of proteins and many commercial kits available. Basically, the gene for the protein of interest is cloned into the appropriate site in a vector (an expression vector), which is then transformed into a bacterium, usually E. coli. By cloning in frame with a known sequence at the N or C terminus of the protein of interest, that protein can be ‘tagged’–which facilitates its purification. The main problems with bacterial expression of plant viral proteins are:
-
1.
They are expressed in a prokaryotic system and will not be modified (say phosphorylated or glycosylated) in the manner that they would be in a eukaryotic system.
-
2.
They may be processed by prokaryotic enzymes (e.g. proteases) in a manner not found in eukaryotic systems.
-
3.
They may prove toxic to the bacterium.
There are two eukaryotic systems commonly used for the expression of plant viral gene products. A frequently used vector is derived from the Autographica californiea nuclear polyhedrosis virus (AcMNPV), which is a member of the Baculoviridae, a large family of occluded viruses pathogenic to arthropods. Baculoviruses occlude their virions in large protein crystals, the matrix of which is composed primarily of polyhedrin, a protein of about 29 kDa. In the baculovirus expression system, the polyhedrin gene, which is not required for viral replication, is replaced by the gene of interest (Smith et al., 1983; Lucknow and Summers, 1988). The foreign gene is expressed from the polyhedrin promoter on infection of an insect cell line, such as Sf21 derived from the moth Spodoptera frugiperda. There are many variants on these baculovirus-based expression systems; for more details see King and Possee (1992).
The other eukaryotic system involves yeast. There is a large pool of information available on the classical and molecular genetics of Saccharomyces cerevisiae (see Botstein and Fink, 1988; Ausubel et al., 1998) and an increasing amount on Schizosaccharonyces pombe. Vector systems are available for the expression of foreign genes in yeasts. As described in Chapter 8 (Section III.A.6), yeast systems are used for the analysis of interactions between proteins and also for unraveling details of viral replication (see Chapter 8, Section III.A.6).
f. Hybrid arrest and hybrid selection procedures
Hybrid arrest and hybrid selection procedures can be used to demonstrate that a particular cDNA clone contains the gene for a particular protein. In hybrid arrest, the cloned cDNA is hybridized to mRNAs, and the mRNAs are translated in an in vitro system. The hybrid will not be translated. Identification of the missing polypeptide defines the gene on the cDNA.
In the hybrid selection procedure, the cDNA-mRNA hybrid is isolated and dissociated. The mRNA is translated in vitro to define the encoded protein. In appropriate circumstances, these procedures can be used to identify gene function. For example, Hellman et al. (1988) used a modified hybrid arrest procedure to obtain evidence identifying the protease gene in TVMV.
g. Sequence comparison with genes of known function
As noted in Section III.B.2.d, sequence comparisons can be used to obtain evidence that a particular ORF may be functional. The same information may also give strong indications as to actual function. For example, the gene for an RNA-dependent RNA polymerase (RdRp) was identified in poliovirus. The study by Kamer and Argos (1984) revealed amino acid sequence similarities between this poliovirus protein and proteins coded for by several plant viruses. This similarity implied quite strongly that these plant viral-coded proteins also have a polymerase function. The conserved amino acid sequences (motifs) of RdRps and many other viral gene products are described at the appropriate places in this book (e.g. for RdRps, see Chapter 8, Section IV.B.1).
h. Functional regions within a gene
Spontaneous mutations and deletions can be used to identify important functional regions within a gene. However, mutants obtained by site-directed mutagenesis, and deletions constructed in vitro can give similar information in a more systematic and controlled manner. For example, the construction and transcription of cDNA representing various portions of the TEV genome, and translation in vitro and testing of the polypeptide products, showed that the proteolytic activity of the 49-kDa viral proteinase lies in the 3’ terminal region. The amino acid sequence in this region suggested that it is a thiol protease related in mechanism to papain (Carrington and Dougherty, 1987a). Proteinases are further described in Section V.B.1.a.
However, care must be taken with this approach. Many functions depend upon the three-dimensional structure of the protein, and mutations not at the active site may have a secondary effect on the protein structure.
IV. SYNTHESIS OF mRNAs
As noted earlier (Fig. 7.1), Baltimore (1971) pointed out that the expression of all viruses has to pass through an mRNA stage. The (+)-sense ssRNAs of many genera of plant viruses can act as mRNAs directly on entry into the host cell. For viruses with other types of genome, mRNAs have to be synthesized at some stage of the infection cycle.
A. Negative-sense single-stranded RNA viruses
All viruses with a (–)-sense ssRNA genome carry the viral RdRp in their virus particles. Thus, one of the early events on entry into a host cell is the transcription of the viral genome to (+)-sense RNA required for both translation of the viral genetic information and as an intermediate for replication. Replication of such viruses is described in Chapter 8 (Section V). Here I will discuss how the mRNAs are formed.
1. Plant Rhabdoviridae
Plant rhabdoviruses, like those infecting vertebrates, possess a genome consisting of a single piece of (–)-sense ssRNA, with a length in the range 11–13 kb and encoding six proteins, one more than animal rhabdoviruses (see Chapter 6, Section VII.A).
From patterns of hybridization with cDNA clones, Heaton et al. (1989a) showed that the SYNV genome is transcribed into a short 3’-terminal ‘leader’ RNA and six mRNAs. Thus, the plant rhabdoviruses appear to be expressed in a manner similar to animal rhabdoviruses such as vesicular stomatitis virus (VSV), which has been studied much more extensively (reviewed by Rodriguez and Nichol, 1999). For vesicular stomatitis virus (VSV), the active transcribing complex consists of the RNA genome tightly associated with the N protein, and the polymerase made up of the phosphoprotein (P) and the large (L) protein. This complex starts transcribing (+)-sense RNA at a single entry site at the 3’-end of the genome and transcribes the leader RNA that is transported to the nucleus where it inhibits host cell transcription. The complex then transcribes the mRNA for the N protein, which is capped during synthesis by the polymerase. At the end of the N gene, and of all genes, is the sequence 5’-AGUUUUUUU-3’ (element I) which signals termination and polyadenylation of the mRNA. This intergenic sequence also comprises a short untranscribed sequence (element II) and the start site for transcription of the next mRNA (element III). Similar sequences are found in plant rhabdoviruses (Fig. 7.10 ) (see Jackson et al., 1999).
Fig. 7.10.

Alignment of the intergenic regions of selected plant and animal rhabdoviruses. The rhabdovirus consensus sequence is shown at the top followed by the sequences of SYVV, LNTV, vesicular stomatitis virus (VSV) and rabies virus (RV). The intergenic sequences (‘gene-junction’ sequences) are separated into three elements: element I constitutes the poly(U) tract at the 3’-end of each gene on the genomic RNA; element II is a short sequence that is not transcribed during mRNA synthesis; element III constitutes the start site for transcription of each mRNA. The bold type in the viral sequences indicates the consensus nucleotides. P indicates pyrimidine, (N)x corresponds to a variable number of nucleotides.
From Jackson et al. (1999), with permission.
© 2002
Thus, the viral genes are transcribed separately from the 3’-end and they are transcribed in decreasing amounts (N>P>sc4>M>G>L) (Wagner and Jackson, 1997). This is an efficient way of regulating gene expression, as the genes that are located at the 3’-end are those that are required in greatest amounts.
2. Tospoviruses
As described in Chapter 6 (Section VII.B.1), the genomes of tospoviruses comprise three ssRNA segments. L RNA is (–)-sense and monocistronic encoding the viral RdRp. The mRNA is transcribed from the virion RNA by the virion-associated polymerase.
The other two RNAs have an ambisense gene arrangement (see Fig. 6.11) with one ORF in the viral strand and one in the complementary strand. The two ORFs are separated by an AU-rich intergenic region of variable length. For both RNAs the virion-sense ORF is expressed from an sgRNA transcribed from the complementary RNA and the complementary-sense ORF from an sgRNA transcribed from the virion RNA (see Fig. 5.42) (de Haan et al., 1990; Kormelink et al., 1992b, 1994).
The intergenic region between the ambisense ORFs is predicted to form stable hairpin structures that are suggested to control the termination of transcription of sgRNAs. However, as noted in the next section this should be considered with circumspection.
As described in Section V.C.4, formation of tospovirus mRNAs involves cap-snatching.
3. Tenuiviruses (reviewed by Falk and Tsai, 1998)
The genome organization of tenuiviruses is described in Chapter 6 (Section VII.B.2). Members of this genus have genomes divided between four or more ssRNA. As with the tospoviruses, the largest RNA is (–)-sense and monocistronic and is considered to be expressed from transcripts made using the virion-associated, virus-encoded RdRp.
Most of the other species in this genus have three other RNAs, each containing two ORFs in an ambisense arrangement (see previous section, and Fig. 6.12). When the MSpV and RHBV virion RNAs are translated in vitro, only a few proteins, including the NCP and N proteins, are detectable (Falk et al., 1987; Ramirez et al., 1992). RNAs corresponding to, but shorter than, RNAs 2, 3 and 4 are found in infected plants and insects. Northern hybridization analysis using strand-specific probes show that these RNA correspond in size and polarity to the ORFs on the ambisense RNAs (Falk et al., 1989; Huiet et al., 1991; Huiet et al., 1992; Estabrook et al., 1996). As at least some of these RNAs are found associated with polyribosomes (Estabrook et al., 1996), they are interpreted as being sgRNAs that arise from transcription in a manner similar to that described above for tospoviruses (Fig. 5.42).
The ambisense ORFs are separated by an AU-rich intergenic region of varying length (see de Miranda et al., 1994, 1995a). It has been suggested that the intergenic regions of RNAs with ambisense ORF arrangement fold into hairpin structures that function in transcriptional termination(Emery and Bishop, 1987; Kakutani et al., 1991); but de Miranda et al. (1994) found that the predicted folding for the RHBV RNA3 intergenic region differed according to what was being analyzed. Stable hairpin structures could be predicted if the computer-assisted folding was performed on the intergenic region alone but not if it was on the whole RNA.
As described in Section V.C.4, the formation of MSpV sgRNA involves cap-snatching. Cap-snatching has been demonstrated for the tospovirus TSWV and tenuivirus MSpV (Estabrook et al., 1998). It is likely that other tenuiviruses also cap-snatch.
B. Double-stranded RNA viruses
1. Plant Reoviridae
Plant members of the Reoviridae family are placed in three genera: Phytoreovirus with 12 dsRNA genome segments, and Fijivirus and Oryzavirus each with 10 dsRNA genome segments. The genome organizations of these genera are shown in Chapter 6 (Section VI). Most of the dsRNA segments are monocistronic but the Fijiviruses RBSDV segments 7 and 9, MRDV segments 6 and 8, OSDV segments 7 and 10, the Phytoreovirus RDV segment 11 and the Oryzavirus segment 4 are bicistronic; RDV segment 12 possibly has three ORFs. However, there is no evidence of these downstream ORFs being expressed.
The plant reoviruses, like their counterparts infecting vertebrates and insects, contain a transcriptase that uses the RNA in the particle as template to produce ssRNA copies. In animal reoviruses, this occurs in subviral particles comprising part of the capsid, the polymerase and the dsRNAs (reviewed by Joklik, 1999; Lawton et al., 2000). Early in infection only (+)-sense ssRNAs are synthesized which act as mRNAs. Later, (–)-sense strands are synthesized leading to viral replication (see Chapter 8, Section VI.A). It is likely that a similar series of events occurs in the plant reoviruses, especially when they multiply in their insect vectors.
C. DNA viruses
The synthesis of mRNAs from either the dsDNA members of the Caulimoviridae or the ssDNA members of the Geminiviridae and the nanoviruses does not involve a virus-coded enzyme but is performed by the host DNA-dependent RNA polymerase II located in the nucleus. This synthesis is initiated by viral promoter sequences, and so in this section I will consider these sequences in the plant DNA viruses. Plant viral DNA promoter sequences have been used widely in gene vectors in plants (see Chapter 16, Section IX.B.1).
1. Caulimoviridae
The genome organizations of the Caulimoviridae genera are described in Chapter 6 (Section IV). Most of the detailed studies have been performed on CaMV and these observations most likely pertain to all members of this family.
As described in Chapter 8 (Section VI.B), there are two phases in the nucleic acid replication cycle of CaMV, the nuclear phase of transcription and the cytoplasmic phase of gene expression and reverse transcription. In the first, the dsDNA of the infecting particle moves to the cell nucleus, where the overlapping nucleotides at the gaps are removed, and the gaps are covalently closed to form a fully dsDNA. These mini-chromosomes form the template used by the host DNA-dependent RNA polymerase to transcribe two RNAs of 19S and 35S, as indicated in Fig. 6.1. As well as promoters for these two mRNAs, the viral DNA also has signals for the polyadenylated termination of transcription.
a. The 35S promoter (reviewed by Hull et al., 2000a)
The identification of a promoter involves mapping the 5’-end of the transcript on to the viral genome. This has been performed for the 35S RNAs of CaMV (Odell et al., 1985), CsMV (Verdaguer et al., 1996), FMV (termed the 34S promoter) (Sanger et al., 1990; Maiti et al., 1997), MMV (Day and Maiti, 1999), PClSV (Maiti and Shepherd, 1998), SoyCMV (Hasegawa et al., 1989), SVBV (Wang et al., 2000), CoYMV (Medberry et al., 1992), RTBV (Bhattacharyya-Pakrasi et al., 1993; Bao and Hull, 1993a; Chen et al., 1994),and SCBV (Tzafrir et al., 1998; Schenk et al., 1999). The approach to studying these promoters involves transgenic or transient expression of constructs comprising the promoter region coupled to a reporter gene, usually the uidA gene expressing β-glucuronidase (GUS). The promoter region usually consisted of several hundred nucleotides upstream of, and up to one hundred nucleotides downstream of, the transcription start site. Mutagenesis and deletion analysis was then used to dissect the regions responsible for the strength and tissue specificity of the promoter.
These studies show that the promoter sequences comprise the core promoter upstream of the transcription start site and various control elements both upstream and downstream of the start site. The core promoter is characterized by what is termed a ‘TATA box’ about 25 nucleotides upstream of the start site (Table 7.1 ).
TABLE 7.1.
Defined and putative promoter sequences of some caulimoviruses and badnaviruses
| as-1 sequence | TATA sequences | Transcription start | Poly(A) signal | |||||
|---|---|---|---|---|---|---|---|---|
| CaMV | 7850 | cacTGACGtaagggaTGACGcac | 34 | ctcTATATAAgca | 21 | ACACGCG | 154 | atcAATAAAttt |
| CVMV | 7380 | tgaAGACGtaagcacTGACGaca | 34 | tccTATATAAgga | 24 | AAGAAAA | ||
| FMV | 6857 | gtaTGACGaacgcacTGACGacc | 13 | ctcTATATAAgaa | ||||
| MMV | aaaTGACGtaagccaTGACGtct | 21 | tccTATATAAgga | 15 | GAAGAGA | 186 | atcAATAAAata | |
| BSV | 7083 | tagTCACGcacga–TGACCttt | 181 | ctcTATATAAgga | 20 | ACACGCA | ||
| RTBV | 7370 | cagTATATAAgga | 21 | TCATCGA | 184 | atcAATAAAgct |
In the transcription start sequences, the nucleotide indicated in ‘outline' is +1 of the transcript. Gaps are where reliable information is not available.
Data from: Sanger et al. (1990); Bao et al. (1993a); Verdaguer et al. (1996); Harper and Hull (1998); Dey and Maiti (1999); all with permission.
© 2002
A detailed analysis of the CaMV promoter revealed that it had a modular nature (reviewed by Benfey and Chua, 1990) with subdomains conferring patterns of tissue-specific expression. Two major domains were identified, domain A (−90 to +8) (numbering relative to transcription start site at +1) which is important for root-specific expression, and domain B (–343 to −90) mainly involved in expression in the aerial parts of the plant. The region of the A domain between −83 and −63 contains an as-1 (activation sequence-1)-like element that is important for the root-specific expression. The as-1 element is present in several non-viral promoters and can be recognized in many of the caulimovirus promoters where it is important. The B domain was dissected further into five subdomains, B1 to B5, each conferring specific expression patterns in developing and mature leaves. Thus, in the full promoter these domains and subdomains act co-ordinately and synergistically to give the constitutive expression of the CaMV 35S promoter. Plant nuclear factors have been identified that bind to various regulatory regions in this promoter (Benfey and Chua, 1990; Hohn and Fütterer, 1992; Sanfaçon, 1992) and also to the RTBV promoter (Yin and Beachy, 1995). Other caulimovirus promoters also show similar modular structures. For instance, three overlapping regions have been identified in the CsVMV promoter (Verdaguer et al., 1998), −222 to −173 controlling expression in green tissues and root tips, −178 to −63 giving vascular-specific expression, and −149 to −63 controlling expression in mesophyll tissues. The promoters of some viruses require sequences downstream of the transcription start site for maximum expression. An example of this is MMV promoter for which the region of +33 to +63 is essential for maximum expression (Day and Maiti, 1999). Similarly, efficient transcription from the RTBV promoter requires an enhancer located in the first 90 nucleotides of the transcript (Chen et al., 1996). Two sub-elements were identified in this enhancer region, one being independent of position and orientation and the other being position-dependent.
The RTBV promoter has been analyzed in detail (Yin and Beachy, 1995; Yin et al., 1997a,b; Klöti et al., 1999) and shown to comprise several elements (Fig. 7.11 ). Some of these elements are upstream of the transcription initiation site but others are downstream (Chen et al., 1996).
Fig. 7.11.

Organization of elements influencing expression from the RTBV promoter. The location of regions containing different elements with apparent activity in transcription control that can be deduced from Klöti et al. (1999) are indicated below a schematic presentation of the transcription unit; stippled lines indicate supposed activity. Sequence elements defined by Yin and Beachy (1995) and Yin et al. (1997a,b) are indicated above. Positions are given relative to the transcription start site. GAGA-like elements are indicated by *; SD, splice donor; SA splice acceptor; lppc+, regions stimulating assembly of low-processivity polymerase complexes.
From Klöti et al. (1999) with kind permission of the copyright holder, © Kluwer Academic Publishers.
© 2002
Some promoters are specific to the vascular tissue. In that of RTBV the region between −164 and −100 is essential for vascular tissue expression (Klöti et al., 1999) and deletion leads to specificity in the epidermis. This tissue specificity is not surprising for RTBV as the virus itself is phloem-limited. However, the CoYMV promoter gives expression primarily in the phloem (Medberry et al., 1992) but the parent virus infects most tissues.
Most of the caulimovirus promoters act in both monocot and dicot plant species even though the parent virus is restricted in host range. The CaMV promoter has been used for the expression of transgenes in many dicot and monocot plant species and is considered a good strong constitutive promoter. It has also been shown to be active in bacteria (Assad and Signer, 1990), in yeast (Pobjecky et al., 1990), in animal HeLa cells (Zahm et al., 1989) and in Xenopus oocytes (Ballas et al., 1989).
b. The 19S promoter
The caulimovirus 19S promoter (reviewed by Rothnie et al., 1994) has been much less studied. Only that of CaMV has been analyzed and shown to be weaker than the 35S promoter when tested in transgenic constructs. For example, expression of the α-subunit of β-conglycinin in petunia plants under control of the 35S promoter was 10–50 times greater than from the 19S promoter (Lawton et al., 1987). The 35S promoter was also found to be 10–30 times more effective than the nopaline synthase promoter from Agrobacterium tumefaciens (García et al., 1987). This is in contrast to virus infections leading to comparable levels of the 35S and 19S RNAs and the product of the gene encoded by the 19S RNA, the ORF VI product, being the most abundant viral protein. The core 19S promoter can be strongly activated by the 35S promoter enhancer elements but no enhancer elements have been detected for the promoter itself.
c. The polyadenylation signal (reviewed by Rothnie et al., 1994)
The caulimovirus 35S and 19S RNAs are 3’ co-terminal and share a polyadenylation signal. The signal motif, AAUAAA, is found upstream of the transcription termination or cleavage site but downstream of the transcription initiation site (Table 7.1). It can be seen from Table 7.1 and Fig. 6.1 that, to generate the more-than-genome length 35S transcript, the transcription termination and polyadenylation signal has not to be effective the first time that it is passed. Sanfaçon and Hohn (1990) suggested that it is the proximity to the transcription start signal that occludes the polyadenylation signal on the first passage. However, sequences upstream of the transcription start site are not required for the initiation of polyadenylation and all the information for efficient polyadenylation are in the repeated region. The bypass is not 100% efficient and short-stop transcripts can be detected arising from first-pass processing. Notable amounts of short-stop transcripts are found in RTBV infections (Klöti et al., 1999). The RTBV polyadenylation signals are very similar to those of CaMV (Rothnie et al., 2001).
2. Geminiviridae
The circular ssDNA genomes of members of the Geminiviridae have ORFs both in the virion-sense and complementary-sense orientations (see Figs. 6.5–6.8 Fig. 6.6 Fig. 6.7 Fig. 6.8). All geminiviruses employ the same basic strategy to transcribe their genomes in that it is bi-directional from at or near the common region (see Chapter 6, Section V.A, for description of the common region), terminating diametrically opposite. However, there are differences between the genera in the details of transcription that are reviewed by depth by Hanley-Bowdoin et al. (1999). Here I will summarize the transcription, and for original references the reader is referred to the review.
a. Begomoviruses
The most detailed studies on begomoviruses have been on TGMV, the DNAA of which is transcribed into six polyadenylated RNAs and DNA B into four polyadenylated RNAs (Fig. 7.12 ).
Fig. 7.12.
Diagram of transcription of begomoviruses. (a) TGMV; (b) TLCV. Genome maps are described in Fig. 6.7. The outer arcs represent the transcripts with the arrowheads indicating the 3’-ends.
Data from Sunter and Bisaro (1989) and Mullineaux et al. (1993), with permission.
© 2002
The polyadenylation sites for the virion- and complementary-sense RNAs overlap so they share a few 3’ nucleotides. Each genome component gives rise to a single virion-sense RNA that is translated into either the coat protein from DNA A or BV1 from DNA B. Complementary-sense transcription is much more complex and gives rise to multiple overlapping RNAs with common 3’-ends but differing in their 5’-ends. The three complementary-sense RNAs from TGMV B DNA all translate to give protein BC1, whereas those from DNA A have different coding capacities. The largest transcript, AC61 (transcripts are designated according to their 5’-ends so AC61 is from the complementary side of DNA A and starts at nucleotide 61), encodes the entire left side of DNA A and is the only RNA giving full-length Rep protein (see Chapter 8, Section VIII.D.4, for Rep proteins). AC2540 and AC2515 may express ORF AC4, and the smallest RNAs (AC1938 and AC1629) specify AC2 from their first ORF and AC3 from their second ORF.
Upstream of the TGMV mRNAs are characteristic eukaryotic RNA polymerase II promoter sequences (reviewed by Hanley-Bowdoin et al., 1999). Transcription of each of four RNAs initiates 20–30 bp downstream of the TATA box motifs, whereas the other RNAs have sequences resembling initiator elements overlapping their 5’-ends. The promoters for the complementary-sense AC61 and AC1629 mRNAs and for the virion-sense AV1 and BV1 RNAs have been studied in some detail. The AC 61 promoter maps to the TGMV A common region and supports high levels of transcription. Deletion mutagenesis showed that most of its activity resided in the 60 bp immediately preceding the AC61 transcription start site, a region that overlaps the origin of (+)-strand DNA synthesis (see Chapter 8, Section VIII.D.3, and Fig. 8.27). These mutations showed the close interactions between transcription and replication. Mutations in both the host factor binding sequences and in the G-box reduced promoter function. The AC61 promoter is autoregulated through the Rep binding site, the repression being specific for the homologous Rep protein and is thought to involve active interference with the transcription apparatus and not just steric hindrance. The AC4 protein also negatively regulates this promoter, the cis element involved being upstream of the G-box and being distinct from the Rep binding site. The analogous promoters of most other begomoviruses are probably regulated by similar mechanisms, but those of some (e.g. ACMV) differ in detail (reviewed by Hanley-Bowdoin et al., 1999). The BC1 promoter sequences are similar to those of the AC61 promoter but differ in the transcription start site and in that the Rep protein does not regulate it.
The virion-sense promoters of TGMV are not as well characterized as those described above for the complementary-sense RNAs. The promoter regions include the common regions and the downstream sequences containing the TATA boxes. The virion-sense promoters require the AC2 protein in trans, the activation being independent of replication. Study of the TGMV AV1 promoter in transgenic plants indicated that its regulation is complex and is controlled differently in different tissues (Sunter and Bisaro, 1997). The promoter for the AC2 gene, which lies upstream of the AC1629 transcription start site, is not responsive to Rep, AC2 or AC3 (reviewed by Hanley-Bowdoin et al., 1999). The regulation of this promoter is unclear and possibilities are discussed by Hanley-Bowdoin et al. (1999).
Some of the begomoviruses have monopartite genomes the transcription of which has been much less studied than that of the bipartite members. Four major transcripts were identified and mapped to the TLCV genome (Mullineaux et al., 1993) (Fig. 7.12). One transcript spanned the C1, C2 and C3 ORFs and a second covered C2 and C3; the C4 ORF, located within C1, was not recognized at that time. On the virion-sense side, there are two 3’ co-terminal transcripts, the 5’-ends of which map either side of the first in-frame AUG codon of the V1 ORF.
To analyze the regulation of expression of TLCV, Dry et al. (2000) studied the expression of fusions of the ORFs with the GUS reporter gene in both stably and transiently transformed Nicotiana tabacum tissues. They showed that the C2 ORF curtailed the expression of the V2 ORF, indicating that the C2 protein is involved in transactivation of virion-sense gene expression. The TLCV ORF-GUS constructs had distinctive tissue expression patterns in transgenic tobacco plants: C1, C4 and V2ΔC (deletion at C terminus of V2 ORF) were constitutive; C2 and C3 were predominantly vascular; V1ΔC reduced expression in cells associated with vascular bundles.
b. Curtoviruses
The single-component curtoviruses produce six or seven proteins in infected plants (Fig. 7.13 ). Frischmuth et al. (1993) identified an abundant virion-sense population of polyadenylated RNA and four complementary-sense polyadenylated RNAs in BCTV-infected plants. The population of virion-sense RNAs comprises 3’ co-terminal overlapping transcripts, the 5’ termini of which are positioned so that they express ORFs V1, V2 and V3. There are two consensus TATA boxes at the appropriate position upstream of the larger two virion-sense RNAs, but there is no detailed information on these or any other promoters in curtoviruses.
Fig. 7.13.
Diagram of transcription map of BCTV. Genome map is described in Fig. 6.6. The outer arcs represent the transcripts with the arrowheads indicating the 3’-ends. The dotted arc is a minor transcript but fits with being that for V1, the coat protein. It possibly was a minor transcript at the sampling time but a major one at another time. The complementary sense transcript(s) were not mapped because deletions complicated their analysis.
Data from Frischmuth et al. (1993), with permission.
© 2002
c. Mastreviruses
The mastreviruses have a single component of ss DNA that codes for four proteins (Fig. 7.14 ), V1 and V2 being expressed from transcripts in the virion sense, while C1 and C2 are expressed from transcripts in the complementary sense.
Fig. 7.14.
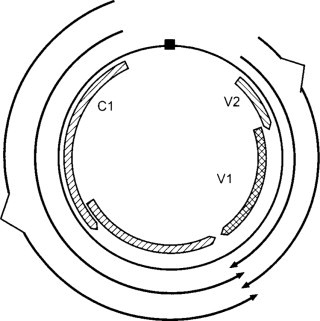
Diagram of transcription map of MSV. Genome map is described in Fig. 6.5. The outer arcs represent the transcripts with the arrowhead indicating the 3’-end. The breaks in the outer arcs linked by angled lines show the site of introns spliced out during processing of transcripts.
Data from Wright et al. (1997), with kind permission of the copyright holder, © Blackwell Science Ltd.
© 2002
The bi-directional transcripts have multiple initiation sites and terminate at overlapping polyadenylation signals. However, unlike the other two geminivirus genera, the production of mRNAs from both the virion and complementary senses involves splicing. The C transcripts are of low abundance and a splicing event fusing C1 to C2 has been found for MSV, DSV and TYDV (Mullineaux et al., 1990; Dekker et al., 1991; Morris et al., 1992). In MSV about 20% of the C transcript is spliced (Wright et al., 1997). For MSV there are two V-sense transcripts, the most abundant starting one nucleotide upstream of the V1 ORF, and the longer, least abundant one initiating 141 nucleotides upstream of V1 (see Wright et al., 1997). About 50% of the major transcript is spliced opening up the V2 ORF and about 10% of the longer transcript is spliced to give an mRNA for V1 (Wright et al., 1997). The products of C1 and C2 are early functions involved with viral replication and the C1:C2 fusion product is essential. The V1 product (movement protein) and V2 product (coat protein) are late functions, the coat protein being required in much greater amounts than the products from the other ORFs. Thus, the splicing is important in both the temporal and quantitative expression of mastreviruses.
Three TATA promoter consensus sequences may be involved in complementary-sense transcription of the MSV genome (Boulton et al., 1991b). As described in Chapter 10 (Section III.O.1.d), mutation of one of these promoters affected both symptoms and host range possibly by indirectly influencing viral replication by influencing the synthesis of the Rep protein. Using a maize protoplast transient expression system, Fenoll et al. (1988) defined further the structure of the MSV virion-sense promoter that drives transcription of the RNA for coat protein. They identified a 122-bp sequence upstream from the start site for transcription that enhances promoter activity. The 122-bp sequence activates the MSV core promoter in a position-dependent, but orientation-independent fashion. The activating sequence specifically binds proteins in maize nuclear extracts. This ‘upstream activating sequence’ lies in the large intergenic region and includes the common region and was mapped to two GC-rich boxes on the distal side of the origin hairpin motif relative to the transcription start site (Fenoll et al., 1990). These GC-rich boxes and the TGMV G-box element are positioned similarly with respect to the hairpins of the respective viruses, implying similar functions (Arguello-Astorga et al., 1994). Thus, mastreviruses may encode a transactivator of virion-sense transcription analogous to the AL2/C2 proteins of begomoviruses.
All the MSV promoters show cell-cycle specificity (Nicovics et al., 2001). The coat protein promoter had highest activity in early G2, whereas the C-sense promoter sequences produced two peaks of activity, in the S and G2 cell-cycle phases.
d. Polyadenylation
As can be seen from Fig. 7.12, Fig. 7.13, Fig. 7.14, the transcripts of geminiviruses terminate on the opposite side of the genome to the promoters. This is an AT-rich region that contains putative signals for polyadenylation. However, no detailed analysis has yet been made of poly(A) signals in these viruses.
3. Nanoviruses
As described in Chapter 6 (Section V.B), the genomes of nanoviruses are divided between six or more circular ssDNA molecules of about 1 kb. Each genome segment encodes at least one protein. The promoters of these genome segments have been studied in most detail in BBTV. Beetham et al. (1997) showed that two mRNAs are transcribed from BBTV DNA-1, one covering the major ORF and the other mapped to an ORF for a 5-kDa protein completely within the major ORF. The promoter activities of all six BBTV DNAs were analyzed in tobacco cell suspensions, banana embryonic cells and transgenic tobacco and banana plants (Dugdale et al., 1998) (Fig. 7.15 ).
Fig. 7.15.
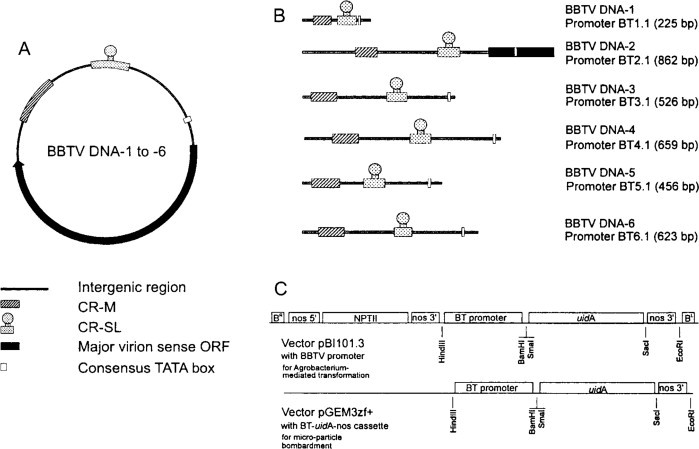
Schematic presentation of (A) general BBTV circular ssDNA genome organization, (B) BBTV DNA 1–6 promoter fragments, and (C) cloning strategy. Promoter fragments incorporating the intergenic regions of BBTV DNA 1–6 were isolated by PCR or restriction digestion from cloned components and inserted upstream of the uidA reporter gene in pBI101.3 for Agrobacterium-mediated transformation of tobacco. The BT-uidA-nos cassettes from each construction were subsequently cloned into pGEM-zf+ for microparticle bombardment transient assays.
From Dugdale et al. (1998), with permission.
© 2002
In these experiments the intergenic region of each genome segment that contains three regions of homology were fused to the uidA (GUS) and the green fluorescent protein (GFP) reporter genes. Two of the three homologous regions are associated with replication (see Chapter 8, Section VIII.E) and one contains the potential TATA box of the promoter. Dugdale et al. (1998) showed that (1) the intergenic regions of all six BBTV DNAs have promoter activity, (2) the activities of the different intergenic regions vary considerably, and (3) the relative activities of the different intergenic regions vary between tobacco and banana. In tobacco cell suspensions, transient expression from promoters of DNAs 2 and 6 was greater than that from the other promoters. In transgenic tobacco, the weak expression from each promoter was phloem-associated, that of the promoter of DNA 6 being the strongest. However, expression from the DNA 6 promoter became high in callus derived from transgenic tobacco. The sensitive GFP reporter could detect activity from these promoters only in banana embryonic cells, where the promoters of DNAs 4 and 5 gave the highest levels of transient activity. In transgenic banana, activity from the DNA 6 promoter was restricted to the phloem of leaves and roots, stomata and root meristems. Deletion analysis of the DNA 6 promoter suggested that the elements required for strong expression were within 239 nucleotides upstream of the translational start site.
V. PLANT VIRAL GENOME STRATEGIES
A. The eukaryotic protein-synthesizing system
It is generally accepted that the eukaryotic protein-synthesizing system translates the information from viral RNAs. This translation system has various features and controls (reviewed by Gallie, 1996):
-
1.
Plant cellular mRNAs have a cap (reviewed by Furuichi and Slatkin, 2000), an inverted and methylated GTP at the 5’ terminus [m7G(5’)ppp(5’)N] and a poly(A) tail at the 3’ terminus.
-
2.
In most circumstances, mRNAs contain a single open reading frame (ORF).
-
3.
Translation is initiated at an AUG start codon, the context of which controls the efficiency of initiation (Kozak, 1989, 1992).
-
4.
The cap, 5’ untranslated region, the coding region, the 3’ untranslated region and the poly(A) tail all have potential to influence translational efficiency and mRNA stability.
In the scanning model for translation (Kozak, 1989, 1992) the 40S ribosomal subunit binds to the 5’ cap (see Fig. 7.23 ), translocates to first AUG in a suitable context where it forms the 80S ribosome which translates only that ORF immediately downstream from the 5’ region of an mRNA; at the stop codon of this ORF the ribosomes dissociate. Thus, ORFs beyond this point normally remain untranslated. Viral genomes, except those of satellite viruses, encode two or more proteins, and therefore are presented with a problem of how to express their downstream proteins in the eukaryotic system. Much of the variation in the way gene products are translated from viral RNA genomes appears to have evolved to meet this constraint and at the same time to provide differential control of expression of the various ORFs.
Fig. 7.23.

Regulation of mRNA expression. (A) Schematic of a typical mRNA and the proteins that bind to the terminal regulatory elements. The initiation factors, eIF-4F, eIF-4A and eIF4B are shown associated with the 5’-terminal cap structure and the poly(A)-binding (PAB) protein is shown bound to the poly(A) tail at the 3’ terminus. After initiator factor binding, the 40S ribosomal subunit at or close to the 5’ terminus scans down the 5’ leader in search for the AUG initiation codon. (B) The co-dependent model of translation, the eIF-4F and eIF-4B are shown bound to both the 5’-terminal cap structure and the poly(A) tail. Protein-protein contacts between eIF-4F/eIF-4B, eIF-4A/PAB and eIF-4B/PAB that stabilize protein/mRNA binding are shown as the multiple thick lines between the proteins. This stable complex maintains the close physical proximity of the termini of the mRNA, which allows the efficient recycling of ribosomes. The 60S subunit is shown dissociating from the mRNA upon translation termination whereas the 40S subunit recycles back to the 5’ terminus to anticipate another round of initiation.
From Gallie (1996), with kind permission of the copyright holder, © Kluwer Academic Publishers.
© 2002
As noted above, the nucleotide sequence surrounding the AUG start codon is important in the efficiency of initiation of translation. For plant systems, the favourable context is AACAA UGG (Lehto et al., 1990a) with a purine at the −3 position and/or a guanine at the +4 positions (position numbering is relative to the A of the AUG codon which is designated +1) playing essential roles.
B. Virus strategies to overcome eukaryotic translation constraints
On current knowledge, there are at least 12 strategies by which RNA viral genomes and transcripts from DNA viruses ensure that all their genes are accessible to the eukaryotic protein-synthesizing system; see Fütterer and Hohn (1996), Gallie (1996) and Drugeon et al. (1999) for reviews. The strategies fall into three groups:
-
1.
Making the viral genomic RNA or segment thereof effectively monocistronic by bringing any downstream AUG to the 5’-end. This is done by either having a single ORF expressing a polyprotein that is subsequently cleaved to give the functional proteins (strategy 1), or by dividing up the viral genome to give monocistronic RNAs either during expression (strategies 2, 11 and 12) or permanently (strategy 3).
-
2.
Avoiding the constraints of the 5’ AUG. There are various strategies to do this (strategies 3 to 8).
-
3.
Maximizing the information expressed from a viral RNA by bringing together two adjacent ORFs to give two proteins, one from the 5’ ORF and the other from both ORFs. Thus the second protein comprises the upstream protein in its N-terminal region, the C-terminal region being from the downstream ORF (strategies 9 and 10).
Strategies 8, 9 and 10 have been termed ‘recoding’ (reviewed by Gesteland and Atkins, 1996).
1. Strategy 1: Polyproteins
Here the coding capacity of the RNA for more than one protein, and sometimes for the whole genome, is translated from a single ORF. The polyprotein is then cleaved at specific sites by a virus-coded proteinase, or proteinases, to give the final gene products. The use of this strategy is exemplified by the potyviruses.
a. Virus-coded proteinases (reviewed by Dougherty and Semler, 1993; Ryan and Flint, 1997; Spall et al., 1997)
Four classes of virus-coded proteinases are currently recognized: serine, cysteine, aspartic and metallo-proteinases named usually after their catalytic site. Three of these four types of proteinase are found among plant viruses (Table 7.2 ).
TABLE 7.2.
Proteinases encoded by plant viruses
| Virus group | Viral proteinase |
|---|---|
| Caulimoviridae | Aspartate |
| Potyviridae | |
| Potyvirus | Serine, cysteine, serine-likea |
| Bymovirus | Cysteine, serine-like |
| Comoviridae | Serine-like |
| Sequiviridae | Serine-like |
| Benyvirus | Cysteine?b |
| Marafivirus | Cysteine |
| Tymovirus | Cysteine |
| Closterovirus | Cysteine, aspartate |
| Polerovirus | Serine |
Serine-like proteinase with cysteine at its active site but with a structure similar to a serine proteinase.
? indicates doubt as to the type of proteinase.
Serine proteinases are also termed 3C proteases (from their expression in picornaviruses) or chymotrypsin-like proteases. Most have a catalytic triad of amino acids His, Asp, Ser, but in some the Ser is replaced by Cys; as the latter have the same overall structure as serine proteinases they are termed ‘serine-like’ proteinases. The serine residue is unusually active and acts as a nucleophile during catalysis by donating an electron to the carbonyl carbon of the peptide bond to be hydrolyzed. An acyl serine is formed and a proton donated to the departing amyl group by the active-site histidine residue. The acyl enzyme is then hydrolyzed, the carboxylic acid product is released, and the active site is regenerated. Serine proteases cleave primarily at Gln–Gly, Gln–Ser, Gin–Ala and Gln–Asn (reviewed by Palmenberg, 1990).
Cysteine proteinases, also known as papain-like or thiol proteinases, have a catalytic dyad comprising Cys and His residues in close proximity that interact with each other. During proteolysis, the Cys sulfhydryl group acts as a nucleophile to initiate attack on the carbonyl carbon of the peptide bond to be hydrolyzed. An acyl enzyme is formed through the carbonyl carbon of the substrate and the sulfhydryl group of the active-site His. The carbonyl carbon is then hydrolyzed from the thiol group of the proteinase and the active-site residues are regenerated.
Aspartic or acid proteinases are composed of a catalytic dyad of two Asp residues. They most likely do not form covalent enzyme-substrate intermediates and are thought to operate by an acid-base catalysis.
Viral proteinases are highly specific for their cognate substrates, a specificity that depends on the three-dimensional structures of both the proteinase and substrate. For instance, CPMV proteinase does not recognize primary translation products of M RNAs from other comoviruses (Goldbach and Krijt, 1982). This substrate specificity is further exemplified by experiments on CPMV reported by Clark et al. (1999) (see Section V.E.8 for processing of CPMV polyprotein). In these experiments, the regions encoding the large (L) and small (S) coat proteins on CPMV M RNA were replaced by the equivalent region of BPMV. These recombinant molecules replicated in cowpea protoplasts in the presence of CPMV B RNA. The junction between the 58/48-kDa and the L coat protein was cleaved. However, there was no processing of that between the L and S coat proteins, and thus no virus particles were formed, even when the sequence at that junction was made CPMV-like. Clark et al. (1999) also translated transcripts from their constructs in the in vitro rabbit reticulocyte system and showed that there was processing in trans, albeit somewhat inefficiently, when the L–S junction was made CPMV-like. This greater specificity in cleavage of the L–S junction in cis than in trans has also been shown for ToRSV (Carrier et al., 1999). It is suggested that the L–S cleavage site is defined by more than just a linear amino acid sequence and probably involves interactions between the L–S loop and the β-barrels of the viral coat proteins.
A fluorometric assay for TuMV proteinase was developed by Yoon et al. (2000). This showed that intramolecularly quenched fluorogenic substrates can be used for the continuous assay of TuMV NIa proteinase (and presumably other proteinases).
b. Potyvirus genus (reviewed by Reichmann et al., 1992; Shukla et al., 1994)
Potyviruses have genomes of approximately 10 kb that contain a single ORF for a polyprotein of about 3000 to 3300 amino acids. Shields and Wilson (1987) found no evidence for the presence of subgenomic RNAs in their preparations of TuMV. Likewise, Dougherty (1983) could find no evidence for authentic subgenomic RNAs in total RNA preparations made from TEV-infected leaves. The polyprotein is cleaved to give 10 proteins (Fig. 7.16 ) using three virus-coded proteases.
Fig. 7.16.

Schematic of the processing of the potyvirus polyprotein. The primary events are probably co-translational and autocatalytic, yielding precursors and mature products. There is little information about the sequential order of these events, nor about the extent, if any, of further C-terminal processing of primary cleavage products.
From Reichmann et al. (1992), with permission.
© 2002
The 35-kDa P1 cleaves itself from the polyprotein at Phe-Ser, the 52-kDa HC-Pro cleaves at its C-terminal Gly–Gly, and the 27-kDa protease domain of the NIa region is a serine protease responsible at most, if not all, the other cleavages at Gln–(Ser/Gly). Although there is not full information on the sequential order of the processing events, there is some evidence for some events taking place before others. For instance, the cleavage at the C terminus of HC-Pro takes place, giving P1-HC-Pro, before P1 is cleaved off (Carrington et al., 1989).
i. Potyviral proteases
P1 is a serine protease that cleaves P1 from the P1-HC-Pro product (reviewed by Reichmann et al., 1992; Shukla et al., 1994). It contains a serine protease active site sequence (HxgDx30 or 31G.x.S.G) in its C-terminal region. This protease has proved to be difficult to analyze, as it does not appear to function in rabbit reticulocyte lysate in vitro translation systems. However, it is active in wheatgerm in vitro translation systems and the cleavage site has been identified as between F and S in both TVMV and TEV. Comparison with homologous regions in other potyviral polyproteins indicates conservation of F or Y at the −1 position and S at the +1 position with preference for Q or H at −2 and M or I at −4. The P1 protein and the P1-HC junction appear not to be present in bymoviruses.
The enzymatic activity responsible for the HC-Pro-P3 cleavage is in a 20-kDa domain in the C-terminal half of the HC-Pro protein (Carrington et al., 1989). This proteinase has cysteine and histidine residues at its active site (Oh and Carrington, 1989) and resembles papain-like cysteine proteinases. The cleavage site specificity has been studied by site-directed mutagenesis and in vitro expression. Positions −4, −2, −1 and +1 of the G:G cleavage site were critical while −5, −3 and +2 were not (Carrington and Herndon, 1992). These sites are conserved in the aphid-transmitted potyviruses but not in the fungus-transmitted bymovirus BaYMV which has G/Y substitution at −4 and S/G substitution at +1 (Shukla et al., 1994).
The small nuclear inclusion protein, NIa, region contains the major proteinase of potyviruses. It has a two-domain structure, the N-terminal genome-linked protein VPg (22 kDa) and the C-terminal protease (27 kDa) (Dougherty and Carrington, 1988). NIa is autocatalytically released from the polyprotein and then catalyses the cleavage of the various junctions to release P3, 6K1, CI 6K2, NIb and CP. The cleavage of the VPg domain from the NIa protein is much less efficient.
Data from experiments using site-directed mutagenesis suggest that the catalytic triad of amino acids in the 49-kDa proteinase of TEV is probably His234, Asp269 and Cys339 (Dougherty et al., 1989). This motif is found in all potyviruses.
The cleavage sites have been identified for several viruses (Table 7.3 ) and the primary sites are considered to be QS, QG and QA with the motif V-X-X-Q/(A, S, G or V) thought to be common to all potyviruses. There is some variation on this motif (Table 7.3) and the site releasing the VPg from the NIa protein obviously differs from most of the others.
TABLE 7.3.
Amino acid sequences of the demonstrated and putative cleavage sites for the NIa and HC-Pro proteinases of four potyviruses and the NIa-like proteinase of BaYMV
| HC-Proa | PPV YLVG/GL | TEV YVVG/GM | TVMV YKVG/GL | PVY YRVG/GV | BaYMV |
|---|---|---|---|---|---|
| A | QVVVHQ/S | EDVLEQ/A | EIVEFQ/A | YDVRHQ/R | PKIVLQ/A |
| B | QAVQHQ/S | EIIYTQ/S | NNVRFQ/S | YEVRHQ/S | ASYGLQ/A |
| C | ECVHHQ/T | ETIYLQ/S | EAVRFQ/S | QFVHHQ/A | |
| D | EEVVHQ/G | EPVYFQ/G | EPVKFQ/G | ETVSHQ/G | |
| E | EFVYTQ/S | ELVYSQ/G | DLVRTQ/G | DVVVEQ/A | DIIHMQ/A |
| F | NWVHQ/A | ENLYFQ/S | ETVRFQ/S | YEVNHQ/A | DEIWLQ/A |
| V | EEVDHE/S | EDLTFE/G | QEVAFE/S | QEVEHE/A |
HC-Pro and letter (A-F and V) refer to sites shown in Fig. 7.16. Amino acids in bold indicate similarities between sites, and / indicates cleavage site.
Data from Reichmann et al. (1992), with permission.
© 2002
A seven-residue motif, E-X-X-Y-X-Q/(S or G), has been identified for TEV (Carrington and Dougherty, 1988). The rate of processing is thought to be controlled by additional virus-specific motifs that include hydrophobic residues at −4, residues other than A, G or S at +1, and residues further from the cleavage site. Controls on the cleavage site appear to cover at least the region of −7 to +2 (reviewed by Shukla et al., 1994).
The properties of the 27-kDa protease have been studied in detail in the cloned and expressed TuMV and TEV proteins. The optimum catalytic activity of the TuMV protein is at about 15°C and pH 8.5 (Kim et al., 1996). The proteins of both viruses are C-terminally processed to a 25-kDa product that has less activity than the 27-kDa protein (Kim et al., 1995; Parks et al., 1995); a second internal cleavage, leading to a 24-kDa product, has been identified for the TuMV protein (Kim et al., 1996). The cleavage site leading to the 25-kDa protein is unusual, being between a Ser and Gly for TuMV and a Met and Ser for TEV; that leading to the 24-kDa TuMV protein is between Thr and Ser. The 25-kDa product has reduced proteolytic activity and the 24-kDa protein has no detectable activity.
ii. In vivo protein synthesis
Some of the proteins found in vivo have been discussed in the preceding section. Donofrio et al. (1986) isolated an RdRp activity from corn infected with MDMV. The activity was solubilized and attributed to a ∼160-kDa protein. The subunit structure of the polymerase has not been characterized. Vance and Beachy (1984) found a genomic-length RNA associated specifically with active polyribosomes in extracts of soybean leaves infected with SMV. They concluded that this RNA is the only viral RNA translated in vivo. Full-length viral RNA, the complementary strand, and a ds viral RNA have been found associated with the chloroplast fraction in tissue extracts (Gadh and Hari, 1986). However, there was no evidence that viral RNA synthesis took place in the chloroplasts.
c. Discussion
There are both advantages and disadvantages to the polyprotein processing strategy. Apart from overcoming the non-5’ start codon problem, there are advantages both in the fact that several functional proteins are produced from a minimum of genetic information and also in the potential for regulating the processing pathway. This is shown in the differences in the cleavage sites for the NIa protease and the influence that the surrounding residues can have on the rates of cleavage. Similarly, the requirement for the processing at the C terminus of the HC-Pro before P1 is cleaved from the product most probably represents a control mechanism that is not yet understood.
The major disadvantage is that it is difficult to visualize how the polyprotein strategy of the potyviruses can be efficient. The coat protein gene is at the 3’-end of the genome (see Fig. 7.16). Thus, for every molecule of the 20-kDa coat protein produced by TEV, a molecule of all the other gene products has to be made, totalling about 320 kDa. Since about 2000 molecules of coat protein are needed to encapsidate each virus particle but probably only one replicase molecule to produce it, this appears to be a very inefficient procedure. Indeed large quantities of several gene products, apparently in a non-functional state, accumulate in infected cells (see Chapter 3, Section IV.C). Nevertheless, the potyviruses are a very successful group. There are many member viruses, and they infect a wide range of host plants. Other viruses using polyprotein processing have additional devices that can avoid this problem. Comoviruses have their two coat proteins on a separate genome segment (see Section V.E.8). There does not appear to be any massive accumulation of non-coat gene products in cells infected with these viruses.
2. Strategy 2: Subgenomic RNAs
Subgenomic RNAs (sgRNA) are synthesized during viral replication from a genomic RNA that contains more than one ORF giving 5’-truncated, 3’ co-terminal versions of the genome. This then places the ORFs that were originally downstream at the 5’-end of the mRNA. When several genes are present at the 3’-end of the genomic RNA, a family of 3’ collinear sgRNAs may be produced; CTV has a nested set of at least nine sgRNAs (Karasev et al., 1997). SgRNAs may be encapsidated (e.g. Bromoviridae, SHMV) and can cause uncertainty as to what comprises an infectious genome. The encapsidation of sgRNAs is dependent on the presence of the origin of assembly on that RNA, and this can differ between viruses within a genus or even between strains of a virus. For instance, the origin of assembly is present on the coat protein sgRNA of SHMV but not on the equivalent RNA of other tobamoviruses, such as TMV (see Chapter 5, Sections III.B and IV.A.2.a); the type strain of BaMV does not encapsidate any sgRNAs (Lin et al., 1992) whereas the V strain encapsidates two sgRNAs (Lee et al., 1998b).
a. Synthesis of subgenomic RNAs
At least four models have been proposed for the synthesis of sgRNA from the genomic RNA. These include:
-
1.
De novo internal initiation on the full-length (–) strand of the genome during (+)-strand synthesis.
-
2.
Initiation on the full-length (–) strand primed by a short leader from the 5’-end of the genomic DNA during (+)-strand synthesis. This has been found for coronaviruses (Liao and Lai, 1994).
-
3.
Intramolecular recombination during (–)-strand synthesis in which the replicase jumps from the subgenomic RNA start site on the full-length (+) strand and reinitiates near the 5’-end of the genome. This also has been found for coronaviruses (Sawiki and Sawiki, 1998).
-
4.
Premature termination during (–)-strand synthesis of the genome followed by the use of the truncated nascent RNA as a template for sgRNA synthesis.
The first and fourth mechanisms have been proposed for plant viruses.
b. De novo internal initiation
The simplest model for de novo internal initiation of sgRNAs necessitates the replicase recognizing a sequence upstream of the sgRNA 5’-end. This is termed the subgenomic promoter. Most studies on subgenomic promoters have been made on BMV but subgenomic promoters have been mapped for several other viruses (e.g. CuNV: Johnston and Rochon, 1995; PVX: Kim and Hemenway, 1996).
BMV has a tripartite genome, RNAs 1 and 2 being monocistronic and RNA3 (2114 nucleotides) dicistronic; the downstream ORF on RNA3, that encoding the coat protein, is expressed from an sgRNA, RNA4 (876 nucleotides) (see Fig. 6.13 for genome organization). Since RNAs 3 and 4 are 3’ co-terminal, the 5’-end of RNA4 maps to position 1238 on RNA3. Thus, a subgenomic promoter for RNA4 is likely to be upstream of position 1238, which is in an intergenic region.
The subgenomic promoter of BMV comprises a ‘core’ promoter, which is the smallest region capable of promoting sgRNA synthesis with low accuracy at a basal level, and ‘enhancer’ regions, which provide accuracy of replication initiation and control yields of sgRNAs. The fully functional subgenomic promoter encompasses about 150 nucleotides.
The BMV core promoter is the 20 nucleotides (−20 to +1) upstream of the subgenomic initiation site (the subgenomic initiation site is designated as + 1, and minus numbers are 5’ of that site as read on the positive-strand RNA) (Marsh et al., 1988; French and Ahlquist, 1988). Siegel et al. (1997, 1998) examined the structure of the core promoter by constructing ‘pro-scripts’ which comprised the core promoter and a template and which directed (+)-strand synthesis. By mutagenesis they showed that the nucleotides at positions −17, −14, −13 and −11 were essential for promoter activity. There was some evidence that the −17 nucleotide recognized the RdRp. The +1 and +2 nucleotides (cytidulate and adenylate) are also important for RNA synthesis (Adkins et al., 1998). The core promoter forms a stable hairpin structure (Jaspars, 1998).
In vitro studies identified three enhancer regions in the BMV promoter, the 16 nucleotides downstream of the start site (+1 to +16) which provide accurate initiation, and two upstream domains, the poly(A) stretch present in all bromoviruses (−20 to −37) and a triple repeat of UUA (-38 to −48) (Marsh et al., 1988). However, analysis in vivo indicated that the core region extended downstream and was from −20 to +16, that the poly(A) tract was essential as were three repeats of the sequence AUCUAUGUU extending the complete promoter to a site between −74 and −95 upstream of the start site (French and Ahlquist, 1988). Deletion of the poly(A) tract led to three revertants in which subgenomic RNA synthesis has been restored (Smirnyagina et al., 1994). Two of these revertants were in the subgenomic promoter and gave increased levels of genomic RNA3. In the third revertant, the mutation was upstream of the subgenomic promoter in a sequence, designated box B, which led to decreased levels of RNA3. Box B is an ICR2-like (internal control region 2) sequence similar to those found in cellular RNA polymerase III and in the TψU loop of tRNA as well as at the 5’-ends of BMV RNAs 1 and 2.
Both the BMV and CCMV RdRps recognize the BMV core subgenomic promoter requiring specific functional residues at positions −17, −14, −13 and −11 (Adkins and Kao, 1998). For CCMV sgRNA synthesis, both RdRps require the same nucleotides and four additional nucleotides at positions −20, −16, −15 and −10. The −20 nucleotides are partially responsible for the differential recognition of the two promoters.
The subgenomic promoter of AMV is more complex than that of BMV particularly in vivo, in that: (1) the sgRNA4 is the mRNA for coat protein which is required for (–)-strand replication; and (2) the intergenic region is only 49 nucleotides which is too short to accommodate the promoter. Thus, the promoter extends into the carboxy terminus of the movement protein gene (ORF 3a) which hampers the possibilities of mutational analysis for in vivo studies. However, characterization of the in vivo behaviour of the promoter was achieved making large enough insertions to contain it at the 5’-end of the sgRNA (van der Kuyl et al., 1990). This study showed that the core promoter was located between −26 and +1 and that there were two enhancer regions, one downstream (+1 to +12) and one upstream (-136 to −94). AMV core promoter has a small conserved sequence (AAU), also present in BMV, which mutations show to be important (van der Vossen et al., 1995). As with BMV, the core promoter forms a hairpin structure (Jaspars, 1998) but the structure for AMV (and Ilarviruses) is more stable than that of BMV. It is suggested that in AMV and Ilarviruses, that require coat protein for replication (see Chapter 8, Section IV.G), the coat protein is needed for the polymerase to interact with the core promoter hairpin.
Thus, AMV and BMV have a common spatial organization and some conserved sequences homologous to a consensus sequence derived from all the alphaviruses of plants and animals (Ou et al., 1982).
c. Premature termination of minus strand
Premature termination of (–)-strand synthesis is effected by either cis or trans long-distance interactions between a region just upstream of the subgenomic promoter and another region of the viral nucleic acid. This can either be on the same nucleic acid molecule as that giving rise to the sgRNA (cis interaction) or on another genomic fragment of a split genome virus (trans interaction).
Formation of sgRNAs by cis interactions have been suggested for TBSV and PVX (Miller et al., 1998; Zhang et al., 1999) and by trans interactions for RCNMV (Sit et al., 1998).
The genome of TBSV has five ORFs, the 3’ three of which are expressed from two sgRNAs, sg mRNA1 and sg mRNA2 (see Fig. 6.23 for genome organization). Deletion mutagenesis identified a 12-nucleotide sequence approximately 1 kb upstream of the initiation site for sg mRNA2 that was required specifically for the accumulation of that sgRNA. The 12-nucleotide sequence can potentially base-pair with a sequences just 5’ to the sgRNA2 initiation site and mutagenesis supported this interpretation. Base-pairing sequence regions with similar stability and relative location are found in the genomes of different tombusviruses. It was proposed that the upstream sequence represents a cis-acting element that facilitates sgRNA promoter activity by long-distance RNA-RNA interactions.
The genome of PVX expresses five proteins, the polymerase from the genomic RNA and the triple block movement proteins and the coat protein from two or three sgRNAs (see Fig. 6.39 for genome organization). Upstream of the two promoters is a conserved octanucleotide sequence (GUUAAGUU) that is conserved between potexviruses (Kim and Hemenway, 1997). Mutagenesis indicated that this conserved sequence and its distance from the start site for sgRNA synthesis are critical for the accumulation of the sgRNAs. Other mutagenesis experiments showed that multiple elements in the 5’-end of the genomic RNA are important in both genomic and sgRNA accumulation (Kim and Hemenway, 1996). The conserved sequences upstream of the sgRNA initiation sites are also found in the 5’ region of the genomic RNA and mutation and compensatory mutation suggested that there are longdistance interactions between the 5’ and sgRNA initiation sites (Kim and Hemenway, 1999).
The genome of RCNMV is divided between two RNAs: RNA1 which is bicistronic encoding the viral polymerase and the coat protein, and the monocistronic RNA2 (for genome organization of dianthoviruses see Fig. 6.30). The coat protein gene is expressed from a subgenomic RNA from RNA1, the putative upstream promoter region predicted to form a stable stem-loop structure (Zavriev et al., 1996). The coat protein sgRNA is expressed only in the presence of RNA2 (Vaewhongs and Lommel, 1995). To study the interaction between RNAs 1 and 2 in expressing the sgRNA, Sit et al. (1998) replaced the coat protein gene on RNA1 with the GFP (green fluorescence protein) gene showing that subgenomic GFP (sGFP) was expressed in vivo when RNA2 from RCNMV, and from the related CRSV and SCNMV, were present. Mutagenesis of RNA2 was difficult because identifying the trans-acting element(s) for subgenomic expression could interfere with the cis-acting element(s) for RNA2 replication. To uncouple these elements, an infectious clone of TBSV was used as a vector for fragments of RCNMV RNA2 (Table 7.4 ).
TABLE 7.4.
Experiment to show that expression of a subgenomic RNA (sGFP) from a construct in which the coat protein gene of RCNMV RNA1 is replaced by GFP (R1sGFP) depends on RNA2 sequences
| R1sGFP was co-inoculated with transcripts from the TBSV replicon (pHST2 in which the coat protein region was engineered to accept and express foreign genes from an sgRNA) containing segments from RCNMV RNA2 in order to delimit the minimal trans-activating elements. | |||
|---|---|---|---|
| Construct | Position of RNA2 sequence | Length of RNA2 inserts (nucleotides) | sGFP produceda |
| RCNMV RNA2 | 1–448 | 1448 | + + + |
| pHST2 | − | − | − |
| pHST2-RC2.3 | 708–1031 | 324 | + |
| pHST2-RC2.4 | 1031–708 (-sense) | 324 | − |
| pHST2-ΔBX | 708–916 | 209 | + + |
| pHST2–707 | 707–837 | 121 | + |
| pHST2–828 | 828–918 | 91 | − |
| pHST2-SL2 | 756–789 | 34 | + + + |
| pHST2-TA38 | 792–755 (-sense) | 38 | − |
| pHST2–20 | 762–782 | 21 | + |
| pHST2-LT2 | 767–775 | 9 | − |
sGFP production relative to induction by RNA2: + + + = 100%; + + = 50%; + = 25%; – = not detected. From Sit et al. (1998), with permission.
In this way, a 34-nucleotide segment (756–789) of RNA2 was identified as the trans-acting element. This 34-nucleotide element forms a stem-loop structure that is conserved between the RNA2 of RCNMV, CRSV and SCNMV, and the 8-nucleotide loop is complementary to an 8-nucleotide sequence on RNA1 two nucleotides upstream of the subgenomic RNA initiation site. Mutations of the 8-nucleotide sequence in RNA1 that did not alter the amino acid sequence of the overlapping replicase gene abolished the formation of the subgenomic RNA, but complementary mutation of the 8-nucleotide loop in RNA2 restored the ability to generate subgenomic RNAs. Thus, the 8-nucleotide loop on RNA2 transactivates the synthesis of the subgenomic RNA on RNA1. From these observations, Sit et al. (1998) proposed a model for the formation of RCNMV subgenomic RNAs (Fig. 7.17 ).
Fig. 7.17.
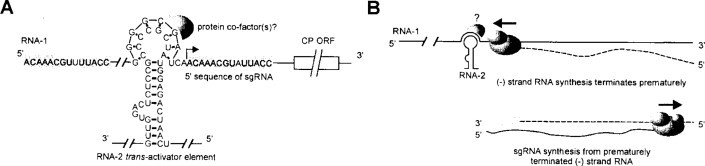
Proposed components and model of RCNMV trans-activation mechanism. (A) Sequences involved in trans-activation. Grey-shaded nucleotides represent conserved sequences between genomic RNA1 and the sgRNA that are likely to be (+)-strand promoters. The loop region of the 34-nucleotide trans-activator is shown base-pairing with the complementary 8-nucleotide element, two nucleotides upstream from the sgRNA start site (right-angle arrow) on RNA1. (B) Model for the generation of sgRNA. Complementary strands are depicted as dashed lines.
From Sit et al. (1998), with kind permission of the copyright holder, © The American Association for the Advancement of Science.
© 2002
In the early phase, RNA 1 and 2 are replicated to high levels. The transactivator element on RNA2 binds to RNA1, preventing the replicase from forming full-length minus strands of that RNA. As the 5’ terminal sequences of genomic RNA1 and the subgenomic RNA are conserved (Fig. 7.17), it is suggested that the truncated complementary RNA serves as a template for the production of positive-sense subgenomic RNA.
d. Discussion
The use of sgRNAs is widespread in plant viruses as a strategy to obviate the limitations of eukaryotic translation (see Table 7.7 ). There appear to be two mechanisms by which these RNAs are synthesized, both of which involve close interlinks with viral replication and both of which have strong controlling systems that are only just beginning to be recognized. The subgenomic promoter may have elements both in intergenic and in coding regions, the latter suggesting that the position may control expression of the promoter.
TABLE 7.7.
Expression strategies of (+)-strand ssRNA plant viruses
| Family | Genus | Termini | Expression strategies |
||||
|---|---|---|---|---|---|---|---|
| 5’ | 3’ | Genome segments | Sub-genomic | Read- Frame-through shift | Proteolytic Other cleavage | ||
| Bromoviridae | Broinovirus | Capa | t | 3 | 1 | ||
| Alfamovirus | Cap | c | 3 | 1 | |||
| Cucumovirus | Cap | t | 3 | 1 | |||
| Ilarvirus | Cap | c | 3 | 1 | |||
| Oleavirus | Cap | c | 3 | 1 | |||
| Comoviridae | Comovirus | VPg | An | 2 | + 2-startb | ||
| Fabavirus | ?VPg | An | 2 | + | |||
| Nepovirus | VPg | An | 2 | + | |||
| Potyviridae | Potyvirus | VPg | An | 1 | + | ||
| Ipomovirus | ?VPg | An | 1 | + | |||
| Macluravirus | ?VPg | An | 1 | + | |||
| Rymovirus | ?VPg | An | 1 | ?+ | |||
| Tritimovirus | ?VPg | An | 1 | + | |||
| Bymovirus | ?VPg | An | 2 | + | |||
| Tombusviridae | Tombusvirus | OH | 1 | 2 | + | ||
| Avenavirus | OH | 1 | 1 | + | |||
| Aureusvirus | OH | 1 | 2 | + | |||
| Carmovirus | OH | 1 | 2 | + | |||
| Dianthovirus | Cap | OH | 2 | 1 | −1 | ||
| Machlomovirus | Cap | OH | 1 | + | + 2 | 2-start | |
| Necrovirus | OH | 1 | 2 | + | |||
| Panicovirus | OH | 1 | 1 | + −1 | |||
| Sequiviridae | Sequivirus | ?VPg | An | 1 | + | ||
| Waikavirus | ?VPg | An | 1 | + | |||
| Closteroviridae | Closterovirus | ?Cap | OH | 1 | 7–10 | + 1 | + |
| Crinivirus | ?Cap | OH | 2 | + | + 1 | ||
| Luteoviridae | Luteovirus | OH | 1 | + | + + | ||
| Polerovirus | VPg | OH | 1 | + | + + | ||
| Enamovirus | VPg | OH | 1 | + | + + | ||
| No family | Tobamovirus | Cap | t | 1 | 2 | + | |
| Tobravirus | Cap | OH | 2 | + | + | ||
| Potexvirus | Cap | 1 | 2 | ||||
| Carlavirus | ?Cap | An | 1 | 2 | |||
| Allexivirus | ?Cap | An | 1 | + | |||
| Capillovirus | ?Cap | An | 1 | 2 | |||
| Fovcavirus | Cap | An | 1 | + | |||
| Trichovirus | Cap | An | 1 | 2 | |||
| Vitivirus | Cap | ||||||
| Furovirus | Cap | t | 2 | + | + | ||
| Pecluvirus | ?Cap | 2 | + | + | |||
| Pomovirus | Cap | t | 3 | + | + | ||
| Benyvirus | Cap | An | 4 | + | + | + 2-start | |
| Hordeivirus | Cap | t | 3 | + | |||
| Sobemovirus | VPg | 1 | + | + CfMV | + | ||
| Marafivirus | Cap | 1 | + | + | |||
| Tymovirus | Cap | t | 1 | 1 | + 2-start | ||
| Ourmiavirus | 3 | ||||||
| Umbravirus | OH | 1 | + | + | |||
An = poly(A); c = conserved 3’ sequence between genome segments; Cap = cap sequence; OH = hydroxyl 3’ terminus; t = tRNA-like sequence; VPg = genomic viral protein.
Two translational starts on same ORF.
Several examples have been noted above of the sgRNA promoter regions being predicted to form strong secondary structures, usually stem-loops. This is predicted for other viruses (e.g. TCV: Wang and Simon, 1997; Wang et al., 1999a).
3. Strategy 3: Multi-partite genomes
Viruses with multi-partite genomes have the information required for the virus infection cycle divided between two or more nucleic acid segments. This is found for both DNA and RNA plant viruses. For the (+)-sense ssRNA viruses, this strategy places the gene at the 5’-end of each RNA segment and thus it is open for translation.
Of the 70 plant virus genera, 28 have multipartite genomes (see Table 7.7 and Appendix 2). In most of these, the genome segments are encapsidated in separate particles, such viruses being termed multi-component. Members of the Reoviridae, and possibly the Partitiviridae, have all their genome segments in one particle.
4. Strategy 4: Internal initiation
In the internal initiation strategy translation is initiated at a site, termed the internal ribosome entry site (IRES) or ribosome landing pad, that is not the 5’ AUG start codon; see Belsham and Sonenberg (1996) for a review of IRESs. It is suggested that the IRES forms a complex secondary/tertiary structure to which ribosomes and transacting factors bind. There are various reports of the downstream ORF of bicistronic RNAs of plant viruses being expressed (Hefferson et al., 1997), but few can be fully attributable to internal initiation. However, the subgenomic mRNA for the coat protein of crucifer TMV (crTMV) and the subgenomic, I2 RNA, mRNAs for the movement proteins of both crTMV and TMV U1 strains are reported to be expressed by internal initiation (Ivanov et al., 1997; Skulachev et al., 1999). These RNAs are uncapped and have long 5’ untranslated regions (UTRs)-148nt for the crTMV coat-protein and 228 nt for the movement-protein mRNAs. The problem is to show that an RNA demonstrating internal initiation in vitro actually does this in vivo. Skulachev et al. (1999) addressed this point and showed good evidence for it happening in vivo.
It is considered that the IRES strategy enables a potentially inefficient mRNA (no cap and long 5’ UTR) to be translated efficiently and might also provide translational control so that gene products such as a movement protein can be expressed at the appropriate time.
5. Strategy 5: Leaky scanning
Leaky scanning is where the 40S ribosomal subunits start scanning from the 5’-end of the RNA but do not all start translation at the first AUG. Some, or all, pass the first AUG and start translation at downstream ORFs. In some cases, the 40S subunits of the 80S ribosomes fail to disengage at a stop codon and they reinitiate translation at a downstream start codon. There are three forms of leaky scanning: (1) two initiation sites on one ORF, (2) overlapping ORFs, and (3) two consecutive ORFs (Fütterer and Hohn, 1996; Maia et al., 1996).
a. Two initiation sites on one ORF (two-start)
The genome of CPMV is divided between two RNA species, the shorter of which, M-RNA, codes for two C-terminal collinear polyproteins of 105 kDa and 95 kDa initiated from two in-frame AUG codons (see Fig. 6.18 for CPMV genome organization). From in vitro translation experiments, it was suggested that there might be internal initiation (Thomas et al., 1991; Verver et al., 1991), but in vivo experiments showed that there was a greater likelihood of leaky scanning being involved (Belsham and Lomonossoff, 1991). This was supported by the fact that the AUG for the 95-kDa protein is in a more favourable context (G at positions −3 and +4) than is that for the 105-kDa protein (A at −3 and U at +4).
b. Overlapping ORFs
The genomes of members of the Luteovirus and Polerovirus genera contain six ORFs, the 3’ ones of which are expressed from sgRNAs (see Fig. 6.35 for genome organizations). ORF 4 (17 kDa) is contained in a different reading frame within ORF 3 (coat protein). The translation of ORF 4 fits very well with leaky scanning from the translation initiation of ORF 3 (Tacke et al. 1990; Dinesh-Kumar and Miller, 1993). The context of the ORF-3 AUG (U at position −3 and A at +4) is unfavourable. There are also interactions between the translation of these two ORFs. Mutations that reduce the initiation efficiency of ORF 4 also reduce initiation at the ORF-3 AUG if the latter's flanking bases are not changed (Dinesh-Kumar and Miller, 1993).
There are two overlapping ORFs on RNA2 of PCV that are translated by leaky scanning (Herzog et al., 1995), and the second and third ORFs of the triple gene block movement protein complexes (see Chapter 9, Section II.D.2.f) of potexviruses, carlaviruses, furoviruses and hordeiviruses are also expressed by a similar leaky scanning mechanism (Zhou and Jackson, 1996; Verchot et al., 1998). The upstream AUG is usually in a poor context and the upstream ORF contains few, if any, other AUGs in any of the three reading frames.
c. Two consecutive ORFs
PCV has a bipartite genome, RNA2 of which has five ORFs (see Fig. 6.47 for genome organization). The 5’ ORF of RNA2 encodes the viral coat protein and overlaps the next ORF (39-kDa protein) by two nucleotides and both are translated from the same RNA. The 620-nucleotide coat-protein ORF is devoid of AUG codons apart from the initiation codon. The insertion of stem structures or of AUG codons in the upstream region inhibited the translation of the 39-kDa protein, suggesting leaky scanning (Herzog et al., 1995).
The expression of ORFs I, II and III of RTBV is another example of leaky scanning due to the lack of AUG codons in upstream regions (see Section V.H.1.b).
d. Discussion
The translation of non-5’ ORFs by leaky scanning usually results from the AUG start codon in the upstream ORF being in a poor context. A lack or dearth of AUG codons in the upstream region can enhance the effect. However, care must be taken in interpreting in vitro translation information as evidence for leaky scanning. Parameters such as translation system and conditions, especially the presence of divalent cations, can affect the expression from non-5’ ORFS.
6. Strategy 6: Non-AUG start codon
Some viral ORFs appear to start with a codon that is not the conventional AUG start codon; the initiation at these ORFs is inefficient.
The genome of RTBV is expressed from a more-than-genome length RNA transcribed from the viral DNA (see Section IV.C).
Computer analyzes of the sequence of this RNA show three conventional ORFs that start with an AUG and finish with a stop codon and one region lacking stop codons but without an AUG. Using mutagenesis and expression of a coupled chlorophenical acetyl transferase (CAT) gene, Fütterer et al. (1996) demonstrated that this region (ORF I) was translated and that the translation was initiated at an AUU codon (Fig. 7.18 ). The efficiency of translation was about 10% that of a gene that had the conventional AUG start codon.
Fig. 7.18.

Diagram of the experiments by Fütterer et al. (1996) showing the non-AUG start codon of RTBV. (A) The top line depicts the genome organization of RTBV displayed in a linear manner with the ORFs shown as boxes. The middle line is an enlargement of the 5’ part of the genome with the 12 short ORFs (sORF) numbered. The bottom line shows the basic construct transiently expressed in rice cells. Transcription is driven by the CaMV 35S promoter and the chloramphenicol acetyl transferase (CAT) gene without its AUG start codon is fused in frame with ORF I. (B) Sequences around the 5’-end of ORF 1 of various constructs. The RTBV sequence is shown in upper case and the CAT sequence in lower case; mutations in the RTBV sequence are shown in lower case or as –. The two ATT codons are underlined. The + and – signs to the right are an estimate of the relative CAT activity, and the percentage figures on the extreme right are the % CAT activity relative to that of constructs in which the CAT gene retained its ATG start codon.
SBWMV has a bipartite genome, RNA2 of which has three ORFs, the coat protein (19 kDa), a read-through ORF from the coat protein, and a 3’ 19-kDa ORF expressed from an sgRNA (see Fig. 6.46 for genome organization). RNA2 also expresses both in vitro and in vivo a 28-kDa protein that reacts with SBWMV coat protein antiserum (Hsu and Brakke, 1985; Shirako and Ehara, 1986). In vitro translation and site-directed mutagenesis indicated that a CUG codon initiated the translation of the 28-kDa protein in a region upstream of, and in frame with, the AUG codon for the 19-kDa coat protein giving a 40-amino-acid N-terminal extension to that protein. (Shirako and Wilson, 1993; Shirako 1998).
7. Strategy 7: Transactivation (reviewed by Fütterer and Hohn, 1996)
The dsDNA genome of CaMV has six closely spaced functional ORFs (I-IV) (see Fig. 6.1 for genome organization) and is transcribed to give a more-than-genome length RNA, the 35S RNA and an mRNA (19S) for ORF VI (see Section IV.C). Although there is some evidence for the use of spliced RNAs for expressing some of the ORFs (see Section V.B.11), it appears that some of the ORFs are expressed from the long RNA. Most of the downstream ORFs are not, or are poorly, expressed in protoplasts or transgenic plants unless the product of ORF VI is present (Bonneville et al., 1989; Fütterer and Hohn, 1991). This gene product is termed a transactivator (TAV) and is thought to facilitate internal initiation. A similar function has been shown for the analogous gene product in FMV (Gowda et al., 1989; Scholthof et al., 1992a,b) and it is likely that all caulimoviruses have a gene with this function in the same relative region of the genome.
Using artificial polycistronic RNAs, Fütterer and Hohn (1991, 1992) examined the parameters that control transactivation. They showed that, for reinitiation at a downstream ORF, there should not be overlapping of long ORFs and that it was particularly efficient when the first ORF was about 30 codons long. The polar effects of the insertion of stem-loop structures, that would inhibit the translocation of ribosomes, into polycistronic mRNAs and the specificity for non-overlapping ORFs indicated that transactivation causes enhanced reinitiation of ribosomes. The optimal 30-codon length for the first ORF is just long enough to emerge from a translating ribosome and suggests that the TAV acts directly or indirectly on the translating or terminating ribosome.
The transactivating function locates to the central one-third of the TAV protein (De Tapia et al., 1993). Using this portion of the gene product (mini-TAV), 100-fold more DNA had to be used to produce normal levels of transactivation in transfected cells. The mini-TAV was only active in Nicotiana plumbaginifolia protoplasts whereas the full-length TAV was active in protoplasts from several dicot and monocot species. There is an RNA-binding activity outside the mini-TAV region that may enhance activity by increasing the TAV concentration near the RNA. TAV associates with polysomes and, as shown by an overlay binding assay, also with an 18-kDa ribosomal or ribosome-associated protein from plants and yeast (Fütterer and Hohn, 1996). It also interacts with the 60S ribosomal subunit protein L18 (RPL18) from Arabidopsis thaliana (Leh et al., 2000), the interacting region being defined as the mini-TAV region.
Using N. edwardsonii protoplasts, Edskes et al. (1996) analyzed the ability of the TAV from three different caulimoviruses to activate viral RNA-based reporter constructs. They found that efficient expression of polycistronic and monocistronic caulimovirus mRNAs in plant cells requires compatible interactions between ORF VI, a translational TAV and a cognate cis-element at the 3’-end of the mRNA.
Expression of the TAV in transgenic plants can give rise to virus-like symptoms (see Chapter 10, Section III.E.6). It is not known whether the transactivation or other activity of this gene causes this.
8. Strategy 8: Translational (ribosome) shunt
Short ORFs (sORFs) (defined arbitrarily as having less than 50 codons and no known function for the product) in a leader sequence can interfere with translation of a downstream ORF. In the translational ribosome shunt mechanism, initially scanning ribosomes are transferred directly from a donor to an acceptor site without linear scanning of the intervening region, thus avoiding sORFs.
The 35S RNAs of plant pararetroviruses, members of the Caulimoviridae, have long and complex leader sequences (Figs 7.18 and 7.19 ). The leader sequences range in length from about 350 to more than 750 nucleotides and contain between three and 19 sORFs. They fold to give complex stem-loop structures (Pooggin et al., 1999). Translational shunting has been proposed as the mechanism by which these constraints to translation are bypassed (Fütterer et al., 1993). For translational shunting there need to be shunt donor and acceptor sites and well-defined structure to bring these together. Most of the studies on these features have been on CaMV.
Fig. 7.19.

(see Plate 7.1) Comparison of the primary structures of the pgRNA leader of plant pararetroviruses. On the left, a phylogenetic tree shows the relationship of their reverse transcriptases (according to Richert-Pöggeler and Shepherd, 1997; note that BSV and DaBV were not included in that comparison). Caulimoviruses and badnaviruses are grouped (as indicated on the right). The leader sequence preceding the first long ORF (ORF VII or ORF I) is depicted as a thick line; the sORFs are indicated by boxes, with internal start codons shown by vertical lines. The numbered genome position of the pgRNA 5’-end is enclosed within an ellipse if mapped by primer extension or not enclosed if putative. The numbering within the leader is from the 5’-end (except for SoyCMV where the latter is unclear). Red arrows under the leader define the complementary sequences that form the base of the large stem-loop structures. The conserved sORFs preceding the structures are also in red. A green triangle indicates a putative or, in the case of CaMV, FMV and RTBV (Rothnie, 1996), mapped poly(A) signal. An arrowhead adjacent to a vertical line (in blue) shows the location of the primer binding site for reverse transcription.
From Pooggin et al. (1999), with permission.
© 2002
The leader sequence of CaMV 35S RNA forms an elongate hairpin conformation that contains a long-range pseudoknot and a dimer (Hemmings-Mieszczak et al., 1997). Chemical modification, enzymatic probing and temperature-gradient electrophoresis showed that the hairpin is made up of three elements–termed stem sections I, II and III (Hemmings-Mieszczak et al., 1997, 1998). Stem section I is the most stable and its structure, rather than its sequence, is important in ribosome shunting (Hemmings-Mieszczak and Hohn, 1999).
To study the effects of the sORFs in the leader sequence, Pooggin et al. (1998) mutated the start codon of each and tested the infectivity of the mutants. Mutation of the 5’ sORF (A) delayed symptom development and the mutant had reverted; infectious mutants in the sORFs located in the stem-loop either reverted or there were compensatory mutations to restore the structure. Thus, sORF A as well as a polypyrimidine stretch at the 3’-end of the hairpin (Hemmings-Mieszczak and Hohn, 1999) are important in stimulating translation via the shunt. The current interpretation of the shunt structure is shown in Fig. 7.20 .
Fig. 7.20.

Non-linear ribosome migration (ribosome shunt) during translation initiation on the CaMV 35S RNA leader. The leader is represented schematically as a combination of unstructured 5’ and 3’ terminal regions flanking an elongated hairpin structure (nucleotide 70–550). Three stem sections of different stability are labeled and the numbers indicate their approximate borders; stem section I (nucleotide 70–130 and 500–550), stem section II (nucleotide 130–180 and 460–500) and stem section III (nucleotide 220–280 and 340–400). The presence of a stable hairpin (stem section 1, black box) promotes translation of a downstream ORF (VII-CAT) via the ribosome shunt mechanism; all essential elements; 5’ cap, sORFa (boxed A), stable segment I, and the pyrimidine-rich sequence around nucleotide 500 (Yn) are indicated in bold. The 5’ and 3’ unidirectional ribosome migration is depicted by a thick black arrow with description below. Sequences preceding and directly following the hairpin are called shunt donor and acceptor sites respectively. The model predicts that, after the cap-dependent initial scanning, the 40S subunit of the ribosome translating sORFA is transferred (shunted) from the shunt donor to the shunt acceptor site, where the scanning is resumed and continues. Then, the 80S ribosome is assembled at the initiation codon of ORF VII, where translation starts.
From Hemmings-Mieszczak et al. (1998), with permission.
© 2002
The ribosome enters at the 5’ cap, which is essential, translocates to sORF A, shunts across the stable hairpin, and then reaches the first true ORF. The sORF MAGDIS from the mammalian AdoMetDC RNA, which conditionally suppresses re-iniation at a downstream ORF, prevented shunting, indicating that in CaMV the shunt involves re-initiation (Ryabova and Hohn, 2000). There are still unknown details as to what happens at sORF A and as to the actual crossing of the shunt junction.
In CaMV the shunt ‘donor’ or ‘take-off’ site is just downstream of sORF A (Dominguez et al., 1998) and the shunt ‘acceptor’ or ‘landing’ site is just upstream of ORF I (Fütterer et al., 1993).
In a study of the effect of the CaMV leader sequence on the expression of a downstream uidA (GUS) reporter gene, Schärer-Hernández and Hohn (1998) showed that the shunt mechanism occurs in planta with an average efficiency of 5% compared with that of a leaderless construct. However, there are some reservations on the use of in planta systems to examine what are essentially artificial constructs as there may be RNA processing, transport and modifications which do not occur in the natural system. Dominguez et al. (1998) have developed an in vitro system that avoids these shortcomings and which supports the above in vivo data. In spite of this there must be some reservations, mentioned above, in the extrapolation of in vitro data to the in vivo situation.
Three cellular proteins that bind to several sites in the CaMV leader sequence have been identified by UV cross-linking assays (Dominguez et al., 1996). p35 binds to RNA non-specifically, p49 binds with low specificity, but p100 interacts specifically with viral sequences. The expression of these proteins is not induced by virus infection.
Translational shunt has also been demonstrated for RTBV with the shunt donor near sORF 1 and the shunt acceptor near the AUU start codon for ORF 1 (Fütterer et al., 1996) (see Section V.H.1.b).
9. Strategy 9: Read-through proteins
The termination codon of the 5’ gene may be ‘leaky’ and allow a proportion of ribosomes to carry on translation to another stop codon downstream from the first, giving rise to a second longer functional polypeptide. This is termed ‘read through’ or ‘stop-codon suppression’ resulting in a protein that has the same sequence as the upstream protein in its N-terminal portion and a unique sequence in its C-terminal portion.
The read-through strategy is found in at least 17 plant virus genera (Table 7.5 ) and is characteristic of all three genera in the Luteoviridae and most genera in the Tombusviridae. The proteins that are produced by read through are either replicases (Tombusviridae) or extensions to the coat protein (luteoviruses) thought to be involved in transmission vector interactions (see Chapter 11, Section III.H.1.a).
TABLE 7.5.
Read-through or stop codons
| Family | Genus | Virus | Read-through stop codon | Read-through protein |
|---|---|---|---|---|
| Tombusviridae | Tombusvirus | TBSV | AAA UAG GGA GGC | Replicase |
| Aureusvirus | PoLV | UAC UAG GGG UGC | Replicase | |
| Avenavirus | OCSV | AAA UAG GGG UGC | Replicase | |
| Carmovirus | CarMV | AAA UAG UUG GAA | Replicase | |
| Machlomovirus | MCMV 1 | AAA UAG GGG UGU | Replicase | |
| MCMV 2 | AAC UGA GCU GGA | Unknown | ||
| Necrovirus | TNV | AAA UAG GGA GGC | Replicase | |
| Panicovirus | PMV | AAG UAG GGG UGU | Replicase | |
| Luteoviridae | Luteovirus | BYDV | AAA UAG GUA GAC | Coat protein extension |
| Polerovirus | PLRV | AAA UAG GUA GAC | Coat protein extension | |
| Enamovirus | PEMV | CUC UGA GGG GAC | Coat protein extension | |
| No family | Benyvirus | BNYVV | CAA UAG CAA UUA | Coat protein extension |
| Furovirus | SBWMV RNA1 | AAA UGA CGG UUU | Replicase | |
| SBWMV RNA2 | AGU UGA CGG GAC | Coat protein extension | ||
| Pechuvirus | PCV RNA1 | AAA UGA CGG UUU | Replicase | |
| Pomovirus | BSBV RNA1 | AAA UGA CGG | Replicase | |
| BSBV RNA2 | GAA UAG CAA UCA | Coat protein extension | ||
| Tobamovirus | TMV | CAA UAG CAA UUA | Replicase | |
| Tobravirus | TRV | UUA UGA CGG UUU | Replicase |
Data from Fütterer and Hohn (1996), amended and updated.
© 2002
In read through, the stop codon is read by a suppressor tRNA instead of the ribosome being released by the eukaryotic release-factor complex. Essentially, there is competition between the release-factor complex and the suppressor tRNAs. Two main factors are involved in read through of stop codons, the context of the stop codon and the suppressor tRNAs involved.
a. Stop codon context
Stop codons have different efficiencies of termination (UAA>UAG>UGA) and the first, and possibly the second, nucleotide 3’ of the stop codon acts as an important efficiency determinant (Stansfield et al., 1995). It can be seen from Table 7.5 that either amber (UAG) or opal (UGA) stop codons are read through; there are no examples of read through of the natural ochre (UAA) stop codon. However, when the suppressible UAG codon of TMV is replaced by a UAA codon the virus can still replicate and produce mature virions (Ishikawa et al., 1986). Unlike with retroviruses, there appears to be no structural requirements for stop codon suppression in plant systems. The context of the TMV amber stop codon at the end of the 126-kDa ORF has been studied in detail by insertion into constructs containing the GUS or other genes which enable read through to be quantitated in protoplasts (Valle et al., 1992). This experimental approach has defined the sequence (C/A)(A/C)A.UAG.CAR.YYA (R = purine, Y = pyrimidine) as the optimal consensus context (Skuzeski et al., 1991; Hamamoto et al., 1993). Efficiently recognized stop codons normally have a purine immediately downstream and avoid having a C residue (Fütterer and Hohn, 1996).
However, an analysis of Table 7.5 shows that the read-through stop codons identified for plant viruses do not necessarily conform to these contexts determined from in vitro systems. As the sequence context differs from that described above, the read through of UAG.G in many of the Tombusviridae and the Luteoviridae suggests that a different mechanism might be involved. It is possible that there may be a requirement for additional cis-acting sequences such as a conserved CCCCA motif or repeated CCXXXX motifs beginning 12 to 21 bases downstream of many of these read-through sites (see Fütterer and Hohn, 1996; Miller et al., 1997a). It may be that other long-distance interactions are involved in the deviations from the experimentally determined optimal contexts for TMV.
b. Suppressor tRNAs
The synthesis of a read-through protein depends primarily on the presence of appropriate suppressors tRNAs. Thus, two normal tRNAstyr from tobacco plants were shown to promote UAG read-through during the translation of TMV RNA in vitro (Beier et al., 1984a). The tRNAtyr must have the appropriate anticodon, shown to be GψA, to allow effective read through. A tRNAtyr from wheatgerm, with an anticodon different from that in tobacco, was ineffective (Beier et al., 1984b). tRNAgln will also suppress the UAG stop codon (Grimm et al., 1998). Various suppressor tRNAs have been found for the UGA stop codon of TRV (Zerfass and Beier, 1992; Urban and Beier, 1995; Baum and Beier, 1998), chloroplast (chl) and cytoplasmic (cyt) tryptophan-specific tRNAs with the anticodon CmCA, and chl and cyt cysteine-specific tRNAs with the anticodon GCA and arginine tRNA isoacceptor with a U*CG anticodon. Interestingly, the chl tRNAtrp suppresses the UGA codon more efficiently than does the cyt tRNAtrp.
The replication of some strains of TRV is associated with mitochondria (see Fig. 8.17) and it is likely that the prokaryote-like tRNAs that are found in chloroplasts will also be found in mitochondria. The tRNAcys isoacceptor from tobacco chloroplasts was more efficient at reading through a UGA stop codon in the TRV (and TMV) context than was the cytoplasmic acceptor (Urban and Beier, 1995).
c. Proportion read through
The proportion of read-through protein produced may be modulated by sequence context of the termination codon (Bouzoubaa et al., 1987; Miller et al., 1988; Brown et al., 1996a), by long-distance effects and by the availability of the suppressor tRNA. Thus, data from in vitro systems may not be fully relevant to the in vivo situation. However, it is reasonable to suggest that about 1–10% of the times that a ribosome reaches a suppressible stop codon result in read through.
d. Discussion
As well as overcoming the constraints of the eukaryotic translation system, read through of stop codons provides a mechanism for the control of the expression of gene products. Transmission helper factors have to be incorporated in the virus capsid but it is probably not necessary to have them on all coat protein subunits. Thus, it could be more efficient for the virus expression system if 1–10% of the coat protein subunits also contain the transmission factor. Similarly, the viral replicase comprises several functional domains (described in Chapter 8, Section IV.B) that are probably required in different amounts and even at different times. The production of two proteins both containing some of the domains and one, the read-through protein, also containing the other domains would give control over the availability of these functions.
10. Strategy 10: Frameshift proteins (reviewed by Brierley, 1995; Farabaugh, 1996a,b)
Another mechanism by which two proteins may commence at the same 5’ AUG is by a switch of reading frame before the termination codon of the 5’ ORF to give a second longer ‘frameshift’ protein. This translational frameshift event allows a ribosome to bypass the stop codon at the 3’-end in one reading frame and switch to another reading frame so that translation can continue to the next stop codon in that reading frame (Hizi et al., 1987). A frameshift is illustrated in Fig. 7.21 .
Fig. 7.21.
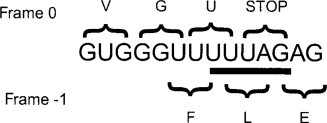
Translational frameshift. The ribosome bypasses the stop codon in frame 0 by switching back one nucleotide to frame −1 at a UUUAG sequence before continuing to read triplets in frame −1 to give the fusion or frameshift protein.
From Matthews (1991).
© 2002
At the frameshift site ribosomes change their reading frame either one nucleotide in the 5’ direction (−1 frameshift) or one nucleotide in the 3’ direction (+1 frameshift). This gives two proteins (the frame and frameshift proteins) identical from the N terminus to the frameshift site but different beyond that point. The frame protein is always produced in greater quantity than the frameshift protein.
Frameshift obviously occurs where ORFs overlap and may be at any place within that overlap.
The frameshift strategy is found in at least nine plant virus genera (Table 7.6 ) and, in all cases that are known, involves the replicase. Most instances of frameshift are in the −1 direction, only those of the Closteroviridae being in the +1 direction.
TABLE 7.6.
Frameshift between open reading frames
| Family | Genus | Virus | Frameshift sequence | Type of frameshift | Signal | Protein(s) |
|---|---|---|---|---|---|---|
| Tombusviridae | Dianthovirus | RCNMV | −1 | Stem-look | Replicase | |
| Panicovirus | PMV | −1 | ND | Unknown | ||
| Closteroviridae | Closterovirus | BYV | + 1 | Pseudoknot | Replicase | |
| Crinivirus | LiYV | + 1 | Replicase | |||
| Luteoviridae | Luteovirus | BYDV | −1 | Stem-loop or pseudoknot | Replicase | |
| Polerovirus | PLRV | −1 | Pseudoknot | Replicase | ||
| Enamovirus | PEMV-1 | −1 | Pseudoknot | Replicase | ||
| No family | Sobeinovirus | SBMV | −1 | Stem-loop | ||
| Umbravirus | PEMV-2 | −1 | Replicase |
a. –1 Frameshifting
For a −1 frameshift three features are needed, a heptanucleotide sequence termed the ‘slippery’ or ‘shifty’ sequence at the frameshift site, a strongly structured region downstream of the frameshift site, and a spacer of four to nine nucleotides between the slippery sequence and the structured region. The involvement of the structure, stability and function of the pseudoknots making up the strongly structured region is reviewed by Giedroc et al. (2000).
The slippery sequence comprises two homopolymeric triplets of the type XXX YYY Z (X = A/G/U; Y = A/U; Z = A/C/U). Upon reaching this heptanucleotide sequence, the two ribosome-bound tRNAs that are in one reading frame (X.XXY.YYZ) shift by one nucleotide in the 5’ direction (XXX.YYY.Z) retaining two out of the three base-paired nucleotides with the viral RNA (Jacks et al., 1988). This mechanism was deduced for retroviruses but the evidence points to a similar mechanism in plant RNA viruses. The slippery sequences listed in Table 7.6 are either indicated from experimentation or predicted from similarities with other viruses.
Mäkinen at al. (1995b) noted a consensus amino acid sequence, WAD/WGD (W = Trp; A = Ala; G = Gly; D = Asp; E = Glu) followed by a D/E-rich domain in front of frameshift sites. The significance of this motif is unknown.
The strongly structured regions are separated by the spacer region from the frameshift point and are either hairpins or pseudoknots (see Chapter 4, Section III.A.3.b, for pseudo-knots). It is considered that the structure causes the ribosome to pause, thereby initiating frameshift. The recent determination of the crystal structure of the pseudoknot in BWYV RNA (Su et al., 1999) should help in gaining a detailed understanding of the mechanics of frameshifting.
The most detailed studies on −1 frameshifting have been on luteoviruses and these observations can be extended to other viruses. The overlap between the two ORFs ranges from 13 nucleotides in BYDV-PAV to several hundred nucleotides in most other luteoviruses. In BYDV-PAV and BWYV there is a stop codon close to the slippery sequence. Mutation analyzes of the slippery sequence and the structured region result in frameshift efficiencies in the range of 1–30% in in vitro translation systems. Slippery sequences containing A or U residues produced higher rates of frameshifting than did those containing G or C residues (reviewed by Fütterer and Hohn, 1996). A stop codon immediately downstream of the frameshift site increases frameshifting probably because it contributes to ribosome pausing.
The kinetics of ribosomal pausing during −1 framshifting have been studied for the Saccharomyces cerevisiae dsRNA virus that has a slippery sequence and a downstream pseudo-knot (Lopinski et al., 2000). About 10% of the ribosomes pause at the slippery site in vitro and some 60% of these continue in the −1 frame. Those that moved to the −1 frame paused for about 10 times longer than it takes to complete a peptide bond in vitro. Thus, there are three ways in which ribosomes pass a frameshift site: (1) without pausing leading to no frameshift, (2) pausing but no frameshift, and (3) pausing and frameshifting. As the features of the frameshift site in the S. cerevisiae viruses are the same as those in viruses of higher plants, presumably these observations also apply to the latter.
Translation of full-length infectious BYDV-PAV transcripts in an in vitro system gave higher rates of frameshift than did shorter RNAs (Di et al., 1993); furthermore, the adjacent stop codon was found not to be necessary for frameshifting (Miller et al., 1997a). Full-length transcripts of RCNMV behave in a similar manner (Kim and Lommel, 1994). For BYDV, sequences located in the 3’ untranslated region of the genome, 4 kb downstream of the frameshift site, were found to be essential (Wang and Miller, 1995). Thus, there is evidence for long-distance interactions but the nature of these has not yet been elucidated.
b. +1 frameshifting
Frameshifting in the +1 direction requires a run of slippery bases and a rare or ‘hungry’ codon or termination codon. A downstream structures region is not necessary but may be found, as has been suggested for BYV (Agranovsky et al., 1994). The following mechanism has been suggested for the +1 frameshift between LIYV ORF la and ORF lb (Klaassen et al., 1995):
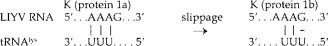
c. Proportion frameshifted
As with the read-through strategy, most of the studies on the proportion of translation events that result in a read-through protein involve in vitro systems. These can give frameshift rates as high as 30%; in vivo, rates of 1–5% are more likely (Brault and Miller, 1992; Prüfer at al., 1992).
11. Strategy 11: Splicing
The production of mRNAs from DNA in eukaryotes involves splicing which removes internal non-coding sequences and can give various versions of an mRNA. Two of the families of plant viruses with DNA genomes, the Caulimoviridae and Geminiviridae, use splicing in the production of mRNAs, a process which, at least in the caulimoviruses, opens up downstream ORFs.
As described above, CaMV produces two major transcripts, the more-than-genome-length 35S RNA and the monocistronic 19S RNA. Several other RNA species and deleted DNA species (suggested to have arisen by reverse transcription of RNA) have been found in CaMV-infected plants and have been interpreted as arising from splicing (Hirochika et al., 1985; Vaden and Melcher, 1990; Scholthof et al., 1991). In an analysis of the potential for splicing in the CaMV genome, Kiss-László et al. (1995) identified splice donor sites in the 35S RNA leader sequence and in ORF I and a splice acceptor site in ORF II. Splicing between the leader sequence and ORF II makes ORF III the 5’ ORF, thus opening it up to conventional translation. Mutants of the splice acceptor site in ORF II are not infectious, indicating that it is essential for the virus infection cycle. It is likely that these and possibly other splicing events are involved in the controlled expression of this group of viruses.
ORF 4 of RTBV is expressed from an mRNA spliced from the 35S RNA (Fütterer et al., 1994). The splice removes an intron of about 6.3 kb and brings ORF 1 into frame with ORF 4. The splice donor and acceptor sequences correspond to the plant splice consensus sequences (Fig. 7.22 ).
Fig. 7.22.
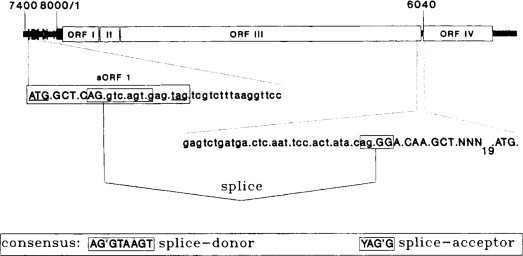
Schematic presentation of splice junction sequences. The sequences around the supposed splice sites are shown and localized schematically on the linear RTBV map. At the 5’-end, the sequence starts with the ATG codon of the first short ORF in the leader (indicated as box; start and stop codons are underlined). At the 3’-end, the sequence ends with the ATG codon of the RTBV ORF IV. The sORF and ORF IV codons are indicated. Sequences found in the cDNA are in uppercase. On the spliced RNA, the ORF initiated at the sORF AUG is in phase with ORF IV. The consensus for splice donor and splice acceptor sites is also shown (‘marks the point of splicing).
From Fütterer et al. (1994), with permission.
© 2002
The circular ssDNA genomes of mastreviruses have four ORFs, two being expressed from transcripts, V1 and V2, in the virion sense and two, C1 and C2, from transcripts in the complementary sense (see Figs 6.5 and 7.14 for genome organization). The C transcripts are of low abundance and a splicing event fusing C1 to C2 has been found for MSV, DSV and TYDV (Mullineaux et al., 1990; Dekker et al., 1991; Morris et al., 1992). Splicing has not yet been found in the other two geminivirus genera and, apart from TYDV, appears to be restricted to geminiviruses that infect monocots.
12. Strategy 12: Translation for both viral and complementary strands (ambisense)
Some of the genome segments of the tospoviruses and tenuiviruses encode two proteins having one ORF in the virion sense and the other in the complementary sense. Thus, one of the proteins is expressed from complementary-sense RNA. This is termed the ambisense expression strategy and is another means by which viruses overcome the eukaryotic translation constraints (see Sections IV.A.2 and 3).
C. Control of translation
Various mechanisms for the control of expression of viral genomes have been described in the section above. These relate primarily to mechanisms that viruses have developed to overcome the problem of the limitation of translation of mRNAs in eukaryotic systems to the 5’ ORF. Among other features that control or regulate the translation of eukaryotic mRNAs are the various non-coding regions (untranslated regions or UTRs) which include the termini of the RNAs, the 5’ terminus being capped (see Section V.A) and the 3’ terminus having a poly(A) tail. Also involved in the control and efficiency of translation are the 5’ leader sequence and the 3’ non-coding region. In eukaryotic mRNAs, there is co-ordinated interaction between the 5’ and 3’ UTRs of these mRNAs (Fig. 7.23) (reviewed by Gallie, 1996, 1998) and even evidence for circularized mRNAs (Wells et al., 1998). Only some plant viral RNAs are capped and have poly(A) tails (see Table 7.7). The majority either have a cap or a poly(A) tail or have neither, yet these may be translated very efficiently.
1. Cap but no poly(A) tail
The genome of TMV is capped but lacks a poly(A) tail. The structure of the 3’ UTR is complex, being composed of five pseudoknots covering a 177 base region (Fig. 7.24B ).
Fig. 7.24.

The sequence and structure of the TMV 5’ leader and 3’ UTR showing the primary sequence required for regulating translation. (A) The 5’ leader sequence with the poly(CAA) region responsible for the enhancing activity of Ω underlined. Three direct repeats are indicated by arrows. (B) The predicted structure of the 3’ UTR. The primary sequence essential for the regulation by the upstream pseudoknot domain (UPD) is shaded and the tRNA-like domain is indicated. The sequence that is absolutely conserved in eight tobamo- and hordei-viruses is shown in the boxed UPD.
From Tanguay and Gallie (1996), with permission.
© 2002
The 3’-terminal two pseudoknots form the tRNA-like structure that is involved in virus replication (see Chapter 8, Section IV.H). The remaining three pseudoknots make up the upstream pseudoknot domain that is conserved in the tobamoviruses and also found in the hordeiviruses. This domain appears to functionally substitute for a poly(A) region in promoting interactions between the 5’ and 3’ termini and enhancing translation initiation in a cap-dependent manner (Leathers et al., 1993). A 102-kDa protein binds to the pseudoknot domain (Fig. 7.24) and also to the 5’ UTR (Tanguay and Gallie, 1996). It is likely that the 102-kDa protein mentioned above is involved in bringing the 5’- and 3’-ends together in the manner shown for eukaryotic mRNAs in Fig. 7.23 above.
2. Poly(A) tail but no cap
Potyviruses are polyadenylated at their 3’ termini but the 5’ terminus is attached to a VPg. The 5’ UTR of TEV confers cap-independent enhancement of translation of reporter genes by interactions between the leader and the poly(A) tail (Carrington and Freed, 1990; Gallie et al., 1995). In a deletion analysis of the 143-nucleotide leader sequence of TEV, Niepel and Gallie (1999) identified two centrally located cap-independent regulatory elements that promote cap-independent translation. Placing the leader sequence into the intercistronic region of a bicistronic construct increased the expression of the second cistron, the enhancement being markedly increased by introducing a stable stem-loop structure upstream of the TEV leader sequence. It was concluded that the two elements in the TEV leader together promote internal initiation and that the function of one element is facilitated by the proximity to the 5’ terminus.
The VPg of the potyvirus TuMV interacts with the eukaryotic translational initiation factor eIF(iso)4E of Arabidopsis thaliana and wheat (Triticum aestivim) (Wittmann et al., 1997; Léonard et al., 2000). eIF(iso)4E binds to cap structures of mRNAs and plays an important role in regulating the initiation of translation (see McEndrick et al., 1999). The interaction domain of TuMV VPGg was mapped to a stretch of 35 amino acids and the substitution of an aspartate residue in this region completely abolished the interaction (Léonard et al., 2000). The cap analog m7GTP, but not GTP, inhibited the VPg–eIF(iso)4E complex formation, suggesting that the VPg and cellular mRNAs compete for eIF(iso)4E binding. The NIa protein of TEV that contains the VPg also binds to eIF4E but in a strain-specific manner (Schaad et al., 2000) with that from the HAT but not from the Oxnard strain interacting with eIF4E from tomato and tobacco.
3. Neither cap nor poly(A) tail
Many plant viruses have neither a 5’ cap nor a 3’ poly(A) tail (see Table 7.7) but these mRNAs are expressed very effectively and thus can be considered to have translation enhancement. There is currently information on three viruses which show some variation in details.
The 4-kb genome of TCV expresses the 5’-proximal two genes from the genomic RNA (by read through) and the other three genes from sgRNAs. Two genes are translated from the sgRNA1 by leaky scanning and the 3’ coat protein gene is expressed from a 1.45-kb sgRNA. Qu and Morris (2000) examined the 5’ UTRs of the genomic and 1.45-kb sgRNAs and their common 3’-ends for roles in translation regulation. Using the firefly luciferase reporter gene, they obtained optimal translation activity when the mRNA contained both 5’ and 3’ UTRs, the synergistic effect being at least 4-fold greater than the sum of the contributions of the individual UTRs. The translational enhancement was cap-independent and was greater for the sgRNA UTRs than for the genomic RNA UTRs.
TBSV has a genome of 4.8 kb with five ORFs of known function and a 3’ sixth small ORF of unknown function (see Fig. 6.23 for genome organization). A 167-nucleotide region (segment 3.5) near the 3’-end was implicated as a determinant of translational efficiency (Oster et al., 1998) and was shown to be involved in cap-independent translation (Wu and White, 1999). Segment 3.5 is part of a larger 3’ cap-independent translational enhancer (termed 3′CITE) and none of the major viral proteins was involved in 3′CITE activity. Unlike the translational enhancers of some other plant small RNA viruses (BYDV and STNV), there was no TBSV 3′CITE-dependent stimulation of translation in an in vitro wheatgerm extract system, which suggests that there might be differences in the enhancement mechanisms (Wu and White, 1999).
The luteovirus BYDV has a genome of 5.6 kb expressing six ORFs from the genomic and three sgRNAs (see Fig. 6.35 for genome organization). Highly efficient cap-independent translation initiation is facilitated by a 3’ translation enhancer sequence (3′TE) (Wang and Miller, 1995; Wang et al., 1997c; Allen et al., 1999). A 109-nucleotide 3’ sequence is sufficient for translational enhancement in vitro, but a larger 3’ region is required for optimal enhancement in vivo (Wang et al., 1999c). The 5’ extremity of this larger region coincides with the 5’ terminus of sgRNA2. Competition studies showed that the 3′TE did not enhance the translation of the genomic RNA as much as that of sgRNA1, this difference being attributable to the different 5’ UTRs of these two RNAs. Thus, the 3′TE stimulates translation in cis but selectively inhibits translation in trans. As the 5’ genes on the genomic RNA, translation of which is stimulated in cis and inhibited in trans, encode an early function, the viral replicase, it is suggested that sgRNA2 is a novel regulatory RNA that switches from early to late gene expression (Fig. 7.25 ) (Wang et al., 1999c) (see also Fig. 7.31 ).
Fig. 7.25.
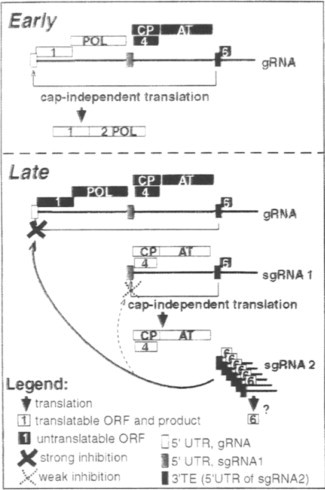
(seePlate 7.2) Translational switch model for trans-regulation of BYDV gene expression by sgRNA2. Open boxes indicate translational ORFs and their translation products (below large arrows). Black boxes indicate ORFs that are not translated. Early: polymerase is translated from the genomic (g)RNA (the only viral RNA at this stage) via the 3′TE (red box) in cis.Late: As abundant sgRNA2 accumulates, it specifically inhibits gRNA (bold cross) in preference to sgRNA1 (dashed cross), via the 3’ TE in trans. This allows almost exclusive translation of late genes from sgRNA1. The different 5’ UTRs of gRNA (gold box) and sgRNA1 (green box) contribute to differential inhibition. The role of ORF 6 encoded by sgRNA2 is unknown (?) but it is not necessary for trans-inhibition.
From Wang et al. (1999c, where the model is discussed in detail), with permission.
© 2002
Fig. 7.31.

Diagram illustrating some mechanisms used in the expression of (A) genus Luteovirus genomes and (B) genus Polerovirus genomes. Mechanisms are indicated by letters: C, cap-independent translation; L, leaky scanning; F, −1 frameshift; R, read through; S, synthesis of an sgRNA. Black boxes represent ORFs and white boxes show the positions of controlling sequences. Dark grey arrows indicate the effects of remote sequences on upstream processes, and pale grey arrows represent the synthesis of an sg mRNA.
From Mayo and Miller (1999), with permission.
© 2002
4. Cap-snatching
Negative-strand RNA viruses with segmented genomes use a mechanism, termed ‘capsnatching’, to initiate transcription of their mRNAs. In this process, cap structures comprising between twelve and twenty 5’ nucleotides are cleaved from host mRNAs by a virus-encoded endonuclease and are then used to prime transcription. Cap-snatching has been demonstrated for the tospovirus TSWV and tenuivirus MSpV (Kormelink et al., 1992a; van Poelwijk et al., 1996; Estabrook et al., 1998). Both viruses can snatch caps from positive-sense RNA viruses (Estabrook et al., 1998; Duijsings et al., 1999).
5. 5′ UTR
As well as being involved in the enhancement of translation initiation, the 5’ UTR of TMV also enhances the efficiency of translation. The 67-nucleotide 5’ UTR, termed the Ω sequence, dramatically enhances translation of downstream genes in both plant and animal cells (reviewed by Gallie, 1996); in constructs in transgenic plants it enhanced translation by 4- to 6-fold (Dowson Day et al., 1993). The Ω sequence has reduced secondary structure (Sleat et al., 1988a) and a 25 base poly(CAA) region (Fig. 7.24A) which mutagenesis indicated is the primary element for in vivo translational enhancement (Gallie and Walbot, 1992).
The 36-base 5’ leader sequence from AMV RNA4 also enhances translation (Jobling and Gehrke, 1987) as does the 84-nucleotide leader sequence of PVX genomic RNAS (Pooggin and Skryabin, 1992).
D. Discussion
The above descriptions show the great diversity of mechanisms that viruses use to express the information required for their function from what are often compact genomes. This diversity is summarized in Table 7.7.
The diversity of mechanisms overcomes constraints imposed by viruses using their host translational machinery. There are two other mechanisms of creating further diversity in gene products, transcriptional editing which is found in paramyxoviruses (Cattaneo, 1991; Niswender, 1998) and protein trans-splicing (reviewed by Perler, 1999), that have not yet been found in plant viruses.
These mechanisms can be viewed in two ways, specifically overcoming the constraints of the eukaryotic system, say in the requirement for a cap for translation initiation and the translation of only the 5’ ORF, and the control of translation so that the right product is in the right place in the right quantity at the right time. The two uses of the mechanisms cannot be separated. For instance, in many cases the frameshift and read-through mechanisms provide different functions of the replication complex, the upstream one from the shorter protein containing the helicase and capping activities and the downstream one the replicase.
The regulation is not only by the recognized ORFs but can be by non-coding sequences and possibly by short ORFs that normally might not be considered. There are many examples of the coat protein gene being expressed more efficiently than that for the replicase; more coat protein is required than the replicase.
Many of the studies on virus expression systems involve the use of in vitro techniques, especially in vitro translation systems. There can be dangers in extrapolating data from these systems to the in vivo situation. The differences between the rabbit reticulocyte and wheatgerm translation system in the processing of the potyviral polyprotein (see Section V.B.1.b) shows that cellular factors and possibly the cellular environment is likely to be involved. Furthermore, there is no real evidence that the RNAs of viruses considered to lack a cap are not capped when in in vivo translation complexes.
Many of the above points occur again in the discussion below of viruses with multiple strategies.
E. Positive-sense ssRNA viruses that have more than one strategy
Most viruses use more than one of the strategies outlined above to express their genetic information. This section describes examples of these multiple strategies.
1. Two strategies: Subgenomic RNAs + read-through protein
a. Tobamovirus genus (reviewed by Okada, 1999)
The genomes of many species of the Tobamovirus genus have been sequenced (van Regenmortel et al., 2000) and the organization of TMV is described in Chapter 6 (Section VIII.H.1). There are several closely packed ORFs, the expression of which is shown in Fig. 7.26 .
Fig. 7.26.

Expression of TMV genome. The genome map is as in Fig. 6.36. The genomic RNA is the template for the 126-kDa and 183-kDa replicase proteins. ORFs III and IV are expressed from two separate 3’ co-terminal sgRNAs giving the 32-kDa movement protein and the 17-kDa coat protein respectively.
An M7Gppp cap is attached to the first nucleotide (guanylic acid). The untranslated leader sequence of 69 nucleotides, the Ω sequence, potentiates efficient translation as described in Section V.C.5. The first ORF, coding for a 126-kDa protein, has a leaky termination codon (UAG) leading to a larger read-through protein (183-kDa). The two smaller ORFs at the 3’-end of the genome are translated from subgenomic RNAs, the 30-kDa from sgRNA I2 and the 17.6-kDa coat protein from the smallest sgRNA. The 3’ untranslated sequences can fold in the terminal region to give a tRNA-like structure that accepts histidine.
A third subgenomic RNA, called I1 RNA, representing approximately the 3’ half of the genome, has been isolated from TMV-infected tissue. SI mapping showed that this RNA species had a distinct 5’ terminus at residue 3405 in the genome (Sulzinski et al., 1985). These workers proposed a model for the translation of I1 RNA. There is an untranslated region of 90 bases followed by an AUG codon initiating a 54-kDa protein terminating at residue 4915. Thus, the amino acid sequence of the 54-kDa protein is the same as the residues at the carboxy terminus of the 183-kDa protein. However, there is no evidence that this sgRNA is involved in viral expression.
The expression of TMV has been studied in great detail and the description below shows how the understanding of the processes involved was developed.
i. Proteins synthesized in vitro
TMV genomic RNA has been translated in several cell-free systems. Two large polypeptides were produced in reticulocyte lysates (Knowland et al., 1975) and in the wheatgerm system (e.g. Bruening et al., 1976), but no coat protein was found by these or later workers. The TMV genome is not large enough to code independently for the two large proteins that were produced. Using a reticulocyte lysate system, Pelham (1978) showed that the synthesis of these two proteins is initiated at the same site, the larger protein being generated by partial read through of a termination codon. The two proteins are read in the same phase, so the amino acid sequence of the smaller protein is also contained within the larger one. Increased production of the larger protein occurred in vitro at lower temperatures (Kurkinen, 1981). The extent of production of the larger protein may also depend on the kind of tRNAtyr present in the extract (Beier et al., 1984a,b) (see Section V.B.9.b).
The 30-kDa and 17.6-kDa proteins are not translated from genomic RNA but from two subgenomic RNAs. The I2 RNA isolated from infected tissue has been studied in in vitro systems by many workers. It is translated to produce a 30-kDa protein. It is uncapped (Joshi et al., 1983) and appears to terminate in 5’ di- and triphosphates (Hunter et al., 1983). The initiation site for transcription has been mapped at residue 1558 from the 3’ terminus (Watanabe et al., 1984a). The I2 RNA also contains the smaller 3’ gene (Fig. 7.26) but this is not translated in in vitro systems.
The smallest TMV gene (the 3’ coat protein gene) is translated in vitro only from the monocistronic subgenomic RNA (Fig. 7.26) (Knowland et al., 1975; Beachy and Zaitlin, 1977). This gene can also be translated efficiently in vitro by prokaryotic protein-synthesizing machinery from E. coli (Glover and Wilson, 1982).
ii. Proteins synthesized in vivo
The data of Siegel et al. (1978) indicated that viral protein synthesis does not suppress total host-cell protein synthesis but occurs in addition to normal synthesis. Two days after infection, viral coat protein synthesis accounted for about 7% of total protein synthesis. Synthesis of the 126-kDa protein was about 1.4% and that of the 183-kDa protein about 0.3% as much as coat protein.
Several workers have reported the association of both full-length TMV RNA and the coat protein mRNA with cytoplasmic polyribosomes in infected tobacco leaves (e.g. Beachy and Zaitlin, 1975). Beachy and Zaitlin also found TMV dsRNAs to be associated with the membrane-bound polyribosome fraction. Confirmation that TMV proteins are synthesized on 80S ribosomes comes from the fact that cycloheximide completely inhibits TMV replication in protoplasts, whereas chloramphenicol does not (Sakai and Takebe, 1970).
Dorokhov et al. (1983, 1984) isolated a ribonucleoprotein fraction from infected tobacco tissue in CsCl density gradients, which had a higher buoyant density than TMV. This material can be released from polyribosomes by EDTA treatment. Genomic, I1, I2 and coat protein RNAs and polypeptides of various sizes were identified as components of the ribonucleoprotein complex.
From studies on viral protein synthesis in tobacco leaves using wild-type virus, a ts mutant held at different temperatures and a protein synthesis inhibitor, Dawson (1983) concluded that the synthesis of 183-kDa, 126-kDa and 17.5-kDa proteins was correlated with dsRNA synthesis rather than that of ssRNAs. It is possible that nascent ssRNAs from replication complexes function as mRNAs. Dawson's results suggest that TMV mRNA is relatively transitory in vivo. However, Dawson and Boyd (1987b) showed that synthesis of TMV proteins in tobacco leaves was not translationally regulated under conditions of heat shock, as were most host proteins. Thus, under appropriate conditions most of the protein being synthesized was virus-coded.
As might be expected, replicative intermediates (RI) and replicative forms (RF) (see Chapter 8, Section IV.A) appear early in infections as does the 126-kDa protein. Coat protein mRNA and genomic RNA are early products. At a later stage, virus production follows closely that of coat protein synthesis. Thus, it appears that the amount of coat protein available may limit the rate at which progeny virus is produced. Watanabe et al. (1984b) first detected the 183-kDa, 126-kDa, 30-kDa and coat proteins 2–4 hours after infection, and before infectious virus could be found. The 183-kDa, 126-kDa and coat protein were synthesized over a period of many hours but synthesis of the 30-kDa protein and its mRNA was detected between 2 and 9 hours after inoculation.
Proteins corresponding approximately in size to the 183-kDa and 126-kDa proteins were found in infected tobacco leaves (e.g. Scalla et al., 1976) and in infected protoplasts (e.g. Sakai and Takebe, 1974). Cyanogen bromide peptide analysis on the 110-kDa protein from infected leaves showed it to be the same as the in vitro translation product of similar size (Scalla et al., 1978).
The 30-kDa protein was detected in both infected tobacco protoplasts (Beier et al., 1980) and leaves (Joshi et al., 1983). Kiberstis et al. (1983) and Ooshika et al. (1984) raised antibodies against a synthetic peptide with the predicted sequence for the 11 or 16 C-terminal amino acids of the 30-kDa protein. The 30-kDa protein from TMV-infected protoplasts was precipitated by these antibodies, positively identifying it as the I2 gene product. Kiberstis et al. (1983) found that the 30-kDa protein was synthesized only between 8 and 16 hours after inoculation of protoplasts. However, in intact leaves, production of the 30-kDa protein continued for some days, but was maximal at around 24 hours (Lehto et al., 1990b).
Many workers have demonstrated the synthesis of TMV coat protein in vivo. Determination of the nucleotide sequence in the 3’ region of the TMV genome readily located the gene for this protein since the full amino acid sequence was already known (Anderer et al., 1960; Tsugita et al., 1960).
iii. Controlling elements in the viral genome
Six controlling elements have been recognized or inferred in TMV RNA:
-
1.
The nucleotide sequence involved in initiating assembly of virus rods, as discussed in Chapter 5 (Section IV.A). This sequence may have other controlling effects. Studies using pH 8.0 treated virus in an in vitro protein-synthesizing system suggest that the strong coat protein-RNA interactions occurring at the origin-of-assembly nucleotide sequence may be a site where translocation of 80S ribosomes is inhibited during uncoating of the virus rod. However, the effects of such a control mechanism would differ with different strains of the virus (Wilson and Watkins, 1985). Furthermore, encapsidation most probably removes the viral genome from the translation system.
-
2.
The replicase recognition site in the 3’ non-coding region (see Chapter 8, Section IV.H.3).
-
3.
The interactions between the 5’ cap and 3’ sequences described in Section V.C.
-
4.
The Ω sequence described above which enhances translation.
-
5.
The start codon sequence context may be one form of translational regulation. The context differs for each of the four known gene products. For example, in strain U1 the contexts are as follows: 126K: ACAAUGG 54K: GAUAUGC 30K: UAGAUGG coat protein: AAUAUGU
The context for the 30-kDa protein is least optimal according to Kozak's model (Kozak, 1981, 1986). This might be considered to be the reason why so little 30-kDa protein is produced compared with coat protein, but changing the start codon context for the 30-kDa AUG to the optimal strong context as defined by Kozak (1986) did not increase expression of the gene in tobacco plants (Lehto and Dawson, 1990). Furthermore, insertion of sequences containing the coat protein subgenomic RNA promoter and leader upstream from the 30-kDa ORF did not lead to increased production of the 30-kDa product (Lehto et al., 1990a). In fact, the production of 30-kDa protein was delayed and virus movement impaired, suggesting that different sequences influence timing of the expression of different genes.
-
6.
RNA promoters presumably have a role in regulating the amounts of subgenomic RNAs produced, but the exact sequences for these subgenomic promoters have not been identified. That for the coat protein lies within 100 nucleotides upstream of the ORF. The regulation of Tobamovirus gene expression is discussed by Dawson and Korhonen-Lehto (1990).
2. Two strategies: Subgenomic RNA + polyprotein
a. Sobemovirus genus (Tamm and Truve, 2000)
The monopartite sobemovirus genomes have four ORFs (see Fig. 6.51 for genome organization). The 3’ ORF, that for coat protein, is expressed from an sgRNA (Rutgers et al., 1980). In vitro translation of SBMV RNAs give four major polypeptides, P1 (100–105 kDa), P2 (60–75 kDa), P3 (28–29 kDa) and P4 (14–25 kDa) (Salerno-Rife et al., 1980; Ghosh et al., 1981; Brisco et al., 1985). It would appear that P4 is from the 5’ ORF but the attribution of the other translation products is not clear. ORF 2 of the cowpea strain of SBMV (now named SCPMV) is translated from scanning ribosomes passing ORF 1, the AUG of which is in a poor context (Sivakumaran and Hacker, 1998). The ORF 2 product is a polyprotein that is cleaved by a virus-encoded protease (Gorbalenya et al., 1988). There is also evidence for frameshift between ORFs 2a and 2b of CfMV (Mäkinen et al., 1995a; Tamm and Truve, 2000), which would suggest that this virus has three strategies.
3. Two strategies: Subgenomic RNA + multipartite genome
a. Bromovirus genus
BMV has a tripartite genome totalling 8243 nucleotides, the organization of which is described in Chapter 6 (Section VIII.A.1). RNAs 1 and 2 are monocistronic and RNA3 is bicistronic, the 3’ ORF, that for the coat protein being translated from a subgenomic RNA (see Fig. 6.13). This subgenomic RNA containing the coat protein gene is found in virus particles.
BMV RNAs are efficient mRNAs in in vitro systems and, in particular, in the wheatgerm system that is derived from a host plant of the virus. In this system, RNA1 directs the synthesis of a single polypeptide of 110 kDa and RNA2 directs a single 105-kDa polypeptide (Shih and Kaesberg, 1976; Davies, 1979). RNA3 directs the synthesis of a 35-kDa protein (Shih and Kaesberg, 1973). The coat protein cistron in RNA3 is not translated. RNA4 directs synthesis of the 20-kDa coat protein very efficiently. It is preferentially translated in the presence of the other viral RNAs in part, at least, because of more efficient binding of ribosomes (Pyne and Hall, 1979). In vitro the coat protein inhibits RNA synthesis by the BMV replicase, in a specific manner, possibly by partial assembly of nucleoprotein (Horikoshi et al., 1987).
Four new proteins were observed in tobacco protoplasts after infection with BMV. These were 20 kDa (coat protein), 35 kDa, 100 kDa and 107 kDa (Sakai et al., 1979). Four BMV-induced proteins with the same molecular weights (within the error of estimation in gels) were also found by Okuno and Furusawa (1979) in infected protoplasts prepared from three plant species–a systemic host, a local lesion host and a non-host. These four proteins account for over 90% of the viral genome. They correspond well in size with the in vitro products noted earlier.
b. Hordeivirus genus
The three genomic RNAs of BSMV are designated α, β and γ and their organization is described in Chapter 6 (Section VIII.H.14). RNAα is monocistronic expressing a 130-kDa protein. RNAβ has five ORFs, the 5’ of which, βa or coat protein, is expressed from the genomic RNA. The second, βb, is expressed from sgRNAβ1 and the 3’ triple gene box (see Chapter 9, Section II.D.2.f) from sgRNAβ2. Genomic RNAγ is bicistronic, the 5’ ORF being expressed from the genomic RNA and the 3’ ORF from sgRNAγ.
4. Two strategies: Multi-partite genome + polyprotein
Species in the bymovirus genus have their genomes divided into two RNA species, each of which encodes a polyprotein (see Fig. 6.22 for genome organization). Essentially, the genome organization is the same as that of potyviruses with the gene products of RNA2 representing the 5’ products from the potyviral genome. It is considered that the polyproteins are processed is a manner similar to potyviruses (see Section V.B.1.b).
5. Three strategies: Subgenomic RNA + multipartite genome + read-through protein
a. Tobravirus genus
Tobraviruses have a bipartite (+)-sense ssRNA genome (see Fig. 6.37).
RNA1 is translated in the rabbit reticulocyte system to give two polypeptides of 170 kDa and 120 kDa (Pelham, 1979). These two polypeptides are also produced together with many smaller products in the wheatgerm system containing added spermidine (Fritsch et al., 1977). These two proteins correspond to the products of ORFs 1 and 2 shown in Fig. 6.37. Similar results have been reported for PEBV (Hughes et al., 1986). A protein product for the 29-kDa ORF is translated from a subgenomic RNA (1A) (Robinson et al., 1983). The 16-kDa protein is also translated from a subgenomic RNA (1B), which is not required for replication or cell-to-cell transport in leaves (Guilford, 1989). A 16-kDa protein product, which was incorporated into a high-molecular-weight cellular component, was found in infected protoplasts (Angenent et al., 1989a).
RNA2 of the PRN strain is translated in vitro to give the coat protein identified by serology, peptide mapping, and specific aggregation with authentic coat protein to form disk aggregates (Fritsch et al., 1977). A second unrelated protein of 31 kDa is also translated. Different strains and viruses appear to differ in the products translated in vitro from RNA2 preparations, perhaps in part due to variable contamination with sgRNAs. No messenger activity has been detected for TRV strain SYM RNA2 in in vitro tests (Robinson et al., 1983). However, a subgenomic mRNA derived from RNA2 was shown to be the mRNA for coat protein.
In summary, RNA1 has three ORFs, the 5’ of which resembles that of TMV. It has a UGA stop codon (TMV has a UAG stop codon) at the end of the 134-kDa product which is read through to give a 194-kDa protein. The 3’ ORFs are expressed from sgRNAs. As noted in Chapter 6 (Section VIII.H.2 and Fig. 6.38), RNA2 varies in length and has variable numbers of ORFs between isolates. The 5’ ORF, that encoding the viral coat protein, is the only consistent one and is expressed from an sgRNA; it is not known how the other ORFs are expressed. However, if coat protein is the only gene product it is not clear why a subgenomic RNA should be required. Certain types of disease symptoms are specified by the short rods even when these give rise to identical coat proteins (e.g. Robinson, 1977). Thus, there may be a second protein coded for by RNA2.
6. Three strategies: Subgenomic RNA + multipartite genome + frameshift protein
a. Dianthovirus genus
The genomes of dianthoviruses are bipartite (+)-sense ssRNAs (Fig. 6.30). RNA1 has three ORFs, the 5’ two of which overlap. The second ORF is expressed by a −1 frameshift from the 5’ ORF (27 kDa) to give a protein of 88 kDa. Both the 27- and 88-kDa proteins are found in vivo and are made by in vitro translation of RNA1 (reviewed by Hamilton and Tremaine, 1996). The 3’ ORF of RNA1 is expressed from a 1.5-kb sgRNA to give the viral coat protein. RNA2 is monocistronic for the viral movement protein.
7. Three strategies: Subgenomic RNA + polyprotein + frameshift protein
a. Closterovirus genus
Closteroviruses have large complex genomes containing up to 12 ORFs (see Fig. 6.33). The first two ORFs overlap and the second is expressed by a +1 frameshift from the first. The first ORF contains a papain-like protease that processes at two sites. The frameshift between the first two ORFs of CTV and the proteolytic processing give rise to nine polypeptides (Fig. 7.27 ) (reviewed by Karasev and Hilf, 1997). The other 10 ORFs are expressed from subgenomic RNAs.
Fig. 7.27.

Map and expression strategy of CTV genome. Boxes represent putative protein products corresponding to the respective ORFs. + 1FS designates the putative ribosomal frameshift. Lines below define the two genomic regions expressed through the proteolytic processing of the polyprotein precursor(s) and through the formation of a nested set of 3’-co-terminal sgRNAs.
From Karasev and Hilf (1997), with permission.
© 2002
A temporal analysis of the expression of BYV genes was undertaken by tagging the genes for HSP70h, the major coat protein and the 20-kDa protein (p20, ORF 7) with the β-glucuronidase (GUS) gene (Hagiwara et al., 1999). This showed that the HSP70h promoter expressed early, followed by the coat protein promoter and later the p20 promoter. The kinetics of other sg promoters was followed by northern blot analysis. These two approaches showed temporal gene expression for BYV with HSP70h, CPm, CP and p21 being expressed early and p64 and p20 later (see Table 6.5 for designation of BYV genes).
8. Three strategies: Multi-partite genome + polyprotein + two-start
a. Comovirus genus (reviewed by Goldbach and Wellink, 1996)
Comoviruses have bipartite genomes, RNA1 also known as B RNA and RNA2 (M RNA), each of which expresses a polyprotein. CPMV, the type member of the Comovirus genus, has been most studied and so this discussion will relate to this virus. The genome organization is described in Chapter 6 (Section VIII.B.1.a).
i. The processing products
The 200-kDa polyprotein of RNA1 is processed at four sites, which should give five products. RNA2 encodes a polyprotein of 105 kDa with two translational start sites and the two cleavage sites giving four products. However, the processing is complex and various intermediate products are also found that have been studied both in vivo (protoplasts) and in vitro (reticulocyte lysate translation system) on cloned expression vectors. In protoplasts inoculated with CPMV and labelled with 35S-methionine, virus-specific proteins of 170, 112, 110, 87, 84, 60, 58, 37, 32 and 23 kDa are readily detected (Rottier et al., 1980; Rezelman et al., 1980). Obviously, the combined molecular weights of these products greatly exceeds the coding capacity of the two RNAs. The 37- and 23-kDa-proteins encoded by RNA2 are the viral capsid proteins. By peptide mapping and immunological analyzes the processing pathway of the polyprotein from RNA1 was determined (Fig. 7.28 ) (reviewed by Goldbach and Wellink, 1996).
Fig. 7.28.
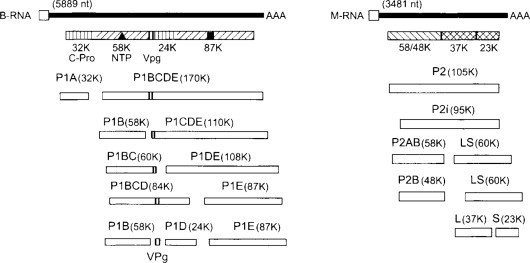
Organization and expression of the genome of CPMV. See Chapter 6, Section VIII.B.1 for description of proteins. All intermediate and final cleavage products have been detected in infected cells.
From Wellink et al. (2000), with permission.
© 2002
The recognition that there were two translation start sites in RNA2 came from in vitro translation and was verified on in vivo samples by the use of antipeptide antibodies. Furthermore, incubation of protoplasts in 2 mM ZnCl2 inhibited the proteinase activity and led to the production of the 105- and 95-kDa polyproteins. From these and other data, the processing pathway of RNA2 products was determined (Fig. 7.28).
In RNA2 there are three AUGs beginning at nucleotides 161, 512 and 524. Using site-directed mutagenesis, Holness et al. (1989) confirmed that the AUG at position 161 is used in vitro to direct the synthesis of a 105-kDa product. Both the 105-kDa protein initiating at nucleotide 161 and a 95 kDa product initiating at nucleotide 512 have been detected in infected protoplasts, as have their cleavage products (Rezelman et al. 1989).
ii. The proteinase
The CPMV proteinase was identified as the 24-kDa protein encoded by RNA1 by two different procedures. Wellink et al. (1987a) synthesized a peptide corresponding to an amino acid sequence in the 200 kDa polyprotein that showed similarity to picornaviral 3C proteases. Antibodies to the peptide reacted with a 24-kDa protein found in CPMV-infected protoplasts and leaves. Verver et al. (1987) constructed a full-length cDNA copy of RNA1. RNA transcribed in vitro could be efficiently translated and proteolytically cleaved. Introduction of an 87-bp deletion into the coding region of the 24-kDa protein abolished cleavage activity, demonstrating this protein to be the viral protease. Vos et al. (1988a,b) extended this work to include a cDNA copy of RNA2. They constructed a series of deletion mutants in the 24-kDa reading frames of RNA1 and the results showed that the 24-kDa protein is the protease responsible for all cleavages in both B and M polyproteins. It cleaves the RNA1 polyprotein most in cis and that of RNA2 in trans. The proteinase has considerable amino acid sequence homology to the picornavirus 3C proteinase (Argos et al., 1984; Franssen et al., 1984c). However, unlike the cellular serine proteinases that the viral ones also show homology to, the viral proteinases have a cysteine instead of a serine at their active site (see Section V.B.1.a). The catalytic site of CPMV proteinase is formed by His40, Glu75 and Cys166 (Dessens and Lomonossoff, 1991).
The 32-kDa protein is released rapidly from the 200-kDa polyprotein, occurring even as soon as ribosome finish translation of the 24-kDa coding region (Franssen et al., 1984a). The processing of the remaining 170-kDa protein occurs slowly both in vitro and in vivo. However, on in vitro translation of mutants lacking the 32-kDa protein the processing of the 170-kDa protein is rapid (Peters et al., 1992). Thus, the 32-kDa protein controls the processing of the 170-kDa product.
For efficient cleavage of the glutamine-methionine site in the RNA2 polyprotein, a second B-encoded protein (32 kDa) is essential, although this protein does not itself have proteolytic activity.
iii. The cleavage sites
N-terminal sequence analyzes have shown that cleavage of the CPMV polyproteins occurs at Gln/Met (two sites), Gln/Ser (two sites) and at Gln/Gly (two sites). Ala or Pro were present at position −2 and five of the six sites have Ala at position −4 (Wellink et al., 1986). Mutational analyzes show that some of the sites have greater constraints on the surrounding residues than do others. For example, when the Gly at position +1 of the Gln/Gly cleavage site between the two coat proteins was changed to Ala, Ser or Met (amino acids present at this position in other sites), cleavage was almost abolished (Vos et al., 1988b).
9: Three strategies: Subgenomic RNA + polyprotein + two-start
a. Tymovirus genus
The genome organization of the tymovirus TYMV is described in Chapter 6 (Section VIII.H.17). The organization is very compact. For example, with the European TYMV only 192 (3%) of the 6318 nucleotides are non-coding. The 5’ ORF runs from the first AUG codon on the RNA (beginning at nucleotide 88) and terminates with a UGA codon at position 1972 (69 kDa). The largest ORF initiates at position 95 and ends at position 5627 with a UAG codon (to give a protein of 206 kDa). Thus, it overlaps the 69-kDa gene over all its length. The 3’ terminal gene encodes the coat protein. There is a 105-nucleotide non-coding 3’ region, which contains a tRNA-like structure.
i. Subgenomic RNA
A small subgenomic RNA is packaged with the genomic RNA and in a series of partially filled particles (e.g. Pleij et al., 1977). No dsRNA corresponding in length to this subgenomic RNA could be detected in vivo. Gargouri et al. (1989) detected nascent subgenomic coat protein (+)-strand RNAs on dsRNA of genomic length. Thus, the coat protein mRNA is synthesized in vivo by internal initiation on (–) strands of genomic length. Ding et al. (1990) compared the available Tymovirus nucleotide sequences around the initiation site for the subgenomic mRNA. They found two conserved regions, one at the initiation site and a 16-nucleotide sequence on the 5’ side of it (Fig. 7.29 ). This longer sequence, which they called the tymobox, may be an important component of the promoter for subgenomic RNA synthesis. The elements that make up this subgenomic promoter have recently been studied in detail (Schirawski et al., 2000).
Fig. 7.29.

Aligned nucleotide sequences of tymoviral genomic RNAs in a region surrounding the initiation site (indicated by arrows in the initiation box) of subgenomic RNA transcription. The positions of these segments in their genomic RNA are shown in brackets where complete genomic sequences have been determined. The two conserved sequences are boxed. The stop codon of the replicase protein gene is underlined and the start codon of the coat protein is in bold. The consensus sequence is that of the subgenomic promoter of BMV and the possible subgenomic promoter of alphaviruses (Gargouri et al., 1989).
From Ding et al. (1990), with kind permission of the copyright holder, © Oxford University Press.
© 2002
The sg promoter sequence of TYMV has been located to a 494-nucleotide fragment that contains the tymobox (Schirawski et al., 2000). Duplication of this fragment into the coat protein ORF led to the in vivo production of a second sgRNA, and mutagenesis of the tymobox showed that it was an essential part of the promoter. The tymobox region can be folded into a hairpin formation, a feature found in other sg promoters (see Section V.B.2).
ii. In vitro translation studies
The small subgenomic RNA is very efficiently translated in in vitro systems to give coat protein (e.g. Pleij et al., 1977; Higgins et al., 1978). The coat protein gene in the genomic RNA is not translated in vitro.
Weiland and Dreher (1989) obtained infectious TYMV RNA transcripts from cloned full-length cDNA. By making mutants in the initiation codons they showed that the 69-kDa protein is expressed from the first AUG beginning at nucleotide 88, while the much larger gene product is expressed from an AUG beginning at nucleotide 95. Antibodies raised from a synthetic peptide corresponding to the C terminus of the shorter ORF are specific for the 69-kDa protein, demonstrating in vitro expression (C. Bozarth, J. Weiland and T. Dreher, personal communication to R. E. F. Matthews).
The large ORF with the AUG beginning at nucleotide 95 appears to be expressed in a variety of in vitro systems and is then cleaved in vitro to give a larger 5’ and a smaller 3’ product (Morch et al., 1982, 1988, 1989; Zagorski et al., 1983).
iii. Viral proteins synthesized in vivo
Coat protein. Biochemical studies indicate that coat protein is synthesized on 80S cytoplasmic ribosomes. Renaudin et al. (1975) showed that viral protein synthesis in Chinese cabbage protoplasts is inhibited by cycloheximide but not by chloramphenicol. The cytoplasmic site for viral protein synthesis has been confirmed by cytological evidence, but viral protein also accumulates in the nucleus.
The uracil analog, 2-thiouracil, blocks TYMV RNA synthesis but not protein synthesis (Francki and Matthews, 1962), implying that the mRNA for coat protein is a relatively stable molecule. The kinetics of labelling with [35S] methionine of empty protein shells and viral nucleoprotein in infected protoplasts shows that these two protein shells are assembled from different pools of protein subunits (Sugimura and Matthews, 1981).
Other proteins. The 69-kDa protein has been detected in vivo in both Chinese cabbage and Arabidopsis thaliana. It is expressed at a 500x lower level than coat protein and appears to be an early nonstructural protein. The 70-kDa C-terminal fragment shown is also found in vivo (C. Bozarth, J. Weiland and T. Dreher, personal communication to R. E. F. Matthews).
Mouches et al. (1984) showed that the TYMV replicase consisted of viral-coded 115-kDa polypeptide and a host-coded protein of 45 kDa. Using immunoblotting, Candresse et al. (1987) showed that the virus-coded replicase subunit appears very soon after inoculation of plants or protoplasts.
iv. Polyamine synthesis
In protoplasts derived from infected leaves, or in healthy protoplasts infected in vitro, newly formed virus particles contained predominantly newly synthesized spermidine and spermine (Balint and Cohen, 1985a,b). When a specific inhibitor of spermidine was present, there was increased synthesis of spermine and an increase in the spermine content of virus particles. Thus, there is some flexibility in the way in which the positive charge contributed by the polyamines is conserved. The biosynthesis of polyamines and their possible roles in plants are discussed in Smith (1985).
10. Four strategies: Subgenomic RNA + read-through protein + frameshift protein + internal initiation
All three genera of the Luteoviridae have similar expression strategies but differ in detail (reviewed by Miller et al., 1997a). The genomes of the Luteovirus and Polerovirus genera contains six ORFs and that of the Enamovirus genus five ORFs. Their expression strategies are shown in Fig. 7.30 .
Fig. 7.30.
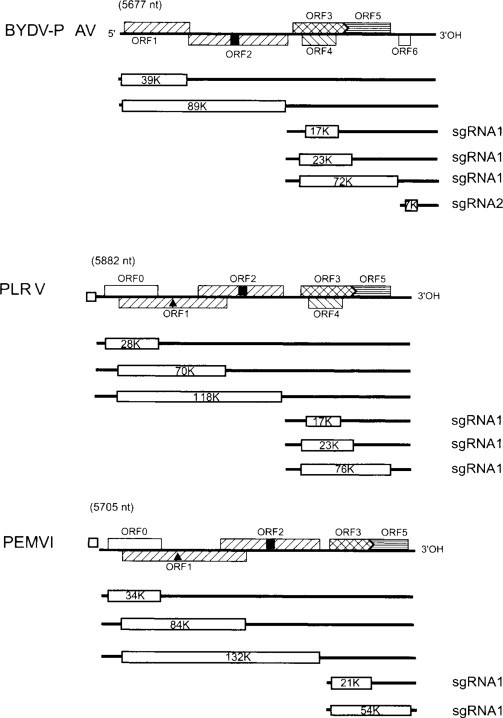
Diagram of the genome (from Fig. 6.35) and map of the translation products typical of viruses in each genus of the family Luteoviridae. Solid lines represent RNA; boxes represent ORFs; thinner boxes represent translation boxes; circles represent VPgs.
From D'Arcy et al. (2000), with permission.
© 2002
The 5’ two ORFs overlap and the second ORF is expressed by a −1 frameshift from the 5’ ORF (see Section V.B.10.a). The product from PLRV ORF 1 is processed by a serine proteinase that it contains (Li et al., 2000). ORFs 3, 4 and 5 are expressed from sgRNA1. ORF 3, which encodes the coat protein, ends in a UAG stop codon that is read through to give read-through protein with ORF 5. ORF 4 is translated from sgRNA1 by internal initiation. ORF 6 is expressed from sgRNA2. A third sgRNA has been suggested for the putative ORF 7 of PLRV (Ashoub et al., 1998). The VPg of poleroviruses is thought to be released from the ORF 1 product by a serine proteinase (see Sadowy et al., 2001b)
The 5’ terminus of BYDV sgRNA1 has been mapped to nucleotide 2670 on the genomic RNA (Koev et al., 1999), a hotspot region of recombination. The promoter for this sgRNA maps between nucleotides 2595 and 2692 and computer predictions reveal two stem-loop structures on the (–) strand of this region.
Results from in vitro and in vivo studies show that the genomic and subgenomic leader sequences of PLRV do not function as translational enhancers (Juszczuk et al., 2000). In fact, deletion analyzes show that both leader sequences not only decrease translation of downstream genes but also alter the ratio of the expressed proteins.
The mechanisms involved in the expression of luteovirus and polerovirus genomes are summarized in Fig. 7.31. From this it can be seen that there are complex control mechanisms involved in the expression of these viruses which were discussed in Section V.C.3.
11. Five strategies: Multi-partite genome + subgenomic RNA + read-through protein + polyprotein + two-start
As described in Chapter 6 (Section VIII.H. 13), the virions in plants naturally infected with BNYVV contain four, or sometimes five, (+)-sense ssRNA species, but only two of them, RNAs 1 and 2, are required for infection. RNA1 has a single ORF (see Fig. 6.49), the translation of which can be initiated at two sites: an AUG at position 154, which gives a 237-kDa product, and an AUG at position 496, which gives a 220-kDa product. The 220-kDa protein is processed by a papain-like protease encoded within this protein to give a 150-kDa and a 66-kDa protein (Hehn et al., 1997). BNYVV RNA2 has six ORFs, the 5’ two of which are in the same frame and separated by a suppressible UAG stop codon. The 5’ ORF encodes the 19-kDa coat protein, the stop codon of which is read through to give a 75-kDa protein. This read-through protein can be detected by immunogold labeling near the extremity of the virus particles and is important in fungal transmission of the virus (see Chapter 11, Section XII) (Haberlé et al., 1994; Tamada et al., 1996). The other four ORFs on RNA2 are expressed from sgRNAs, two of them, sgRNA a and sgRNA c, being monocistronic for a 42-kDa and 14-kDa protein respectively, and sgRNA b being bicistronic for a 13-kDa and 15-kDa protein. The expression of the 42-kDa, 13-kDa and 15-kDa is similar to that of the triple gene block of PVX.
12. Discussion
The above examples show that most plant viruses with (+)-sense ssRNA genomes adopt more than one strategy to overcome the constraints that their host places on them when expressing their genetic information. However, as noted above this bypassing of constraints is also used to control their gene expression, and there is often an interlink between the strategies to give temporal and quantitative control.
F. Negative-sense single-stranded RNA viruses
The ways by which plant viruses with (–)-sense ssRNA genomes derive their mRNAs are described in Section IV.A. The Rhabdovirus genome is transcribed to give a series of monocistronic sgRNAs that differ from those of (+)-strand RNA viruses in being sequential along the genome and not 3’ co-terminal. The major control of genome expression appears to be by the sequential transcription giving decreasing amounts of the mRNAs along the genome.
As described in Sections IV.A.2 and 3, the tospoviruses and tenuiviruses use two strategies; their largest RNA segment is transcribed to give a monocistronic mRNA and their other RNA genomic segments have an ambisense arrangement. Thus, mRNAs are transcribed from both the genomic- and complementary-sense RNAs. There is little information on any control mechanisms that may be involved.
G. Double-stranded RNA viruses
Transcription of the dsRNA genome segments of plant Reoviruses gives monocistronic mRNAs in most cases (see Section IV.B). Some of the genome segments potentially contain two ORFs but there is no evidence that the downstream ORF is expressed.
H. DNA viruses
1. Caulimoviridae
The transcription of the dsDNA genomes of members of the Caulimoviridae, and the promoters involved, are described in Section IV.C. For all genera in this family the transcription yields a more-than-genome-length RNA, the 35S or 34S RNA, that is both the template for expression of some of the gene products and the template for the reverse transcription phase of replication. For some of the genera other RNAs are transcribed from the viral DNA. Most of the studies on genome expression have been on CaMV and RTBV and these will be described in detail.
a. Cauliflower mosaic virus
The CaMV genome is transcribed from two promoters, one giving the 35S RNA and the other the 19S RNA (see Fig. 6.1). The 19S RNA is the monocistronic mRNA for ORF VI; ORFs I–V are expressed from the 35S RNA or products thereof. There have been suggestions of other transcripts such as a separate one for ORF V (Plant et al., 1985; Schulze et al., 1990) and for ORFs I and IV (Kobayashi et al., 1998) but these have not been substantiated.
There are two major problems to be faced in translating the information from ORFs I–V in the 35S RNA. Firstly, the RNA has a long leader sequence of 600 nucleotides or more; and secondly, these ORFs are downstream of the putative ORF VII and several sORFs and therefore should be closed to the eukaryotic translation system. Two unusual mechanisms are proposed to overcome these problems together with the probability that at least some of the downstream ORFs are opened up by splicing.
The first of the unusual mechanisms is used to bypass the long leader sequence up to ORF I that contains not only ORF VII but also several sORFs. This mechanism, termed ‘ribosome shunting’, is described in Section V.B.8 and involves the ribosome passing from a donor to acceptor site in the highly structured leader sequence. It is considered that ribosome shunting enables much of the leader sequence to be bypassed and delivers the 40S ribosome subunit to the start codon of ORF I where it forms an 80S ribosome and starts translation.
ORFs I–V are closely appressed to each other, either overlapping by a few nucleotides or being separated by a few nucleotides. At the termination codon of ORF I it is considered that the second mechanism, that of trans-activation by the product of ORF VI, takes over. This has been termed the ‘relay race’ model for the translation of the 35S RNA (Dixon and Hohn, 1984). In this model, a ribosome binds first to the 5’-end of the RNA and translates to the first termination codon. At this point, it does not completely leave the RNA but re-initiates protein synthesis at the nearest AUG whether just downstream or upstream from the termination. Support for the model came from site-directed mutagenesis studies in ORF VII and the region between ORF VII and ORF I, regions that are not essential for infectivity under laboratory conditions. Insertion of an AUG into either of these regions rendered the viral DNA noninfectious unless the inserted AUG was followed by an in-frame termination codon (Dixon and Hohn, 1984).
Transactivation is discussed in Section V.B.7 and is considered to prevent the ribosome completely disengaging at the stop codon and to enable it to commence translation again at the next start codon. The transactivation of CaMV is fully described in Rothnie et al. (1994) and in Hohn and Fütterer (1997).
As noted in Section V.B.11, there is increasing evidence for splicing in CaMV. It is difficult to identify spliced RNAs by northern blotting techniques of RNAs extracted from CaMV-infected tissues as there is always a smear of hybridizing RNA below the 35S RNA; this most likely arises from degradation of the 35S RNA during reverse transcription.
Using protoplasts transfected with 35S RNA-driven and promoter-less ORF I- and ORF IV-3-glucuronidase fusion constructs, Kobayashi et al. (1998) obtained results that they interpreted as suggesting that sgRNAs were involved in the expression of ORFs I and IV.
However, no such sgRNAs have been found in CaMV infections.
b. Rice tungro bacilliform virus
The genome of RTBV contains four ORFs (see Fig. 6.4). The dsDNA genome is transcribed to give a 35S RNA which is spliced (as described in Section V.B.11) to form the mRNA for ORF IV. Thus, the expression of ORFs I–III faces similar problems to those described for CaMV above. RTBV has a long leader sequence (more than 600 nucleotides) that contains 12 sORFs. ORF I has an AUU start codon (see Fig. 7.18) and the next approximately 1000 nucleotides have only two AUG codons in any reading frame, those for ORFs II and III. ORFs I, II and III each overlap the next by one nucleotide having a ‘stop/start’ signal of AUGA (Hay et al., 1991).
This has led to the development of the model shown in Fig. 7.32 for the expression of RTBV ORFs I-III. As noted in Section V.B.8, RTBV resembles CaMV in that much of the long leader sequence is bypassed by a ribosome shunt mechanism. This places the 40S ribosome subunits at the AUU start codon of ORF I, a start codon that is about 10% as efficient as an AUG codon. It is suggested that only some of the 40S ribosome subunits initiate translation at the ORF I start codon, the rest translocating to the next AUG, which is the start codon for ORF II but is in a poor context. The model suggests that only some of these 40S ribosome subunits initiate here, the remainder passing to the next AUG, which is at the start of ORF III and in a good context.
Fig. 7.32.

Model for the expression of the first three ORFs of RTBV. The top line depicts the RTBV DNA genome (thick line) and ORFs I–III. The bottom line shows the genome with, above it, the stem loop formed by the 5’ region of the 35S RNA, ORFs I–III and depiction of ribosomes translating the genetic information. Note that only a proportion of ribosomes initiate at the 5’ termini of ORFs I and II, whereas initiation on ORF III is more efficient. Below the line are the sequences at the initiation sites of the three ORFs with the protein sequence below. The assembly of 80S ribosomes and disassembly into ribosomal subunits are depicted.
From data reviewed in Hull (1996).
© 2002
2. Geminiviridae
The synthesis and regulation of mRNAs from the ssDNA genomes of geminiviruses are described in Section IV.C.2.
VI. DISCUSSION
Plant viruses have evolved strategies for effectively expressing their genetic information from a minimum of genetic material (Drugeon et al., 1999). In doing so they have to overcome various constraints imposed by their presence in eukaryotic cells. These constraints are outlined in Sections V.A and V.B. Some emerging ideas on new mechanisms of initiating translation in eukaryotes have been reviewed by Kosak (2001). As well as overcoming these constraints, the viral mRNA(s) has to compete with host mRNAs for the translational machinery and also has to express the various gene products in the right amount, at the right time and in the right place. Thus, these strategies have a sophistication that we are only just beginning to understand.
Little is known about how viruses compete with host mRNAs. They are, in many cases, expressed to much higher levels that many plant genes. Two of the virus groups (the tospoviruses and tenuiviruses) undertake ‘capsnatching’ which removes the 5’ cap structure from host mRNAs (see Section V.C.4) and is presumed to give the viral messengers an advantage over the host messengers. But, what about the plant viral mRNAs that have caps and those that do not? Several viruses that have capped mRNAs express some of their proteins to very high levels. For instance, TMV coat protein can be one of the most highly expressed proteins in infected cells. Other uncapped viral RNAs also express proteins to a high level. To a certain extent, the level of expression is a reflection of the turnover rate of the various proteins as the comparisons of viral and host protein amounts are made on their steady-state levels.
However, in spite of these considerations, presumably there are factors that make viral mRNAs more efficient at sequestering the host translational machinery or more effective at doing so. In the latter case, it is likely that the fact they are present at much higher levels than host mRNAs that is important. It is probable that the more efficient expression of viral RNAs is due to the influence(s) of regions distant from the sites of ribosome binding and translation initiation. We are just beginning to identify these sites and to identify the interactions involved. The findings described in Chapter 9 (Section III.B.3) show that, in at least some viral infections, there is a differential effect on host gene expression with some being turned off, others enhanced and yet others not affected. It will be interesting to know if this is a widespread phenomenon and to gain an understanding of the mechanism of it.
As well as host macromolecules, such as ribosomes, being involved in the expression of viruses, there is an increasing number of host proteins and factors being recognized as being used. It is likely that the avidity of viral mRNAs for these proteins and factors is, in part, responsible for the relative efficiency of viral expression. On the other hand, the interactions between viral sequences and these proteins and factors could play a part in controlling virus expression.
It is obvious that viral gene products are required in different amounts and at different times during the infection cycle. As noted in Section V.B.1.c, just one or, at the most, a very few copies of the replicase protein(s) are required to produce a new copy of the viral genome, but 60 (for spherical viruses with T = 1 structure) to several thousand (for long rod-shaped viruses) copies of the coat protein to encapsidate it. Furthermore, the replicase is required early in the infection cycle whereas the coat protein is required relatively late. The other gene products, such as those required for cell-to-cell movement or interactions with vectors, are also required in different amount at different times. Viral expression strategies that involve subgenomic RNAs would give control of both the timing and amount of gene products. It is of interest to note that, for many viruses, the coat protein gene lies at the 3’-end of the genome, and possibly this is effective in producing coat protein relatively late in infection. As noted in Section V.B.1.c, the use of the polyprotein strategy does pose some questions about its efficiency. In the viruses that express their information via a polyprotein but from a divided genome (e.g. comoviruses, bymoviruses) the ‘early’ genes appear to be on one genome component and the ‘late’ genes on another; this would allow temporal differentiation of expression.
The strategies that involve frameshift from one ORF to an overlapping one, or read through a weak stop codon, control the relative amount of the product from the downstream ORF; in vitro studies indicate that the frameshift or read through occurs on about 5–10% of the occasions that the upstream ORF is translated. Most, if not all, the products of these two strategies are involved either in viral replication or interaction with the virus vector. The read-through or frameshift products from the coat protein gene that are involved in virus vector interactions are incorporated into the viral capsid and it is obvious that they are required in a smaller amount than the coat protein itself. The requirement for differential expression of the replication proteins is less well understood.
It may be that, as we gather more information about the replication of viruses that our concepts of what is required early and late in infection will change. For instance, coat protein is required for the replication of AMV (see Chapter 8, Section IV.G) and thus cannot be considered to be a ‘late’ gene.
As discussed above, viruses compete very effectively with the host mRNA for their expression. However, there has to be control to prevent viral expression causing irreparable damage to the host cell, as the virus is totally dependent on maintaining the integrity of the cell and host plant. Thus, encapsidation plays an important role in sequestering the viral genome from the translation machinery. However, this raises a further question as to how re-initiation of virus uncoating is prevented. As discussed in Section II, for uncoating, the virus particle has to be destabilized by factors such as pH and removal of divalent cations. It would seem likely that the newly encapsidated virions are placed in a cellular compartment where this cannot occur.