

Abstract
Fluoride ions are highly reactive, and their incorporation in forming dental enamel at low concentrations promotes mineralization. In contrast, excessive fluoride intake causes dental fluorosis, visually recognizable enamel defects that can increase the risk of caries. To investigate the molecular bases of dental fluorosis, we analyzed the effects of fluoride exposure in enamel cells to assess its impact on Ca2+ signaling. Primary enamel cells and an enamel cell line (LS8) exposed to fluoride showed decreased internal Ca2+ stores and store-operated Ca2+ entry (SOCE). RNA- sequencing analysis revealed changes in gene expression suggestive of endoplasmic reticulum (ER) stress in fluoride- treated LS8 cells. Fluoride exposure did not alter Ca2+ homeostasis or increase the expression of ER stress–associated genes in HEK-293 cells. In enamel cells, fluoride exposure affected the functioning of the ER-localized Ca2+ channel IP3R and the activity of the sarco-endoplasmic reticulum Ca2+-ATPase (SERCA) pump during Ca2+ refilling of the ER. Fluoride negatively affected mitochondrial respiration, elicited mitochondrial membrane depolarization, and disrupted mitochondrial morphology. Together, these data provide a potential mechanism underlying dental fluorosis.
One-sentence summary:
The mechanisms by which excessive fluoride causes defects in tooth enamel mineralization are revealed.
Editor’s Summary:How too much fluoride derails dentition
Excessive fluoride ingestion during childhood results in defective tooth enamel mineralization, which can lead to dental problems later in life. Aulestia et al. investigated the molecular mechanisms underlying fluorosis in enamel-forming cells isolated from rats and in an enamel cell line. Exposure of enamel cells to fluoride resulted in decreases in ER Ca2+ content and store-operated Ca2+ entry into the ER, reduced the expression of genes encoding ER stress–response proteins, and resulted in mitochondrial dysfunction. These effects were not seen in HEK-293 cells (which are derived from kidney epithelium). These data may explain how fluorosis affects Ca2+ homeostasis in enamel-forming cells and highlight cell type–specific stress responses.
INTRODUCTION
Fluoride is abundant in the environment, readily ingested, and found in serum at low micromolar concentrations (1). The main sources of fluoride intake are drinking water and toothpaste. When epidemiological studies reported that fluoride intake was an important factor in caries prevention, drinking water was supplemented in many areas of the world (2, 3). Fluoride ions are highly reactive, and their incorporation in dental enamel during the development phase at low concentrations promotes mineralization and decreases the solubility of enamel (3, 4). Enamel formed by fluoroapatite is more resistant to acid attack (5).
Enamel crystals develop in specialized extracellular compartments modulated by the activities of epithelial cells, known as ameloblasts, during the secretory and maturation stages of enamel development (6–8). Ameloblasts coordinate the transport of ions required for the growth of crystal (7, 8). The effects of fluoride incorporation during enamel development are reversed when excessive fluoride intake occurs, posing a health problem known as dental fluorosis (DF) (3, 9, 10). Rather than strengthening the bonds between enamel crystals, excessive fluoride disrupts mineralization, resulting in pitted enamel with white opaque surfaces and hypomineralization (3, 9, 11, 12). DF is exclusively a developmental defect and has a major effect worldwide: ~30% of the U.S. population and ~60 million people in India are affected by DF with varying degrees of severity (2, 13). Therefore, the current recommendation for daily fluoride intake is less than 1.0 ppm (parts per million), with water fluoridation not exceeding 0.7 ppm (0.7 mg/kg) (14).
The mechanisms by which fluoride causes DF are complex. Variables affecting the impact of fluoride include its concentration, duration of exposure, and whether fluoride intake occurs during the formative (or secretory) or mineralizing (or maturation) stages of enamel development (3, 10, 12, 15). It may also have a genetic component given the variable impact of excessive fluoride intake on different mouse strains (16). Fluoride is primarily excreted in urine, which may also affect DF models. DF induction in rodents requires a higher fluoride dosage than in humans, likely because fluoride excretion is faster in rodents (7, 17).
Unlike bone, enamel does not remodel once formed, and there- fore, developmental defects such as DF cannot be reversed, leading to studies of the effects of excessive fluoride intake on the formation of enamel crystals in the extracellular milieu (3, 12, 15, 18). Excess fluoride leads to retention of enamel matrix proteins, irregular crystal formation, and hypomineralization (12, 15, 19–21). Despite decades of research on DF, the cellular mechanisms directly responsible for this disease remain poorly understood (22). In primary enamel cells or cell lines, fluoride causes protein misfolding, induces endoplasmic reticulum (ER) stress, and increases the unfolded protein response (UPR) (23–26). The UPR enables cells to cope with misfolding of proteins in the ER (27, 28). These effects suggest that fluoride could interfere with ER Ca2+ concentration ([Ca2+]ER), though this has not yet been explored. The ER is the main cellular hub for protein folding, requiring the presence of luminal ER Ca2+ ([Ca2+]ER) (~500 μM) to allow chaperones to perform their protein-folding functions (29, 30). Thus, disruptions in [Ca2+]ER mediated by fluoride could be a cause for reported protein misfolding and UPR in enamel cells.
Fluoride also affects Ca2+ transport because it reduces Ca2+ levels in the enamel fluid, the compartment where crystals are formed, of fluoride-treated rats (31). An important modulator of Ca2+ homeostasis in enamel cells is store-operated Ca2+ entry (SOCE), a mechanism that enables sustained Ca2+ influx (32, 33). SOCE is mediated by the ER-localized Ca2+ sensors STIM1 and STIM2, which interact with the pore of the ORAI1 to ORAI3 channels found in the plasma membrane (34, 35). The activation of SOCE is initiated after the loss of luminal Ca2+ in the ER, stimulating the accumulation of STIM1- forming punctae, a prerequisite for the gating of ORAI1 (36, 37).
These reports on UPR and abnormal Ca2+ homeostasis in enamel cells suggest a possible connection between fluoride exposure and abnormal ER Ca2+. Here, we address these questions, focusing on the possible interaction of fluoride with molecular elements associated with Ca2+ signaling and homeostasis in the cell membrane and endomembranes. We report that fluoride-treated enamel cells showed abnormal ER Ca2+ loading and up-regulation of ER stress markers, in part because the refilling of ER by SERCA (sarco-endoplasmic reticulum Ca2+–adenosine triphosphatase) was affected. Fluoride disrupted the activity of inositol 1,4,5-trisphosphate receptor (IP3R) and affected cellular bioenergetics. RNA-sequencing (RNASeq) analysis revealed that fluoride affected LS8 and human embryonic kidney (HEK)–293 cells differently. These data provide a mechanism on the intracellular effects of fluoride in enamel cells, which help to explain the molecular mechanisms that cause DF.
RESULTS
Enamel cells, but not HEK-293 cells, show low Ca2+ after fluoride treatment
To investigate the possibility that fluoride affects Ca2+ homeostasis in enamel cells, we isolated rat primary enamel organ (EO) cells from secretory and maturation stages as described previously (38, 39). Because primary EO cells are notoriously difficult to maintain in culture (6, 40), we also used the well-known ameloblast cell line, LS8 cells (41), and HEK-293 cells. Cells were treated for 24 hours with NaF (1 mM) before they were loaded with the cytosolic Ca2+ dye Fura-2 AM (1 μM). We used 1 mM NaF (~19 ppm) because dose-response experiments showed that LS8 cells exposed at 1 mM had the greatest defects in peak release and SOCE (fig. S1). This concentration is higher than fluoride levels in serum (~8 μM), but serum [F−] can rise several fold once fluoride is ingested including through the consumption of water with ppm values higher than those used here (42–44). Moreover, 1 mM NaF has been reported to cause cell stress in enamel cells in vitro (26) and affects rat incisor enamel in vivo (45). The internal Ca2+ stores were depleted passively using the SERCA inhibitor thapsigargin (1 μM) or tBHQ (5 μM) (2,5-di-tert-butyl- 1,4-benzohydroquinone) before external Ca2+ containing Ringer solution was re-added to stimulate SOCE, as previously described (32). Cells exposed to thapsigargin or tBHQ (5 μM) showed similar release kinetics (fig. S2). After SERCA inhibition, all three enamel cell types (secretory, maturation, and LS8) showed lower internal Ca2+ and SOCE compared to control cells, effects not seen in HEK-293 cells (Fig. 1, A to F). These data combined indicate that the effects of fluoride on internal Ca2+ stores and SOCE are not generalized features in all cells because only enamel cells were affected.
Fig. 1. Fluoride leads to loss Ca2+ in internal stores only in enamel cells.
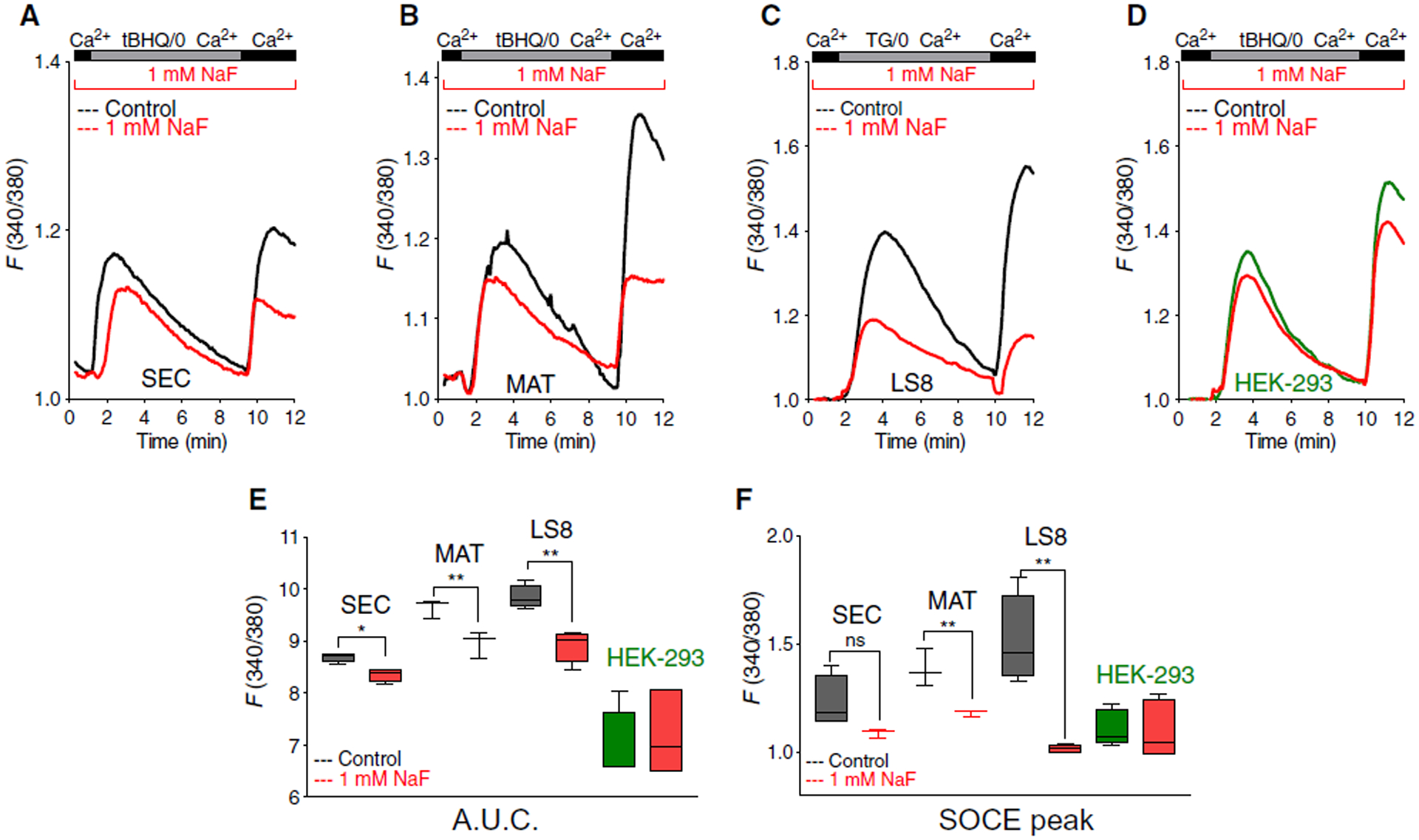
(A to C) Ca2+ levels in internal stores and SOCE upon re-addition of external Ca2+ in isolated primary enamel organ (EO) cells from secretory (SEC) (A) and maturation (MAT) (B) stages and LS8 cells (C) incubated with NaF (1 mM) for 24 hours and treated with the SERCA inhibitor thapsigargin (TG) or tBHQ. (D) Ca2+ levels in internal stores and SOCE upon re-addition of external Ca2+ in fluoride-treated HEK-293 incubated with NaF (1 mM) for 24 hours and treated with the SERCA inhibitor tBHQ. (E and F) Quantification of area under the curve (A.U.C.) (E) (between 2 and 9 min) and peak of SOCE (F) from (A) to (D). Data in (A) and (B) represent the mean ± SEM of three independent experiments analyzed using unpaired Student’s t test (*P < 0.01 and **P < 0.001). EO cells were obtained from six rats. Data in (C) and (D) represent the mean ± SEM of four to six independent experiments using unpaired Student’s t test (**P < 0.001); ns, nonsignificant.
Fluoride, but not bromide or chloride, affects [Ca2+] in enamel cells
To directly determine the effects of fluoride on [Ca2+]ER, we measured [Ca2+]ER with the genetically encoded ER-targeted Ca2+ indicator R-CEPIA1-er in cells treated with NaF. Depletion of ER Ca2+ by thapsigargin resulted in loss of Ca2+ after 30 min of stimulation, with additional decreases 1 and 2 hours after treatment (Fig. 2A). We analyzed [Ca2+]ER in secretory cells because these cells are abundant in ~4-week-old rat incisors. Analysis with the ER Ca2+ probe Mag-Fura-2 revealed a decrease in ER Ca2+ levels after NaF incubation (Fig. 2B). To determine whether physiological agonists such as adenosine triphosphate (ATP) could mobilize Ca2+ from internal stores in fluoride-treated cells, we transiently stimulated Fura-2 AM-loaded LS8 cells with ATP after incubation with NaF but measured only a small response (fig. S3). Together, these data suggest that fluoride decreases ER Ca2+ pools in enamel cells.
Fig. 2. Fluoride, but not bromide, affects ER Ca2+.
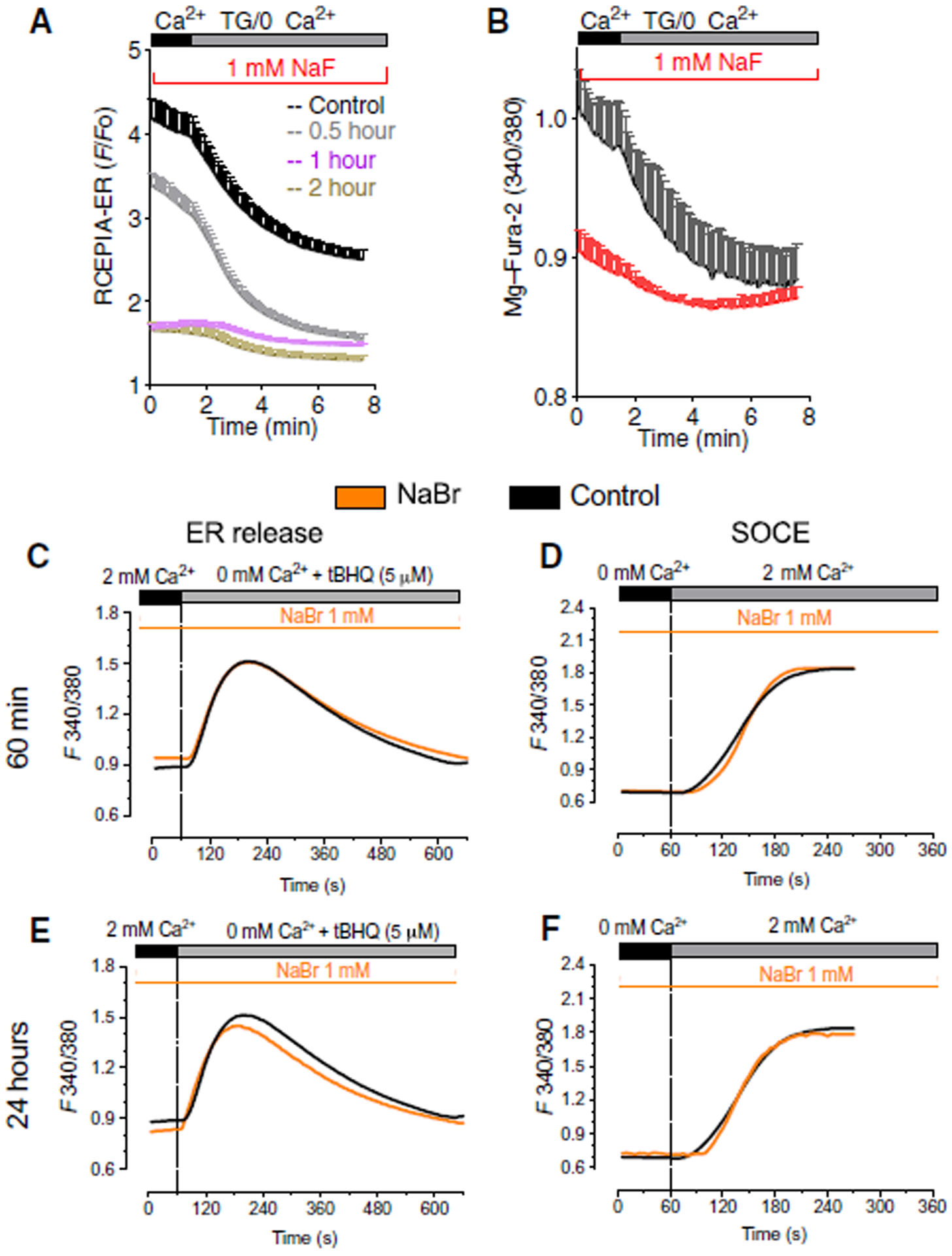
(A) ER Ca2+ in LS8 cells transfected with the genetically encoded ER Ca2+ probe CEPIA-red stimulated with thapsigargin and exposed to fluoride for 30 min (gray trace), 1 hour (blue trace), and 2 hours (green trace). Data represent the mean ± SEM of three independent experiments analyzed using Dunnett’s multiple comparisons test (**P < 0.001) of 30 min, 1 hour, and 2 hours compared to controls. (B) ER Ca2+ in primary secretory cells treated with fluoride (24 hours, 1 mM NaF) loaded with the ER Ca2+ probe Mag-Fura-2 and stimulated with thapsigargin. Data represent the mean ± SEM of three independent experiments analyzed using Student’s t test (black compared to red: ***P < 0.001). EO cells obtained from three rats. (C) ER Ca2+ levels in untreated LS8 cells (black trace) or cells treated for 1 hour with NaBr (1 mM) (orange trace). (D) SOCE in untreated LS8 (black trace) or cells treated with NaBr (1 mM) (orange trace) for 1 hour after maximally depleting the stores with tBHQ (5 μM) before re-addition of 2 mM Ca2+. (E and F) Same as (C) and (D) with cells treated for 24 hours. Data in (C) to (F) represent the mean ± SEM of three independent experiments.
Because of the unusual effects of fluoride on [Ca2+], we next asked whether other anions would induce similar responses in LS8 cells. NaBr (1 mM) did not elicit changes in ER Ca2+ release or SOCE after 1 or 24 hours of exposure (Fig. 2, C to F). Similarly, treatment with NaCl (1 mM) did not affect ER Ca2+ or SOCE (fig. S4). These data suggest that fluoride was responsible for changes in ER Ca2+ release and SOCE in enamel cells.
RNASeq analysis identifies distinct effects of fluoride in HEK-293 and enamel cells
Because LS8 and HEK-293 cells showed different responses to fluoride treatment, we used RNASeq to analyze global patterns of gene expression changes and pathways involved in this response. We first performed a flow cytometry analysis of annexin V–positive cells, which did not show an increase in cell death in fluoride-treated LS8 cells after 24 hours (fig. S5).
RNASeq analyses identified 2743 differentially expressed genes [DEGs; false discovery rate (FDR) < 1%] in fluoride-treated LS8 cells and 1723 DEGs (FDR < 1%) in HEK-293 cells (Fig. 3, A and B, and tables S1 and S2). Because fluoride exposure can have detrimental and stressful effects on cells, we analyzed the DEGs to determine whether ER stress and the UPR were induced, as is the case when [Ca2+]ER is abnormally low (1, 17, 28, 46). Genes encoding factors associated with cell stress and UPR were up-regulated in LS8 cells (Fig. 3C) but not in HEK-293 cells (fig. S6), as was also confirmed by quantitative real-time polymerase chain reaction (qRT-PCR) (Fig. 3D and fig. S7). Numerous genes encoding factors involved in the biogenesis of the mitochondrial complexes I, II, and V and coenzyme Q were significantly down-regulated (FDR < 1%) (Fig. 3E) together with mitochondrial ribosomal proteins (MRPs), with 35 MRP elements out of 78 down-regulated (Fig. 3F and table S1). Mitochondrial ribosomes (mitoribosomes) contain 78 genes in the family (MRPL-long; MRPS-short) and almost exclusively synthesize the protein complexes of the respiratory chain (47). The expression of Stim1, Stim2, or Orai genes was not changed in fluoride-treated LS8 cells, but Saraf (which encodes store-operated Ca2+ entry–associated regulatory factor, a negative modulator of SOCE) was up-regulated (table S1).
Fig. 3. RNASeq analyses of fluoride-treated LS8 and HEK-293 cells.
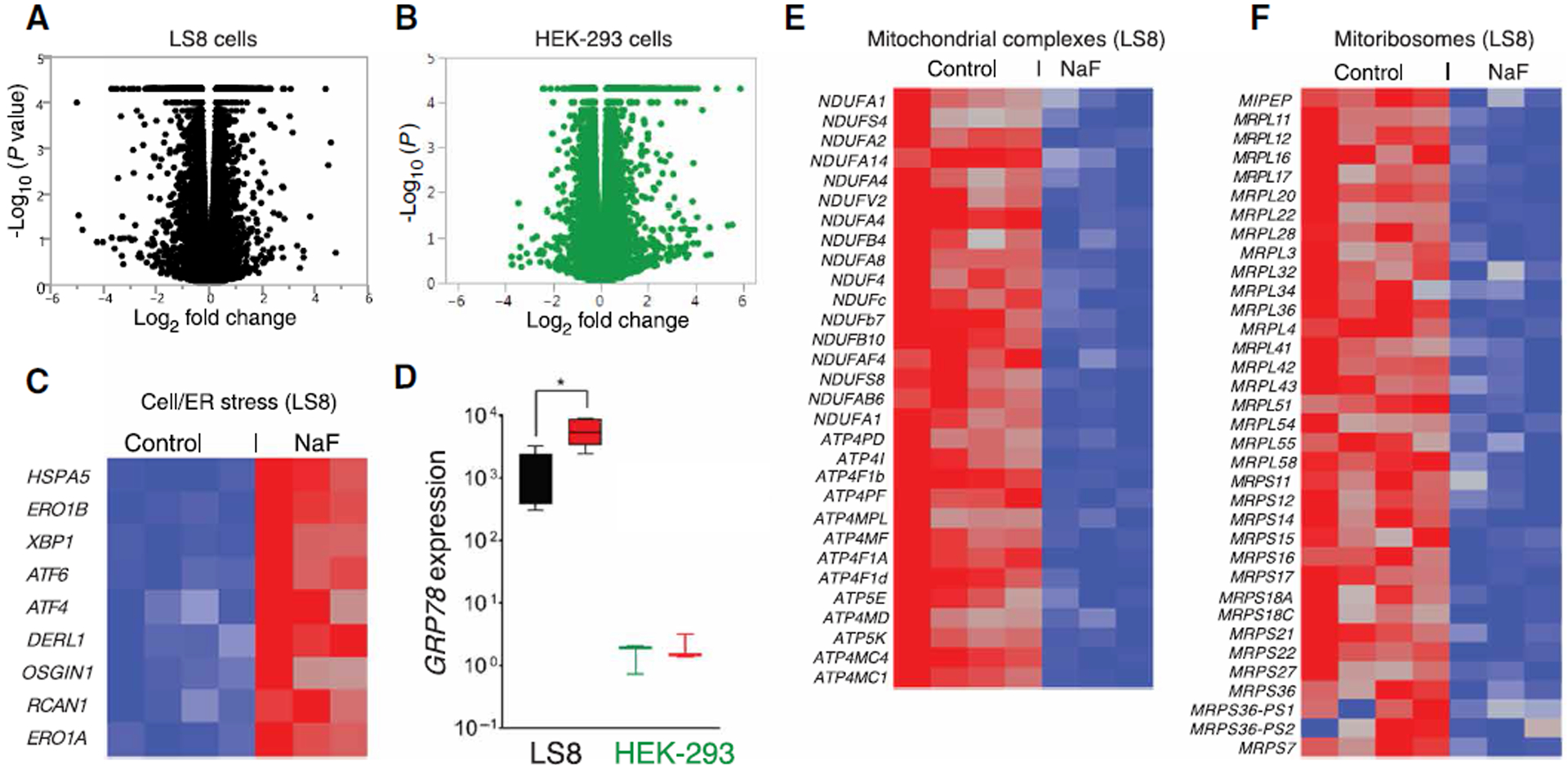
(A and B) Volcano plots of statistical significance (shown as the negative log base 10 of the P value on the y axis) compared to the magnitude of differential gene expression (shown as the log base 2 of magnitude of mean expression difference on the x axis) between control and NaF (1 mM) treatment for murine LS8 (A) and HEK-293 (B) cells. Data in (A) and (B) were obtained from four sets of untreated and three sets of NaF-treated cells. (C) Heat map of transcript abundance using one-way hierarchical clustering of untreated and 1 mM NaF–treated LS8 cells for 24 hours, showing the differential expression of genes encoding proteins associated with cell stress and UPR. Red indicates up-regulation. (D) RT-PCR showing the mRNA expression of the ER stress marker GRP78 in LS8 and HEK-293 cells in response to NaF (red boxes). Data represent the mean ± SEM of three independent experiments (*P < 0.05, ANOVA). (E) Differential expression of genes encoding proteins associated with the biosynthesis of mitochondrial complexes in LS8 cells. (F) Differential expression of genes encoding proteins associated with mitoribosome biosynthesis in HEK-293 cells. (E and F) Blue indicates down-regulation.
In contrast, HEK-293 showed extensive down-regulation (~4% of 1723 DEGs) in genes encoding factors associated with the biogenesis of ribosomal proteins (RPs), with ~82% (66 of 80) small subunit (SSU) or large subunit (LSU) RPs significantly down-regulated (FDR < 1%) (fig. S8 and table S2). RPs are key components of ribosome biogenesis, and they are a limiting rate in protein translation. Humans have 47 LSU RPs and 33 SSU RPs (48). This large-scale down-regulation of the RPs of the LSU and SSU in HEK-293 cells suggests suppression of protein translation, thus preventing UPRs because it averts an increase in misfolded proteins. Similar to LS8 cells, the expression of STIM1, STIM2, or ORAI genes did not change, but in contrast, SARAF expression was down-regulated (table S2).
IP3Rs mediate ER Ca2+ release in enamel cells in a manner altered by fluoride
Low Ca2+ ER content in fluoride-treated enamel cells suggests that the release mechanism of ER Ca2+ or SERCA-mediated refilling might have been affected. Our previous work reported that the inositol receptors (IP3Rs) were the likely ER Ca2+ release channels in LS8 and primary enamel cells (38, 49), although this conclusion was based on gene and protein expression patterns of the IP3R subtypes. Using two approaches, we analyzed whether IP3Rs were functional channels in enamel cells. First, saponin-permeabilized primary secretory and maturation and LS8 cells loaded with the ER Ca2+ dye Mag-Fura-2 were stimulated with IP3 (100 nM), which resulted in a marked drop in ER Ca2+ content (Fig. 4, A to C). To confirm these results by a different approach in intact cells, we loaded secretory EO cells simultaneously with caged IP3 and the cytosolic Ca2+ dye Fluo-8, as previously reported (50). Secretory EO cells were stimulated with two pulses of ultraviolet (UV) light to uncage IP3, resulting in increased [Ca2+]cyt (fig. S9). These data indicate that enamel cells express functional IP3Rs.
Fig. 4. IP3R and SERCA functions are modified by fluoride in enamel cells.
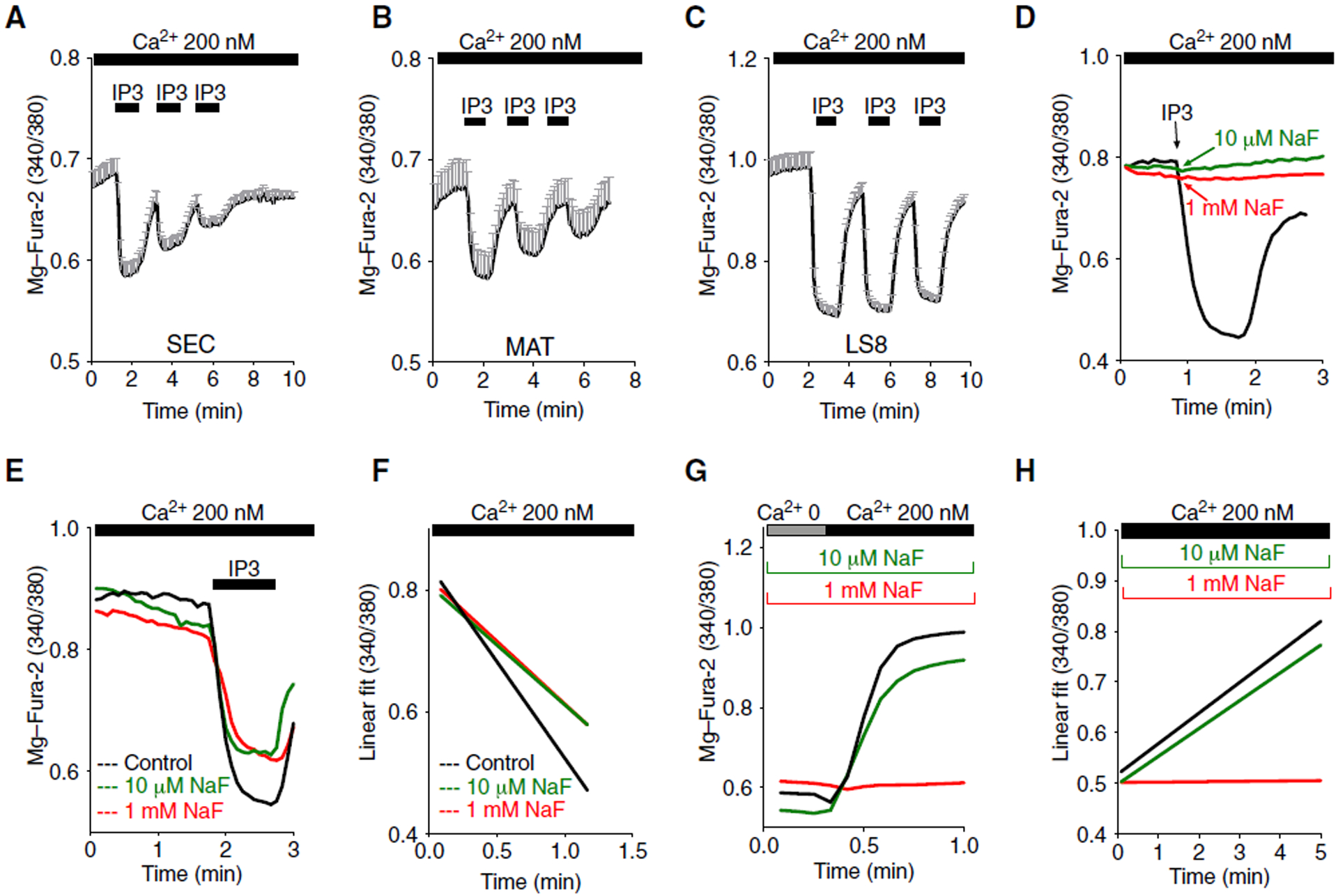
(A to C) Permeabilized LS8 cells (A), primary secretory (B), and maturation (C) stage cells loaded with the ER Ca2+ indicator Mag-Fura-2 were stimulated with IP3 to induce Ca2+ release from the ER. Each stimulus resulted in ER Ca2+ decrease. Representative tracings of ~100 cells per cell type. (D) Release from ER Ca2+ pools in permeabilized LS8 cells loaded with the ER Ca2+ dye Mag-Fura-2 and transiently stimulated with fluoride at 10 μM (green trace) or 1 mM (red trace). Representative of ~140 cells per group. (E) ER Ca2+ release by IP3 stimulation was measured in permeabilized LS8 cells loaded with Mag-Fura-2 pretreated with 10 μM NaF (green trace) and 1 mM NaF (red trace). Representative of ~140 cells per group. (F) Kinetics of release from ER Ca2+ pools in cells in (E). Data represent the mean ± SEM of three independent experiments using Tukey’s multiple comparisons test (black compared to red: **P < 0.0001; black compared to green: **P < 0.001; red compared to green: ns). (G and H) Kinetics of SERCA refilling measured in permeabilized untreated LS8 cells after tBHQ stimulation (black trace) and exposure to NaF (10 μM; green trace, and 1 mM; red trace). Data represent the mean ± SEM of three independent experiments using Tukey’s multiple comparisons test (black compared to red: ****P < 0.0001; black compared to green: *P < 0.0235; red compared to green: ****P < 0.0001).
As expected, IP3 uncaging in fluoride-treated secretory EO cells did not elicit changes in [Ca2+]cyt (fig. S9), suggesting that fluoride affects ER Ca2+ release by modulating IP3Rs. To determine the speed at which fluoride affected IP3Rs, we loaded permeabilized LS8 cells with Mag-Fura-2 and transiently stimulated the cells with NaF. Neither of the NaF concentrations used (10 μM and 1 mM) mobilized ER Ca2+ pools (Fig. 4D), suggesting that the effects of fluoride on IP3Rs did not occur immediately. Similarly in intact cells, transient application of various concentrations of NaF failed to elicit changes in [Ca2+]cyt (fig. S10, A to D). To address whether fluoride treatment altered the IP3-IP3R interaction, we loaded LS8 cells with Mag-Fura-2 to assess the kinetics of the release of IP3 -sensitive ER Ca2+ pools in the presence and absence of fluoride (Fig. 4E). The release rate of IP3 -sensitive Ca2+ pools was significantly faster in untreated control cells than in fluoride-treated cells (for 24 hours) (Fig. 4F). These data suggest that fluoride affects the IP3 -induced Ca2+ release.
ER refilling by SERCA is modified by fluoride
Because [Ca2+]ER depends on the interplay between Ca2+ release and refilling by SERCA, we also investigated the refilling capacity of these pumps. We treated LS8 cells with NaF 1 mM (24 hours) and used the reversible SERCA inhibitor tBHQ (5 μM). In saponin- permeabilized, NaF-treated cells, tBHQ depleted ER Ca2+ stores. We then switched the solution to an intracellular-like medium to allow ER Ca2+ refilling as previously reported (33) to measure the kinetics of ER refilling. Fluoride-treated cells showed slower refilling kinetics by SERCA compared to control cells (Fig. 4, G and H) without changes in SERCA2 abundance (fig. S11), indicating that, in addition to its effects on ER Ca2+ release, fluoride also affected SERCA activity.
Fluoride alters mitochondrial Ca2+ function and morphology
Because RNASeq analyses of fluoride-treated LS8 cells revealed down-regulation of mitochondrial genes, we further investigated the effects of fluoride on mitochondria. Stimulation of Rhod-2–loaded (2 μM) LS8 cells with ATP resulted in a marked increase in mitochondrial Ca2+ (Fig. 5A). Although fluoride-treated cells were able to take up Ca2+ in mitochondria under the same conditions, this increase was diminished compared to untreated cells (Fig. 5A). Because fluoride affects the mitochondrial membrane potential (51), we analyzed this effect in LS8 and in primary secretory EO cells loaded with the fluorescent probe TMRM (tetramethylrhodamine methyl ester) to monitor the membrane potential of mitochondria. Transient application of the protonophore FCCP (carbonyl cyanide-p-trifluoromethoxyphenylhydrazone) or fluoride depolarized mitochondria in LS8 and in primary secretory EO cells (Fig. 5, B and C). We next determined the potential effects of fluoride on mitochondrial function by measuring oxygen consumption rate (OCR), an indicator of mitochondrial respiration (52), in LS8 cells treated with NaF for 4 hours, a time period that minimized cell detachment during experimental manipulations and during which initial effects of NaF could be detected (Fig. 2A). Compared to control cells, fluoride pretreatment resulted in decreased basal and maximal respiration and in decreased ATP turnover rate (Fig. 5D). We next investigated the ultrastructural characteristics of mitochondria in fluoride-treated cells by transmission electron microscopy (TEM). In many of these cells, mitochondria showed the presence of white, non–electron-dense matrix spaces and a lack of cristae compared to control cells (Fig. 5, E to G). Together, these data indicate that fluoride affects mitochondrial morphology and function in enamel cells.
Fig. 5. Fluoride modified mitochondrial function and SOCE.
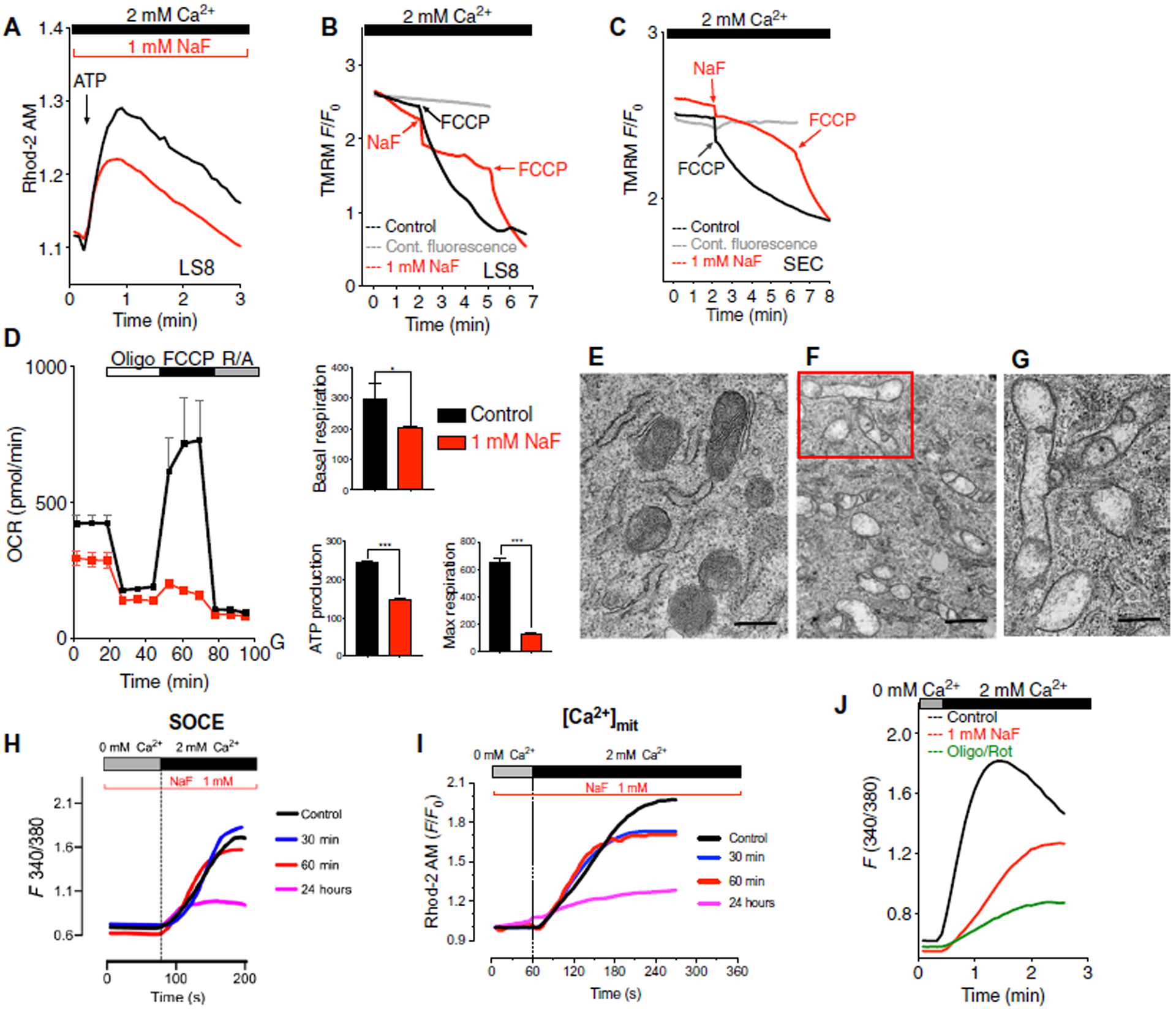
(A) Rhod-2–loaded control (untreated), LS8 (black trace), and fluoride-treated (24 hours) (red trace) cells transiently stimulated with ATP. Data represent the mean ± SEM of three independent experiments using Student’s t test (black compared to red: ****P < 0.0001). (B) Mitochondrial membrane potential measured in LS8 cells loaded with TMRM that were untreated and stimulated with FCCP (1.5 μM; black trace) or transiently stimulated with fluoride (1 mM) and FCCP (red trace). TMRM fluorescence was also monitored in cells not treated with FCCP (gray traces). Representative of 150, 100, and 120 cells. (C) Primary secretory cells analyzed as in (B). Representative of 90 untreated and stimulated with FCCP (black traces), 65 fluoride-and FCCP-treated cells (red traces), and 80 untreated cells (gray traces). (D) Oxygen consumption rate (OCR), basal respiration, ATP production, and maximal respiration in LS8 cells after 4 hours of NaF (1 mM) pretreatment. Data represent the mean ± SEM of three independent experiments using Student’s t test. (*P < 0.05 and ***P < 0.001). (E to G) Transmission electron micrographs of control LS8 cells (E) and fluoride-treated cells (F) (1 mM, 24 hours). Close-up of mitochondria (G) from (F) showing mitochondrial matrix with non–electron-dense matrix lacking cristae. Scale bars, 1 μm (E and F) and 0.5 μm (G). (H) SOCE measured after treating cells for 20 min with thapsigargin in untreated LS8 cells (black trace) or cells treated with NaF (1 mM) after 30 min, 60 min, and 24 hours. (I) [Ca2+] mit measured in Rhod-2–loaded LS8 cells after stimulation of SOCE as in (A). (J) SOCE measured after treating cells for 20 min with thapsigargin in untreated LS8 cells (black trace) or fluoride-treated (1 mM) cells (red trace) and cells treated with oligomycin (1 μM) and rotenone (2 μM) (green trace). Data in (H) to (J) represent the mean ± SEM of three independent experiments.
Disrupted ETC complexes affect SOCE
Our data above provide a basis for interpreting the effects of fluoride on ER Ca2+ homeostasis. However, the reasons why SOCE is also decreased in fluoride-treated cells remains unclear. In some cells, the capacity of mitochondria to sequester Ca2+ in their matrix is an important element in the functioning of SOCE (53–55). We found that fluoride treatment decreased SOCE and [Ca2+]mit, although [Ca2+]mit was affected before SOCE was decreased (Fig. 5, H and I). To test whether SOCE is dependent on healthy mitochondria, we measured SOCE in LS8 cells with and without oligomycin and rotenone to disrupt the mitochondrial electron transport chain (ETC) complexes (Fig. 5J). We found that disrupting the ETC complexes markedly decreased SOCE compared to control cells (Fig. 5J and fig. S12), indicating that, in enamel cells, properly functioning mitochondria are required for normal SOCE.
DISCUSSION
Fluoride alters ameloblast function (12), yet the mechanisms associated with DF remain poorly understood despite an increase in DF prevalence (56). Here, we approached the study of DF by analyzing intracellular Ca2+ signaling and associated effects in enamel cells exposed to fluoride. Our rationale was based on previous reports of enamel cells treated with fluoride, highlighting ER stress and abnormal protein synthesis (24–26, 57) and low Ca2+ concentration in the enamel fluid of fluoride-treated rodents (31). We hypothesized that these effects were caused by abnormal Ca2+ homeostasis.
We showed that fluoride treatment resulted in lower intra- cellular Ca2+ pools in primary secretory and maturation stage EO cells of rat incisors and in LS8 cells. Furthermore, SOCE was abnormally low in all fluoride-treated enamel cells (Fig. 1, A and B). However, HEK-293 cells did not show alterations in intracellular Ca2+ pools after fluoride exposure (Fig. 1D), demonstrating that fluoride does not affect Ca2+ homeostasis uniformly. Our data also showed that the internal Ca2+ stores affected by fluoride were the ER pools (Fig. 2, A and B). Moreover, cells were not affected by exposure to Br− or increased Cl− levels, pointing to F− as the mediator of changes in Ca2+ signaling (Fig. 2, C to F, and fig. S4).
RNASeq analyses provided insight into the processes altered by fluoride. UPR-associated genes were up-regulated in LS8 cells, including GRP78, a marker of ER stress (58), likely in response to low ER Ca2+. Our study revealed that mitochondrial-associated processes were affected, as seen by down-regulation of genes associated with mitochondrial biogenesis as well as components of the mitochondrial complexes of the ETC. Seventeen genes encoding factors associated with the biogenesis of NADH (reduced form of nicotinamide adenine dinucleotide) (complex I) and 13 genes encoding ATP synthase (complex V) were down-regulated in LS8 cells (Fig. 3E and table S1), suggesting an attenuation of the mitochondrial respiratory chain. In contrast, the effects of fluoride on HEK-293 cells were dominated by a bulk down-regulation of genes encoding RPs (~82% of all ribosomal genes) (fig. S8 and table S2). The cause of the difference in gene expression changes between LS8 and HEK-293 cells is unclear, although one possibility is that the factors necessary for ribosome biogenesis, which are cell and tissue specific and are correlated with ribosome abundance and the requirements for protein synthesis (59, 60), differ between LS8 and HEK-293 cells. Further work is needed to address this issue.
Fluoride treatment affected [Ca 2+]ER in enamel cells, raising at least two possibilities: Fluoride stimulates the release of ER Ca2+ or it affects the role of SERCA in the refilling of luminal ER Ca2+. With regard to this first point, we have previously reported that IP3Rs, rather than ryanodine receptors, are the likely Ca2+ release channels in the ER of enamel cells (38). In the present study, we confirmed that IP3Rs were active components in ER Ca2+ release in permeabilized primary EO and LS8 cells (Fig. 4, A to C) and in intact primary secretory cells (fig. S9).
We found that prolonged treatment but not transient treatment of LS8 cells with fluoride (1 mM or 10 μM) slowed the release of IP3-sensitive ER Ca2+ pools. However, because IP3 can be stimulated by the activation of G proteins (61) and fluoride stimulates this pathway (62), it is not unreasonable to consider that fluoride stimulated the IP3-mediated Ca2+ release in enamel cells by activating G proteins. Fluoride interactions with G proteins require the presence of Al3+, which results in the formation of AlF4−. However, not all G proteins can interact with AlF4− (61). As expected, ATP, which stimulates IP3 production, elicited elevations in [Ca2+]cyt in LS8 cells (fig. S10). However, intact LS8 cells transiently stimulated with various concentrations of fluoride did not show a rise in [Ca2+]cyt, suggesting that, in LS8 cells, either fluoride does not stimulate IP3 production or Al3+ is not present in our solutions. If fluoride slows the release of ER Ca2+, how is the [Ca2+]ER so diminished in fluoride-treated cells? [Ca2+]ER results from an interplay between release by IP3R and refilling by SERCA. Fluoride can interfere with SERCA activity in pancreatic acinar cells (63). Fluoride-treated LS8 cells in which ER Ca2+ was depleted showed limited refilling activity, suggesting that fluoride negatively affects the pumping action of SERCA. Over time, this limited action of SERCA could have detrimental effects on ER function.
The tight connections between ER and mitochondria are of interest because disruptions in the flow of Ca2+ between these two key organelles can underlie disease pathogenesis (64). We found that stimulation with ATP of fluoride-treated cells resulted in Ca2+ up- take by mitochondria, albeit at a significantly decreased amount compared to control cells (Fig. 5A). Because the components of the mitochondrial complexes were down-regulated as shown in the RNASeq analyses (Fig. 3E), we expected a decrease in oxidative phosphorylation. Fluoride-treated LS8 cells showed substantially decreased basal respiration and ATP production after inhibition of the Fo portion of the ATP synthase (complex V) with oligomycin. Maximal respiration elicited by FCCP, which forces H+ across the inner mitochondrial membrane instead of through the ATP synthase, was also substantially lower in fluoride-treated cells, indicating that fluoride modified cellular bioenergetics. The requirement of ATP for endocytosis (65) may also explain why fluoride affects endocytosis in enamel cells (66) and why fluorosed enamel retains proteins (15). Moreover, we found that transient application of fluoride induced mitochondrial depolarization in LS8 cells. This effect was not as marked as depolarization induced by FCCP, but fluoride moderately modified ΔΨm. In addition to these functional defects, TEM micrographs of fluoride-treated LS8 cells demonstrated that many cells and, in many regions of these cells, mitochondria showed non–electron-dense spaces in the matrix, unlike in control cells where most regions of the cells showed mitochondria with well-organized cristae (Fig. 5, E to G).
The data discussed above described the effects of fluoride on ER Ca2+ homeostasis. Why SOCE decreased was unclear, although various possibilities can be assessed. The magnitude of SOCE generally correlates with the amount of Ca2+ released by the ER. However, we showed that the magnitude in the decrease in ER Ca2+ release and SOCE was not comparable in the three types of enamel cells analyzed (Fig. 1, A to C). Mitochondria contribute to SOCE activity (54, 55, 67), and mitochondrial depolarization decreases SOCE (67). Enamel cells transiently exposed to fluoride showed a decrease in ΔΨm (Fig. 5, B and C). In these cells with disrupted ETC, SOCE was significantly decreased compared to control cells (Fig. 5J), suggesting that healthy mitochondria are important for SOCE in enamel cells. In addition, the up-regulation in NaF-treated LS8 cells of Saraf, which encodes a negative modulator of SOCE (68), could also play a role. Last, CRAC channel function is linked with the availability of intracellular ATP (69, 70), and the substantial down-regulation of ATP production in NaF-treated LS8 cells may have also negatively affected SOCE. Together, these data indicate that fluoride may disrupt SOCE by affecting multiple pathways.
In summary, this study investigated how fluoride affected the intracellular milieu of enamel cells. The effects of fluoride were wide ranging and complex. Fluoride affected Ca2+ homeostasis but not in all cells. Because transient application of fluoride disrupted ΔΨm but did not immediately affect IP3 R-mediated ER Ca2+ release, we suggest that the dysregulation of Ca2+ homeostasis by fluoride in enamel cells is initiated in the mitochondria (Fig. 6). The subsequent disruption of the transmembrane potential for hydrogen ions, which is required for ATP synthesis, results in decreased ATP levels. Such a decrease in ATP limits SERCA function, and as a result, there is a decline in ER Ca2+ content. These detrimental defects are compounded by a down-regulation of MRPs. SOCE is attenuated because of low ATP levels, up-regulation of Saraf, or mitochondrial dysfunction. Together, our data provide a potential mechanism for DF.
Fig. 6. Model for the effects of high fluoride in enamel cells.
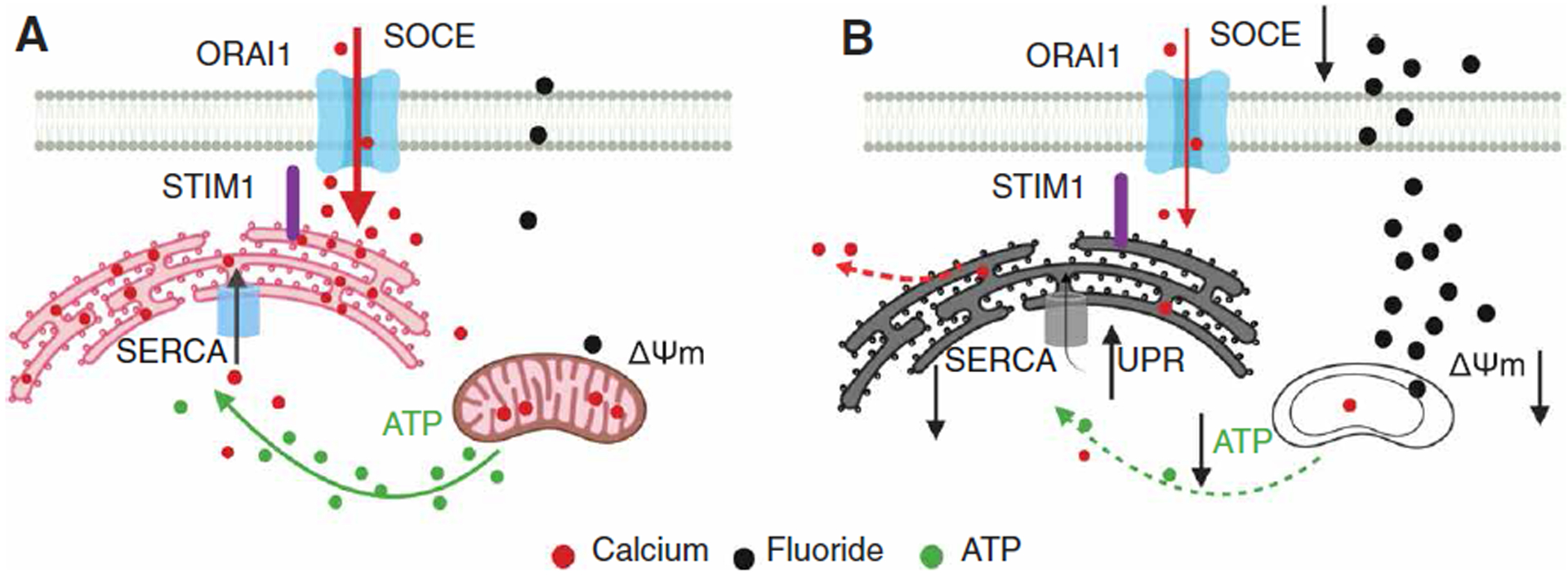
(A) In a healthy cell exposed to low fluoride, mitochondrial membrane potential (ΔΨm) and ATP production are normal, enabling SERCA to maintain its pumping function. SOCE is normal. (B) High fluoride exposure results in a decrease in the ΔΨm disrupting the proton motive force required to generate ATP. Decreased levels of ATP limit the capacity of SERCA to transfer Ca2+ into the ER lumen, resulting in increased Ca2+ leak. Cells respond by activating the UPR that allows the cells to survive, albeit in a low metabolic state. SOCE may be decreased because of low ATP or dys-functional mitochondria.
MATERIALS AND METHODS
Cell culture
Primary EO cells were isolated from the lower incisors of Sprague-Dawley rats (100 to 120 g). Secretory and maturation EO cells were isolated as previously described using a molar reference line (38, 39). EO cells were digested with Liberase (0.25 mg/ml; Roche) for 30 min at 37°C, washed in Hanks’ balanced salt solution, and plated onto CellTak-coated (Corning) coverslips in X-Vivo15 medium (Lonza) supplemented with 10% fetal bovine serum (FBS), 1% penicillin/ streptomycin, and 1% glutamine. Isolated EO cells were used within 24 hours after dissection. LS8 cells are an immortalized murine- derived enamel cell line (37). LS8 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% FBS at 37°C with 5% CO2.
[Ca2+]i measurements
Measurements of [Ca2+]i of single cells were performed as described (32). Briefly, single cells were plated overnight on a round microscope cover glass in X-Vivo15 or DMEM medium supplemented with 10% FBS. Cells were loaded with 1 μM Fura 2-AM for 60 min at room temperature, 1 μM Mag-Fura 2-AM for 60 min at room temperature, or 2 μM Rhod 2-AM for 20 min at 37°C, and washed in Ca2+ Ringer solution [2 mM CaCl2, 155 mM NaCl, 4.5 mM KCl, 1 mM MgCl2, 10 mM Na-Hepes, and 10 mM D-glucose (pH 7.4)]. To increase the cell purity in primary EO cells, we labeled fibroblasts using a PE-conjugated anti-CD90 antibody (1:500 dilution; BioLegend) as described (39) and excluded from further analysis. ER store depletion was stimulated with either 1 μM thapsigargin (Sigma) or 5 μM tBHQ (Sigma) in nominal Ca2+-free Ringer solution, followed by re-addition of 2 mM extracellular Ca2+ Ringer solution to stimulate SOCE. Alternatively, SOCE was stimulated by maximally depleting the ER stores with a preincubation for 20 min with thapsigargin (1.25 μM) or tBHQ (5 μM) of Fura-2 AM–loaded (1 μM) cells before re-addition of 2 mM Ca2+. Fluorescence intensities at 510 nm were recorded every 5 s after excitation at 340 and 380 nm for Fura-2 AM or 555 nm for Rhod-2 using a Nikon 2000U Eclipse microscope or a FlexStation III plate reader. The ratio of fluorescence 340 and 380 values correlating with [Ca2+]i were calculated and graphed.
Cell death
Analysis of cell viability was performed with the APC Annexin V Apoptosis Detection Kit (BioLegend) and propidium iodide (PI; Sigma-Aldrich), and staining was performed according to the manufacturer’s protocol. Flow cytometry analysis was carried out in an LSRII flow cytometer using FACSDiva software (BD Biosciences), and data were further analyzed with FlowJo (Tree Star). As a positive control for apoptosis induction, cells were treated with 1 μM staurosporine (Tocris) in parallel with NaF treatments.
Cell transfections
LS8 cells were plated overnight on a round microscope cover glass in X-Vivo15 or DMEM medium supplemented with 10% FBS. Cells were washed with Opti-MEM medium (OM; Thermo Fisher) and kept in OM (1 ml). OM was replaced with 800 μl (per well) of transfection reaction [1 μg of er-RedCEPIA plasmid and Lipofectamine 2000 (Invitrogen) with a 1:3 ratio in OM]. The cells were incubated with the transfection mix over 4 hours before switching to DMEM + 10% serum.
IP3R stimulation
Cells were plated on 25-mm optical borosilicate poly-L-lysine–coated sterile glass covers (Sigma) at 80% confluence and loaded with 1 μM Mag-Fura-2AM in Ringer solution for 60 min at 37°C in warm water. For permeabilization, cells were perfused (5 ml/min) with intracellular-like medium containing 130 mM KCl, 1 mM KH2PO4, 1 mM MgCl2, 1 mM ATP, 5 mM sodium succinate, 5 mM sodium pyruvate, 20 mM sodium Hepes (pH 7.0), Ca2+ buffered at 200 nM (with titrated 0.2 mM EGTA and EGTA-Ca2+ solutions), and 0.01 saponin for 1 min. The solution was then switched to the intracellular-like medium without saponin for 3 min to allow ER Ca2+ refilling. When the ER refilling reached the steady state (plateau), the IP3R stimulation was done in the same 200 nM Ca2+ medium supplemented with 100 nM IP3. Fluorescence intensities were recorded as indicated above.
IP3 uncaging
IP3 uncaging was performed as described previously (50). Briefly, LS8 and secretory cells were treated with NaF (1 mM) for 24 hours before loading them with the Ca2+ indicator Fluo-4 and with a cell-permeable form of caged inositol trisphosphate (ci-IP3/PM) (Tocris Bioscience), simultaneously, for 30 min. ci-IP3/PM is a cell mem- brane diffusible compound that is hydrolyzed by cellular esterases. Once the PM group is removed, ci-IP3 is liberated from the cage via UV photo-stimulus acting in a similar manner to InsP3 at InsP3R, releasing Ca2+. An additional period of ~30 min was allowed for de- esterification of the dye and cage. Cells were then illuminated at 488 ± 10 nm, and fluorescence was collected through a 525 ± 25–nm band-pass filter and captured using the Till Photonics imaging suite. These traces are displayed as % ΔF/Fo, where F is the recorded fluorescence and Fo is the mean of the initial 10 sequential frames. Photo- lytic release was performed by using a pulsed xenon arc lamp (Till Photonics). A high-intensity (0.5- to 5-ms duration; 80 J) discharge of UV light (360 ± 7.5 nm) was reflected onto the plane of focus by using a DM400 dichroic mirror and Nikon 40× oil immersion objective, 1.3 numerical aperture (NA).
SERCA refilling
Cells were plated as indicated above and loaded with 1 μM Mag-Fura-2AM. For permeabilization, cells were perfused (5 ml/min) with 2 mM Ca2+ Ringer solution for 1 min followed by perfusion with 0.01 saponin for 1 min in intracellular-like medium containing 2 mM EGTA plus 5 μM of the reversible SERCA inhibitor tBHQ. To allow ER Ca2+ refilling, we switched to intracellular-like medium constaining 130 mM KCl, 1 mM KH2PO4,1 mM MgCl2, 1 mM ATP, 5 mM sodium succinate, 5 mM sodium pyruvate, 20 mM sodium Hepes (pH 7.0), and Ca2+ buffered at 100 nM (with titrated 0.2 mM EGTA and EGTA-Ca2+ solutions). Fluorescence intensities were recorded as indicated above. The initial SERCA pumping speed was calculated as the slope of the refilling after the re-addition of 200 nM Ca2+ between the first second after the re-addition and when the plateau of the curve is reached. This parameter was obtained using the mean values.
Real-time PCR
Total RNA was isolated using the RNeasy Micro Kit (Qiagen) as indicated by the manufacturer followed by reverse transcription using the iScript cDNA Synthesis Kit (Bio-Rad). For quantitative real- time PCR, we used the SsoAdvanced Universal SYBR Green qPCR Supermix (Bio-Rad) and performed the experiments in a CFX Connect Thermocycler (Bio-Rad). Gapdh was used as a housekeeping gene. Primers for GRP78 were reported in (32) and were used at a concentration of 0.25 nM. Relative quantification of gene expression was determined by the 2–ΔΔCT method unless indicated otherwise.
Western blotting
Samples were transferred to a microcentrifuge tube containing 50 μl of nonreducing SDS–polyacrylamide gel electrophoresis containing protease inhibitors and homogenized before heating at 90°C for 2 min, centrifuged, and loaded on 10% mini gels. Electrophoresis was carried out at 200 V and transferred onto nitrocellulose membrane at 80 V for 1 hour. Blots were blocked overnight in 5% nonfat milk powder (Bio-Rad, no. 170–6404) in tris-buffered saline containing 0.05% Tween (TTBS) at 4°C. Membranes were probed with the SERCA antibody (Abcam) diluted at 1:1000 and actin (1:2000; Santa Cruz Biotechnology) in TTBS for 2 hours at room temperature. Blots were washed in TTBS and incubated in anti-rabbit immunoglobulin G peroxidase conjugate (Sigma, no. A6154) for 1 hour (room temperature), washed, and developed using a 3,3′-diaminobenzidine (DAB) staining kit (Sigma, no. D0426) in accordance with the manufacturer’s instructions. Blots were documented (Bio-Rad ChemiDoc MP) and then incubated with anti–β-tubulin horseradish peroxidase conjugate (Abcam, no. ab21058) diluted 1:2000 at room temperature for 1 hour and developed using the DAB staining kit.
Mitochondrial respiration
We used the Mitochondrial Stress Test Kit (Agilent) to analyze mitochondrial oxygen consumption in LS8 cells following the manufacturer’s instructions. Briefly, LS8 cells were seeded 24 hours in an XFe24-well microplate (Agilent) at 2500 cells per well in complete DMEM (10% FBS, 1% penicillin/streptomycin, and 1% glutamine). In parallel, a cartridge plate was hydrated with XF Calibrant (1 ml per well; Agilent) and kept overnight in a non-CO2 incubator. The following day, XF base medium (Agilent) containing 1 mM sodium pyruvate, 2 mM L-glutamine, and 10 mM glucose (pH7.4) was pre- pared, and cells were washed several times with this medium. Each well was refilled with exactly 500 μl of completed XF base medium, and cells were equilibrated for 1 hour in a non-CO2 incubator. The ATP synthase inhibitor oligomycin, the mitochondrial uncoupler FCCP, and complex I and III inhibitors (rotenone/antimycin A) were serially added in a Seahorse XFe24 Analyzer. All compounds including oligomycin, FCCP, and rotenone/antimycin A (Agilent) were prepared in stock solutions and loaded into the compound plate. Cell plate and compound plate were loaded into a Seahorse XFe Analyzer, and OCR was analyzed by sequential additions of oligomycin, FCCP, and rotenone/antimycin A. After each run, protein content of each well was analyzed via bicinchoninic acid, and data were normalized before analyzing basal respiration, ATP production, maximal respiration, and respiratory reserve.
Mitochondrial membrane potential
Cells were plated on 25-mm optical borosilicate poly-L-lysine–coated sterile glass covers (Sigma) at 80% confluence and loaded with 60 nM TMRM in Ringer solution for 30 min at 37°C in warm water. To maintain the balance of Nernst, the rest of the solutions that were applied in the experiments contained 20 nM TMRM. Images were obtained using a Nikon Eclipse 2000TE coupled with a light-emitting diode light to stimulate cells at 555 nm.
SOCE measurements after inhibiting ETC
To determine the effects of inhibiting mitochondrial function on SOCE in LS8 cells, we maximally depleted the ER stores by preincubating cells for 20 min with thapsigargin (1.25 μM). Cells were also loaded with Fura 2-AM (1 μM). To disrupt mitochondrial complexes of the ETC, we treated cells for 10 min with rotenone (2 μM) and oligomycin (1 μM) to block complexes I and V, respectively, before the re-addition of 2 mM Ca2+ to induce SOCE.
RNASeq data
LS8 and HEK-293 cells were treated with NaF (1 mM) for 24 hours in six-well plates at a density of ~80%. RNA was isolated using the RNeasy Micro Kit (Qiagen). RNA quality and quantity were analyzed on an Agilent Bioanalyzer nano chip. RNASeq libraries were prepared using the TruSeq Stranded mRNA Kit (Illumina), starting from 500 ng of deoxyribonuclease (DNase) I (Qiagen)–treated total RNA, following the manufacturer’s protocol. The amplified libraries (10 PCR cycles) were purified using AMPure beads (Beckman Coulter), quantified with a Qubit 2.0 fluorometer (Life Technologies), and visualized in an Agilent 2200 TapeStation system. The libraries were pooled equimolarly, loaded on an S1100 flow cell, and run as paired-end 50-nucleotide reads on a NovaSeq 6000. Raw paired-end sequencing reads from both human and mouse samples were quality controlled using the FASTQC tool (www.bioinformatics.babraham.ac.uk/projects/fastqc/). Poor-quality reads/bases, adapters, and barcodes were removed from the data using Trimmomatic v0.32 (41, 42). The high-quality reads were aligned to either the human genome reference (Ensembl release 84-GRCh38) or the mouse genome (Ensembl release 95-GRCm38) using the STAR aligner (STAR_2.5.2a) and default parameters. Transcript abundance estimation and differential expression analysis were performed using Cufflinks v2.2.1 and Cuffdiff v2.2.1, respectively. Hierarchical clustering using the Ward method and data visualization were done using JMP Genomics v8 (SAS Institute).
Statistics
Statistical analyses of data were done using Prism7 (GraphPad Software). Normality tests were performed before comparing the means. Two-tailed unpaired Student’s t test or two-way analysis of variance (ANOVA) was used to compare the means. Where appropriate, post hoc tests were performed (Dunnett’s or Tukey’s multiple comparisons). Differences with P values of <0.05 were considered significant: *P < 0.05, **P < 0.005, and ***P < 0.001. For RNASeq differential expression analysis, an FDR (the Benjamini-Hochberg procedure) of 1% was used as threshold for statistical significance.
Supplementary Material
stke.sciencemag.org/cgi/content/full/13/619/eaay0086/DC1
Fig. S1. ER Ca2+ release and SOCE in LS8 cells treated with various concentrations of NaF.
Fig. S2. Comparison of ER Ca2+ release by thapsigargin and tBHQ.
Fig. S3. ATP stimulation of fluoride-treated LS8 cells.
Fig. S4. Fluoride, but not chloride, affects Ca2+ homeostasis.
Fig. S5. Cell death analyses.
Fig. S6. Heat map of cell and ER stress genes in HEK-293 cells.
Fig. S7. Expression of the ER stress marker GRP78 in cells exposed to various NaF concentrations.
Fig. S8. Heat map of RP genes in HEK-293 cells.
Fig. S9. IP3 uncaging in primary secretory stage enamel cells.
Fig. S10. Transient fluoride application does not elicit changes in [Ca2+]cyt.
Fig. S11. Western blot of SERCA in LS8 cells treated with fluoride.
Fig. S12. Altering mitochondrial function by rotenone/oligomycin disrupts SOCE in LS8 cells.
Table S1. DEGs in fluoride-treated LS8 cells.
Table S2. DEGs in fluoride-treated HEK-293 cells.
Acknowledgments:
We thank J. Bartlett for discussions, A. Heguy and P. Meyn (Genome Technology Center, NYU School of Medicine) for the RNASeq preparation, and N. Drou and the NYUAD Bioinformatics Core team for assistance with RNASeq data processing.
Funding: This work was funded by the National Institute of Dental and Craniofacial Research (NIH/NIDCR) (R01DE025639 and R01DE027679 to R.S.L. and R01DE014756 to D.I.Y.). Y.I. was supported by New York University Abu Dhabi research grant AD105. The NYU Genome Technology Center is partially supported by Cancer Center Support grant P30CA016087 at the Laura and Isaac Perlmutter Cancer Center.
Footnotes
Competing interests: The other authors declare that they have no competing interests.
Data and materials availability: The RNASeq data have been deposited in NCBI Gene Expression Omnibus (GEO) under the accession number GSE143536. All other data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.
REFERENCES AND NOTES
- 1.Barbier O, Arreola-Mendoza L, Del Razo LM, Molecular mechanisms of fluoride toxicity. Chem. Biol. Interact 188, 319–333 (2010). [DOI] [PubMed] [Google Scholar]
- 2.Ajiboye AS, Dawson DR III, Fox CH, Committee ASI, American associationfor dental research policy statement on community water fluoridation. J. Dent. Res 97, 1293–1296 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Aoba T, Fejerskov O, Dental fluorosis: Chemistry and biology. Crit. Rev. Oral Biol. Med 13, 155–170 (2002). [DOI] [PubMed] [Google Scholar]
- 4.Simmer JP, Fincham AG, Molecular mechanisms of dental enamel formation. Crit. Rev. Oral Biol. Med 6, 84–108 (1995). [DOI] [PubMed] [Google Scholar]
- 5.Fejerskov O, Changing paradigms in concepts on dental caries: Consequences for oral health care. Caries Res. 38, 182–191 (2004). [DOI] [PubMed] [Google Scholar]
- 6.Lacruz RS, Enamel: Molecular identity of its transepithelial ion transport system. Cell Calcium 65, 1–7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lacruz RS, Habelitz S, Wright JT, Paine ML, Dental enamel formation and implications for oral health and disease. Physiol. Rev 97, 939–993 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smith CE, Cellular and chemical events during enamel maturation. Crit. Rev. Oral Biol. Med 9, 128–161 (1998). [DOI] [PubMed] [Google Scholar]
- 9.Angmar-Mansson B, Whitford GM, Single fluoride doses and enamel fluorosis in the rat. Caries Res. 19, 145–152 (1985). [DOI] [PubMed] [Google Scholar]
- 10.DenBesten PK, Biological mechanisms of dental fluorosis relevant to the use of fluoride supplements. Community Dent. Oral Epidemiol 27, 41–47 (1999). [DOI] [PubMed] [Google Scholar]
- 11.DenBesten P, Li W, Chronic fluoride toxicity: Dental fluorosis. Monogr. Oral Sci 22, 81–96 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bronckers ALJJ, Lyaruu DM, DenBesten PK, The impact of fluoride on ameloblasts and the mechanisms of enamel fluorosis. J. Dent. Res 88, 877–893 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Perumal E, Paul V, Govindarajan V, Panneerselvam L, A brief review on experimental fluorosis. Toxicol. Lett 223, 236–251 (2013). [DOI] [PubMed] [Google Scholar]
- 14.Department of Health US and Human Services Federal Panel on Community Water Fluoridation, U.S. Public Health Service recommendation for fluoride concentration in drinking water for the prevention of dental caries. Public Health Rep 130, 318–331 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Robinson C, Connell S, Kirkham J, Brookes SJ, Shore RC, Smith AM, The effect of fluoride on the developing tooth. Caries Res. 38, 268–276 (2004). [DOI] [PubMed] [Google Scholar]
- 16.Everett ET, Yin Z, Yan D, Zou F, Fine mapping of dental fluorosis quantitative trait loci in mice. Eur. J. Oral Sci 119 (suppl. 1), 8–12 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Buzalaf MAR, Whitford GM, Fluoride metabolism. Monogr. Oral Sci 22, 20–36 (2011). [DOI] [PubMed] [Google Scholar]
- 18.Lyaruu DM, Medina JF, Sarvide S, Bervoets TJM, Everts V, Denbesten P, Smith CE, Bronckers ALJJ, Barrier formation: Potential molecular mechanism of enamel fluorosis. J. Dent. Res 93, 96–102 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.DenBesten PK, Crenshaw MA, The effects of chronic high fluoride levels on forming enamel in the rat. Arch. Oral Biol 29, 675–679 (1984). [DOI] [PubMed] [Google Scholar]
- 20.Chen H, Czajka-Jakubowska A, Spencer NJ, Mansfield JF, Robinson C, Clarkson BH, Effects of systemic fluoride and in vitro fluoride treatment on enamel crystals. J. Dent. Res 85, 1042–1045 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Riksen EA, Kalvik A, Brookes S, Hynne A, Snead ML, Lyngstadaas SP, Reseland JE, Fluoride reduces the expression of enamel proteins and cytokines in an ameloblast- derived cell line. Arch. Oral Biol 56, 324–330 (2011). [DOI] [PubMed] [Google Scholar]
- 22.Bartlett JD, Dwyer SE, Beniash E, Skobe Z, Payne-Ferreira TL, Fluorosis: A new model and new insights. J. Dent. Res 84, 832–836 (2005). [DOI] [PubMed] [Google Scholar]
- 23.Suzuki M, Bandoski C, Bartlett JD, Fluoride induces oxidative damage and SIRT1/ autophagy through ROS-mediated JNK signaling. Free Radic. Biol. Med 89, 369–378 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sierant ML, Bartlett JD, Stress response pathways in ameloblasts: Implications for amelogenesis and dental fluorosis. Cell 1, 631–645 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sharma R, Tsuchiya M, Tannous BA, Bartlett JD, Measurement of fluoride-induced endoplasmic reticulum stress using Gaussia luciferase. Methods Enzymol. 491, 111–125 (2011). [DOI] [PubMed] [Google Scholar]
- 26.Sharma R, Tsuchiya M, Bartlett JD, Fluoride induces endoplasmic reticulum stress and inhibits protein synthesis and secretion. Environ. Health Perspect 116, 1142–1146 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Corbett EF, Oikawa K, Francois P, Tessier DC, Kay C, Bergeron JJM, Thomas DY, Krause KH, Michalak M, Ca2+ regulation of interactions between endoplasmic reticulum chaperones. J. Biol. Chem 274, 6203–6211 (1999). [DOI] [PubMed] [Google Scholar]
- 28.Lin JH, Walter P, Yen TS, Endoplasmic reticulum stress in disease pathogenesis. Annu. Rev. Pathol 3, 399–425 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vincenz-Donnelly L, Hipp MS, The endoplasmic reticulum: A hub of protein quality control in health and disease. Free Radic. Biol. Med 108, 383–393 (2017). [DOI] [PubMed] [Google Scholar]
- 30.van Anken E, Braakman I, Versatility of the endoplasmic reticulum protein folding factory. Crit. Rev. Biochem. Mol. Biol 40, 191–228 (2005). [DOI] [PubMed] [Google Scholar]
- 31.Crenshaw MA, Bawden JW, Proteolytic activity in embryonic bovine secretory enamel, in Tooth Enamel IV, Fearnhead RW, Suga S, Eds. (Elsevier Science, 1984) pp. 109–113. [Google Scholar]
- 32.Eckstein M, Vaeth M, Fornai C, Vinu M, Bromage TG, Nurbaeva MK, Sorge JL, Coelho PG, Idaghdour Y, Feske S, Lacruz RS, Store-operated Ca2+ entry controls ameloblast cell function and enamel development. JCI Insight 2, e91166 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eckstein M, Vaeth M, Aulestia FJ, Costiniti V, Kassam SN, Bromage TG, Pedersen P, Issekutz T, Idaghdour Y, Moursi AM, Feske S, Lacruz RS, Differential regulation of Ca2+ influx by ORAI channels mediates enamel mineralization. Sci. Signal 12, eaav4663 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Prakriya M, Lewis RS, Store-operated calcium channels. Physiol. Rev 95, 1383–1436 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Putney JW, The physiological function of store-operated calcium entry. Neurochem. Res 36, 1157–1165 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Parekh AB, Putney JW Jr., Store-operated calcium channels. Physiol. Rev 85, 757–810 (2005). [DOI] [PubMed] [Google Scholar]
- 37.Wu MM, Buchanan J, Luik RM, Lewis RS, Ca2+ store depletion causes STIM1to accumulate in ER regions closely associated with the plasma membrane. J. Cell Biol 174, 803–813 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nurbaeva MK, Eckstein M, Concepcion AR, Smith CE, Srikanth S, Paine ML, Gwack Y, Hubbard MJ, Feske S, Lacruz RS, Dental enamel cells express functional SOCE channels. Sci. Rep 5, 15803 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nurbaeva MK, Eckstein M, Devotta A, Saint-Jeannet JP, Yule DI, Hubbard MJ, Lacruz RS, Evidence that calcium entry into calcium-transporting dental enamel cells is regulated by cholecystokinin, acetylcholine and ATP. Front. Physiol 9, 801 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Klein O, Duverger O, Shaw W, Lacruz RS, Joester D, Moradian-Oldak J, Pugach MK, Wright JT, Millar SE, Kulkarni AB, Bartlett JD, Diekwisch TG, DenBesten P, Simmer JP, Meeting report: A hard look at the state of enamel research. Int. J. Oral Sci 9, e3 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen LS, Couwenhoven RI, Hsu D, Luo W, Snead ML, Maintenance of amelogenin gene expression by transformed epithelial cells of mouse enamel organ. Arch. Oral Biol 37, 771–778 (1992). [DOI] [PubMed] [Google Scholar]
- 42.Whitford GM, The metabolism and toxicity of fluoride. Monogr. Oral Sci 16, 1–153 (1996). [PubMed] [Google Scholar]
- 43.Bronckers ALJJ, Lyaruu DM, Guo J, Bijvelds MJC, Bervoets TJM, Zandieh-Doulabi B, Medina JF, Li Z, Zhang Y, DenBesten PK, Composition of mineralizing incisor enamel in cystic fibrosis transmembrane conductance regulator-deficient mice. Eur. J. Oral Sci 123, 9–16 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Taves DR, Normal human serum fluoride concentrations. Nature 211, 192–193 (1966). [DOI] [PubMed] [Google Scholar]
- 45.Denbesten PK, Crenshaw MA, Wilson MH, Changes in the fluoride-induced modulation of maturation stage ameloblasts of rats. J. Dent. Res 64, 1365–1370 (1985). [DOI] [PubMed] [Google Scholar]
- 46.Mekahli D, Bultynck G, Parys JB, De Smedt H, Missiaen L, Endoplasmic-reticulum calcium depletion and disease. Cold Spring Harb. Perspect. Biol 3, a004317 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Greber BJ, Ban N, Structure and function of the mitochondrial ribosome. Annu. Rev. Biochem 85, 103–132 (2016). [DOI] [PubMed] [Google Scholar]
- 48.Xie X, Guo P, Yu H, Wang Y, Chen G, Ribosomal proteins: Insight into molecular roles and functions in hepatocellular carcinoma. Oncogene 37, 277–285 (2018). [DOI] [PubMed] [Google Scholar]
- 49.Nurbaeva MK, Eckstein M, Snead ML, Feske S, Lacruz RS, Store-operated Ca2+ Entry Modulates the Expression of Enamel Genes. J. Dent. Res 94, 1471–1477 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wagner II LE, Li W-H, Joseph SK, Yule DI, Functional consequencesof phosphomimetic mutations at key cAMP-dependent protein kinase phosphorylation sites in the type 1 inositol 1,4,5-trisphosphate receptor. J. Biol. Chem 279, 46242–46252 (2004). [DOI] [PubMed] [Google Scholar]
- 51.Yan X, Yang X, Hao X, Ren Q, Gao J, Wang Y, Chang N, Qiu Y, Song G, Sodium Fluoride Induces Apoptosis in H9c2 Cardiomyocytes by Altering Mitochondrial Membrane Potential and Intracellular ROS level. Biol. Trace Elem. Res 166, 210–215 (2015). [DOI] [PubMed] [Google Scholar]
- 52.Pelletier M, Billingham LK, Ramaswamy M, Siegel RM, Extracellular flux analysisto monitor glycolytic rates and mitochondrial oxygen consumption. Methods Enzymol. 542, 125–149 (2014). [DOI] [PubMed] [Google Scholar]
- 53.Parekh AB, Store-operated Ca2+ entry: Dynamic interplay between endoplasmic reticulum, mitochondria and plasma membrane. J. Physiol 547 (Pt. 2), 333–348 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Watson R, Parekh AB, Mitochondrial regulation of CRAC channel-driven cellular responses. Cell Calcium 52, 52–56 (2012). [DOI] [PubMed] [Google Scholar]
- 55.Hoth M, Fanger CM, Lewis RS, Mitochondrial regulation of store-operated calcium signaling in T lymphocytes. J. Cell Biol 137, 633–648 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Suzuki M, Everett ET, Whitford GM, Bartlett JD, 4-Phenylbutyrate mitigates fluoride-induced cytotoxicity in ALC cells. Front. Physiol 8, 302 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Suzuki M, Sierant ML, Antone JV, Everett ET, Whitford GM, Bartlett JD, Uncoupling protein-2 is an antioxidant that is up-regulated in the enamel organ of fluoride-treated rats. Connect. Tissue Res 55 (suppl. 1), 25–28 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Brostrom MA, Brostrom CO, Calcium dynamics and endoplasmic reticular function in the regulation of protein synthesis: Implications for cell growth and adaptability. Cell Calcium 34, 345–363 (2003). [DOI] [PubMed] [Google Scholar]
- 59.Mills EW, Green R, Ribosomopathies: There’s strength in numbers. Science 358, eaan2755 (2017). [DOI] [PubMed] [Google Scholar]
- 60.Farley-Barnes KI, Ogawa LM, Baserga SJ, Ribosomopathies: Old concepts, new controversies. Trends Genet. 35, 754–767 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bigay J, Deterre P, Pfister C, Chabre M, Fluoride complexes of aluminium or beryllium act on G-proteins as reversibly bound analogues of the gamma phosphate of GTP. EMBO J. 6, 2907–2913 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mertz LM, Horn VJ, Baum BJ, Ambudkar IS, Calcium entry in rat parotid acini: Activation by carbachol and aluminum fluoride. Am. J. Physiol 258 (4 Pt. 1), C654–C661 (1990). [DOI] [PubMed] [Google Scholar]
- 63.Chong SA, Hong SY, Moon SJ, Park JW, Hong JH, An JM, Lee SI, Shin DM, Seo JT, Partial inhibition of SERCA is responsible for extracellular Ca2+ dependence of AlF-4-induced [Ca2+]i oscillations in rat pancreatic. Am. J. Physiol. Cell Physiol 285, C1142–C1149 (2003). [DOI] [PubMed] [Google Scholar]
- 64.Pinton P, Mitochondria-associated membranes (MAMs) and pathologies. Cell Death Dis. 9, 413 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schmid SL, Carter LL, ATP is required for receptor-mediated endocytosis in intact cells. J. Cell Biol 111 (6 Pt 1), 2307–2318 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Duan X, Mao Y, Wen X, Yang T, Xue Y, Excess fluoride interferes with chloride-channel- dependent endocytosis in ameloblasts. J. Dent. Res 90, 175–180 (2011). [DOI] [PubMed] [Google Scholar]
- 67.Singaravelu K, Nelson C, Bakowski D, de Brito OM, Ng SW, di Capite J, Powell T, Scorrano L, Parekh AB, Mitofusin 2 regulates STIM1 migration from the Ca2+ store to the plasma membrane in cells with depolarized mitochondria. J. Biol. Chem 286, 12189–12201 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jha A, Ahuja M, Maléth J, Moreno CM, Yuan JP, Kim MS, Muallem S, The STIM1 CTID domain determines access of SARAF to SOAR to regulate Orai1 channel function. J. Cell Biol 202, 71–79 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chvanov M, Walsh CM, Haynes LP, Voronina SG, Lur G, Gerasimenko OV, Barraclough R, Rudland PS, Petersen OH, Burgoyne RD, Tepikin AV, ATP depletion induces translocation of STIM1 to puncta and formation of STIM1-ORAI1 clusters: Translocation and re-translocation of STIM1 does not require ATP. Pflugers Arch. 457, 505–517 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Innocenti B, Pozzan T, Fasolato C, Intracellular ADP modulates the Ca2+ release-activated Ca2+ current in a temperature-and Ca2+−dependent Way. J. Biol. Chem 271, 8582–8587 (1996). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
stke.sciencemag.org/cgi/content/full/13/619/eaay0086/DC1
Fig. S1. ER Ca2+ release and SOCE in LS8 cells treated with various concentrations of NaF.
Fig. S2. Comparison of ER Ca2+ release by thapsigargin and tBHQ.
Fig. S3. ATP stimulation of fluoride-treated LS8 cells.
Fig. S4. Fluoride, but not chloride, affects Ca2+ homeostasis.
Fig. S5. Cell death analyses.
Fig. S6. Heat map of cell and ER stress genes in HEK-293 cells.
Fig. S7. Expression of the ER stress marker GRP78 in cells exposed to various NaF concentrations.
Fig. S8. Heat map of RP genes in HEK-293 cells.
Fig. S9. IP3 uncaging in primary secretory stage enamel cells.
Fig. S10. Transient fluoride application does not elicit changes in [Ca2+]cyt.
Fig. S11. Western blot of SERCA in LS8 cells treated with fluoride.
Fig. S12. Altering mitochondrial function by rotenone/oligomycin disrupts SOCE in LS8 cells.
Table S1. DEGs in fluoride-treated LS8 cells.
Table S2. DEGs in fluoride-treated HEK-293 cells.