

Abstract
Background
Perphenazine is an old phenothiazine antipsychotic with a potency similar to haloperidol. It has been used for many years and is popular in the northern European countries and Japan.
Objectives
To examine the clinical effects and safety of perphenazine for those with schizophrenia and schizophrenia‐like psychoses.
Search methods
We updated our original search using the Cochrane Schizophrenia Group's register (September 2013), references of all included studies and contacted pharmaceutical companies and authors of included studies in order to identify further trials.
Selection criteria
We included all randomised controlled trials that compared perphenazine with other treatments for people with schizophrenia and/or schizophrenia‐like psychoses. We excluded trials of depot formulations of perphenazine.
Data collection and analysis
Two review authors independently inspected citations and, where possible, abstracts. We ordered papers, inspected and quality assessed them. We extracted data, again working independently. If loss to follow‐up was greater than 50% we considered results as 'prone to bias'. For dichotomous data, we calculated risk ratios (RR) and for continuous data we calculated mean differences (MD), both with the 95% confidence intervals (CI). We assessed quality of data using the GRADE (Grading of Recommendations Assessment, Development and Evaluationtool) and assessed risk of bias for included studies.
Main results
Thirty‐one studies fulfilled the inclusion criteria, with a total of 4662 participants (of which 4522 were receiving the drugs relevant to our comparison) and presented data that could be used for at least one comparison. The trial centres were located in Europe (especially Scandinavia), Japan and Northern America.
When comparing perphenazine with placebo, for our primary outcome of clinical response, results favoured perphenazine with significantly more people receiving placebo rated as either 'no better or deterioration' for global state than people receiving perphenazine (1 RCT, n = 61 RR 0.32 CI 0.13 to 0.78, very low quality evidence). More people receiving placebo relapsed, although not a statistically significant number (1 RCT, n = 48, RR 0.14 CI 0.02 to 1.07, very low quality evidence). Death was not reported in the perphenazine versus placebo comparison. Experiences of dystonia were equivocal between groups (1 RCT, n = 48, RR 1.00 CI 0.07 to 15.08, very low quality evidence); other outcomes not reported in this comparison include serious adverse events, economic outcomes, and service use and hospitalisation.
For the comparison of perphenazine versus any other antipsychotic drugs, no real differences in effect between the drugs were found. There was no significant difference between groups for those considered 'no better or deterioration' (17 RCTs, n = 1879, RR 1.04 CI 0.91 to 1.17, very low quality evidence). For mental state outcome of 'no effect' of the study drug, there was again no significant difference between groups (4 RCTs, n = 383, RR 1.24 CI 0.61 to 2.52, very low quality evidence). Death was not reported in any of the included studies. There was no significant difference in rates of dystonia with perphenazine versus any other antipsychotic drugs (4 RCTs, n = 416, RR 1.36 CI 0.23 to 8.16, very low quality evidence), nor was there a significant difference between groups for serious adverse events (2 RCTs, n = 1760, RR 0.98 CI 0.68 to 1.41, very low quality evidence).
Authors' conclusions
Although perphenazine has been used in randomised trials for more than 50 years, incomplete reporting and the variety of comparators used make it impossible to draw clear conclusions. All data for the main outcomes in this review were of very low quality evidence. At best we can say that perphenazine showed similar effects and adverse events as several of the other antipsychotic drugs. Since perphenazine is a relatively inexpensive and frequently used compound, further trials are justified to clarify the properties of this classical antipsychotic drug.
Keywords: Humans, Antipsychotic Agents, Antipsychotic Agents/adverse effects, Antipsychotic Agents/therapeutic use, Mental Disorders, Mental Disorders/drug therapy, Perphenazine, Perphenazine/adverse effects, Perphenazine/therapeutic use, Randomized Controlled Trials as Topic, Schizophrenia, Schizophrenia/drug therapy
Plain language summary
Perphenazine for schizophrenia
People with schizophrenia often hear voices or see things (hallucinations) and have strange beliefs (delusions). The main treatment for these symptoms of schizophrenia is antipsychotic drugs. Perphenazine is an old antipsychotic drug (typical or first‐generation) formulated in the 1950s with an effectiveness similar to another antipsychotic drug called haloperidol. Perphenazine is particularly popular in Northern European countries and Japan.
The aim of the review was to examine the effectiveness and safety of perphenazine for schizophrenia. A search for relevant randomised studies was run in September 2013. The review authors included 31 studies that randomised people with schizophrenia to receive either perphenazine or placebo or another antipsychotic drug. A total of 4662 people participated in these studies. The quality of evidence presented by the trials was rated by the review authors to be very low quality. It was found that perphenazine was no better or worse than other older antipsychotic drugs in treating the symptoms of schizophrenia, and like other older antipsychotic drugs, the side effects of perphenazine included tremors, uncontrollable shaking, the inability to sit still and feeling restless.
Although perphenazine has been used for more than 50 years, poor studies with bad reporting of information mean that it is difficult to draw more clear findings and conclusions as to the effectiveness and safety of perphenazine. However, perphenazine is inexpensive and a popular antipsychotic drug in many countries, so further research and trials on its effectiveness and safety are much needed.
Ben Gray, Senior Peer Researcher, McPin Foundation.http://mcpin.org/
Summary of findings
Summary of findings for the main comparison. PERPHENAZINE compared with PLACEBO for schizophrenia.
| PERPHENAZINE compared with PLACEBO for schizophrenia | ||||||
| Patient or population: patients with schizophrenia Settings: hospitals, USA and Canada Intervention: PERPHENAZINE Comparison: PLACEBO | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| PLACEBO | PERPHENAZINE | |||||
| Global state: Change over time ‐ no better or deterioration ‐ short term clinically defined Follow‐up: 6 weeks | 500 per 10001 | 160 per 1000 (65 to 390) | RR 0.32 (0.13 to 0.78) | 61 (1 study) | ⊕⊝⊝⊝ very low2,3 | |
| Mental state: Relapse ‐ short term clinical diagnosis Follow‐up: 12 weeks | 292 per 10004 | 41 per 1000 (6 to 312) | RR 0.14 (0.02 to 1.07) | 48 (1 study) | ⊕⊝⊝⊝ very low5,6,7 | |
| Adverse events: death ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | No study reported this outcome |
| Adverse events: extrapyramidal adverse effects ‐ dystonia ‐ short term Follow‐up: 12 weeks | 42 per 10008 | 42 per 1000 (3 to 628) | RR 1 (0.07 to 15.08) | 48 (1 study) | ⊕⊝⊝⊝ very low5,7,8 | |
| Adverse events: serious adverse events ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | No study reported this outcome |
| Economic outcomes ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | No study reported this outcome |
| Service use and hospitalisation: admission ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | No study reported this outcome |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Control risk: mean baseline risk presented for single study (50%) 2 Risk of bias: rated 'very serious' ‐ randomisation methods not described in the single included study; data for participants who did not complete were excluded from data analysis; data to support this outcome taken from single study (n = 238), with n = 61 participants in the perphenazine vs placebo comparison 3 Indirectness: rated 'serious' ‐ single included had a total of eight drug comparisons 4 Control risk: mean baseline risk presented for single study (29.2%) 5 Risk of bias: rated 'very serious' ‐ randomisation methods not described in the single included study; selective reporting detected; data to support this outcome taken from single study with n=48 participants 6 Indirectness: rated 'serious' ‐ single included had a total of four drug comparisons 7 Imprecision: rated 'serious' ‐ 95% confidence intervals for best estimate of effect include both 'no effect' and appreciable benefit/harm. 8 Control risk: mean baseline risk presented for single study (4.2%)
Summary of findings 2. PERPHENAZINE compared with ANY ANTIPSYCHOTICS for schizophrenia.
| PERPHENAZINE compared with ANY ANTIPSYCHOTICS for schizophrenia | ||||||
| Patient or population: patients with schizophrenia Settings: multi‐centre and single‐centre; international Intervention: PERPHENAZINE Comparison: ANY ANTIPSYCHOTICS | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| ANY ANTIPSYCHOTICS | PERPHENAZINE | |||||
| Global state: Change over time ‐ no better or deterioration ‐ short and medium term as defined in each study Follow‐up: mean 8 weeks | Low1 | RR 1.04 (0.91 to 1.17) | 1879 (17 studies) | ⊕⊝⊝⊝ very low2,3,4 | ||
| 50 per 1000 | 52 per 1000 (46 to 58) | |||||
| Moderate1 | ||||||
| 450 per 1000 | 468 per 1000 (410 to 526) | |||||
| High1 | ||||||
| 900 per 1000 | 936 per 1000 (819 to 1000) | |||||
| Mental state: State ‐ 'no effect' BPRS score reduction Follow‐up: mean 8 weeks | 216 per 10005 | 267 per 1000 (132 to 543) | RR 1.24 (0.61 to 2.52) | 383 (4 studies) | ⊕⊝⊝⊝ very low4,6,7,8 | |
| Death ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | No study reported this outcome |
| Adverse events: extrapyramidal adverse effects dystonia ‐ short term Follow‐up: mean 10 weeks | 33 per 10005 | 59 per 1000 (21 to 166) | RR 1.36 (0.23 to 8.16) | 416 (4 studies) | ⊕⊝⊝⊝ very low4,8,9,10 | |
| Other adverse events: any serious adverse event ‐ short and long term Follow‐up: mean 39 weeks | Study population | RR 0.98 (0.68 to 1.41) | 1760 (2 studies) | ⊕⊝⊝⊝ very low4,12,13 | ||
| 109 per 100011 | 108 per 1000 (81 to 147) | |||||
| Moderate | ||||||
| 152 per 100011 | 150 per 1000 (112 to 204) | |||||
| Economic outcomes ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | No study reported this outcome |
| Service use and hospitalisation: admission ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | No study reported this outcome |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Control risk: three risks presented based on control risk in included studies; 'moderate' equates with total control group risk (47.8%) 2 Risk of bias: rated 'serious' ‐ only four studies of seventeen adequately reported randomisation methods; most studies rated 'high' on at least one risk of bias domain, including detected selective reporting and attrition bias 3 Indirectness: rated 'serious' ‐ each included study involved different antipsychotic drugs as comparators (sulpiride, clopenthixol, benperidol, loxapine, risperidone, zotepine, clothiapine, thiothixene, methyperidol, penfluridol, aripiprazole, clocapramine, prochlorperazine, trifluopromazine, mepazine, promethazine, zuclopenthixol, timiperone) 4 Imprecision: rated 'serious' ‐ 95% confidence intervals for best estimate of effect include both 'no effect' and appreciable benefit/harm. 5 Control risk: mean baseline risk presented; considerable heterogeneity in results to be wary of 6 Risk of bias: rated 'serious' ‐ no studies adequately explained randomisation; two studies rated as 'high' on at least one risk of bias domain, including attrition bias and detected selective reporting 7 Inconsistency: rated 'serious' ‐ heterogeneity was considerable (Chi² = 5.25, df=3 (P = 0.15); I² = 43%). Suspected heterogeneity due to different comparator drugs used in all four studies (quetiapine; risperidone; aripiprazole; clozapine and clozapine + perphenazine) 8 Indirectness: rated 'serious' ‐ each included study involved different antipsychotic drugs as comparators 9 Risk of bias: rated 'serious' ‐ three out of four studies did not adequately explain randomisation methods; three studies rated 'high' on at least one risk of bias domain, including selective reporting and incomplete outcome data 10 Inconsistency: rated 'serious' ‐ heterogeneity was considerable (Chi² = 6.48, df = 3 (P = 0.09); I² = 54%). Suspected heterogeneity due to different comparator drugs used in all four studies (quetiapine; risperidone; timiperone; loxapine) 11 Control risk: median control group risk presented from the studies 12 Risk of bias: rated 'serious' ‐ randomisation methods not described in one out of the two studies (Kane 2003), this same study was funded by pharmaceutical companies, with all authors affiliated with either company 13 Indirectness: rated 'serious' ‐ one included study had five treatment arms (CATIE 2005)
Background
Description of the condition
The term 'schizophrenia was first used in 1908 by Paul Eugen Bleuler to describe a state of splitting of the functions between thought, memory, personality and perception (Bleuler 1908). It has since been recognised as a chronic, serious mental health condition that alters a person's perceptions, behaviours and mental state; it is experienced differently between individuals who develop the illness. It is typically characterised by the presence of both 'positive' symptoms (hallucinations, delusions and disturbance of thought) and 'negative' symptoms (deficits of normal response and thought processes, including poverty of speech, apathy, lack of motivation and avolition). Schizophrenia affects roughly 24 million people worldwide (WHO 2014), with a high prevalence of between 0.4% to 1.4% due to chronicity but, a low incidence rate of approximately 0.11 per 1000 (Cannon 1996; NICE 2010).
Description of the intervention
Perphenazine was formulated in the 1950s. It was first marketed as an oral preparation in late 1957, and as an injectable form for intravenous or intramuscular treatment of acutely ill people in the 1970s (Parfitt 1999). Furthermore, two long‐acting depot formulations (perphenazine enanthate and decanoate) have been developed. Perphenazine's main indication is the treatment of schizophrenia, but it is also used for other psychiatric disorders such as mania, agitated behaviour and severe anxiety. Outside psychiatry, it is said to be effective for the management of post‐operative or chemotherapy‐induced nausea and vomiting, and for the treatment of intractable hiccup. Perphenazine is available in many countries. Although we have not found precise data on how much perphenazine is used, it seems to be especially popular in the Nordic states of Europe, and in Japan.
How the intervention might work
Perphenazine is a phenothiazine antipsychotic with a piperazine side‐chain (Parfitt 1999). It has a neuroleptic potency similar to that of haloperidol, so extrapyramidal adverse effects are said to be common (Benkert 1996). There is a substantial first‐pass effect and its bioavailability is only about 40%. The main metabolites of perphenazine are glucuronides and sulphoxides found in the urine. Only two per cent of the absorbed perphenazine is excreted non‐metabolised by the urine. Its elimination half‐life period is about 20 hours and it is able to pass the placenta, although the concentrations found in the foetus and amnion are low. Perphenazine is also distributed into breast milk where similar concentrations to those reported in the blood can be found (Bundesverband 2001).
Why it is important to do this review
In terms of the costs of schizophrenia, this was estimated at about £6.7 billion in England in 2004/05, of which the direct costs were £2 million, while the indirect costs accounted for the rest (Mangalore 2007). The cost of perphenazine itself is expensive compared to other typical antipsychotic drugs, at £34.25 for 100 tablets of 4 mg each. The maximum daily dose of perphenazine is 24 mg per day, which costs £2.1 per day, or £63 per month (BNF 2012). The newer, atypical antipsychotic drugs in comparison are more expensive than typical antipsychotic drugs, with olanzapine available at £13.11 for 28 5 mg tablets, and clozapine (Clozaril) at £21.56 for 28 100 mg tablets.
It is essential to complement the clinical effectiveness of perphenazine with its cost‐effectiveness. Davies 2007 conducted a study on cost‐effectiveness of first‐generation antipsychotic drugs (i.e. flupentixol, trifluoperazine, chlorpromazine) and the second‐generation antipsychotic drugs (i.e. risperidone, olanzapine, amisulpiride). The study findings argue that there is no evidence to suggest that atypical (second‐generation) antipsychotic drugs are more cost‐effective than typical (first‐generation) antipsychotic drugs.
Objectives
To examine the effects of perphenazine for treatment of schizophrenia or related psychoses in comparison with placebo, no treatment or other antipsychotic medication.
Methods
Criteria for considering studies for this review
Types of studies
We included all relevant randomised controlled trials. We included trials that were described as double‐blind, but that did not mention whether the study was randomised, in a sensitivity analysis. If there was no substantive difference within the primary outcomes (see 'Types of outcome measures') when these studies were added, then we included them in the final analysis. If there was a substantive difference, we used only clearly randomised trials and described the results of the sensitivity analysis in the text. We excluded quasi‐randomised studies, such as those allocating by using alternate days of the week.
Types of participants
People with schizophrenia and schizophrenia‐like disorders such as schizophreniform disorder, delusional disorder or schizoaffective disorder, diagnosed by any criteria. We also included studies involving people with 'serious/chronic mental illness' or 'psychotic illness'. If possible we excluded people with dementing illnesses, depression and primarily problems associated with substance misuse.
Types of interventions
1. Perphenazine
Any dose and route of administration (however, we excluded the depot formulations perphenazine enanthate and decanoate, because these were the topic of another review, David 2005) against one or more of the following:
2. Placebo or no treatment
3. Other antipsychotic drugs
Any dose or pattern of administration.
Types of outcome measures
We divided all outcomes into short‐term (less than three months), medium‐term (three to six months) and long‐term follow‐up (longer than six months).
Primary outcomes
1. Clinical response (Global or mental state)
1.1 Clinically significant response in global state ‐ as defined by each of the studies 1.2 Clinically significant response in mental state ‐ as defined by each of the studies
Secondary outcomes
1. Leaving the study early
2. Clinical response (Global or mental state)
2.1 Clinically significant response in global state ‐ as defined by each of the studies 2.2 Average score/change in global state 2.3 Clinically significant response in mental state ‐ as defined by each of the studies 2.4 Average score/change in mental state 2.5 Clinically significant response on positive symptoms ‐ as defined by each of the studies 2.6 Average score/change in positive symptoms 2.7 Clinically significant response on negative symptoms ‐ as defined by each of the studies 2.8 Average score/change in negative symptoms
3. Behaviour
3.1 Clinically significant response in behaviour (e.g. aggressive behaviour, behaviour on the ward etc) ‐ as defined by each of the studies 3.2 Average score/change in behaviour
4. Adverse events ‐ movement disorders
4.1 Incidence of use of antiparkinson drugs 4.2 Clinically significant extrapyramidal side effects ‐ as defined by each of the studies 4.3 Average score/change in extrapyramidal side effects
5. Other adverse effects, general and specific
5.1 Anticholinergic 5.2 Arousal 5.3 At least one adverse event 5.4 Cardiovascular 5.5 Central nervous system 5.6 Endocrine 5.7 Gastrointestinal 5.8 Genitourinary 5.9 Haematology 5.10 Skin 5.11 Others
6. Economic
6.1 Average change in total cost of medical and mental health care 6.2 Total indirect and direct costs 6.3 Direct resource use: 6.3.1 Outpatients ‐ number of contacts (GP consultation, psychiatrist, psychologists, psychiatric nurse, counsellor, social worker) 6.3.2 Hospitalisation (taking battery of tests, patients’ physical, psychiatric and psychological profile and psychological assessment, number of days, relapse) 6.3.3 Medication (different types of antipsychotic drugs to include dose and frequency, treatment of side effects) 6.3.4 Psychological therapies (different types of psychological therapies to include session numbers and frequency) 6.3.5 Other resources (day centres, night shelter) and transportation for medical care visits 6.4 Indirect resource use: 6.4.1 Family, relative and friends resources 6.4.2 Police, criminal justice system 6.4.3 Benefits paid, social security payments 6.4.4 Employment agency workers, absence from work, loss of productivity 6.5 Cost‐effectiveness ratios represented by ICER (incremental cost‐effectiveness ratio) 6.6 Cost‐utilities represented by incremental costs per QALY (quality‐adjusted life year) or DALYs (disability‐adjusted life year) 6.7 Cost benefit represented by net Benefit Ratio, others.
7. Service use and hospitalisation
7.1 Hospital admission 7.2 Medication use 7.3 Engagement with health services
8. Satisfaction
8.1 Satisfaction with treatment 8.2 Satisfaction with care 8.3 Quality of life outcomes 8.4 Employment status
9. 'Summary of findings' tables
We used the GRADE approach to interpret findings (Schünemann 2008) and the GRADE profiler (GRADEPRO) to import data from RevMan 5 (Review Manager) to create 'Summary of findings' tables. These tables provide outcome‐specific information concerning the overall quality of evidence from each included study in the comparison, the magnitude of effect of the interventions examined, and the sum of available data on all outcomes we rated as important to patient‐care and decision making. We selected the following main outcomes for inclusion in the 'Summary of findings' tables.
Clinical response in global state (as defined by each of the studies)
Clinical response in mental state (as defined by each of the studies)
Adverse events: death
Adverse events: extrapyramidal adverse effects (dystonia; akathisia; Parkinsonism; tardive dyskinesia)
Adverse events: serious adverse events (as defined by each of the studies)
Economic outcomes
Service use and hospitalisation: admission
Search methods for identification of studies
Electronic searches
For this update, we searched the Cochrane Schizophrenia Group's old MeerKat MS Access database [MK060213] (September 2013) searched by query ‘[(Title like ‘*Perphenazine*’) or (Abstract like ‘*Perphenazine*’)] and [Keyword not like ‘*Perphenazine*’].
Searching other resources
1. Reference searching
Review authors (SS and BH) inspected the reference lists of all identified studies, including existing reviews, for relevant citations.
2. Personal contact
Review authors (SS and BH) contacted the first author of each relevant study for information on unpublished trials. We also consulted experts in the area of schizophrenia and weight gain. We contacted authors and experts by email or post to establish missing details in the methods and results sections of the written reports and to determine their knowledge of or involvement in any current work in the area. We also asked contacts at major pharmaceutical companies if they have conducted or were currently undertaking any weight‐related interventions in relation to schizophrenia (including representatives from Janssen Pharmaceuticals, Pfizer Inc, Eli Lilly and Company, Astra Zeneca, and Bristol‐Myers Squibb). Responses to our letters are noted in notes of Characteristics of included studies.
Data collection and analysis
Selection of studies
For this 2013 update, SS independently inspected citations from the new electronic search and identified relevant abstracts. SS also inspected full articles of the abstracts meeting inclusion criteria. CEA carried out the reliability check of all citations from the new electronic search.
Data extraction and management
1. Extraction
For this 2013 update, SS extracted data from included studies. We extracted data presented only in graphs and figures whenever possible. When further information was necessary, we contacted authors of studies in order to obtain missing data or for clarification. It was not possible to extract data relevant to each component centre separately of the identified multi‐centre studies.
2. Management
2.1 Forms
We extracted data onto standard, simple forms.
2.2 Scale‐derived data
We included continuous data from rating scales only if: a. the psychometric properties of the measuring instrument have been described in a peer‐reviewed journal (Marshall 2000); and b. the measuring instrument has not been written or modified by one of the trialists for that particular trial.
Ideally, the measuring instrument should either be i. a self‐report or ii. completed by an independent rater or relative (not the therapist). We realise that this is not often reported clearly; we have noted whether or not this is the case in Description of studies.
2.3 Endpoint versus change data
There are advantages of both endpoint and change data. Change data can remove a component of between‐person variability from the analysis. On the other hand, calculation of change needs two assessments (baseline and endpoint), which can be difficult in unstable and difficult to measure conditions such as schizophrenia. We decided primarily to use endpoint data, and only use change data if the former were not available. We combined endpoint and change data in the analysis as we used mean differences (MD) rather than standardised mean differences throughout (Higgins 2011, Chapter 9.4.5.2).
2.4 Skewed data
Continuous data on clinical and social outcomes are often not normally distributed. To avoid the pitfall of applying parametric tests to non‐parametric data, we aimed to apply the following standards to all data before inclusion: a) standard deviations (SDs) and means are reported in the paper or obtainable from the authors; b) when a scale starts from the finite number zero, the SD, when multiplied by two, is less than the mean (as otherwise the mean is unlikely to be an appropriate measure of the centre of the distribution (Altman 1996)); c) if a scale started from a positive value (such as PANSS which can have values from 30 to 210), we modified the calculation described above to take the scale starting point into account. In these cases skew is present if 2SD > (S‐S min), where S is the mean score and S min is the minimum score. Endpoint scores on scales often have a finite start and end point and these rules can be applied. We entered skewed endpoint data from studies of fewer than 200 participants in 'other tables' within the data and analyses section rather than into a statistical analysis. Skewed data pose less of a problem when looking at the mean if the sample size is large; we entered such data from studies with over 200 participants into syntheses. When continuous data are presented on a scale that includes a possibility of negative values (such as change data), it is difficult to tell whether data are skewed or not; we entered skewed change data into the analyses.
2.5 Common measure
Had we identified such data, to facilitate comparison between trials, we intended to convert variables that can be reported in different metrics, such as days in hospital (mean days per year, per week or per month) to a common metric (e.g. mean days per month). However, we did not identify such data.
2.6 Conversion of continuous to binary
Where possible, we made efforts to convert outcome measures to dichotomous data. This can be done by identifying cut‐off points on rating scales and dividing participants accordingly into 'clinically improved' or 'not clinically improved'. It is generally assumed that if there is a 50% reduction in a scale‐derived score such as the Brief Psychiatric Rating Scale (BPRS, Overall 1962) or the Positive and Negative Syndrome Scale (PANSS, Kay 1986), this could be considered as a clinically significant response (Leucht 2005; Leucht 2005a). If data based on these thresholds were not available, we used the primary cut‐off presented by the original authors. Some included studies provided a definition of response as a reduction in PANSS or Clinical Global Impression (CGI) scores, in which case we employed the dichotomous data provided from the primary study report.
2.7 Direction of graphs
Where possible, we entered data in such a way that the area to the left of the line of no effect indicated a favourable outcome for perphenazine.
Assessment of risk of bias in included studies
For this 2013 update, review author SS worked independently by using criteria described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011) to assess trial quality. This new set of criteria is based on evidence of associations between overestimate of effect and high risk of bias of the article such as sequence generation, allocation concealment, blinding, incomplete outcome data and selective reporting.
Where inadequate details of randomisation and other characteristics of trials were provided, we contacted authors of the studies in order to obtain additional information.
We have noted the level of risk of bias in both the text of the review and in the Table 1; Table 2.
Measures of treatment effect
1. Binary data
For binary outcomes we calculated a standard estimation of the risk ratio (RR) and its 95% confidence interval (CI). It has been shown that RR is more intuitive (Boissel 1999) than odds ratios and that odds ratios tend to be interpreted as RR by clinicians (Deeks 2000). The Number Needed to Treat/Harm (NNT/H) statistic with its confidence intervals is intuitively attractive to clinicians but is problematic both in its accurate calculation in meta‐analyses and interpretation (Hutton 2009). For binary data presented in the 'Summary of findings' table/s, where possible, we calculated illustrative comparative risks.
2. Continuous data
For continuous outcomes we estimated mean difference (MD) between groups.
Unit of analysis issues
1. Cluster trials
We did not identify any cluster‐randomised studies. In future versions of this review, we remain aware that studies increasingly employ 'cluster randomisation' (such as randomisation by clinician or practice), but analysis and pooling of clustered data poses problems. Authors often fail to account for intra‐class correlation in clustered studies, leading to a 'unit of analysis' error (Divine 1992), whereby P values are spuriously low, confidence intervals unduly narrow and statistical significance overestimated. This causes type I errors (Bland 1997; Gulliford 1999).
Where clustering is not accounted for in primary studies, we will present data in a table, with a (*) symbol to indicate the presence of a probable unit of analysis error. In subsequent versions of this review we will seek to contact first authors of studies to obtain intra‐class correlation coefficients (ICCs) for their clustered data and to adjust for this by using accepted methods (Gulliford 1999). Where clustering had been incorporated into the analysis of primary studies, we will present these data as if from a non‐cluster randomised study, but will adjust for the clustering effect.
We have sought statistical advice and have been advised that the binary data as presented in a report should be divided by a 'design effect'. This is calculated using the mean number of participants per cluster (m) and the ICC [Design effect = 1+(m‐1)*ICC] (Donner 2002). If the ICC is not reported, it will be assumed to be 0.1 (Ukoumunne 1999).
If cluster studies have been appropriately analysed taking into account ICCs and relevant data documented in the report, synthesis with other studies will be possible using the generic inverse variance technique.
2. Cross‐over trials
A major concern of cross‐over trials is the carry‐over effect. It occurs if an effect (e.g. pharmacological, physiological or psychological) of the treatment in the first phase is carried over to the second phase. As a consequence, on entry to the second phase the participants can differ systematically from their initial state despite a wash‐out phase. For the same reason cross‐over trials are not appropriate if the condition of interest is unstable (Elbourne 2002). As both effects are very likely in severe mental illness, we planned only to use data of the first phase of cross‐over studies. However, we did not identify any cross‐over studies in our search.
3. Studies with multiple treatment groups
Where a study involves more than two treatment arms, if relevant, we presented the additional treatment arms in comparisons. If data were binary, we simply added these and combined within the two‐by‐two table. If data were continuous, we combined data following the formula in section 7.7.3.8 (Combining groups) of the Handbook (Higgins 2011). Where the additional treatment arms were not relevant, we did not reproduce these data. Five studies had multiple treatment arms, each of which were relevant to this review and subsequently presented in relevant comparisons (Bennett 1961; CATIE 2005; Hanlon 1965; Kurland 1961; Sun 2000); four studies involved treatment arms that were not relevant to this review (mainly anti‐depressants) and therefore those particular treatment arms were excluded (Chouinard 1975; Chouinard 1977; Collins 1967; Hanlon 1964). One study had multiple treatment arms that were relevant, as well as treatment arms that were not (including phenobarbital); relevant treatment arms were included in the relevant comparisons (Kurland 1961).
Dealing with missing data
1. Overall loss of credibility
At some degree of loss of follow‐up, data must lose credibility (Xia 2009). We chose that, for any particular outcome, should more than 50% of data be unaccounted for, we would not reproduce these data or use them within analyses. Had we identified such levels of attrition where more than 50% of those in one arm of a study were lost, but the total loss was less than 50%, we would have addressed this within the 'Summary of findings' table/s by down‐rating quality.
2. Binary
In the case where attrition for a binary outcome was between 0% and 50% and where these data were not clearly described, we presented data on a 'once‐randomised‐always‐analyse' basis (an intention‐to‐treat analysis). Those leaving the study early were all assumed to have the same rates of negative outcome as those who completed, with the exception of the outcome of death and adverse effects. For these outcomes, the rate of those who stayed in the study ‐ in that particular arm of the trial ‐ were used for those who did not. We undertook a sensitivity analysis to test how prone the primary outcomes were to change when 'completer' data only were compared to the intention‐to‐treat analysis using the above assumptions (see Effects of interventions).
3. Continuous
3.1 Attrition
In the case where attrition for a continuous outcome was between 0% and 50% and completer‐only data were reported, we reproduced these.
3.2 Standard deviations
We did not impute standard deviations (SD) in any included study. In future versions of this review where we may identify such data, if SDs are not reported, we will first try to obtain the missing values from the authors. If not available, where there are missing measures of variance for continuous data, but an exact standard error (SE) and confidence intervals available for group means, and either P value or T value available for differences in mean, we can calculate them according to the rules described in the Handbook (Higgins 2011): When only the SE is reported, SDs are calculated by the formula SD = SE * square root (n). Chapters 7.7.3 and 16.1.3 of the Handbook (Higgins 2011) present detailed formulae for estimating SDs from P values, T or F values, confidence intervals, ranges or other statistics. If these formulae do not apply, we would calculate the SDs according to a validated imputation method based on the SDs of the other included studies (Furukawa 2006). Although some of these imputation strategies can introduce error, the alternative would be to exclude a given study’s outcome and thus to lose information. We nevertheless will examine the validity of the imputations in a sensitivity analysis excluding imputed values.
3.3 Last observation carried forward
We anticipated that in some studies the method of last observation carried forward (LOCF) would be employed within the study report. As with all methods of imputation to deal with missing data, LOCF introduces uncertainty about the reliability of the results (Leucht 2007). Therefore, where LOCF data have been used in the trial, if less than 50% of the data have been assumed, we have reproduced these data and indicated that they are the product of LOCF assumptions.
Assessment of heterogeneity
1. Clinical heterogeneity
We considered all included studies initially, without seeing comparison data, to judge clinical heterogeneity. We simply inspected all studies for clearly outlying people or situations which we had not predicted would arise. When such situations or participant groups arose, we fully discussed these.
2. Methodological heterogeneity
We considered all included studies initially, without seeing comparison data, to judge methodological heterogeneity. We simply inspected all studies for clearly outlying methods which we had not predicted would arise. When such methodological outliers arose, we fully discussed these.
3. Statistical heterogeneity
3.1 Visual inspection
We visually inspected graphs to investigate the possibility of statistical heterogeneity.
3.2 Employing the I2 statistic
We investigated heterogeneity between studies by considering the I2 method alongside the Chi2 P value. The I2 provides an estimate of the percentage of inconsistency thought to be due to chance (Higgins 2003). The importance of the observed value of I2 depends on i. magnitude and direction of effects and ii. strength of evidence for heterogeneity (e.g. P value from Chi2 test, or a confidence interval for I2). An I2 estimate greater than or equal to around 50% accompanied by a statistically significant Chi2 statistic was interpreted as evidence of substantial levels of heterogeneity (Higgins 2011). When substantial levels of heterogeneity were found in the primary outcome, we explored reasons for heterogeneity (Subgroup analysis and investigation of heterogeneity).
Assessment of reporting biases
Reporting biases arise when the dissemination of research findings is influenced by the nature and direction of results (Egger 1997). These are described in Section 10 of the Handbook (Higgins 2011). We are aware that funnel plots may be useful in investigating reporting biases but are of limited power to detect small‐study effects. We did not use funnel plots for outcomes where there were 10 or fewer studies, or where all studies were of similar sizes. In other cases, where funnel plots are possible, we sought statistical advice in their interpretation. We used a funnel plot for our primary outcome in the comparison of perphenazine versus any antipsychotic (see Figure 1).
1.
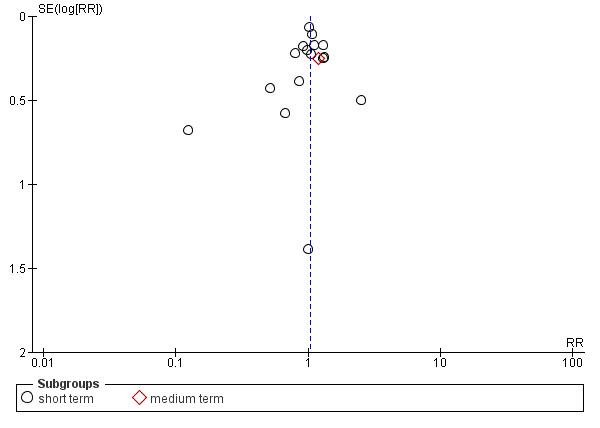
Funnel plot of comparison: 2 PERPHENAZINE vs ANY ANTIPSYCHOTICS, outcome: 2.4 Global state: 1. Change over time ‐ no better or deterioration (ITT).
Data synthesis
We understand that there is no closed argument for preference for use of fixed‐effect or random‐effects models. The random‐effects method incorporates an assumption that the different studies are estimating different, yet related, intervention effects. This often seems to be true to us and the random‐effects model takes into account differences between studies even if there is no statistically significant heterogeneity. There is, however, a disadvantage to the random‐effects model: it puts added weight onto small studies, which often are the most biased ones. Depending on the direction of effect, these studies can either inflate or deflate the effect size. We chose the random‐effects model for all analyses. The reader is, however, able to choose to inspect the data using the fixed‐effect model.
Subgroup analysis and investigation of heterogeneity
1. Subgroup analyses ‐ only primary outcomes
1.1 Clinical state, stage or problem
We proposed to undertake this review and provide an overview of the effects of perphenazine for people with schizophrenia in general. In addition, however, we tried to report data on subgroups of people in the same clinical state, stage and with similar problems. In particular, we included data for people in acute phase of schizophrenia.
2. Investigation of heterogeneity
If inconsistency was high, we have reported this. First, we investigated whether data had been entered correctly. Second, if data were correct, we visually inspected the graph and successively removed outlying to see if homogeneity was restored. For this review, we decided that should this occur with data contributing to the summary finding of no more than around 10% of the total weighting, we would present data. If not, then we did not pool data and discussed issues. We know of no supporting research for this 10% cut‐off, but we use prediction intervals as an alternative to this unsatisfactory state.
When unanticipated clinical or methodological heterogeneity was obvious, we simply stated hypotheses regarding these for future reviews or versions of this review. We do not anticipate undertaking analyses relating to these.
Sensitivity analysis
We applied all sensitivity analyses to the primary outcomes of this review.
1. Implication of randomisation
We aimed to include trials in a sensitivity analysis if they were described in some way so as to imply randomisation. For the primary outcomes, we included these studies and if there was no substantive difference when the implied randomised studies were added to those with better description of randomisation, then we entered all data from these studies.
2. Assumptions for lost binary data
Where assumptions had to be made regarding people lost to follow‐up and missing SDs data (see Dealing with missing data), we compared the findings on primary outcomes when we used our assumption compared with complete data only. We undertook a sensitivity analysis to test how prone results were to change when 'completer' data only were compared to the imputed data using the above assumption. If there was a substantial difference, we reported results and discussed them, but continued to employ our assumption.
3. Risk of bias
We analysed the effects of excluding trials that were judged to be at high risk of bias across one or more of the domains of randomisation (implied as randomised with no further details available): allocation concealment, blinding and outcome reporting for the meta‐analysis of the primary outcome. If the exclusion of trials at high risk of bias did not substantially alter the direction of effect or the precision of the effect estimates, then we included data from these trials in the analysis.
4. Imputed values
We planned to undertake a sensitivity analysis to assess the effects of including data from trials if we used imputed values for ICC in calculating the design effect in cluster‐randomised trials.
Had we noted substantial differences in the direction or precision of effect estimates in any of the sensitivity analyses listed above, we would not have pooled data from the excluded trials with the other trials contributing to the outcome, but presented them separately.
5. Fixed‐effect and random‐effects
We synthesised data using a random‐effects model, however synthesised data for primary outcomes using a fixed‐effect model to investigate whether this altered the estimate of the effect.
Results
Description of studies
For substantive descriptions of studies please see Characteristics of included studies and Characteristics of excluded studies tables.
Results of the search
In the 2013 update search, we identified 618 records and screened 615 after duplicates were removed (See Appendix 3 for original results of search). Of these 334 records were excluded based on title and abstract, with the remaining 187 full‐text references assessed for eligibility by inspecting their full citations. A total of 108 studies were excluded with reasons, with 14 awaiting classification for either inclusion or exclusion. This current review includes a total of 31 studies in meta‐analyses (see Figure 2). One study that was awaiting assessment from the previous version of this review has now been included since the update study search identified newer references with published results (Naukkarinen 2000). Three more studies that were previously awaiting assessment have now been excluded: Akimoto 1966 because there was no control group; Eklund 1976 because the study was judged to not be adequately randomised, and Galdi 1988 because it was found that perphenazine was not used as a study drug, nor were all the included participants diagnosed with schizophrenia. Conversely, two studies that were previously included have now been excluded: Nahunek 1967 because it was described as an 'open cross‐over' study, with no description of randomisation; and Whittaker 1963 because participants were 'arbitrarily allocated', which was judged to not amount to adequate randomisation.
2.
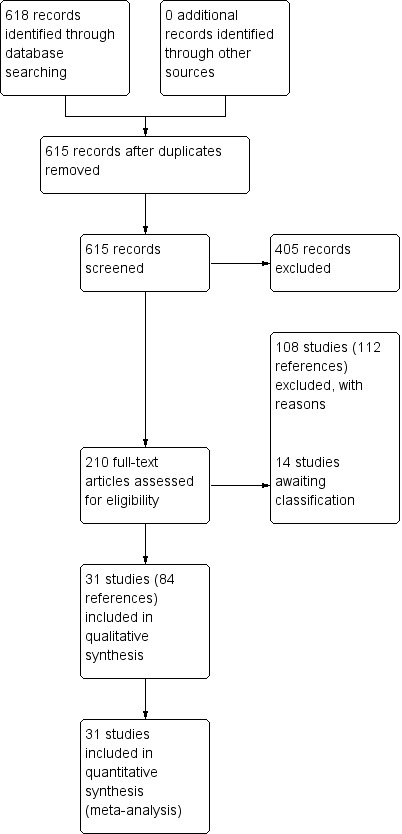
Study flow diagram: 2013 update search
Included studies
Thirty‐one studies fulfilled the inclusion criteria, with a total of n = 4662 (of which n = 4522 were receiving the drugs relevant to our comparison) and presented data that could be used for at least one comparison. The trial centres were located in Europe (especially Scandinavia), Japan and Northern America.
1. Length of trials
The duration of included trials covered a range from 10 days to 18 months, with the most common being 12 weeks (mean 11 weeks). We classified 28 of the studies as 'short term' and only one study (Lepola 1989) fell into the 'medium‐term' category. The longest study was 18 months and classified as 'long term' for this systematic review (CATIE 2005). One study did not specify its length (Itoh 1969 II).
2. Design
All trials used a parallel‐group design. Only seven of the included studies adequately described randomisation methods (Amakusa 1973; CATIE 2005; Itoh 1969; Itoh 1969 II; Itoh 1969 III; Naukkarinen 2000; Takahashi 1982).
3. Participants
Only 11 studies described the diagnostic criteria used: Hoyberg 1993 used the Diagnostic and Statistical Manual of Mental disorders, Third edition, Revised (DSM‐III‐R), and four studies (CATIE 2005; Kane 2003; Naukkarinen 2000; Wang 2008b) used the Diagnostic and Statistical Manual, Fourth Edition (DSM‐IV). Eckmann 1984 used the International Classification of Diseases, ninth revision (ICD‐9) criteria; Lepola 1989 used Research Diagnostic Criteria (RDC) criteria and four Chinese studies used Chinese Classification of Mental Disorders (CCMD) criteria, including CCMD‐2‐R (Sun 2000) and CCMD‐3 (Wang 2008a; Wang 2008c; Zhang 2010). The remaining studies probably used clinical criteria to diagnose study participants. In Hanlon 1964, there was a special inclusion criterion that the participants had to be "anergic and withdrawn" people with schizophrenia. Only four studies also included some participants who did not suffer from schizophrenia: Fruensgaard 1978 included people with acute schizophreniform psychotic episodes, acute exacerbation of a chronic schizophrenic process and psychogenic psychoses. Among the 115 participants in Hanlon 1964, 99 had a diagnosis of schizophrenia, the other diagnoses were not specified. Hanlon 1965 included 52 participants with a neurotic or a personality disorder and 270 with schizophrenia. Kurland 1961 described the included participants as having "predominantly" schizophrenia. These four studies were included because the majority of randomised participants had schizophrenia. Seven studies included participants with acute schizophrenia, and were included in an acute subgroup in meta‐analysis (Eckmann 1984; Fruensgaard 1978; Hoyberg 1993; Kurland 1961; Lepola 1989; Remvig 1987; Van Praag 1976).
In the studies that gave information on the sex of participants, there were 2442 males and 1345 females. Among the studies in which the participants' age was indicated, these ages ranged from 13 to 70 years.
4. Setting
Seventeen studies were multi‐centre, with the remaining studies single‐centre. Most of the included studies were undertaken in Japan, followed by North America, UK, Germany, Denmark and Findland.
5. Study size
Twenty‐eight per cent of the studies included more than 100 people. The number of people in the included studies ranged from 28 (Van Praag 1976) to 1493 (CATIE 2005). Only around 10% of the studies had 40 or less participants, with a mean sample size of around 150. A total of 4662 people participated in the 31 trials, 4551 of whom had been randomised to interventions that were relevant for the review.
6. Interventions
6.1 perphenazine
A total of 1457 people were given perphenazine in the included studies. Doses varied from a minimum of 2 to 4 mg (Zhang 2010) to a maximum of 90 mg/day (Itoh 1969). The mode of administration and dose in Itoh 1969 II is unclear. Six studies used a fixed dose (Bennett 1961; Chouinard 1975; Chouinard 1977; Collins 1967; Hanlon 1964; Woggon 1978), whilst the remaining studies used a flexible dose, most often according to the judgement of each physician in each individual study.
6.2 Control interventions
| Control Intervention | Average dose (control) | Studies | Average dose (perphenazine) |
| vs Placebo | to match study drug | Chouinard 1975; Chouinard 1977;Collins 1967;Hanlon 1964 | mean 13 mg/day |
| vs Aripiprazole | mean 26.8 mg/day | Kane 2003; Zhang 2010 | mean 25.15 mg/day |
| vs Benperidol | range 6 or 12 mg/day | Eckmann 1984 | range 12 or 24 mg/day |
| vs Bromperidol | mean 6 mg/day | Woggon 1978 | mean 20 mg/day |
| vs Chlorpromazine | mean 487.63 mg/day | Bennett 1961; Hanlon 1965; Kurland 1961 | mean 40.91 mg/day |
| vs Clocapramine | range 75 to 150 mg/day | Kurihara 1983 | range 9 to 18 mg/day |
| vs Clopenthixol | range 25 to 250 mg/day | Dehnel 1968 | range 8 to 80 mg/day |
| vs Clothiapine | range 12 mg/day increased to 24 mg/day | Itoh 1969 | range 45 mg/day increased to 90 mg/day |
| vs Clozapine | range 0 to 600 mg/day | Sun 2000; Van Praag 1976 | range 0 to 60 mg/day |
| vs Clozapine + perphenazine | range 32 to 50 mg + 100 to 300 mg/mg/day | Sun 2000 | range 8 to 60 mg/day |
| vs Fluphenazine | mean 5.92 mg/day | Hanlon 1965 | mean 38.61 mg/day |
| vs Haloperdiol | range 3 to 6 mg/day | Kurihara 1983 | range 9 to 18 mg/day |
| vs Loxapine | range 20 mg/day to max 150 mg/day | Fruensgaard 1978 | range 16 mg/day to max 120 mg/day |
| vs Mepazine | mean 151 mg/day | Bennett 1961; Kurland 1961 | mean 42 mg/day |
| vs Methyperidol | range 15 mg to 30 mg/day | Itoh 1969 III | range 9 mg/day to 18 mg/day |
| vs Olanzapine | mean 16.5 mg/day | CATIE 2005; Naukkarinen 2000; Wang 2008b | mean 24.9 mg/day |
| vs Penfluridol | 2 mg/day the flexible up to max 100 mg/day | Itoh 1976 | 12 mg/day then flexible dose age up to max 60 mg/day |
| vs Prochlorperazine | mean 59.9 mg/day | Bennett 1961; Hanlon 1965; Kurland 1961 | mean 40.91 mg/day |
| vs Quetiapine | mean 401.16 mg/day | CATIE 2005; Wang 2008c | mean 22.65 mg/day |
| vs Promazine | mean 438.92 mg/day | Kurland 1961 | mean 30.83 mg/day |
| vs Risperidone | mean 5.3 mg/day | CATIE 2005; Hoyberg 1993; Wang 2008a | mean 25.4 mg/day |
| vs Sulpiride | mean 900 mg/day | Amakusa 1973; Lepola 1989 | mean 20.5 mg/day |
| vs Thioproprazate | mean 20.83 mg/day | Hanlon 1965 | mean 38.61 mg/day |
| vs Thioridazine | mean 193.46 mg/day | Hanlon 1965 | mean 38.61 mg/day |
| vs Thiothixene | no details | Itoh 1969 II | no details |
| vs Timiperone | range 2 mg/day up to max of 12 mg/day | Takahashi 1982 | range 8 mg/day up to max of 48 mg/day |
| vs Trifluoperazine | mean 11.49 mg/day | Hanlon 1965 | mean 38.61 mg/day |
| vs Trifluopromazine | mean 124.13 mg/day | Bennett 1961; Hanlon 1965; Kurland 1961 | mean 40.9 mg/day |
| vs Ziprasidone | mean 112.8 mg/day | CATIE 2005 | mean 20.8 mg/day |
| vs Zotepine | range 75 mg/day to max 150 mg/day | Imai 1980 | range 12 mg/day to max 24 mg/day |
| vs Zuclopenthixol | mean 37 mg/day | Remvig 1987 | mean 30 mg/day |
The number of comparators is greater than the total number of included studies because several studies used more than one comparison group (Bennett 1961; CATIE 2005; Chouinard 1975; Chouinard 1977; Collins 1967; Hanlon 1964; Hanlon 1965; Kurihara 1983; Kurland 1961; Sun 2000).
7. Outcomes
7.1 General remarks
There was a relatively high degree of inconsistency in terms of the outcome measures used in the included trials. In addition, many trials presented results in graphical form, which made it difficult to extract data. The use of continuous data were often not possible because standard deviations were not indicated, and in other cases because the data seemed to be skewed. For skewed data, results are shown in the 'other data tables'. The outcomes that could be used most frequently were 'global clinical state', 'leaving the study early' and 'adverse events' (movement disorders and others).
7.2 Acceptability and efficacy
We could extract the number of people 'leaving the studies early', which can be used as a measure of acceptability of treatment, from 28 studies; data were not available for Wang 2008a; Wang 2008b; Zhang 2010. The number of people who showed no improvement or deteriorated according to the Clinical Global Impression (CGI) was given in 17 studies. The Brief Psychiatric Rating Scale (BPRS) is one of the most widely used rating scales to monitor the general mental state of those with schizophrenia, but only Hoyberg 1993; Lepola 1989; Naukkarinen 2000; Sun 2000; Wang 2008c; Zhang 2010 provided usable data. Hoyberg 1993; Kane 2003; Naukkarinen 2000; Wang 2008a; Wang 2008b; Zhang 2010 were the only studies that used the Positive and Negative Symptom Scale (PANSS) to evaluate symptoms. Behavioural changes were assessed in the three studies by Itoh (Itoh 1969; Itoh 1969 II; Itoh 1969 III) using the 'Behaviour Scale', a Japanese scale about which we were not able to obtain further information.
7.3 Adverse events
The number of participants with at least one adverse event could be extracted from many trials. Extrapyramidal adverse events (EPS) were most intensively monitored in the studies. An outcome related to EPS ‐ the number of participants who needed antiparkinsonian medication at least once ‐ was also frequently recorded. The occurrence of adverse events other than EPS was much less consistently indicated.
7.4 Outcome scales
Details of scales that provided usable data are shown below. Reasons for the exclusion of data from other instruments are given under 'Outcomes' in the 'Included studies' section.
7.4.1 Global state
i. Clinical Global Impression ‐ CGI (Guy 1976) This is a rating instrument that enables clinicians to quantify severity of illness and overall clinical improvement during therapy. A seven‐point scoring system is usually used with low scores indicating decreased severity and/or greater recovery. Only one included study provided continuous data using this scale (Kane 2003).
7.4.2 Mental state
i. Brief Psychiatric Rating Scale ‐ BPRS (Overall 1962) This scale is used to assess the severity of abnormal mental states. The original scale has 16 items, but a revised 18‐item scale is commonly used. Each item is defined on a seven‐point scale varying from 'not present' to 'extremely severe', scoring from 0 ‐ 6 or 1 ‐ 7. Scores can range from 0 ‐ 108 or 18 ‐ 126, respectively. High scores indicate more severe symptoms. The BPRS‐positive cluster comprises four items, which are conceptual disorganisation, suspiciousness, hallucinatory behaviour and unusual thought content. The BPRS‐negative cluster comprises only three items, which are emotional withdrawal, motor retardation, and blunted affect. Four included studies provided dichotomised data using this scale (Hoyberg 1993; Sun 2000; Wang 2008c; Zhang 2010); only one study provided continuous data using this scale (Wang 2008c); while two studies provided skew data (Lepola 1989; Naukkarinen 2000).
ii. Positive and Negative Symptom Scale ‐ PANSS (Kay 1987) The positive and negative syndrome scale was originated as a method for evaluating positive, negative and other symptom dimensions in schizophrenia. The scale has 30 items, and each item can be rated on a seven‐point scoring system varying from one (absent) to seven (extreme). This scale can be divided into three subscales for measuring the severity of general psychopathology, positive symptoms (PANSS‐P) and negative symptoms (PANSS‐N). A low score indicates low levels of symptoms. Three included studies reported dichotomised data using this scale (Hoyberg 1993; Wang 2008a; Wang 2008b); three studies reported continuous data using this scale (Hoyberg 1993; Wang 2008b; Zhang 2010); and two studies reported skew data using this scale (Kane 2003; Naukkarinen 2000).
7.4.3. Behaviour
i. Wing‐Scale B (Wing 1961)
This is a 12‐item rating scale, measuring such behaviours as 'slowness of movement', 'underactivity', 'conversation', 'laughing and talking to self', 'threatening violent behaviour', and others including 'personal hygiene'. The items are rated 0 to 2, (0 = no particular behaviour present, and 2 = extreme behaviour present). Only one study provided data using this scale (Collins 1967).
7.4.4 Adverse events
i. TESS (ICH E3 1995)
The ICH E3 1995 guideline has stated that 'treatment‐emergent signs and symptoms' (TESS) are to be defined as "events not seen at baseline and events that worsened even if present at baseline". It can be difficult to document this accurately taking into account variables in time, dosage, adverse events and severity. Generally, TESS scores for particular adverse events are categorised as 'mild', 'moderate', or 'severe', with an appropriate action taken (e.g. none; discontinued; dose changed' hospitalisation; additional medication given. It is not often that studies publish continuous data from these measurements, with the more common presentation of dichotomised TESS ratings. Only one study provided continuous data using this rating (Zhang 2010).
7.5 Missing outcomes
Many important outcomes such as economic costs, issues of hospital admission, death and satisfaction with care were not addressed in the included studies.
Excluded studies
In this updated review, we excluded a total of 19 studies in addition to those already excluded from the previous review, totaling 108 excluded studies. Several of these studies were not randomised (Affleck 1969; Eklund 1976; Opjordsmoen 2000; Sharpley 1964). Other studies were excluded because they did not compare appropriate interventions (perphenazine enanthate or decanoate; perphenazine in combination with other drugs; perphenazine versus perphenazine; perphenazine versus unlicensed neuroleptics). Eight studies did not use a placebo or neuroleptic comparator group. Sixteen studies were randomised controlled trials, but we had to exclude them because outcome data were no longer available. With the exception of Levine 1997, all of the latter studies were undertaken by the research group under Professor Svestka (Brno, Czech Republic) who kindly informed us that the data are no longer available. Other exclusions included comparisons of perphenazine combined with psychological therapy versus perphenazine alone (Zhu 2009), and perphenazine versus 1‐stepholidine (Wang 2001; Zhong 2001), both are topics which would be interesting and appropriate in another review.
Studies awaiting assessment
We have obtained 14 publications but cannot use them without further information from authors and have written letters to the primary authors requesting more information. Answers from these authors are still due but have become unlikely. We hope that someone may provide us with the missing information in the future, and therefore, we have retained these studies in the category 'awaiting assessment'.
Ongoing studies
We identified no ongoing studies.
Risk of bias in included studies
For a graphical overview, see Figure 3; Figure 4.
3.
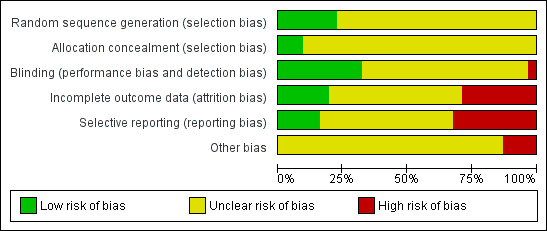
'Risk of bias' graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
4.
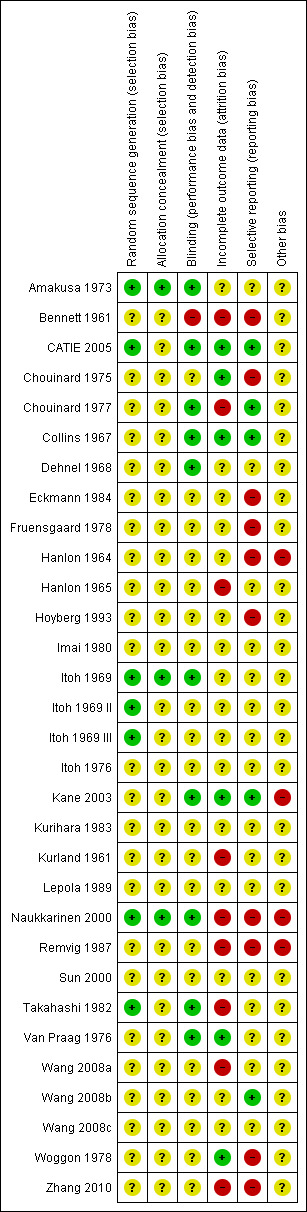
'Risk of bias' summary: review authors' judgements about each risk of bias item for each included study.
Allocation
Although all trials were described as 'randomised', only seven publications presented adequate information about sequence generation (Amakusa 1973; CATIE 2005; Itoh 1969; Itoh 1969 II; Itoh 1969 III; Naukkarinen 2000; Takahashi 1982). All other 24 included studies were rated as 'unclear', because it was unknown exactly how the randomisation had been undertaken.
Blinding
All studies apart from one (Bennett 1961) were stated to have been conducted in a double‐blind fashion. How blindness was assured in this one study is unclear, with possible single‐blinding implied, and therefore rated as a 'high' risk of bias. Ten studies were rated as a 'low' risk of bias for providing adequate information as to blinding with most studies using capsules identical in appearance to assist in maintaining blindness (Amakusa 1973; CATIE 2005; Chouinard 1977; Collins 1967; Dehnel 1968; Itoh 1969; Kane 2003; Naukkarinen 2000; Takahashi 1982; Van Praag 1976), with the remaining studies rated as 'unclear.
Incomplete outcome data
Nine studies were rated as a 'high' risk of bias for failing to describe participants who may have discontinued or left the studies early (Bennett 1961; Chouinard 1977; Hanlon 1965; Kurland 1961; Naukkarinen 2000; Remvig 1987; Takahashi 1982; Wang 2008a; Zhang 2010). These studies either had a high attrition rate (of more than 50%), or did not adequately explain which data were included in the final data analysis. Six studies were rated as a 'low' risk of bias, for either having no losses (Collins 1967), or for clearly explaining all participants who discontinued early and including them in full analysis on either ITT or LOCF (CATIE 2005; Chouinard 1975; Kane 2003; Van Praag 1976; Woggon 1978). All of the remaining studies were rated as an 'unclear' risk for inadequate explanation.
Selective reporting
Ten studies were rated as a 'high' risk for reporting bias (Bennett 1961; Chouinard 1975; Eckmann 1984; Fruensgaard 1978; Hanlon 1964; Hoyberg 1993; Naukkarinen 2000; Remvig 1987; Woggon 1978; Zhang 2010), mainly due to stated outcomes either not being reported at all in results, or data rendered unusable ‐ specifically continuous data, which in some studies were not reported in their entirety (i.e. including mean, standard deviation, reported by group). Five studies were rated as a 'low' risk (CATIE 2005; Chouinard 1977; Collins 1967; Kane 2003; Wang 2008b). All remaining studies were rated as an 'unclear' risk for inadequate information provided in the study reports. We constructed a funnel plot for our primary outcome of global state: change over time ‐ no better or deterioration (see Figure 1), which demonstrates asymmetry ‐ publication bias (selective reporting or analysis reporting) is one possible explanation, however poor methodological design is another factor to consider.
Other potential sources of bias
The vast majority of studies were rated as 'unclear' for other potential sources of bias, as these can be difficult to detect ‐ particularly because the reporting standards of included studies were generally quite low due to the age of included studies (ranging from the years 1961 to 2005). We rated four studies as a 'high' risk on this domain (Hanlon 1964; Kane 2003; Naukkarinen 2000; Remvig 1987), largely due to pharmaceutical industry support in each of these studies, as well as potential protocol deviation detected (Remvig 1987).
Effects of interventions
For dichotomous outcomes we calulated risk ratios (RR) and for continuous outcomes we calculated mean differences (MD), both with the associated 95% confidence intervals (CI).
COMPARISON 1: PERPHENAZINE versus PLACEBO
1.1 Leaving the study early: 1. Any reason
1.1.1 short term
In this subgroup we found four relevant trials (n = 203). There was no significant difference between perphenazine and placebo (RR 0.26 CI 0.01 to 4.92, Analysis 1.1). This subgroup had important levels of heterogeneity (Chi2 = 25.5; df = 2; P < 0.00001; I2 = 92%).
1.1. Analysis.
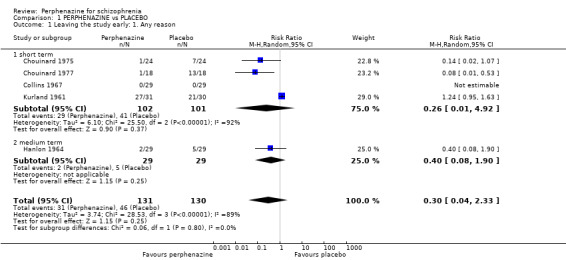
Comparison 1 PERPHENAZINE vs PLACEBO, Outcome 1 Leaving the study early: 1. Any reason.
1.1.2 medium term
In this subgroup we only found one relevant trial (n = 58) (Hanlon 1964). There was no significant difference between perphenazine and placebo (RR 0.40 CI 0.08 to 1.90, Analysis 1.1).
1.2 Leaving the study early: 2. Due to adverse events
1.2.1 short term
In this subgroup we only found one relevant trial (n = 61) (Kurland 1961). There was a statistically significant difference (P = 0.05) favouring placebo over perphenazine (RR 16.47 CI 0.99 to 273.29, Analysis 1.2).
1.2. Analysis.
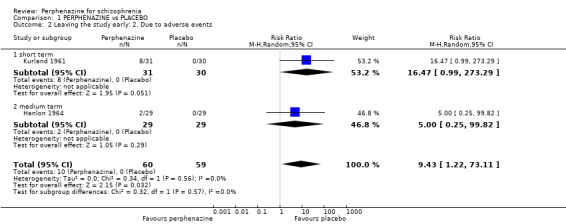
Comparison 1 PERPHENAZINE vs PLACEBO, Outcome 2 Leaving the study early: 2. Due to adverse events.
1.2.2 medium term
In this subgroup we only found one relevant trial (n = 58) (Hanlon 1964). There was no significant difference between perphenazine and placebo (RR 5.00 CI 0.25 to 99.82, Analysis 1.2).
Overall, there was a statistically significant difference (P = 0.03) favouring placebo in the short and medium term (2 RCTS, n = 119, RR 9.43 CI 1.22 to 73.11).
1.3 Leaving the study early: 3. Due to relapse/worsening or no improvement
1.3.1 short term
In this subgroup we found two relevant trials (n = 84). There was a statistically significant difference (P = 0.001) favouring perphenazine over placebo (RR 0.10 CI 0.03 to 0.42, Analysis 1.3).
1.3. Analysis.
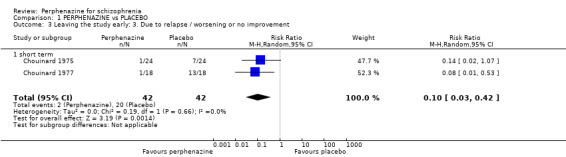
Comparison 1 PERPHENAZINE vs PLACEBO, Outcome 3 Leaving the study early: 3. Due to relapse / worsening or no improvement.
1.4 Global state: 1. Change over time ‐ no better or deterioration (ITT)
1.4.1 short term
In this subgroup we only found one relevant trial (n = 61) (Kurland 1961). There was a statistically significant (P = 0.01) difference favouring perphenazine over placebo (RR 0.32 CI 0.13 to 0.78, Analysis 1.4).
1.4. Analysis.
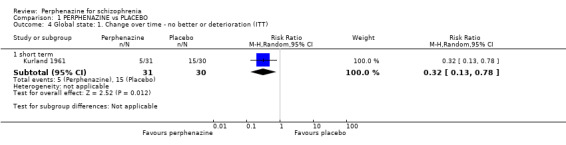
Comparison 1 PERPHENAZINE vs PLACEBO, Outcome 4 Global state: 1. Change over time ‐ no better or deterioration (ITT).
1.5 Mental state: 1. Relapse (clinical diagnosis)
1.5.1 short term
In this subgroup we only found one relevant trial (n = 48) (Chouinard 1975). There was no significant difference between perphenazine and placebo (RR 0.14 CI 0.02 to 1.07, Analysis 1.5).
1.5. Analysis.
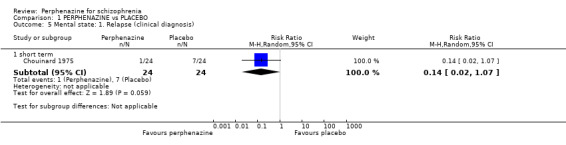
Comparison 1 PERPHENAZINE vs PLACEBO, Outcome 5 Mental state: 1. Relapse (clinical diagnosis).
1.6 Behaviour: 1a. Wing‐Scale B: social withdrawal ‐ mean change from baseline to endpoint
1.6.1 short term
In this subgroup we only found one relevant trial (n = 58) (Collins 1967). There was no significant difference between perphenazine and placebo (MD 0.76 CI ‐0.51 to 2.03, Analysis 1.6).
1.6. Analysis.
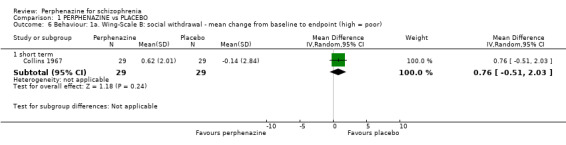
Comparison 1 PERPHENAZINE vs PLACEBO, Outcome 6 Behaviour: 1a. Wing‐Scale B: social withdrawal ‐ mean change from baseline to endpoint (high = poor).
1.7 Behaviour: 1b. Wing‐Scale B: socially embarrassing behaviour ‐ mean change from baseline to endpoint
1.7.1 short term
In this subgroup we only found one relevant trial (n = 58) (Collins 1967). There was no significant difference between perphenazine and placebo (MD 0.10 CI ‐0.54 to 0.74, Analysis 1.7).
1.7. Analysis.
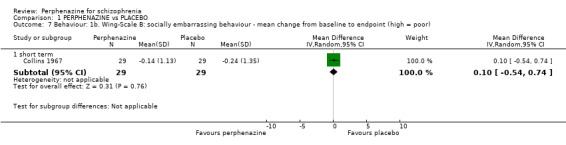
Comparison 1 PERPHENAZINE vs PLACEBO, Outcome 7 Behaviour: 1b. Wing‐Scale B: socially embarrassing behaviour ‐ mean change from baseline to endpoint (high = poor).
1.8 Adverse events: Movement disorders
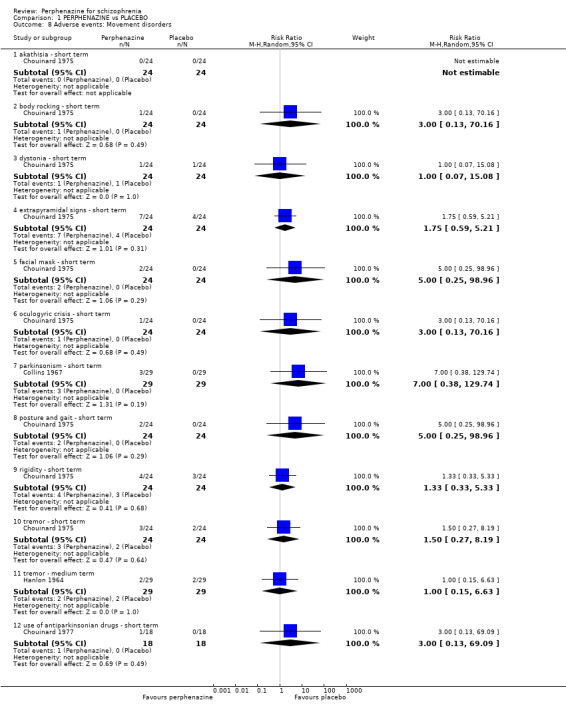
1.8.1 akathisia ‐ short term
In this subgroup we only found one relevant trial (n = 48) (Chouinard 1975). There was no significant difference between perphenazine and placebo, with no events reported in either group (Analysis 1.8).
1.8. Analysis.
Comparison 1 PERPHENAZINE vs PLACEBO, Outcome 8 Adverse events: Movement disorders.
1.8.2 body rocking ‐ short term
In this subgroup we only found one relevant trial (n = 48) (Chouinard 1975). There was no significant difference between perphenazine and placebo (RR 3.00 CI 0.13 to 70.16, Analysis 1.8).
1.8.3 dystonia ‐ short term
In this subgroup we only found one relevant trial (n = 48) (Chouinard 1975). There was no significant difference between perphenazine and placebo (RR 1.00 CI 0.07 to 15.08, Analysis 1.8).
1.8.4 extrapyramidal signs ‐ short term
In this subgroup we only found one relevant trial (n = 48) (Chouinard 1975). There was no significant difference between perphenazine and placebo (RR 1.75 CI 0.59 to 5.21, Analysis 1.8).
1.8.5 facial mask ‐ short term
In this subgroup we only found one relevant trial (n = 48) (Chouinard 1975). There was no significant difference between perphenazine and placebo (RR 5.00 CI 0.25 to 98.96, Analysis 1.8).
1.8.6 oculogyric crisis ‐ short term
In this subgroup we only found one relevant trial (n = 48) (Chouinard 1975). There was no significant difference between perphenazine and placebo (RR 3.00 CI 0.13 to 70.16, Analysis 1.8).
1.8.7 parkinsonism ‐ short term
In this subgroup we only found one relevant trial (n = 58) (Collins 1967). There was no significant difference between perphenazine and placebo (RR 7.00 CI 0.38 to 129.74, Analysis 1.8).
1.8.8 posture and gait ‐ short term
In this subgroup we only found one relevant trial (n = 48) (Chouinard 1975). There was no significant difference between perphenazine and placebo (RR 5.00 CI 0.25 to 98.96, Analysis 1.8).
1.8.9 rigidity ‐ short term
In this subgroup we only found one relevant trial (n = 48) (Chouinard 1975). There was no significant difference between perphenazine and placebo (RR 1.33 CI 0.33 to 5.33, Analysis 1.8).
1.8.10 tremor ‐ short term
In this subgroup we only found one relevant trial (n = 48) (Chouinard 1975). There was no significant difference between perphenazine and placebo (RR 1.50 CI 0.27 to 8.19, Analysis 1.8).
1.8.11 tremor ‐ medium term
In this subgroup we only found one relevant trial (n = 58) (Hanlon 1964). There was no significant difference between perphenazine and placebo (RR 1.00 CI 0.15 to 6.63, Analysis 1.8).
1.8.12 use of antiparkinsonian drugs ‐ short term
In this subgroup we only found one relevant trial (n = 36) (Chouinard 1977). There was no significant difference between perphenazine and placebo (RR 3.00 CI 0.13 to 69.09, Analysis 1.8).
1.9 Other adverse events: 1. Anticholinergic
1.9.1 blurred vision ‐ short term
In this subgroup we only found one relevant trial (n = 48) (Chouinard 1975). There was no significant difference between perphenazine and placebo, with no events reported in either group (Analysis 1.9).
1.9. Analysis.
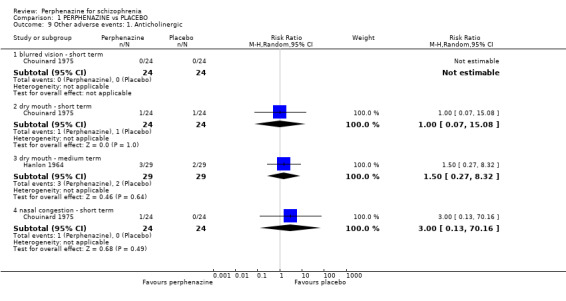
Comparison 1 PERPHENAZINE vs PLACEBO, Outcome 9 Other adverse events: 1. Anticholinergic.
1.9.2 dry mouth ‐ short term
In this subgroup we only found one relevant trial (n = 48) (Chouinard 1975). There was no significant difference between perphenazine and placebo (RR 1.00 CI 0.07 to 15.08, Analysis 1.9).
1.9.3 dry mouth ‐ medium term
In this subgroup we only found one relevant trial (n = 58) (Hanlon 1964). There was no significant difference between perphenazine and placebo (RR 1.50 CI 0.27 to 8.32, Analysis 1.9).
1.9.4 nasal congestion ‐ short term
In this subgroup we only found one relevant trial (n = 48) (Chouinard 1975). There was no significant difference between perphenazine and placebo (RR 3.00 CI 0.13 to 70.16, Analysis 1.9).
1.10 Other adverse events: 2. Arousal
1.10.1 agitation ‐ medium term
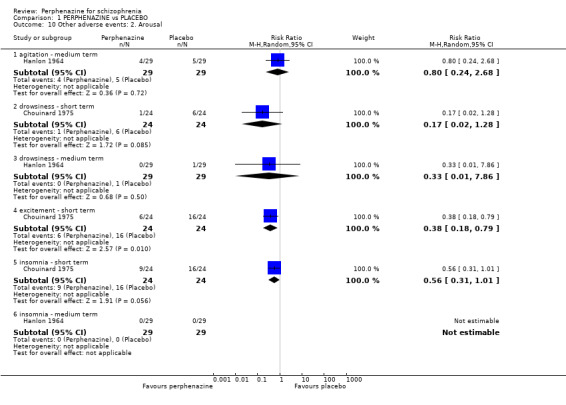
In this subgroup we only found one relevant trial (n = 58) (Hanlon 1964). There was no significant difference between perphenazine and placebo (RR 0.80 CI 0.24 to 2.68, Analysis 1.10).
1.10. Analysis.
Comparison 1 PERPHENAZINE vs PLACEBO, Outcome 10 Other adverse events: 2. Arousal.
1.10.2 drowsiness ‐ short term
In this subgroup we only found one relevant trial (n = 48) (Chouinard 1975). There was no significant difference between perphenazine and placebo (RR 0.17 CI 0.02 to 1.28, Analysis 1.10).
1.10.3 drowsiness ‐ medium term
In this subgroup we only found one relevant trial (n = 58) (Hanlon 1964). There was no significant difference between perphenazine and placebo (RR 0.33 CI 0.01 to 7.86, Analysis 1.10).
1.10.4 excitement ‐ short term
In this subgroup we only found one relevant trial (n = 48) (Chouinard 1975). There was a statistically significant difference (P = 0.01) favouring perphenazine over placebo (RR 0.38 CI 0.18 to 0.79, Analysis 1.10).
1.10.5 insomnia ‐ short term
In this subgroup we only found one relevant trial (n = 48) (Chouinard 1975). There was no significant difference between perphenazine and placebo (RR 0.56 CI 0.31 to 1.01, Analysis 1.10).
1.10.6 insomnia ‐ medium term
In this subgroup we only found one relevant trial (n = 58) (Hanlon 1964). There was no significant difference between perphenazine and placebo, with no events reported in either group (Analysis 1.10).
1.11 Other adverse events: 3. At least one
1.11.1 short term
In this subgroup we found two relevant trials (n = 94). There was no significant difference between perphenazine and placebo (RR 1.50 CI 0.47 to 4.76, Analysis 1.11).
1.11. Analysis.
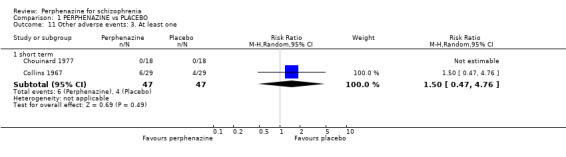
Comparison 1 PERPHENAZINE vs PLACEBO, Outcome 11 Other adverse events: 3. At least one.
1.12 Other adverse events: 4. Cardiovascular
1.12.1 ECG abnormalities ‐ short term
In this subgroup we only found one relevant trial (n = 36) (Chouinard 1977). There was no significant difference between perphenazine and placebo (RR 2.00 CI 0.42 to 9.58, Analysis 1.12).
1.12. Analysis.
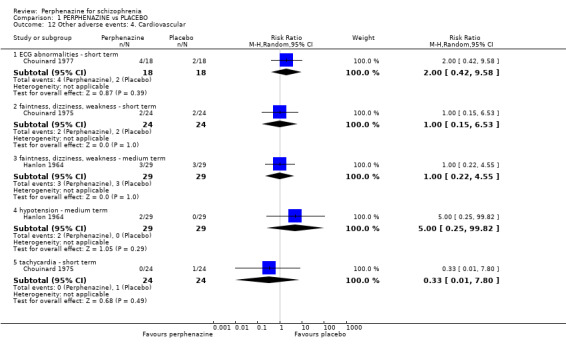
Comparison 1 PERPHENAZINE vs PLACEBO, Outcome 12 Other adverse events: 4. Cardiovascular.
1.12.2 faintness, dizziness, weakness ‐ short term
In this subgroup we only found one relevant trial (n = 48) (Chouinard 1975). There was no significant difference between perphenazine and placebo (RR 1.00 CI 0.15 to 6.53, Analysis 1.12).
1.12.3 faintness, dizziness, weakness ‐ medium term
In this subgroup we only found one relevant trial (n = 58) (Hanlon 1964). There was no significant difference between perphenazine and placebo (RR 1.00 CI 0.22 to 4.55, Analysis 1.12).
1.12.4 hypotension ‐ medium term
In this subgroup we only found one relevant trial (n = 58) (Hanlon 1964). There was no significant difference between perphenazine and placebo (RR 5.00 CI 0.25 to 99.82, Analysis 1.12).
1.12.5 tachycardia ‐ short term
In this subgroup we only found one relevant trial (n = 48) (Chouinard 1975). There was no significant difference between perphenazine and placebo (RR 0.33 CI 0.01 to 7.80, Analysis 1.12).
1.13 Other adverse events: 5. Central nervous system
1.13.1 headache ‐ short term
In this subgroup we only found one relevant trial (n = 48) (Chouinard 1975). There was no significant difference between perphenazine and placebo (RR 0.75 CI 0.19 to 3.00, Analysis 1.13).
1.13. Analysis.
Comparison 1 PERPHENAZINE vs PLACEBO, Outcome 13 Other adverse events: 5. Central nervous system.
1.14 Other adverse events: 6. Endocrine
1.14.1 lactation ‐ short term
In this subgroup we only found one relevant trial (n = 48) (Chouinard 1975). There was no significant difference between perphenazine and placebo, with no events reported in either group (Analysis 1.14).
1.14. Analysis.
Comparison 1 PERPHENAZINE vs PLACEBO, Outcome 14 Other adverse events: 6. Endocrine.
1.15 Other adverse events: 7. Gastrointestinal
1.15.1 constipation ‐ short term
In this subgroup we only found one relevant trial (n = 48) (Chouinard 1975). There was no significant difference between perphenazine and placebo (RR 0.80 CI 0.24 to 2.62, Analysis 1.15).
1.15. Analysis.
Comparison 1 PERPHENAZINE vs PLACEBO, Outcome 15 Other adverse events: 7. Gastrointestinal.
1.15.2 diarrhoea ‐ short term
In this subgroup we only found one relevant trial (n = 48) (Chouinard 1975). There was no significant difference between perphenazine and placebo (RR 0.33 CI 0.01 to 7.80, Analysis 1.15).
1.15.3 nausea or vomiting ‐ short term
In this subgroup we only found one relevant trial (n = 48) (Chouinard 1975). There was no significant difference between perphenazine and placebo (RR 0.60 CI 0.16 to 2.23, Analysis 1.15).
1.15.4 nausea or vomiting ‐ medium term
In this subgroup we only found one relevant trial (n = 58) (Hanlon 1964). There was no significant difference between perphenazine and placebo (RR 5.00 CI 0.25 to 99.82, Analysis 1.15).
1.16 Other adverse events: 8. Haematology ‐ abnormal laboratory results
1.16.1 short term
In this subgroup we only found one relevant trial (n = 48) (Chouinard 1975). There was no significant difference between perphenazine and placebo (RR 1.50 CI 0.48 to 4.65, Analysis 1.16).
1.16. Analysis.
Comparison 1 PERPHENAZINE vs PLACEBO, Outcome 16 Other adverse events: 8. Haematology ‐ abnormal laboratory results.
1.17 Other adverse events: 9. Skin
1.17.1 dermatitis ‐ short term
In this subgroup we only found one relevant trial (n = 48) (Chouinard 1975). There was no significant difference between perphenazine and placebo (RR 0.33 CI 0.01 to 7.80, Analysis 1.17).
1.17. Analysis.
Comparison 1 PERPHENAZINE vs PLACEBO, Outcome 17 Other adverse events: 9. Skin.
1.17.2 oedema ‐ medium term
In this subgroup we only found one relevant trial (n = 58) (Hanlon 1964). There was no significant difference between perphenazine and placebo (RR 0.33 CI 0.01 to 7.86, Analysis 1.17).
1.18 Other adverse events: 10. Others
1.18.1 salivation increased ‐ short term
In this subgroup we only found one relevant trial (n = 48) (Chouinard 1975). There was no significant difference between perphenazine and placebo (RR 2.00 CI 0.19 to 20.61, Analysis 1.18).
1.18. Analysis.
Comparison 1 PERPHENAZINE vs PLACEBO, Outcome 18 Other adverse events: 10. Others.
1.18.2 tenseness ‐ medium term
In this subgroup we only found one relevant trial (n = 58) (Hanlon 1964). There was no significant difference between perphenazine and placebo (RR 0.50 CI 0.05 to 5.21, Analysis 1.18).
COMPARISON 2: PERPHENAZINE versus ANY ANTIPSYCHOTIC
2.1 Leaving the study early: 1. Any reason
2.1.1 short term
In this subgroup we found 21 relevant trials (n = 2500). There was no significant difference between perphenazine and any antipsychotic drugs (RR 1.09 CI 0.83 to 1.42, Analysis 2.1). This subgroup had moderate levels of heterogeneity (Chi2 = 34.32; df = 17; P = 0.008; I2 = 50%).
2.1. Analysis.
Comparison 2 PERPHENAZINE vs ANY ANTIPSYCHOTIC, Outcome 1 Leaving the study early: 1. Any reason.
2.1.2 medium term
In this subgroup we only found one relevant trial (n = 47) (Lepola 1989). There was no significant difference between perphenazine and any antipsychotic drugs (RR 1.92 CI 0.86 to 4.25, Analysis 2.1).
2.1.3 long term
In this subgroup we found two relevant trials (n = 1520). There was no significant difference between perphenazine and any antipsychotic drugs (RR 1.01 CI 0.94 to 1.09, Analysis 2.1).
Across all short, medium and long term, there was no significant difference between perphenazine and any other antipsychotic drugs (24 RCTs, n = 4067, RR 1.10 CI 0.92 to 1.31).
2.2 Leaving the study early: 2. Due to adverse events
2.2.1 short term
In this subgroup we found nine relevant trials (n = 1661). There was no significant difference between perphenazine and any antipsychotic drugs (RR 1.13 CI 0.70 to 1.83, Analysis 2.2). This subgroup had moderate levels of heterogeneity (Chi2 = 12.82; df = 8; P = 0.12; I2 = 38%).
2.2. Analysis.
Comparison 2 PERPHENAZINE vs ANY ANTIPSYCHOTIC, Outcome 2 Leaving the study early: 2. Due to adverse events.
2.2.2 medium term
In this subgroup we only found one relevant trial (n = 47) (Lepola 1989). There was no significant difference between perphenazine and any antipsychotic drugs (RR 2.88 CI 0.12 to 67.29, Analysis 2.2).
2.2.3 long term
In this subgroup we found two relevant trials (n = 1506). There was no significant difference between perphenazine and any antipsychotic drugs (RR 1.07 CI 0.79 to 1.47, Analysis 2.2).
Across all short, medium and long term, there was no significant difference between perphenazine and any other antipsychotic drugs (12 RCTs, n = 3214, RR 1.11 CI 0.82 to 1.52).
2.3 Leaving the study early: 3. Due to relapse/worsening or no improvement
2.3.1 short term
In this subgroup we found seven relevant trials (n = 979). There was no significant difference between perphenazine and any antipsychotic drugs (RR 1.26 CI 0.78 to 2.03, Analysis 2.3).
2.3. Analysis.
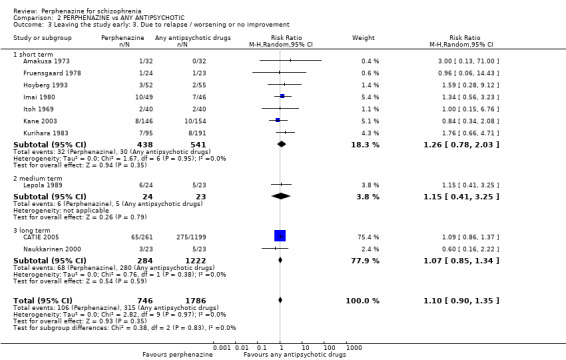
Comparison 2 PERPHENAZINE vs ANY ANTIPSYCHOTIC, Outcome 3 Leaving the study early: 3. Due to relapse / worsening or no improvement.
2.3.2 medium term
In this subgroup we only found one relevant trial (n = 47) (Lepola 1989). There was no significant difference between perphenazine and any antipsychotic drugs (RR 1.15 CI 0.41 to 3.25, Analysis 2.3).
2.3.3 long term
In this subgroup we found two relevant trials (n = 1506). There was no significant difference between perphenazine and any antipsychotic drugs (RR 1.07 CI 0.85 to 1.34, Analysis 2.3).
Across all short, medium and long term, there was no significant difference between perphenazine and any other antipsychotic drugs (10 RCTs, n = 2532, RR 1.10 CI 0.90 to 1.35).
2.4 Global state: 1. Change over time ‐ no better or deterioration (ITT)
2.4.1 short term
In this subgroup we found 16 relevant trials (n = 1803). There was no significant difference between perphenazine and any antipsychotic drugs (RR 1.03 CI 0.9 to 1.17, Analysis 2.4). This subgroup had moderate levels of heterogeneity (Chi2 = 22.17; df = 15; P = 0.10; I2 = 32%).
2.4. Analysis.
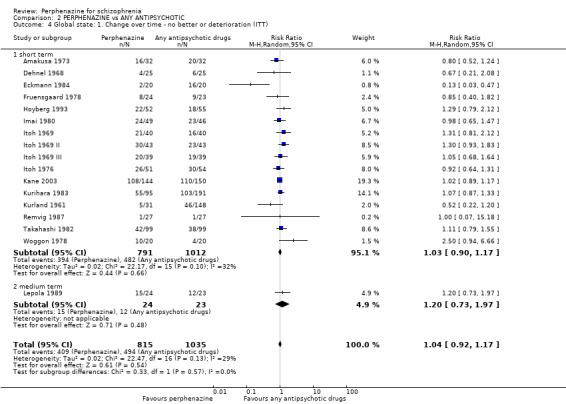
Comparison 2 PERPHENAZINE vs ANY ANTIPSYCHOTIC, Outcome 4 Global state: 1. Change over time ‐ no better or deterioration (ITT).
2.4.2 medium term
In this subgroup we only found one relevant trial (n = 47) (Lepola 1989). There was no significant difference between perphenazine and any antipsychotic drugs (RR 1.20 CI 0.73 to 1.97, Analysis 2.4).
Across both short and medium term, there was no significant difference between perphenazine and any other antipsychotic drugs (17 RCTs, n = 1850 RR 1.04 CI 0.92 to 1.17).
2.5 Global state: 2. Average endpoint score (CGI‐S)
2.5.1 short term
In this subgroup we only found one relevant trial (n = 294) (Kane 2003). There was no significant difference between perphenazine and any antipsychotic drugs (MD ‐0.20 CI ‐0.56 to 0.16, Analysis 2.5).
2.5. Analysis.
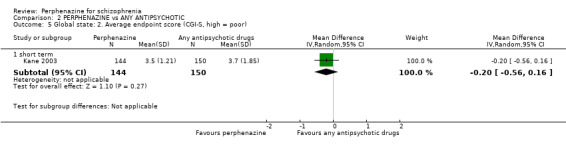
Comparison 2 PERPHENAZINE vs ANY ANTIPSYCHOTIC, Outcome 5 Global state: 2. Average endpoint score (CGI‐S, high = poor).
2.6 Mental state: 2a. State ‐ BPRS score reduction ('no effect')
For mental state outcome of 'no effect' of the study drug (BPRS score) reduction, there was again no significant difference between groups (4 RCTs, n = 383, RR 1.24 CI 0.61 to 2.52, Analysis 2.6)
2.6. Analysis.
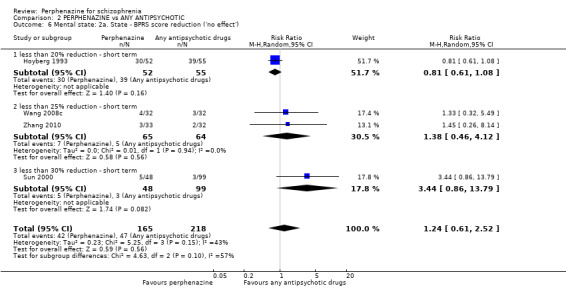
Comparison 2 PERPHENAZINE vs ANY ANTIPSYCHOTIC, Outcome 6 Mental state: 2a. State ‐ BPRS score reduction ('no effect').
2.6.1 less than 20% reduction ‐ short term
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was no significant difference between perphenazine and any antipsychotic drugs (RR 0.81 CI 0.61 to 1.08).
2.6.2 less than 25% reduction ‐ short term
In this subgroup we found two relevant trials (n = 129). There was no significant difference between perphenazine and any antipsychotic drugs (RR 1.38 CI 0.46 to 4.12).
2.6.3 less than 30% reduction ‐ short term
In this subgroup we only found one relevant trial (n = 147) (Sun 2000). There was no significant difference between perphenazine and any antipsychotic drugs (RR 3.44 CI 0.86 to 13.79).
2.7 Mental state: 2b. State ‐ BPRS total score (high = poor)
2.7.1 short term
In this subgroup we only found one relevant trial (n = 64) (Wang 2008c). There was no significant difference between perphenazine and any antipsychotic drugs (MD 1.10 CI ‐1.26 to 3.46, Analysis 2.7).
2.7. Analysis.
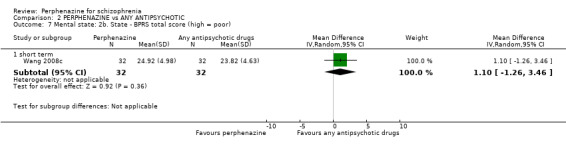
Comparison 2 PERPHENAZINE vs ANY ANTIPSYCHOTIC, Outcome 7 Mental state: 2b. State ‐ BPRS total score (high = poor).
2.8 Mental state: 2c. State ‐ BPRS end score (high = poor, skewed data)
2.8.1 medium term
Data for this outcome are skewed and are best inspected by viewing Analysis 2.8.
2.8. Analysis.
Comparison 2 PERPHENAZINE vs ANY ANTIPSYCHOTIC, Outcome 8 Mental state: 2c. State ‐ BPRS end score (high = poor, skewed data).
| Mental state: 2c. State ‐ BPRS end score (high = poor, skewed data) | |||||
|---|---|---|---|---|---|
| Study | Intervention | Subgroup | Mean | SD | N |
| medium term | |||||
| Lepola 1989 | Perphenazine | Acute schizophrenia | 15.5 | 23.8 | 7 |
| Lepola 1989 | Perphenazine | Chronic schizophrenia | 30.5 | 14.4 | 17 |
| Lepola 1989 | Sulpiride | Acute schizophrenia | 4.0 | 6.3 | 10 |
| Lepola 1989 | Sulpiride | Chronic schizophrenia | 26.0 | 14.4 | 13 |
2.9 Mental state: 2c. State ‐ BPRS end score (high = poor, skewed data)
2.9.1 long term
Data for this outcome are skewed and are best inspected by viewing Analysis 2.9.
2.9. Analysis.
Comparison 2 PERPHENAZINE vs ANY ANTIPSYCHOTIC, Outcome 9 Mental state: 2c. State ‐ BPRS end score (high = poor, skewed data).
| Mental state: 2c. State ‐ BPRS end score (high = poor, skewed data) | ||||
|---|---|---|---|---|
| Study | Intervention: | Mean | SD | N |
| long term | ||||
| Naukkarinen 2000 | Perphenazine | 12.5 | 12.6 | 23 |
| Naukkarinen 2000 | Olanzapine | 11.7 | 12.6 | 23 |
2.10 Mental state: 2d. State ‐ PANSS score reduction ('no effect')
2.10.1 less than 20% reduction ‐ short term
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was no significant difference between perphenazine and any antipsychotic drugs (RR 0.86 CI 0.64 to 1.15, Analysis 2.10).
2.10. Analysis.
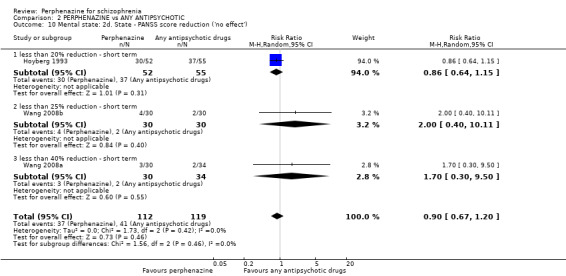
Comparison 2 PERPHENAZINE vs ANY ANTIPSYCHOTIC, Outcome 10 Mental state: 2d. State ‐ PANSS score reduction ('no effect').
2.10.2 less than 25% reduction ‐ short term
In this subgroup we only found one relevant trial (n = 60) (Wang 2008b). There was no significant difference between perphenazine and any antipsychotic drugs (RR 2.00 CI 0.40 to 10.11, Analysis 2.10).
2.10.3 less than 40% reduction ‐ short term
In this subgroup we only found one relevant trial (n = 64) (Wang 2008a). There was no significant difference between perphenazine and any antipsychotic drugs (RR 1.70 CI 0.30 to 9.50, Analysis 2.10).
2.11 Mental state: 2e. State ‐ PANSS: mean change/endpoint from baseline (high = poor)
2.11.1 total ‐ short term
In this subgroup we found three relevant trials (n = 232). There was a statistically significant difference (P < 0.0001) favouring any antipsychotic drugs over perphenazine (MD 3.65 CI 1.86 to 5.43, Analysis 2.11).
2.11. Analysis.
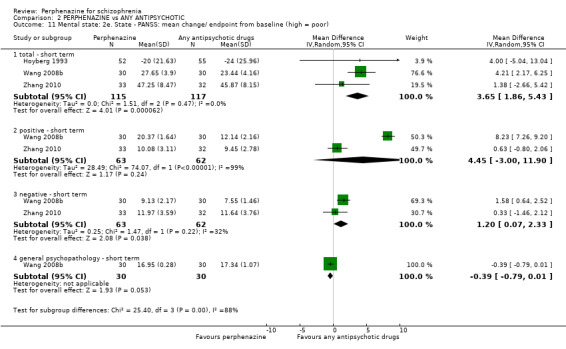
Comparison 2 PERPHENAZINE vs ANY ANTIPSYCHOTIC, Outcome 11 Mental state: 2e. State ‐ PANSS: mean change/ endpoint from baseline (high = poor).
2.11.2 positive ‐ short term
In this subgroup we found two relevant trials (n = 125). There was no significant difference between perphenazine and any antipsychotic drugs (MD 4.45 CI ‐3.00 to 11.90, Analysis 2.11). This subgroup had important high levels of heterogeneity (Chi2 = 74.07; df = 1; P = 0.0; I2 = 99%).
2.11.3 negative ‐ short term
In this subgroup we found two relevant trials (n = 125). There was a statistically significant difference (P = 0.04) favouring any antipsychotic drugs over perphenazine (MD 1.20 CI 0.07 to 2.33, Analysis 2.11). This subgroup had moderate levels of heterogeneity (Chi2 = 1.47; df = 1; P = 0.225; I2 = 32%).
2.11.4 general psychopathology ‐ short term
In this subgroup we only found one relevant trial (n = 60) (Wang 2008b). There was a statistically significant difference (P = 0.05) favouring perphenazine over any antipsychotic drugs (MD ‐0.39 CI ‐0.79 to 0.01, Analysis 2.11).
2.12 Mental state: 2f. State ‐ PANSS total change score (high = poor, skewed data)
2.12.1 short term
Data for this outcome are skewed and are best inspected by viewing Analysis 2.12.
2.12. Analysis.
Comparison 2 PERPHENAZINE vs ANY ANTIPSYCHOTIC, Outcome 12 Mental state: 2f. State ‐ PANSS total change score (high = poor, skewed data).
| Mental state: 2f. State ‐ PANSS total change score (high = poor, skewed data) | ||||
|---|---|---|---|---|
| Study | Intervention | Mean | SD | N |
| short term | ||||
| Kane 2003 | Perphenazine | ‐10.5 | 21.24 | 144 |
| Kane 2003 | Aripiprazole | ‐9.8 | 21.07 | 150 |
2.13 Mental state: 2g. State ‐ PANSS total endpoint score (high = poor, skewed data)
2.13.1 long term
Data for this outcome are skewed and are best inspected by viewing Analysis 2.13.
2.13. Analysis.
Comparison 2 PERPHENAZINE vs ANY ANTIPSYCHOTIC, Outcome 13 Mental state: 2g. State ‐ PANSS total endpoint score (high = poor, skewed data).
| Mental state: 2g. State ‐ PANSS total endpoint score (high = poor, skewed data) | ||||
|---|---|---|---|---|
| Study | Intervention | Mean | SD | N |
| long term | ||||
| Naukkarinen 2000 | Perphenazine | 54.5 | 23.9 | 23 |
| Naukkarinen 2000 | Olanzapine | 53.7 | 23.9 | 23 |
2.14 Behaviour: 1. Behaviour subscale of the Psychiatric Rating Scale: no better or worse
For this outcome we found three relevant trials (n = 244). There was no significant difference between perphenazine and any antipsychotic drugs (RR 1.03 CI 0.86 to 1.23, Analysis 2.14).
2.14. Analysis.
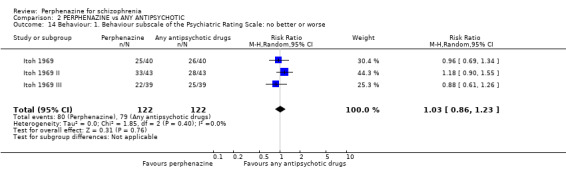
Comparison 2 PERPHENAZINE vs ANY ANTIPSYCHOTIC, Outcome 14 Behaviour: 1. Behaviour subscale of the Psychiatric Rating Scale: no better or worse.
2.15 Adverse events: Movement disorders
2.15.1 akathisia ‐ short term
In this subgroup we found 11 relevant trials (n = 1736). There was no significant difference between perphenazine and any antipsychotic drugs (RR 1.12 CI 0.79 to 1.58, Analysis 2.15). This subgroup had important levels of heterogeneity (Chi2 = 21.74; df = 10; P = 0.016; I2 = 54%).
2.15. Analysis.
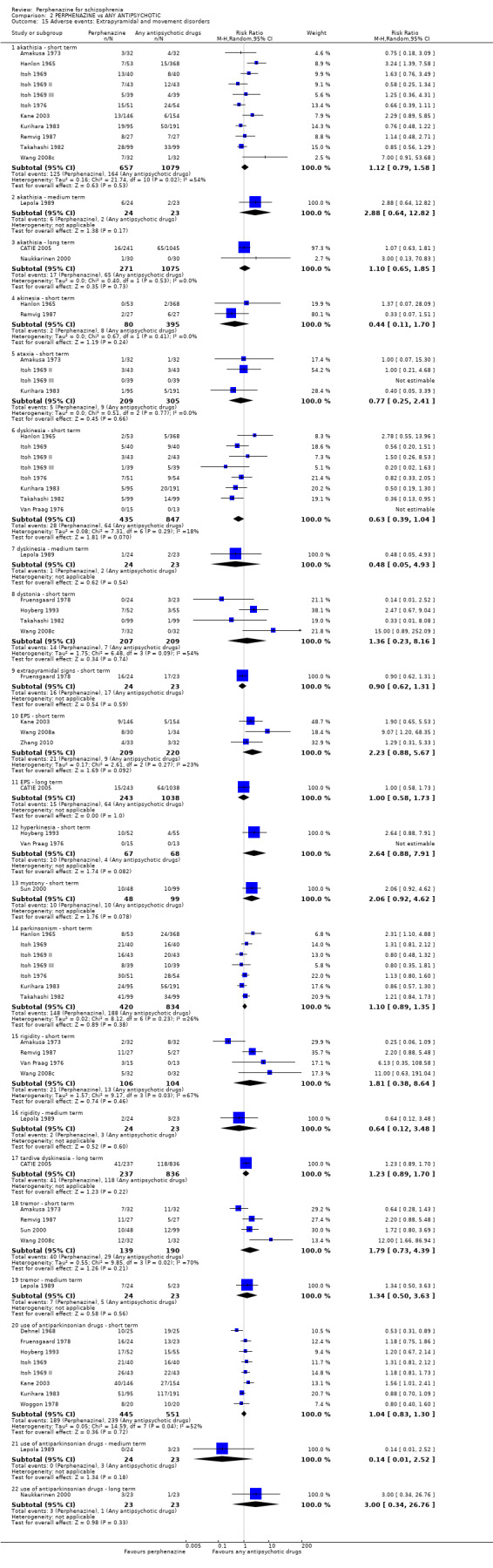
Comparison 2 PERPHENAZINE vs ANY ANTIPSYCHOTIC, Outcome 15 Adverse events: Extrapyramidal and movement disorders.
2.15.2 akathisia ‐ medium term
In this subgroup we only found one relevant trial (n = 47) (Lepola 1989). There was no significant difference between perphenazine and any antipsychotic drugs (RR 2.88 CI 0.64 to 12.82, Analysis 2.15).
2.15.3 akathisia ‐ long term
In this subgroup we found two relevant trials (n = 1346). There was no significant difference between perphenazine and any antipsychotic drugs (RR 1.10 CI 0.65 to 1.85, Analysis 2.15).
2.15.4 akinesia ‐ short term
In this subgroup we found two relevant trials (n = 475). There was no significant difference between perphenazine and any antipsychotic drugs (RR 0.44 CI 0.11 to 1.70, Analysis 2.15).
2.15.5 ataxia ‐ short term
In this subgroup we found four relevant trials (n = 514). There was no significant difference between perphenazine and any antipsychotic drugs (RR 0.77 CI 0.25 to 2.41, Analysis 2.15).
2.15.6 dyskinesia ‐ short term
In this subgroup we found eight relevant trials (n = 1282). There was no significant difference between perphenazine and any antipsychotic drugs (RR 0.63 CI 0.39 to 1.04, Analysis 2.15).
2.15.7 dyskinesia ‐ medium term
In this subgroup we only found one relevant trial (n = 47) (Lepola 1989). There was no significant difference between perphenazine and any antipsychotic drugs (RR 0.48 CI 0.05 to 4.93, Analysis 2.15).
2.15.8 dystonia ‐ short term
In this subgroup we found four relevant trials (n = 416). There was no significant difference between perphenazine and any antipsychotic drugs (RR 1.36 CI 0.23 to 8.16, Analysis 2.15). This subgroup had important levels of heterogeneity (Chi2 = 6.48; df = 3; P = 0.09; I2 = 54%).
2.15.9 extrapyramidal signs ‐ short term
In this subgroup we only found one relevant trial (n = 47) (Fruensgaard 1978). There was no significant difference between perphenazine and any antipsychotic drugs (RR 0.90 CI 0.62 to 1.31, Analysis 2.15).
2.15.10 EPS ‐ short term
In this subgroup we found three relevant trials (n = 429). There was no significant difference between perphenazine and any antipsychotic drugs (RR 2.23 CI 0.88 to 5.67, Analysis 2.15).
2.15.11 EPS ‐ long term
In this subgroup we only found one relevant trial (n = 1281) (CATIE 2005). There was no significant difference between perphenazine and any antipsychotic drugs (RR 1.00 CI 0.58 to 1.73, Analysis 2.15).
2.15.12 hyperkinesia ‐ short term
In this subgroup we found two relevant trials (n = 135). There was no significant difference between perphenazine and any antipsychotic drugs (RR 2.64 CI 0.88 to 7.91, Analysis 2.15).
2.15.13 myotony ‐ short term
In this subgroup we only found one relevant trial (n = 147) (Sun 2000). There was no significant difference between perphenazine and any antipsychotic drugs (RR 2.06 CI 0.92 to 4.62, Analysis 2.15).
2.15.14 parkinsonism ‐ short term
In this subgroup we found seven relevant trials (n = 1254). There was no significant difference between perphenazine and any antipsychotic drugs (RR 1.10 CI 0.89 to 1.35, Analysis 2.15).
2.15.15 rigidity ‐ short term
In this subgroup we found four relevant trials (n = 210). There was no significant difference between perphenazine and any antipsychotic drugs (RR 1.81 CI 0.38 to 8.64, Analysis 2.15). This subgroup had important levels of heterogeneity (Chi2 = 9.17; df = 3; P = 0.027; I2 = 67%).
2.15.16 rigidity ‐ medium term
In this subgroup we only found one relevant trial (n = 47) (Lepola 1989). There was no significant difference between perphenazine and any antipsychotic drugs (RR 0.64 CI 0.12 to 3.48, Analysis 2.15).
2.15.17 tardive dyskinesia ‐ long term
In this subgroup we only found one relevant trial (n = 1073) (CATIE 2005). There was no significant difference between perphenazine and any antipsychotic drugs (RR 1.23 CI 0.89 to 1.70, Analysis 2.15).
2.15.18 tremor ‐ short term
In this subgroup we found four relevant trials (n = 329). There was no significant difference between perphenazine and any antipsychotic drugs (RR 1.79 CI 0.73 to 4.39, Analysis 2.15). This subgroup had important levels of heterogeneity (Chi2 = 9.85; df = 3; P = 0.02; I2 = 70%).
2.15.19 tremor ‐ medium term
In this subgroup we only found one relevant trial (n = 47) (Lepola 1989). There was no significant difference between perphenazine and any antipsychotic drugs (RR 1.34 CI 0.50 to 3.63, Analysis 2.15).
2.15.20 use of antiparkinsonian drugs ‐ short term
In this subgroup we found eight relevant trials (n = 996). There was no significant difference between perphenazine and any antipsychotic drugs (RR 1.04 CI 0.83 to 1.30, Analysis 2.15). This subgroup had important levels of heterogeneity (Chi2 = 14.59; df = 7; P = 0.042; I2 = 52%).
2.15.21 use of antiparkinsonian drugs ‐ medium term
In this subgroup we only found one relevant trial (n = 47) (Lepola 1989). There was no significant difference between perphenazine and any antipsychotic drugs (RR 0.14 CI 0.01 to 2.52, Analysis 2.15).
2.15.22 use of antiparkinsonian drugs ‐ long term
In this subgroup we only found one relevant trial (n = 46) (Naukkarinen 2000). There was no significant difference between perphenazine and any antipsychotic drugs (RR 3.00 CI 0.34 to 26.76, Analysis 2.15).
2.16 Adverse events: Movement disorders ‐ TESS total score (high = poor, skewed data)
2.16.1 short term
Data for this outcome are skewed and are best inspected by viewing Analysis 2.16.
2.16. Analysis.
Comparison 2 PERPHENAZINE vs ANY ANTIPSYCHOTIC, Outcome 16 Adverse events: Movement disorders ‐ TESS total score (high = poor, skewed data).
| Adverse events: Movement disorders ‐ TESS total score (high = poor, skewed data) | ||||
|---|---|---|---|---|
| Study | Intervention | Mean | SD | N |
| short term | ||||
| Zhang 2010 | Perphenazine | 5.13 | 4.21 | 33 |
| Zhang 2010 | Aripiprazole | 4.26 | 3.75 | 32 |
2.17 Other adverse events: 1. Anticholinergic
2.17.1 blurred vision ‐ short term
In this subgroup we found eight relevant trials (n = 1028). There was no significant difference between perphenazine and any antipsychotic drugs (RR 0.80 CI 0.40 to 1.61, Analysis 2.17). This subgroup had moderate levels of heterogeneity (Chi2 = 10.85; df = 7; P = 0.145; I2 = 35%).
2.17. Analysis.
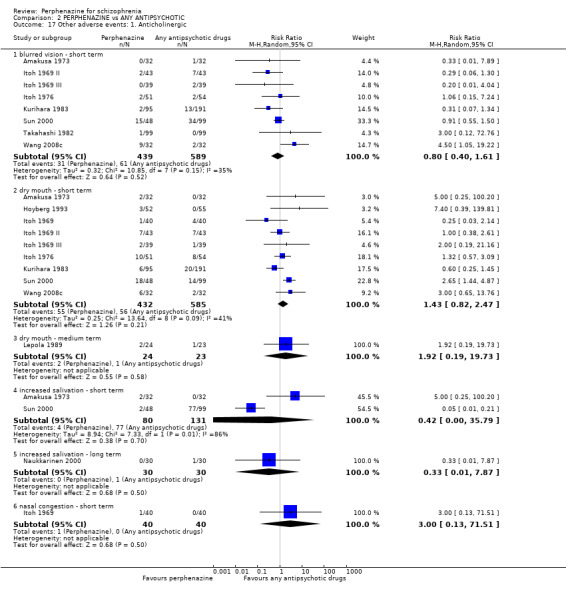
Comparison 2 PERPHENAZINE vs ANY ANTIPSYCHOTIC, Outcome 17 Other adverse events: 1. Anticholinergic.
2.17.2 dry mouth ‐ short term
In this subgroup we found nine relevant trials (n = 1017). There was no significant difference between perphenazine and any antipsychotic drugs (RR 1.43 CI 0.82 to 2.47, Analysis 2.17). This subgroup had moderate levels of heterogeneity (Chi2 = 13.64; df = 8; P = 0.092; I2 = 41%).
2.17.3 dry mouth ‐ medium term
In this subgroup we only found one relevant trial (n = 47) (Lepola 1989). There was no significant difference between perphenazine and any antipsychotic drugs (RR 1.92 CI 0.19 to 19.73, Analysis 2.17).
2.17.4 increased salivation ‐ short term
In this subgroup we found two relevant trials (n = 211). There was no significant difference between perphenazine and any antipsychotic drugs (RR 0.42 CI 0.00 to 35.79, Analysis 2.17). This subgroup had important levels of heterogeneity (Chi2 = 7.33; df = 1; P = 0.007; I2 = 86%).
2.17.5 increased salivation ‐ long term
In this subgroup we only found one relevant trial (n = 60) (Naukkarinen 2000). There was no significant difference between perphenazine and any antipsychotic drugs (RR 0.33 CI 0.01 to 7.87, Analysis 2.17).
2.17.6 nasal congestion ‐ short term
In this subgroup we only found one relevant trial (n = 80) (Itoh 1969). There was no significant difference between perphenazine and any antipsychotic drugs (RR 3.00 CI 0.13 to 71.51, Analysis 2.17).
2.18 Other adverse events: 2. Arousal
2.18.1 agitation ‐ short term
In this subgroup we only found one relevant trial (n = 300) (Kane 2003). There was no significant difference between perphenazine and any antipsychotic drugs (RR 1.05 CI 0.63 to 1.77, Analysis 2.18).
2.18. Analysis.
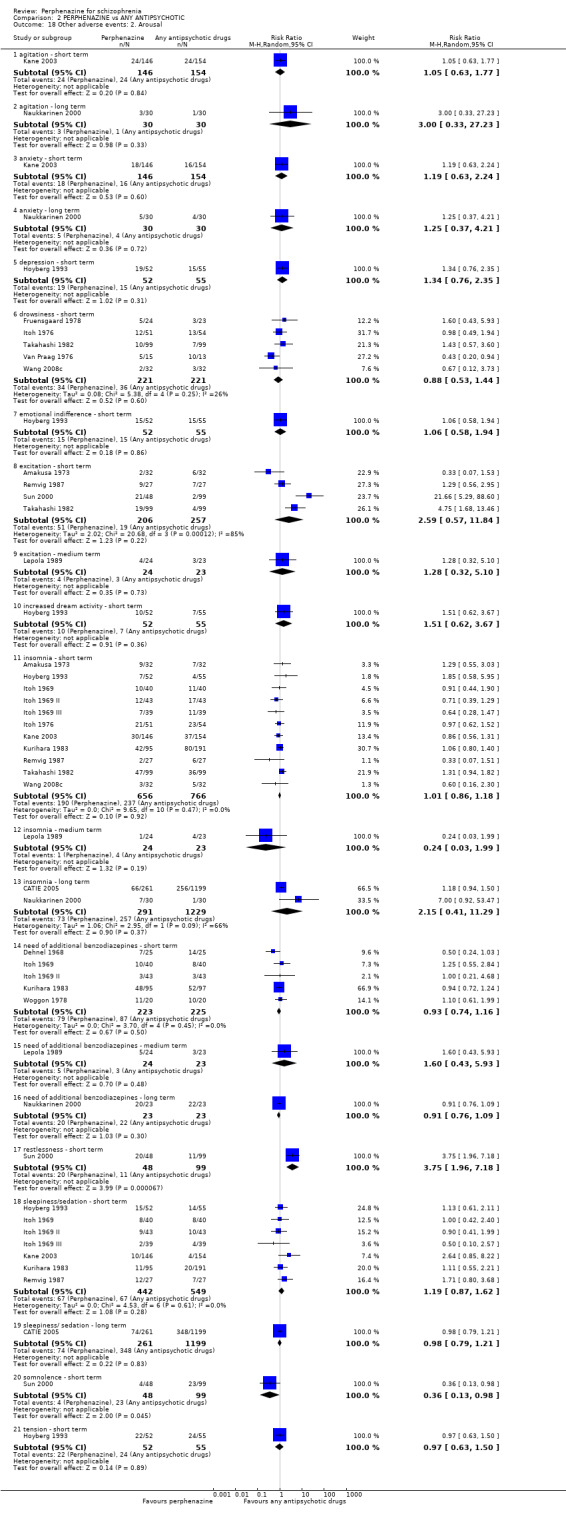
Comparison 2 PERPHENAZINE vs ANY ANTIPSYCHOTIC, Outcome 18 Other adverse events: 2. Arousal.
2.18.2 agitation ‐ long term
In this subgroup we only found one relevant trial (n = 60) (Naukkarinen 2000). There was no significant difference between perphenazine and any antipsychotic drugs (RR 3.00 CI 0.33 to 27.23, Analysis 2.18).
2.18.3 anxiety ‐ short term
In this subgroup we only found one relevant trial (n = 300) (Kane 2003). There was no significant difference between perphenazine and any antipsychotic drugs (RR 1.19 CI 0.63 to 2.24, Analysis 2.18).
2.18.4 anxiety ‐ long term
In this subgroup we only found one relevant trial (n = 60) (Naukkarinen 2000). There was no significant difference between perphenazine and any antipsychotic drugs (RR 1.25 CI 0.37 to 4.21, Analysis 2.18).
2.18.5 depression ‐ short term
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was no significant difference between perphenazine and any antipsychotic drugs (RR 1.34 CI 0.76 to 2.35, Analysis 2.18).
2.18.6 drowsiness ‐ short term
In this subgroup we found five relevant trials (n = 442). There was no significant difference between perphenazine and any antipsychotic drugs (RR 0.88 CI 0.53 to 1.44, Analysis 2.18).
2.18.7 emotional indifference ‐ short term
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was no significant difference between perphenazine and any antipsychotic drugs (RR 1.06 CI 0.58 to 1.94, Analysis 2.18).
2.18.8 excitation ‐ short term
In this subgroup we found four relevant trials (n = 463). There was no significant difference between perphenazine and any antipsychotic drugs (RR 2.59 CI 0.57 to 11.84, Analysis 2.18). This subgroup had important levels of heterogeneity (Chi2 = 20.68; df = 3; P = 0.0; I2 = 85%).
2.18.9 excitation ‐ medium term
In this subgroup we only found one relevant trial (n = 47) (Lepola 1989). There was no significant difference between perphenazine and any antipsychotic drugs (RR 1.28 CI 0.32 to 5.10, Analysis 2.18).
2.18.10 increased dream activity ‐ short term
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was no significant difference between perphenazine and any antipsychotic drugs (RR 1.51 CI 0.62 to 3.67, Analysis 2.18).
2.18.11 insomnia ‐ short term
In this subgroup we found 11 relevant trials (n = 1422). There was no significant difference between perphenazine and any antipsychotic drugs (RR 1.01 CI 0.86 to 1.18, Analysis 2.18).
2.18.12 insomnia ‐ medium term
In this subgroup we only found one relevant trial (n = 47) (Lepola 1989). There was no significant difference between perphenazine and any antipsychotic drugs (RR 0.24 CI 0.03 to 1.99, Analysis 2.18).
2.18.13 insomnia ‐ long term
In this subgroup we found two relevant trials (n = 1520). There was no significant difference between perphenazine and any antipsychotic drugs (RR 2.15 CI 0.41 to 11.29, Analysis 2.18). This subgroup had important levels of heterogeneity (Chi2 = 2.95; df = 1; P = 0.086; I2 = 66%).
2.18.14 need of additional benzodiazepines ‐ short term
In this subgroup we found five relevant trials (n = 448). There was no significant difference between perphenazine and any antipsychotic drugs (RR 0.93 CI 0.74 to 1.16, Analysis 2.18).
2.18.15 need of additional benzodiazepines ‐ medium term
In this subgroup we only found one relevant trial (n = 47) (Lepola 1989). There was no significant difference between perphenazine and any antipsychotic drugs (RR 1.60 CI 0.43 to 5.93, Analysis 2.18).
2.18.16 need of additional benzodiazepines ‐ long term
In this subgroup we only found one relevant trial (n = 46) (Naukkarinen 2000). There was no significant difference between perphenazine and any antipsychotic drugs (RR 0.91 CI 0.76 to 1.09, Analysis 2.18).
2.18.17 restlessness ‐ short term
In this subgroup we only found one relevant trial (n = 147) (Sun 2000). There was a statistically significant difference (P < 0.0001) favouring any antipsychotic drugs over perphenazine (RR 3.75 CI 1.96 to 7.18, Analysis 2.18).
2.18.18 sleepiness/sedation ‐ short term
In this subgroup we found seven relevant trials (n = 991). There was no significant difference between perphenazine and any antipsychotic drugs (RR 1.19 CI 0.87 to 1.62, Analysis 2.18).
2.18.19 sleepiness/sedation ‐ long term
In this subgroup we only found one relevant trial (n = 1460) (CATIE 2005). There was no significant difference between perphenazine and any antipsychotic drugs (RR 0.98 CI 0.79 to 1.21, Analysis 2.18).
2.18.20 somnolence ‐ short term
In this subgroup we only found one relevant trial (n = 147) (Sun 2000). There was a statistically significant difference (P = 0.05) favouring perphenazine over any antipsychotic drugs (RR 0.36 CI 0.13 to 0.98, Analysis 2.18).
2.18.21 tension ‐ short term
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was no significant difference between perphenazine and any antipsychotic drugs (RR 0.97 CI 0.63 to 1.50, Analysis 2.18).
2.19 Other adverse events: 3. At least one
2.19.1 short term
In this subgroup we found nine relevant trials (n = 974). There was no significant difference between perphenazine and any antipsychotic drugs (RR 1.02 CI 0.95 to 1.09, Analysis 2.19).
2.19. Analysis.
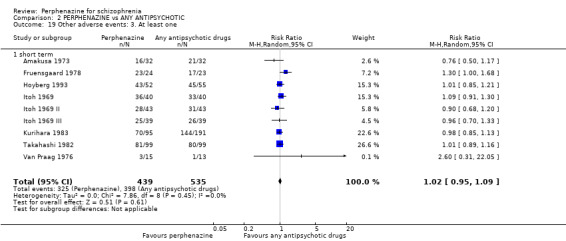
Comparison 2 PERPHENAZINE vs ANY ANTIPSYCHOTIC, Outcome 19 Other adverse events: 3. At least one.
2.20 Other adverse events: 4. Cardiovascular
2.20.1 angina pectoris ‐ short term
In this subgroup we found two relevant trials (n = 391). There was no significant difference between perphenazine and any antipsychotic drugs (RR 0.57 CI 0.26 to 1.26, Analysis 2.20).
2.20. Analysis.
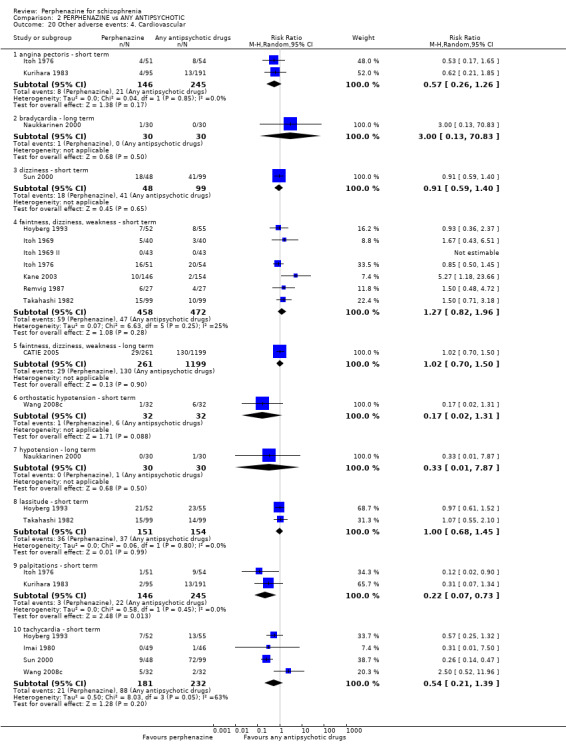
Comparison 2 PERPHENAZINE vs ANY ANTIPSYCHOTIC, Outcome 20 Other adverse events: 4. Cardiovascular.
2.20.2 bradycardia ‐ long term
In this subgroup we only found one relevant trial (n = 60) (Naukkarinen 2000). There was no significant difference between perphenazine and any antipsychotic drugs (RR 3.00 CI 0.13 to 70.83, Analysis 2.20).
2.20.3 dizziness ‐ short term
In this subgroup we only found one relevant trial (n = 147) (Sun 2000). There was no significant difference between perphenazine and any antipsychotic drugs (RR 0.91 CI 0.59 to 1.40, Analysis 2.20).
2.20.4 faintness, dizziness, weakness ‐ short term
In this subgroup we found seven relevant trials (n = 930). There was no significant difference between perphenazine and any antipsychotic drugs (RR 1.27 CI 0.82 to 1.96, Analysis 2.20).
2.20.5 faintness, dizziness, weakness ‐ long term
In this subgroup we only found one relevant trial (n = 1460) (CATIE 2005). There was no significant difference between perphenazine and any antipsychotic drugs (RR 1.02 CI 0.70 to 1.50, Analysis 2.20).
2.20.6 orthostatic hypotension ‐ short term
In this subgroup we only found one relevant trial (n = 64) (Wang 2008c). There was no significant difference between perphenazine and any antipsychotic drugs (RR 0.17 CI 0.02 to 1.31, Analysis 2.20).
2.20.7 hypotension ‐ long term
In this subgroup we only found one relevant trial (n = 60) (Naukkarinen 2000). There was no significant difference between perphenazine and any antipsychotic drugs (RR 0.33 CI 0.01 to 7.87, Analysis 2.20).
2.20.8 lassitude ‐ short term
In this subgroup we found two relevant trials (n = 305). There was no significant difference between perphenazine and any antipsychotic drugs (RR 1.00 CI 0.68 to 1.45, Analysis 2.20).
2.20.9 palpitations ‐ short term
In this subgroup we found two relevant trials (n = 391). There was a statistically significant difference (P = 0.01) favouring perphenazine over any antipsychotic drugs (RR 0.22 CI 0.07 to 0.73, Analysis 2.20).
2.20.10 tachycardia ‐ short term
In this subgroup we found four relevant trials (n = 413). There was no significant difference between perphenazine and any antipsychotic drugs (RR 0.54 CI 0.21 to 1.39, Analysis 2.20). This subgroup had important levels of heterogeneity (Chi2 = 8.03; df = 3; P = 0.045; I2 = 63%).
2.21 Other adverse events: 5. Central nervous system
2.21.1 confusion ‐ short term
In this subgroup we only found one relevant trial (n = 198) (Takahashi 1982). There was no significant difference between perphenazine and any antipsychotic drugs (RR 0.67 CI 0.11 to 3.90, Analysis 2.21).
2.21. Analysis.
Comparison 2 PERPHENAZINE vs ANY ANTIPSYCHOTIC, Outcome 21 Other adverse events: 5. Central nervous system.
2.21.2 difficulty in concentration ‐ short term
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was no significant difference between perphenazine and any antipsychotic drugs (RR 1.06 CI 0.71 to 1.58, Analysis 2.21).
2.21.3 disturbance of consciousness ‐ short term
In this subgroup we found four relevant trials (n = 648). There was no significant difference between perphenazine and any antipsychotic drugs (RR 0.84 CI 0.10 to 6.78, Analysis 2.21).
2.21.4 dysarticulation ‐ short term
In this subgroup we only found one relevant trial (n = 198) (Takahashi 1982). There was no significant difference between perphenazine and any antipsychotic drugs (RR 0.83 CI 0.38 to 1.84, Analysis 2.21).
2.21.5 electroencephalogram (increase) ‐ short term
In this subgroup we only found one relevant trial (n = 25) (Bennett 1961). There was no significant difference between perphenazine and any antipsychotic drugs (RR 0.57 CI 0.09 to 3.64, Analysis 2.21).
2.21.6 headache ‐ short term
In this subgroup we found nine relevant trials (n = 1304). There was no significant difference between perphenazine and any antipsychotic drugs (RR 0.72 CI 0.50 to 1.03, Analysis 2.21).
2.21.7 headache ‐ long term
In this subgroup we only found one relevant trial (n = 60) (Naukkarinen 2000). There was no significant difference between perphenazine and any antipsychotic drugs (RR 0.71 CI 0.25 to 2.00, Analysis 2.21).
2.21.8 hypertonia ‐ long term
In this subgroup we only found one relevant trial (n = 60) (Naukkarinen 2000). There was no significant difference between perphenazine and any antipsychotic drugs (RR 3.00 CI 0.13 to 70.83, Analysis 2.21).
2.21.9 paraesthesia ‐ short term
In this subgroup we found six relevant trials (n = 726). There was no significant difference between perphenazine and any antipsychotic drugs (RR 1.37 CI 0.57 to 3.26, Analysis 2.21).
2.21.10 seizure ‐ short term
In this subgroup we only found one relevant trial (n = 198) (Takahashi 1982). There was no significant difference between perphenazine and any antipsychotic drugs, with no events reported in either group (Analysis 2.21).
2.21.11 vertigo ‐ short term
In this subgroup we found two relevant trials (n = 364). There was no significant difference between perphenazine and any antipsychotic drugs (RR 0.87 CI 0.44 to 1.74, Analysis 2.21).
2.22 Other adverse events: 6. Endocrine
2.22.1 amenorrhoea (women only) ‐ short term
In this subgroup we only found one relevant trial (n = 30) (Hoyberg 1993). There was no significant difference between perphenazine and any antipsychotic drugs (RR 3.00 CI 0.13 to 68.26, Analysis 2.22).
2.22. Analysis.
Comparison 2 PERPHENAZINE vs ANY ANTIPSYCHOTIC, Outcome 22 Other adverse events: 6. Endocrine.
2.22.2 amenorrhoea ‐ medium term
In this subgroup we only found one relevant trial (n = 47) (Lepola 1989). There was no significant difference between perphenazine and any antipsychotic drugs (RR 0.32 CI 0.04 to 2.85, Analysis 2.22).
2.22.3 gynaecomastia ‐ short term
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was no significant difference between perphenazine and any antipsychotic drugs (RR 0.15 CI 0.01 to 2.85, Analysis 2.22).
2.22.4 lactation ‐ short term
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was no significant difference between perphenazine and any antipsychotic drugs (RR 0.21 CI 0.01 to 4.30, Analysis 2.22).
2.22.5 lactation ‐ medium term
In this subgroup we only found one relevant trial (n = 47) (Lepola 1989). There was no significant difference between perphenazine and any antipsychotic drugs (RR 0.19 CI 0.01 to 3.80, Analysis 2.22).
2.22.6 menorrhagia (women only) ‐ short term
In this subgroup we only found one relevant trial (n = 30) (Hoyberg 1993). There was no significant difference between perphenazine and any antipsychotic drugs (RR 3.00 CI 0.13 to 68.26, Analysis 2.22).
2.23 Other adverse events: 7. Gastrointestinal
2.23.1 constipation ‐ short term
In this subgroup we found nine relevant trials (n = 1017). There was no significant difference between perphenazine and any antipsychotic drugs (RR 0.71 CI 0.40 to 1.28, Analysis 2.23). This subgroup had important levels of heterogeneity (Chi2 = 18.83; df = 8; P = 0.016; I2 = 58%).
2.23. Analysis.
Comparison 2 PERPHENAZINE vs ANY ANTIPSYCHOTIC, Outcome 23 Other adverse events: 7. Gastrointestinal.
2.23.2 diarrhoea ‐ short term
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was no significant difference between perphenazine and any antipsychotic drugs (RR 0.15 CI 0.01 to 2.85, Analysis 2.23).
2.23.3 diarrhoea ‐ long term
In this subgroup we only found one relevant trial (n = 60) (Naukkarinen 2000). There was no significant difference between perphenazine and any antipsychotic drugs (RR 1.00 CI 0.22 to 4.56, Analysis 2.23).
2.23.4 dyspepsia ‐ short term
In this subgroup we only found one relevant trial (n = 300) (Kane 2003). There was no significant difference between perphenazine and any antipsychotic drugs (RR 0.59 CI 0.27 to 1.30, Analysis 2.23).
2.23.5 gastroenteritis ‐ long term
In this subgroup we only found one relevant trial (n = 60) (Naukkarinen 2000). There was no significant difference between perphenazine and any antipsychotic drugs (RR 3.00 CI 0.13 to 70.83, Analysis 2.23).
2.23.6 icterus ‐ short term
In this subgroup we found three relevant trials (n = 450). There was no significant difference between perphenazine and any antipsychotic drugs (RR 0.33 CI 0.01 to 7.96, Analysis 2.23).
2.23.7 loss of appetite ‐ short term
In this subgroup we found six relevant trials (n = 699). There was no significant difference between perphenazine and any antipsychotic drugs (RR 0.96 CI 0.69 to 1.33, Analysis 2.23).
2.23.8 nausea and vomiting ‐ short term
In this subgroup we found seven relevant trials (n = 806). There was no significant difference between perphenazine and any antipsychotic drugs (RR 1.12 CI 0.75 to 1.67, Analysis 2.23).
2.23.9 nausea and vomiting ‐ medium term
In this subgroup we only found one relevant trial (n = 47) (Lepola 1989). There was no significant difference between perphenazine and any antipsychotic drugs (RR 0.32 CI 0.04 to 2.85, Analysis 2.23).
2.23.10 nausea and vomiting ‐ long term
In this subgroup we only found one relevant trial (n = 60) (Naukkarinen 2000). There was no significant difference between perphenazine and any antipsychotic drugs (RR 1.00 CI 0.07 to 15.26, Analysis 2.23).
2.23.11 weight loss ‐ short term
In this subgroup we found two relevant trials (n = 171). There was no significant difference between perphenazine and any antipsychotic drugs (RR 3.03 CI 0.75 to 12.20, Analysis 2.23).
2.23.12 weight gain ‐ short term
In this subgroup we found three relevant trials (n = 235). There was no significant difference between perphenazine and any antipsychotic drugs (RR 1.53 CI 0.29 to 8.10, Analysis 2.23). This subgroup had important levels of heterogeneity (Chi2 = 6.33; df = 2; P = 0.042; I2 = 68%).
2.23.13 weight gain ‐ long term
In this subgroup we only found one relevant trial (n = 1316) (CATIE 2005). There was a statistically significant difference (P = 0.02) favouring perphenazine over any antipsychotic drugs (RR 0.66 CI 0.46 to 0.95, Analysis 2.23).
2.24 Other adverse events: 8. Skin
2.24.1 cellulitis ‐ long term
In this subgroup we only found one relevant trial (n = 60) (Naukkarinen 2000). There was no significant difference between perphenazine and any antipsychotic drugs (RR 0.33 CI 0.01 to 7.87, Analysis 2.24).
2.24. Analysis.
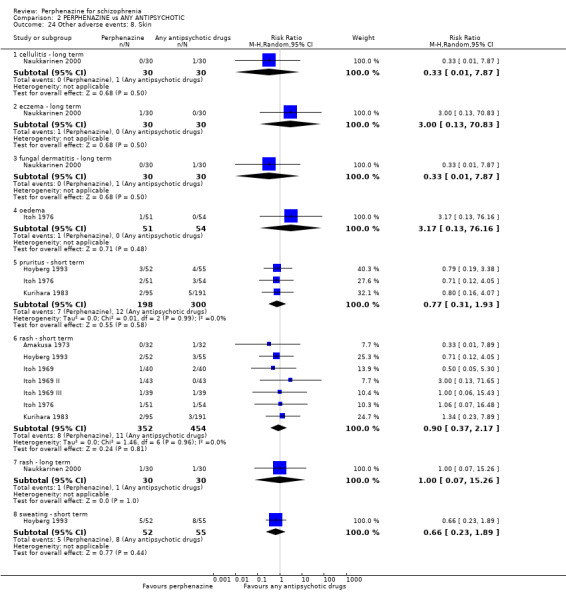
Comparison 2 PERPHENAZINE vs ANY ANTIPSYCHOTIC, Outcome 24 Other adverse events: 8. Skin.
2.24.2 eczema ‐ long term
In this subgroup we only found one relevant trial (n = 60) (Naukkarinen 2000). There was no significant difference between perphenazine and any antipsychotic drugs (RR 3.00 CI 0.13 to 70.83, Analysis 2.24).
2.24.3 fungal dermatitis ‐ long term
In this subgroup we only found one relevant trial (n = 60) (Naukkarinen 2000). There was no significant difference between perphenazine and any antipsychotic drugs (RR 0.33 CI 0.01 to 7.87, Analysis 2.24).
2.24.4 oedema
In this subgroup we only found one relevant trial (n = 105) (Itoh 1976). There was no significant difference between perphenazine and any antipsychotic drugs (RR 3.17 CI 0.13 to 76.16, Analysis 2.24).
2.24.5 pruritus ‐ short term
In this subgroup we found three relevant trials (n = 498). There was no significant difference between perphenazine and any antipsychotic drugs (RR 0.77 CI 0.31 to 1.93, Analysis 2.24).
2.24.6 rash ‐ short term
In this subgroup we found seven relevant trials (n = 806). There was no significant difference between perphenazine and any antipsychotic drugs (RR 0.90 CI 0.37 to 2.17, Analysis 2.24).
2.24.7 rash ‐ long term
In this subgroup we only found one relevant trial (n = 60) (Naukkarinen 2000). There was no significant difference between perphenazine and any antipsychotic drugs (RR 1.00 CI 0.07 to 15.26, Analysis 2.24).
2.24.8 sweating ‐ short term
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was no significant difference between perphenazine and any antipsychotic drugs (RR 0.66 CI 0.23 to 1.89, Analysis 2.24).
2.25 Other adverse events: 9. Genitourinary
2.25.1 diminished sexual desire
In this subgroup we found four relevant trials (n = 1958). There was no significant difference between perphenazine and any antipsychotic drugs (RR 1.03 CI 0.82 to 1.30, Analysis 2.25).
2.25. Analysis.
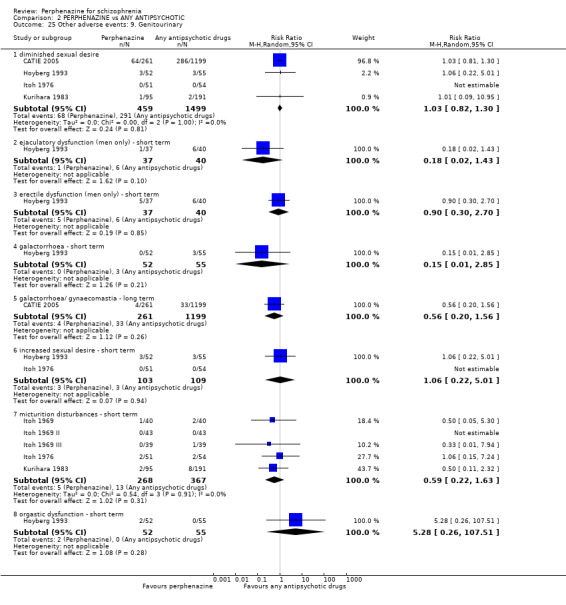
Comparison 2 PERPHENAZINE vs ANY ANTIPSYCHOTIC, Outcome 25 Other adverse events: 9. Genitourinary.
2.25.2 ejaculatory dysfunction (men only) ‐ short term
In this subgroup we only found one relevant trial (n = 77) (Hoyberg 1993). There was no significant difference between perphenazine and any antipsychotic drugs (RR 0.18 CI 0.02 to 1.43, Analysis 2.25).
2.25.3 erectile dysfunction (men only) ‐ short term
In this subgroup we only found one relevant trial (n = 77) (Hoyberg 1993). There was no significant difference between perphenazine and any antipsychotic drugs (RR 0.90 CI 0.30 to 2.70, Analysis 2.25).
2.25.4 galactorrhoea ‐ short term
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was no significant difference between perphenazine and any antipsychotic drugs (RR 0.15 CI 0.01 to 2.85, Analysis 2.25).
2.25.5 galactorrhoea/gynaecomastia ‐ long term
In this subgroup we only found one relevant trial (n = 1460) (CATIE 2005). There was no significant difference between perphenazine and any antipsychotic drugs (RR 0.56 CI 0.20 to 1.56, Analysis 2.25).
2.25.6 increased sexual desire ‐ short term
In this subgroup we found two relevant trials (n = 212). There was no significant difference between perphenazine and any antipsychotic drugs (RR 1.06 CI 0.22 to 5.01, Analysis 2.25).
2.25.7 micturition disturbances ‐ short term
In this subgroup we found five relevant trials (n = 635). There was no significant difference between perphenazine and any antipsychotic drugs (RR 0.59 CI 0.22 to 1.63, Analysis 2.25).
2.25.8 orgastic dysfunction ‐ short term
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was no significant difference between perphenazine and any antipsychotic drugs (RR 5.28 CI 0.26 to 107.51, Analysis 2.25).
2.26 Other adverse events: 10. Others
2.26.1 abdominal pain ‐ medium term
In this subgroup we only found one relevant trial (n = 47) (Lepola 1989). There was no significant difference between perphenazine and any antipsychotic drugs (RR 0.14 CI 0.01 to 2.52, Analysis 2.26).
2.26. Analysis.
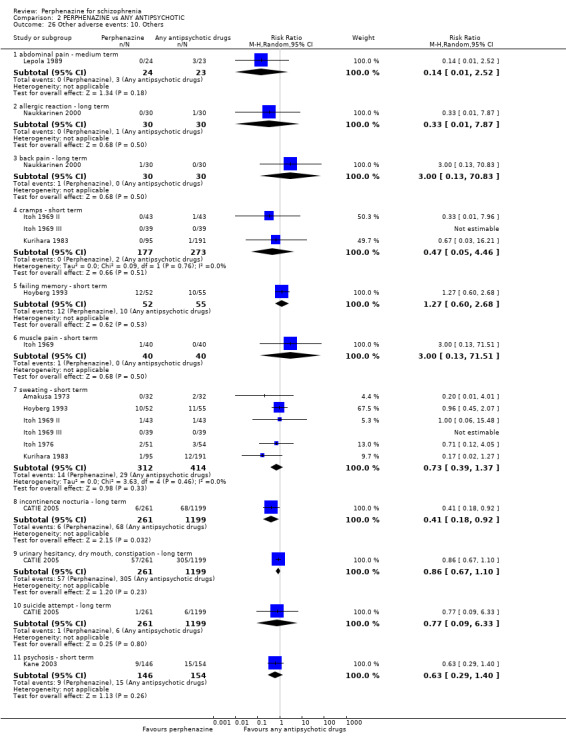
Comparison 2 PERPHENAZINE vs ANY ANTIPSYCHOTIC, Outcome 26 Other adverse events: 10. Others.
2.26.2 allergic reaction ‐ long term
In this subgroup we only found one relevant trial (n = 60) (Naukkarinen 2000). There was no significant difference between perphenazine and any antipsychotic drugs (RR 0.33 CI 0.01 to 7.87, Analysis 2.26).
2.26.3 back pain ‐ long term
In this subgroup we only found one relevant trial (n = 60) (Naukkarinen 2000). There was no significant difference between perphenazine and any antipsychotic drugs (RR 3.00 CI 0.13 to 70.83, Analysis 2.26).
2.26.4 cramps ‐ short term
In this subgroup we found three relevant trials (n = 450). There was no significant difference between perphenazine and any antipsychotic drugs (RR 0.47 CI 0.05 to 4.46, Analysis 2.26).
2.26.5 failing memory ‐ short term
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was no significant difference between perphenazine and any antipsychotic drugs (RR 1.27 CI 0.60 to 2.68, Analysis 2.26).
2.26.6 muscle pain ‐ short term
In this subgroup we only found one relevant trial (n = 80) (Itoh 1969). There was no significant difference between perphenazine and any antipsychotic drugs (RR 3.00 CI 0.13 to 71.51, Analysis 2.26).
2.26.7 sweating ‐ short term
In this subgroup we found six relevant trials (n = 726). There was no significant difference between perphenazine and any antipsychotic drugs (RR 0.73 CI 0.39 to 1.37, Analysis 2.26).
2.26.8 incontinence nocturia ‐ long term
In this subgroup we only found one relevant trial (n = 1460) (CATIE 2005). There was a statistically significant difference (P = 0.03) favouring perphenazine over any antipsychotic drugs (RR 0.41 CI 0.18 to 0.92, Analysis 2.26).
2.26.9 urinary hesitancy, dry mouth, constipation ‐ long term
In this subgroup we only found one relevant trial (n = 1460) (CATIE 2005). There was no significant difference between perphenazine and any antipsychotic drugs (RR 0.86 CI 0.67 to 1.10, Analysis 2.26).
2.26.10 suicide attempt ‐ long term
In this subgroup we only found one relevant trial (n = 1460) (CATIE 2005). There was no significant difference between perphenazine and any antipsychotic drugs (RR 0.77 CI 0.09 to 6.33, Analysis 2.26).
2.26.11 psychosis ‐ short term
In this subgroup we only found one relevant trial (n = 300) (Kane 2003). There was no significant difference between perphenazine and any antipsychotic drugs (RR 0.63 CI 0.29 to 1.40, Analysis 2.26).
2.27 Other adverse events: 11. Lab data
2.27.1 abnormal EKG/ECG ‐ short term
In this subgroup we found two relevant trials (n = 211). There was no significant difference between perphenazine and any antipsychotic drugs (RR 2.44 CI 0.62 to 9.59, Analysis 2.27). This subgroup had important levels of heterogeneity (Chi2 = 2.06; df = 1; P = 0.151; I2 = 51%).
2.27. Analysis.
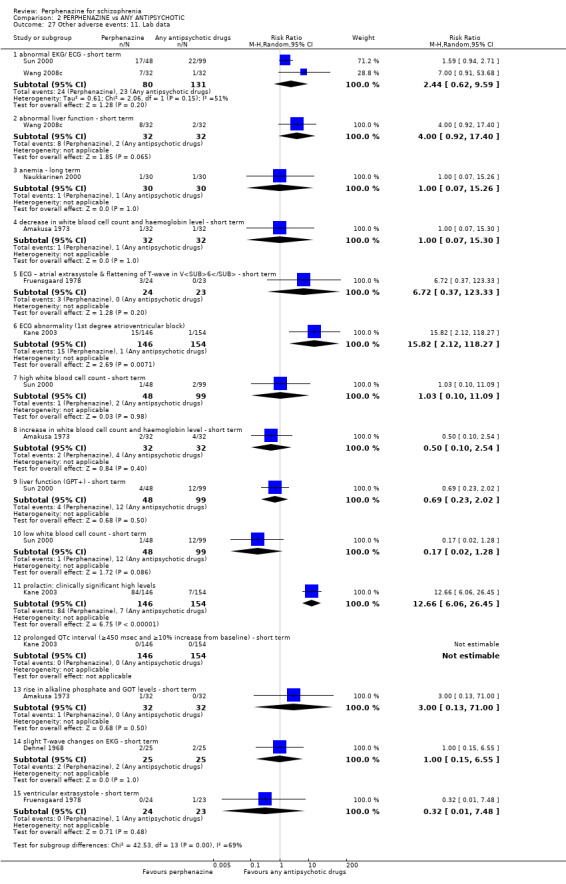
Comparison 2 PERPHENAZINE vs ANY ANTIPSYCHOTIC, Outcome 27 Other adverse events: 11. Lab data.
2.27.2 abnormal liver function ‐ short term
In this subgroup we only found one relevant trial (n = 64) (Wang 2008c). There was no significant difference between perphenazine and any antipsychotic drugs (RR 4.00 CI 0.92 to 17.40, Analysis 2.27).
2.27.3 anaemia ‐ long term
In this subgroup we only found one relevant trial (n = 60) (Naukkarinen 2000). There was no significant difference between perphenazine and any antipsychotic drugs (RR 1.00 CI 0.07 to 15.26, Analysis 2.27).
2.27.4 decrease in white blood cell count and haemoglobin level ‐ short term
In this subgroup we only found one relevant trial (n = 64) (Amakusa 1973). There was no significant difference between perphenazine and any antipsychotic drugs (RR 1.00 CI 0.07 to 15.30, Analysis 2.27).
2.27.5 ECG ‐ atrial extrasystole & flattening of T‐wave in V6 ‐ short term
In this subgroup we only found one relevant trial (n = 47) (Fruensgaard 1978). There was no significant difference between perphenazine and any antipsychotic drugs (RR 6.72 CI 0.37 to 123.33, Analysis 2.27).
2.27.6 ECG abnormality (1st degree atrioventricular block)
In this subgroup we only found one relevant trial (n = 300) (Kane 2003). There was a statistically significant difference (P = 0.007) favouring perphenazine over any antipsychotic drugs (RR 15.82 CI 2.12 to 118.27, Analysis 2.27).
2.27.7 high white blood cell count ‐ short term
In this subgroup we only found one relevant trial (n = 147) (Sun 2000). There was no significant difference between perphenazine and any antipsychotic drugs (RR 1.03 CI 0.10 to 11.09, Analysis 2.27).
2.27.8 increase in white blood cell count and haemoglobin level ‐ short term
In this subgroup we only found one relevant trial (n = 64) (Amakusa 1973). There was no significant difference between perphenazine and any antipsychotic drugs (RR 0.50 CI 0.10 to 2.54, Analysis 2.27).
2.27.9 liver function (GPT+) ‐ short term
In this subgroup we only found one relevant trial (n = 147) (Sun 2000). There was no significant difference between perphenazine and any antipsychotic drugs (RR 0.69 CI 0.23 to 2.02, Analysis 2.27).
2.27.10 low white blood cell count ‐ short term
In this subgroup we only found one relevant trial (n = 147) (Sun 2000). There was no significant difference between perphenazine and any antipsychotic drugs (RR 0.17 CI 0.02 to 1.28, Analysis 2.27).
2.27.11 prolactin: clinically significant high levels
In this subgroup we only found one relevant trial (n = 300) (Kane 2003). There was a statistically significant difference (P < 0.00001) favouring any antipsychotic drugs over perphenazine (RR 12.66 CI 6.06 to 26.45, Analysis 2.27).
2.27.12 prolonged QTc interval (≥450 msec and ≥10% increase from baseline) ‐ short term
In this subgroup we only found one relevant trial (n = 300) (Kane 2003). There was no significant difference between perphenazine and any antipsychotic drugs, with no events reported in either group (Analysis 2.27).
2.27.13 rise in alkaline phosphate and GOT levels ‐ short term
In this subgroup we only found one relevant trial (n = 64) (Amakusa 1973). There was no significant difference between perphenazine and any antipsychotic drugs (RR 3.00 CI 0.13 to 71.00, Analysis 2.27).
2.27.14 slight T‐wave changes on EKG ‐ short term
In this subgroup we only found one relevant trial (n = 50) (Dehnel 1968). There was no significant difference between perphenazine and any antipsychotic drugs (RR 1.00 CI 0.15 to 6.55, Analysis 2.27).
2.27.15 ventricular extrasystole ‐ short term
In this subgroup we only found one relevant trial (n = 47) (Fruensgaard 1978). There was no significant difference between perphenazine and any antipsychotic drugs (RR 0.32 CI 0.01 to 7.48, Analysis 2.27).
2.28 Other adverse events: 11a. Lab data (skew)
2.28.1 weight change
Data for this outcome are skewed and are best inspected by viewing Analysis 2.28.
2.28. Analysis.
Comparison 2 PERPHENAZINE vs ANY ANTIPSYCHOTIC, Outcome 28 Other adverse events: 11a. Lab data (skew).
| Other adverse events: 11a. Lab data (skew) | ||||
|---|---|---|---|---|
| Study | Intervention | Mean | SD | N |
| weight change | ||||
| Naukkarinen 2000 | Perphenazine | 0.37 | 3.19 | 23 |
| Naukkarinen 2000 | Olanzapine | 2.63 | 4.97 | 23 |
2.29 Other adverse events: 12. Any serious adverse event
Overall there was no significant difference between groups for serious adverse events (2 RCTs, n = 1760, RR 0.98 CI 0.68 to 1.41 (Analysis 2.29).
2.29. Analysis.
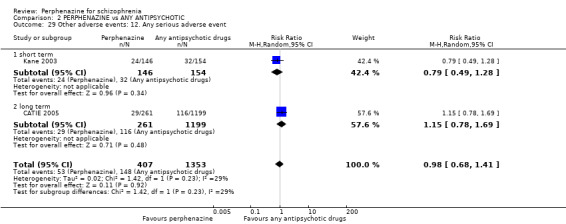
Comparison 2 PERPHENAZINE vs ANY ANTIPSYCHOTIC, Outcome 29 Other adverse events: 12. Any serious adverse event.
2.29.1 short term
In this subgroup we only found one relevant trial (n = 300) (Kane 2003). There was no significant difference between perphenazine and any antipsychotic drugs (RR 0.79 CI 0.49 to 1.28).
2.29.2 long term
In this subgroup we only found one relevant trial (n = 1460) (CATIE 2005). There was no significant difference between perphenazine and any antipsychotic drugs (RR 1.15 CI 0.78 to 1.69).
COMPARISON 3: PERPHENAZINE versus ANY ANTIPSYCHOTICS (ACUTE)
3.1 Dichotomous outcomes for acute participants
3.1.1 Leaving the study early: 1. Any reason
In this subgroup we found seven relevant trials (n = 447). There was a statistically significant difference (P = 0.007) between perphenazine and any antipsychotic drugs (acute) (RR 1.25 CI 1.06 to 1.47, Analysis 3.1).
3.1. Analysis.
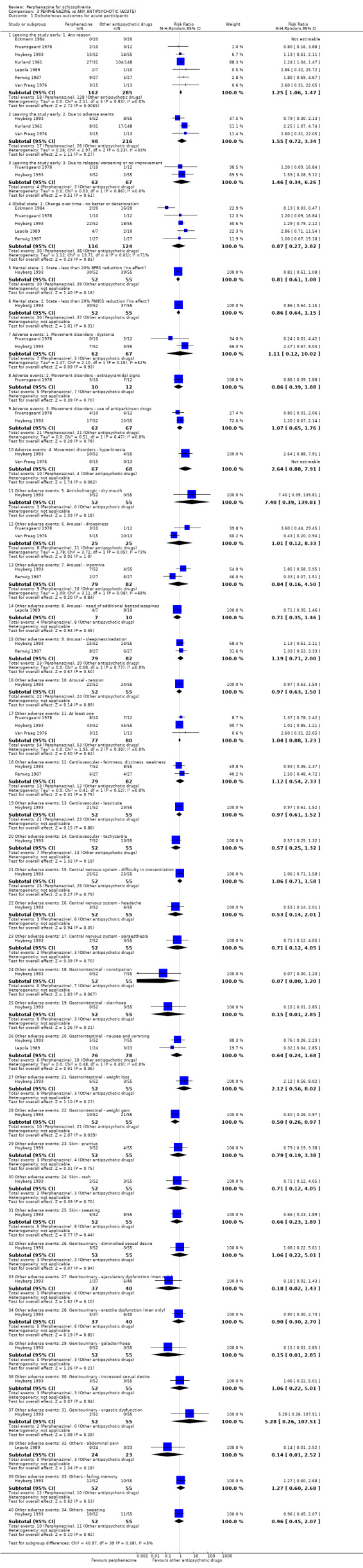
Comparison 3 PERPHENAZINE vs ANY ANTIPSYCHOTIC (ACUTE), Outcome 1 Dichotomous outcomes for acute participants.
3.1.2 Leaving the study early: 2. Due to adverse events
In this subgroup we found three relevant trials (n = 314). There was no significant difference between perphenazine and any antipsychotic drugs (acute) (RR 1.55 CI 0.72 to 3.34, Analysis 3.1). This subgroup had moderate levels of heterogeneity (Chi2 = 2.97; df = 2; P = 0.226; I2 = 33%).
3.1.3 Leaving the study early: 3. Due to relapse/worsening or no improvement
In this subgroup we found two relevant trials (n = 129). There was no significant difference between perphenazine and any antipsychotic drugs (acute) (RR 1.46 CI 0.34 to 6.26, Analysis 3.1).
3.1.4 Global state: 1. Change over time ‐ no better or deterioration
In this subgroup we found five relevant trials (n = 240). There was no significant difference between perphenazine and any antipsychotic drugs (acute) (RR 0.87 CI 0.27 to 2.82, Analysis 3.1). This subgroup had important levels of heterogeneity (Chi2 = 13.71; df = 4; P = 0.008; I2 = 71%).
3.1.5 Mental state: 1. State ‐ less than 20% BPRS reduction ('no effect')
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was no significant difference between perphenazine and any antipsychotic drugs (acute) (RR 0.81 CI 0.61 to 1.08, Analysis 3.1).
3.1.6 Mental state: 2. State ‐ less than 20% PANSS reduction ('no effect')
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was no significant difference between perphenazine and any antipsychotic drugs (acute) (RR 0.86 CI 0.64 to 1.15, Analysis 3.1).
3.1.7 Adverse events: 1. Movement disorders ‐ dystonia
In this subgroup we found two relevant trials (n = 129). There was no significant difference between perphenazine and any antipsychotic drugs (acute) (RR 1.11 CI 0.12 to 10.02, Analysis 3.1). This subgroup had important levels of heterogeneity (Chi2 = 2.1; df = 1; P = 0.147; I2 = 52%).
3.1.8 Adverse events: 2. Movement disorders ‐ extrapyramidal signs
In this subgroup we only found one relevant trial (n = 22) (Fruensgaard 1978). There was no significant difference between perphenazine and any antipsychotic drugs (acute) (RR 0.86 CI 0.39 to 1.88, Analysis 3.1).
3.1.9 Adverse events: 3. Movement disorders ‐ use of antiparkinson drugs
In this subgroup we found two relevant trials (n = 129). There was no significant difference between perphenazine and any antipsychotic drugs (acute) (RR 1.07 CI 0.65 to 1.76, Analysis 3.1).
3.1.10 Adverse events: 4. Movement disorders ‐ hyperkinesia
In this subgroup we found two relevant trials (n = 135). There was no significant difference between perphenazine and any antipsychotic drugs (acute) (RR 2.64 CI 0.88 to 7.91, Analysis 3.1).
3.1.11 Other adverse events: 5. Anticholinergic ‐ dry mouth
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was no significant difference between perphenazine and any antipsychotic drugs (acute) (RR 7.40 CI 0.39 to 139.81, Analysis 3.1).
3.1.12 Other adverse events: 6. Arousal ‐ drowsiness
In this subgroup we found two relevant trials (n = 50). There was no significant difference between perphenazine and any antipsychotic drugs (acute) (RR 1.01 CI 0.12 to 8.33, Analysis 3.1). This subgroup had important levels of heterogeneity (Chi2 = 3.72; df = 1; P = 0.054; I2 = 73%).
3.1.13 Other adverse events: 7. Arousal ‐ insomnia
In this subgroup we found two relevant trials (n = 161). There was no significant difference between perphenazine and any antipsychotic drugs (acute) (RR 0.84 CI 0.16 to 4.50, Analysis 3.1). This subgroup had important levels of heterogeneity (Chi2 = 3.11; df = 1; P = 0.078; I2 = 68%).
3.1.14 Other adverse events: 8. Arousal ‐ need of additional benzodiazepines
In this subgroup we only found one relevant trial (n = 17) (Lepola 1989). There was no significant difference between perphenazine and any antipsychotic drugs (acute) (RR 0.71 CI 0.35 to 1.46, Analysis 3.1).
3.1.15 Other adverse events: 9. Arousal ‐ sleepiness/sedation
In this subgroup we found two relevant trials (n = 161). There was no significant difference between perphenazine and any antipsychotic drugs (acute) (RR 1.19 CI 0.71 to 2.00, Analysis 3.1).
3.1.16 Other adverse events: 10. Arousal ‐ tension
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was no significant difference between perphenazine and any antipsychotic drugs (acute) (RR 0.97 CI 0.63 to 1.50, Analysis 3.1).
3.1.17 Other adverse events: 11. At least one
In this subgroup we found three relevant trials (n = 157). There was no significant difference between perphenazine and any antipsychotic drugs (acute) (RR 1.04 CI 0.88 to 1.23, Analysis 3.1).
3.1.18 Other adverse events: 12. Cardiovascular ‐ faintness, dizziness, weakness
In this subgroup we found two relevant trials (n = 161). There was no significant difference between perphenazine and any antipsychotic drugs (acute) (RR 1.12 CI 0.54 to 2.33, Analysis 3.1).
3.1.19 Other adverse events: 13. Cardiovascular ‐ lassitude
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was no significant difference between perphenazine and any antipsychotic drugs (acute) (RR 0.97 CI 0.61 to 1.52, Analysis 3.1).
3.1.20 Other adverse events: 14. Cardiovascular ‐ tachycardia
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was no significant difference between perphenazine and any antipsychotic drugs (acute) (RR 0.57 CI 0.25 to 1.32, Analysis 3.1).
3.1.21 Other adverse events: 15. Central nervous system ‐ difficulty in concentration
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was no significant difference between perphenazine and any antipsychotic drugs (acute) (RR 1.06 CI 0.71 to 1.58, Analysis 3.1).
3.1.22 Other adverse events: 16. Central nervous system ‐ headache
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was no significant difference between perphenazine and any antipsychotic drugs (acute) (RR 0.53 CI 0.14 to 2.01, Analysis 3.1).
3.1.23 Other adverse events: 17. Central nervous system ‐ paraesthesia
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was no significant difference between perphenazine and any antipsychotic drugs (acute) (RR 0.71 CI 0.12 to 4.05, Analysis 3.1).
3.1.24 Other adverse events: 18. Gastrointestinal ‐ constipation
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was no significant difference between perphenazine and any antipsychotic drugs (acute) (RR 0.07 CI 0.00 to 1.20, Analysis 3.1).
3.1.25 Other adverse events: 19. Gastrointestinal ‐ diarrhoea
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was no significant difference between perphenazine and any antipsychotic drugs (acute) (RR 0.15 CI 0.01 to 2.85, Analysis 3.1).
3.1.26 Other adverse events: 20. Gastrointestinal ‐ nausea and vomiting
In this subgroup we found two relevant trials (n = 154). There was no significant difference between perphenazine and any antipsychotic drugs (acute) (RR 0.64 CI 0.24 to 1.68, Analysis 3.1).
3.1.27 Other adverse events: 21. Gastrointestinal ‐ weight loss
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was no significant difference between perphenazine and any antipsychotic drugs (acute) (RR 2.12 CI 0.56 to 8.02, Analysis 3.1).
3.1.28 Other adverse events: 22. Gastrointestinal ‐ weight gain
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was a statistically significant difference (P = 0.04) favouring perphenazine over any antipsychotic drugs (acute) (RR 0.50 CI 0.26 to 0.97, Analysis 3.1).
3.1.29 Other adverse events: 23. Skin ‐ pruritus
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was no significant difference between perphenazine and any antipsychotic drugs (acute) (RR 0.79 CI 0.19 to 3.38, Analysis 3.1).
3.1.30 Other adverse events: 24. Skin ‐ rash
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was no significant difference between perphenazine and any antipsychotic drugs (acute) (RR 0.71 CI 0.12 to 4.05, Analysis 3.1).
3.1.31 Other adverse events: 25. Skin ‐ sweating
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was no significant difference between perphenazine and any antipsychotic drugs (acute) (RR 0.66 CI 0.23 to 1.89, Analysis 3.1).
3.1.32 Other adverse events: 26. Genitourinary ‐ diminished sexual desire
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was no significant difference between perphenazine and any antipsychotic drugs (acute) (RR 1.06 CI 0.22 to 5.01, Analysis 3.1).
3.1.33 Other adverse events: 27. Genitourinary ‐ ejaculatory dysfunction (men only)
In this subgroup we only found one relevant trial (n = 77) (Hoyberg 1993). There was no significant difference between perphenazine and any antipsychotic drugs (acute) (RR 0.18 CI 0.02 to 1.43, Analysis 3.1).
3.1.34 Other adverse events: 28. Genitourinary ‐ erectile dysfunction (men only)
In this subgroup we only found one relevant trial (n = 77) (Hoyberg 1993). There was no significant difference between perphenazine and any antipsychotic drugs (acute) (RR 0.90 CI 0.30 to 2.70, Analysis 3.1).
3.1.35 Other adverse events: 29. Genitourinary ‐ galactorrhoea
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was no significant difference between perphenazine and any antipsychotic drugs (acute) (RR 0.15 CI 0.01 to 2.85, Analysis 3.1).
3.1.36 Other adverse events: 30. Genitourinary ‐ increased sexual desire
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was no significant difference between perphenazine and any antipsychotic drugs (acute) (RR 1.06 CI 0.22 to 5.01, Analysis 3.1).
3.1.37 Other adverse events: 31. Genitourinary ‐ orgastic dysfunction
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was no significant difference between perphenazine and any antipsychotic drugs (acute) (RR 5.28 CI 0.26 to 107.51, Analysis 3.1).
3.1.38 Other adverse events: 32. Others ‐ abdominal pain
In this subgroup we only found one relevant trial (n = 47) (Lepola 1989). There was no significant difference between perphenazine and any antipsychotic drugs (acute) (RR 0.14 CI 0.01 to 2.52, Analysis 3.1).
3.1.39 Other adverse events: 33. Others ‐ failing memory
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was no significant difference between perphenazine and any antipsychotic drugs (acute) (RR 1.27 CI 0.60 to 2.68, Analysis 3.1).
3.1.40 Other adverse events: 34. Others ‐ sweating
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was no significant difference between perphenazine and any antipsychotic drugs (acute) (RR 0.96 CI 0.45 to 2.07, Analysis 3.1).
3.2 Continuous outcomes for acute participants
3.2.1 Mental state: 1. State ‐ PANSS: mean change from baseline to endpoint
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was no significant difference between perphenazine and any antipsychotic drugs (acute) (MD 4.00 CI ‐5.04 to 13.04, Analysis 3.2).
3.2. Analysis.
Comparison 3 PERPHENAZINE vs ANY ANTIPSYCHOTIC (ACUTE), Outcome 2 Continuous outcomes for acute participants.
COMPARISON 4: PERPHENAZINE versus ARIPIPRAZOLE
4.1 Leaving the study early: 1. Any reason
4.1.1 short term
In this subgroup we only found one relevant trial (n = 300) (Kane 2003). There was no significant difference between perphenazine and aripiprazole (RR 0.74 CI 0.50 to 1.11, Analysis 4.1).
4.1. Analysis.
Comparison 4 PERPHENAZINE vs ARIPIPRAZOLE, Outcome 1 Leaving the study early: 1. Any reason.
4.2 Leaving the study early: 2. Due to adverse events
4.2.1 short term
In this subgroup we only found one relevant trial (n = 300) (Kane 2003). There was no significant difference between perphenazine and aripiprazole (RR 0.53 CI 0.27 to 1.05, Analysis 4.2).
4.2. Analysis.
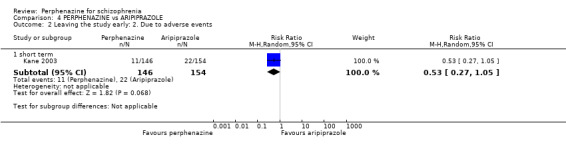
Comparison 4 PERPHENAZINE vs ARIPIPRAZOLE, Outcome 2 Leaving the study early: 2. Due to adverse events.
4.3 Leaving the study early: 3. Due to relapse/worsening or no improvement
4.3.1 short term
In this subgroup we only found one relevant trial (n = 300) (Kane 2003). There was no significant difference between perphenazine and aripiprazole (RR 0.84 CI 0.34 to 2.08, Analysis 4.3).
4.3. Analysis.
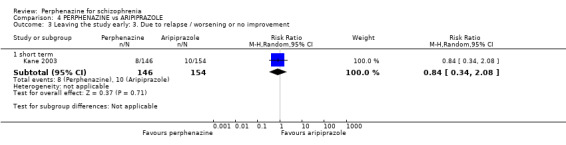
Comparison 4 PERPHENAZINE vs ARIPIPRAZOLE, Outcome 3 Leaving the study early: 3. Due to relapse / worsening or no improvement.
4.4 Global state: 1. Change over time ‐ no better or deterioration (ITT)
4.4.1 short term
In this subgroup we only found one relevant trial (n = 294) (Kane 2003). There was no significant difference between perphenazine and aripiprazole (RR 1.02 CI 0.89 to 1.17, Analysis 4.4).
4.4. Analysis.
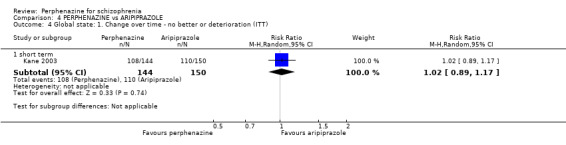
Comparison 4 PERPHENAZINE vs ARIPIPRAZOLE, Outcome 4 Global state: 1. Change over time ‐ no better or deterioration (ITT).
4.5 Global state: 2. Average endpoint score (CGI‐S)
4.5.1 short term
In this subgroup we only found one relevant trial (n = 294) (Kane 2003). There was no significant difference between perphenazine and aripiprazole (MD ‐0.20 CI ‐0.56 to 0.16, Analysis 4.5).
4.5. Analysis.
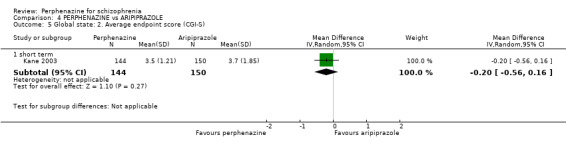
Comparison 4 PERPHENAZINE vs ARIPIPRAZOLE, Outcome 5 Global state: 2. Average endpoint score (CGI‐S).
4.6 Mental state: 2a. State ‐ BPRS score reduction ('no effect')
4.6.1 less than 25% reduction ‐ short term
In this subgroup we only found one relevant trial (n = 65) (Zhang 2010). There was no significant difference between perphenazine and aripiprazole (RR 1.45 CI 0.26 to 8.14, Analysis 4.6).
4.6. Analysis.
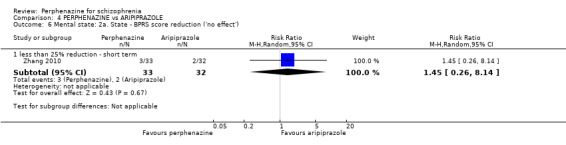
Comparison 4 PERPHENAZINE vs ARIPIPRAZOLE, Outcome 6 Mental state: 2a. State ‐ BPRS score reduction ('no effect').
4.7 Mental state: 2b. State ‐ PANSS: mean endpoint from baseline (high = poor)
4.7.1 total ‐ short term
In this subgroup we only found one relevant trial (n = 65) (Zhang 2010). There was no significant difference between perphenazine and aripiprazole (MD 1.38 CI ‐2.66 to 5.42, Analysis 4.7).
4.7. Analysis.
Comparison 4 PERPHENAZINE vs ARIPIPRAZOLE, Outcome 7 Mental state: 2b. State ‐ PANSS: mean endpoint from baseline (high = poor).
4.7.2 positive ‐ short term
In this subgroup we only found one relevant trial (n = 65) (Zhang 2010). There was no significant difference between perphenazine and aripiprazole (MD 0.63 CI ‐0.80 to 2.06, Analysis 4.7).
4.7.3 negative ‐ short term
In this subgroup we only found one relevant trial (n = 65) (Zhang 2010). There was no significant difference between perphenazine and aripiprazole (MD 0.33 CI ‐1.46 to 2.12, Analysis 4.7).
4.8 Mental state: 2c. State ‐ PANSS total change score (high = poor, skewed data)
4.8.1 short term
Data for this outcome are skewed and are best inspected by viewing Analysis 4.8.
4.8. Analysis.
Comparison 4 PERPHENAZINE vs ARIPIPRAZOLE, Outcome 8 Mental state: 2c. State ‐ PANSS total change score (high = poor, skewed data).
| Mental state: 2c. State ‐ PANSS total change score (high = poor, skewed data) | ||||
|---|---|---|---|---|
| Study | Intervention | Mean | SD | N |
| short term | ||||
| Kane 2003 | Perphenazine | ‐10.5 | 21.24 | 144 |
| Kane 2003 | Aripiprazole | ‐9.8 | 21.07 | 150 |
4.9 Adverse events: Movement disorders
4.9.1 akathisia ‐ short term
In this subgroup we only found one relevant trial (n = 300) (Kane 2003). There was no significant difference between perphenazine and aripiprazole (RR 2.29 CI 0.89 to 5.85, Analysis 4.9).
4.9. Analysis.
Comparison 4 PERPHENAZINE vs ARIPIPRAZOLE, Outcome 9 Adverse events: Movement disorders.
4.9.2 EPS ‐ short term
In this subgroup we found two relevant trials (n = 365). There was no significant difference between perphenazine and aripiprazole (RR 1.65 CI 0.70 to 3.88, Analysis 4.9).
4.9.3 use of antiparkinsonian drugs ‐ short term
In this subgroup we only found one relevant trial (n = 300) (Kane 2003). There was a statistically significant difference (P = 0.04) favouring aripiprazole over perphenazine (RR 1.56 CI 1.01 to 2.41, Analysis 4.9).
4.10 Adverse events: Movement disorders ‐ TESS total score (high = poor, skewed data)
4.10.1 short term
Data for this outcome are skewed and are best inspected by viewing Analysis 4.10.
4.10. Analysis.
Comparison 4 PERPHENAZINE vs ARIPIPRAZOLE, Outcome 10 Adverse events: Movement disorders ‐ TESS total score (high = poor, skewed data).
| Adverse events: Movement disorders ‐ TESS total score (high = poor, skewed data) | ||||
|---|---|---|---|---|
| Study | Intervention | Mean | SD | N |
| short term | ||||
| Zhang 2010 | Perphenazine | 5.13 | 4.21 | 33 |
| Zhang 2010 | Aripiprazole | 4.26 | 3.75 | 32 |
4.11 Other adverse events: 1. Arousal
4.11.1 agitation ‐ short term
In this subgroup we only found one relevant trial (n = 300) (Kane 2003). There was no significant difference between perphenazine and aripiprazole (RR 1.05 CI 0.63 to 1.77, Analysis 4.11).
4.11. Analysis.
Comparison 4 PERPHENAZINE vs ARIPIPRAZOLE, Outcome 11 Other adverse events: 1. Arousal.
4.11.2 anxiety ‐ short term
In this subgroup we only found one relevant trial (n = 300) (Kane 2003). There was no significant difference between perphenazine and aripiprazole (RR 1.19 CI 0.63 to 2.24, Analysis 4.11).
4.11.3 insomnia ‐ short term
In this subgroup we only found one relevant trial (n = 300) (Kane 2003). There was no significant difference between perphenazine and aripiprazole (RR 0.86 CI 0.56 to 1.31, Analysis 4.11).
4.11.4 sleepiness/sedation ‐ short term
In this subgroup we only found one relevant trial (n = 300) (Kane 2003). There was no significant difference between perphenazine and aripiprazole (RR 2.64 CI 0.85 to 8.22, Analysis 4.11).
4.12 Other adverse events: 2. Cardiovascular
4.12.1 faintness, dizziness, weakness ‐ short term
In this subgroup we only found one relevant trial (n = 300) (Kane 2003). There was a statistically significant difference (P = 0.03) favouring aripiprazole over perphenazine (RR 5.27 CI 1.18 to 23.66, Analysis 4.12).
4.12. Analysis.
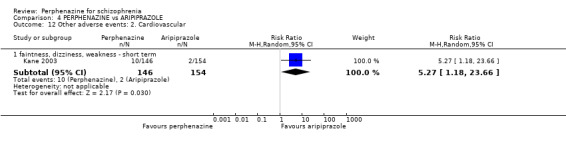
Comparison 4 PERPHENAZINE vs ARIPIPRAZOLE, Outcome 12 Other adverse events: 2. Cardiovascular.
4.13 Other adverse events: 3. Central nervous system
4.13.1 headache ‐ short term
In this subgroup we only found one relevant trial (n = 300) (Kane 2003). There was no significant difference between perphenazine and aripiprazole (RR 0.55 CI 0.29 to 1.03, Analysis 4.13).
4.13. Analysis.
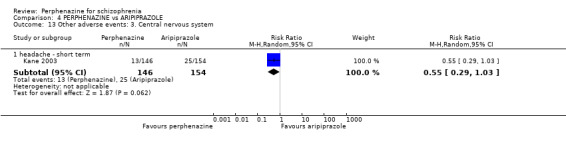
Comparison 4 PERPHENAZINE vs ARIPIPRAZOLE, Outcome 13 Other adverse events: 3. Central nervous system.
4.14 Other adverse events: 4. Gastrointestinal
4.14.1 dyspepsia ‐ short term
In this subgroup we only found one relevant trial (n = 300) (Kane 2003). There was no significant difference between perphenazine and aripiprazole (RR 0.59 CI 0.27 to 1.30, Analysis 4.14).
4.14. Analysis.
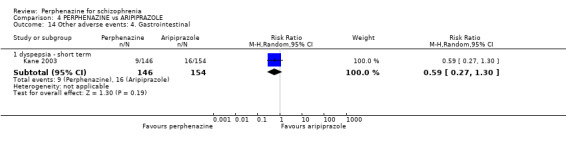
Comparison 4 PERPHENAZINE vs ARIPIPRAZOLE, Outcome 14 Other adverse events: 4. Gastrointestinal.
4.15 Other adverse events: 5. lab data
4.15.1 ECG abnormality (1st degree atrioventricular block)
In this subgroup we only found one relevant trial (n = 300) (Kane 2003). There was a statistically significant difference (P = 0.007) favouring aripiprazole over perphenazine (RR 15.82 CI 2.12 to 118.27, Analysis 4.15).
4.15. Analysis.
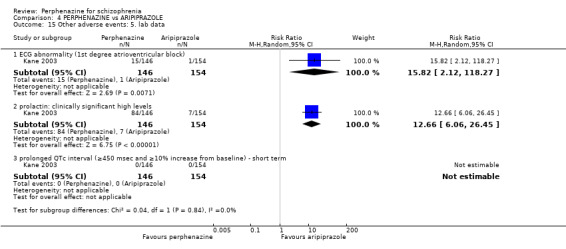
Comparison 4 PERPHENAZINE vs ARIPIPRAZOLE, Outcome 15 Other adverse events: 5. lab data.
4.15.2 prolactin: clinically significant high levels
In this subgroup we only found one relevant trial (n = 300) (Kane 2003). There was a statistically significant difference (P < 0.00001) favouring aripiprazole over perphenazine (RR 12.66 CI 6.06 to 26.45, Analysis 4.15).
4.15.3 prolonged QTc interval (≥ 450 msec and ≥ 10% increase from baseline) ‐ short term
In this subgroup we only found one relevant trial (n = 300) (Kane 2003). There was no significant difference between perphenazine and aripiprazole, with no events reported in either group (Analysis 4.15).
4.16 Other adverse events: 6. any serious adverse event
4.16.1 short term
In this subgroup we only found one relevant trial (n = 300) (Kane 2003). There was no significant difference between perphenazine and aripiprazole (RR 0.79 CI 0.49 to 1.28, Analysis 4.16).
4.16. Analysis.
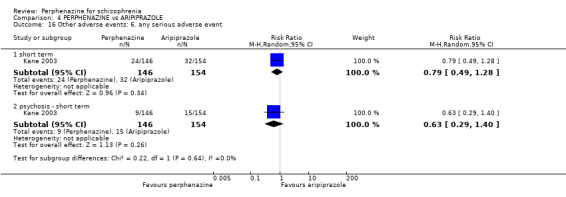
Comparison 4 PERPHENAZINE vs ARIPIPRAZOLE, Outcome 16 Other adverse events: 6. any serious adverse event.
4.16.2 psychosis ‐ short term
In this subgroup we only found one relevant trial (n = 300) (Kane 2003). There was no significant difference between perphenazine and aripiprazole (RR 0.63 CI 0.29 to 1.40, Analysis 4.16).
COMPARISON 5: PERPHENAZINE versus BENPERIDOL
5.1 Leaving the study early: 1. Any reason
5.1.1 short term
In this subgroup we only found one relevant trial (n = 40) (Eckmann 1984). There was no significant difference between perphenazine and benperidol, with no events reported in either group (Analysis 5.1).
5.1. Analysis.
Comparison 5 PERPHENAZINE vs BENPERIDOL, Outcome 1 Leaving the study early: 1. Any reason.
5.2 Global state: 1. Change over time ‐ no better or deterioration (ITT)
5.2.1 short term
In this subgroup we only found one relevant trial (n = 40) (Eckmann 1984). There was a statistically significant difference (P = 0.002) favouring perphenazine over benperidol (RR 0.13 CI 0.03 to 0.47, Analysis 5.2).
5.2. Analysis.
Comparison 5 PERPHENAZINE vs BENPERIDOL, Outcome 2 Global state: 1. Change over time ‐ no better or deterioration (ITT).
COMPARISON 6: PERPHENAZINE versus BROMPERIDOL
6.1 Leaving the study early: 1. Any reason
6.1.1 short term
In this subgroup we only found one relevant trial (n = 40) (Woggon 1978). There was no significant difference between perphenazine and bromperidol, with no events reported in either group (Analysis 6.1).
6.1. Analysis.
Comparison 6 PERPHENAZINE vs BROMPERIDOL, Outcome 1 Leaving the study early: 1. Any reason.
6.2 Global state: 1. Change over time ‐ no better or deterioration (ITT)
6.2.1 short term
In this subgroup we only found one relevant trial (n = 40) (Woggon 1978). There was no significant difference between perphenazine and bromperidol (RR 2.50 CI 0.94 to 6.66, Analysis 6.2).
6.2. Analysis.
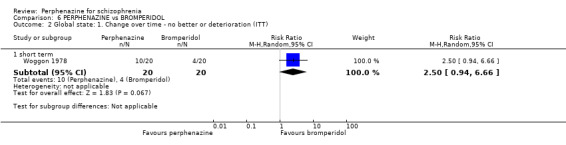
Comparison 6 PERPHENAZINE vs BROMPERIDOL, Outcome 2 Global state: 1. Change over time ‐ no better or deterioration (ITT).
6.3 Adverse events: Movement disorders
6.3.1 use of antiparkinsonian drugs ‐ short term
In this subgroup we only found one relevant trial (n = 40) (Woggon 1978). There was no significant difference between perphenazine and bromperidol (RR 0.80 CI 0.40 to 1.60, Analysis 6.3).
6.3. Analysis.
Comparison 6 PERPHENAZINE vs BROMPERIDOL, Outcome 3 Adverse events: Movement disorders.
6.4 Other adverse events: 1. Arousal
6.4.1 need of additional benzodiazepines ‐ short term
In this subgroup we only found one relevant trial (n = 40) (Woggon 1978). There was no significant difference between perphenazine and bromperidol (RR 1.10 CI 0.61 to 1.99, Analysis 6.4).
6.4. Analysis.
Comparison 6 PERPHENAZINE vs BROMPERIDOL, Outcome 4 Other adverse events: 1. Arousal.
COMPARISON 7: PERPHENAZINE versus CHLORPROMAZINE
7.1 Leaving the study early: 1. Any reason
7.1.1 short term
In this subgroup we found three relevant trials (n = 178). There was no significant difference between perphenazine and chlorpromazine (RR 1.10 CI 0.89 to 1.35, Analysis 7.1).
7.1. Analysis.
Comparison 7 PERPHENAZINE vs CHLORPROMAZINE, Outcome 1 Leaving the study early: 1. Any reason.
7.2 Leaving the study early: 2. Due to adverse events
7.2.1 short term
In this subgroup we found two relevant trials (n = 168). There was a statistically significant difference (P = 0.0002) favouring perphenazine over chlorpromazine (RR 0.33 CI 0.18 to 0.59, Analysis 7.2).
7.2. Analysis.
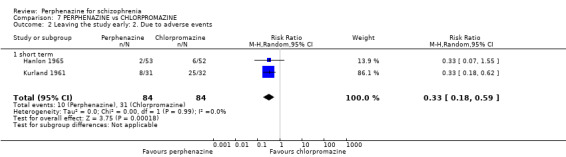
Comparison 7 PERPHENAZINE vs CHLORPROMAZINE, Outcome 2 Leaving the study early: 2. Due to adverse events.
7.3 Global state: 1. Change over time ‐ no better or deterioration (ITT)
7.3.1 short term
In this subgroup we only found one relevant trial (n = 63) (Kurland 1961). There was no significant difference between perphenazine and chlorpromazine (RR 0.57 CI 0.22 to 1.52, Analysis 7.3).
7.3. Analysis.
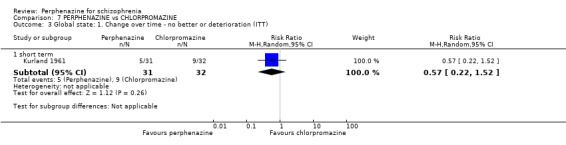
Comparison 7 PERPHENAZINE vs CHLORPROMAZINE, Outcome 3 Global state: 1. Change over time ‐ no better or deterioration (ITT).
7.4 Adverse events: Movement disorders
7.4.1 akathisia ‐ short term
In this subgroup we only found one relevant trial (n = 105) (Hanlon 1965). There was no significant difference between perphenazine and chlorpromazine (RR 6.87 CI 0.88 to 53.89, Analysis 7.4).
7.4. Analysis.
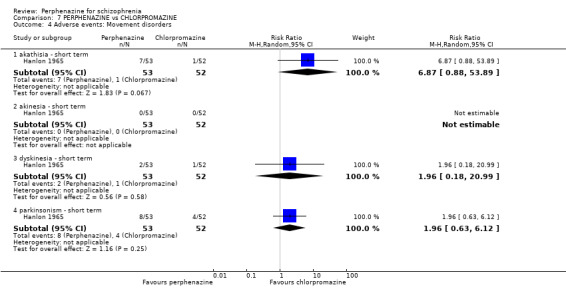
Comparison 7 PERPHENAZINE vs CHLORPROMAZINE, Outcome 4 Adverse events: Movement disorders.
7.4.2 akinesia ‐ short term
In this subgroup we only found one relevant trial (n = 105) (Hanlon 1965). There was no significant difference between perphenazine and chlorpromazine, with no events reported in either group (Analysis 7.4).
7.4.3 dyskinesia ‐ short term
In this subgroup we only found one relevant trial (n = 105) (Hanlon 1965). There was no significant difference between perphenazine and chlorpromazine (RR 1.96 CI 0.18 to 20.99, Analysis 7.4).
7.4.4 parkinsonism ‐ short term
In this subgroup we only found one relevant trial (n = 105) (Hanlon 1965). There was no significant difference between perphenazine and chlorpromazine (RR 1.96 CI 0.63 to 6.12, Analysis 7.4).
7.5 Other adverse events: 1. Central nervous system
7.5.1 electroencephalogram (increase) ‐ short term
In this subgroup we only found one relevant trial (n = 10) (Bennett 1961). There was no significant difference between perphenazine and chlorpromazine (RR 0.33 CI 0.05 to 2.21, Analysis 7.5).
7.5. Analysis.
Comparison 7 PERPHENAZINE vs CHLORPROMAZINE, Outcome 5 Other adverse events: 1. Central nervous system.
COMPARISON 8: PERPHENAZINE versus CLOCAPRAMINE
8.1 Leaving the study early: 1. Any reason
8.1.1 short term
In this subgroup we only found one relevant trial (n = 192) (Kurihara 1983). There was no significant difference between perphenazine and clocapramine (RR 1.40 CI 0.59 to 3.34, Analysis 8.1).
8.1. Analysis.
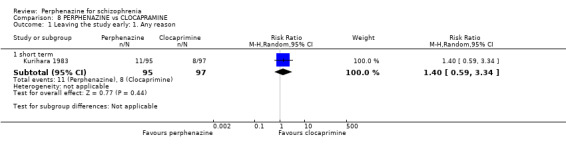
Comparison 8 PERPHENAZINE vs CLOCAPRAMINE, Outcome 1 Leaving the study early: 1. Any reason.
8.2 Leaving the study early: 2. Due to adverse events
8.2.1 short term
In this subgroup we only found one relevant trial (n = 192) (Kurihara 1983). There was no significant difference between perphenazine and clocapramine (RR 0.61 CI 0.15 to 2.49, Analysis 8.2).
8.2. Analysis.
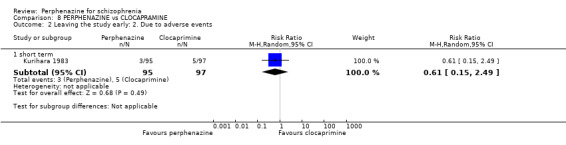
Comparison 8 PERPHENAZINE vs CLOCAPRAMINE, Outcome 2 Leaving the study early: 2. Due to adverse events.
8.3 Leaving the study early: 3. Due to relapse/worsening or no improvement
8.3.1 short term
In this subgroup we only found one relevant trial (n = 192) (Kurihara 1983). There was no significant difference between perphenazine and clocapramine (RR 2.38 CI 0.63 to 8.94, Analysis 8.3).
8.3. Analysis.
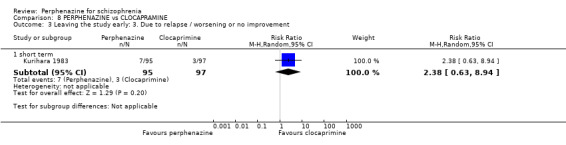
Comparison 8 PERPHENAZINE vs CLOCAPRAMINE, Outcome 3 Leaving the study early: 3. Due to relapse / worsening or no improvement.
8.4 Global state: 1. Change over time ‐ no better or deterioration (ITT)
8.4.1 short term
In this subgroup we only found one relevant trial (n = 192) (Kurihara 1983). There was no significant difference between perphenazine and clocapramine (RR 1.15 CI 0.88 to 1.49, Analysis 8.4).
8.4. Analysis.
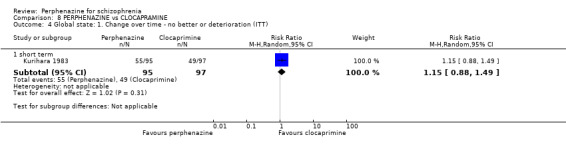
Comparison 8 PERPHENAZINE vs CLOCAPRAMINE, Outcome 4 Global state: 1. Change over time ‐ no better or deterioration (ITT).
8.5 Adverse events: Movement disorders
8.5.1 akathisia ‐ short term
In this subgroup we only found one relevant trial (n = 192) (Kurihara 1983). There was no significant difference between perphenazine and clocapramine (RR 0.84 CI 0.49 to 1.44, Analysis 8.5).
8.5. Analysis.
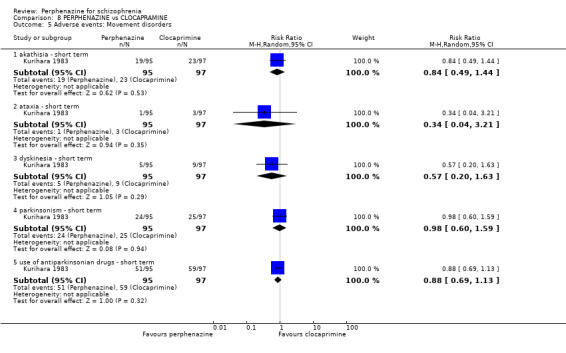
Comparison 8 PERPHENAZINE vs CLOCAPRAMINE, Outcome 5 Adverse events: Movement disorders.
8.5.2 ataxia ‐ short term
In this subgroup we only found one relevant trial (n = 192) (Kurihara 1983). There was no significant difference between perphenazine and clocapramine (RR 0.34 CI 0.04 to 3.21, Analysis 8.5).
8.5.3 dyskinesia ‐ short term
In this subgroup we only found one relevant trial (n = 192) (Kurihara 1983). There was no significant difference between perphenazine and clocapramine (RR 0.57 CI 0.20 to 1.63, Analysis 8.5).
8.5.4 parkinsonism ‐ short term
In this subgroup we only found one relevant trial (n = 192) (Kurihara 1983). There was no significant difference between perphenazine and clocapramine (RR 0.98 CI 0.60 to 1.59, Analysis 8.5).
8.5.5 use of antiparkinsonian drugs ‐ short term
In this subgroup we only found one relevant trial (n = 192) (Kurihara 1983). There was no significant difference between perphenazine and clocapramine (RR 0.88 CI 0.69 to 1.13, Analysis 8.5).
8.6 Other adverse events: 1. Anticholinergic
8.6.1 blurred vision ‐ short term
In this subgroup we only found one relevant trial (n = 192) (Kurihara 1983). There was no significant difference between perphenazine and clocapramine (RR 0.41 CI 0.08 to 2.05, Analysis 8.6).
8.6. Analysis.
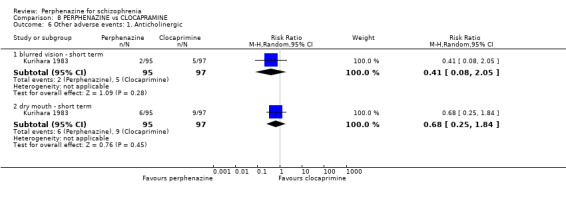
Comparison 8 PERPHENAZINE vs CLOCAPRAMINE, Outcome 6 Other adverse events: 1. Anticholinergic.
8.6.2 dry mouth ‐ short term
In this subgroup we only found one relevant trial (n = 192) (Kurihara 1983). There was no significant difference between perphenazine and clocapramine (RR 0.68 CI 0.25 to 1.84, Analysis 8.6).
8.7 Other adverse events: 2. Arousal
8.7.1 insomnia ‐ short term
In this subgroup we only found one relevant trial (n = 192) (Kurihara 1983). There was no significant difference between perphenazine and clocapramine (RR 1.16 CI 0.83 to 1.63, Analysis 8.7).
8.7. Analysis.
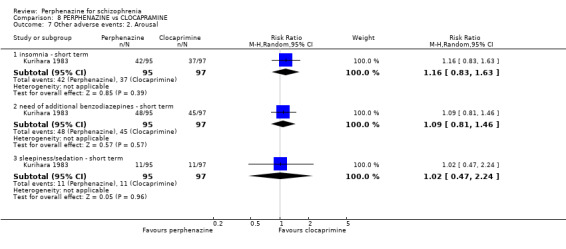
Comparison 8 PERPHENAZINE vs CLOCAPRAMINE, Outcome 7 Other adverse events: 2. Arousal.
8.7.2 need of additional benzodiazepines ‐ short term
In this subgroup we only found one relevant trial (n = 192) (Kurihara 1983). There was no significant difference between perphenazine and clocapramine (RR 1.09 CI 0.81 to 1.46, Analysis 8.7).
8.7.3 sleepiness/sedation ‐ short term
In this subgroup we only found one relevant trial (n = 192) (Kurihara 1983). There was no significant difference between perphenazine and clocapramine (RR 1.02 CI 0.47 to 2.24, Analysis 8.7).
8.8 Other adverse events: 3. At least one
8.8.1 short term
In this subgroup we only found one relevant trial (n = 192) (Kurihara 1983). There was no significant difference between perphenazine and clocapramine (RR 0.92 CI 0.78 to 1.07, Analysis 8.8).
8.8. Analysis.
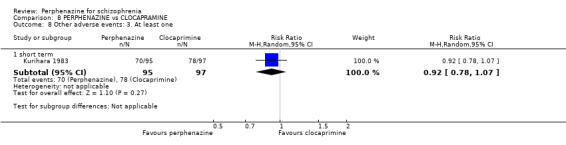
Comparison 8 PERPHENAZINE vs CLOCAPRAMINE, Outcome 8 Other adverse events: 3. At least one.
8.9 Other adverse events: 4. Cardiovascular
8.9.1 angina pectoris ‐ short term
In this subgroup we only found one relevant trial (n = 192) (Kurihara 1983). There was no significant difference between perphenazine and clocapramine (RR 0.68 CI 0.20 to 2.34, Analysis 8.9).
8.9. Analysis.
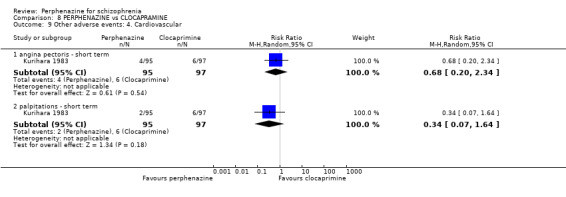
Comparison 8 PERPHENAZINE vs CLOCAPRAMINE, Outcome 9 Other adverse events: 4. Cardiovascular.
8.9.2 palpitations ‐ short term
In this subgroup we only found one relevant trial (n = 192) (Kurihara 1983). There was no significant difference between perphenazine and clocapramine (RR 0.34 CI 0.07 to 1.64, Analysis 8.9).
8.10 Other adverse events: 5. Central nervous system
8.10.1 disturbance of consciousness ‐ short term
In this subgroup we only found one relevant trial (n = 192) (Kurihara 1983). There was no significant difference between perphenazine and clocapramine, with no events reported in either group (Analysis 8.10).
8.10. Analysis.
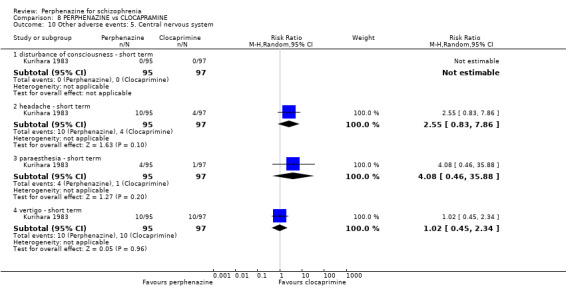
Comparison 8 PERPHENAZINE vs CLOCAPRAMINE, Outcome 10 Other adverse events: 5. Central nervous system.
8.10.2 headache ‐ short term
In this subgroup we only found one relevant trial (n = 192) (Kurihara 1983). There was no significant difference between perphenazine and clocapramine (RR 2.55 CI 0.83 to 7.86, Analysis 8.10).
8.10.3 paraesthesia ‐ short term
In this subgroup we only found one relevant trial (n = 192) (Kurihara 1983). There was no significant difference between perphenazine and clocapramine (RR 4.08 CI 0.46 to 35.88, Analysis 8.10).
8.10.4 vertigo ‐ short term
In this subgroup we only found one relevant trial (n = 192) (Kurihara 1983). There was no significant difference between perphenazine and clocapramine (RR 1.02 CI 0.45 to 2.34, Analysis 8.10).
8.11 Other adverse events: 6. Gastrointestinal
8.11.1 constipation ‐ short term
In this subgroup we only found one relevant trial (n = 192) (Kurihara 1983). There was no significant difference between perphenazine and clocapramine (RR 1.25 CI 0.54 to 2.87, Analysis 8.11).
8.11. Analysis.
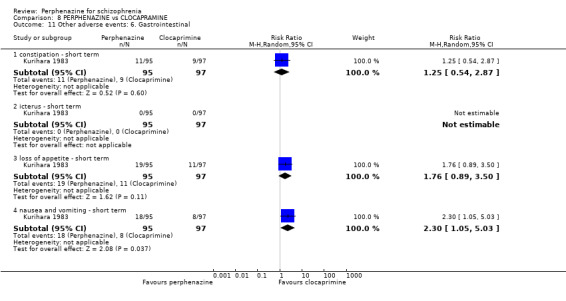
Comparison 8 PERPHENAZINE vs CLOCAPRAMINE, Outcome 11 Other adverse events: 6. Gastrointestinal.
8.11.2 icterus ‐ short term
In this subgroup we only found one relevant trial (n = 192) (Kurihara 1983). There was no significant difference between perphenazine and clocapramine, with no events reported in either group (Analysis 8.11).
8.11.3 loss of appetite ‐ short term
In this subgroup we only found one relevant trial (n = 192) (Kurihara 1983). There was no significant difference between perphenazine and clocapramine (RR 1.76 CI 0.89 to 3.50, Analysis 8.11).
8.11.4 nausea and vomiting ‐ short term
In this subgroup we only found one relevant trial (n = 192) (Kurihara 1983). There was a statistically significant difference (P = 0.04) favouring clocapramine over perphenazine (RR 2.30 CI 1.05 to 5.03, Analysis 8.11).
8.12 Other adverse events: 7. Skin
8.12.1 pruritus ‐ short term
In this subgroup we only found one relevant trial (n = 192) (Kurihara 1983). There was no significant difference between perphenazine and clocapramine (RR 1.02 CI 0.15 to 7.10, Analysis 8.12).
8.12. Analysis.
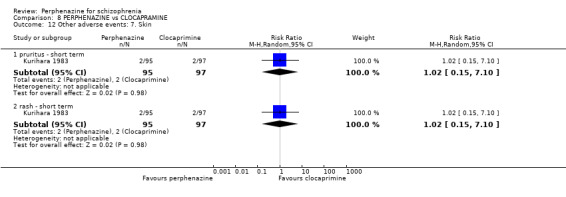
Comparison 8 PERPHENAZINE vs CLOCAPRAMINE, Outcome 12 Other adverse events: 7. Skin.
8.12.2 rash ‐ short term
In this subgroup we only found one relevant trial (n = 192) (Kurihara 1983). There was no significant difference between perphenazine and clocapramine (RR 1.02 CI 0.15 to 7.10, Analysis 8.12).
8.13 Other adverse events: 8. Genitourinary
8.13.1 diminished sexual desire
In this subgroup we only found one relevant trial (n = 192) (Kurihara 1983). There was no significant difference between perphenazine and clocapramine (RR 1.02 CI 0.06 to 16.09, Analysis 8.13).
8.13. Analysis.
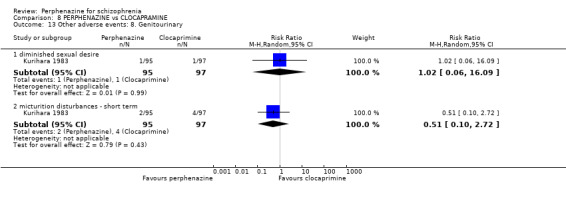
Comparison 8 PERPHENAZINE vs CLOCAPRAMINE, Outcome 13 Other adverse events: 8. Genitourinary.
8.13.2 micturition disturbances ‐ short term
In this subgroup we only found one relevant trial (n = 192) (Kurihara 1983). There was no significant difference between perphenazine and clocapramine (RR 0.51 CI 0.10 to 2.72, Analysis 8.13).
8.14 Other adverse events: 9. Others
8.14.1 cramps ‐ short term
In this subgroup we only found one relevant trial (n = 192) (Kurihara 1983). There was no significant difference between perphenazine and clocapramine, with no events reported in either group (Analysis 8.14).
8.14. Analysis.
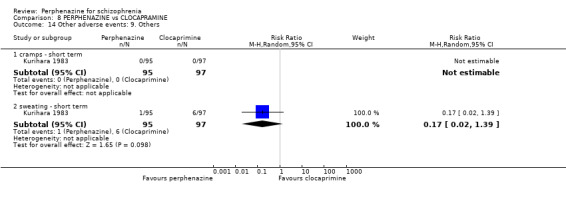
Comparison 8 PERPHENAZINE vs CLOCAPRAMINE, Outcome 14 Other adverse events: 9. Others.
8.14.2 sweating ‐ short term
In this subgroup we only found one relevant trial (n = 192) (Kurihara 1983). There was no significant difference between perphenazine and clocapramine (RR 0.17 CI 0.02 to 1.39, Analysis 8.14).
COMPARISON 9: PERPHENAZINE versus CLOPENTHIXOL
9.1 Leaving the study early: 1. Any reason
9.1.1 short term
In this subgroup we only found one relevant trial (n = 50) (Dehnel 1968). There was no significant difference between perphenazine and clopenthixol (RR 1.00 CI 0.15 to 6.55, Analysis 9.1).
9.1. Analysis.
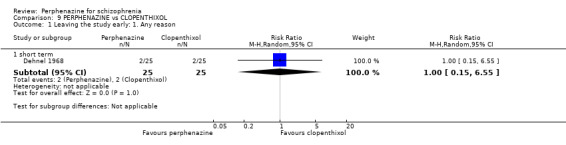
Comparison 9 PERPHENAZINE vs CLOPENTHIXOL, Outcome 1 Leaving the study early: 1. Any reason.
9.2 Global state: 1. Change over time ‐ no better or deterioration (ITT)
9.2.1 short term
In this subgroup we only found one relevant trial (n = 50) (Dehnel 1968). There was no significant difference between perphenazine and clopenthixol (RR 0.67 CI 0.21 to 2.08, Analysis 9.2).
9.2. Analysis.
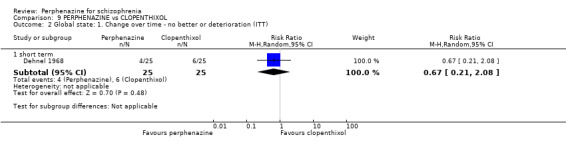
Comparison 9 PERPHENAZINE vs CLOPENTHIXOL, Outcome 2 Global state: 1. Change over time ‐ no better or deterioration (ITT).
9.3 Adverse events: Movement disorders
9.3.1 use of antiparkinsonian drugs ‐ short term
In this subgroup we only found one relevant trial (n = 50) (Dehnel 1968). There was a statistically significant difference (P = 0.02) favouring perphenazine over clopenthixol (RR 0.53 CI 0.31 to 0.89, Analysis 9.3).
9.3. Analysis.
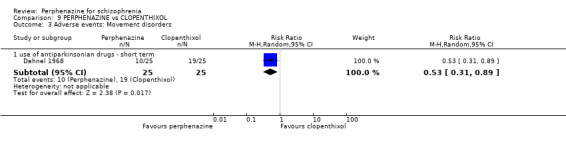
Comparison 9 PERPHENAZINE vs CLOPENTHIXOL, Outcome 3 Adverse events: Movement disorders.
9.4 Other adverse events: 1. Arousal
9.4.1 need of additional benzodiazepines ‐ short term
In this subgroup we only found one relevant trial (n = 50) (Dehnel 1968). There was no significant difference between perphenazine and clopenthixol (RR 0.50 CI 0.24 to 1.03, Analysis 9.4).
9.4. Analysis.
Comparison 9 PERPHENAZINE vs CLOPENTHIXOL, Outcome 4 Other adverse events: 1. Arousal.
9.5 Other adverse events: 2. lab data
9.5.1 slight T‐wave changes on EKG ‐ short term
In this subgroup we only found one relevant trial (n = 50) (Dehnel 1968). There was no significant difference between perphenazine and clopenthixol (RR 1.00 CI 0.15 to 6.55, Analysis 9.5).
9.5. Analysis.
Comparison 9 PERPHENAZINE vs CLOPENTHIXOL, Outcome 5 Other adverse events: 2. lab data.
COMPARISON 10: PERPHENAZINE versus CLOTHIAPINE
10.1 Leaving the study early: 1. Any reason
10.1.1 short term
In this subgroup we only found one relevant trial (n = 80) (Itoh 1969). There was no significant difference between perphenazine and clothiapine (RR 1.00 CI 0.21 to 4.66, Analysis 10.1).
10.1. Analysis.
Comparison 10 PERPHENAZINE vs CLOTHIAPINE, Outcome 1 Leaving the study early: 1. Any reason.
10.2 Leaving the study early: 2. Due to relapse/worsening or no improvement
10.2.1 short term
In this subgroup we only found one relevant trial (n = 80) (Itoh 1969). There was no significant difference between perphenazine and clothiapine (RR 1.00 CI 0.15 to 6.76, Analysis 10.2).
10.2. Analysis.
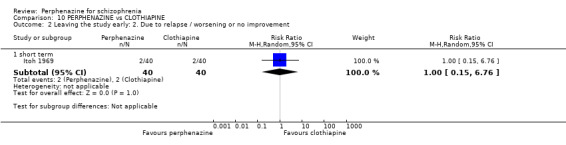
Comparison 10 PERPHENAZINE vs CLOTHIAPINE, Outcome 2 Leaving the study early: 2. Due to relapse / worsening or no improvement.
10.3 Global state: 1. Change over time ‐ no better or deterioration (ITT)
10.3.1 short term
In this subgroup we only found one relevant trial (n = 80) (Itoh 1969). There was no significant difference between perphenazine and clothiapine (RR 1.31 CI 0.81 to 2.12, Analysis 10.3).
10.3. Analysis.
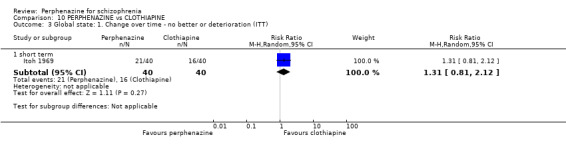
Comparison 10 PERPHENAZINE vs CLOTHIAPINE, Outcome 3 Global state: 1. Change over time ‐ no better or deterioration (ITT).
10.4 Behaviour: 1. Behaviour subscale of the Psychiatric Rating Scale: no better or worse
For this outcome we only found one relevant trial (n = 80) (Itoh 1969). There was no significant difference between perphenazine and clothiapine (RR 0.96 CI 0.69 to 1.34, Analysis 10.4).
10.4. Analysis.
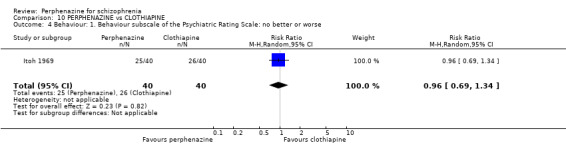
Comparison 10 PERPHENAZINE vs CLOTHIAPINE, Outcome 4 Behaviour: 1. Behaviour subscale of the Psychiatric Rating Scale: no better or worse.
10.5 Adverse events: Movement disorders
10.5.1 akathisia ‐ short term
In this subgroup we only found one relevant trial (n = 80) (Itoh 1969). There was no significant difference between perphenazine and clothiapine (RR 1.63 CI 0.76 to 3.49, Analysis 10.5).
10.5. Analysis.
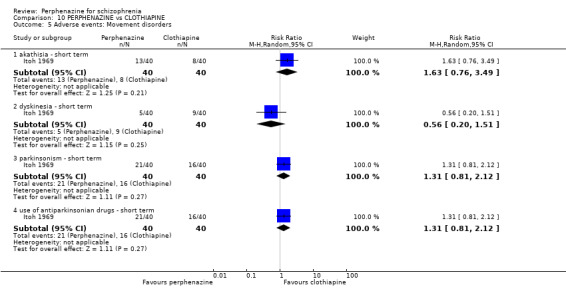
Comparison 10 PERPHENAZINE vs CLOTHIAPINE, Outcome 5 Adverse events: Movement disorders.
10.5.2 dyskinesia ‐ short term
In this subgroup we only found one relevant trial (n = 80) (Itoh 1969). There was no significant difference between perphenazine and clothiapine (RR 0.56 CI 0.20 to 1.51, Analysis 10.5).
10.5.3 parkinsonism ‐ short term
In this subgroup we only found one relevant trial (n = 80) (Itoh 1969). There was no significant difference between perphenazine and clothiapine (RR 1.31 CI 0.81 to 2.12, Analysis 10.5).
10.5.4 use of antiparkinsonian drugs ‐ short term
In this subgroup we only found one relevant trial (n = 80) (Itoh 1969). There was no significant difference between perphenazine and clothiapine (RR 1.31 CI 0.81 to 2.12, Analysis 10.5).
10.6 Other adverse events: 1. Anticholinergic
10.6.1 dry mouth ‐ short term
In this subgroup we only found one relevant trial (n = 80) (Itoh 1969). There was no significant difference between perphenazine and clothiapine (RR 0.25 CI 0.03 to 2.14, Analysis 10.6).
10.6. Analysis.
Comparison 10 PERPHENAZINE vs CLOTHIAPINE, Outcome 6 Other adverse events: 1. Anticholinergic.
10.6.2 nasal congestion ‐ short term
In this subgroup we only found one relevant trial (n = 80) (Itoh 1969). There was no significant difference between perphenazine and clothiapine (RR 3.00 CI 0.13 to 71.51, Analysis 10.6).
10.7 Other adverse events: 2. Arousal
10.7.1 insomnia ‐ short term
In this subgroup we only found one relevant trial (n = 80) (Itoh 1969). There was no significant difference between perphenazine and clothiapine (RR 0.91 CI 0.44 to 1.90, Analysis 10.7).
10.7. Analysis.
Comparison 10 PERPHENAZINE vs CLOTHIAPINE, Outcome 7 Other adverse events: 2. Arousal.
10.7.2 need of additional benzodiazepines ‐ short term
In this subgroup we only found one relevant trial (n = 80) (Itoh 1969). There was no significant difference between perphenazine and clothiapine (RR 1.25 CI 0.55 to 2.84, Analysis 10.7).
10.7.3 sleepiness/sedation ‐ short term
In this subgroup we only found one relevant trial (n = 80) (Itoh 1969). There was no significant difference between perphenazine and clothiapine (RR 1.00 CI 0.42 to 2.40, Analysis 10.7).
10.8 Other adverse events: 3. At least one
10.8.1 short term
In this subgroup we only found one relevant trial (n = 80) (Itoh 1969). There was no significant difference between perphenazine and clothiapine (RR 1.09 CI 0.91 to 1.30, Analysis 10.8).
10.8. Analysis.
Comparison 10 PERPHENAZINE vs CLOTHIAPINE, Outcome 8 Other adverse events: 3. At least one.
10.9 Other adverse events: 4. Cardiovascular
10.9.1 faintness, dizziness, weakness ‐ short term
In this subgroup we only found one relevant trial (n = 80) (Itoh 1969). There was no significant difference between perphenazine and clothiapine (RR 1.67 CI 0.43 to 6.51, Analysis 10.9).
10.9. Analysis.
Comparison 10 PERPHENAZINE vs CLOTHIAPINE, Outcome 9 Other adverse events: 4. Cardiovascular.
10.10 Other adverse events: 5. Central nervous system
10.10.1 headache ‐ short term
In this subgroup we only found one relevant trial (n = 80) (Itoh 1969). There was no significant difference between perphenazine and clothiapine (RR 0.33 CI 0.01 to 7.95, Analysis 10.10).
10.10. Analysis.
Comparison 10 PERPHENAZINE vs CLOTHIAPINE, Outcome 10 Other adverse events: 5. Central nervous system.
10.11 Other adverse events: 6. Gastrointestinal
10.11.1 constipation ‐ short term
In this subgroup we only found one relevant trial (n = 80) (Itoh 1969). There was no significant difference between perphenazine and clothiapine (RR 0.25 CI 0.06 to 1.11, Analysis 10.11).
10.11. Analysis.
Comparison 10 PERPHENAZINE vs CLOTHIAPINE, Outcome 11 Other adverse events: 6. Gastrointestinal.
10.11.2 loss of appetite ‐ short term
In this subgroup we only found one relevant trial (n = 80) (Itoh 1969). There was no significant difference between perphenazine and clothiapine (RR 1.33 CI 0.32 to 5.58, Analysis 10.11).
10.11.3 nausea and vomiting ‐ short term
In this subgroup we only found one relevant trial (n = 80) (Itoh 1969). There was no significant difference between perphenazine and clothiapine (RR 3.00 CI 0.13 to 71.51, Analysis 10.11).
10.12 Other adverse events: 7. Skin
10.12.1 rash ‐ short term
In this subgroup we only found one relevant trial (n = 80) (Itoh 1969). There was no significant difference between perphenazine and clothiapine (RR 0.50 CI 0.05 to 5.30, Analysis 10.12).
10.12. Analysis.
Comparison 10 PERPHENAZINE vs CLOTHIAPINE, Outcome 12 Other adverse events: 7. Skin.
10.13 Other adverse events: 8. Genitourinary
10.13.1 micturition disturbances ‐ short term
In this subgroup we only found one relevant trial (n = 80) (Itoh 1969). There was no significant difference between perphenazine and clothiapine (RR 0.50 CI 0.05 to 5.30, Analysis 10.13).
10.13. Analysis.
Comparison 10 PERPHENAZINE vs CLOTHIAPINE, Outcome 13 Other adverse events: 8. Genitourinary.
10.14 Other adverse events: 9. Others
10.14.1 muscle pain ‐ short term
In this subgroup we only found one relevant trial (n = 80) (Itoh 1969). There was no significant difference between perphenazine and clothiapine (RR 3.00 CI 0.13 to 71.51, Analysis 10.14).
10.14. Analysis.
Comparison 10 PERPHENAZINE vs CLOTHIAPINE, Outcome 14 Other adverse events: 9. Others.
COMPARISON 11: PERPHENAZINE versus CLOZAPINE
11.1 Leaving the study early: 1. Any reason
11.1.1 short term
In this subgroup we found two relevant trials (n = 130). There was no significant difference between perphenazine and clozapine (RR 1.87 CI 0.48 to 7.23, Analysis 11.1).
11.1. Analysis.
Comparison 11 PERPHENAZINE vs CLOZAPINE, Outcome 1 Leaving the study early: 1. Any reason.
11.2 Leaving the study early: 2. Due to adverse events
11.2.1 short term
In this subgroup we only found one relevant trial (n = 28) (Van Praag 1976). There was no significant difference between perphenazine and clozapine (RR 2.60 CI 0.31 to 22.05, Analysis 11.2).
11.2. Analysis.
Comparison 11 PERPHENAZINE vs CLOZAPINE, Outcome 2 Leaving the study early: 2. Due to adverse events.
11.3 Mental state: 1. State ‐ less than 30% BPRS reduction ('no effect')
11.3.1 short term
In this subgroup we only found one relevant trial (n = 97) (Sun 2000). There was no significant difference between perphenazine and clozapine (RR 2.55 CI 0.52 to 12.52, Analysis 11.3).
11.3. Analysis.
Comparison 11 PERPHENAZINE vs CLOZAPINE, Outcome 3 Mental state: 1. State ‐ less than 30% BPRS reduction ('no effect').
11.4 Mental state: 2. State ‐ BPRS end score (high = poor)
11.4.1 total score ‐ short term
In this subgroup we only found one relevant trial (n = 102) (Sun 2000). There was no significant difference between perphenazine and clozapine (MD 0.94 CI ‐1.49 to 3.37, Analysis 11.4).
11.4. Analysis.
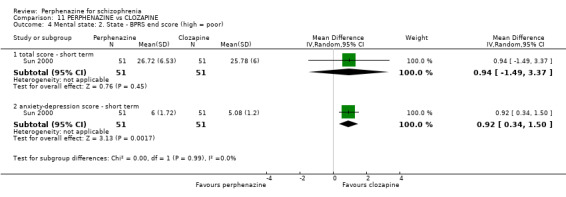
Comparison 11 PERPHENAZINE vs CLOZAPINE, Outcome 4 Mental state: 2. State ‐ BPRS end score (high = poor).
11.4.2 anxiety‐depression score ‐ short term
In this subgroup we only found one relevant trial (n = 102) (Sun 2000). There was a significant difference (P = 0.002) favouring clozapine over perphenazine (MD 0.92 CI 0.34 to 1.50, Analysis 11.4).
11.5 Adverse events: Movement disorders
11.5.1 dyskinesia ‐ short term
In this subgroup we only found one relevant trial (n = 28) (Van Praag 1976). There was no significant difference between perphenazine and clozapine, with no events reported in either group (Analysis 11.5).
11.5. Analysis.
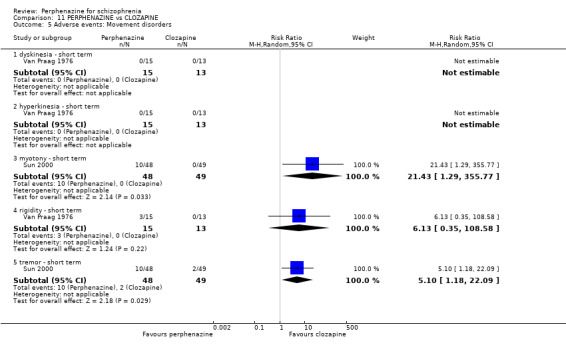
Comparison 11 PERPHENAZINE vs CLOZAPINE, Outcome 5 Adverse events: Movement disorders.
11.5.2 hyperkinesia ‐ short term
In this subgroup we only found one relevant trial (n = 28) (Van Praag 1976). There was no significant difference between perphenazine and clozapine, with no events reported in either group (Analysis 11.5).
11.5.3 myotony ‐ short term
In this subgroup we only found one relevant trial (n = 97) (Sun 2000). There was a statistically significant difference (P = 0.03) favouring clozapine over perphenazine (RR 21.43 CI 1.29 to 355.77, Analysis 11.5).
11.5.4 rigidity ‐ short term
In this subgroup we only found one relevant trial (n = 28) (Van Praag 1976). There was no significant difference between perphenazine and clozapine (RR 6.13 CI 0.35 to 108.58, Analysis 11.5).
11.5.5 tremor ‐ short term
In this subgroup we only found one relevant trial (n = 97) (Sun 2000). There was a statistically significant difference (P = 0.03) favouring clozapine over perphenazine (RR 5.10 CI 1.18 to 22.09, Analysis 11.5).
11.6 Other adverse events: 1. Anticholinergic
11.6.1 blurred vision ‐ short term
In this subgroup we only found one relevant trial (n = 97) (Sun 2000). There was no significant difference between perphenazine and clozapine (RR 0.85 CI 0.49 to 1.49, Analysis 11.6).
11.6. Analysis.
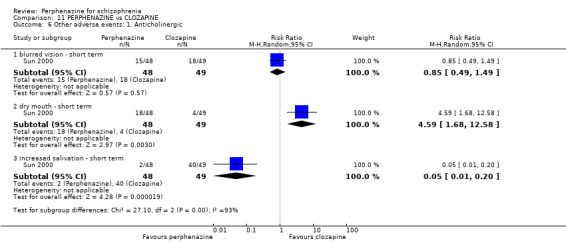
Comparison 11 PERPHENAZINE vs CLOZAPINE, Outcome 6 Other adverse events: 1. Anticholinergic.
11.6.2 dry mouth ‐ short term
In this subgroup we only found one relevant trial (n = 97) (Sun 2000). There was a statistically significant difference (P = 0.003) favouring clozapine over piperazine (RR 4.59 CI 1.68 to 12.58, Analysis 11.6).
11.6.3 increased salivation ‐ short term
In this subgroup we only found one relevant trial (n = 97) (Sun 2000). There was a statistically significant difference (P < 0.0001) favouring perphenazine over clozapine (RR 0.05 CI 0.01 to 0.20, Analysis 11.6).
11.7 Other adverse events: 2. Arousal
11.7.1 drowsiness ‐ short term
In this subgroup we only found one relevant trial (n = 28) (Van Praag 1976). There was a statistically significant difference (P = 0.03) favouring perphenazine and over clozapine (RR 0.43 CI 0.20 to 0.94, Analysis 11.7).
11.7. Analysis.
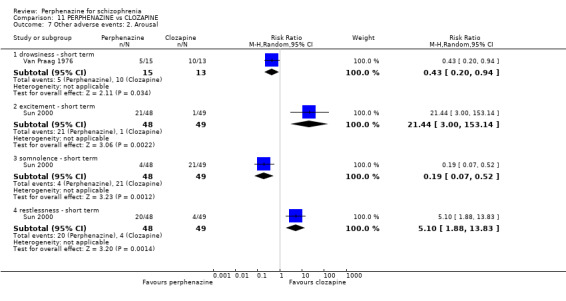
Comparison 11 PERPHENAZINE vs CLOZAPINE, Outcome 7 Other adverse events: 2. Arousal.
11.7.2 excitement ‐ short term
In this subgroup we only found one relevant trial (n = 97) (Sun 2000). There was a statistically significant difference (P = 0.002) favouring clozapine over perphenazine (RR 21.44 CI 3.00 to 153.14, Analysis 11.7).
11.7.3 somnolence ‐ short term
In this subgroup we only found one relevant trial (n = 97) (Sun 2000). There was a statistically significant difference (P = 0.001) favouring perphenazine and over clozapine (RR 0.19 CI 0.07 to 0.52, Analysis 11.7).
11.7.4 restlessness ‐ short term
In this subgroup we only found one relevant trial (n = 97) (Sun 2000). There was a statistically significant difference (P = 0.001) favouring clozapine over perphenazine (RR 5.10 CI 1.88 to 13.83, Analysis 11.7).
11.8 Other adverse events: 3. At least one
11.8.1 short term
In this subgroup we only found one relevant trial (n = 28) (Van Praag 1976). There was no significant difference between perphenazine and clozapine (RR 2.60 CI 0.31 to 22.05, Analysis 11.8).
11.8. Analysis.
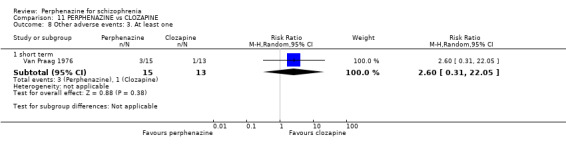
Comparison 11 PERPHENAZINE vs CLOZAPINE, Outcome 8 Other adverse events: 3. At least one.
11.9 Other adverse events: 4. Cardiovascular
11.9.1 dizziness ‐ short term
In this subgroup we only found one relevant trial (n = 97) (Sun 2000). There was no significant difference between perphenazine and clozapine (RR 0.92 CI 0.56 to 1.51, Analysis 11.9).
11.9. Analysis.
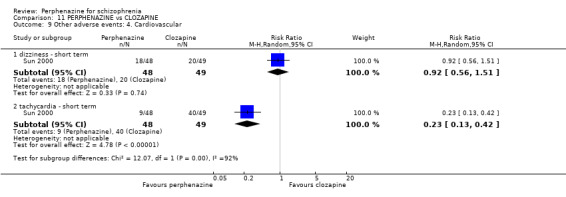
Comparison 11 PERPHENAZINE vs CLOZAPINE, Outcome 9 Other adverse events: 4. Cardiovascular.
11.9.2 tachycardia ‐ short term
In this subgroup we only found one relevant trial (n = 97) (Sun 2000). There was a statistically significant difference between (P < 0.00001) favouring perphenazine over clozapine (RR 0.23 CI 0.13 to 0.42, Analysis 11.9).
11.10 Other adverse events: 5. Gastrointestinal
11.10.1 constipation ‐ short term
In this subgroup we only found one relevant trial (n = 97) (Sun 2000). There was a statistically significant difference (P < 0.00001) favouring perphenazine over clozapine (RR 0.33 CI 0.20 to 0.54, Analysis 11.10).
11.10. Analysis.
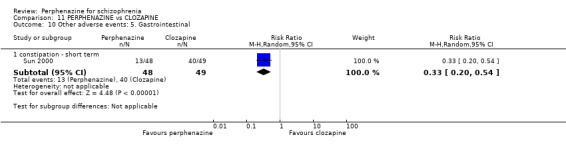
Comparison 11 PERPHENAZINE vs CLOZAPINE, Outcome 10 Other adverse events: 5. Gastrointestinal.
11.11 Other adverse events: 6. Lab data
11.11.1 abnormal EKG ‐ short term
In this subgroup we only found one relevant trial (n = 97) (Sun 2000). There was no significant difference between perphenazine and clozapine (RR 1.33 CI 0.73 to 2.44, Analysis 11.11).
11.11. Analysis.
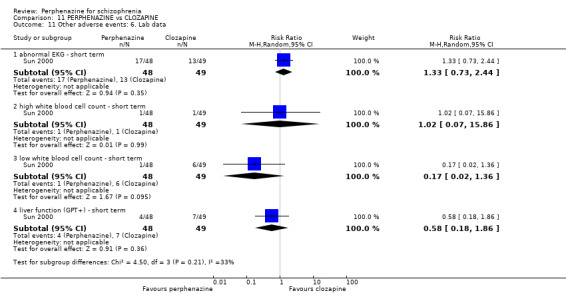
Comparison 11 PERPHENAZINE vs CLOZAPINE, Outcome 11 Other adverse events: 6. Lab data.
11.11.2 high white blood cell count ‐ short term
In this subgroup we only found one relevant trial (n = 97) (Sun 2000). There was no significant difference between perphenazine and clozapine (RR 1.02 CI 0.07 to 15.86, Analysis 11.11).
11.11.3 low white blood cell count ‐ short term
In this subgroup we only found one relevant trial (n = 97) (Sun 2000). There was no significant difference between perphenazine and clozapine (RR 0.17 CI 0.02 to 1.36, Analysis 11.11).
11.11.4 liver function (GPT+) ‐ short term
In this subgroup we only found one relevant trial (n = 97) (Sun 2000). There was no significant difference between perphenazine and clozapine (RR 0.58 CI 0.18 to 1.86, Analysis 11.11).
11.12 Other adverse events: 7. Average endpoint score (TESS, skew)
11.12.1 short term
Data for this outcome are skewed and are best inspected by viewing Analysis 11.12.
11.12. Analysis.
Comparison 11 PERPHENAZINE vs CLOZAPINE, Outcome 12 Other adverse events: 7. Average endpoint score (TESS, skew).
| Other adverse events: 7. Average endpoint score (TESS, skew) | ||||
|---|---|---|---|---|
| Study | Intervention | Mean | SD | N |
| short term | ||||
| Sun 2000 | Perphenazine | 3.38 | 2.19 | 51 |
| Sun 2000 | Clozapine | 5.37 | 2.67 | 51 |
COMPARISON 12: PERPHENAZINE versus CLOZAPINE + PERPHENAZINE
12.1 Leaving the study early: 1. Any reason
12.1.1 short term
In this subgroup we only found one relevant trial (n = 102) (Sun 2000). There was no significant difference between perphenazine and clozapine + perphenazine (RR 3.00 CI 0.32 to 27.89, Analysis 12.1).
12.1. Analysis.
Comparison 12 PERPHENAZINE vs CLOZAPINE + PERPHENAZINE, Outcome 1 Leaving the study early: 1. Any reason.
12.2 Mental state: 1. State ‐ less than 30% BPRS reduction ('no effect')
12.2.1 short term
In this subgroup we only found one relevant trial (n = 147) (Sun 2000). There was no significant difference between perphenazine and clozapine + perphenazine (RR 3.44 CI 0.86 to 13.79, Analysis 12.2).
12.2. Analysis.
Comparison 12 PERPHENAZINE vs CLOZAPINE + PERPHENAZINE, Outcome 2 Mental state: 1. State ‐ less than 30% BPRS reduction ('no effect').
12.3 Mental state: 1a. State ‐ BPRS end score (high = poor)
12.3.1 total score ‐ short term
In this subgroup we only found one relevant trial (n = 102) (Sun 2000). There was no significant difference between perphenazine and clozapine + perphenazine (MD 0.74 CI ‐1.72 to 3.20, Analysis 12.3).
12.3. Analysis.
Comparison 12 PERPHENAZINE vs CLOZAPINE + PERPHENAZINE, Outcome 3 Mental state: 1a. State ‐ BPRS end score (high = poor).
12.3.2 anxiety‐depression score ‐ short term
In this subgroup we only found one relevant trial (n = 102) (Sun 2000). There was no significant difference between perphenazine and clozapine + perphenazine (MD 0.51 CI ‐0.10 to 1.12, Analysis 12.3).
12.4 Adverse events: Movement disorders
12.4.1 myotony ‐ short term
In this subgroup we only found one relevant trial (n = 98) (Sun 2000). There was no significant difference between perphenazine and clozapine + perphenazine (RR 1.04 CI 0.48 to 2.28, Analysis 12.4).
12.4. Analysis.
Comparison 12 PERPHENAZINE vs CLOZAPINE + PERPHENAZINE, Outcome 4 Adverse events: Movement disorders.
12.4.2 tremor ‐ short term
In this subgroup we only found one relevant trial (n = 98) (Sun 2000). There was no significant difference between perphenazine and clozapine + perphenazine (RR 1.04 CI 0.48 to 2.28, Analysis 12.4).
12.5 Other adverse events: 1. Anticholinergic
12.5.1 blurred vision ‐ short term
In this subgroup we only found one relevant trial (n = 98) (Sun 2000). There was no significant difference between perphenazine and clozapine + perphenazine (RR 0.98 CI 0.55 to 1.75, Analysis 12.5).
12.5. Analysis.
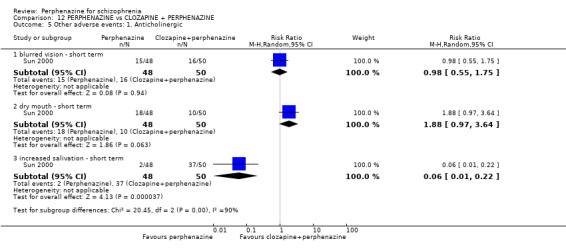
Comparison 12 PERPHENAZINE vs CLOZAPINE + PERPHENAZINE, Outcome 5 Other adverse events: 1. Anticholinergic.
12.5.2 dry mouth ‐ short term
In this subgroup we only found one relevant trial (n = 98) (Sun 2000). There was no significant difference between perphenazine and clozapine + perphenazine (RR 1.88 CI 0.97 to 3.64, Analysis 12.5).
12.5.3 increased salivation ‐ short term
In this subgroup we only found one relevant trial (n = 98) (Sun 2000). There was a statistically significant difference (P < 0.0001) favouring perphenazine over clozapine + perphenazine (RR 0.06 CI 0.01 to 0.22, Analysis 12.5).
12.6 Other adverse events: 2. Arousal
12.6.1 excitement ‐ short term
In this subgroup we only found one relevant trial (n = 98) (Sun 2000). There was a statistically significant difference (P = 0.002) favouring clozapine + perphenazine over perphenazine alone (RR 21.88 CI 3.06 to 156.33, Analysis 12.6).
12.6. Analysis.
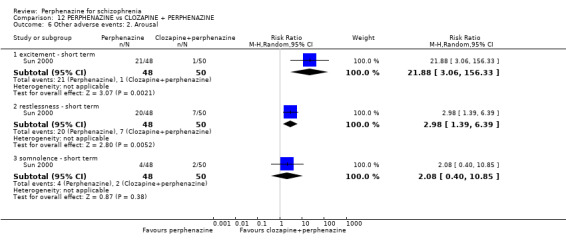
Comparison 12 PERPHENAZINE vs CLOZAPINE + PERPHENAZINE, Outcome 6 Other adverse events: 2. Arousal.
12.6.2 restlessness ‐ short term
In this subgroup we only found one relevant trial (n = 98) (Sun 2000). There was a statistically significant difference (P = 0.005) favouring clozapine + perphenazine over perphenazine alone (RR 2.98 CI 1.39 to 6.39, Analysis 12.6).
12.6.3 somnolence ‐ short term
In this subgroup we only found one relevant trial (n = 98) (Sun 2000). There was no significant difference between perphenazine and clozapine + perphenazine (RR 2.08 CI 0.0 to 10.85, Analysis 12.6).
12.7 Other adverse events: 3. Cardiovascular
12.7.1 dizziness ‐ short term
In this subgroup we only found one relevant trial (n = 98) (Sun 2000). There was no significant difference between perphenazine and clozapine + perphenazine (RR 0.89 CI 0.55 to 1.46, Analysis 12.7).
12.7. Analysis.
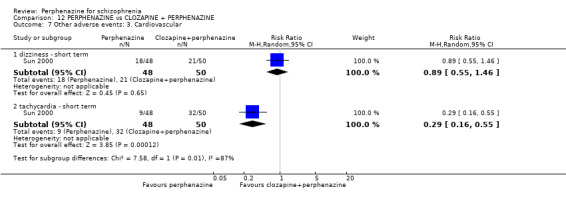
Comparison 12 PERPHENAZINE vs CLOZAPINE + PERPHENAZINE, Outcome 7 Other adverse events: 3. Cardiovascular.
12.7.2 tachycardia ‐ short term
In this subgroup we only found one relevant trial (n = 98) (Sun 2000). There was a statistically significant difference (P = 0.0001) favouring perphenazine alone over clozapine + perphenazine (RR 0.29 CI 0.16 to 0.55, Analysis 12.7).
12.8 Other adverse events: 4. Gastrointestinal
12.8.1 constipation ‐ short term
In this subgroup we only found one relevant trial (n = 98) (Sun 2000). There was a statistically significant difference (P = 0.002) favouring perphenazine alone over clozapine + perphenazine (RR 0.44 CI 0.26 to 0.73, Analysis 12.8).
12.8. Analysis.
Comparison 12 PERPHENAZINE vs CLOZAPINE + PERPHENAZINE, Outcome 8 Other adverse events: 4. Gastrointestinal.
12.9 Other adverse events: 5. Lab data
12.9.1 abnormal EKG ‐ short term
In this subgroup we only found one relevant trial (n = 98) (Sun 2000). There was no significant difference between perphenazine and clozapine + perphenazine (RR 1.97 CI 0.97 to 3.98, Analysis 12.9).
12.9. Analysis.
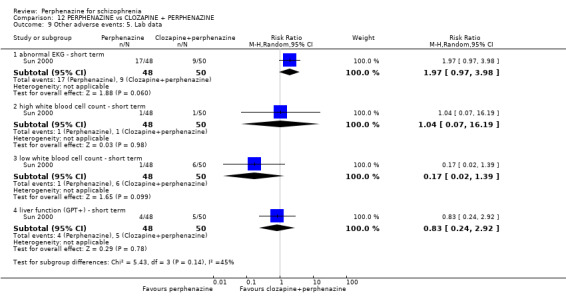
Comparison 12 PERPHENAZINE vs CLOZAPINE + PERPHENAZINE, Outcome 9 Other adverse events: 5. Lab data.
12.9.2 high white blood cell count ‐ short term
In this subgroup we only found one relevant trial (n = 98) (Sun 2000). There was no significant difference between perphenazine and clozapine + perphenazine (RR 1.04 CI 0.07 to 16.19, Analysis 12.9).
12.9.3 low white blood cell count ‐ short term
In this subgroup we only found one relevant trial (n = 98) (Sun 2000). There was no significant difference between perphenazine and clozapine + perphenazine (RR 0.17 CI 0.02 to 1.39, Analysis 12.9).
12.9.4 liver function (GPT+) ‐ short term
In this subgroup we only found one relevant trial (n = 98) (Sun 2000). There was no significant difference between perphenazine and clozapine + perphenazine (RR 0.83 CI 0.24 to 2.92, Analysis 12.9).
12.10 Other adverse events: 6. Average endpoint score (TESS, skewed)
12.10.1 short term
Data for this outcome are skewed and are best inspected by viewing Analysis 12.10.
12.10. Analysis.
Comparison 12 PERPHENAZINE vs CLOZAPINE + PERPHENAZINE, Outcome 10 Other adverse events: 6. Average endpoint score (TESS, skewed).
| Other adverse events: 6. Average endpoint score (TESS, skewed) | ||||
|---|---|---|---|---|
| Study | Intervention | Mean | SD | N |
| short term | ||||
| Sun 2000 | Perphenazine | 3.38 | 2.19 | 51 |
| Sun 2000 | Clozapine + perphenazine | 5.65 | 3.53 | 51 |
COMPARISON 13: PERPHENAZINE versus FLUPHENAZINE
13.1 Leaving the study early: 1. Any reason
13.1.1 short term
In this subgroup we only found one relevant trial (n = 106) (Hanlon 1965). There was a statistically significant difference (P = 0.05) favouring perphenazine over fluphenazine (RR 0.62 CI 0.38 to 1.01, Analysis 13.1).
13.1. Analysis.
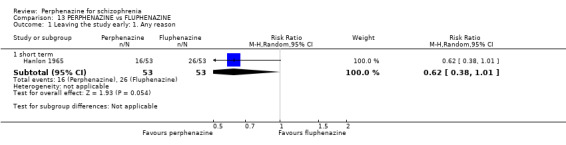
Comparison 13 PERPHENAZINE vs FLUPHENAZINE, Outcome 1 Leaving the study early: 1. Any reason.
13.2 Leaving the study early: 2. Due to adverse events
13.2.1 short term
In this subgroup we only found one relevant trial (n = 106) (Hanlon 1965). There was no significant difference between perphenazine and fluphenazine (RR 1.00 CI 0.15 to 6.84, Analysis 13.2).
13.2. Analysis.
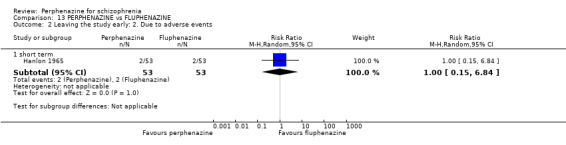
Comparison 13 PERPHENAZINE vs FLUPHENAZINE, Outcome 2 Leaving the study early: 2. Due to adverse events.
13.3 Adverse events: Movement disorders
13.3.1 akathisia ‐ short term
In this subgroup we only found one relevant trial (n = 106) (Hanlon 1965). There was no significant difference between perphenazine and fluphenazine (RR 1.17 CI 0.42 to 3.24, Analysis 13.3).
13.3. Analysis.
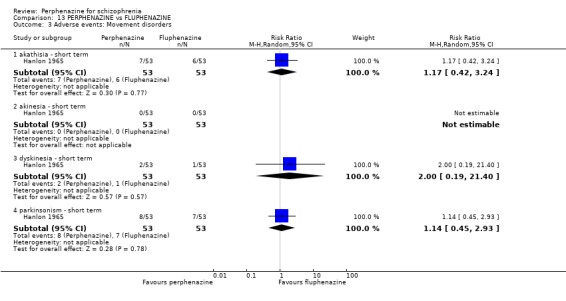
Comparison 13 PERPHENAZINE vs FLUPHENAZINE, Outcome 3 Adverse events: Movement disorders.
13.3.2 akinesia ‐ short term
In this subgroup we only found one relevant trial (n = 106) (Hanlon 1965). There was no significant difference between perphenazine and fluphenazine, with no events reported in either group (Analysis 13.3).
13.3.3 dyskinesia ‐ short term
In this subgroup we only found one relevant trial (n = 106) (Hanlon 1965). There was no significant difference between perphenazine and fluphenazine (RR 2.00 CI 0.19 to 21.40, Analysis 13.3).
13.3.4 parkinsonism ‐ short term
In this subgroup we only found one relevant trial (n = 106) (Hanlon 1965). There was no significant difference between perphenazine and fluphenazine (RR 1.14 CI 0.45 to 2.93, Analysis 13.3).
COMPARISON 14: PERPHENAZINE versus HALOPERIDOL
14.1 Leaving the study early: 1. Any reason
14.1.1 short term
In this subgroup we only found one relevant trial (n = 189) (Kurihara 1983). There was no significant difference between perphenazine and haloperidol (RR 0.84 CI 0.40 to 1.77, Analysis 14.1).
14.1. Analysis.
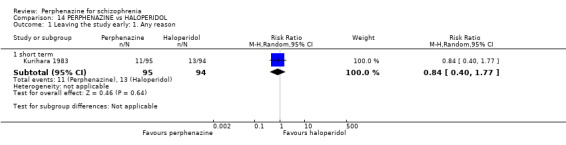
Comparison 14 PERPHENAZINE vs HALOPERIDOL, Outcome 1 Leaving the study early: 1. Any reason.
14.2 Leaving the study early: 2. Due to adverse events
14.2.1 short term
In this subgroup we only found one relevant trial (n = 189) (Kurihara 1983). There was no significant difference between perphenazine and haloperidol (RR 0.49 CI 0.13 to 1.92, Analysis 14.2).
14.2. Analysis.
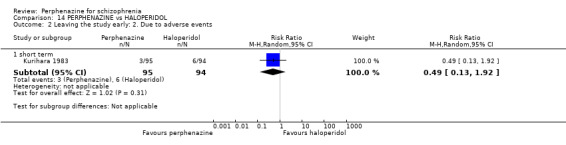
Comparison 14 PERPHENAZINE vs HALOPERIDOL, Outcome 2 Leaving the study early: 2. Due to adverse events.
14.3 Leaving the study early: 3. Due to relapse/worsening or no improvement
14.3.1 short term
In this subgroup we only found one relevant trial (n = 189) (Kurihara 1983). There was no significant difference between perphenazine and haloperidol (RR 1.39 CI 0.46 to 4.21, Analysis 14.3).
14.3. Analysis.
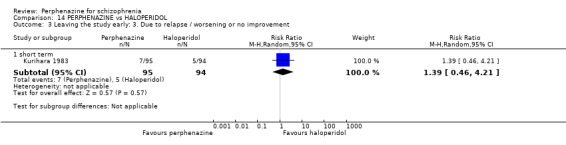
Comparison 14 PERPHENAZINE vs HALOPERIDOL, Outcome 3 Leaving the study early: 3. Due to relapse / worsening or no improvement.
14.4 Global state: 1. Change over time ‐ no better or deterioration (ITT)
14.4.1 short term
In this subgroup we only found one relevant trial (n = 189) (Kurihara 1983). There was no significant difference between perphenazine and haloperidol (RR 1.01 CI 0.79 to 1.29, Analysis 14.4).
14.4. Analysis.
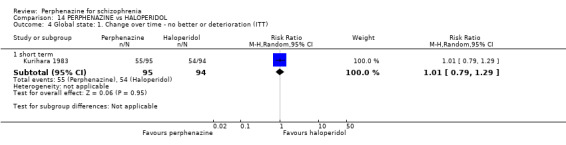
Comparison 14 PERPHENAZINE vs HALOPERIDOL, Outcome 4 Global state: 1. Change over time ‐ no better or deterioration (ITT).
14.5 Adverse events: Movement disorders
14.5.1 akathisia ‐ short term
In this subgroup we only found one relevant trial (n = 189) (Kurihara 1983). There was no significant difference between perphenazine and haloperidol (RR 0.70 CI 0.42 to 1.16, Analysis 14.5).
14.5. Analysis.
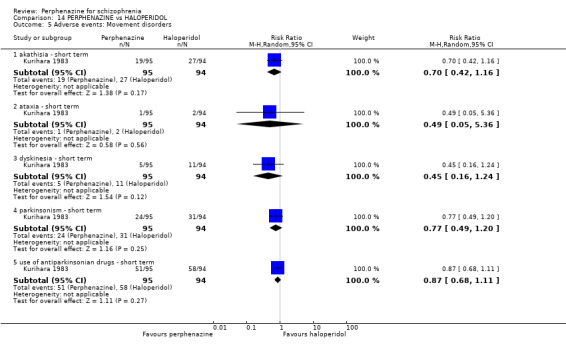
Comparison 14 PERPHENAZINE vs HALOPERIDOL, Outcome 5 Adverse events: Movement disorders.
14.5.2 ataxia ‐ short term
In this subgroup we only found one relevant trial (n = 189) (Kurihara 1983). There was no significant difference between perphenazine and haloperidol (RR 0.49 CI 0.05 to 5.36, Analysis 14.5).
14.5.3 dyskinesia ‐ short term
In this subgroup we only found one relevant trial (n = 189) (Kurihara 1983). There was no significant difference between perphenazine and haloperidol (RR 0.45 CI 0.16 to 1.24, Analysis 14.5).
14.5.4 parkinsonism ‐ short term
In this subgroup we only found one relevant trial (n = 189) (Kurihara 1983). There was no significant difference between perphenazine and haloperidol (RR 0.77 CI 0.49 to 1.20, Analysis 14.5).
14.5.5 use of antiparkinsonian drugs ‐ short term
In this subgroup we only found one relevant trial (n = 189) (Kurihara 1983). There was no significant difference between perphenazine and haloperidol (RR 0.87 CI 0.68 to 1.11, Analysis 14.5).
14.6 Other adverse events: 1. Anticholinergic
14.6.1 blurred vision ‐ short term
In this subgroup we only found one relevant trial (n = 189) (Kurihara 1983). There was no significant difference between perphenazine and haloperidol (RR 0.25 CI 0.05 to 1.13, Analysis 14.6).
14.6. Analysis.
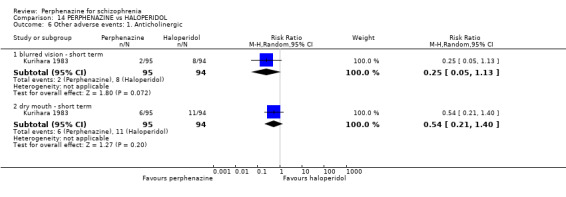
Comparison 14 PERPHENAZINE vs HALOPERIDOL, Outcome 6 Other adverse events: 1. Anticholinergic.
14.6.2 dry mouth ‐ short term
In this subgroup we only found one relevant trial (n = 189) (Kurihara 1983). There was no significant difference between perphenazine and haloperidol (RR 0.54 CI 0.21 to 1.40, Analysis 14.6).
14.7 Other adverse events: 2. Arousal
14.7.1 insomnia ‐ short term
In this subgroup we only found one relevant trial (n = 189) (Kurihara 1983). There was no significant difference between perphenazine and haloperidol (RR 0.97 CI 0.71 to 1.32, Analysis 14.7).
14.7. Analysis.
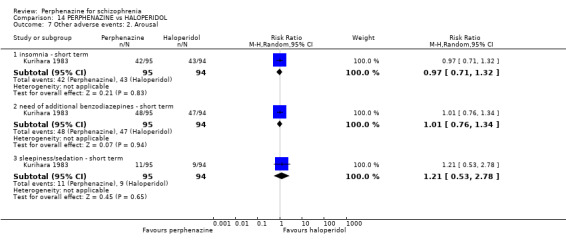
Comparison 14 PERPHENAZINE vs HALOPERIDOL, Outcome 7 Other adverse events: 2. Arousal.
14.7.2 need of additional benzodiazepines ‐ short term
In this subgroup we only found one relevant trial (n = 189) (Kurihara 1983). There was no significant difference between perphenazine and haloperidol (RR 1.01 CI 0.76 to 1.34, Analysis 14.7).
14.7.3 sleepiness/sedation ‐ short term
In this subgroup we only found one relevant trial (n = 189) (Kurihara 1983). There was no significant difference between perphenazine and haloperidol (RR 1.21 CI 0.53 to 2.78, Analysis 14.7).
14.8 Other adverse events: 3. At least one
14.8.1 short term
In this subgroup we only found one relevant trial (n = 189) (Kurihara 1983). There was no significant difference between perphenazine and haloperidol (RR 1.05 CI 0.88 to 1.25, Analysis 14.8).
14.8. Analysis.
Comparison 14 PERPHENAZINE vs HALOPERIDOL, Outcome 8 Other adverse events: 3. At least one.
14.9 Other adverse events: 4. Cardiovascular
14.9.1 angina pectoris ‐ short term
In this subgroup we only found one relevant trial (n = 189) (Kurihara 1983). There was no significant difference between perphenazine and haloperidol (RR 0.57 CI 0.17 to 1.87, Analysis 14.9).
14.9. Analysis.
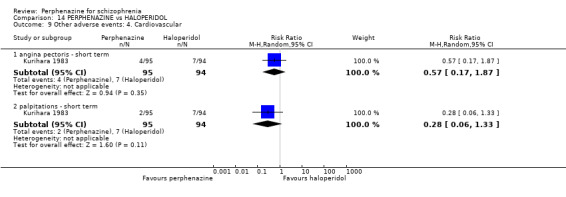
Comparison 14 PERPHENAZINE vs HALOPERIDOL, Outcome 9 Other adverse events: 4. Cardiovascular.
14.9.2 palpitations ‐ short term
In this subgroup we only found one relevant trial (n = 189) (Kurihara 1983). There was no significant difference between perphenazine and haloperidol (RR 0.28 CI 0.06 to 1.33, Analysis 14.9).
14.10 Other adverse events: 5. Central nervous system
14.10.1 disturbance of consciousness ‐ short term
In this subgroup we only found one relevant trial (n = 189) (Kurihara 1983). There was no significant difference between perphenazine and haloperidol (RR 0.33 CI 0.01 to 8.00, Analysis 14.10).
14.10. Analysis.
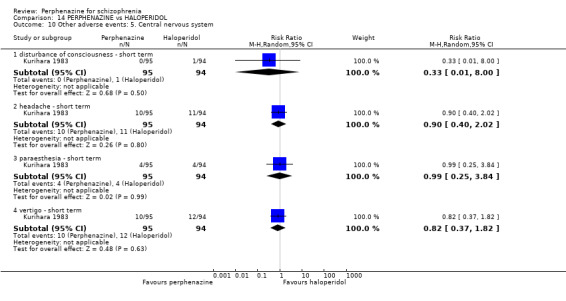
Comparison 14 PERPHENAZINE vs HALOPERIDOL, Outcome 10 Other adverse events: 5. Central nervous system.
14.10.2 headache ‐ short term
In this subgroup we only found one relevant trial (n = 189) (Kurihara 1983). There was no significant difference between perphenazine and haloperidol (RR 0.90 CI 0.40 to 2.02, Analysis 14.10).
14.10.3 paraesthesia ‐ short term
In this subgroup we only found one relevant trial (n = 189) (Kurihara 1983). There was no significant difference between perphenazine and haloperidol (RR 0.99 CI 0.25 to 3.84, Analysis 14.10).
14.10.4 vertigo ‐ short term
In this subgroup we only found one relevant trial (n = 189) (Kurihara 1983). There was no significant difference between perphenazine and haloperidol (RR 0.82 CI 0.37 to 1.82, Analysis 14.10).
14.11 Other adverse events: 6. Gastrointestinal
14.11.1 constipation ‐ short term
In this subgroup we only found one relevant trial (n = 189) (Kurihara 1983). There was no significant difference between perphenazine and haloperidol (RR 0.84 CI 0.40 to 1.77, Analysis 14.11).
14.11. Analysis.
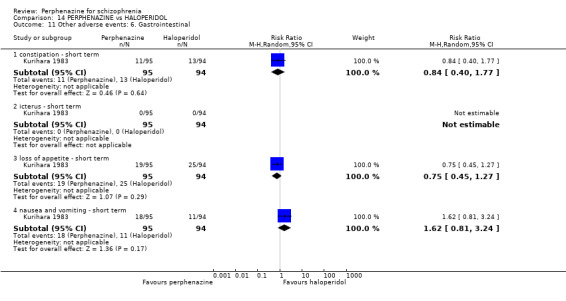
Comparison 14 PERPHENAZINE vs HALOPERIDOL, Outcome 11 Other adverse events: 6. Gastrointestinal.
14.11.2 icterus ‐ short term
In this subgroup we only found one relevant trial (n = 189) (Kurihara 1983). There was no significant difference between perphenazine and haloperidol, with no events reported in either group (Analysis 14.11).
14.11.3 loss of appetite ‐ short term
In this subgroup we only found one relevant trial (n = 189) (Kurihara 1983). There was no significant difference between perphenazine and haloperidol (RR 0.75 CI 0.45 to 1.27, Analysis 14.11).
14.11.4 nausea and vomiting ‐ short term
In this subgroup we only found one relevant trial (n = 189) (Kurihara 1983). There was no significant difference between perphenazine and haloperidol (RR 1.62 CI 0.81 to 3.24, Analysis 14.11).
14.12 Other adverse events: 7. Skin
14.12.1 pruritus ‐ short term
In this subgroup we only found one relevant trial (n = 189) (Kurihara 1983). There was no significant difference between perphenazine and haloperidol (RR 0.66 CI 0.11 to 3.86, Analysis 14.12).
14.12. Analysis.
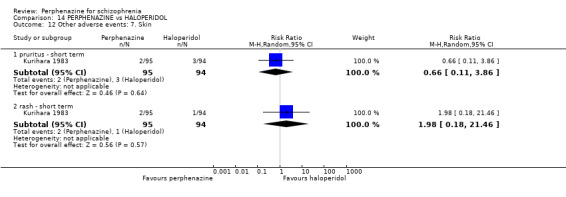
Comparison 14 PERPHENAZINE vs HALOPERIDOL, Outcome 12 Other adverse events: 7. Skin.
14.12.2 rash ‐ short term
In this subgroup we only found one relevant trial (n = 189) (Kurihara 1983). There was no significant difference between perphenazine and haloperidol (RR 1.98 CI 0.18 to 21.46, Analysis 14.12).
14.13 Other adverse events: 8. Genitourinary
14.13.1 diminished sexual desire
In this subgroup we only found one relevant trial (n = 189) (Kurihara 1983). There was no significant difference between perphenazine and haloperidol (RR 0.99 CI 0.06 to 15.59, Analysis 14.13).
14.13. Analysis.
Comparison 14 PERPHENAZINE vs HALOPERIDOL, Outcome 13 Other adverse events: 8. Genitourinary.
14.13.2 micturition disturbances ‐ short term
In this subgroup we only found one relevant trial (n = 189) (Kurihara 1983). There was no significant difference between perphenazine and haloperidol (RR 0.49 CI 0.09 to 2.64, Analysis 14.13).
14.14 Other adverse events: 9. Others
14.14.1 cramps ‐ short term
In this subgroup we only found one relevant trial (n = 189) (Kurihara 1983). There was no significant difference between perphenazine and haloperidol (RR 0.33 CI 0.01 to 8.00, Analysis 14.14).
14.14. Analysis.
Comparison 14 PERPHENAZINE vs HALOPERIDOL, Outcome 14 Other adverse events: 9. Others.
14.14.2 sweating ‐ short term
In this subgroup we only found one relevant trial (n = 189) (Kurihara 1983). There was no significant difference between perphenazine and haloperidol (RR 0.16 CI 0.02 to 1.34, Analysis 14.14).
COMPARISON 15: PERPHENAZINE versus LOXAPINE
15.1 Leaving the study early: 1. Any reason
15.1.1 short term
In this subgroup we only found one relevant trial (n = 47) (Fruensgaard 1978). There was no significant difference between perphenazine and loxapine (RR 0.96 CI 0.32 to 2.88, Analysis 15.1).
15.1. Analysis.
Comparison 15 PERPHENAZINE vs LOXAPINE, Outcome 1 Leaving the study early: 1. Any reason.
15.2 Leaving the study early: 2. Due to relapse/worsening or no improvement
15.2.1 short term
In this subgroup we only found one relevant trial (n = 47) (Fruensgaard 1978). There was no significant difference between perphenazine and loxapine (RR 0.96 CI 0.06 to 14.43, Analysis 15.2).
15.2. Analysis.
Comparison 15 PERPHENAZINE vs LOXAPINE, Outcome 2 Leaving the study early: 2. Due to relapse / worsening or no improvement.
15.3 Global state: 1. Change over time ‐ no better or deterioration (ITT)
15.3.1 short term
In this subgroup we only found one relevant trial (n = 47) (Fruensgaard 1978). There was no significant difference between perphenazine and loxapine (RR 0.85 CI 0.40 to 1.82, Analysis 15.3).
15.3. Analysis.
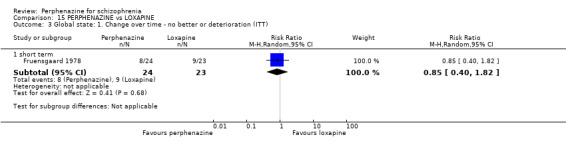
Comparison 15 PERPHENAZINE vs LOXAPINE, Outcome 3 Global state: 1. Change over time ‐ no better or deterioration (ITT).
15.4 Adverse events: Movement disorders
15.4.1 dystonia ‐ short term
In this subgroup we only found one relevant trial (n = 47) (Fruensgaard 1978). There was no significant difference between perphenazine and loxapine (RR 0.14 CI 0.01 to 2.52, Analysis 15.4).
15.4. Analysis.
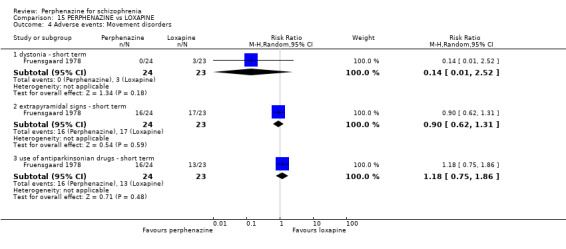
Comparison 15 PERPHENAZINE vs LOXAPINE, Outcome 4 Adverse events: Movement disorders.
15.4.2 extrapyramidal signs ‐ short term
In this subgroup we only found one relevant trial (n = 47) (Fruensgaard 1978). There was no significant difference between perphenazine and loxapine (RR 0.90 CI 0.62 to 1.31, Analysis 15.4).
15.4.3 use of antiparkinsonian drugs ‐ short term
In this subgroup we only found one relevant trial (n = 47) (Fruensgaard 1978). There was no significant difference between perphenazine and loxapine (RR 1.18 CI 0.75 to 1.86, Analysis 15.4).
15.5 Other adverse events: 1. Arousal
15.5.1 drowsiness ‐ short term
In this subgroup we only found one relevant trial (n = 47) (Fruensgaard 1978). There was no significant difference between perphenazine and loxapine (RR 1.60 CI 0.43 to 5.93, Analysis 15.5).
15.5. Analysis.
Comparison 15 PERPHENAZINE vs LOXAPINE, Outcome 5 Other adverse events: 1. Arousal.
15.6 Other adverse events: 2. At least one
15.6.1 short term
In this subgroup we only found one relevant trial (n = 47) (Fruensgaard 1978). There was a statistically significant difference (P = 0.05) favouring loxapine over perphenazine (RR 1.30 CI 1.00 to 1.68, Analysis 15.6).
15.6. Analysis.
Comparison 15 PERPHENAZINE vs LOXAPINE, Outcome 6 Other adverse events: 2. At least one.
15.7 Other adverse events: 3. lab data
15.7.1 ECG – atrial extrasystole & flattening of T‐wave in V6 ‐ short term
In this subgroup we only found one relevant trial (n = 47) (Fruensgaard 1978). There was no significant difference between perphenazine and loxapine (RR 6.72 CI 0.37 to 123.33, Analysis 15.7).
15.7. Analysis.
Comparison 15 PERPHENAZINE vs LOXAPINE, Outcome 7 Other adverse events: 3. lab data.
15.7.2 ventricular extrasystole ‐ short term
In this subgroup we only found one relevant trial (n = 47) (Fruensgaard 1978). There was no significant difference between perphenazine and loxapine (RR 0.32 CI 0.01 to 7.48, Analysis 15.7).
COMPARISON 16: PERPHENAZINE versus MEPAZINE
16.1 Leaving the study early: 1. Any reason
16.1.1 short term
In this subgroup we found two relevant trials (n = 68). There was no significant difference between perphenazine and mepazine (RR 1.18 CI 0.91 to 1.53, Analysis 16.1).
16.1. Analysis.
Comparison 16 PERPHENAZINE vs MEPAZINE, Outcome 1 Leaving the study early: 1. Any reason.
16.2 Leaving the study early: 2. Due to adverse events
16.2.1 short term
In this subgroup we only found one relevant trial (n = 58) (Kurland 1961). There was no significant difference between perphenazine and mepazine (RR 6.97 CI 0.93 to 52.20, Analysis 16.2).
16.2. Analysis.
Comparison 16 PERPHENAZINE vs MEPAZINE, Outcome 2 Leaving the study early: 2. Due to adverse events.
16.3 Global state: 1. Change over time ‐ no better or deterioration (ITT)
16.3.1 short term
In this subgroup we only found one relevant trial (n = 58) (Kurland 1961). There was a statistically significant difference (P = 0.05) favouring perphenazine over mepazine (RR 0.40 CI 0.16 to 1.00, Analysis 16.3).
16.3. Analysis.
Comparison 16 PERPHENAZINE vs MEPAZINE, Outcome 3 Global state: 1. Change over time ‐ no better or deterioration (ITT).
16.4 Other adverse events: 1. Central nervous system
16.4.1 electroencephalogram (increase) ‐ short term
In this subgroup we only found one relevant trial (n = 10) (Bennett 1961). There was no significant difference between perphenazine and mepazine (RR 1.00 CI 0.08 to 11.93, Analysis 16.4).
16.4. Analysis.
Comparison 16 PERPHENAZINE vs MEPAZINE, Outcome 4 Other adverse events: 1. Central nervous system.
COMPARISON 17: PERPHENAZINE versus METHYPERIDOL
17.1 Leaving the study early: 1. Any reason
17.1.1 short term
In this subgroup we only found one relevant trial (n = 78) (Itoh 1969 III). There was no significant difference between perphenazine and methyperidol (RR 1.00 CI 0.35 to 2.83, Analysis 17.1).
17.1. Analysis.
Comparison 17 PERPHENAZINE vs METHYPERIDOL, Outcome 1 Leaving the study early: 1. Any reason.
17.2 Leaving the study early: 2. Due to adverse events
17.2.1 short term
In this subgroup we only found one relevant trial (n = 78) (Itoh 1969 III). There was no significant difference between perphenazine and methyperidol (RR 4.00 CI 0.47 to 34.20, Analysis 17.2).
17.2. Analysis.
Comparison 17 PERPHENAZINE vs METHYPERIDOL, Outcome 2 Leaving the study early: 2. Due to adverse events.
17.3 Global state: 1. Change over time ‐ no better or deterioration (ITT)
17.3.1 short term
In this subgroup we only found one relevant trial (n = 78) (Itoh 1969 III). There was no significant difference between perphenazine and methyperidol (RR 1.05 CI 0.68 to 1.64, Analysis 17.3).
17.3. Analysis.
Comparison 17 PERPHENAZINE vs METHYPERIDOL, Outcome 3 Global state: 1. Change over time ‐ no better or deterioration (ITT).
17.4 Behaviour: 1. Behaviour subscale of the Psychiatric Rating Scale: no better or worse
For this outcome we only found one relevant trial (n = 78) (Itoh 1969 III). There was no significant difference between perphenazine and methyperidol (RR 0.88 CI 0.61 to 1.26, Analysis 17.4).
17.4. Analysis.
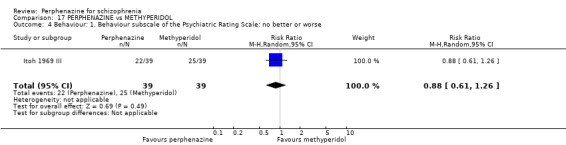
Comparison 17 PERPHENAZINE vs METHYPERIDOL, Outcome 4 Behaviour: 1. Behaviour subscale of the Psychiatric Rating Scale: no better or worse.
17.5 Adverse events: Movement disorders
17.5.1 akathisia ‐ short term
In this subgroup we only found one relevant trial (n = 78) (Itoh 1969 III). There was no significant difference between perphenazine and methyperidol (RR 1.25 CI 0.36 to 4.31, Analysis 17.5).
17.5. Analysis.
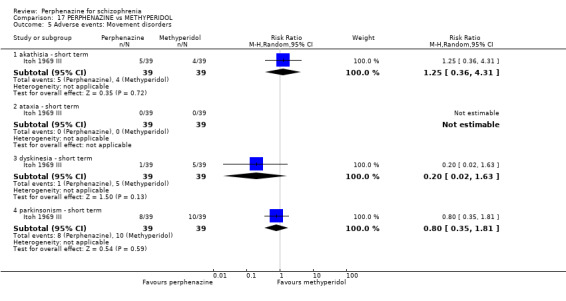
Comparison 17 PERPHENAZINE vs METHYPERIDOL, Outcome 5 Adverse events: Movement disorders.
17.5.2 ataxia ‐ short term
In this subgroup we only found one relevant trial (n = 78) (Itoh 1969 III). There was no significant difference between perphenazine and methyperidol, with no events reported in either group (Analysis 17.5).
17.5.3 dyskinesia ‐ short term
In this subgroup we only found one relevant trial (n = 78) (Itoh 1969 III). There was no significant difference between perphenazine and methyperidol (RR 0.20 CI 0.02 to 1.63, Analysis 17.5).
17.5.4 parkinsonism ‐ short term
In this subgroup we only found one relevant trial (n = 78) (Itoh 1969 III). There was no significant difference between perphenazine and methyperidol (RR 0.80 CI 0.35 to 1.81, Analysis 17.5).
17.6 Other adverse events: 1. Anticholinergic
17.6.1 blurred vision ‐ short term
In this subgroup we only found one relevant trial (n = 78) (Itoh 1969 III). There was no significant difference between perphenazine and methyperidol (RR 0.20 CI 0.01 to 4.04, Analysis 17.6).
17.6. Analysis.
Comparison 17 PERPHENAZINE vs METHYPERIDOL, Outcome 6 Other adverse events: 1. Anticholinergic.
17.6.2 dry mouth ‐ short term
In this subgroup we only found one relevant trial (n = 78) (Itoh 1969 III). There was no significant difference between perphenazine and methyperidol (RR 2.00 CI 0.19 to 21.16, Analysis 17.6).
17.7 Other adverse events: 2. Arousal
17.7.1 insomnia ‐ short term
In this subgroup we only found one relevant trial (n = 78) (Itoh 1969 III). There was no significant difference between perphenazine and methyperidol (RR 0.64 CI 0.28 to 1.47, Analysis 17.7).
17.7. Analysis.
Comparison 17 PERPHENAZINE vs METHYPERIDOL, Outcome 7 Other adverse events: 2. Arousal.
17.7.2 sleepiness/sedation ‐ short term
In this subgroup we only found one relevant trial (n = 78) (Itoh 1969 III). There was no significant difference between perphenazine and methyperidol (RR 0.50 CI 0.10 to 2.57, Analysis 17.7).
17.8 Other adverse events: 3. At least one
17.8.1 short term
In this subgroup we only found one relevant trial (n = 78) (Itoh 1969 III). There was no significant difference between perphenazine and methyperidol (RR 0.96 CI 0.70 to 1.33, Analysis 17.8).
17.8. Analysis.
Comparison 17 PERPHENAZINE vs METHYPERIDOL, Outcome 8 Other adverse events: 3. At least one.
17.9 Other adverse events: 4. Central nervous system
17.9.1 disturbance of consciousness ‐ short term
In this subgroup we only found one relevant trial (n = 78) (Itoh 1969 III). There was no significant difference between perphenazine and methyperidol, with no events reported in either group (Analysis 17.9).
17.9. Analysis.
Comparison 17 PERPHENAZINE vs METHYPERIDOL, Outcome 9 Other adverse events: 4. Central nervous system.
17.9.2 headache ‐ short term
In this subgroup we only found one relevant trial (n = 78) (Itoh 1969 III). There was no significant difference between perphenazine and methyperidol (RR 1.00 CI 0.06 to 15.43, Analysis 17.9).
17.9.3 paraesthesia ‐ short term
In this subgroup we only found one relevant trial (n = 78) (Itoh 1969 III). There was no significant difference between perphenazine and methyperidol, with no events reported in either group (Analysis 17.9).
17.9.4 vertigo ‐ short term
In this subgroup we only found one relevant trial (n = 78) (Itoh 1969 III). There was no significant difference between perphenazine and methyperidol (RR 0.33 CI 0.01 to 7.94, Analysis 17.9).
17.10 Other adverse events: 5. Gastrointestinal
17.10.1 constipation ‐ short term
In this subgroup we only found one relevant trial (n = 78) (Itoh 1969 III). There was no significant difference between perphenazine and methyperidol (RR 0.33 CI 0.01 to 7.94, Analysis 17.10).
17.10. Analysis.
Comparison 17 PERPHENAZINE vs METHYPERIDOL, Outcome 10 Other adverse events: 5. Gastrointestinal.
17.10.2 icterus ‐ short term
In this subgroup we only found one relevant trial (n = 78) (Itoh 1969 III). There was no significant difference between perphenazine and methyperidol, with no events reported in either group (Analysis 17.10).
17.10.3 loss of appetite ‐ short term
In this subgroup we only found one relevant trial (n = 78) (Itoh 1969 III). There was no significant difference between perphenazine and methyperidol (RR 0.60 CI 0.15 to 2.34, Analysis 17.10).
17.10.4 nausea and vomiting ‐ short term
In this subgroup we only found one relevant trial (n = 78) (Itoh 1969 III). There was no significant difference between perphenazine and methyperidol (RR 1.50 CI 0.27 to 8.49, Analysis 17.10).
17.11 Other adverse events: 6. Skin
17.11.1 rash ‐ short term
In this subgroup we only found one relevant trial (n = 78) (Itoh 1969 III). There was no significant difference between perphenazine and methyperidol (RR 1.00 CI 0.06 to 15.43, Analysis 17.11).
17.11. Analysis.
Comparison 17 PERPHENAZINE vs METHYPERIDOL, Outcome 11 Other adverse events: 6. Skin.
17.12 Other adverse events: 7. Genitourinary
17.12.1 micturition disturbances ‐ short term
In this subgroup we only found one relevant trial (n = 78) (Itoh 1969 III). There was no significant difference between perphenazine and methyperidol (RR 0.33 CI 0.01 to 7.94, Analysis 17.12).
17.12. Analysis.
Comparison 17 PERPHENAZINE vs METHYPERIDOL, Outcome 12 Other adverse events: 7. Genitourinary.
17.13 Other adverse events: 8. Others
17.13.1 cramps ‐ short term
In this subgroup we only found one relevant trial (n = 78) (Itoh 1969 III). There was no significant difference between perphenazine and methyperidol, with no events reported in either group (Analysis 17.13).
17.13. Analysis.
Comparison 17 PERPHENAZINE vs METHYPERIDOL, Outcome 13 Other adverse events: 8. Others.
17.13.2 sweating ‐ short term
In this subgroup we only found one relevant trial (n = 78) (Itoh 1969 III). There was no significant difference between perphenazine and methyperidol, with no events reported in either group (Analysis 17.13).
COMPARISON 18: PERPHENAZINE versus OLANZAPINE
18.1 Leaving the study early: 1. Any reason
18.1.1 long term
In this subgroup we found two relevant trials (n = 657). There was a statistically significant difference (P = 0.004) favouring olanzapine over perphenazine (RR 1.16 CI 1.05 to 1.29, Analysis 18.1).
18.1. Analysis.
Comparison 18 PERPHENAZINE vs OLANZAPINE, Outcome 1 Leaving the study early: 1. Any reason.
18.2 Leaving the study early: 2. Due to adverse events
18.2.1 long term
In this subgroup we found two relevant trials (n = 643). There was no significant difference between perphenazine and olanzapine (RR 0.85 CI 0.59 to 1.21, Analysis 18.2).
18.2. Analysis.
Comparison 18 PERPHENAZINE vs OLANZAPINE, Outcome 2 Leaving the study early: 2. Due to adverse events.
18.3 Leaving the study early: 3. Due to relapse/worsening or no improvement
18.3.1 long term
In this subgroup we found two relevant trials (n = 643). There was no significant difference between perphenazine and olanzapine (RR 1.24 CI 0.47 to 3.29, Analysis 18.3). This subgroup had important levels of heterogeneity (Chi2 = 2.39; df = 1; P = 0.122; I2 = 58%).
18.3. Analysis.
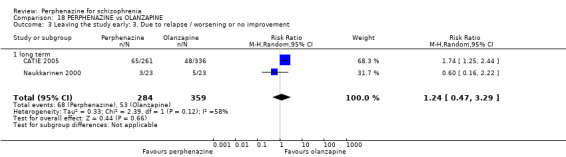
Comparison 18 PERPHENAZINE vs OLANZAPINE, Outcome 3 Leaving the study early: 3. Due to relapse / worsening or no improvement.
18.4 Mental state: 1a. State ‐ BPRS end score (high = poor, skewed data)
18.4.1 long term
Data for this outcome are skewed, and are best inspected by viewing Analysis 18.4.
18.4. Analysis.
Comparison 18 PERPHENAZINE vs OLANZAPINE, Outcome 4 Mental state: 1a. State ‐ BPRS end score (high = poor, skewed data).
| Mental state: 1a. State ‐ BPRS end score (high = poor, skewed data) | ||||
|---|---|---|---|---|
| Study | Intervention: | Mean | SD | N |
| long term | ||||
| Naukkarinen 2000 | Perphenazine | 12.5 | 12.6 | 23 |
| Naukkarinen 2000 | Olanzapine | 11.7 | 12.6 | 23 |
18.5 Mental state: 1b. State ‐ PANSS total endpoint score (high = poor, skewed data)
18.5.1 long term
Data for this outcome are skewed, and are best inspected by viewing Analysis 18.5.
18.5. Analysis.
Comparison 18 PERPHENAZINE vs OLANZAPINE, Outcome 5 Mental state: 1b. State ‐ PANSS total endpoint score (high = poor, skewed data).
| Mental state: 1b. State ‐ PANSS total endpoint score (high = poor, skewed data) | ||||
|---|---|---|---|---|
| Study | Intervention | Mean | SD | N |
| long term | ||||
| Naukkarinen 2000 | Perphenazine | 54.5 | 23.9 | 23 |
| Naukkarinen 2000 | Olanzapine | 53.7 | 23.9 | 23 |
18.6 Mental state: 1c. State ‐ PANSS: mean endpoint score from baseline (high = poor)
18.6.1 total ‐ short term
In this subgroup we found one relevant trial (n = 60). There was a statistically significant difference (P < 0.0001) favouring olanzapine over perphenazine (MD 4.2 CI 2.17 to 6.25, Analysis 18.6).
18.6. Analysis.
Comparison 18 PERPHENAZINE vs OLANZAPINE, Outcome 6 Mental state: 1c. State ‐ PANSS: mean endpoint score from baseline (high = poor).
18.6.2 positive ‐ short term
In this subgroup we only found one relevant trial (n = 60) (Wang 2008b). There was a statistically significant difference (P < 0.00001) favouring olanzapine over perphenazine (MD 8.23 CI 7.26 to 9.20, Analysis 18.6).
18.6.3 negative ‐ short term
In this subgroup we only found one relevant trial (n = 60) (Wang 2008b). There was a statistically significant difference (P = 0.0009) favouring olanzapine over perphenazine (MD 1.58 CI 0.64 to 2.52, Analysis 18.6).
18.6.4 general psychopathology ‐ short term
In this subgroup we only found one relevant trial (n = 60) (Wang 2008b). There was a statistically significant difference (P = 0.05) favouring perphenazine over olanzapine (MD ‐0.39 CI ‐0.79 to 0.01, Analysis 18.6).
18.7 Mental state: 1d. State ‐ less than 25% PANSS reduction ('no effect')
18.7.1 short term
In this subgroup we only found one relevant trial (n = 60) (Wang 2008b). There was no significant difference between perphenazine and olanzapine (RR 2.00 CI 0.40 to 10.11, Analysis 18.7).
18.7. Analysis.
Comparison 18 PERPHENAZINE vs OLANZAPINE, Outcome 7 Mental state: 1d. State ‐ less than 25% PANSS reduction ('no effect').
18.8 Adverse events: Movement disorders
18.8.1 akathisia ‐ long term
In this subgroup we found two relevant trials (n = 591). There was no significant difference between perphenazine and olanzapine (RR 1.33 CI 0.68 to 2.60, Analysis 18.8).
18.8. Analysis.
Comparison 18 PERPHENAZINE vs OLANZAPINE, Outcome 8 Adverse events: Movement disorders.
18.8.2 EPS ‐ long term
In this subgroup we only found one relevant trial (n = 539) (CATIE 2005). There was no significant difference between perphenazine and olanzapine (RR 0.79 CI 0.42 to 1.49, Analysis 18.8).
18.8.3 tardive dyskinesia ‐ long term
In this subgroup we only found one relevant trial (n = 473) (CATIE 2005). There was no significant difference between perphenazine and olanzapine (RR 1.28 CI 0.83 to 1.95, Analysis 18.8).
18.8.4 use of antiparkinsonian drugs ‐ long term
In this subgroup we only found one relevant trial (n = 46) (Naukkarinen 2000). There was no significant difference between perphenazine and olanzapine (RR 3.00 CI 0.34 to 26.76, Analysis 18.8).
18.9 Other adverse events: 1. Anticholinergic
18.9.1 increased salivation ‐ long term
In this subgroup we only found one relevant trial (n = 60) (Naukkarinen 2000). There was no significant difference between perphenazine and olanzapine (RR 0.33 CI 0.01 to 7.87, Analysis 18.9).
18.9. Analysis.
Comparison 18 PERPHENAZINE vs OLANZAPINE, Outcome 9 Other adverse events: 1. Anticholinergic.
18.10 Other adverse events: 2. Arousal
18.10.1 agitation ‐ long term
In this subgroup we only found one relevant trial (n = 60) (Naukkarinen 2000). There was no significant difference between perphenazine and olanzapine (RR 3.00 CI 0.33 to 27.23, Analysis 18.10).
18.10. Analysis.
Comparison 18 PERPHENAZINE vs OLANZAPINE, Outcome 10 Other adverse events: 2. Arousal.
18.10.2 anxiety ‐ long term
In this subgroup we only found one relevant trial (n = 60) (Naukkarinen 2000). There was no significant difference between perphenazine and olanzapine (RR 1.25 CI 0.37 to 4.21, Analysis 18.10).
18.10.3 insomnia ‐ long term
In this subgroup we found two relevant trials (n = 657). There was no significant difference between perphenazine and olanzapine (RR 2.34 CI 0.62 to 8.89, Analysis 18.10). This subgroup had important levels of heterogeneity (Chi2 = 2.11; df = 1; P = 0.146; I2 = 53%).
18.10.4 sleepiness/sedation ‐ long term
In this subgroup we only found one relevant trial (n = 597) (CATIE 2005). There was no significant difference between perphenazine and olanzapine (RR 0.92 CI 0.71 to 1.18, Analysis 18.10).
18.11 Other adverse events: 3. Cardiovascular
18.11.1 bradycardia ‐ long term
In this subgroup we only found one relevant trial (n = 60) (Naukkarinen 2000). There was no significant difference between perphenazine and olanzapine (RR 3.00 CI 0.13 to 70.83, Analysis 18.11).
18.11. Analysis.
Comparison 18 PERPHENAZINE vs OLANZAPINE, Outcome 11 Other adverse events: 3. Cardiovascular.
18.11.2 faintness, dizziness, weakness ‐ long term
In this subgroup we only found one relevant trial (n = 597) (CATIE 2005). There was no significant difference between perphenazine and olanzapine (RR 1.20 CI 0.75 to 1.95, Analysis 18.11).
18.11.3 hypotension ‐ long term
In this subgroup we only found one relevant trial (n = 60) (Naukkarinen 2000). There was no significant difference between perphenazine and olanzapine (RR 0.33 CI 0.01 to 7.87, Analysis 18.11).
18.12 Other adverse events: 4. Central nervous system
18.12.1 headache ‐ long term
In this subgroup we only found one relevant trial (n = 60) (Naukkarinen 2000). There was no significant difference between perphenazine and olanzapine (RR 0.71 CI 0.25 to 2.00, Analysis 18.12).
18.12. Analysis.
Comparison 18 PERPHENAZINE vs OLANZAPINE, Outcome 12 Other adverse events: 4. Central nervous system.
18.12.2 hypertonia ‐ long term
In this subgroup we only found one relevant trial (n = 60) (Naukkarinen 2000). There was no significant difference between perphenazine and olanzapine (RR 3.00 CI 0.13 to 70.83, Analysis 18.12).
18.13 Other adverse events: 5. Gastrointestinal
18.13.1 gastroenteritis ‐ long term
In this subgroup we only found one relevant trial (n = 60) (Naukkarinen 2000). There was no significant difference between perphenazine and olanzapine (RR 3.00 CI 0.13 to 70.83, Analysis 18.13).
18.13. Analysis.
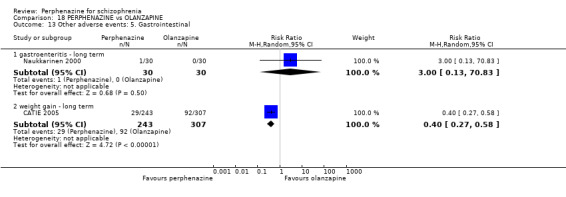
Comparison 18 PERPHENAZINE vs OLANZAPINE, Outcome 13 Other adverse events: 5. Gastrointestinal.
18.13.2 weight gain ‐ long term
In this subgroup we only found one relevant trial (n = 550) (CATIE 2005). There was a statistically significant difference (P < 0.00001) favouring perphenazine over olanzapine (RR 0.40 CI 0.27 to 0.58, Analysis 18.13).
18.14 Other adverse events: 6. Skin
18.14.1 cellulitis ‐ long term
In this subgroup we only found one relevant trial (n = 60) (Naukkarinen 2000). There was no significant difference between perphenazine and olanzapine (RR 0.33 CI 0.01 to 7.87, Analysis 18.14).
18.14. Analysis.
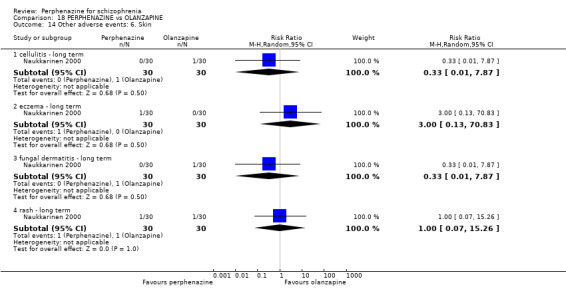
Comparison 18 PERPHENAZINE vs OLANZAPINE, Outcome 14 Other adverse events: 6. Skin.
18.14.2 eczema ‐ long term
In this subgroup we only found one relevant trial (n = 60) (Naukkarinen 2000). There was no significant difference between perphenazine and olanzapine (RR 3.00 CI 0.13 to 70.83, Analysis 18.14).
18.14.3 fungal dermatitis ‐ long term
In this subgroup we only found one relevant trial (n = 60) (Naukkarinen 2000). There was no significant difference between perphenazine and olanzapine (RR 0.33 CI 0.01 to 7.87, Analysis 18.14).
18.14.4 rash ‐ long term
In this subgroup we only found one relevant trial (n = 60) (Naukkarinen 2000). There was no significant difference between perphenazine and olanzapine (RR 1.00 CI 0.07 to 15.26, Analysis 18.14).
18.15 Other adverse events: 7. Genitourinary
18.15.1 diminished sexual desire ‐ long term
In this subgroup we only found one relevant trial (n = 597) (CATIE 2005). There was no significant difference between perphenazine and olanzapine (RR 0.91 CI 0.69 to 1.19, Analysis 18.15).
18.15. Analysis.
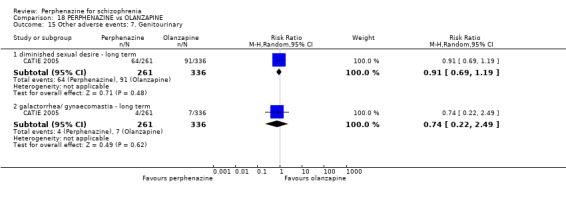
Comparison 18 PERPHENAZINE vs OLANZAPINE, Outcome 15 Other adverse events: 7. Genitourinary.
18.15.2 galactorrhoea/gynaecomastia ‐ long term
In this subgroup we only found one relevant trial (n = 597) (CATIE 2005). There was no significant difference between perphenazine and olanzapine (RR 0.74 CI 0.22 to 2.49, Analysis 18.15).
18.16 Other adverse events: 8. Others
18.16.1 allergic reaction ‐ long term
In this subgroup we only found one relevant trial (n = 60) (Naukkarinen 2000). There was no significant difference between perphenazine and olanzapine (RR 0.33 CI 0.01 to 7.87, Analysis 18.16).
18.16. Analysis.
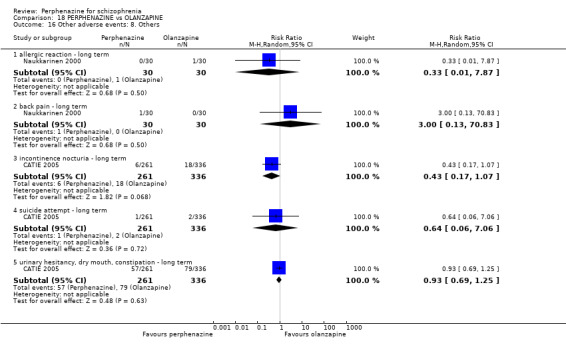
Comparison 18 PERPHENAZINE vs OLANZAPINE, Outcome 16 Other adverse events: 8. Others.
18.16.2 back pain ‐ long term
In this subgroup we only found one relevant trial (n = 60) (Naukkarinen 2000). There was no significant difference between perphenazine and olanzapine (RR 3.00 CI 0.13 to 70.83, Analysis 18.16).
18.16.3 incontinence nocturia ‐ long term
In this subgroup we only found one relevant trial (n = 597) (CATIE 2005). There was no significant difference between perphenazine and olanzapine (RR 0.43 CI 0.17 to 1.07, Analysis 18.16).
18.16.4 suicide attempt ‐ long term
In this subgroup we only found one relevant trial (n = 597) (CATIE 2005). There was no significant difference between perphenazine and olanzapine (RR 0.64 CI 0.06 to 7.06, Analysis 18.16).
18.16.5 urinary hesitancy, dry mouth, constipation ‐ long term
In this subgroup we only found one relevant trial (n = 597) (CATIE 2005). There was no significant difference between perphenazine and olanzapine (RR 0.93 CI 0.69 to 1.25, Analysis 18.16).
18.17 Other adverse events: 9. any serious adverse event
18.17.1 long term
In this subgroup we only found one relevant trial (n = 597) (CATIE 2005). There was no significant difference between perphenazine and olanzapine (RR 1.17 CI 0.72 to 1.88, Analysis 18.17).
18.17. Analysis.
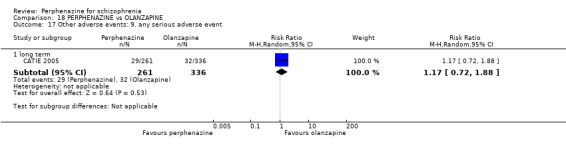
Comparison 18 PERPHENAZINE vs OLANZAPINE, Outcome 17 Other adverse events: 9. any serious adverse event.
18.18 Other adverse events: 10. lab data
18.18.1 anaemia ‐ long term
In this subgroup we only found one relevant trial (n = 60) (Naukkarinen 2000). There was no significant difference between perphenazine and olanzapine (RR 1.00 CI 0.07 to 15.26, Analysis 18.18).
18.18. Analysis.
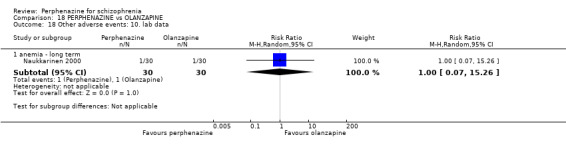
Comparison 18 PERPHENAZINE vs OLANZAPINE, Outcome 18 Other adverse events: 10. lab data.
18.19 Other adverse events: 11a. lab data
18.19.1 mean weight change (lb) ‐ long term
In this subgroup we only found one relevant trial (n = 597) (CATIE 2005). There was a statistically significant difference (P < 0.00001) favouring perphenazine over olanzapine (MD ‐11.40 CI ‐14.19 to ‐8.61, Analysis 18.19).
18.19. Analysis.
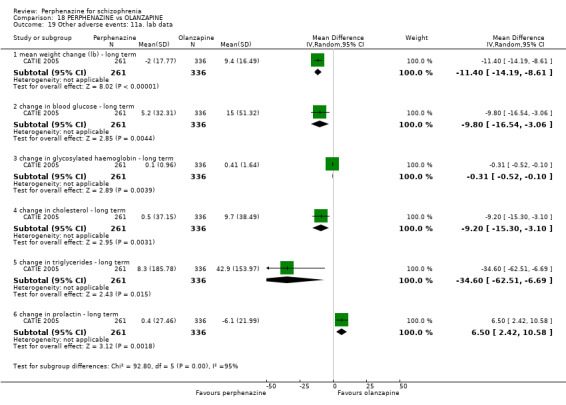
Comparison 18 PERPHENAZINE vs OLANZAPINE, Outcome 19 Other adverse events: 11a. lab data.
18.19.2 change in blood glucose ‐ long term
In this subgroup we only found one relevant trial (n = 597) (CATIE 2005). There was a statistically significant difference (P = 0.004) favouring perphenazine over olanzapine (MD ‐9.80 CI ‐16.54 to ‐3.06, Analysis 18.19).
18.19.3 change in glycosylated haemoglobin ‐ long term
In this subgroup we only found one relevant trial (n = 597) (CATIE 2005). There was a statistically significant difference (P = 0.004) favouring perphenazine over olanzapine (MD ‐0.31 CI ‐0.52 to ‐0.10, Analysis 18.19).
18.19.4 change in cholesterol ‐ long term
In this subgroup we only found one relevant trial (n = 597) (CATIE 2005). There was a statistically significant difference (P = 0.003) favouring perphenazine over olanzapine (MD ‐9.20 CI ‐15.30 to ‐3.10, Analysis 18.19).
18.19.5 change in triglycerides ‐ long term
In this subgroup we only found one relevant trial (n = 597) (CATIE 2005). There was a statistically significant difference (P = 0.002) favouring perphenazine over olanzapine (MD ‐34.60 CI ‐62.51 to ‐6.69, Analysis 18.19).
18.19.6 change in prolactin ‐ long term
In this subgroup we only found one relevant trial (n = 597) (CATIE 2005). There was a statistically significant difference (P = 0.002) favouring olanzapine over perphenazine (MD 6.50 CI 2.42 to 10.58, Analysis 18.19).
18.20 Other adverse events: 11b. lab data (skewed)
18.20.1 weight change
Data for this outcome are skewed and are best inspected by viewing Analysis 18.20.
18.20. Analysis.
Comparison 18 PERPHENAZINE vs OLANZAPINE, Outcome 20 Other adverse events: 11b. lab data (skewed).
| Other adverse events: 11b. lab data (skewed) | ||||
|---|---|---|---|---|
| Study | Intervention | Mean | SD | N |
| weight change | ||||
| Naukkarinen 2000 | Perphenazine | 0.37 | 3.19 | 23 |
| Naukkarinen 2000 | Olanzapine | 2.63 | 4.97 | 23 |
COMPARISON 19: PERPHENAZINE versus PENFLURIDOL
19.1 Leaving the study early: 1. Any reason
19.1.1 short term
In this subgroup we only found one relevant trial (n = 105) (Itoh 1976). There was no significant difference between perphenazine and penfluridol (RR 0.49 CI 0.20 to 1.19, Analysis 19.1).
19.1. Analysis.
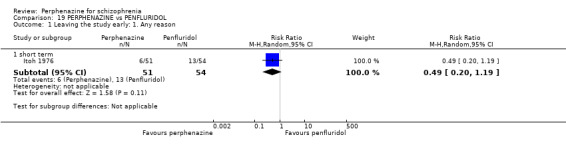
Comparison 19 PERPHENAZINE vs PENFLURIDOL, Outcome 1 Leaving the study early: 1. Any reason.
19.2 Global state: 1. Change over time ‐ no better or deterioration (ITT)
19.2.1 short term
In this subgroup we only found one relevant trial (n = 105) (Itoh 1976). There was no significant difference between perphenazine and penfluridol (RR 0.92 CI 0.64 to 1.31, Analysis 19.2).
19.2. Analysis.
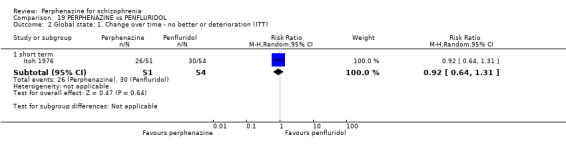
Comparison 19 PERPHENAZINE vs PENFLURIDOL, Outcome 2 Global state: 1. Change over time ‐ no better or deterioration (ITT).
19.3 Adverse events: Movement disorders
19.3.1 akathisia ‐ short term
In this subgroup we only found one relevant trial (n = 105) (Itoh 1976). There was no significant difference between perphenazine and penfluridol (RR 0.66 CI 0.39 to 1.11, Analysis 19.3).
19.3. Analysis.
Comparison 19 PERPHENAZINE vs PENFLURIDOL, Outcome 3 Adverse events: Movement disorders.
19.3.2 dyskinesia ‐ short term
In this subgroup we only found one relevant trial (n = 105) (Itoh 1976). There was no significant difference between perphenazine and penfluridol (RR 0.82 CI 0.33 to 2.05, Analysis 19.3).
19.3.3 parkinsonism ‐ short term
In this subgroup we only found one relevant trial (n = 105) (Itoh 1976). There was no significant difference between perphenazine and penfluridol (RR 1.13 CI 0.80 to 1.60, Analysis 19.3).
19.4 Other adverse events: 1. Anticholinergic
19.4.1 blurred vision ‐ short term
In this subgroup we only found one relevant trial (n = 105) (Itoh 1976). There was no significant difference between perphenazine and penfluridol (RR 1.06 CI 0.15 to 7.24, Analysis 19.4).
19.4. Analysis.
Comparison 19 PERPHENAZINE vs PENFLURIDOL, Outcome 4 Other adverse events: 1. Anticholinergic.
19.4.2 dry mouth ‐ short term
In this subgroup we only found one relevant trial (n = 105) (Itoh 1976). There was no significant difference between perphenazine and penfluridol (RR 1.32 CI 0.57 to 3.09, Analysis 19.4).
19.5 Other adverse events: 2. Arousal
19.5.1 drowsiness ‐ short term
In this subgroup we only found one relevant trial (n = 105) (Itoh 1976). There was no significant difference between perphenazine and penfluridol (RR 0.98 CI 0.49 to 1.94, Analysis 19.5).
19.5. Analysis.
Comparison 19 PERPHENAZINE vs PENFLURIDOL, Outcome 5 Other adverse events: 2. Arousal.
19.5.2 insomnia ‐ short term
In this subgroup we only found one relevant trial (n = 105) (Itoh 1976). There was no significant difference between perphenazine and penfluridol (RR 0.97 CI 0.62 to 1.52, Analysis 19.5).
19.6 Other adverse events: 3. Cardiovascular
19.6.1 angina pectoris ‐ short term
In this subgroup we only found one relevant trial (n = 105) (Itoh 1976). There was no significant difference between perphenazine and penfluridol (RR 0.53 CI 0.17 to 1.65, Analysis 19.6).
19.6. Analysis.
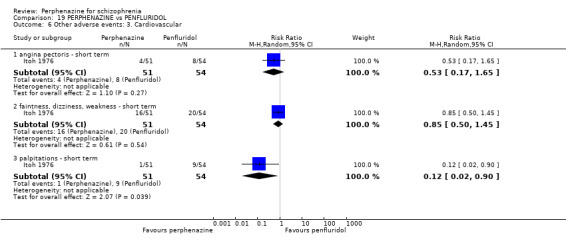
Comparison 19 PERPHENAZINE vs PENFLURIDOL, Outcome 6 Other adverse events: 3. Cardiovascular.
19.6.2 faintness, dizziness, weakness ‐ short term
In this subgroup we only found one relevant trial (n = 105) (Itoh 1976). There was no significant difference between perphenazine and penfluridol (RR 0.85 CI 0.50 to 1.45, Analysis 19.6).
19.6.3 palpitations ‐ short term
In this subgroup we only found one relevant trial (n = 105) (Itoh 1976). There was a statistically significant difference (P = 0.04) favouring perphenazine over penfluridol (RR 0.12 CI 0.02 to 0.90, Analysis 19.6).
19.7 Other adverse events: 4. Central nervous system
19.7.1 headache ‐ short term
In this subgroup we only found one relevant trial (n = 105) (Itoh 1976). There was no significant difference between perphenazine and penfluridol (RR 0.79 CI 0.37 to 1.72, Analysis 19.7).
19.7. Analysis.
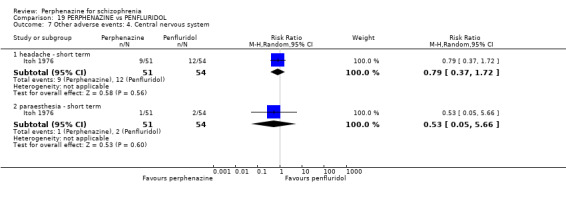
Comparison 19 PERPHENAZINE vs PENFLURIDOL, Outcome 7 Other adverse events: 4. Central nervous system.
19.7.2 paraesthesia ‐ short term
In this subgroup we only found one relevant trial (n = 105) (Itoh 1976). There was no significant difference between perphenazine and penfluridol (RR 0.53 CI 0.05 to 5.66, Analysis 19.7).
19.8 Other adverse events: 5. Gastrointestinal
19.8.1 constipation ‐ short term
In this subgroup we only found one relevant trial (n = 105) (Itoh 1976). There was no significant difference between perphenazine and penfluridol (RR 1.38 CI 0.66 to 2.86, Analysis 19.8).
19.8. Analysis.
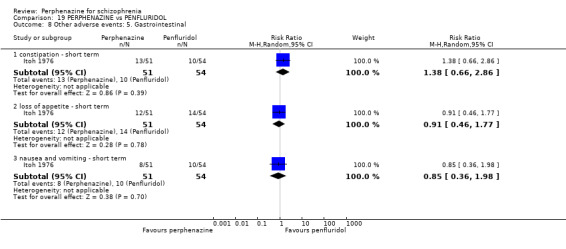
Comparison 19 PERPHENAZINE vs PENFLURIDOL, Outcome 8 Other adverse events: 5. Gastrointestinal.
19.8.2 loss of appetite ‐ short term
In this subgroup we only found one relevant trial (n = 105) (Itoh 1976). There was no significant difference between perphenazine and penfluridol (RR 0.91 CI 0.46 to 1.77, Analysis 19.8).
19.8.3 nausea and vomiting ‐ short term
In this subgroup we only found one relevant trial (n = 105) (Itoh 1976). There was no significant difference between perphenazine and penfluridol (RR 0.85 CI 0.36 to 1.98, Analysis 19.8).
19.9 Other adverse events: 6. Skin
19.9.1 oedema
In this subgroup we only found one relevant trial (n = 105) (Itoh 1976). There was no significant difference between perphenazine and penfluridol (RR 3.17 CI 0.13 to 76.16, Analysis 19.9).
19.9. Analysis.
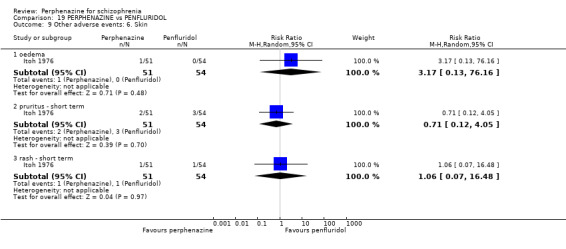
Comparison 19 PERPHENAZINE vs PENFLURIDOL, Outcome 9 Other adverse events: 6. Skin.
19.9.2 pruritus ‐ short term
In this subgroup we only found one relevant trial (n = 105) (Itoh 1976). There was no significant difference between perphenazine and penfluridol (RR 0.71 CI 0.12 to 4.05, Analysis 19.9).
19.9.3 rash ‐ short term
In this subgroup we only found one relevant trial (n = 105) (Itoh 1976). There was no significant difference between perphenazine and penfluridol (RR 1.06 CI 0.07 to 16.48, Analysis 19.9).
19.10 Other adverse events: 7. Genitourinary
19.10.1 diminished sexual desire
In this subgroup we only found one relevant trial (n = 105) (Itoh 1976). There was no significant difference between perphenazine and penfluridol, with no events reported in either group (Analysis 19.10).
19.10. Analysis.
Comparison 19 PERPHENAZINE vs PENFLURIDOL, Outcome 10 Other adverse events: 7. Genitourinary.
19.10.2 increased sexual desire ‐ short term
In this subgroup we only found one relevant trial (n = 105) (Itoh 1976). There was no significant difference between perphenazine and penfluridol, with no events reported in either group (Analysis 19.10).
19.10.3 micturition disturbances ‐ short term
In this subgroup we only found one relevant trial (n = 105) (Itoh 1976). There was no significant difference between perphenazine and penfluridol (RR 1.06 CI 0.15 to 7.24, Analysis 19.10).
19.11 Other adverse events: 8. Others
19.11.1 sweating ‐ short term
In this subgroup we only found one relevant trial (n = 105) (Itoh 1976). There was no significant difference between perphenazine and penfluridol (RR 0.71 CI 0.12 to 4.05, Analysis 19.11).
19.11. Analysis.
Comparison 19 PERPHENAZINE vs PENFLURIDOL, Outcome 11 Other adverse events: 8. Others.
COMPARISON 20: PERPHENAZINE versus PROCHLORPERAZINE
20.1 Leaving the study early: 1. Any reason
20.1.1 short term
In this subgroup we found three relevant trials (n = 175). There was no significant difference between perphenazine and prochlorperazine (RR 0.90 CI 0.37 to 2.19, Analysis 20.1). This subgroup had important levels of heterogeneity (Chi2 = 9.87; df = 1; P = 0.002; I2 = 90%).
20.1. Analysis.
Comparison 20 PERPHENAZINE vs PROCHLORPERAZINE, Outcome 1 Leaving the study early: 1. Any reason.
20.2 Leaving the study early: 2. Due to adverse events
20.2.1 short term
In this subgroup we found two relevant trials (n = 165). There was no significant difference between perphenazine and prochlorperazine (RR 2.37 CI 0.80 to 7.05, Analysis 20.2).
20.2. Analysis.
Comparison 20 PERPHENAZINE vs PROCHLORPERAZINE, Outcome 2 Leaving the study early: 2. Due to adverse events.
20.3 Global state: 1. Change over time ‐ no better or deterioration (ITT)
20.3.1 short term
In this subgroup we only found one relevant trial (n = 60) (Kurland 1961). There was no significant difference between perphenazine and prochlorperazine (RR 0.67 CI 0.24 to 1.87, Analysis 20.3).
20.3. Analysis.
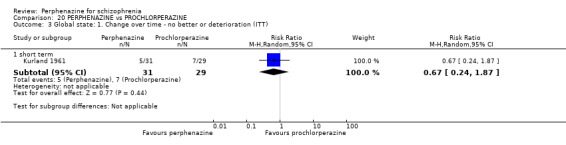
Comparison 20 PERPHENAZINE vs PROCHLORPERAZINE, Outcome 3 Global state: 1. Change over time ‐ no better or deterioration (ITT).
20.4 Adverse events: Movement disorders
20.4.1 akathisia ‐ short term
In this subgroup we only found one relevant trial (n = 105) (Hanlon 1965). There was no significant difference between perphenazine and prochlorperazine (RR 2.29 CI 0.63 to 8.38, Analysis 20.4).
20.4. Analysis.
Comparison 20 PERPHENAZINE vs PROCHLORPERAZINE, Outcome 4 Adverse events: Movement disorders.
20.4.2 akinesia ‐ short term
In this subgroup we only found one relevant trial (n = 105) (Hanlon 1965). There was no significant difference between perphenazine and prochlorperazine (RR 0.33 CI 0.01 to 7.85, Analysis 20.4).
20.4.3 dyskinesia ‐ short term
In this subgroup we only found one relevant trial (n = 105) (Hanlon 1965). There was no significant difference between perphenazine and prochlorperazine (RR 1.96 CI 0.18 to 20.99, Analysis 20.4).
20.4.4 parkinsonism ‐ short term
In this subgroup we only found one relevant trial (n = 105) (Hanlon 1965). There was no significant difference between perphenazine and prochlorperazine (RR 1.96 CI 0.63 to 6.12, Analysis 20.4).
20.5 Other adverse events: 1. Central nervous system
20.5.1 electroencephalogram (increase) ‐ short term
In this subgroup we only found one relevant trial (n = 10) (Bennett 1961). There was no significant difference between perphenazine and prochlorperazine (RR 0.33 CI 0.05 to 2.21, Analysis 20.5).
20.5. Analysis.
Comparison 20 PERPHENAZINE vs PROCHLORPERAZINE, Outcome 5 Other adverse events: 1. Central nervous system.
COMPARISON 21: PERPHENAZINE versus QUETIAPINE
21.1 Leaving the study early: 1. Any reason
21.1.1 short term
In this subgroup we only found one relevant trial (n = 64) (Wang 2008c). There was no significant difference between perphenazine and quetiapine (RR 3.00 CI 0.33 to 27.33, Analysis 21.1).
21.1. Analysis.
Comparison 21 PERPHENAZINE vs QUETIAPINE, Outcome 1 Leaving the study early: 1. Any reason.
21.1.2 long term
In this subgroup we only found one relevant trial (n = 598) (CATIE 2005). There was a statistically significant difference (P = 0.04) favouring perphenazine over quetiapine (RR 0.91 CI 0.84 to 1.00, Analysis 21.1).
21.2 Leaving the study early: 2. Due to adverse events
21.2.1 long term
In this subgroup we only found one relevant trial (n = 598) (CATIE 2005). There was no significant difference between perphenazine and quetiapine (RR 1.05 CI 0.72 to 1.55, Analysis 21.2).
21.2. Analysis.
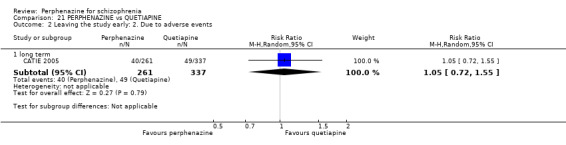
Comparison 21 PERPHENAZINE vs QUETIAPINE, Outcome 2 Leaving the study early: 2. Due to adverse events.
21.3 Leaving the study early: 3. Due to relapse/worsening or no improvement
21.3.1 long term
In this subgroup we only found one relevant trial (n = 598) (CATIE 2005). There was no significant difference between perphenazine and quetiapine (RR 0.91 CI 0.69 to 1.20, Analysis 21.3).
21.3. Analysis.
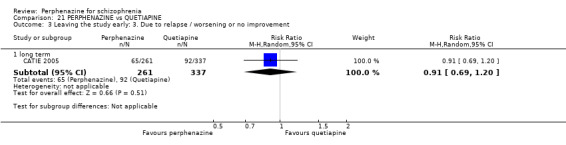
Comparison 21 PERPHENAZINE vs QUETIAPINE, Outcome 3 Leaving the study early: 3. Due to relapse / worsening or no improvement.
21.4 Mental state: 2a. State ‐ BPRS score reduction ('no effect')
21.4.1 less than 25% reduction ‐ short term
In this subgroup we only found one relevant trial (n = 64) (Wang 2008c). There was no significant difference between perphenazine and quetiapine (RR 1.33 CI 0.32 to 5.49, Analysis 21.4).
21.4. Analysis.
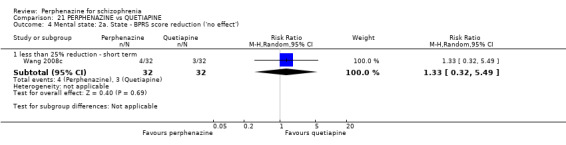
Comparison 21 PERPHENAZINE vs QUETIAPINE, Outcome 4 Mental state: 2a. State ‐ BPRS score reduction ('no effect').
21.5 Mental state: 2b. State ‐ BPRS total score (high = worse)
21.5.1 short term
In this subgroup we only found one relevant trial (n = 64) (Wang 2008c). There was no significant difference between perphenazine and quetiapine (MD 1.10 CI ‐1.26 to 3.46, Analysis 21.5).
21.5. Analysis.
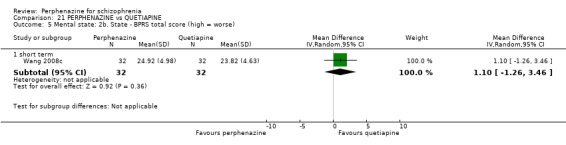
Comparison 21 PERPHENAZINE vs QUETIAPINE, Outcome 5 Mental state: 2b. State ‐ BPRS total score (high = worse).
21.6 Adverse events: Movement disorders
21.6.1 akathisia ‐ short term
In this subgroup we only found one relevant trial (n = 64) (Wang 2008c). There was no significant difference between perphenazine and quetiapine (RR 7.00 CI 0.91 to 53.68, Analysis 21.6).
21.6. Analysis.
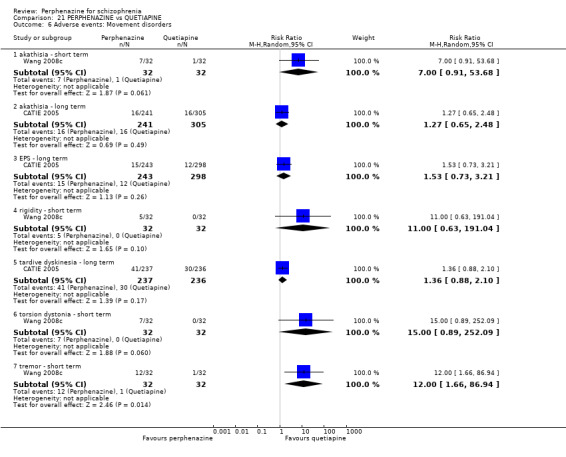
Comparison 21 PERPHENAZINE vs QUETIAPINE, Outcome 6 Adverse events: Movement disorders.
21.6.2 akathisia ‐ long term
In this subgroup we only found one relevant trial (n = 546) (CATIE 2005). There was no significant difference between perphenazine and quetiapine (RR 1.27 CI 0.65 to 2.48, Analysis 21.6).
21.6.3 EPS ‐ long term
In this subgroup we only found one relevant trial (n = 541) (CATIE 2005). There was no significant difference between perphenazine and quetiapine (RR 1.53 CI 0.73 to 3.21, Analysis 21.6).
21.6.4 rigidity ‐ short term
In this subgroup we only found one relevant trial (n = 64) (Wang 2008c). There was no significant difference between perphenazine and quetiapine (RR 11.00 CI 0.63 to 191.04, Analysis 21.6).
21.6.5 tardive dyskinesia ‐ long term
In this subgroup we only found one relevant trial (n = 473) (CATIE 2005). There was no significant difference between perphenazine and quetiapine (RR 1.36 CI 0.88 to 2.10, Analysis 21.6).
21.6.6 torsion dystonia ‐ short term
In this subgroup we only found one relevant trial (n = 64) (Wang 2008c). There was no significant difference between perphenazine and quetiapine (RR 15.00 CI 0.89 to 252.09, Analysis 21.6).
21.6.7 tremor ‐ short term
In this subgroup we only found one relevant trial (n = 64) (Wang 2008c). There was a statistically significant difference (P = 0.01) favouring perphenazine over quetiapine (RR 12.00 CI 1.66 to 86.94, Analysis 21.6).
21.7 Other adverse events: 1. Anticholinergic
21.7.1 blurred vision ‐ short term
In this subgroup we only found one relevant trial (n = 64) (Wang 2008c). There was a statistically significant difference (P = 0.04) favouring quetiapine over perphenazine (RR 4.50 CI 1.05 to 19.22, Analysis 21.7).
21.7. Analysis.
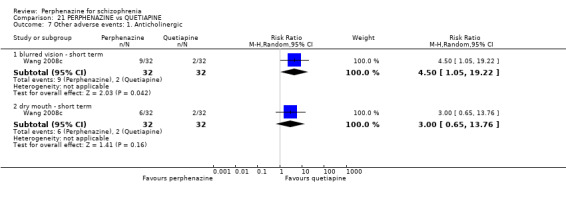
Comparison 21 PERPHENAZINE vs QUETIAPINE, Outcome 7 Other adverse events: 1. Anticholinergic.
21.7.2 dry mouth ‐ short term
In this subgroup we only found one relevant trial (n = 64) (Wang 2008c). There was no significant difference between perphenazine and quetiapine (RR 3.00 CI 0.65 to 13.76, Analysis 21.7).
21.8 Other adverse events: 2. Arousal
21.8.1 drowsiness ‐ short term
In this subgroup we only found one relevant trial (n = 64) (Wang 2008c). There was no significant difference between perphenazine and quetiapine (RR 0.67 CI 0.12 to 3.73, Analysis 21.8).
21.8. Analysis.
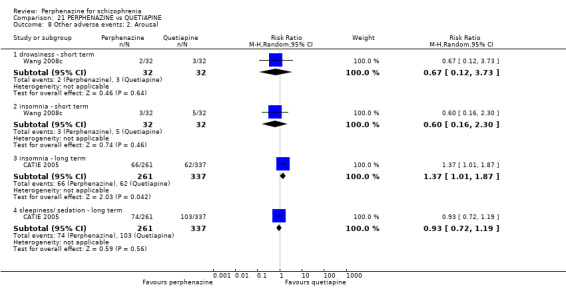
Comparison 21 PERPHENAZINE vs QUETIAPINE, Outcome 8 Other adverse events: 2. Arousal.
21.8.2 insomnia ‐ short term
In this subgroup we only found one relevant trial (n = 64) (Wang 2008c). There was no significant difference between perphenazine and quetiapine (RR 0.60 CI 0.16 to 2.30, Analysis 21.8).
21.8.3 insomnia ‐ long term
In this subgroup we only found one relevant trial (n = 598) (CATIE 2005). There was a statistically significant difference (P = 0.04) favouring quetiapine over perphenazine (RR 1.37 CI 1.01 to 1.87, Analysis 21.8).
21.8.4 sleepiness/sedation ‐ long term
In this subgroup we only found one relevant trial (n = 598) (CATIE 2005). There was no significant difference between perphenazine and quetiapine (RR 0.93 CI 0.72 to 1.19, Analysis 21.8).
21.9 Other adverse events: 3. Cardiovascular
21.9.1 faintness, dizziness, weakness ‐ long term
In this subgroup we only found one relevant trial (n = 598) (CATIE 2005). There was no significant difference between perphenazine and quetiapine (RR 0.99 CI 0.62 to 1.55, Analysis 21.9).
21.9. Analysis.
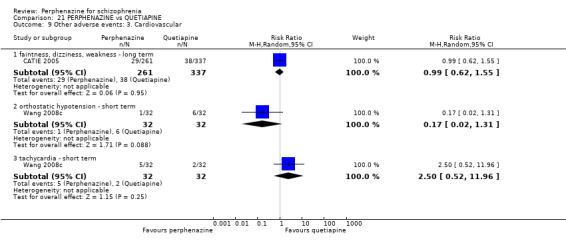
Comparison 21 PERPHENAZINE vs QUETIAPINE, Outcome 9 Other adverse events: 3. Cardiovascular.
21.9.2 orthostatic hypotension ‐ short term
In this subgroup we only found one relevant trial (n = 64) (Wang 2008c). There was no significant difference between perphenazine and quetiapine (RR 0.17 CI 0.02 to 1.31, Analysis 21.9).
21.9.3 tachycardia ‐ short term
In this subgroup we only found one relevant trial (n = 64) (Wang 2008c). There was no significant difference between perphenazine and quetiapine (RR 2.50 CI 0.52 to 11.96, Analysis 21.9).
21.10 Other adverse events: 4. Gastrointestinal
21.10.1 constipation ‐ short term
In this subgroup we only found one relevant trial (n = 64) (Wang 2008c). There was no significant difference between perphenazine and quetiapine (RR 2.50 CI 0.52 to 11.96, Analysis 21.10).
21.10. Analysis.
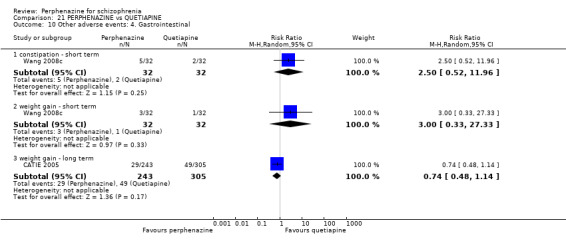
Comparison 21 PERPHENAZINE vs QUETIAPINE, Outcome 10 Other adverse events: 4. Gastrointestinal.
21.10.2 weight gain ‐ short term
In this subgroup we only found one relevant trial (n = 64) (Wang 2008c). There was no significant difference between perphenazine and quetiapine (RR 3.00 CI 0.33 to 27.33, Analysis 21.10).
21.10.3 weight gain ‐ long term
In this subgroup we only found one relevant trial (n = 548) (CATIE 2005). There was no significant difference between perphenazine and quetiapine (RR 0.74 CI 0.48 to 1.14, Analysis 21.10).
21.11 Other adverse events: 5. Genitourinary
21.11.1 diminished sexual desire ‐ long term
In this subgroup we only found one relevant trial (n = 598) (CATIE 2005). There was no significant difference between perphenazine and quetiapine (RR 1.20 CI 0.89 to 1.62, Analysis 21.11).
21.11. Analysis.
Comparison 21 PERPHENAZINE vs QUETIAPINE, Outcome 11 Other adverse events: 5. Genitourinary.
21.11.2 galactorrhoea/gynaecomastia ‐ long term
In this subgroup we only found one relevant trial (n = 598) (CATIE 2005). There was no significant difference between perphenazine and quetiapine (RR 0.86 CI 0.25 to 3.02, Analysis 21.11).
21.12 Other adverse events: 6. Others
21.12.1 urinary hesitancy, dry mouth, constipation ‐ long term
In this subgroup we only found one relevant trial (n = 598) (CATIE 2005). There was a statistically significant difference (P = 0.01) favouring perphenazine over quetiapine (RR 0.70 CI 0.53 to 0.93, Analysis 21.12).
21.12. Analysis.
Comparison 21 PERPHENAZINE vs QUETIAPINE, Outcome 12 Other adverse events: 6. Others.
21.12.2 suicide attempt ‐ long term
In this subgroup we only found one relevant trial (n = 598) (CATIE 2005). There was no significant difference between perphenazine and quetiapine (RR 1.29 CI 0.08 to 20.55, Analysis 21.12).
21.12.3 incontinence nocturia ‐ long term
In this subgroup we only found one relevant trial (n = 598) (CATIE 2005). There was no significant difference between perphenazine and quetiapine (RR 0.52 CI 0.20 to 1.31, Analysis 21.12).
21.13 Other adverse events: 7. any serious adverse event
21.13.1 long term
In this subgroup we only found one relevant trial (n = 598) (CATIE 2005). There was no significant difference between perphenazine and quetiapine (RR 1.17 CI 0.73 to 1.88, Analysis 21.13).
21.13. Analysis.
Comparison 21 PERPHENAZINE vs QUETIAPINE, Outcome 13 Other adverse events: 7. any serious adverse event.
21.14 Other adverse events: 8a. lab data
21.14.1 abnormal ECG ‐ short term
In this subgroup we only found one relevant trial (n = 64) (Wang 2008c). There was no significant difference between perphenazine and quetiapine (RR 7.00 CI 0.91 to 53.68, Analysis 21.14).
21.14. Analysis.
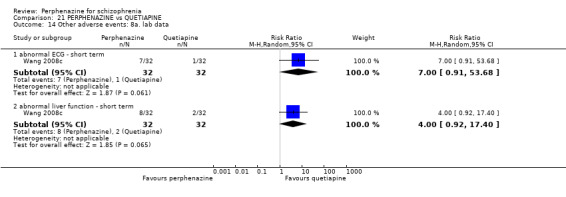
Comparison 21 PERPHENAZINE vs QUETIAPINE, Outcome 14 Other adverse events: 8a. lab data.
21.14.2 abnormal liver function ‐ short term
In this subgroup we only found one relevant trial (n = 64) (Wang 2008c). There was no significant difference between perphenazine and quetiapine (RR 4.00 CI 0.92 to 17.40, Analysis 21.14).
21.15 Other adverse events: 8b. lab data
21.15.1 mean weight change (lb) ‐ long term
In this subgroup we only found one relevant trial (n = 598) (CATIE 2005). There was a statistically significant difference (P = 0.03) favouring perphenazine over quetiapine (MD ‐3.10 CI ‐5.89 to ‐0.31, Analysis 21.15).
21.15. Analysis.
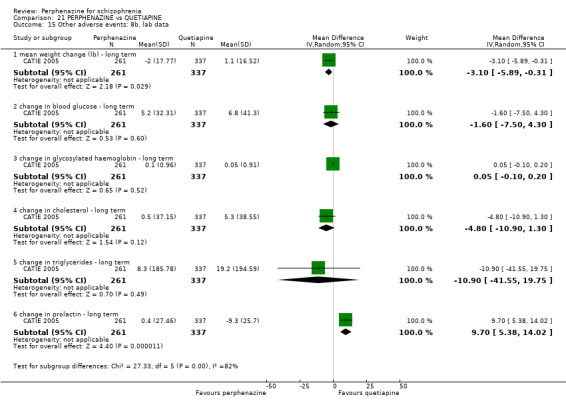
Comparison 21 PERPHENAZINE vs QUETIAPINE, Outcome 15 Other adverse events: 8b. lab data.
21.15.2 change in blood glucose ‐ long term
In this subgroup we only found one relevant trial (n = 598) (CATIE 2005). There was no significant difference between perphenazine and quetiapine (MD ‐1.60 CI ‐7.50 to 4.30, Analysis 21.15).
21.15.3 change in glycosylated haemoglobin ‐ long term
In this subgroup we only found one relevant trial (n = 598) (CATIE 2005). There was a statistically significant difference between perphenazine and quetiapine (MD 0.05 CI ‐0.10 to 0.20, Analysis 21.15).
21.15.4 change in cholesterol ‐ long term
In this subgroup we only found one relevant trial (n = 598) (CATIE 2005). There was no significant difference between perphenazine and quetiapine (MD ‐4.80 CI ‐10.90 to 1.30, Analysis 21.15).
21.15.5 change in triglycerides ‐ long term
In this subgroup we only found one relevant trial (n = 598) (CATIE 2005). There was no significant difference between perphenazine and quetiapine (MD ‐10.90 CI ‐41.55 to 19.75, Analysis 21.15).
21.15.6 change in prolactin ‐ long term
In this subgroup we only found one relevant trial (n = 598) (CATIE 2005). There was a statistically significant difference (P < 0.0001) favouring quetiapine over perphenazine (MD 9.70 CI 5.38 to 14.02, Analysis 21.15).
COMPARISON 22: PERPHENAZINE versus PROMAZINE
22.1 Leaving the study early: 1. Any reason
22.1.1 short term
In this subgroup we only found one relevant trial (n = 60) (Kurland 1961). There was no significant difference between perphenazine and promazine (RR 1.20 CI 0.93 to 1.56, Analysis 22.1).
22.1. Analysis.
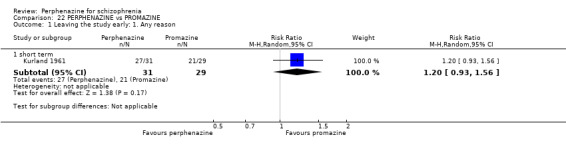
Comparison 22 PERPHENAZINE vs PROMAZINE, Outcome 1 Leaving the study early: 1. Any reason.
22.2 Leaving the study early: 2. Due to adverse events
22.2.1 short term
In this subgroup we only found one relevant trial (n = 60) (Kurland 1961). There was no significant difference between perphenazine and promazine (RR 2.49 CI 0.73 to 8.50, Analysis 22.2).
22.2. Analysis.
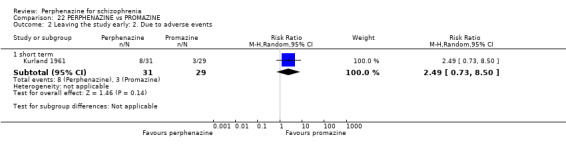
Comparison 22 PERPHENAZINE vs PROMAZINE, Outcome 2 Leaving the study early: 2. Due to adverse events.
22.3 Global state: 1. Change over time ‐ no better or deterioration (ITT)
22.3.1 short term
In this subgroup we only found one relevant trial (n = 60) (Kurland 1961). There was a statistically significant difference (P = 0.04) favouring perphenazine over promazine (RR 0.39 CI 0.16 to 0.97, Analysis 22.3).
22.3. Analysis.
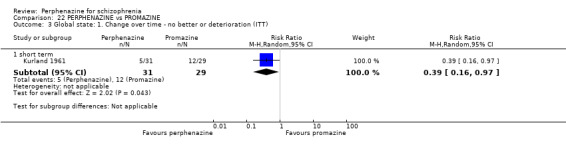
Comparison 22 PERPHENAZINE vs PROMAZINE, Outcome 3 Global state: 1. Change over time ‐ no better or deterioration (ITT).
COMPARISON 23: PERPHENAZINE versus RISPERIDONE
23.1 Leaving the study early: 1. Any reason
23.1.1 short term
In this subgroup we found two relevant trials (n = 709). There was no significant difference between perphenazine and risperidone (RR 1.01 CI 0.92 to 1.11, Analysis 23.1).
23.1. Analysis.
Comparison 23 PERPHENAZINE vs RISPERIDONE, Outcome 1 Leaving the study early: 1. Any reason.
23.2 Leaving the study early: 2. Due to adverse events
23.2.1 short term
In this subgroup we found two relevant trials (n = 709). There was no significant difference between perphenazine and risperidone (RR 1.29 CI 0.73 to 2.29, Analysis 23.2). This subgroup had moderate levels of heterogeneity (Chi2 = 1.45; df = 1; P = 0.229; I2 = 31%).
23.2. Analysis.
Comparison 23 PERPHENAZINE vs RISPERIDONE, Outcome 2 Leaving the study early: 2. Due to adverse events.
23.3 Leaving the study early: 3. Due to relapse/worsening or no improvement
23.3.1 short term
In this subgroup we found two relevant trials (n = 709). There was no significant difference between perphenazine and risperidone (RR 0.95 CI 0.72 to 1.24, Analysis 23.3).
23.3. Analysis.
Comparison 23 PERPHENAZINE vs RISPERIDONE, Outcome 3 Leaving the study early: 3. Due to relapse / worsening or no improvement.
23.4 Global state: 1. Change over time ‐ no better or deterioration (ITT)
23.4.1 short term
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was no significant difference between perphenazine and risperidone (RR 1.29 CI 0.79 to 2.12, Analysis 23.4).
23.4. Analysis.
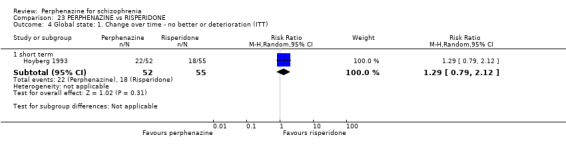
Comparison 23 PERPHENAZINE vs RISPERIDONE, Outcome 4 Global state: 1. Change over time ‐ no better or deterioration (ITT).
23.5 Mental state: 1a. State ‐ less than 20% BPRS reduction ('no effect')
23.5.1 short term
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was no significant difference between perphenazine and risperidone (RR 0.81 CI 0.61 to 1.08, Analysis 23.5).
23.5. Analysis.
Comparison 23 PERPHENAZINE vs RISPERIDONE, Outcome 5 Mental state: 1a. State ‐ less than 20% BPRS reduction ('no effect').
23.6 Mental state: 1b. State ‐ PANSS reduction ('no effect')
23.6.1 less than 20% reduction ‐ short term
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was no significant difference between perphenazine and risperidone (RR 0.86 CI 0.64 to 1.15, Analysis 23.6).
23.6. Analysis.
Comparison 23 PERPHENAZINE vs RISPERIDONE, Outcome 6 Mental state: 1b. State ‐ PANSS reduction ('no effect').
23.6.2 less than 40% reduction ‐ short term
In this subgroup we only found one relevant trial (n = 64) (Wang 2008a). There was no significant difference between perphenazine and risperidone (RR 1.70 CI 0.30 to 9.50, Analysis 23.6).
23.7 Mental state: 1c. State ‐ PANSS: mean change from baseline to endpoint
23.7.1 short term
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was no significant difference between perphenazine and risperidone (MD 4.00 CI ‐5.04 to 13.04, Analysis 23.7).
23.7. Analysis.
Comparison 23 PERPHENAZINE vs RISPERIDONE, Outcome 7 Mental state: 1c. State ‐ PANSS: mean change from baseline to endpoint.
23.8 Adverse events: Movement disorders
23.8.1 akathisia ‐ long term
In this subgroup we only found one relevant trial (n = 399) (CATIE 2005). There was no significant difference between perphenazine and risperidone (RR 0.75 CI 0.38 to 1.49, Analysis 23.8).
23.8. Analysis.
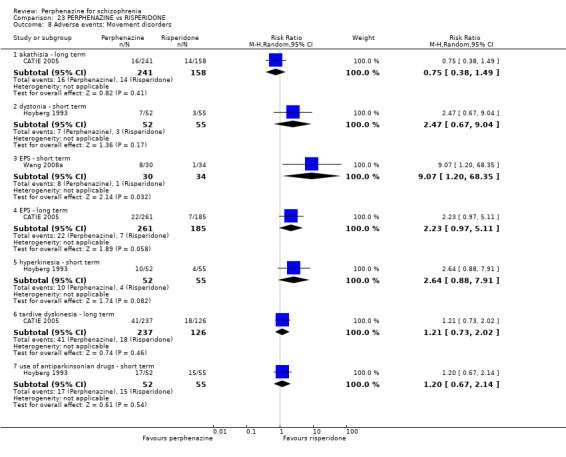
Comparison 23 PERPHENAZINE vs RISPERIDONE, Outcome 8 Adverse events: Movement disorders.
23.8.2 dystonia ‐ short term
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was no significant difference between perphenazine and risperidone (RR 2.47 CI 0.67 to 9.04, Analysis 23.8).
23.8.3 EPS ‐ short term
In this subgroup we only found one relevant trial (n = 64) (Wang 2008a). There was a statistically significant difference (P = 0.03) favouring risperidone over perphenazine (RR 9.07 CI 1.20 to 68.35, Analysis 23.8).
23.8.4 EPS ‐ long term
In this subgroup we only found one relevant trial (n = 446) (CATIE 2005). There was no significant difference between perphenazine and risperidone (RR 2.23 CI 0.97 to 5.11, Analysis 23.8).
23.8.5 hyperkinesia ‐ short term
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was no significant difference between perphenazine and risperidone (RR 2.64 CI 0.88 to 7.91, Analysis 23.8).
23.8.6 tardive dyskinesia ‐ long term
In this subgroup we only found one relevant trial (n = 363) (CATIE 2005). There was no significant difference between perphenazine and risperidone (RR 1.21 CI 0.73 to 2.02, Analysis 23.8).
23.8.7 use of antiparkinsonian drugs ‐ short term
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was no significant difference between perphenazine and risperidone (RR 1.20 CI 0.67 to 2.14, Analysis 23.8).
23.9 Other adverse events: 1. Anticholinergic
23.9.1 dry mouth ‐ short term
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was no significant difference between perphenazine and risperidone (RR 7.40 CI 0.39 to 139.81, Analysis 23.9).
23.9. Analysis.
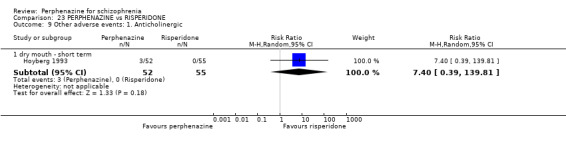
Comparison 23 PERPHENAZINE vs RISPERIDONE, Outcome 9 Other adverse events: 1. Anticholinergic.
23.10 Other adverse events: 2. Arousal
23.10.1 depression ‐ short term
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was no significant difference between perphenazine and risperidone (RR 1.34 CI 0.76 to 2.35, Analysis 23.10).
23.10. Analysis.
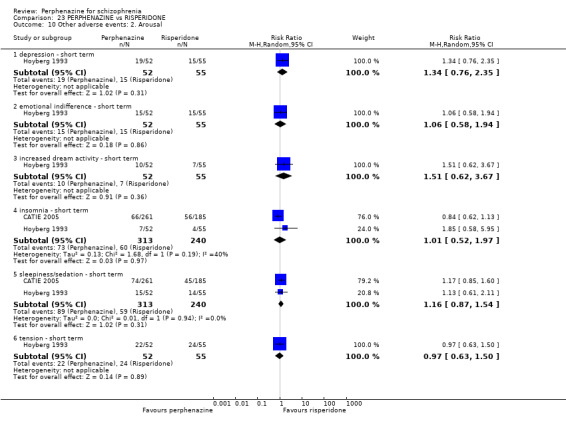
Comparison 23 PERPHENAZINE vs RISPERIDONE, Outcome 10 Other adverse events: 2. Arousal.
23.10.2 emotional indifference ‐ short term
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was no significant difference between perphenazine and risperidone (RR 1.06 CI 0.58 to 1.94, Analysis 23.10).
23.10.3 increased dream activity ‐ short term
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was no significant difference between perphenazine and risperidone (RR 1.51 CI 0.62 to 3.67, Analysis 23.10).
23.10.4 insomnia ‐ short term
In this subgroup we found two relevant trials (n = 553). There was no significant difference between perphenazine and risperidone (RR 1.01 CI 0.52 to 1.97, Analysis 23.10). This subgroup had moderate levels of heterogeneity (Chi2 = 1.68; df = 1; P = 0.195; I2 = 40%).
23.10.5 sleepiness/sedation ‐ short term
In this subgroup we found two relevant trials (n = 553). There was no significant difference between perphenazine and risperidone (RR 1.16 CI 0.87 to 1.54, Analysis 23.10).
23.10.6 tension ‐ short term
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was no significant difference between perphenazine and risperidone (RR 0.97 CI 0.63 to 1.50, Analysis 23.10).
23.11 Other adverse events: 3. At least one
23.11.1 short term
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was no significant difference between perphenazine and risperidone (RR 1.01 CI 0.85 to 1.21, Analysis 23.11).
23.11. Analysis.
Comparison 23 PERPHENAZINE vs RISPERIDONE, Outcome 11 Other adverse events: 3. At least one.
23.12 Other adverse events: 4. Cardiovascular
23.12.1 faintness, dizziness, weakness ‐ short term
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was no significant difference between perphenazine and risperidone (RR 0.93 CI 0.36 to 2.37, Analysis 23.12).
23.12. Analysis.
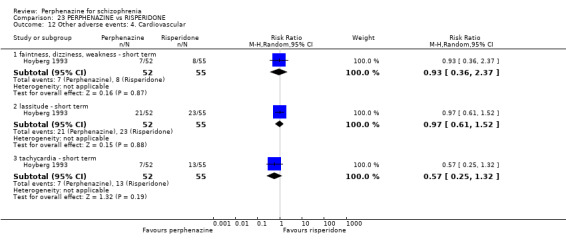
Comparison 23 PERPHENAZINE vs RISPERIDONE, Outcome 12 Other adverse events: 4. Cardiovascular.
23.12.2 lassitude ‐ short term
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was no significant difference between perphenazine and risperidone (RR 0.97 CI 0.61 to 1.52, Analysis 23.12).
23.12.3 tachycardia ‐ short term
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was no significant difference between perphenazine and risperidone (RR 0.57 CI 0.25 to 1.32, Analysis 23.12).
23.13 Other adverse events: 5. Central nervous system
23.13.1 difficulty in concentration ‐ short term
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was no significant difference between perphenazine and risperidone (RR 1.06 CI 0.71 to 1.58, Analysis 23.13).
23.13. Analysis.
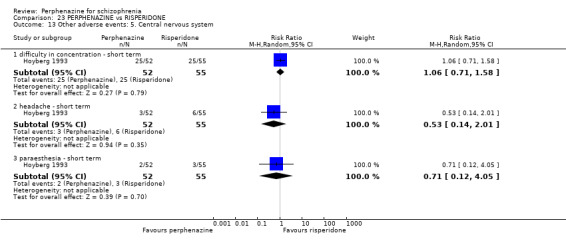
Comparison 23 PERPHENAZINE vs RISPERIDONE, Outcome 13 Other adverse events: 5. Central nervous system.
23.13.2 headache ‐ short term
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was no significant difference between perphenazine and risperidone (RR 0.53 CI 0.14 to 2.01, Analysis 23.13).
23.13.3 paraesthesia ‐ short term
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was no significant difference between perphenazine and risperidone (RR 0.71 CI 0.12 to 4.05, Analysis 23.13).
23.14 Other adverse events: 6. Endocrine
23.14.1 amenorrhoea (women only) ‐ short term
In this subgroup we only found one relevant trial (n = 30) (Hoyberg 1993). There was no significant difference between perphenazine and risperidone (RR 3.00 CI 0.13 to 68.26, Analysis 23.14).
23.14. Analysis.
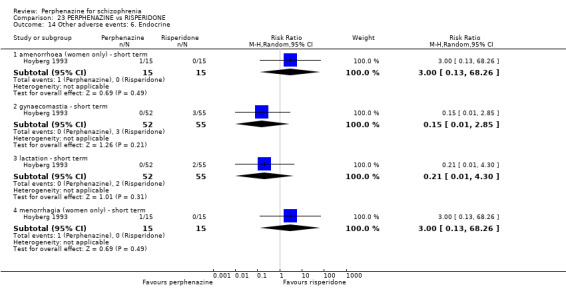
Comparison 23 PERPHENAZINE vs RISPERIDONE, Outcome 14 Other adverse events: 6. Endocrine.
23.14.2 gynaecomastia ‐ short term
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was no significant difference between perphenazine and risperidone (RR 0.15 CI 0.01 to 2.85, Analysis 23.14).
23.14.3 lactation ‐ short term
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was no significant difference between perphenazine and risperidone (RR 0.21 CI 0.01 to 4.30, Analysis 23.14).
23.14.4 menorrhagia (women only) ‐ short term
In this subgroup we only found one relevant trial (n = 30) (Hoyberg 1993). There was no significant difference between perphenazine and risperidone (RR 3.00 CI 0.13 to 68.26, Analysis 23.14).
23.15 Other adverse events: 7. Gastrointestinal
23.15.1 constipation ‐ short term
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was no significant difference between perphenazine and risperidone (RR 0.07 CI 0.00 to 1.20, Analysis 23.15).
23.15. Analysis.
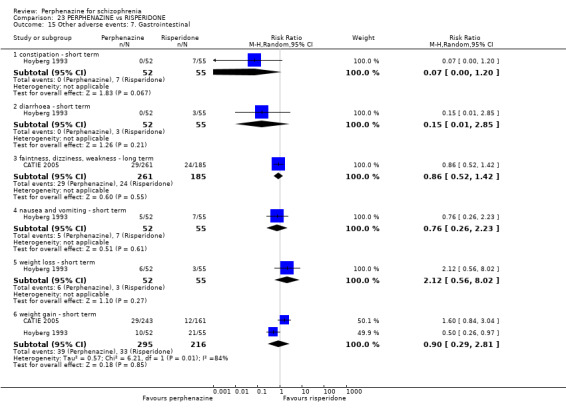
Comparison 23 PERPHENAZINE vs RISPERIDONE, Outcome 15 Other adverse events: 7. Gastrointestinal.
23.15.2 diarrhoea ‐ short term
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was no significant difference between perphenazine and risperidone (RR 0.15 CI 0.01 to 2.85, Analysis 23.15).
23.15.3 faintness, dizziness, weakness ‐ long term
In this subgroup we only found one relevant trial (n = 446) (CATIE 2005). There was no significant difference between perphenazine and risperidone (RR 0.86 CI 0.52 to 1.42, Analysis 23.15).
23.15.4 nausea and vomiting ‐ short term
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was no significant difference between perphenazine and risperidone (RR 0.76 CI 0.26 to 2.23, Analysis 23.15).
23.15.5 weight loss ‐ short term
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was no significant difference between perphenazine and risperidone (RR 2.12 CI 0.56 to 8.02, Analysis 23.15).
23.15.6 weight gain ‐ short term
In this subgroup we found two relevant trials (n = 511). There was no significant difference between perphenazine and risperidone (RR 0.90 CI 0.29 to 2.81, Analysis 23.15). This subgroup had important levels of heterogeneity (Chi2 = 6.21; df = 1; P = 0.013; I2 = 84%).
23.16 Other adverse events: 8. Skin
23.16.1 pruritus ‐ short term
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was no significant difference between perphenazine and risperidone (RR 0.79 CI 0.19 to 3.38, Analysis 23.16).
23.16. Analysis.
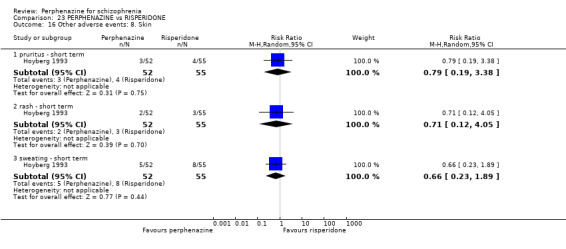
Comparison 23 PERPHENAZINE vs RISPERIDONE, Outcome 16 Other adverse events: 8. Skin.
23.16.2 rash ‐ short term
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was no significant difference between perphenazine and risperidone (RR 0.71 CI 0.12 to 4.05, Analysis 23.16).
23.16.3 sweating ‐ short term
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was no significant difference between perphenazine and risperidone (RR 0.66 CI 0.23 to 1.89, Analysis 23.16).
23.17 Other adverse events: 9. Genitourinary
23.17.1 diminished sexual desire
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was no significant difference between perphenazine and risperidone (RR 1.06 CI 0.22 to 5.01, Analysis 23.17).
23.17. Analysis.
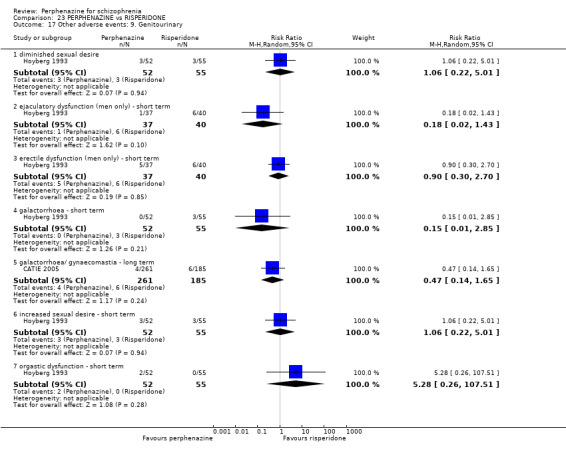
Comparison 23 PERPHENAZINE vs RISPERIDONE, Outcome 17 Other adverse events: 9. Genitourinary.
23.17.2 ejaculatory dysfunction (men only) ‐ short term
In this subgroup we only found one relevant trial (n = 77) (Hoyberg 1993). There was no significant difference between perphenazine and risperidone (RR 0.18 CI 0.02 to 1.43, Analysis 23.17).
23.17.3 erectile dysfunction (men only) ‐ short term
In this subgroup we only found one relevant trial (n = 77) (Hoyberg 1993). There was no significant difference between perphenazine and risperidone (RR 0.90 CI 0.30 to 2.70, Analysis 23.17).
23.17.4 galactorrhoea ‐ short term
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was no significant difference between perphenazine and risperidone (RR 0.15 CI 0.01 to 2.85, Analysis 23.17).
23.17.5 galactorrhoea/gynaecomastia ‐ long term
In this subgroup we only found one relevant trial (n = 446) (CATIE 2005). There was no significant difference between perphenazine and risperidone (RR 0.47 CI 0.14 to 1.65, Analysis 23.17).
23.17.6 increased sexual desire ‐ short term
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was no significant difference between perphenazine and risperidone (RR 1.06 CI 0.22 to 5.01, Analysis 23.17).
23.17.7 orgastic dysfunction ‐ short term
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was no significant difference between perphenazine and risperidone (RR 5.28 CI 0.26 to 107.51, Analysis 23.17).
23.18 Other adverse events: 10. Others
23.18.1 failing memory ‐ short term
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was no significant difference between perphenazine and risperidone (RR 1.27 CI 0.60 to 2.68, Analysis 23.18).
23.18. Analysis.
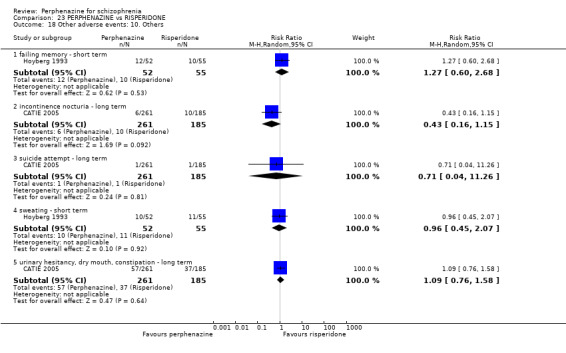
Comparison 23 PERPHENAZINE vs RISPERIDONE, Outcome 18 Other adverse events: 10. Others.
23.18.2 incontinence nocturia ‐ long term
In this subgroup we only found one relevant trial (n = 446) (CATIE 2005). There was no significant difference between perphenazine and risperidone (RR 0.43 CI 0.16 to 1.15, Analysis 23.18).
23.18.3 suicide attempt ‐ long term
In this subgroup we only found one relevant trial (n = 446) (CATIE 2005). There was no significant difference between perphenazine and risperidone (RR 0.71 CI 0.04 to 11.26, Analysis 23.18).
23.18.4 sweating ‐ short term
In this subgroup we only found one relevant trial (n = 107) (Hoyberg 1993). There was no significant difference between perphenazine and risperidone (RR 0.96 CI 0.45 to 2.07, Analysis 23.18).
23.18.5 urinary hesitancy, dry mouth, constipation ‐ long term
In this subgroup we only found one relevant trial (n = 446) (CATIE 2005). There was no significant difference between perphenazine and risperidone (RR 1.09 CI 0.76 to 1.58, Analysis 23.18).
23.19 Other adverse events: 11. any serious adverse event
23.19.1 long term
In this subgroup we only found one relevant trial (n = 446) (CATIE 2005). There was no significant difference between perphenazine and risperidone (RR 1.08 CI 0.63 to 1.87, Analysis 23.19).
23.19. Analysis.
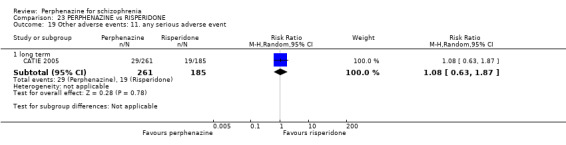
Comparison 23 PERPHENAZINE vs RISPERIDONE, Outcome 19 Other adverse events: 11. any serious adverse event.
23.20 Other adverse events: 12. lab data
23.20.1 mean weight change (lb) ‐ long term
In this subgroup we only found one relevant trial (n = 602) (CATIE 2005). There was a statistically significant difference (P = 0.05) favouring perphenazine over risperidone (MD ‐2.80 CI ‐5.58 to ‐0.02, Analysis 23.20).
23.20. Analysis.
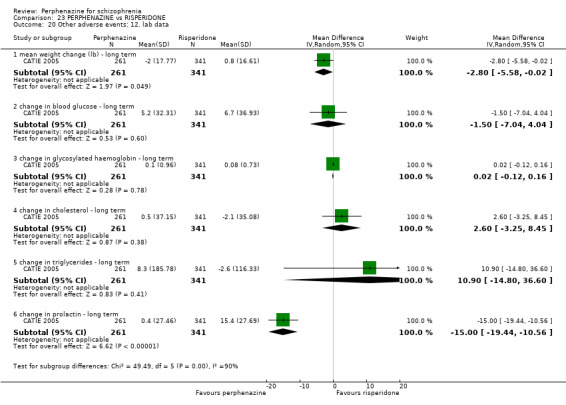
Comparison 23 PERPHENAZINE vs RISPERIDONE, Outcome 20 Other adverse events: 12. lab data.
23.20.2 change in blood glucose ‐ long term
In this subgroup we only found one relevant trial (n = 602) (CATIE 2005). There was no significant difference between perphenazine and risperidone (MD ‐1.50 CI ‐7.04 to 4.04, Analysis 23.20).
23.20.3 change in glycosylated haemoglobin ‐ long term
In this subgroup we only found one relevant trial (n = 602) (CATIE 2005). There was a statistically significant difference between perphenazine and risperidone (MD 0.02 CI ‐0.12 to 0.16, Analysis 23.20).
23.20.4 change in cholesterol ‐ long term
In this subgroup we only found one relevant trial (n = 602) (CATIE 2005). There was no significant difference between perphenazine and risperidone (MD 2.60 CI ‐3.25 to 8.45, Analysis 23.20).
23.20.5 change in triglycerides ‐ long term
In this subgroup we only found one relevant trial (n = 602) (CATIE 2005). There was no significant difference between perphenazine and risperidone (MD 10.90 CI ‐14.80 to 36.60, Analysis 23.20).
23.20.6 change in prolactin ‐ long term
In this subgroup we only found one relevant trial (n = 602) (CATIE 2005). There was a statistically significant difference (P < 0.00001) favouring perphenazine over risperidone (MD ‐15.0 CI ‐19.44 to ‐10.56, Analysis 23.20).
COMPARISON 24: PERPHENAZINE versus SULPIRIDE
24.1 Leaving the study early: 1. Any reason
24.1.1 short term
In this subgroup we only found one relevant trial (n = 64) (Amakusa 1973). There was no significant difference between perphenazine and sulpiride (RR 0.80 CI 0.24 to 2.71, Analysis 24.1).
24.1. Analysis.
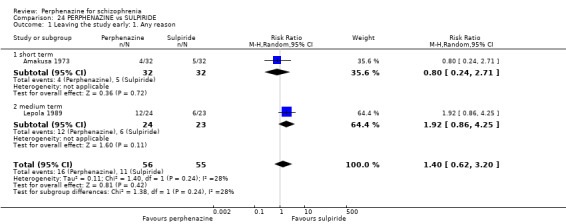
Comparison 24 PERPHENAZINE vs SULPIRIDE, Outcome 1 Leaving the study early: 1. Any reason.
24.1.2 medium term
In this subgroup we only found one relevant trial (n = 47) (Lepola 1989). There was no significant difference between perphenazine and sulpiride (RR 1.92 CI 0.86 to 4.25, Analysis 24.1).
24.2 Leaving the study early: 2. Due to adverse events
24.2.1 short term
In this subgroup we only found one relevant trial (n = 64) (Amakusa 1973). There was no significant difference between perphenazine and sulpiride (RR 3.00 CI 0.13 to 71.00, Analysis 24.2).
24.2. Analysis.
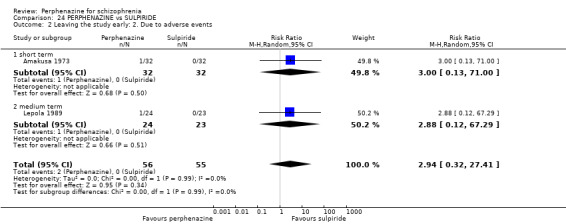
Comparison 24 PERPHENAZINE vs SULPIRIDE, Outcome 2 Leaving the study early: 2. Due to adverse events.
24.2.2 medium term
In this subgroup we only found one relevant trial (n = 47) (Lepola 1989). There was no significant difference between perphenazine and sulpiride (RR 2.88 CI 0.12 to 67.29, Analysis 24.2).
24.3 Leaving the study early: 3. Due to relapse/worsening or no improvement
24.3.1 short term
In this subgroup we only found one relevant trial (n = 64) (Amakusa 1973). There was no significant difference between perphenazine and sulpiride (RR 3.00 CI 0.13 to 71.00, Analysis 24.3).
24.3. Analysis.
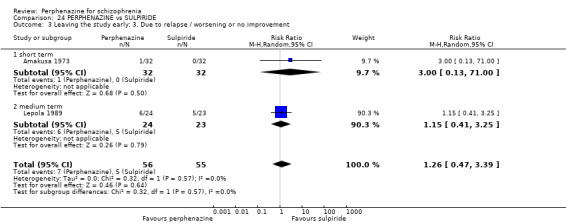
Comparison 24 PERPHENAZINE vs SULPIRIDE, Outcome 3 Leaving the study early: 3. Due to relapse / worsening or no improvement.
24.3.2 medium term
In this subgroup we only found one relevant trial (n = 47) (Lepola 1989). There was no significant difference between perphenazine and sulpiride (RR 1.15 CI 0.41 to 3.25, Analysis 24.3).
24.4 Global state: 1. Change over time ‐ no better or deterioration (ITT)
24.4.1 short term
In this subgroup we only found one relevant trial (n = 64) (Amakusa 1973). There was no significant difference between perphenazine and sulpiride (RR 0.80 CI 0.52 to 1.24, Analysis 24.4).
24.4. Analysis.
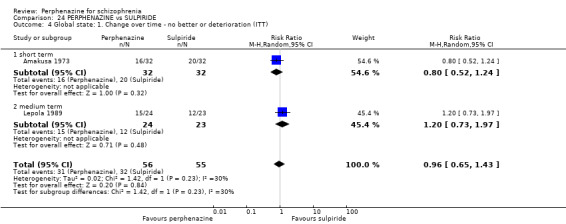
Comparison 24 PERPHENAZINE vs SULPIRIDE, Outcome 4 Global state: 1. Change over time ‐ no better or deterioration (ITT).
24.4.2 medium term
In this subgroup we only found one relevant trial (n = 47) (Lepola 1989). There was no significant difference between perphenazine and sulpiride (RR 1.20 CI 0.73 to 1.97, Analysis 24.4).
24.5 Mental state: 1. State ‐ BPRS end score (high = poor, skewed data)
24.5.1 medium term
Data for this outcome are skewed and are best inspected by viewing Analysis 24.5.
24.5. Analysis.
Comparison 24 PERPHENAZINE vs SULPIRIDE, Outcome 5 Mental state: 1. State ‐ BPRS end score (high = poor, skewed data).
| Mental state: 1. State ‐ BPRS end score (high = poor, skewed data) | |||||
|---|---|---|---|---|---|
| Study | Intervention | Subgroup | Mean | SD | N |
| medium term | |||||
| Lepola 1989 | Perphenazine | Acute schizophrenia | 15.5 | 23.8 | 7 |
| Lepola 1989 | Perphenazine | Chronic schizophrenia | 30.5 | 14.4 | 17 |
| Lepola 1989 | Sulpiride | Acute schizophrenia | 4.0 | 6.3 | 10 |
| Lepola 1989 | Sulpiride | Chronic schizophrenia | 26.0 | 14.4 | 13 |
24.6 Adverse events: Movement disorders
24.6.1 akathisia ‐ short term
In this subgroup we only found one relevant trial (n = 64) (Amakusa 1973). There was no significant difference between perphenazine and sulpiride (RR 0.75 CI 0.18 to 3.09, Analysis 24.6).
24.6. Analysis.
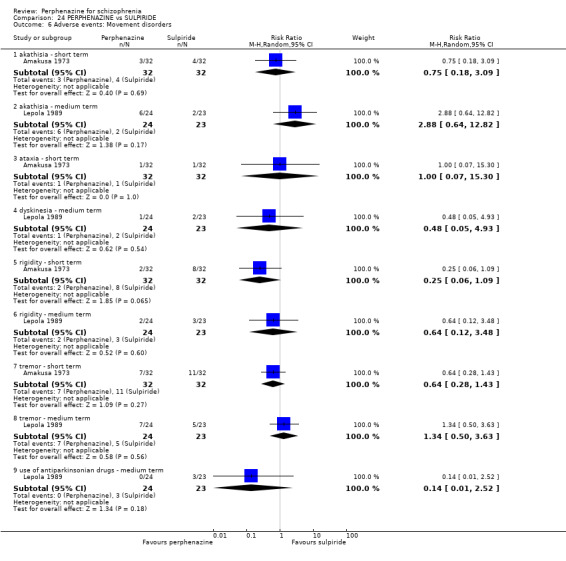
Comparison 24 PERPHENAZINE vs SULPIRIDE, Outcome 6 Adverse events: Movement disorders.
24.6.2 akathisia ‐ medium term
In this subgroup we only found one relevant trial (n = 47) (Lepola 1989). There was no significant difference between perphenazine and sulpiride (RR 2.88 CI 0.64 to 12.82, Analysis 24.6).
24.6.3 ataxia ‐ short term
In this subgroup we only found one relevant trial (n = 64) (Amakusa 1973). There was no significant difference between perphenazine and sulpiride (RR 1.00 CI 0.07 to 15.30, Analysis 24.6).
24.6.4 dyskinesia ‐ medium term
In this subgroup we only found one relevant trial (n = 47) (Lepola 1989). There was no significant difference between perphenazine and sulpiride (RR 0.48 CI 0.05 to 4.93, Analysis 24.6).
24.6.5 rigidity ‐ short term
In this subgroup we only found one relevant trial (n = 64) (Amakusa 1973). There was no significant difference between perphenazine and sulpiride (RR 0.25 CI 0.06 to 1.09, Analysis 24.6).
24.6.6 rigidity ‐ medium term
In this subgroup we only found one relevant trial (n = 47) (Lepola 1989). There was no significant difference between perphenazine and sulpiride (RR 0.64 CI 0.12 to 3.48, Analysis 24.6).
24.6.7 tremor ‐ short term
In this subgroup we only found one relevant trial (n = 64) (Amakusa 1973). There was no significant difference between perphenazine and sulpiride (RR 0.64 CI 0.28 to 1.43, Analysis 24.6).
24.6.8 tremor ‐ medium term
In this subgroup we only found one relevant trial (n = 47) (Lepola 1989). There was no significant difference between perphenazine and sulpiride (RR 1.34 CI 0.50 to 3.63, Analysis 24.6).
24.6.9 use of antiparkinsonian drugs ‐ medium term
In this subgroup we only found one relevant trial (n = 47) (Lepola 1989). There was no significant difference between perphenazine and sulpiride (RR 0.14 CI 0.01 to 2.52, Analysis 24.6).
24.7 Other adverse events: 1. Anticholinergic
24.7.1 blurred vision ‐ short term
In this subgroup we only found one relevant trial (n = 64) (Amakusa 1973). There was no significant difference between perphenazine and sulpiride (RR 0.33 CI 0.01 to 7.89, Analysis 24.7).
24.7. Analysis.
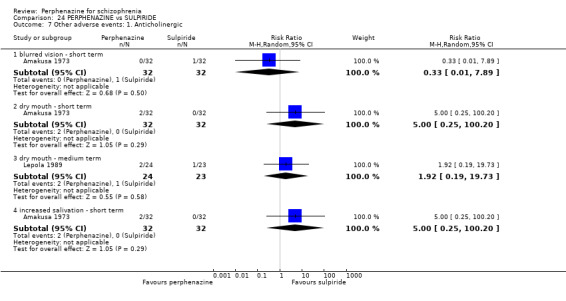
Comparison 24 PERPHENAZINE vs SULPIRIDE, Outcome 7 Other adverse events: 1. Anticholinergic.
24.7.2 dry mouth ‐ short term
In this subgroup we only found one relevant trial (n = 64) (Amakusa 1973). There was no significant difference between perphenazine and sulpiride (RR 5.0 CI 0.25 to 100.2, Analysis 24.7).
24.7.3 dry mouth ‐ medium term
In this subgroup we only found one relevant trial (n = 47) (Lepola 1989). There was no significant difference between perphenazine and sulpiride (RR 1.92 CI 0.19 to 19.73, Analysis 24.7).
24.7.4 increased salivation ‐ short term
In this subgroup we only found one relevant trial (n = 64) (Amakusa 1973). There was no significant difference between perphenazine and sulpiride (RR 5.00 CI 0.25 to 100.20, Analysis 24.7).
24.8 Other adverse events: 2. Arousal
24.8.1 excitation ‐ short term
In this subgroup we only found one relevant trial (n = 64) (Amakusa 1973). There was no significant difference between perphenazine and sulpiride (RR 0.33 CI 0.07 to 1.53, Analysis 24.8).
24.8. Analysis.
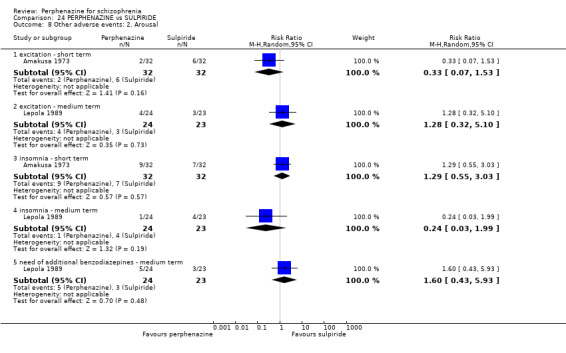
Comparison 24 PERPHENAZINE vs SULPIRIDE, Outcome 8 Other adverse events: 2. Arousal.
24.8.2 excitation ‐ medium term
In this subgroup we only found one relevant trial (n = 47) (Lepola 1989). There was no significant difference between perphenazine and sulpiride (RR 1.28 CI 0.32 to 5.10, Analysis 24.8).
24.8.3 insomnia ‐ short term
In this subgroup we only found one relevant trial (n = 64) (Amakusa 1973). There was no significant difference between perphenazine and sulpiride (RR 1.29 CI 0.55 to 3.03, Analysis 24.8).
24.8.4 insomnia ‐ medium term
In this subgroup we only found one relevant trial (n = 47) (Lepola 1989). There was no significant difference between perphenazine and sulpiride (RR 0.24 CI 0.03 to 1.99, Analysis 24.8).
24.8.5 need of additional benzodiazepines ‐ medium term
In this subgroup we only found one relevant trial (n = 47) (Lepola 1989). There was no significant difference between perphenazine and sulpiride (RR 1.60 CI 0.43 to 5.93, Analysis 24.8).
24.9 Other adverse events: 3. At least one
24.9.1 short term
In this subgroup we only found one relevant trial (n = 64) (Amakusa 1973). There was no significant difference between perphenazine and sulpiride (RR 0.76 CI 0.50 to 1.17, Analysis 24.9).
24.9. Analysis.
Comparison 24 PERPHENAZINE vs SULPIRIDE, Outcome 9 Other adverse events: 3. At least one.
24.10 Other adverse events: 4. Central nervous system
24.10.1 headache ‐ short term
In this subgroup we only found one relevant trial (n = 64) (Amakusa 1973). There was no significant difference between perphenazine and sulpiride (RR 0.50 CI 0.05 to 5.24, Analysis 24.10).
24.10. Analysis.
Comparison 24 PERPHENAZINE vs SULPIRIDE, Outcome 10 Other adverse events: 4. Central nervous system.
24.10.2 paraesthesia ‐ short term
In this subgroup we only found one relevant trial (n = 64) (Amakusa 1973). There was no significant difference between perphenazine and sulpiride (RR 5.00 CI 0.25 to 100.20, Analysis 24.10).
24.11 Other adverse events: 5. Endocrine
24.11.1 amenorrhoea ‐ medium term
In this subgroup we only found one relevant trial (n = 47) (Lepola 1989). There was no significant difference between perphenazine and sulpiride (RR 0.32 CI 0.04 to 2.85, Analysis 24.11).
24.11. Analysis.
Comparison 24 PERPHENAZINE vs SULPIRIDE, Outcome 11 Other adverse events: 5. Endocrine.
24.11.2 lactation ‐ medium term
In this subgroup we only found one relevant trial (n = 47) (Lepola 1989). There was no significant difference between perphenazine and sulpiride (RR 0.19 CI 0.01 to 3.80, Analysis 24.11).
24.12 Other adverse events: 6. Gastrointestinal
24.12.1 constipation ‐ short term
In this subgroup we only found one relevant trial (n = 64) (Amakusa 1973). There was no significant difference between perphenazine and sulpiride (RR 0.50 CI 0.05 to 5.24, Analysis 24.12).
24.12. Analysis.
Comparison 24 PERPHENAZINE vs SULPIRIDE, Outcome 12 Other adverse events: 6. Gastrointestinal.
24.12.2 loss of appetite ‐ short term
In this subgroup we only found one relevant trial (n = 64) (Amakusa 1973). There was no significant difference between perphenazine and sulpiride (RR 0.80 CI 0.24 to 2.71, Analysis 24.12).
24.12.3 nausea and vomiting ‐ short term
In this subgroup we only found one relevant trial (n = 64) (Amakusa 1973). There was no significant difference between perphenazine and sulpiride (RR 0.40 CI 0.08 to 1.91, Analysis 24.12).
24.12.4 nausea and vomiting ‐ medium term
In this subgroup we only found one relevant trial (n = 47) (Lepola 1989). There was no significant difference between perphenazine and sulpiride (RR 0.32 CI 0.04 to 2.85, Analysis 24.12).
24.12.5 weight loss ‐ short term
In this subgroup we only found one relevant trial (n = 64) (Amakusa 1973). There was no significant difference between perphenazine and sulpiride (RR 11.00 CI 0.63 to 191.04, Analysis 24.12).
24.12.6 weight gain ‐ short term
In this subgroup we only found one relevant trial (n = 64) (Amakusa 1973). There was no significant difference between perphenazine and sulpiride (RR 5.00 CI 0.62 to 40.44, Analysis 24.12).
24.13 Other adverse events: 7. Skin
24.13.1 rash ‐ short term
In this subgroup we only found one relevant trial (n = 64) (Amakusa 1973). There was no significant difference between perphenazine and sulpiride (RR 0.33 CI 0.01 to 7.89, Analysis 24.13).
24.13. Analysis.
Comparison 24 PERPHENAZINE vs SULPIRIDE, Outcome 13 Other adverse events: 7. Skin.
24.14 Other adverse events: 8. Others
24.14.1 abdominal pain ‐ medium term
In this subgroup we only found one relevant trial (n = 47) (Lepola 1989). There was no significant difference between perphenazine and sulpiride (RR 0.14 CI 0.01 to 2.52, Analysis 24.14).
24.14. Analysis.
Comparison 24 PERPHENAZINE vs SULPIRIDE, Outcome 14 Other adverse events: 8. Others.
24.14.2 sweating ‐ short term
In this subgroup we only found one relevant trial (n = 64) (Amakusa 1973). There was no significant difference between perphenazine and sulpiride (RR 0.20 CI 0.01 to 4.01, Analysis 24.14).
24.15 Other adverse events: 9. lab data
24.15.1 decrease in white blood cell count and haemoglobin level ‐ short term
In this subgroup we only found one relevant trial (n = 64) (Amakusa 1973). There was no significant difference between perphenazine and sulpiride (RR 1.00 CI 0.07 to 15.30, Analysis 24.15).
24.15. Analysis.
Comparison 24 PERPHENAZINE vs SULPIRIDE, Outcome 15 Other adverse events: 9. lab data.
24.15.2 increase in white blood cell count and haemoglobin level ‐ short term
In this subgroup we only found one relevant trial (n = 64) (Amakusa 1973). There was no significant difference between perphenazine and sulpiride (RR 0.50 CI 0.10 to 2.54, Analysis 24.15).
24.15.3 rise in alkaline phosphate and GOT levels ‐ short term
In this subgroup we only found one relevant trial (n = 64) (Amakusa 1973). There was no significant difference between perphenazine and sulpiride (RR 3.00 CI 0.13 to 71.00, Analysis 24.15).
COMPARISON 25: PERPHENAZINE versus THIOPROPRAZATE
25.1 Leaving the study early: 1. Any reason
25.1.1 short term
In this subgroup we only found one relevant trial (n = 106) (Hanlon 1965). There was no significant difference between perphenazine and thioproprazate (RR 0.73 CI 0.43 to 1.22, Analysis 25.1).
25.1. Analysis.
Comparison 25 PERPHENAZINE vs THIOPROPRAZATE, Outcome 1 Leaving the study early: 1. Any reason.
25.2 Leaving the study early: 2. Due to adverse events
25.2.1 short term
In this subgroup we only found one relevant trial (n = 106) (Hanlon 1965). There was no significant difference between perphenazine and thioproprazate (RR 2.00 CI 0.19 to 21.40, Analysis 25.2).
25.2. Analysis.
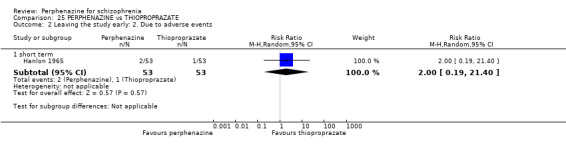
Comparison 25 PERPHENAZINE vs THIOPROPRAZATE, Outcome 2 Leaving the study early: 2. Due to adverse events.
25.3 Adverse events: Movement disorders
25.3.1 akathisia ‐ short term
In this subgroup we only found one relevant trial (n = 106) (Hanlon 1965). There was no significant difference between perphenazine and thioproprazate (RR 7.00 CI 0.89 to 54.94, Analysis 25.3).
25.3. Analysis.
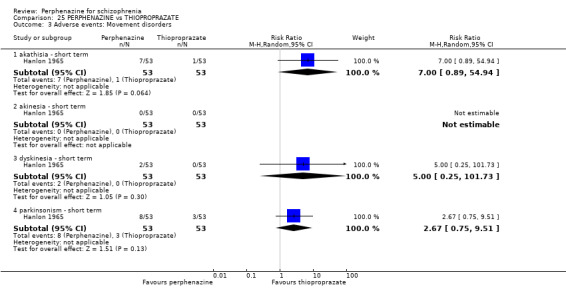
Comparison 25 PERPHENAZINE vs THIOPROPRAZATE, Outcome 3 Adverse events: Movement disorders.
25.3.2 akinesia ‐ short term
In this subgroup we only found one relevant trial (n = 106) (Hanlon 1965). There was no significant difference between perphenazine and thioproprazate, with no events reported in either group (Analysis 25.3).
25.3.3 dyskinesia ‐ short term
In this subgroup we only found one relevant trial (n = 106) (Hanlon 1965). There was no significant difference between perphenazine and thioproprazate (RR 5.00 CI 0.25 to 101.73, Analysis 25.3).
25.3.4 parkinsonism ‐ short term
In this subgroup we only found one relevant trial (n = 106) (Hanlon 1965). There was no significant difference between perphenazine and thioproprazate (RR 2.67 CI 0.75 to 9.51, Analysis 25.3).
COMPARISON 26: PERPHENAZINE versus THIORIDAZINE
26.1 Leaving the study early: 1. Any reason
26.1.1 short term
In this subgroup we only found one relevant trial (n = 106) (Hanlon 1965). There was no significant difference between perphenazine and thioridazine (RR 0.73 CI 0.43 to 1.22, Analysis 26.1).
26.1. Analysis.
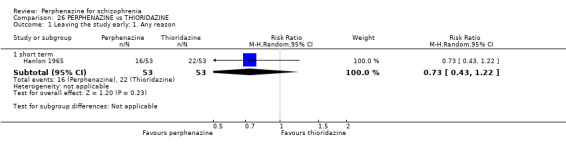
Comparison 26 PERPHENAZINE vs THIORIDAZINE, Outcome 1 Leaving the study early: 1. Any reason.
26.2 Leaving the study early: 2. Due to adverse events
26.2.1 short term
In this subgroup we only found one relevant trial (n = 106) (Hanlon 1965). There was no significant difference between perphenazine and thioridazine (RR 2.00 CI 0.19 to 21.40, Analysis 26.2).
26.2. Analysis.
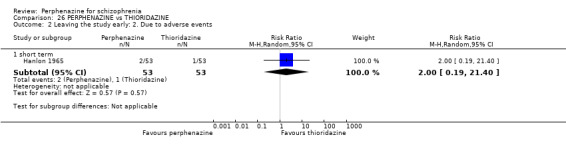
Comparison 26 PERPHENAZINE vs THIORIDAZINE, Outcome 2 Leaving the study early: 2. Due to adverse events.
26.3 Adverse events: Movement disorders
26.3.1 akathisia ‐ short term
In this subgroup we only found one relevant trial (n = 106) (Hanlon 1965). There was no significant difference between perphenazine and thioridazine (RR 15.00 CI 0.88 to 256.16, Analysis 26.3).
26.3. Analysis.
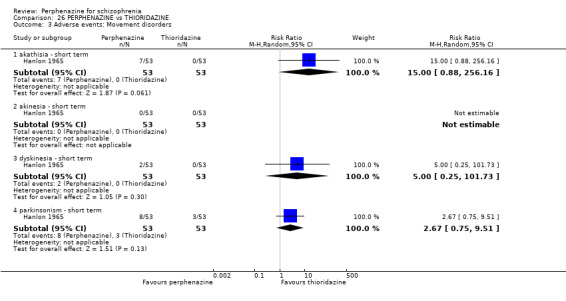
Comparison 26 PERPHENAZINE vs THIORIDAZINE, Outcome 3 Adverse events: Movement disorders.
26.3.2 akinesia ‐ short term
In this subgroup we only found one relevant trial (n = 106) (Hanlon 1965). There was no significant difference between perphenazine and thioridazine, with no events reported in either group (Analysis 26.3).
26.3.3 dyskinesia ‐ short term
In this subgroup we only found one relevant trial (n = 106) (Hanlon 1965). There was no significant difference between perphenazine and thioridazine (RR 5.00 CI 0.25 to 101.73, Analysis 26.3).
26.3.4 parkinsonism ‐ short term
In this subgroup we only found one relevant trial (n = 106) (Hanlon 1965). There was no significant difference between perphenazine and thioridazine (RR 2.67 CI 0.75 to 9.51, Analysis 26.3).
COMPARISON 27: PERPHENAZINE versus THIOTHIXENE
27.1 Leaving the study early: 1. Any reason
27.1.1 short term
In this subgroup we only found one relevant trial (n = 86) (Itoh 1969 II). There was no significant difference between perphenazine and thiothixene (RR 1.00 CI 0.06 to 15.48, Analysis 27.1).
27.1. Analysis.
Comparison 27 PERPHENAZINE vs THIOTHIXENE, Outcome 1 Leaving the study early: 1. Any reason.
27.2 Global state: 1. Change over time ‐ no better or deterioration (ITT)
27.2.1 short term
In this subgroup we only found one relevant trial (n = 86) (Itoh 1969 II). There was no significant difference between perphenazine and thiothixene (RR 1.30 CI 0.93 to 1.83, Analysis 27.2).
27.2. Analysis.
Comparison 27 PERPHENAZINE vs THIOTHIXENE, Outcome 2 Global state: 1. Change over time ‐ no better or deterioration (ITT).
27.3 Behaviour: 1. Behaviour subscale of the Psychiatric Rating Scale: no better or worse
For this outcome we only found one relevant trial (n = 86) (Itoh 1969 II). There was no significant difference between perphenazine and thiothixene (RR 1.18 CI 0.90 to 1.55, Analysis 27.3).
27.3. Analysis.
Comparison 27 PERPHENAZINE vs THIOTHIXENE, Outcome 3 Behaviour: 1. Behaviour subscale of the Psychiatric Rating Scale: no better or worse.
27.4 Adverse events: Movement disorders
27.4.1 akathisia ‐ short term
In this subgroup we only found one relevant trial (n = 86) (Itoh 1969 II). There was no significant difference between perphenazine and thiothixene (RR 0.58 CI 0.25 to 1.34, Analysis 27.4).
27.4. Analysis.
Comparison 27 PERPHENAZINE vs THIOTHIXENE, Outcome 4 Adverse events: Movement disorders.
27.4.2 ataxia ‐ short term
In this subgroup we only found one relevant trial (n = 86) (Itoh 1969 II). There was no significant difference between perphenazine and thiothixene (RR 1.00 CI 0.21 to 4.68, Analysis 27.4).
27.4.3 dyskinesia ‐ short term
In this subgroup we only found one relevant trial (n = 86) (Itoh 1969 II). There was no significant difference between perphenazine and thiothixene (RR 1.50 CI 0.26 to 8.53, Analysis 27.4).
27.4.4 parkinsonism ‐ short term
In this subgroup we only found one relevant trial (n = 86) (Itoh 1969 II). There was no significant difference between perphenazine and thiothixene (RR 0.80 CI 0.48 to 1.32, Analysis 27.4).
27.4.5 use of antiparkinsonian drugs ‐ short term
In this subgroup we only found one relevant trial (n = 86) (Itoh 1969 II). There was no significant difference between perphenazine and thiothixene (RR 1.18 CI 0.81 to 1.73, Analysis 27.4).
27.5 Other adverse events: 1. Anticholinergic
27.5.1 blurred vision ‐ short term
In this subgroup we only found one relevant trial (n = 86) (Itoh 1969 II). There was no significant difference between perphenazine and thiothixene (RR 0.29 CI 0.06 to 1.30, Analysis 27.5).
27.5. Analysis.
Comparison 27 PERPHENAZINE vs THIOTHIXENE, Outcome 5 Other adverse events: 1. Anticholinergic.
27.5.2 dry mouth ‐ short term
In this subgroup we only found one relevant trial (n = 86) (Itoh 1969 II). There was no significant difference between perphenazine and thiothixene (RR 1.00 CI 0.38 to 2.61, Analysis 27.5).
27.6 Other adverse events: 2. Arousal
27.6.1 insomnia ‐ short term
In this subgroup we only found one relevant trial (n = 86) (Itoh 1969 II). There was no significant difference between perphenazine and thiothixene (RR 0.71 CI 0.39 to 1.29, Analysis 27.6).
27.6. Analysis.
Comparison 27 PERPHENAZINE vs THIOTHIXENE, Outcome 6 Other adverse events: 2. Arousal.
27.6.2 need of additional benzodiazepines ‐ short term
In this subgroup we only found one relevant trial (n = 86) (Itoh 1969 II). There was no significant difference between perphenazine and thiothixene (RR 1.00 CI 0.21 to 4.68, Analysis 27.6).
27.6.3 sleepiness/sedation ‐ short term
In this subgroup we only found one relevant trial (n = 86) (Itoh 1969 II). There was no significant difference between perphenazine and thiothixene (RR 0.90 CI 0.41 to 1.99, Analysis 27.6).
27.7 Other adverse events: 3. At least one
27.7.1 short term
In this subgroup we only found one relevant trial (n = 86) (Itoh 1969 II). There was no significant difference between perphenazine and thiothixene (RR 0.90 CI 0.68 to 1.20, Analysis 27.7).
27.7. Analysis.
Comparison 27 PERPHENAZINE vs THIOTHIXENE, Outcome 7 Other adverse events: 3. At least one.
27.8 Other adverse events: 4. Cardiovascular
27.8.1 faintness, dizziness, weakness ‐ short term
In this subgroup we only found one relevant trial (n = 86) (Itoh 1969 II). There was no significant difference between perphenazine and thiothixene, with no events reported in either group (Analysis 27.8).
27.8. Analysis.
Comparison 27 PERPHENAZINE vs THIOTHIXENE, Outcome 8 Other adverse events: 4. Cardiovascular.
27.9 Other adverse events: 5. Central nervous system
27.9.1 disturbance of consciousness ‐ short term
In this subgroup we only found one relevant trial (n = 86) (Itoh 1969 II). There was no significant difference between perphenazine and thiothixene, with no events reported in either group (Analysis 27.9).
27.9. Analysis.
Comparison 27 PERPHENAZINE vs THIOTHIXENE, Outcome 9 Other adverse events: 5. Central nervous system.
27.9.2 headache ‐ short term
In this subgroup we only found one relevant trial (n = 86) (Itoh 1969 II). There was no significant difference between perphenazine and thiothixene (RR 2.00 CI 0.19 to 21.24, Analysis 27.9).
27.9.3 paraesthesia ‐ short term
In this subgroup we only found one relevant trial (n = 86) (Itoh 1969 II). There was no significant difference between perphenazine and thiothixene (RR 5.00 CI 0.25 to 101.18, Analysis 27.9).
27.10 Other adverse events: 6. Gastrointestinal
27.10.1 constipation ‐ short term
In this subgroup we only found one relevant trial (n = 86) (Itoh 1969 II). There was no significant difference between perphenazine and thiothixene (RR 1.20 CI 0.40 to 3.64, Analysis 27.10).
27.10. Analysis.
Comparison 27 PERPHENAZINE vs THIOTHIXENE, Outcome 10 Other adverse events: 6. Gastrointestinal.
27.10.2 icterus ‐ short term
In this subgroup we only found one relevant trial (n = 86) (Itoh 1969 II). There was no significant difference between perphenazine and thiothixene (RR 0.33 CI 0.01 to 7.96, Analysis 27.10).
27.10.3 loss of appetite ‐ short term
In this subgroup we only found one relevant trial (n = 86) (Itoh 1969 II). There was no significant difference between perphenazine and thiothixene (RR 0.88 CI 0.35 to 2.20, Analysis 27.10).
27.10.4 nausea and vomiting ‐ short term
In this subgroup we only found one relevant trial (n = 86) (Itoh 1969 II). There was no significant difference between perphenazine and thiothixene, with no events reported in either group (Analysis 27.10).
27.11 Other adverse events: 7. Skin
27.11.1 rash ‐ short term
In this subgroup we only found one relevant trial (n = 86) (Itoh 1969 II). There was no significant difference between perphenazine and thiothixene (RR 3.00 CI 0.13 to 71.65, Analysis 27.11).
27.11. Analysis.
Comparison 27 PERPHENAZINE vs THIOTHIXENE, Outcome 11 Other adverse events: 7. Skin.
27.12 Other adverse events: 8. Genitourinary
27.12.1 micturition disturbances ‐ short term
In this subgroup we only found one relevant trial (n = 86) (Itoh 1969 II). There was no significant difference between perphenazine and thiothixene, with no events reported in either group (Analysis 27.12).
27.12. Analysis.
Comparison 27 PERPHENAZINE vs THIOTHIXENE, Outcome 12 Other adverse events: 8. Genitourinary.
27.13 Other adverse events: 9. Others
27.13.1 cramps ‐ short term
In this subgroup we only found one relevant trial (n = 86) (Itoh 1969 II). There was no significant difference between perphenazine and thiothixene (RR 0.33 CI 0.01 to 7.96, Analysis 27.13).
27.13. Analysis.
Comparison 27 PERPHENAZINE vs THIOTHIXENE, Outcome 13 Other adverse events: 9. Others.
27.13.2 sweating ‐ short term
In this subgroup we only found one relevant trial (n = 86) (Itoh 1969 II). There was no significant difference between perphenazine and thiothixene (RR 1.00 CI 0.06 to 15.48, Analysis 27.13).
COMPARISON 28: PERPHENAZINE versus TIMIPERONE
28.1 Leaving the study early: 1. Any reason
28.1.1 short term
In this subgroup we only found one relevant trial (n = 198) (Takahashi 1982). There was no significant difference between perphenazine and timiperone (RR 1.33 CI 0.67 to 2.67, Analysis 28.1).
28.1. Analysis.
Comparison 28 PERPHENAZINE vs TIMIPERONE, Outcome 1 Leaving the study early: 1. Any reason.
28.2 Leaving the study early: 2. Due to adverse events
28.2.1 short term
In this subgroup we only found one relevant trial (n = 198) (Takahashi 1982). There was no significant difference between perphenazine and timiperone (RR 1.33 CI 0.67 to 2.67, Analysis 28.2).
28.2. Analysis.
Comparison 28 PERPHENAZINE vs TIMIPERONE, Outcome 2 Leaving the study early: 2. Due to adverse events.
28.3 Global state: 1. Change over time ‐ no better or deterioration (ITT)
28.3.1 short term
In this subgroup we only found one relevant trial (n = 198) (Takahashi 1982). There was no significant difference between perphenazine and timiperone (RR 1.11 CI 0.79 to 1.55, Analysis 28.3).
28.3. Analysis.
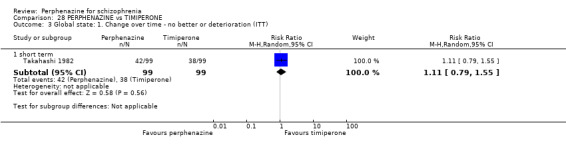
Comparison 28 PERPHENAZINE vs TIMIPERONE, Outcome 3 Global state: 1. Change over time ‐ no better or deterioration (ITT).
28.4 Adverse events: Movement disorders
28.4.1 akathisia ‐ short term
In this subgroup we only found one relevant trial (n = 198) (Takahashi 1982). There was no significant difference between perphenazine and timiperone (RR 0.85 CI 0.56 to 1.29, Analysis 28.4).
28.4. Analysis.
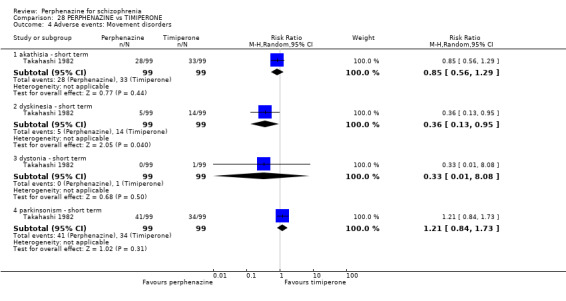
Comparison 28 PERPHENAZINE vs TIMIPERONE, Outcome 4 Adverse events: Movement disorders.
28.4.2 dyskinesia ‐ short term
In this subgroup we only found one relevant trial (n = 198) (Takahashi 1982). There was a statistically significant difference (P = 0.04) favouring perphenazine over timiperone (RR 0.36 CI 0.13 to 0.95, Analysis 28.4).
28.4.3 dystonia ‐ short term
In this subgroup we only found one relevant trial (n = 198) (Takahashi 1982). There was no significant difference between perphenazine and timiperone (RR 0.33 CI 0.01 to 8.08, Analysis 28.4).
28.4.4 parkinsonism ‐ short term
In this subgroup we only found one relevant trial (n = 198) (Takahashi 1982). There was no significant difference between perphenazine and timiperone (RR 1.21 CI 0.84 to 1.73, Analysis 28.4).
28.5 Other adverse events: 1. Anticholinergic
28.5.1 blurred vision ‐ short term
In this subgroup we only found one relevant trial (n = 198) (Takahashi 1982). There was no significant difference between perphenazine and timiperone (RR 3.00 CI 0.12 to 72.76, Analysis 28.5).
28.5. Analysis.
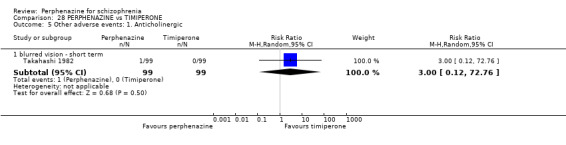
Comparison 28 PERPHENAZINE vs TIMIPERONE, Outcome 5 Other adverse events: 1. Anticholinergic.
28.6 Other adverse events: 2. Arousal
28.6.1 drowsiness ‐ short term
In this subgroup we only found one relevant trial (n = 198) (Takahashi 1982). There was no significant difference between perphenazine and timiperone (RR 1.43 CI 0.57 to 3.60, Analysis 28.6).
28.6. Analysis.
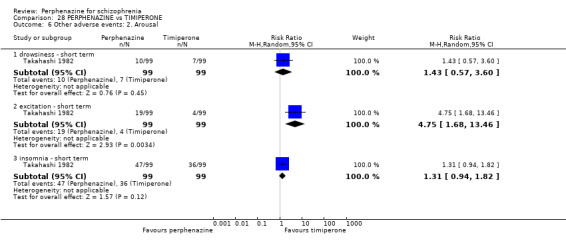
Comparison 28 PERPHENAZINE vs TIMIPERONE, Outcome 6 Other adverse events: 2. Arousal.
28.6.2 excitation ‐ short term
In this subgroup we only found one relevant trial (n = 198) (Takahashi 1982). There was a statistically significant difference (P = 0.003) favouring timiperone over perphenazine (RR 4.75 CI 1.68 to 13.46, Analysis 28.6).
28.6.3 insomnia ‐ short term
In this subgroup we only found one relevant trial (n = 198) (Takahashi 1982). There was no significant difference between perphenazine and timiperone (RR 1.31 CI 0.94 to 1.82, Analysis 28.6).
28.7 Other adverse events: 3. At least one
28.7.1 short term
In this subgroup we only found one relevant trial (n = 198) (Takahashi 1982). There was no significant difference between perphenazine and timiperone (RR 1.01 CI 0.89 to 1.16, Analysis 28.7).
28.7. Analysis.
Comparison 28 PERPHENAZINE vs TIMIPERONE, Outcome 7 Other adverse events: 3. At least one.
28.8 Other adverse events: 4. Cardiovascular
28.8.1 faintness, dizziness, weakness ‐ short term
In this subgroup we only found one relevant trial (n = 198) (Takahashi 1982). There was no significant difference between perphenazine and timiperone (RR 1.50 CI 0.71 to 3.18, Analysis 28.8).
28.8. Analysis.
Comparison 28 PERPHENAZINE vs TIMIPERONE, Outcome 8 Other adverse events: 4. Cardiovascular.
28.8.2 lassitude ‐ short term
In this subgroup we only found one relevant trial (n = 198) (Takahashi 1982). There was no significant difference between perphenazine and timiperone (RR 1.07 CI 0.55 to 2.10, Analysis 28.8).
28.9 Other adverse events: 5. Central nervous system
28.9.1 confusion ‐ short term
In this subgroup we only found one relevant trial (n = 198) (Takahashi 1982). There was no significant difference between perphenazine and timiperone (RR 0.67 CI 0.11 to 3.90, Analysis 28.9).
28.9. Analysis.
Comparison 28 PERPHENAZINE vs TIMIPERONE, Outcome 9 Other adverse events: 5. Central nervous system.
28.9.2 disturbance of consciousness ‐ short term
In this subgroup we only found one relevant trial (n = 198) (Takahashi 1982). There was no significant difference between perphenazine and timiperone (RR 1.00 CI 0.06 to 15.76, Analysis 28.9).
28.9.3 dysarticulation ‐ short term
In this subgroup we only found one relevant trial (n = 198) (Takahashi 1982). There was no significant difference between perphenazine and timiperone (RR 0.83 CI 0.38 to 1.84, Analysis 28.9).
28.9.4 headache ‐ short term
In this subgroup we only found one relevant trial (n = 198) (Takahashi 1982). There was no significant difference between perphenazine and timiperone (RR 0.33 CI 0.09 to 1.19, Analysis 28.9).
28.9.5 seizure ‐ short term
In this subgroup we only found one relevant trial (n = 198) (Takahashi 1982). There was no significant difference between perphenazine and timiperone, with no events reported in either group (Analysis 28.9).
COMPARISON 29: PERPHENAZINE versus TRIFLUOPERAZINE
29.1 Leaving the study early: 1. Any reason
29.1.1 short term
In this subgroup we only found one relevant trial (n = 105) (Hanlon 1965). There was no significant difference between perphenazine and trifluoperazine (RR 0.63 CI 0.38 to 1.03, Analysis 29.1).
29.1. Analysis.
Comparison 29 PERPHENAZINE vs TRIFLUOPERAZINE, Outcome 1 Leaving the study early: 1. Any reason.
29.2 Leaving the study early: 2. Due to adverse events
29.2.1 short term
In this subgroup we only found one relevant trial (n = 105) (Hanlon 1965). There was no significant difference between perphenazine and trifluoperazine (RR 0.65 CI 0.11 to 3.76, Analysis 29.2).
29.2. Analysis.
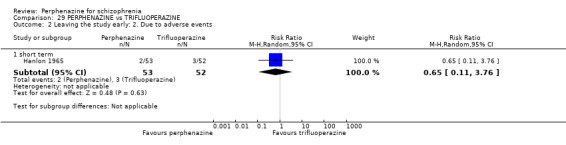
Comparison 29 PERPHENAZINE vs TRIFLUOPERAZINE, Outcome 2 Leaving the study early: 2. Due to adverse events.
29.3 Adverse events: Movement disorders
29.3.1 akathisia ‐ short term
In this subgroup we only found one relevant trial (n = 105) (Hanlon 1965). There was no significant difference between perphenazine and trifluoperazine (RR 2.29 CI 0.63 to 8.38, Analysis 29.3).
29.3. Analysis.
Comparison 29 PERPHENAZINE vs TRIFLUOPERAZINE, Outcome 3 Adverse events: Movement disorders.
29.3.2 akinesia ‐ short term
In this subgroup we only found one relevant trial (n = 105) (Hanlon 1965). There was no significant difference between perphenazine and trifluoperazine (RR 0.33 CI 0.01 to 7.85, Analysis 29.3).
29.3.3 dyskinesia ‐ short term
In this subgroup we only found one relevant trial (n = 105) (Hanlon 1965). There was no significant difference between perphenazine and trifluoperazine (RR 0.98 CI 0.14 to 6.71, Analysis 29.3).
29.3.4 parkinsonism ‐ short term
In this subgroup we only found one relevant trial (n = 105) (Hanlon 1965). There was no significant difference between perphenazine and trifluoperazine (RR 2.62 CI 0.73 to 9.32, Analysis 29.3).
COMPARISON 30: PERPHENAZINE versus TRIFLUOPROMAZINE
30.1 Leaving the study early: 1. Any reason
30.1.1 short term
In this subgroup we found three trials (n = 178). There was no significant difference between perphenazine and trifluopromazine (RR 1.14 CI 0.66 to 1.98, Analysis 30.1). There were important levels of heterogeneity (Chi2 = 3.19; df = 1; P = 0.07; I2 = 69%).
30.1. Analysis.
Comparison 30 PERPHENAZINE vs TRIFLUOPROMAZINE, Outcome 1 Leaving the study early: 1. Any reason.
30.2 Leaving the study early: 2. Due to adverse events
30.2.1 short term
In this subgroup we found two trials (n = 168). There was no significant difference between perphenazine and trifluopromazine (RR 2.00 CI 0.74 to 5.39, Analysis 30.2).
30.2. Analysis.
Comparison 30 PERPHENAZINE vs TRIFLUOPROMAZINE, Outcome 2 Leaving the study early: 2. Due to adverse events.
30.3 Global state: 1. Change over time ‐ no better or deterioration (ITT)
30.3.1 short term
In this subgroup we found one trial (n = 62) (Kurland 1961). There was no significant difference between perphenazine and trifluopromazine (RR 0.71 CI 0.25 to 2.01, Analysis 30.3)
30.3. Analysis.
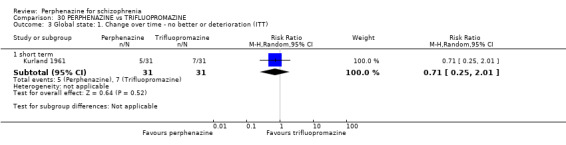
Comparison 30 PERPHENAZINE vs TRIFLUOPROMAZINE, Outcome 3 Global state: 1. Change over time ‐ no better or deterioration (ITT).
30.4 Adverse events: Movement disorders
30.4.1 akathisia ‐ short term
In this subgroup we found one trial (n = 106) (Hanlon 1965). There was no significant difference between perphenazine and trifluopromazine (RR 7.00 CI 0.89 to 54.94, Analysis 30.4).
30.4. Analysis.
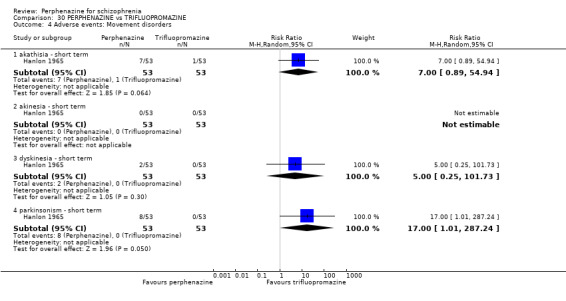
Comparison 30 PERPHENAZINE vs TRIFLUOPROMAZINE, Outcome 4 Adverse events: Movement disorders.
30.4.2 akinesia ‐ short term
In this subgroup we found one trial (n = 106) (Hanlon 1965). There was no significant difference between perphenazine and trifluopromazine, with no events reported in either group (Analysis 30.4).
30.4.3 dyskinesia ‐ short term
In this subgroup we found one trial (n = 106) (Hanlon 1965). There was no significant difference between perphenazine and trifluopromazine (RR 5.00 CI 0.25 to 101.73, Analysis 30.4).
30.4.4 parkinsonism ‐ short term
In this subgroup we found one trial (n = 106) (Hanlon 1965). There was a statistically significant difference (P = 0.05) favouring trifluopromazine over perphenazine (RR 17.00 CI 1.01 to 287.24, Analysis 30.4).
30.5 Other adverse events: 1. Central nervous system
30.5.1 electroencephalogram (increase) ‐ short term
In this subgroup we found one trial (n = 10) (Bennett 1961). There was a no significant difference between perphenazine and trifluopromazine (RR 1.00 CI 0.08 to 11.93, Analysis 30.5).
30.5. Analysis.
Comparison 30 PERPHENAZINE vs TRIFLUOPROMAZINE, Outcome 5 Other adverse events: 1. Central nervous system.
COMPARISON 31: PERPHENAZINE versus ZIPRASIDONE
31.1 Leaving the study early: 1. Any reason
31.1.1 long term
In this subgroup we found one trial (n = 446). There was a no significant difference between perphenazine and ziprasidone (RR 0.95 CI 0.85 to 1.05, Analysis 31.1).
31.1. Analysis.
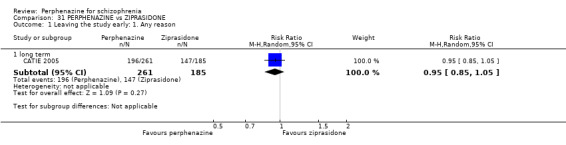
Comparison 31 PERPHENAZINE vs ZIPRASIDONE, Outcome 1 Leaving the study early: 1. Any reason.
31.2 Leaving the study early: 2. Due to adverse events
31.2.1 long term
In this subgroup we found one trial (n = 446) (CATIE 2005). There was a no significant difference between perphenazine and ziprasidone (RR 1.01 CI 0.65 to 1.58, Analysis 31.2).
31.2. Analysis.
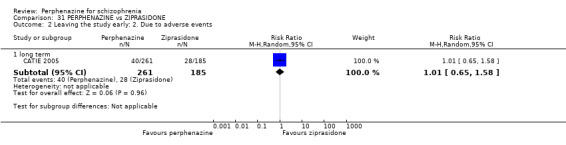
Comparison 31 PERPHENAZINE vs ZIPRASIDONE, Outcome 2 Leaving the study early: 2. Due to adverse events.
31.3 Leaving the study early: 3. Due to relapse/worsening or no improvement
31.3.1 long term
In this subgroup we found one trial (n = 446) (CATIE 2005). There was a no significant difference between perphenazine and ziprasidone (RR 1.05 CI 0.75 to 1.46, Analysis 31.3).
31.3. Analysis.
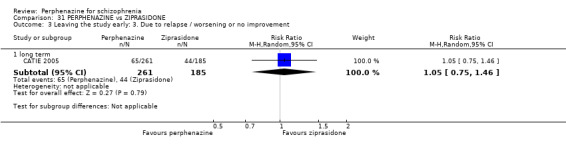
Comparison 31 PERPHENAZINE vs ZIPRASIDONE, Outcome 3 Leaving the study early: 3. Due to relapse / worsening or no improvement.
31.4 Adverse events: Movement disorders (completer‐only data)
31.4.1 akathisia ‐ long term
In this subgroup we found one trial (n = 399) (CATIE 2005). There was a no significant difference between perphenazine and ziprasidone (RR 0.75 CI 0.38 to 1.49, Analysis 31.4).
31.4. Analysis.
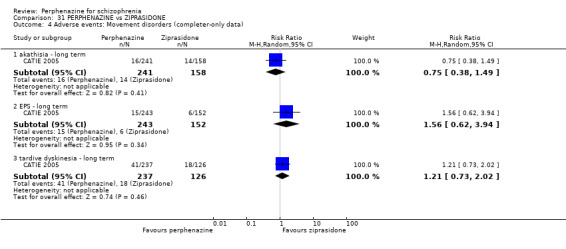
Comparison 31 PERPHENAZINE vs ZIPRASIDONE, Outcome 4 Adverse events: Movement disorders (completer‐only data).
31.4.2 EPS ‐ long term
In this subgroup we found one trial (n = 395) (CATIE 2005). There was a no significant difference between perphenazine and ziprasidone (RR 1.56 CI 0.62 to 3.94, Analysis 31.4).
31.4.3 tardive dyskinesia ‐ long term
In this subgroup we found one trial (n = 363) (CATIE 2005). There was a no significant difference between perphenazine and ziprasidone (RR 1.21 CI 0.73 to 2.02, Analysis 31.4).
31.5 Other adverse events: 1. Arousal
31.5.1 insomnia ‐ long term
In this subgroup we found one trial (n = 446) (CATIE 2005). There was a no significant difference between perphenazine and ziprasidone (RR 0.84 CI 0.62 to 1.13, Analysis 31.5).
31.5. Analysis.
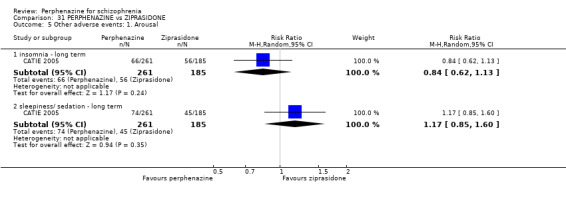
Comparison 31 PERPHENAZINE vs ZIPRASIDONE, Outcome 5 Other adverse events: 1. Arousal.
31.5.2 sleepiness/sedation ‐ long term
In this subgroup we found one trial (n = 446) (CATIE 2005). There was a no significant difference between perphenazine and ziprasidone (RR 1.17 CI 0.85 to 1.60, Analysis 31.5).
31.6 Other adverse events: 2. Cardiovascular
31.6.1 faintness, dizziness, weakness ‐ long term
In this subgroup we found one trial (n = 446) (CATIE 2005). There was a no significant difference between perphenazine and ziprasidone (RR 0.86 CI 0.52 to 1.42, Analysis 31.6).
31.6. Analysis.
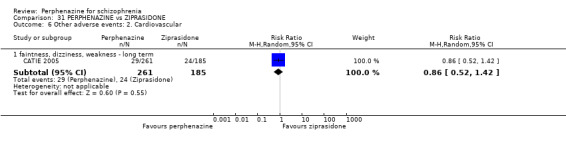
Comparison 31 PERPHENAZINE vs ZIPRASIDONE, Outcome 6 Other adverse events: 2. Cardiovascular.
31.7 Other adverse events: 3. Gastrointestinal
31.7.1 weight gain ‐ long term
In this subgroup we found one trial (n = 404) (CATIE 2005). There was a no significant difference between perphenazine and ziprasidone (RR 1.60 CI 0.84 to 3.04, Analysis 31.7).
31.7. Analysis.
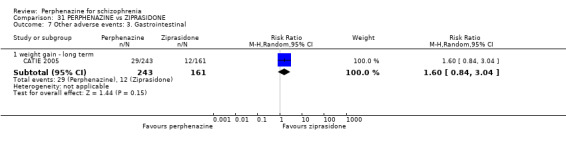
Comparison 31 PERPHENAZINE vs ZIPRASIDONE, Outcome 7 Other adverse events: 3. Gastrointestinal.
31.8 Other adverse events: 4. Genitourinary
31.8.1 diminished sexual desire ‐ long term
In this subgroup we found one trial (n = 446) (CATIE 2005). There was a no significant difference between perphenazine and ziprasidone (RR 1.30 CI 0.90 to 1.87, Analysis 31.8.
31.8. Analysis.
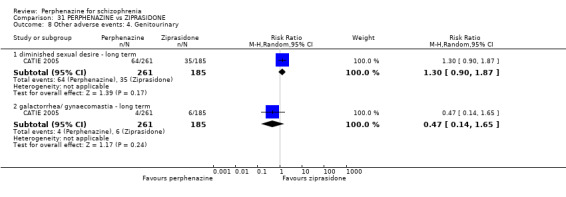
Comparison 31 PERPHENAZINE vs ZIPRASIDONE, Outcome 8 Other adverse events: 4. Genitourinary.
31.8.2 galactorrhoea/gynaecomastia ‐ long term
In this subgroup we found one trial (n = 446) (CATIE 2005). There was a no significant difference between perphenazine and ziprasidone (RR 0.47 CI 0.14 to 1.65, Analysis 31.8.,
31.9 Other adverse events: 5. Others
31.9.1 urinary hesitancy, dry mouth, constipation ‐ long term
In this subgroup we found one trial (n = 446) (CATIE 2005). There was a no significant difference between perphenazine and ziprasidone (RR 1.09 CI 0.76, 1.58, Analysis 31.9).
31.9. Analysis.
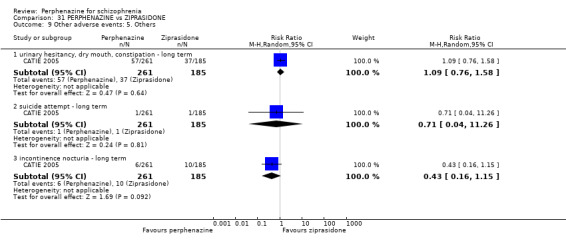
Comparison 31 PERPHENAZINE vs ZIPRASIDONE, Outcome 9 Other adverse events: 5. Others.
31.9.2 suicide attempt ‐ long term
In this subgroup we found one trial (n = 446) (CATIE 2005). There was a no significant difference between perphenazine and ziprasidone (RR 0.71 CI 0.04, 11.26, Analysis 31.9).
31.9.3 incontinence nocturia ‐ long term
In this subgroup we found one trial (n = 446) (CATIE 2005). There was a no significant difference between perphenazine and ziprasidone (RR 0.43 CI 0.16, 1.15, Analysis 31.9).
31.10 Other adverse events: 6. any serious adverse event
31.10.1 long term
In this subgroup we found one trial (n = 446) (CATIE 2005). There was a no significant difference between perphenazine and ziprasidone (RR 1.08 CI 0.63, 1.87, Analysis 31.10).
31.10. Analysis.
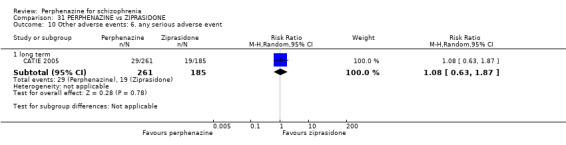
Comparison 31 PERPHENAZINE vs ZIPRASIDONE, Outcome 10 Other adverse events: 6. any serious adverse event.
31.11 Other adverse events: 7. lab data
31.11.1 mean weight change (lb) ‐ long term
In this subgroup we found one trial (n = 446) (CATIE 2005). There was a no significant difference between perphenazine and ziprasidone (RR ‐0.40 CI ‐3.45 to 2.65, Analysis 31.11).
31.11. Analysis.
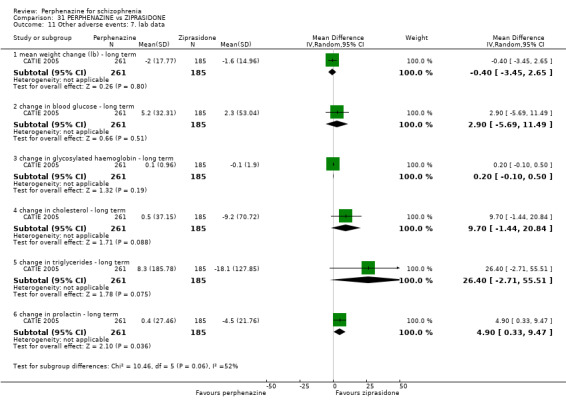
Comparison 31 PERPHENAZINE vs ZIPRASIDONE, Outcome 11 Other adverse events: 7. lab data.
31.11.2 change in blood glucose ‐ long term
In this subgroup we found one trial (n = 446) (CATIE 2005). There was a no significant difference between perphenazine and ziprasidone (RR 2.90 CI ‐5.69 to 11.49, Analysis 31.11).
31.11.3 change in glycosylated haemoglobin ‐ long term
In this subgroup we found one trial (n = 446) (CATIE 2005). There was a no significant difference between perphenazine and ziprasidone (RR 0.20 CI ‐0.10 to 0.50, Analysis 31.11).
31.11.4 change in cholesterol ‐ long term
In this subgroup we found one trial (n = 446) (CATIE 2005). There was a no significant difference between perphenazine and ziprasidone (RR 9.70 CI ‐1.44 to 20.84, Analysis 31.11).
31.11.5 change in triglycerides ‐ long term
In this subgroup we found one trial (n = 446) (CATIE 2005). There was a no significant difference between perphenazine and ziprasidone (RR 26.40 CI ‐2.71 to 55.51, Analysis 31.11).
31.11.6 change in prolactin ‐ long term
In this subgroup we found one trial (n = 446) (CATIE 2005). There was a statistically significant difference (P = 0.04) favouring ziprasidone over perphenazine (RR 4.90 CI 0.33 to 9.47, Analysis 31.11).
COMPARISON 32: PERPHENAZINE versus ZOTEPINE
32.1 Leaving the study early: 1. Any reason
32.1.1 short term
In this subgroup we found one trial (n = 95) (Imai 1980). There was a no significant difference between perphenazine and zotepine (RR 1.48 CI 0.63 to 3.48, Analysis 32.1).
32.1. Analysis.
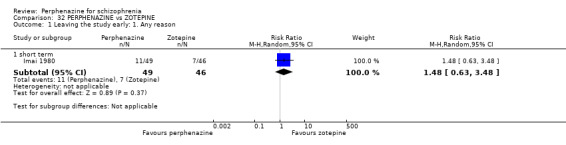
Comparison 32 PERPHENAZINE vs ZOTEPINE, Outcome 1 Leaving the study early: 1. Any reason.
32.2 Leaving the study early: 2. Due to relapse/worsening or no improvement
32.2.1 short term
In this subgroup we found one trial (n = 95) (Imai 1980). There was a no significant difference between perphenazine and zotepine (RR 1.34 CI 0.56 to 3.23, Analysis 32.2).
32.2. Analysis.
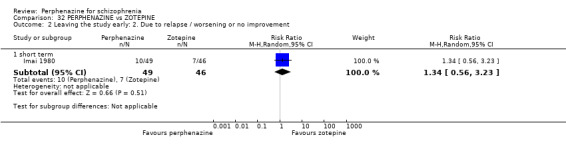
Comparison 32 PERPHENAZINE vs ZOTEPINE, Outcome 2 Leaving the study early: 2. Due to relapse / worsening or no improvement.
32.3 Global state: 1. Change over time ‐ no better or deterioration (ITT)
32.3.1 short term
In this subgroup we found one trial (n = 95) (Imai 1980). There was a no significant difference between perphenazine and zotepine (RR 0.98 CI 0.65 to 1.47, Analysis 32.3).
32.3. Analysis.
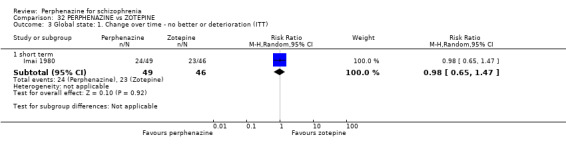
Comparison 32 PERPHENAZINE vs ZOTEPINE, Outcome 3 Global state: 1. Change over time ‐ no better or deterioration (ITT).
32.4 Other adverse events: 1. Cardiovascular
32.4.1 tachycardia ‐ short term
In this subgroup we found one trial (n = 95) (Imai 1980). There was a no significant difference between perphenazine and zotepine (RR 0.31 CI 0.01 to 7.50, Analysis 32.4).
32.4. Analysis.
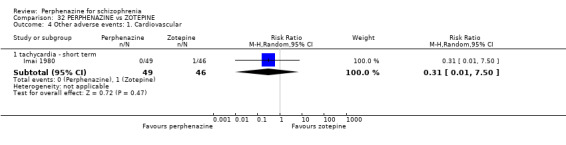
Comparison 32 PERPHENAZINE vs ZOTEPINE, Outcome 4 Other adverse events: 1. Cardiovascular.
COMPARISON 33: PERPHENAZINE versus ZUCLOPENTHIXOL
33.1 Leaving the study early: 1. Any reason
33.1.1 short term
In this subgroup we found one trial (n = 54) (Remvig 1987). There was a no significant difference between perphenazine and zuclupenthixol (RR 1.80 CI 0.69 to 4.67, Analysis 33.1).
33.1. Analysis.
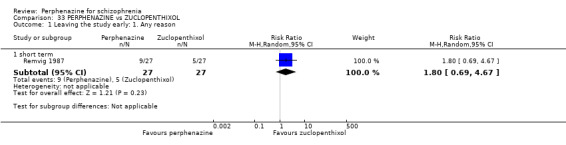
Comparison 33 PERPHENAZINE vs ZUCLOPENTHIXOL, Outcome 1 Leaving the study early: 1. Any reason.
33.2 Global state: 1. Change over time ‐ no better or deterioration (ITT)
33.2.1 short term
In this subgroup we found one trial (n = 54) (Remvig 1987). There was a no significant difference between perphenazine and zuclupenthixol (RR 1.00 CI 0.07 to 15.18, Analysis 33.2).
33.2. Analysis.
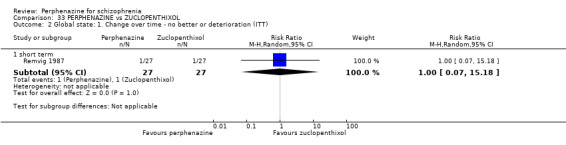
Comparison 33 PERPHENAZINE vs ZUCLOPENTHIXOL, Outcome 2 Global state: 1. Change over time ‐ no better or deterioration (ITT).
33.3 Adverse events: Movement disorders
33.3.1 akathisia ‐ short term
In this subgroup we found one trial (n = 54) (Remvig 1987). There was a no significant difference between perphenazine and zuclupenthixol (RR 1.14 CI 0.48 to 2.71, Analysis 33.3).
33.3. Analysis.
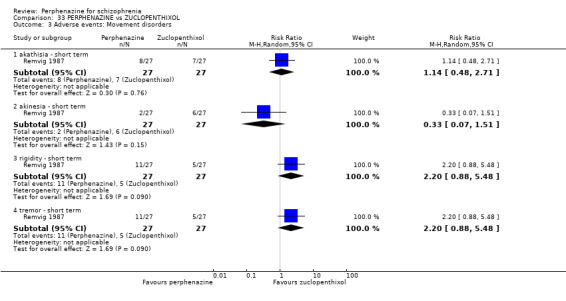
Comparison 33 PERPHENAZINE vs ZUCLOPENTHIXOL, Outcome 3 Adverse events: Movement disorders.
33.3.2 akinesia ‐ short term
In this subgroup we found one trial (n = 54) (Remvig 1987). There was a no significant difference between perphenazine and zuclupenthixol (RR 0.33 CI 0.07 to 1.51, Analysis 33.3).
33.3.3 rigidity ‐ short term
In this subgroup we found one trial (n = 54) (Remvig 1987). There was a no significant difference between perphenazine and zuclupenthixol (RR 2.20 CI 0.88 to 5.48, Analysis 33.3).
33.3.4 tremor ‐ short term
In this subgroup we found one trial (n = 54) (Remvig 1987). There was a no significant difference between perphenazine and zuclupenthixol (RR 2.20 CI 0.88 to 5.48, Analysis 33.3).
33.4 Other adverse events: 1. Arousal
33.4.1 excitation ‐ short term
In this subgroup we found one trial (n = 54) (Remvig 1987). There was a no significant difference between perphenazine and zuclupenthixol (RR 1.29 CI 0.56 to 2.95, Analysis 33.4).
33.4. Analysis.
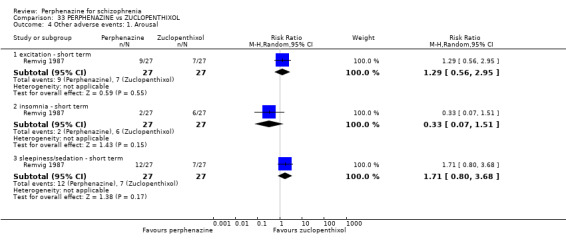
Comparison 33 PERPHENAZINE vs ZUCLOPENTHIXOL, Outcome 4 Other adverse events: 1. Arousal.
33.4.2 insomnia ‐ short term
In this subgroup we found one trial (n = 54) (Remvig 1987). There was a no significant difference between perphenazine and zuclupenthixol (RR 0.33 CI 0.07 to 1.51, Analysis 33.4).
33.4.3 sleepiness/sedation ‐ short term
In this subgroup we found one trial (n = 54) (Remvig 1987). There was a no significant difference between perphenazine and zuclupenthixol (RR 1.71 CI 0.80 to 3.68, Analysis 33.4).
33.5 Other adverse events: 2. Cardiovascular
33.5.1 faintness, dizziness, weakness ‐ short term
In this subgroup we found one trial (n = 54) (Remvig 1987). There was a no significant difference between perphenazine and zuclupenthixol (RR 1.50 CI 0.48 to 4.72, Analysis 33.5).
33.5. Analysis.
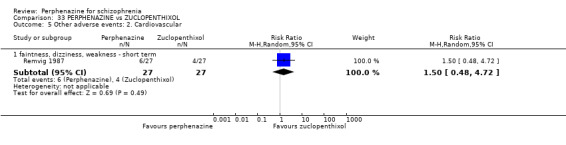
Comparison 33 PERPHENAZINE vs ZUCLOPENTHIXOL, Outcome 5 Other adverse events: 2. Cardiovascular.
34. SENSITIVITY ANALYSIS
1. Implication of randomisation
For the primary outcome of clinical response (as defined in each study), there was no difference in the estimate of the effect when all studies that did not fully describe randomisation methods (this included all studies rated as 'unclear' risk of bias, with only those rated as a 'low' risk remaining, including Amakusa 1973; Itoh 1976; Itoh 1969 II; Itoh 1969 III; Takahashi 1982) were excluded from meta‐analysis, with 'no better or deterioration' in the comparison of perphenazine versus any antipsychotic remaining as no significant difference between groups (5 RCTs, n = 531, RR 1.05 CI 0.88 to 1.24, Analysis 2.4). Only one study provided data for this primary outcome in the perphenazine versus placebo comparison (Kurland 1961, Analysis 1.4), which did not adequately explain randomisation methods; subsequently, the exclusion of this study left us with no data to include in the sensitivity analysis.
2. Assumptions for lost binary data
Where assumptions had to be made regarding people lost to follow‐up and missing SDs data (see Dealing with missing data), we compared the findings on primary outcomes when we used our assumption compared with completer data only. We undertook a sensitivity analysis to test how prone results were to change when 'completer' data only were compared to the imputed data using the above assumption with our primary outcome of global effect: change over time ‐ no better or deterioration. For the perphenazine versus placebo comparison we did not assume any of the outcome data as intention‐to‐treat was used in the study. For the comparison of perphenazine versus any antipsychotic drugs, assumptions were made in five of the 17 studies providing data for the primary outcome of global state: change over time ‐ no better or worse (Amakusa 1973; Fruensgaard 1978; Hoyberg 1993; Kane 2003; Lepola 1989). After replacing our assumptions with completer‐only data, there was no substantial alteration of the estimate of effect (17 RCTs, n = 1771, RR 1.05 CI 0.91 to 1.22, Analysis 2.4). We ultimately continued to employ our assumption.
3. Risk of bias
Sixteen included studies rated as a 'high' risk of bias across at least one risk of bias domain; this included the one study in the placebo comparison for our primary outcome of 'global effect: change over time ‐ no better or deterioration' (Kurland 1961, Analysis 1.4), again, the exclusion of this study left us with no data to compare. With the perphenazine versus any antipsychotic drugs comparison, again there was no difference (9 RCTs, n = 891, RR 1.06 CI 0.94 to 1.20, Analysis 2.4) in the estimate of effect after removing all included studies rated as a 'high' risk across on one or more risk of bias domain (including Eckmann 1984; Fruensgaard 1978; Hoyberg 1993; Kane 2003; Kurland 1961; Remvig 1987; Takahashi 1982; Woggon 1978).
We analysed the effects of excluding trials that were judged to be at high risk of bias across one or more of the domains of randomisation (implied as randomised with no further details available): allocation concealment, blinding and outcome reporting for the meta‐analysis of the primary outcome. If the exclusion of trials at high risk of bias did not substantially alter the direction of effect or the precision of the effect estimates, then we included data from these trials in the analysis.
4. Imputed values
We had planned to undertake a sensitivity analysis to assess the effects of including data from trials where we used imputed values for intra‐class correlation coefficients (ICCs) in calculating the design effect in cluster‐randomised trials. However, no cluster‐randomised studies were included, therefore we did not undertake this part of the sensitivity analysis.
5. Fixed‐effect and random‐effects
For the perphenazine versus placebo comparison, with only one included study, there was no difference in the estimate of effect for global state: change over time ‐ no better or deterioration when using a fixed‐effect model in place of the random‐effects model (Kurland 1961, Analysis 1.4). This was also the case when comparing the same outcome for the perphenazine versus any antipsychotic comparison when using a fixed‐effect model (17 RCTs, n = 1879, RR 1.01 CI 0.92 to 1.11, Analysis 2.4).
Discussion
Summary of main results
1. PERPHENAZINE versus PLACEBO
In general, due to the low number of studies and participants included, we can draw no firm conclusions concerning the effects of perphenazine compared to placebo.
2.1 Acceptability of treatment (leaving the studies early)
When we assessed the acceptability of treatment indirectly from the number of people leaving the studies early for any reason, perphenazine did not appear to be more acceptable to those with schizophrenia than placebo. However, more participants in the placebo groups left the studies prematurely due to relapse or insufficient response. This underlines the fact that perphenazine seems an effective treatment for schizophrenia.
2.2 Global state
Data from one small study (Kurland 1961) indicate that perphenazine is preferable over placebo for improvement, with more people receiving placebo rated as 'no better or deteriorated'; however, due to the size of this study, results need interpreting with caution (n = 61).
2.3 Mental state
Findings on relapse rates are again compatible with perphenazine being more effective than placebo in improving mental state. As only n = 87 participants from two trials (Chouinard 1975; Whittaker 1963) contributed to this finding, the result must be considered with caution.
2.5 Behaviour
Based on only one small study (Collins 1967), we found no significant effects of perphenazine on behaviour. Again, no conclusion can be drawn on such little data.
2.4 Adverse events
Although a number of outcomes related to tolerability were analysed (at least one adverse event, movement disorders, anticholinergic events, arousal, cardiovascular events, central nervous system related problems, endocrine events, gastrointestinal events, abnormal laboratory results, skin problems), the results were never based on more than two studies. Therefore, it is not surprising that we found no significant differences between groups, not even for movement disorders, although perphenazine is well known to induce this troublesome adverse effect in clinical routine.
2. PERPHENAZINE versus OTHER ANTIPSYCHOTIC DRUG
Again, for this comparison, incomplete reporting of the trials clearly limited our analysis. In addition, many different comparators were used. On the one hand, this diversity may improve applicability to a certain extent. For example, most of the newer 'atypical' antipsychotic drugs, such as risperidone, olanzapine or quetiapine, have mainly been compared with haloperidol, an agent that is associated with a high risk for extrapyramidal side effects (EPS). Therefore, it can only be concluded from such trials that new antipsychotic drugs induce fewer EPS than haloperidol, whereas we do not know how the new drugs compare with other old drugs. On the other hand, the variability in terms of comparators found for perphenazine is problematic, because antipsychotic drugs have very different side‐effect profiles and, if pooled in a meta‐analysis, differences may cancel each other out. In this review update, we have presented all drug comparisons separately in order to complement the overall pooled analysis, with a total of 30 drug comparators.
2.1 Leaving the study early
There is no evidence that perphenazine is associated with lower overall tolerability as measured by the numbers of people leaving due to adverse events or with lower efficacy as measured by attrition due to inefficacy of treatment. There were important levels of heterogeneity in the results for leaving the study early for any reason (Chi² = 34.32, df = 17 (P = 0.008); I² = 50%) and due to adverse events (Chi² = 12.82, df = 8 (P = 0.12); I² = 38%), no doubt attributable to the variability in comparisons and varying dosages of either drug used in the individual studies. For leaving the study early due to any reason, the source of heterogeneity was found with Kurland 1961, which compared perphenazine with prochlorperazine, triflupromazine, mepazine, chlorpromazine and promazine; as per our protocol, we combined all comparator treatment arms for the 'perphenazine versus any antipsychotic drugs' comparison, which may well explain this heterogeneity.
2.2 Global state
A relatively large number of people (n = 1850) contributed to the global efficacy outcome 'change over time ‐ no better or worse'. Around 50% of the participants were considered unimproved or worse, but these people were distributed relatively evenly between perphenazine and other antipsychotic drugs. Only Kane 2003 presented a usable CGI end score. There was no significant difference between groups. With only one study providing data for this outcome, it is impossible to generalise such a result.
2.3 Mental state
2.3.1 General
Data were too poorly reported to allow any reasonable assessment of this outcome. Only four studies provided dichotomised data using the BPRS of < 20% reduction in score (Hoyberg 1993), < 25% reduction (Wang 2008c; Zhang 2010), and < 30% reduction (Sun 2000). They found no significant differences between perphenazine and risperidone, quetiapine, aripiprazole or clozapine, respectively. There was a high amount of skewed data reported between studies using both the BPRS and PANSS. Only one statistically significant result was found for mental state using the PANSS total score, which found that significantly more people receiving any other antipsychotic drugs (risperidone, quetiapine, aripiprazole) scored lower on the scale than those receiving perphenazine.
2.3.2 Specific
Results for all other outcomes using the various PANSS subscales were equivocal, making it difficult to generalise results. There was a significant degree of heterogeneity in results for the PANSS positive subscale (Chi² = 74.07, df = 1 (P < 0.00001); I2 = 99%), which we suspect is attributable to the differing drug comparisons between studies. (Wang 2008b used olanzapine as a comparator; Zhang 2010 used aripiprazole), as well as the difference in the permitted maximum dose of perphenazine between the studies or 16 mg/day (Zhang 2010) or 60 mg/day (Wang 2008b).
2.4 Behaviour
Only the three early studies published by Itoh in 1969 reported on behaviour in general, but they found no difference between perphenazine and the comparators clotiapine, thiothixene and methylperidol.
2.5 Tolerability
2.5.1 Movement disorder
According to the 22 studies that provided data on movement disorder, perphenazine showed a similar risk of inducing such adverse effects as other antipsychotic drugs. However, the results appeared on the whole to be relatively heterogeneous, particularly with akathisia, dystonia, rigidity, tremor and use of anti‐parkinsonian drugs. This heterogeneity may be partly due to the diversity of comparators used. More studies using the single comparators would be needed to allow a finer grained analysis of the extrapyramidal symptoms (EPS) risk associated with perphenazine and other single antipsychotic drugs.
2.5.2 Other adverse events
On the whole, there were no convincing differences between perphenazine and other antipsychotic in terms of other adverse events such as anticholinergic, cardiovascular, dermatological, endocrine, gastrointestinal or genitourinary problems. According to a small number of studies, palpitations were significantly less frequent in the perphenazine group.
2.6 Missing data
It is disappointing, and remarkable, that even after the 2013 update search, despite considerable investment in clinical trials, no data are available on social functioning, employment status, cognitive functioning, satisfaction with care, hospital admission, family burden and economic costs.
3. PERPHENAZINE versus OTHER ANTIPSYCHOTICS (ACUTE)
We pooled data for participants in the seven studies that included those in the acute stage of psychosis (Eckmann 1984; Fruensgaard 1978; Hoyberg 1993; Kurland 1961; Lepola 1989; Remvig 1987; Van Praag 1976).
3.1 Leaving the study early
Data demonstrate that significantly more people receiving perphenazine left the study early due to any reason when compared with people receiving any other type of antipsychotic medication. This may suggest that perphenazine is less tolerable and subsequently not as appropriate for people in the acute stage of illness as compared to other antipsychotic drugs, including risperidone, loxapine, sulpiride, zuclopenthixol, and clozapine. However, when assessed individually, there is no difference in the estimate of effect with the occurrence of leaving the study early with 'any other antipsychotic' versus perphenazine.
3.2 Global state
As regards global state, there was no difference in people considered 'no better or deteriorated'; this outcome did present high degrees of heterogeneity (Chi² = 13.71, df = 4 (P = 0.008); I² = 71%); the source of this was found to be Eckmann 1984, interventions of which included perphenazine 12 mg or 24 mg/day versus benperidol 6 mg or 12 mg/day ‐ benperidol is a highly potent butyrophenone derivative that is not commonly prescribed in practice. This may go some way to explaining the inconsistency in the results that include other, less potent comparisons.
3.3 Mental state
In the individual study that included acute participants and provided mental state outcomes using the BPRS and PANSS (Hoyberg 1993, n = 107), there was no significant difference between drugs.
3.4 Adverse events
Across all adverse events that were reported, the only significant results was that of weight gain, which was seen more in people receiving risperidone than those receiving perphenazine (Hoyberg 1993, n = 107). Again, this finding is based on one small study ‐ more evidence is needed to derive power from this results.
4. PERPHENAZINE versus SPECIFIC‐NAMED ANTIPSYCHOTICS
All antipsychotic drugs that were used as a comparator have been presented separately from the meta‐analysis in comparison two; however, there was never more than three RCTs included in these other comparisons, making a meaningful interpretation of the evidence relating to each specific drug difficult. We have, however, aimed to make available and accessible all specific comparisons, and have summarised the results to these below.
4.1 Leaving the study early
In the main comparison of 'perphenazine versus any antipsychotic drugs', the high degree of heterogeneity in the results for leaving the study early for any reason (Chi² = 34.32, df = 17 (P = 0.008); I² = 50%) was found to be attributable to Kurland 1961, which compared perphenazine with prochlorperazine, triflupromazine, mepazine, chlorpromazine and promazine; again, as per our protocol, we combined all comparator treatment arms for the 'perphenazine versus any antipsychotic drugs' comparison, which may well explain this heterogeneity. As suspected, when we inspected these drug comparators individually, we identified no significant differences between perphenazine and prochlorperazine (Analysis 20.1), trifluopromazine (Analysis 30.1), mepazine (Analysis 16.1), chlorpromazine (Analysis 7.1), and promazine (Analysis 23.1). The addition of CATIE 2005 to the body of the evidence has highlighted that people receiving perphenazine were more likely to leave the study early when compared to olanzapine, however with more leaving early if receiving quetiapine, and no difference with risperidone or ziprasidone.
4.2 Global state
For the outcome of global state ('no better or deterioration', which was defined by each individual study), results were significantly in favour of perphenazine when comparing with benperidol (Analysis 5.2), mepazine (Analysis 16.3) and promazine (Analysis 22.3). However, these findings are based on the results of two studies providing data for these outcomes, with less than n = 60 participants overall (Eckmann 1984; Kurland 1961), therefore, it is not possible to draw meaningful conclusions from these results. All other comparisons demonstrated no difference when compared with perphenazine.
4.3 Mental state
Generally, there was no difference between specific‐named antipsychotic drugs versus perphenazine for both dichotomous and continuous outcomes. However, one small study (Wang 2008b, n = 60) found a statistically significant difference between groups for higher scores (worse mental state) on the PANSS total, positive and negative scores, when comparing perphenazine with olanzapine (Analysis 18.6). Another study (Sun 2000) found a statistically significant difference between groups for higher scores (worse mental state) on the BPRS anxiety‐depression score when comparing perphenazine with clozapine. Again, these data need backing‐up with further studies making these specific comparisons in order to draw meaningful conclusions.
4.4 Behaviour
Only three studies overall provided data for behaviour outcomes (Itoh 1969; Itoh 1969 II; Itoh 1969 III); each comparator demonstrated no difference (clothiapine, methyperidol and thiothixene) when compared with perphenazine. All other studies, disappointingly, did not provide any useable data relating to behavioural outcomes.
4.5 Tolerability
as regards movement disorders, there was little light between studies comparing specific antipsychotic drugs with perphenazine; the only statistically significant differences to be found were with instances of EPS, which were more prominent in people receiving perphenazine compared to risperidone (CATIE 2005). Perphenazine was also less favourable with participants. with significantly more people experiencing tremor than those receiving either clozapine (Sun 2000) or quetiapine (Wang 2008c). More people receiving perphenazine required EPS drugs compared to those receiving aripiprazole (Kane 2003), a significant finding. However, it was found that more people receiving clopenthixol required EPS drugs compared to those receiving perphenazine (Dehnel 1968). These are all findings that need further evidence from research in order to confirm the differences expected between the various comparator drugs.
Overall completeness and applicability of evidence
1. Applicability
Eleven studies out of the included 31 used operational criteria to diagnose participants. Since the diagnosis of schizophrenia has changed over time, the use of simple clinical judgement may have led to the inclusion of some people who would nowadays not be considered to have schizophrenia. On the other hand, the studies may reflect clinical practice where diagnostic instruments are rarely used to meticulously‐classify patients. Indeed, the participants appear to be broadly similar to those in clinical practice in terms of duration and characteristics of illness. Furthermore, the centres of the trials covered a relatively wide range of geographic regions that included Japan, Europe (especially Scandinavia) and North America. One problem is, however, that the vast majority of studies were undertaken in hospitals. Many people with schizophrenia are treated in the community, but not many outpatient studies on perphenazine are available.
The included studies mainly fell into the 'short‐term' category. The lack of longer‐term studies is unfortunate because schizophrenia is typically a chronic illness. Only two trials had a duration of more than six months (CATIE 2005; Naukkarinen 2000). Little can therefore be concluded regarding the long‐term effects of perphenazine and more long‐term trials are needed.
Regarding interventions and doses, the studies in this review presented strategies that are similar to those used in daily clinical practice. In most of the studies, doses between 8 mg and 24 mg were given. However, in some studies, doses could be increased to up to 120 mg daily (Fruensgaard 1978). Perphenazine was compared with placebo and a wide variety of 30 other antipsychotic drugs. This high variability of comparators may in part explain why the meta‐analytic combination of the trials revealed no convincing differences between perphenazine and control antipsychotic drugs. However, even with analysis of perphenazine versus specific antipsychotic drugs, little difference was found between either groups. Finally, it frequently appeared likely that the patients' therapists made the assessments with scales, although clear statements were frequently not made. Many of the data must therefore be considered as prone to bias. The addition of the CATIE 2005 study was timely, as it provided the review with evidence from an important RCT concerning the effectiveness of older antipsychotic drugs as compared to the newer 'atypicals', the results of which our review largely concurs.
Quality of the evidence
The overall quality of the evidence was 'very low'; only seven publications presented adequate information about allocation concealment. All other 24 included studies were rated as 'unclear', because it was unknown exactly how the randomisation had been undertaken. This is a potential fundamental source of bias.
Funnel plots did not suggest evidence for publication bias in the sense that small trials with no beneficial effects for perphenazine might not have been published. However, in most comparisons it was possible to include only a few studies, which clearly limits the power of this method to detect publication bias (if fewer than eight studies were available, funnel‐plots were not even considered).
Potential biases in the review process
The review protocol and process of study selection were strictly adhered to throughout the entire review process, and the process for searching for studies was thorough and data were extracted independently; however, this was made difficult due to numerical inconsistencies in parts of the reported data. Review authors adhered to the guidance from the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011) and the Methodological Expectations for Cochrane Intervention Reviews (MECIR). Furthermore, authors of included studies were contacted to obtain details of ongoing or unpublished studies, but there remains a possibility that other unpublished trials of the intervention exist to which the review authors do not currently have access. As a consequence, publication bias may have been perpetuated as we worked predominantly with published reports.
Agreements and disagreements with other studies or reviews
For other formulations of perphenazine that were not included in this review (depot perphenazine decanoate and enanthate), the currently‐published Cochrane review demonstrates again little differences between the depot or enanthate compounds and other antipsychotic drugs with similar administration (David 2005). As with our review, the body of evidence and the quality was either poor or unclear, with more call for further high quality, large RCTs that will give greater, clearer indication of the benefits or harms of these compounds versus others. The addition of the CATIE 2005 study added to the body of evidence that perphenazine seems to be as effective as other antipsychotic drugs for the treatment of schizophrenia.
Authors' conclusions
Implications for practice.
1. For people with schizophrenia
Perphenazine seems to be more effective than placebo and as effective as other antipsychotic drugs for the treatment of schizophrenia. The adverse effect profile of the drug could not be differentiated from that of other antipsychotic drugs, but this is, in part, due to incomplete reporting of data.
2. For clinicians
Most of the data about perphenazine relate to a short treatment period. We found no clear differences in terms either of efficacy or adverse effects compared to other antipsychotic compounds. We believe that this result was, in part, related to a high variability of comparators used, which may have blurred differences compared to specific groups of antipsychotic drugs, e.g. high/low‐potency compounds or 'atypical' antipsychotic drugs.
3. For managers or policy makers
Included studies reported no data relating to service utilisation or functioning in the community. No good data are available even for simple clinical questions such as optimum dose. On the other hand, perphenazine is a relatively cheap drug compared to new, 'atypical' antipsychotic drugs. Therefore, investing some money in research on this drug could be cost‐saving in the long run. CATIE 2005 performed a cost‐effectiveness evaluation of the drugs used in the study, and we anticipate a more complete economic evaluation in a future update of this review.
Implications for research.
1. General
The vast majority of the studies took place at least 30 years ago. This explains why reporting may not be to modern standards (Begg 1996; Moher 2001) and thus limiting possibilities of summarising data in a meta‐analysis and arriving at clear conclusions. We therefore believe that studies are still warranted, since perphenazine is an inexpensive drug that has been used by physicians over decades. Generally, this review has shown that there is very little difference between perphenazine and other (either 'typical' or 'atypical') antipsychotic medication, and that the decision regarding which antipsychotic will be best for a person should be judged between clinician and patient, according to the latter's individual needs.
2. Specific
Future trials should be longer and better‐reported than the currently available studies. Such studies should address not only pure 'efficacy', but also clinically relevant outcomes such as relapse, hospital admission, satisfaction with care, social functioning, family burden and employment. The optimum dose of perphenazine needs to be evaluated in dose finding studies. Other objectives might be to compare perphenazine with 'atypical' antipsychotic drugs and with high/low‐potency antipsychotic drugs to better identify the profile of the compound.
What's new
| Date | Event | Description |
|---|---|---|
| 2 December 2014 | New citation required but conclusions have not changed | Six new trials added after 2013 update search, results from these trials did not change overall conclusions of the review. |
| 4 September 2013 | New search has been performed | Update search run in September 2013 |
History
Protocol first published: Issue 1, 2002 Review first published: Issue 1, 2005
| Date | Event | Description |
|---|---|---|
| 22 October 2004 | New citation required and conclusions have changed | Substantive amendment |
Acknowledgements
The review authors would like to thank the editorial team of the Cochrane Schizophrenia Group for all their help. The Group produces and maintains standard text for use in the methods sections of their reviews. We have used this text as the basis for our protocol and adapted it as required. We would also like to thank Kristian Wahlbeck for translating Povlsen 1987. We would also like to thank Noriko Yamashita for her help with translating included Japanese studies and Makoto Wada for extracting the data from the Japanese studies. Laux Gerd helped develop the protocol and provided access to further reports on perphenazine, we thank him for his help.
Many letters were sent by the review authors to authors, asking for extra information about their trials. We are very grateful that Profs. Rimon, Levine, van Praag, Shalev and Svestka were kind enough to respond.
Ricardo Harripaul kindly peer reviewed this version.
Appendices
Appendix 1. Previous searches
1. Electronic searching We searched Cochrane Schizophrenia Group's Register (June 2001) with the phrase:
[aethaperazin or apo‐perphenazine or decentan or fentazin or f‐mon or peratsin or perfenazine or perfenazin leciva or pms‐perphenazine or trilafon or trilifan or trilafon prolongatum or trilafon repetabs or 10‐{3‐[4‐(2‐hydroxyethyl)‐1‐piperazinyl]‐propyl}‐2‐chlor‐phenothiazin]
2. Reference searching We inspected references of all identified studies for more studies.
3. Personal contact We contacted the first author of each included study for more information regarding unpublished trials.
4. Drug company We contacted the German manufacturers of perphenazine (Merck, Germany) for additional data.
Appendix 2. Previous data collection and analyses
1. Selection of trials We (Benno Hartung BH, Stefan Leucht SL) independently inspected all citations of studies identified by the search. Where disagreement occurred we resolved this by discussion, or, when there was still doubt, we acquired the full article for further inspection. Once we had obtained the full articles BH decided whether they met review criteria and this was checked by SL. Once again, we resolved disagreement by discussion, but if doubt remained, we put the study on the list of those awaiting assessment, pending acquisition of more information. These rules could not be respected for the Japanese studies. They were analysed by Makoto Wada (MW)who had to explain his analysis to one of the other reviewers.
2. Quality assessment We used criteria in the Cochrane Collaboration Handbook (Clarke 2000) to assess trial quality. This simple set of criteria is based on evidence of strong association between overestimation of effect and poor concealment of allocation, and is defined as follows:
A. Low risk of bias (adequate allocation concealment) B. Moderate risk of bias (some doubt about the results) C. High risk of bias (inadequate allocation concealment)
The Jadad scale (Jadad 1996) measures a wider range of factors that impact on the quality of a trial. The scale includes three items:
1. Was the study described as randomised? 2. Was the study described as double blind? 3. Was there a description of withdrawals and dropouts?
Each item scores one point if the answer is positive and an additional point for the first two items if the means of randomisation/blinding is described. In addition, a point can be deducted if either the randomisation or the blinding/masking described were inadequate.
For the purpose of analysis of this review, we included studies if they met criteria A and B of the Handbook. We gave studies not described as randomised by the authors a 'C' rating and excluded them from analyses. We did not use the Jadad scale to exclude studies, but for a further description of the quality of included trials.
3. Data extraction 3.1 Reliable extraction BH extracted data from selected trials and SL checked all data extraction. When disputes arose we attempted resolution by discussion. If doubt remained and further information was necessary to resolve the dilemma, we did not enter data but contacted the authors of the studies for further information. This rule could not be respected for the Japanese studies. These were analysed by MW who had to explain his analysis to the reviewers.
3.2 Multiple doses If several doses of perphenazine were compared with one dose of the control drug, we pooled different dose groups of perphenazine. For example, for dichotomous data we considered all patients treated with perphenazine as a single group. For continuous data we used the mean of all patients on perphenazine in the comparisons.
4. Data synthesis 4.1 Data types Outcomes are assessed using continuous (for example, average changes on a behaviour scale), categorical (for example, one of three categories on a behaviour scale, such as 'little change', 'moderate change' or 'much change') or dichotomous measures (for example, either 'no important changes' or 'important changes' in a person's behaviour). Currently the RevMan software does not support categorical data, so we presented them only in the text of the review.
4.2 Incomplete data With the exception of the outcome of leaving the study early, we included trial outcomes of studies with a remark 'prone to bias' if more than 50% of people in short and medium‐term studies or 60% of people in long‐term studies were not reported in the final analysis. We felt that with such a great degree of attrition, great assumptions would have to be made. We checked whether the exclusion of these results made a substantial difference in a sensitivity analysis.
4.3 Dichotomous/binary data 4.3.1 Defining cut offs: we recorded in RevMan where the original authors of the studies gave outcomes such as 'clinically improved' or 'not clinically improved', based on their clinical judgement, predetermined criteria, or any scale. We only included data from a rater not clearly stated to be independent if they did not change the results.Otherwise we presented such data separately with a label 'prone to bias'. We made efforts to convert relevant categorical or continuous outcome measures to dichotomous data by identifying cut off points on rating scales and dividing people accordingly into groups. For example, for end of study data, we used the cut off points 'moderate or severe impairment' and 'no better or worse' for change data. For another example, the Brief Psychiatric Rating Scale (BPRS ‐ Overall 1962) is frequently used as a measure of change of symptoms in studies. We defined a 50% change on this particular scale as clinically important, although it was recognised that for many people, especially those with chronic or severe illnesses, a less rigorous definition of important improvement would be equally valid. If individual patient data had been available, the 50% cut off would also have been used in the case of non‐chronically ill people and 20% for those with chronic illness.
4.3.2 Intention‐to‐treat analysis: the review presents an intention to treat analysis. As long as more than 50% of people in short and medium term studies and 40% in long term studies completed the trial, we counted everyone allocated to the intervention whether or not they completed follow up. It was assumed that those who left the study early had no change in their outcome.
4.3.3 Summary statistic: we used relative risk (RR) and 95% confidence interval (CI) based on the random effects model. This method takes into account differences between studies, even if heterogeneity is not statistically significant. It has also been shown that RR is more intuitive and that odds ratios tend to be interpreted as RR by clinicians (Deeks 2000). This misinterpretation then leads to an overestimate of the impression of the effect. Where possible, reviewers estimated the number needed to treat (NNT) using an on‐line calculator (http://www.nntonline.net/).
4.4 Continuous data 4.4.1 Intention‐to‐treat versus completer analyses: in the case of continuous data we supposed that in many cases an intention‐to‐treat analysis would not be available, so presented a completer analysis.
4.4.2 Rating scales: a wide range of instruments is available to measure mental health outcomes. These instruments vary in quality and many are not valid, or even ad hoc. For outcome instruments some minimum standards have to be set. We included continuous data from rating scales only if the measuring instrument had been described in a peer‐reviewed journal (Marshall 2000), the instrument was either a self report or completed by an independent rater or relative (not the therapist), and the instrument could be considered a global assessment of an area of functioning. However, as it was expected that therapists would frequently also be the rater, such data were commented on with 'prone to bias'.
4.4.3 Normal distribution of data: mental health continuous data are often not normally distributed. Most statistics assume a normal distribution. To avoid including non‐normally distributed data in the statistical analysis, the following criteria are applied to all data before inclusion: a. Standard deviations and means were reported or derivable from data in the paper, or were obtainable from the authors. b. When a scale starts from zero, the standard deviation, when multiplied by two, is less than the mean (as otherwise the mean is unlikely to be an appropriate measure of the centre of the distribution ‐ Altman 1996). Endpoint scores on scales often have a finite start and end point and this rule can be applied. c. When continuous data are presented on a scale which includes a possibility of negative values (such as change on a scale) it is impossible to tell whether data are non‐normally distributed (skewed) or not. It is thus preferable to use scale end point data, which typically cannot have negative values. If end point data were not available, reviewers chose to use change data, because the statistics used in Metaview are rather robust towards skew. d. If a scale starts from a positive value (such as PANSS, which can have values from 30‐210) the calculation described above in (b) should be modified to take the scale starting point into account. In these cases skewness is present if 2SD>(S‐Smin), where S is the mean score and Smin is the minimum score.
4.4.4 Endpoint versus change data: endpoint scale‐derived data are finite, ranging from one score to another. Change data are more problematic and for them the rule described above does not hold. Although most change scores are likely to be skewed, this cannot be proven so we presented them in MetaView. Where both endpoint and change were available for the same outcome, we presented only the former.
4.4.5 Summary statistic: for continuous outcomes, a weighted mean difference (WMD) between groups was estimated. Again, a random effects model was used.
4.5 Cluster trials Studies increasingly employ 'cluster randomisation' (such as randomisation by clinician or practice) but analysis and pooling of clustered data poses problems. Firstly, authors often fail to account for intra class correlation in clustered studies, leading to a 'unit of analysis' error (Divine 1992) whereby p values are spuriously low, confidence intervals unduly narrow and statistical significance overestimated. This causes type I errors (Bland 1997, Gulliford 1999).
Where clustering was not accounted for in primary studies, we presented the data in a table, with a (*) symbol to indicate the presence of a probable unit of analysis error. In subsequent versions of this review we will seek to contact first authors of studies to obtain intra class correlation co‐efficients of their clustered data and to adjust for this by using accepted methods (Gulliford 1999). Where clustering has been incorporated into the analysis of primary studies, we will also present these data as if from a non‐cluster randomised study, but adjusted for the clustering effect.
We have sought statistical advice and have been advised that the binary data as presented in a report should be divided by a 'design effect'. This is calculated using the mean number of participants per cluster (m) and the intra‐class correlation co‐efficient (ICC) Design effect=1+(m‐1)*ICC (Donner 2002). If the ICC was not reported it was assumed to be 0.1 (Ukoumunne 1999).
If cluster studies had been appropriately analysed taking into account intra‐class correlation co‐efficients and relevant data documented in the report, synthesis with other studies would have been possible using the generic inverse variance technique.
4.6 Sensitivity analyses For binary data, we compared results for intention‐to‐treat analysis with those using completer only data. If analyses using completer data did result in a substantive change in the estimate of effect, we discussed this in the section Discussion.
5. Heterogeneity Firstly, we considered all the included studies within any comparison to judge clinical heterogeneity. Then we visually inspected graphs to investigate the possibility of statistical heterogeneity. This was supplemented, primarily, by employing the I‐squared statistic. This provides an estimate of the percentage of inconsistency thought to be due to chance. Where the I‐squared estimate was greater than or equal to 75%, this was interpreted as evidence of high levels of heterogeneity (Higgins 2003). If inconsistency was high, we did not summate the data, but presented them separately and investigated reasons for heterogeneity.
6. Addressing publication bias We entered data from all identified and selected trials into a funnel graph (trial effect versus trial size) in an attempt to investigate overt publication bias (Egger 1997).
7. Tables and figures Where possible, we entered data into RevMan so the area to the left of the line of no effect indicated a favourable outcome for perphenazine.
Appendix 3. Previous results of search
Results of the search
The original searches yielded 133 electronic records and we inspected the abstract of each identified reference. We then ordered and inspected 90 possibly relevant reports. During this inspection process, we examined the papers' reference lists and identified a further six references as being relevant. Personal mail contact with the first authors of each included trial revealed no additional reports. One unpublished manuscript was provided by the manufacturers (Eckmann 1984). All these papers were considered to match the review's inclusion criteria closely enough to be mentioned in either the 'Included studies' (25 references) or 'Excluded studies' (88 references) section. Fourteen remaining reports are still 'awaiting assessment' for the reasons mentioned above.
Data and analyses
Comparison 1. PERPHENAZINE vs PLACEBO.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Leaving the study early: 1. Any reason | 5 | 261 | Risk Ratio (M‐H, Random, 95% CI) | 0.30 [0.04, 2.33] |
| 1.1 short term | 4 | 203 | Risk Ratio (M‐H, Random, 95% CI) | 0.26 [0.01, 4.92] |
| 1.2 medium term | 1 | 58 | Risk Ratio (M‐H, Random, 95% CI) | 0.40 [0.08, 1.90] |
| 2 Leaving the study early: 2. Due to adverse events | 2 | 119 | Risk Ratio (M‐H, Random, 95% CI) | 9.43 [1.22, 73.11] |
| 2.1 short term | 1 | 61 | Risk Ratio (M‐H, Random, 95% CI) | 16.47 [0.99, 273.29] |
| 2.2 medium term | 1 | 58 | Risk Ratio (M‐H, Random, 95% CI) | 5.0 [0.25, 99.82] |
| 3 Leaving the study early: 3. Due to relapse / worsening or no improvement | 2 | 84 | Risk Ratio (M‐H, Random, 95% CI) | 0.10 [0.03, 0.42] |
| 3.1 short term | 2 | 84 | Risk Ratio (M‐H, Random, 95% CI) | 0.10 [0.03, 0.42] |
| 4 Global state: 1. Change over time ‐ no better or deterioration (ITT) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 short term | 1 | 61 | Risk Ratio (M‐H, Random, 95% CI) | 0.32 [0.13, 0.78] |
| 5 Mental state: 1. Relapse (clinical diagnosis) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 short term | 1 | 48 | Risk Ratio (M‐H, Random, 95% CI) | 0.14 [0.02, 1.07] |
| 6 Behaviour: 1a. Wing‐Scale B: social withdrawal ‐ mean change from baseline to endpoint (high = poor) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 6.1 short term | 1 | 58 | Mean Difference (IV, Random, 95% CI) | 0.76 [‐0.51, 2.03] |
| 7 Behaviour: 1b. Wing‐Scale B: socially embarrassing behaviour ‐ mean change from baseline to endpoint (high = poor) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 7.1 short term | 1 | 58 | Mean Difference (IV, Random, 95% CI) | 0.10 [‐0.54, 0.74] |
| 8 Adverse events: Movement disorders | 4 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 8.1 akathisia ‐ short term | 1 | 48 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 8.2 body rocking ‐ short term | 1 | 48 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.13, 70.16] |
| 8.3 dystonia ‐ short term | 1 | 48 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.07, 15.08] |
| 8.4 extrapyramidal signs ‐ short term | 1 | 48 | Risk Ratio (M‐H, Random, 95% CI) | 1.75 [0.59, 5.21] |
| 8.5 facial mask ‐ short term | 1 | 48 | Risk Ratio (M‐H, Random, 95% CI) | 5.0 [0.25, 98.96] |
| 8.6 oculogyric crisis ‐ short term | 1 | 48 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.13, 70.16] |
| 8.7 parkinsonism ‐ short term | 1 | 58 | Risk Ratio (M‐H, Random, 95% CI) | 7.0 [0.38, 129.74] |
| 8.8 posture and gait ‐ short term | 1 | 48 | Risk Ratio (M‐H, Random, 95% CI) | 5.0 [0.25, 98.96] |
| 8.9 rigidity ‐ short term | 1 | 48 | Risk Ratio (M‐H, Random, 95% CI) | 1.33 [0.33, 5.33] |
| 8.10 tremor ‐ short term | 1 | 48 | Risk Ratio (M‐H, Random, 95% CI) | 1.5 [0.27, 8.19] |
| 8.11 tremor ‐ medium term | 1 | 58 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.15, 6.63] |
| 8.12 use of antiparkinsonian drugs ‐ short term | 1 | 36 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.13, 69.09] |
| 9 Other adverse events: 1. Anticholinergic | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 9.1 blurred vision ‐ short term | 1 | 48 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 9.2 dry mouth ‐ short term | 1 | 48 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.07, 15.08] |
| 9.3 dry mouth ‐ medium term | 1 | 58 | Risk Ratio (M‐H, Random, 95% CI) | 1.5 [0.27, 8.32] |
| 9.4 nasal congestion ‐ short term | 1 | 48 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.13, 70.16] |
| 10 Other adverse events: 2. Arousal | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 10.1 agitation ‐ medium term | 1 | 58 | Risk Ratio (M‐H, Random, 95% CI) | 0.8 [0.24, 2.68] |
| 10.2 drowsiness ‐ short term | 1 | 48 | Risk Ratio (M‐H, Random, 95% CI) | 0.17 [0.02, 1.28] |
| 10.3 drowsiness ‐ medium term | 1 | 58 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 7.86] |
| 10.4 excitement ‐ short term | 1 | 48 | Risk Ratio (M‐H, Random, 95% CI) | 0.38 [0.18, 0.79] |
| 10.5 insomnia ‐ short term | 1 | 48 | Risk Ratio (M‐H, Random, 95% CI) | 0.56 [0.31, 1.01] |
| 10.6 insomnia ‐ medium term | 1 | 58 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 11 Other adverse events: 3. At least one | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 11.1 short term | 2 | 94 | Risk Ratio (M‐H, Random, 95% CI) | 1.5 [0.47, 4.76] |
| 12 Other adverse events: 4. Cardiovascular | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 12.1 ECG abnormalities ‐ short term | 1 | 36 | Risk Ratio (M‐H, Random, 95% CI) | 2.0 [0.42, 9.58] |
| 12.2 faintness, dizziness, weakness ‐ short term | 1 | 48 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.15, 6.53] |
| 12.3 faintness, dizziness, weakness ‐ medium term | 1 | 58 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.22, 4.55] |
| 12.4 hypotension ‐ medium term | 1 | 58 | Risk Ratio (M‐H, Random, 95% CI) | 5.0 [0.25, 99.82] |
| 12.5 tachycardia ‐ short term | 1 | 48 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 7.80] |
| 13 Other adverse events: 5. Central nervous system | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 13.1 headache ‐ short term | 1 | 48 | Risk Ratio (M‐H, Random, 95% CI) | 0.75 [0.19, 3.00] |
| 14 Other adverse events: 6. Endocrine | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 14.1 lactation ‐ short term | 1 | 48 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 15 Other adverse events: 7. Gastrointestinal | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 15.1 constipation ‐ short term | 1 | 48 | Risk Ratio (M‐H, Random, 95% CI) | 0.8 [0.24, 2.62] |
| 15.2 diarrhoea ‐ short term | 1 | 48 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 7.80] |
| 15.3 nausea or vomiting ‐ short term | 1 | 48 | Risk Ratio (M‐H, Random, 95% CI) | 0.6 [0.16, 2.23] |
| 15.4 nausea or vomiting ‐ medium term | 1 | 58 | Risk Ratio (M‐H, Random, 95% CI) | 5.0 [0.25, 99.82] |
| 16 Other adverse events: 8. Haematology ‐ abnormal laboratory results | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 16.1 short term | 1 | 48 | Risk Ratio (M‐H, Random, 95% CI) | 1.5 [0.48, 4.65] |
| 17 Other adverse events: 9. Skin | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 17.1 dermatitis ‐ short term | 1 | 48 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 7.80] |
| 17.2 oedema ‐ medium term | 1 | 58 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 7.86] |
| 18 Other adverse events: 10. Others | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 18.1 salivation increased ‐ short term | 1 | 48 | Risk Ratio (M‐H, Random, 95% CI) | 2.0 [0.19, 20.61] |
| 18.2 tenseness ‐ medium term | 1 | 58 | Risk Ratio (M‐H, Random, 95% CI) | 0.5 [0.05, 5.21] |
Comparison 2. PERPHENAZINE vs ANY ANTIPSYCHOTIC.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Leaving the study early: 1. Any reason | 24 | 4067 | Risk Ratio (M‐H, Random, 95% CI) | 1.10 [0.92, 1.31] |
| 1.1 short term | 21 | 2500 | Risk Ratio (M‐H, Random, 95% CI) | 1.09 [0.83, 1.42] |
| 1.2 medium term | 1 | 47 | Risk Ratio (M‐H, Random, 95% CI) | 1.92 [0.86, 4.25] |
| 1.3 long term | 2 | 1520 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.94, 1.09] |
| 2 Leaving the study early: 2. Due to adverse events | 12 | 3214 | Risk Ratio (M‐H, Random, 95% CI) | 1.11 [0.82, 1.52] |
| 2.1 short term | 9 | 1661 | Risk Ratio (M‐H, Random, 95% CI) | 1.13 [0.70, 1.83] |
| 2.2 medium term | 1 | 47 | Risk Ratio (M‐H, Random, 95% CI) | 2.88 [0.12, 67.29] |
| 2.3 long term | 2 | 1506 | Risk Ratio (M‐H, Random, 95% CI) | 1.07 [0.79, 1.47] |
| 3 Leaving the study early: 3. Due to relapse / worsening or no improvement | 10 | 2532 | Risk Ratio (M‐H, Random, 95% CI) | 1.10 [0.90, 1.35] |
| 3.1 short term | 7 | 979 | Risk Ratio (M‐H, Random, 95% CI) | 1.26 [0.78, 2.03] |
| 3.2 medium term | 1 | 47 | Risk Ratio (M‐H, Random, 95% CI) | 1.15 [0.41, 3.25] |
| 3.3 long term | 2 | 1506 | Risk Ratio (M‐H, Random, 95% CI) | 1.07 [0.85, 1.34] |
| 4 Global state: 1. Change over time ‐ no better or deterioration (ITT) | 17 | 1850 | Risk Ratio (M‐H, Random, 95% CI) | 1.04 [0.92, 1.17] |
| 4.1 short term | 16 | 1803 | Risk Ratio (M‐H, Random, 95% CI) | 1.03 [0.90, 1.17] |
| 4.2 medium term | 1 | 47 | Risk Ratio (M‐H, Random, 95% CI) | 1.20 [0.73, 1.97] |
| 5 Global state: 2. Average endpoint score (CGI‐S, high = poor) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 5.1 short term | 1 | 294 | Mean Difference (IV, Random, 95% CI) | ‐0.20 [‐0.56, 0.16] |
| 6 Mental state: 2a. State ‐ BPRS score reduction ('no effect') | 4 | 383 | Risk Ratio (M‐H, Random, 95% CI) | 1.24 [0.61, 2.52] |
| 6.1 less than 20% reduction ‐ short term | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 0.81 [0.61, 1.08] |
| 6.2 less than 25% reduction ‐ short term | 2 | 129 | Risk Ratio (M‐H, Random, 95% CI) | 1.38 [0.46, 4.12] |
| 6.3 less than 30% reduction ‐ short term | 1 | 147 | Risk Ratio (M‐H, Random, 95% CI) | 3.44 [0.86, 13.79] |
| 7 Mental state: 2b. State ‐ BPRS total score (high = poor) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 7.1 short term | 1 | 64 | Mean Difference (IV, Random, 95% CI) | 1.10 [‐1.26, 3.46] |
| 8 Mental state: 2c. State ‐ BPRS end score (high = poor, skewed data) | Other data | No numeric data | ||
| 8.1 medium term | Other data | No numeric data | ||
| 9 Mental state: 2c. State ‐ BPRS end score (high = poor, skewed data) | Other data | No numeric data | ||
| 9.1 long term | Other data | No numeric data | ||
| 10 Mental state: 2d. State ‐ PANSS score reduction ('no effect') | 3 | 231 | Risk Ratio (M‐H, Random, 95% CI) | 0.90 [0.67, 1.20] |
| 10.1 less than 20% reduction ‐ short term | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 0.86 [0.64, 1.15] |
| 10.2 less than 25% reduction ‐ short term | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 2.0 [0.40, 10.11] |
| 10.3 less than 40% reduction ‐ short term | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 1.7 [0.30, 9.50] |
| 11 Mental state: 2e. State ‐ PANSS: mean change/ endpoint from baseline (high = poor) | 3 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 11.1 total ‐ short term | 3 | 232 | Mean Difference (IV, Random, 95% CI) | 3.65 [1.86, 5.43] |
| 11.2 positive ‐ short term | 2 | 125 | Mean Difference (IV, Random, 95% CI) | 4.45 [‐1.00, 11.90] |
| 11.3 negative ‐ short term | 2 | 125 | Mean Difference (IV, Random, 95% CI) | 1.20 [0.07, 2.33] |
| 11.4 general psychopathology ‐ short term | 1 | 60 | Mean Difference (IV, Random, 95% CI) | ‐0.39 [‐0.79, 0.01] |
| 12 Mental state: 2f. State ‐ PANSS total change score (high = poor, skewed data) | Other data | No numeric data | ||
| 12.1 short term | Other data | No numeric data | ||
| 13 Mental state: 2g. State ‐ PANSS total endpoint score (high = poor, skewed data) | Other data | No numeric data | ||
| 13.1 long term | Other data | No numeric data | ||
| 14 Behaviour: 1. Behaviour subscale of the Psychiatric Rating Scale: no better or worse | 3 | 244 | Risk Ratio (M‐H, Random, 95% CI) | 1.03 [0.86, 1.23] |
| 15 Adverse events: Extrapyramidal and movement disorders | 22 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 15.1 akathisia ‐ short term | 11 | 1736 | Risk Ratio (M‐H, Random, 95% CI) | 1.12 [0.79, 1.58] |
| 15.2 akathisia ‐ medium term | 1 | 47 | Risk Ratio (M‐H, Random, 95% CI) | 2.88 [0.64, 12.82] |
| 15.3 akathisia ‐ long term | 2 | 1346 | Risk Ratio (M‐H, Random, 95% CI) | 1.10 [0.65, 1.85] |
| 15.4 akinesia ‐ short term | 2 | 475 | Risk Ratio (M‐H, Random, 95% CI) | 0.44 [0.11, 1.70] |
| 15.5 ataxia ‐ short term | 4 | 514 | Risk Ratio (M‐H, Random, 95% CI) | 0.77 [0.25, 2.41] |
| 15.6 dyskinesia ‐ short term | 8 | 1282 | Risk Ratio (M‐H, Random, 95% CI) | 0.63 [0.39, 1.04] |
| 15.7 dyskinesia ‐ medium term | 1 | 47 | Risk Ratio (M‐H, Random, 95% CI) | 0.48 [0.05, 4.93] |
| 15.8 dystonia ‐ short term | 4 | 416 | Risk Ratio (M‐H, Random, 95% CI) | 1.36 [0.23, 8.16] |
| 15.9 extrapyramidal signs ‐ short term | 1 | 47 | Risk Ratio (M‐H, Random, 95% CI) | 0.90 [0.62, 1.31] |
| 15.10 EPS ‐ short term | 3 | 429 | Risk Ratio (M‐H, Random, 95% CI) | 2.23 [0.88, 5.67] |
| 15.11 EPS ‐ long term | 1 | 1281 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.58, 1.73] |
| 15.12 hyperkinesia ‐ short term | 2 | 135 | Risk Ratio (M‐H, Random, 95% CI) | 2.64 [0.88, 7.91] |
| 15.13 myotony ‐ short term | 1 | 147 | Risk Ratio (M‐H, Random, 95% CI) | 2.06 [0.92, 4.62] |
| 15.14 parkinsonism ‐ short term | 7 | 1254 | Risk Ratio (M‐H, Random, 95% CI) | 1.10 [0.89, 1.35] |
| 15.15 rigidity ‐ short term | 4 | 210 | Risk Ratio (M‐H, Random, 95% CI) | 1.81 [0.38, 8.64] |
| 15.16 rigidity ‐ medium term | 1 | 47 | Risk Ratio (M‐H, Random, 95% CI) | 0.64 [0.12, 3.48] |
| 15.17 tardive dyskinesia ‐ long term | 1 | 1073 | Risk Ratio (M‐H, Random, 95% CI) | 1.23 [0.89, 1.70] |
| 15.18 tremor ‐ short term | 4 | 329 | Risk Ratio (M‐H, Random, 95% CI) | 1.79 [0.73, 4.39] |
| 15.19 tremor ‐ medium term | 1 | 47 | Risk Ratio (M‐H, Random, 95% CI) | 1.34 [0.50, 3.63] |
| 15.20 use of antiparkinsonian drugs ‐ short term | 8 | 996 | Risk Ratio (M‐H, Random, 95% CI) | 1.04 [0.83, 1.30] |
| 15.21 use of antiparkinsonian drugs ‐ medium term | 1 | 47 | Risk Ratio (M‐H, Random, 95% CI) | 0.14 [0.01, 2.52] |
| 15.22 use of antiparkinsonian drugs ‐ long term | 1 | 46 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.34, 26.76] |
| 16 Adverse events: Movement disorders ‐ TESS total score (high = poor, skewed data) | Other data | No numeric data | ||
| 16.1 short term | Other data | No numeric data | ||
| 17 Other adverse events: 1. Anticholinergic | 12 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 17.1 blurred vision ‐ short term | 8 | 1028 | Risk Ratio (M‐H, Random, 95% CI) | 0.80 [0.40, 1.61] |
| 17.2 dry mouth ‐ short term | 9 | 1017 | Risk Ratio (M‐H, Random, 95% CI) | 1.43 [0.82, 2.47] |
| 17.3 dry mouth ‐ medium term | 1 | 47 | Risk Ratio (M‐H, Random, 95% CI) | 1.92 [0.19, 19.73] |
| 17.4 increased salivation ‐ short term | 2 | 211 | Risk Ratio (M‐H, Random, 95% CI) | 0.42 [0.00, 35.79] |
| 17.5 increased salivation ‐ long term | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 7.87] |
| 17.6 nasal congestion ‐ short term | 1 | 80 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.13, 71.51] |
| 18 Other adverse events: 2. Arousal | 19 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 18.1 agitation ‐ short term | 1 | 300 | Risk Ratio (M‐H, Random, 95% CI) | 1.05 [0.63, 1.77] |
| 18.2 agitation ‐ long term | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.33, 27.23] |
| 18.3 anxiety ‐ short term | 1 | 300 | Risk Ratio (M‐H, Random, 95% CI) | 1.19 [0.63, 2.24] |
| 18.4 anxiety ‐ long term | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 1.25 [0.37, 4.21] |
| 18.5 depression ‐ short term | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 1.34 [0.76, 2.35] |
| 18.6 drowsiness ‐ short term | 5 | 442 | Risk Ratio (M‐H, Random, 95% CI) | 0.88 [0.53, 1.44] |
| 18.7 emotional indifference ‐ short term | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 1.06 [0.58, 1.94] |
| 18.8 excitation ‐ short term | 4 | 463 | Risk Ratio (M‐H, Random, 95% CI) | 2.59 [0.57, 11.84] |
| 18.9 excitation ‐ medium term | 1 | 47 | Risk Ratio (M‐H, Random, 95% CI) | 1.28 [0.32, 5.10] |
| 18.10 increased dream activity ‐ short term | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 1.51 [0.62, 3.67] |
| 18.11 insomnia ‐ short term | 11 | 1422 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.86, 1.18] |
| 18.12 insomnia ‐ medium term | 1 | 47 | Risk Ratio (M‐H, Random, 95% CI) | 0.24 [0.03, 1.99] |
| 18.13 insomnia ‐ long term | 2 | 1520 | Risk Ratio (M‐H, Random, 95% CI) | 2.15 [0.41, 11.29] |
| 18.14 need of additional benzodiazepines ‐ short term | 5 | 448 | Risk Ratio (M‐H, Random, 95% CI) | 0.93 [0.74, 1.16] |
| 18.15 need of additional benzodiazepines ‐ medium term | 1 | 47 | Risk Ratio (M‐H, Random, 95% CI) | 1.60 [0.43, 5.93] |
| 18.16 need of additional benzodiazepines ‐ long term | 1 | 46 | Risk Ratio (M‐H, Random, 95% CI) | 0.91 [0.76, 1.09] |
| 18.17 restlessness ‐ short term | 1 | 147 | Risk Ratio (M‐H, Random, 95% CI) | 3.75 [1.96, 7.18] |
| 18.18 sleepiness/sedation ‐ short term | 7 | 991 | Risk Ratio (M‐H, Random, 95% CI) | 1.19 [0.87, 1.62] |
| 18.19 sleepiness/ sedation ‐ long term | 1 | 1460 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.79, 1.21] |
| 18.20 somnolence ‐ short term | 1 | 147 | Risk Ratio (M‐H, Random, 95% CI) | 0.36 [0.13, 0.98] |
| 18.21 tension ‐ short term | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 0.97 [0.63, 1.50] |
| 19 Other adverse events: 3. At least one | 9 | 974 | Risk Ratio (M‐H, Random, 95% CI) | 1.02 [0.95, 1.09] |
| 19.1 short term | 9 | 974 | Risk Ratio (M‐H, Random, 95% CI) | 1.02 [0.95, 1.09] |
| 20 Other adverse events: 4. Cardiovascular | 13 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 20.1 angina pectoris ‐ short term | 2 | 391 | Risk Ratio (M‐H, Random, 95% CI) | 0.57 [0.26, 1.26] |
| 20.2 bradycardia ‐ long term | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.13, 70.83] |
| 20.3 dizziness ‐ short term | 1 | 147 | Risk Ratio (M‐H, Random, 95% CI) | 0.91 [0.59, 1.40] |
| 20.4 faintness, dizziness, weakness ‐ short term | 7 | 930 | Risk Ratio (M‐H, Random, 95% CI) | 1.27 [0.82, 1.96] |
| 20.5 faintness, dizziness, weakness ‐ long term | 1 | 1460 | Risk Ratio (M‐H, Random, 95% CI) | 1.02 [0.70, 1.50] |
| 20.6 orthostatic hypotension ‐ short term | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 0.17 [0.02, 1.31] |
| 20.7 hypotension ‐ long term | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 7.87] |
| 20.8 lassitude ‐ short term | 2 | 305 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.68, 1.45] |
| 20.9 palpitations ‐ short term | 2 | 391 | Risk Ratio (M‐H, Random, 95% CI) | 0.22 [0.07, 0.73] |
| 20.10 tachycardia ‐ short term | 4 | 413 | Risk Ratio (M‐H, Random, 95% CI) | 0.54 [0.21, 1.39] |
| 21 Other adverse events: 5. Central nervous system | 11 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 21.1 confusion ‐ short term | 1 | 198 | Risk Ratio (M‐H, Random, 95% CI) | 0.67 [0.11, 3.90] |
| 21.2 difficulty in concentration ‐ short term | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 1.06 [0.71, 1.58] |
| 21.3 disturbance of consciousness ‐ short term | 4 | 648 | Risk Ratio (M‐H, Random, 95% CI) | 0.84 [0.10, 6.78] |
| 21.4 dysarticulation ‐ short term | 1 | 198 | Risk Ratio (M‐H, Random, 95% CI) | 0.83 [0.38, 1.84] |
| 21.5 electroencephalogram (increase) ‐ short term | 1 | 25 | Risk Ratio (M‐H, Random, 95% CI) | 0.57 [0.09, 3.64] |
| 21.6 headache ‐ short term | 9 | 1304 | Risk Ratio (M‐H, Random, 95% CI) | 0.72 [0.50, 1.03] |
| 21.7 headache ‐ long term | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 0.71 [0.25, 2.00] |
| 21.8 hypertonia ‐ long term | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.13, 70.83] |
| 21.9 paraesthesia ‐ short term | 6 | 726 | Risk Ratio (M‐H, Random, 95% CI) | 1.37 [0.57, 3.26] |
| 21.10 seizure ‐ short term | 1 | 198 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 21.11 vertigo ‐ short term | 2 | 364 | Risk Ratio (M‐H, Random, 95% CI) | 0.87 [0.44, 1.74] |
| 22 Other adverse events: 6. Endocrine | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 22.1 amenorrhoea (women only) ‐ short term | 1 | 30 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.13, 68.26] |
| 22.2 amenorrhoea ‐ medium term | 1 | 47 | Risk Ratio (M‐H, Random, 95% CI) | 0.32 [0.04, 2.85] |
| 22.3 gynaecomastia ‐ short term | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 0.15 [0.01, 2.85] |
| 22.4 lactation ‐ short term | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 0.21 [0.01, 4.30] |
| 22.5 lactation ‐ medium term | 1 | 47 | Risk Ratio (M‐H, Random, 95% CI) | 0.19 [0.01, 3.80] |
| 22.6 menorrhagia (women only) ‐ short term | 1 | 30 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.13, 68.26] |
| 23 Other adverse events: 7. Gastrointestinal | 13 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 23.1 constipation ‐ short term | 9 | 1017 | Risk Ratio (M‐H, Random, 95% CI) | 0.71 [0.40, 1.28] |
| 23.2 diarrhoea ‐ short term | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 0.15 [0.01, 2.85] |
| 23.3 diarrhoea ‐ long term | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.22, 4.56] |
| 23.4 dyspepsia ‐ short term | 1 | 300 | Risk Ratio (M‐H, Random, 95% CI) | 0.59 [0.27, 1.30] |
| 23.5 gastroenteritis ‐ long term | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.13, 70.83] |
| 23.6 icterus ‐ short term | 3 | 450 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 7.96] |
| 23.7 loss of appetite ‐ short term | 6 | 699 | Risk Ratio (M‐H, Random, 95% CI) | 0.96 [0.69, 1.33] |
| 23.8 nausea and vomiting ‐ short term | 7 | 806 | Risk Ratio (M‐H, Random, 95% CI) | 1.12 [0.75, 1.67] |
| 23.9 nausea and vomiting ‐ medium term | 1 | 47 | Risk Ratio (M‐H, Random, 95% CI) | 0.32 [0.04, 2.85] |
| 23.10 nausea and vomiting ‐ long term | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.07, 15.26] |
| 23.11 weight loss ‐ short term | 2 | 171 | Risk Ratio (M‐H, Random, 95% CI) | 3.03 [0.75, 12.20] |
| 23.12 weight gain ‐ short term | 3 | 235 | Risk Ratio (M‐H, Random, 95% CI) | 1.53 [0.29, 8.10] |
| 23.13 weight gain ‐ long term | 1 | 1316 | Risk Ratio (M‐H, Random, 95% CI) | 0.66 [0.46, 0.95] |
| 24 Other adverse events: 8. Skin | 8 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 24.1 cellulitis ‐ long term | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 7.87] |
| 24.2 eczema ‐ long term | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.13, 70.83] |
| 24.3 fungal dermatitis ‐ long term | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 7.87] |
| 24.4 oedema | 1 | 105 | Risk Ratio (M‐H, Random, 95% CI) | 3.17 [0.13, 76.16] |
| 24.5 pruritus ‐ short term | 3 | 498 | Risk Ratio (M‐H, Random, 95% CI) | 0.77 [0.31, 1.93] |
| 24.6 rash ‐ short term | 7 | 806 | Risk Ratio (M‐H, Random, 95% CI) | 0.90 [0.37, 2.17] |
| 24.7 rash ‐ long term | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.07, 15.26] |
| 24.8 sweating ‐ short term | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 0.66 [0.23, 1.89] |
| 25 Other adverse events: 9. Genitourinary | 7 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 25.1 diminished sexual desire | 4 | 1958 | Risk Ratio (M‐H, Random, 95% CI) | 1.03 [0.82, 1.30] |
| 25.2 ejaculatory dysfunction (men only) ‐ short term | 1 | 77 | Risk Ratio (M‐H, Random, 95% CI) | 0.18 [0.02, 1.43] |
| 25.3 erectile dysfunction (men only) ‐ short term | 1 | 77 | Risk Ratio (M‐H, Random, 95% CI) | 0.90 [0.30, 2.70] |
| 25.4 galactorrhoea ‐ short term | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 0.15 [0.01, 2.85] |
| 25.5 galactorrhoea/ gynaecomastia ‐ long term | 1 | 1460 | Risk Ratio (M‐H, Random, 95% CI) | 0.56 [0.20, 1.56] |
| 25.6 increased sexual desire ‐ short term | 2 | 212 | Risk Ratio (M‐H, Random, 95% CI) | 1.06 [0.22, 5.01] |
| 25.7 micturition disturbances ‐ short term | 5 | 635 | Risk Ratio (M‐H, Random, 95% CI) | 0.59 [0.22, 1.63] |
| 25.8 orgastic dysfunction ‐ short term | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 5.28 [0.26, 107.51] |
| 26 Other adverse events: 10. Others | 11 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 26.1 abdominal pain ‐ medium term | 1 | 47 | Risk Ratio (M‐H, Random, 95% CI) | 0.14 [0.01, 2.52] |
| 26.2 allergic reaction ‐ long term | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 7.87] |
| 26.3 back pain ‐ long term | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.13, 70.83] |
| 26.4 cramps ‐ short term | 3 | 450 | Risk Ratio (M‐H, Random, 95% CI) | 0.47 [0.05, 4.46] |
| 26.5 failing memory ‐ short term | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 1.27 [0.60, 2.68] |
| 26.6 muscle pain ‐ short term | 1 | 80 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.13, 71.51] |
| 26.7 sweating ‐ short term | 6 | 726 | Risk Ratio (M‐H, Random, 95% CI) | 0.73 [0.39, 1.37] |
| 26.8 incontinence nocturia ‐ long term | 1 | 1460 | Risk Ratio (M‐H, Random, 95% CI) | 0.41 [0.18, 0.92] |
| 26.9 urinary hesitancy, dry mouth, constipation ‐ long term | 1 | 1460 | Risk Ratio (M‐H, Random, 95% CI) | 0.86 [0.67, 1.10] |
| 26.10 suicide attempt ‐ long term | 1 | 1460 | Risk Ratio (M‐H, Random, 95% CI) | 0.77 [0.09, 6.33] |
| 26.11 psychosis ‐ short term | 1 | 300 | Risk Ratio (M‐H, Random, 95% CI) | 0.63 [0.29, 1.40] |
| 27 Other adverse events: 11. Lab data | 7 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 27.1 abnormal EKG/ ECG ‐ short term | 2 | 211 | Risk Ratio (M‐H, Random, 95% CI) | 2.44 [0.62, 9.59] |
| 27.2 abnormal liver function ‐ short term | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 4.0 [0.92, 17.40] |
| 27.3 anemia ‐ long term | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.07, 15.26] |
| 27.4 decrease in white blood cell count and haemoglobin level ‐ short term | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.07, 15.30] |
| 27.5 ECG – atrial extrasystole & flattening of T‐wave in V6 ‐ short term | 1 | 47 | Risk Ratio (M‐H, Random, 95% CI) | 6.72 [0.37, 123.33] |
| 27.6 ECG abnormality (1st degree atrioventricular block) | 1 | 300 | Risk Ratio (M‐H, Random, 95% CI) | 15.82 [2.12, 118.27] |
| 27.7 high white blood cell count ‐ short term | 1 | 147 | Risk Ratio (M‐H, Random, 95% CI) | 1.03 [0.10, 11.09] |
| 27.8 increase in white blood cell count and haemoglobin level ‐ short term | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 0.5 [0.10, 2.54] |
| 27.9 liver function (GPT+) ‐ short term | 1 | 147 | Risk Ratio (M‐H, Random, 95% CI) | 0.69 [0.23, 2.02] |
| 27.10 low white blood cell count ‐ short term | 1 | 147 | Risk Ratio (M‐H, Random, 95% CI) | 0.17 [0.02, 1.28] |
| 27.11 prolactin: clinically significant high levels | 1 | 300 | Risk Ratio (M‐H, Random, 95% CI) | 12.66 [6.06, 26.45] |
| 27.12 prolonged QTc interval (≥450 msec and ≥10% increase from baseline) ‐ short term | 1 | 300 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 27.13 rise in alkaline phosphate and GOT levels ‐ short term | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.13, 71.00] |
| 27.14 slight T‐wave changes on EKG ‐ short term | 1 | 50 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.15, 6.55] |
| 27.15 ventricular extrasystole ‐ short term | 1 | 47 | Risk Ratio (M‐H, Random, 95% CI) | 0.32 [0.01, 7.48] |
| 28 Other adverse events: 11a. Lab data (skew) | Other data | No numeric data | ||
| 28.1 weight change | Other data | No numeric data | ||
| 29 Other adverse events: 12. Any serious adverse event | 2 | 1760 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.68, 1.41] |
| 29.1 short term | 1 | 300 | Risk Ratio (M‐H, Random, 95% CI) | 0.79 [0.49, 1.28] |
| 29.2 long term | 1 | 1460 | Risk Ratio (M‐H, Random, 95% CI) | 1.15 [0.78, 1.69] |
Comparison 3. PERPHENAZINE vs ANY ANTIPSYCHOTIC (ACUTE).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Dichotomous outcomes for acute participants | 7 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Leaving the study early: 1. Any reason | 7 | 447 | Risk Ratio (M‐H, Random, 95% CI) | 1.25 [1.06, 1.47] |
| 1.2 Leaving the study early: 2. Due to adverse events | 3 | 314 | Risk Ratio (M‐H, Random, 95% CI) | 1.55 [0.72, 3.34] |
| 1.3 Leaving the study early: 3. Due to relapse/ worsening or no improvement | 2 | 129 | Risk Ratio (M‐H, Random, 95% CI) | 1.46 [0.34, 6.26] |
| 1.4 Global state: 1. Change over time ‐ no better or deterioration | 5 | 240 | Risk Ratio (M‐H, Random, 95% CI) | 0.87 [0.27, 2.82] |
| 1.5 Mental state: 1. State ‐ less than 20% BPRS reduction ('no effect') | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 0.81 [0.61, 1.08] |
| 1.6 Mental state: 2. State ‐ less than 20% PANSS reduction ('no effect') | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 0.86 [0.64, 1.15] |
| 1.7 Adverse events: 1. Movement disorders ‐ dystonia | 2 | 129 | Risk Ratio (M‐H, Random, 95% CI) | 1.11 [0.12, 10.02] |
| 1.8 Adverse events: 2. Movement disorders ‐ extrapyramidal signs | 1 | 22 | Risk Ratio (M‐H, Random, 95% CI) | 0.86 [0.39, 1.88] |
| 1.9 Adverse events: 3. Movement disorders ‐ use of antiparkinson drugs | 2 | 129 | Risk Ratio (M‐H, Random, 95% CI) | 1.07 [0.65, 1.76] |
| 1.10 Adverse events: 4. Movement disorders ‐ hyperkinesia | 2 | 135 | Risk Ratio (M‐H, Random, 95% CI) | 2.64 [0.88, 7.91] |
| 1.11 Other adverse events: 5. Anticholinergic ‐ dry mouth | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 7.40 [0.39, 139.81] |
| 1.12 Other adverse events: 6. Arousal ‐ drowsiness | 2 | 50 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.12, 8.33] |
| 1.13 Other adverse events: 7. Arousal ‐ insomnia | 2 | 161 | Risk Ratio (M‐H, Random, 95% CI) | 0.84 [0.16, 4.50] |
| 1.14 Other adverse events: 8. Arousal ‐ need of additional benzodiazepines | 1 | 17 | Risk Ratio (M‐H, Random, 95% CI) | 0.71 [0.35, 1.46] |
| 1.15 Other adverse events: 9. Arousal ‐ sleepiness/sedation | 2 | 161 | Risk Ratio (M‐H, Random, 95% CI) | 1.19 [0.71, 2.00] |
| 1.16 Other adverse events: 10. Arousal ‐ tension | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 0.97 [0.63, 1.50] |
| 1.17 Other adverse events: 11. At least one | 3 | 157 | Risk Ratio (M‐H, Random, 95% CI) | 1.04 [0.88, 1.23] |
| 1.18 Other adverse events: 12. Cardiovascular ‐ faintness, dizziness, weakness | 2 | 161 | Risk Ratio (M‐H, Random, 95% CI) | 1.12 [0.54, 2.33] |
| 1.19 Other adverse events: 13. Cardiovascular ‐ lassitude | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 0.97 [0.61, 1.52] |
| 1.20 Other adverse events: 14. Cardiovascular ‐ tachycardia | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 0.57 [0.25, 1.32] |
| 1.21 Other adverse events: 15. Central nervous system ‐ difficulty in concentration | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 1.06 [0.71, 1.58] |
| 1.22 Other adverse events: 16. Central nervous system ‐ headache | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 0.53 [0.14, 2.01] |
| 1.23 Other adverse events: 17. Central nervous system ‐ paraesthesia | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 0.71 [0.12, 4.05] |
| 1.24 Other adverse events: 18. Gastrointestinal ‐ constipation | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 0.07 [0.00, 1.20] |
| 1.25 Other adverse events: 19. Gastrointestinal ‐ diarrhoea | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 0.15 [0.01, 2.85] |
| 1.26 Other adverse events: 20. Gastrointestinal ‐ nausea and vomiting | 2 | 154 | Risk Ratio (M‐H, Random, 95% CI) | 0.64 [0.24, 1.68] |
| 1.27 Other adverse events: 21. Gastrointestinal ‐ weight loss | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 2.12 [0.56, 8.02] |
| 1.28 Other adverse events: 22. Gastrointestinal ‐ weight gain | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 0.50 [0.26, 0.97] |
| 1.29 Other adverse events: 23. Skin ‐ pruritus | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 0.79 [0.19, 3.38] |
| 1.30 Other adverse events: 24. Skin ‐ rash | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 0.71 [0.12, 4.05] |
| 1.31 Other adverse events: 25. Skin ‐ sweating | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 0.66 [0.23, 1.89] |
| 1.32 Other adverse events: 26. Genitourinary ‐ diminished sexual desire | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 1.06 [0.22, 5.01] |
| 1.33 Other adverse events: 27. Genitourinary ‐ ejaculatory dysfunction (men only) | 1 | 77 | Risk Ratio (M‐H, Random, 95% CI) | 0.18 [0.02, 1.43] |
| 1.34 Other adverse events: 28. Genitourinary ‐ erectile dysfunction (men only) | 1 | 77 | Risk Ratio (M‐H, Random, 95% CI) | 0.90 [0.30, 2.70] |
| 1.35 Other adverse events: 29. Genitourinary ‐ galactorrhoea | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 0.15 [0.01, 2.85] |
| 1.36 Other adverse events: 30. Genitourinary ‐ increased sexual desire | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 1.06 [0.22, 5.01] |
| 1.37 Other adverse events: 31. Genitourinary ‐ orgastic dysfunction | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 5.28 [0.26, 107.51] |
| 1.38 Other adverse events: 32. Others ‐ abdominal pain | 1 | 47 | Risk Ratio (M‐H, Random, 95% CI) | 0.14 [0.01, 2.52] |
| 1.39 Other adverse events: 33. Others ‐ failing memory | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 1.27 [0.60, 2.68] |
| 1.40 Other adverse events: 34. Others ‐ sweating | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 0.96 [0.45, 2.07] |
| 2 Continuous outcomes for acute participants | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 2.1 Mental state: 1. State ‐ PANSS: mean change from baseline to endpoint | 1 | 107 | Mean Difference (IV, Random, 95% CI) | 4.0 [‐5.04, 13.04] |
Comparison 4. PERPHENAZINE vs ARIPIPRAZOLE.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Leaving the study early: 1. Any reason | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 short term | 1 | 300 | Risk Ratio (M‐H, Random, 95% CI) | 0.74 [0.50, 1.11] |
| 2 Leaving the study early: 2. Due to adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 short term | 1 | 300 | Risk Ratio (M‐H, Random, 95% CI) | 0.53 [0.27, 1.05] |
| 3 Leaving the study early: 3. Due to relapse / worsening or no improvement | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 short term | 1 | 300 | Risk Ratio (M‐H, Random, 95% CI) | 0.84 [0.34, 2.08] |
| 4 Global state: 1. Change over time ‐ no better or deterioration (ITT) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 short term | 1 | 294 | Risk Ratio (M‐H, Random, 95% CI) | 1.02 [0.89, 1.17] |
| 5 Global state: 2. Average endpoint score (CGI‐S) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 5.1 short term | 1 | 294 | Mean Difference (IV, Random, 95% CI) | ‐0.20 [‐0.56, 0.16] |
| 6 Mental state: 2a. State ‐ BPRS score reduction ('no effect') | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 6.1 less than 25% reduction ‐ short term | 1 | 65 | Risk Ratio (M‐H, Random, 95% CI) | 1.45 [0.26, 8.14] |
| 7 Mental state: 2b. State ‐ PANSS: mean endpoint from baseline (high = poor) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 7.1 total ‐ short term | 1 | 65 | Mean Difference (IV, Random, 95% CI) | 1.38 [‐2.66, 5.42] |
| 7.2 positive ‐ short term | 1 | 65 | Mean Difference (IV, Random, 95% CI) | 0.63 [‐0.80, 2.06] |
| 7.3 negative ‐ short term | 1 | 65 | Mean Difference (IV, Random, 95% CI) | 0.33 [‐1.46, 2.12] |
| 8 Mental state: 2c. State ‐ PANSS total change score (high = poor, skewed data) | Other data | No numeric data | ||
| 8.1 short term | Other data | No numeric data | ||
| 9 Adverse events: Movement disorders | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 9.1 akathisia ‐ short term | 1 | 300 | Risk Ratio (M‐H, Random, 95% CI) | 2.29 [0.89, 5.85] |
| 9.2 EPS ‐ short term | 2 | 365 | Risk Ratio (M‐H, Random, 95% CI) | 1.65 [0.70, 3.88] |
| 9.3 use of antiparkinsonian drugs ‐ short term | 1 | 300 | Risk Ratio (M‐H, Random, 95% CI) | 1.56 [1.01, 2.41] |
| 10 Adverse events: Movement disorders ‐ TESS total score (high = poor, skewed data) | Other data | No numeric data | ||
| 10.1 short term | Other data | No numeric data | ||
| 11 Other adverse events: 1. Arousal | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 11.1 agitation ‐ short term | 1 | 300 | Risk Ratio (M‐H, Random, 95% CI) | 1.05 [0.63, 1.77] |
| 11.2 anxiety ‐ short term | 1 | 300 | Risk Ratio (M‐H, Random, 95% CI) | 1.19 [0.63, 2.24] |
| 11.3 insomnia ‐ short term | 1 | 300 | Risk Ratio (M‐H, Random, 95% CI) | 0.86 [0.56, 1.31] |
| 11.4 sleepiness/sedation ‐ short term | 1 | 300 | Risk Ratio (M‐H, Random, 95% CI) | 2.64 [0.85, 8.22] |
| 12 Other adverse events: 2. Cardiovascular | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 12.1 faintness, dizziness, weakness ‐ short term | 1 | 300 | Risk Ratio (M‐H, Random, 95% CI) | 5.27 [1.18, 23.66] |
| 13 Other adverse events: 3. Central nervous system | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 13.1 headache ‐ short term | 1 | 300 | Risk Ratio (M‐H, Random, 95% CI) | 0.55 [0.29, 1.03] |
| 14 Other adverse events: 4. Gastrointestinal | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 14.1 dyspepsia ‐ short term | 1 | 300 | Risk Ratio (M‐H, Random, 95% CI) | 0.59 [0.27, 1.30] |
| 15 Other adverse events: 5. lab data | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 15.1 ECG abnormality (1st degree atrioventricular block) | 1 | 300 | Risk Ratio (M‐H, Random, 95% CI) | 15.82 [2.12, 118.27] |
| 15.2 prolactin: clinically significant high levels | 1 | 300 | Risk Ratio (M‐H, Random, 95% CI) | 12.66 [6.06, 26.45] |
| 15.3 prolonged QTc interval (≥450 msec and ≥10% increase from baseline) ‐ short term | 1 | 300 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 16 Other adverse events: 6. any serious adverse event | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 16.1 short term | 1 | 300 | Risk Ratio (M‐H, Random, 95% CI) | 0.79 [0.49, 1.28] |
| 16.2 psychosis ‐ short term | 1 | 300 | Risk Ratio (M‐H, Random, 95% CI) | 0.63 [0.29, 1.40] |
Comparison 5. PERPHENAZINE vs BENPERIDOL.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Leaving the study early: 1. Any reason | 1 | 40 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 1.1 short term | 1 | 40 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 2 Global state: 1. Change over time ‐ no better or deterioration (ITT) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 short term | 1 | 40 | Risk Ratio (M‐H, Random, 95% CI) | 0.13 [0.03, 0.47] |
Comparison 6. PERPHENAZINE vs BROMPERIDOL.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Leaving the study early: 1. Any reason | 1 | 40 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 1.1 short term | 1 | 40 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 2 Global state: 1. Change over time ‐ no better or deterioration (ITT) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 short term | 1 | 40 | Risk Ratio (M‐H, Random, 95% CI) | 2.5 [0.94, 6.66] |
| 3 Adverse events: Movement disorders | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 use of antiparkinsonian drugs ‐ short term | 1 | 40 | Risk Ratio (M‐H, Random, 95% CI) | 0.8 [0.40, 1.60] |
| 4 Other adverse events: 1. Arousal | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 need of additional benzodiazepines ‐ short term | 1 | 40 | Risk Ratio (M‐H, Random, 95% CI) | 1.1 [0.61, 1.99] |
Comparison 7. PERPHENAZINE vs CHLORPROMAZINE.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Leaving the study early: 1. Any reason | 3 | 178 | Risk Ratio (M‐H, Random, 95% CI) | 1.10 [0.89, 1.35] |
| 1.1 short term | 3 | 178 | Risk Ratio (M‐H, Random, 95% CI) | 1.10 [0.89, 1.35] |
| 2 Leaving the study early: 2. Due to adverse events | 2 | 168 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.18, 0.59] |
| 2.1 short term | 2 | 168 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.18, 0.59] |
| 3 Global state: 1. Change over time ‐ no better or deterioration (ITT) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 short term | 1 | 63 | Risk Ratio (M‐H, Random, 95% CI) | 0.57 [0.22, 1.52] |
| 4 Adverse events: Movement disorders | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 akathisia ‐ short term | 1 | 105 | Risk Ratio (M‐H, Random, 95% CI) | 6.87 [0.88, 53.89] |
| 4.2 akinesia ‐ short term | 1 | 105 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 4.3 dyskinesia ‐ short term | 1 | 105 | Risk Ratio (M‐H, Random, 95% CI) | 1.96 [0.18, 20.99] |
| 4.4 parkinsonism ‐ short term | 1 | 105 | Risk Ratio (M‐H, Random, 95% CI) | 1.96 [0.63, 6.12] |
| 5 Other adverse events: 1. Central nervous system | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 electroencephalogram (increase) ‐ short term | 1 | 10 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.05, 2.21] |
Comparison 8. PERPHENAZINE vs CLOCAPRAMINE.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Leaving the study early: 1. Any reason | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 short term | 1 | 192 | Risk Ratio (M‐H, Random, 95% CI) | 1.40 [0.59, 3.34] |
| 2 Leaving the study early: 2. Due to adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 short term | 1 | 192 | Risk Ratio (M‐H, Random, 95% CI) | 0.61 [0.15, 2.49] |
| 3 Leaving the study early: 3. Due to relapse / worsening or no improvement | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 short term | 1 | 192 | Risk Ratio (M‐H, Random, 95% CI) | 2.38 [0.63, 8.94] |
| 4 Global state: 1. Change over time ‐ no better or deterioration (ITT) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 short term | 1 | 192 | Risk Ratio (M‐H, Random, 95% CI) | 1.15 [0.88, 1.49] |
| 5 Adverse events: Movement disorders | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 akathisia ‐ short term | 1 | 192 | Risk Ratio (M‐H, Random, 95% CI) | 0.84 [0.49, 1.44] |
| 5.2 ataxia ‐ short term | 1 | 192 | Risk Ratio (M‐H, Random, 95% CI) | 0.34 [0.04, 3.21] |
| 5.3 dyskinesia ‐ short term | 1 | 192 | Risk Ratio (M‐H, Random, 95% CI) | 0.57 [0.20, 1.63] |
| 5.4 parkinsonism ‐ short term | 1 | 192 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.60, 1.59] |
| 5.5 use of antiparkinsonian drugs ‐ short term | 1 | 192 | Risk Ratio (M‐H, Random, 95% CI) | 0.88 [0.69, 1.13] |
| 6 Other adverse events: 1. Anticholinergic | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 6.1 blurred vision ‐ short term | 1 | 192 | Risk Ratio (M‐H, Random, 95% CI) | 0.41 [0.08, 2.05] |
| 6.2 dry mouth ‐ short term | 1 | 192 | Risk Ratio (M‐H, Random, 95% CI) | 0.68 [0.25, 1.84] |
| 7 Other adverse events: 2. Arousal | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 7.1 insomnia ‐ short term | 1 | 192 | Risk Ratio (M‐H, Random, 95% CI) | 1.16 [0.83, 1.63] |
| 7.2 need of additional benzodiazepines ‐ short term | 1 | 192 | Risk Ratio (M‐H, Random, 95% CI) | 1.09 [0.81, 1.46] |
| 7.3 sleepiness/sedation ‐ short term | 1 | 192 | Risk Ratio (M‐H, Random, 95% CI) | 1.02 [0.47, 2.24] |
| 8 Other adverse events: 3. At least one | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 8.1 short term | 1 | 192 | Risk Ratio (M‐H, Random, 95% CI) | 0.92 [0.78, 1.07] |
| 9 Other adverse events: 4. Cardiovascular | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 9.1 angina pectoris ‐ short term | 1 | 192 | Risk Ratio (M‐H, Random, 95% CI) | 0.68 [0.20, 2.34] |
| 9.2 palpitations ‐ short term | 1 | 192 | Risk Ratio (M‐H, Random, 95% CI) | 0.34 [0.07, 1.64] |
| 10 Other adverse events: 5. Central nervous system | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 10.1 disturbance of consciousness ‐ short term | 1 | 192 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 10.2 headache ‐ short term | 1 | 192 | Risk Ratio (M‐H, Random, 95% CI) | 2.55 [0.83, 7.86] |
| 10.3 paraesthesia ‐ short term | 1 | 192 | Risk Ratio (M‐H, Random, 95% CI) | 4.08 [0.46, 35.88] |
| 10.4 vertigo ‐ short term | 1 | 192 | Risk Ratio (M‐H, Random, 95% CI) | 1.02 [0.45, 2.34] |
| 11 Other adverse events: 6. Gastrointestinal | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 11.1 constipation ‐ short term | 1 | 192 | Risk Ratio (M‐H, Random, 95% CI) | 1.25 [0.54, 2.87] |
| 11.2 icterus ‐ short term | 1 | 192 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 11.3 loss of appetite ‐ short term | 1 | 192 | Risk Ratio (M‐H, Random, 95% CI) | 1.76 [0.89, 3.50] |
| 11.4 nausea and vomiting ‐ short term | 1 | 192 | Risk Ratio (M‐H, Random, 95% CI) | 2.30 [1.05, 5.03] |
| 12 Other adverse events: 7. Skin | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 12.1 pruritus ‐ short term | 1 | 192 | Risk Ratio (M‐H, Random, 95% CI) | 1.02 [0.15, 7.10] |
| 12.2 rash ‐ short term | 1 | 192 | Risk Ratio (M‐H, Random, 95% CI) | 1.02 [0.15, 7.10] |
| 13 Other adverse events: 8. Genitourinary | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 13.1 diminished sexual desire | 1 | 192 | Risk Ratio (M‐H, Random, 95% CI) | 1.02 [0.06, 16.09] |
| 13.2 micturition disturbances ‐ short term | 1 | 192 | Risk Ratio (M‐H, Random, 95% CI) | 0.51 [0.10, 2.72] |
| 14 Other adverse events: 9. Others | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 14.1 cramps ‐ short term | 1 | 192 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 14.2 sweating ‐ short term | 1 | 192 | Risk Ratio (M‐H, Random, 95% CI) | 0.17 [0.02, 1.39] |
Comparison 9. PERPHENAZINE vs CLOPENTHIXOL.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Leaving the study early: 1. Any reason | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 short term | 1 | 50 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.15, 6.55] |
| 2 Global state: 1. Change over time ‐ no better or deterioration (ITT) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 short term | 1 | 50 | Risk Ratio (M‐H, Random, 95% CI) | 0.67 [0.21, 2.08] |
| 3 Adverse events: Movement disorders | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 use of antiparkinsonian drugs ‐ short term | 1 | 50 | Risk Ratio (M‐H, Random, 95% CI) | 0.53 [0.31, 0.89] |
| 4 Other adverse events: 1. Arousal | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 need of additional benzodiazepines ‐ short term | 1 | 50 | Risk Ratio (M‐H, Random, 95% CI) | 0.5 [0.24, 1.03] |
| 5 Other adverse events: 2. lab data | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 slight T‐wave changes on EKG ‐ short term | 1 | 50 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.15, 6.55] |
Comparison 10. PERPHENAZINE vs CLOTHIAPINE.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Leaving the study early: 1. Any reason | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 short term | 1 | 80 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.21, 4.66] |
| 2 Leaving the study early: 2. Due to relapse / worsening or no improvement | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 short term | 1 | 80 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.15, 6.76] |
| 3 Global state: 1. Change over time ‐ no better or deterioration (ITT) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 short term | 1 | 80 | Risk Ratio (M‐H, Random, 95% CI) | 1.31 [0.81, 2.12] |
| 4 Behaviour: 1. Behaviour subscale of the Psychiatric Rating Scale: no better or worse | 1 | 80 | Risk Ratio (M‐H, Random, 95% CI) | 0.96 [0.69, 1.34] |
| 5 Adverse events: Movement disorders | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 akathisia ‐ short term | 1 | 80 | Risk Ratio (M‐H, Random, 95% CI) | 1.63 [0.76, 3.49] |
| 5.2 dyskinesia ‐ short term | 1 | 80 | Risk Ratio (M‐H, Random, 95% CI) | 0.56 [0.20, 1.51] |
| 5.3 parkinsonism ‐ short term | 1 | 80 | Risk Ratio (M‐H, Random, 95% CI) | 1.31 [0.81, 2.12] |
| 5.4 use of antiparkinsonian drugs ‐ short term | 1 | 80 | Risk Ratio (M‐H, Random, 95% CI) | 1.31 [0.81, 2.12] |
| 6 Other adverse events: 1. Anticholinergic | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 6.1 dry mouth ‐ short term | 1 | 80 | Risk Ratio (M‐H, Random, 95% CI) | 0.25 [0.03, 2.14] |
| 6.2 nasal congestion ‐ short term | 1 | 80 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.13, 71.51] |
| 7 Other adverse events: 2. Arousal | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 7.1 insomnia ‐ short term | 1 | 80 | Risk Ratio (M‐H, Random, 95% CI) | 0.91 [0.44, 1.90] |
| 7.2 need of additional benzodiazepines ‐ short term | 1 | 80 | Risk Ratio (M‐H, Random, 95% CI) | 1.25 [0.55, 2.84] |
| 7.3 sleepiness/sedation ‐ short term | 1 | 80 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.42, 2.40] |
| 8 Other adverse events: 3. At least one | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 8.1 short term | 1 | 80 | Risk Ratio (M‐H, Random, 95% CI) | 1.09 [0.91, 1.30] |
| 9 Other adverse events: 4. Cardiovascular | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 9.1 faintness, dizziness, weakness ‐ short term | 1 | 80 | Risk Ratio (M‐H, Random, 95% CI) | 1.67 [0.43, 6.51] |
| 10 Other adverse events: 5. Central nervous system | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 10.1 headache ‐ short term | 1 | 80 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 7.95] |
| 11 Other adverse events: 6. Gastrointestinal | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 11.1 constipation ‐ short term | 1 | 80 | Risk Ratio (M‐H, Random, 95% CI) | 0.25 [0.06, 1.11] |
| 11.2 loss of appetite ‐ short term | 1 | 80 | Risk Ratio (M‐H, Random, 95% CI) | 1.33 [0.32, 5.58] |
| 11.3 nausea and vomiting ‐ short term | 1 | 80 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.13, 71.51] |
| 12 Other adverse events: 7. Skin | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 12.1 rash ‐ short term | 1 | 80 | Risk Ratio (M‐H, Random, 95% CI) | 0.5 [0.05, 5.30] |
| 13 Other adverse events: 8. Genitourinary | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 13.1 micturition disturbances ‐ short term | 1 | 80 | Risk Ratio (M‐H, Random, 95% CI) | 0.5 [0.05, 5.30] |
| 14 Other adverse events: 9. Others | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 14.1 muscle pain ‐ short term | 1 | 80 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.13, 71.51] |
Comparison 11. PERPHENAZINE vs CLOZAPINE.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Leaving the study early: 1. Any reason | 2 | 130 | Risk Ratio (M‐H, Random, 95% CI) | 1.87 [0.48, 7.23] |
| 1.1 short term | 2 | 130 | Risk Ratio (M‐H, Random, 95% CI) | 1.87 [0.48, 7.23] |
| 2 Leaving the study early: 2. Due to adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 short term | 1 | 28 | Risk Ratio (M‐H, Random, 95% CI) | 2.6 [0.31, 22.05] |
| 3 Mental state: 1. State ‐ less than 30% BPRS reduction ('no effect') | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 short term | 1 | 97 | Risk Ratio (M‐H, Random, 95% CI) | 2.55 [0.52, 12.52] |
| 4 Mental state: 2. State ‐ BPRS end score (high = poor) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 4.1 total score ‐ short term | 1 | 102 | Mean Difference (IV, Random, 95% CI) | 0.94 [‐1.49, 3.37] |
| 4.2 anxiety‐depression score ‐ short term | 1 | 102 | Mean Difference (IV, Random, 95% CI) | 0.92 [0.34, 1.50] |
| 5 Adverse events: Movement disorders | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 dyskinesia ‐ short term | 1 | 28 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 5.2 hyperkinesia ‐ short term | 1 | 28 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 5.3 myotony ‐ short term | 1 | 97 | Risk Ratio (M‐H, Random, 95% CI) | 21.43 [1.29, 355.77] |
| 5.4 rigidity ‐ short term | 1 | 28 | Risk Ratio (M‐H, Random, 95% CI) | 6.13 [0.35, 108.58] |
| 5.5 tremor ‐ short term | 1 | 97 | Risk Ratio (M‐H, Random, 95% CI) | 5.10 [1.18, 22.09] |
| 6 Other adverse events: 1. Anticholinergic | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 6.1 blurred vision ‐ short term | 1 | 97 | Risk Ratio (M‐H, Random, 95% CI) | 0.85 [0.49, 1.49] |
| 6.2 dry mouth ‐ short term | 1 | 97 | Risk Ratio (M‐H, Random, 95% CI) | 4.59 [1.68, 12.58] |
| 6.3 increased salivation ‐ short term | 1 | 97 | Risk Ratio (M‐H, Random, 95% CI) | 0.05 [0.01, 0.20] |
| 7 Other adverse events: 2. Arousal | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 7.1 drowsiness ‐ short term | 1 | 28 | Risk Ratio (M‐H, Random, 95% CI) | 0.43 [0.20, 0.94] |
| 7.2 excitement ‐ short term | 1 | 97 | Risk Ratio (M‐H, Random, 95% CI) | 21.44 [3.00, 153.14] |
| 7.3 somnolence ‐ short term | 1 | 97 | Risk Ratio (M‐H, Random, 95% CI) | 0.19 [0.07, 0.52] |
| 7.4 restlessness ‐ short term | 1 | 97 | Risk Ratio (M‐H, Random, 95% CI) | 5.10 [1.88, 13.83] |
| 8 Other adverse events: 3. At least one | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 8.1 short term | 1 | 28 | Risk Ratio (M‐H, Random, 95% CI) | 2.6 [0.31, 22.05] |
| 9 Other adverse events: 4. Cardiovascular | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 9.1 dizziness ‐ short term | 1 | 97 | Risk Ratio (M‐H, Random, 95% CI) | 0.92 [0.56, 1.51] |
| 9.2 tachycardia ‐ short term | 1 | 97 | Risk Ratio (M‐H, Random, 95% CI) | 0.23 [0.13, 0.42] |
| 10 Other adverse events: 5. Gastrointestinal | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 10.1 constipation ‐ short term | 1 | 97 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.20, 0.54] |
| 11 Other adverse events: 6. Lab data | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 11.1 abnormal EKG ‐ short term | 1 | 97 | Risk Ratio (M‐H, Random, 95% CI) | 1.33 [0.73, 2.44] |
| 11.2 high white blood cell count ‐ short term | 1 | 97 | Risk Ratio (M‐H, Random, 95% CI) | 1.02 [0.07, 15.86] |
| 11.3 low white blood cell count ‐ short term | 1 | 97 | Risk Ratio (M‐H, Random, 95% CI) | 0.17 [0.02, 1.36] |
| 11.4 liver function (GPT+) ‐ short term | 1 | 97 | Risk Ratio (M‐H, Random, 95% CI) | 0.58 [0.18, 1.86] |
| 12 Other adverse events: 7. Average endpoint score (TESS, skew) | Other data | No numeric data | ||
| 12.1 short term | Other data | No numeric data |
Comparison 12. PERPHENAZINE vs CLOZAPINE + PERPHENAZINE.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Leaving the study early: 1. Any reason | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 short term | 1 | 102 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.32, 27.89] |
| 2 Mental state: 1. State ‐ less than 30% BPRS reduction ('no effect') | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 short term | 1 | 147 | Risk Ratio (M‐H, Random, 95% CI) | 3.44 [0.86, 13.79] |
| 3 Mental state: 1a. State ‐ BPRS end score (high = poor) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 3.1 total score ‐ short term | 1 | 102 | Mean Difference (IV, Random, 95% CI) | 0.74 [‐1.72, 3.20] |
| 3.2 anxiety‐depression score ‐ short term | 1 | 102 | Mean Difference (IV, Random, 95% CI) | 0.51 [‐0.10, 1.12] |
| 4 Adverse events: Movement disorders | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 myotony ‐ short term | 1 | 98 | Risk Ratio (M‐H, Random, 95% CI) | 1.04 [0.48, 2.28] |
| 4.2 tremor ‐ short term | 1 | 98 | Risk Ratio (M‐H, Random, 95% CI) | 1.04 [0.48, 2.28] |
| 5 Other adverse events: 1. Anticholinergic | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 blurred vision ‐ short term | 1 | 98 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.55, 1.75] |
| 5.2 dry mouth ‐ short term | 1 | 98 | Risk Ratio (M‐H, Random, 95% CI) | 1.88 [0.97, 3.64] |
| 5.3 increased salivation ‐ short term | 1 | 98 | Risk Ratio (M‐H, Random, 95% CI) | 0.06 [0.01, 0.22] |
| 6 Other adverse events: 2. Arousal | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 6.1 excitement ‐ short term | 1 | 98 | Risk Ratio (M‐H, Random, 95% CI) | 21.88 [3.06, 156.33] |
| 6.2 restlessness ‐ short term | 1 | 98 | Risk Ratio (M‐H, Random, 95% CI) | 2.98 [1.39, 6.39] |
| 6.3 somnolence ‐ short term | 1 | 98 | Risk Ratio (M‐H, Random, 95% CI) | 2.08 [0.40, 10.85] |
| 7 Other adverse events: 3. Cardiovascular | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 7.1 dizziness ‐ short term | 1 | 98 | Risk Ratio (M‐H, Random, 95% CI) | 0.89 [0.55, 1.46] |
| 7.2 tachycardia ‐ short term | 1 | 98 | Risk Ratio (M‐H, Random, 95% CI) | 0.29 [0.16, 0.55] |
| 8 Other adverse events: 4. Gastrointestinal | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 8.1 constipation ‐ short term | 1 | 98 | Risk Ratio (M‐H, Random, 95% CI) | 0.44 [0.26, 0.73] |
| 9 Other adverse events: 5. Lab data | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 9.1 abnormal EKG ‐ short term | 1 | 98 | Risk Ratio (M‐H, Random, 95% CI) | 1.97 [0.97, 3.98] |
| 9.2 high white blood cell count ‐ short term | 1 | 98 | Risk Ratio (M‐H, Random, 95% CI) | 1.04 [0.07, 16.19] |
| 9.3 low white blood cell count ‐ short term | 1 | 98 | Risk Ratio (M‐H, Random, 95% CI) | 0.17 [0.02, 1.39] |
| 9.4 liver function (GPT+) ‐ short term | 1 | 98 | Risk Ratio (M‐H, Random, 95% CI) | 0.83 [0.24, 2.92] |
| 10 Other adverse events: 6. Average endpoint score (TESS, skewed) | Other data | No numeric data | ||
| 10.1 short term | Other data | No numeric data |
Comparison 13. PERPHENAZINE vs FLUPHENAZINE.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Leaving the study early: 1. Any reason | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 short term | 1 | 106 | Risk Ratio (M‐H, Random, 95% CI) | 0.62 [0.38, 1.01] |
| 2 Leaving the study early: 2. Due to adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 short term | 1 | 106 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.15, 6.84] |
| 3 Adverse events: Movement disorders | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 akathisia ‐ short term | 1 | 106 | Risk Ratio (M‐H, Random, 95% CI) | 1.17 [0.42, 3.24] |
| 3.2 akinesia ‐ short term | 1 | 106 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.3 dyskinesia ‐ short term | 1 | 106 | Risk Ratio (M‐H, Random, 95% CI) | 2.0 [0.19, 21.40] |
| 3.4 parkinsonism ‐ short term | 1 | 106 | Risk Ratio (M‐H, Random, 95% CI) | 1.14 [0.45, 2.93] |
Comparison 14. PERPHENAZINE vs HALOPERIDOL.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Leaving the study early: 1. Any reason | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 short term | 1 | 189 | Risk Ratio (M‐H, Random, 95% CI) | 0.84 [0.40, 1.77] |
| 2 Leaving the study early: 2. Due to adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 short term | 1 | 189 | Risk Ratio (M‐H, Random, 95% CI) | 0.49 [0.13, 1.92] |
| 3 Leaving the study early: 3. Due to relapse / worsening or no improvement | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 short term | 1 | 189 | Risk Ratio (M‐H, Random, 95% CI) | 1.39 [0.46, 4.21] |
| 4 Global state: 1. Change over time ‐ no better or deterioration (ITT) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 short term | 1 | 189 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.79, 1.29] |
| 5 Adverse events: Movement disorders | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 akathisia ‐ short term | 1 | 189 | Risk Ratio (M‐H, Random, 95% CI) | 0.70 [0.42, 1.16] |
| 5.2 ataxia ‐ short term | 1 | 189 | Risk Ratio (M‐H, Random, 95% CI) | 0.49 [0.05, 5.36] |
| 5.3 dyskinesia ‐ short term | 1 | 189 | Risk Ratio (M‐H, Random, 95% CI) | 0.45 [0.16, 1.24] |
| 5.4 parkinsonism ‐ short term | 1 | 189 | Risk Ratio (M‐H, Random, 95% CI) | 0.77 [0.49, 1.20] |
| 5.5 use of antiparkinsonian drugs ‐ short term | 1 | 189 | Risk Ratio (M‐H, Random, 95% CI) | 0.87 [0.68, 1.11] |
| 6 Other adverse events: 1. Anticholinergic | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 6.1 blurred vision ‐ short term | 1 | 189 | Risk Ratio (M‐H, Random, 95% CI) | 0.25 [0.05, 1.13] |
| 6.2 dry mouth ‐ short term | 1 | 189 | Risk Ratio (M‐H, Random, 95% CI) | 0.54 [0.21, 1.40] |
| 7 Other adverse events: 2. Arousal | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 7.1 insomnia ‐ short term | 1 | 189 | Risk Ratio (M‐H, Random, 95% CI) | 0.97 [0.71, 1.32] |
| 7.2 need of additional benzodiazepines ‐ short term | 1 | 189 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.76, 1.34] |
| 7.3 sleepiness/sedation ‐ short term | 1 | 189 | Risk Ratio (M‐H, Random, 95% CI) | 1.21 [0.53, 2.78] |
| 8 Other adverse events: 3. At least one | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 8.1 short term | 1 | 189 | Risk Ratio (M‐H, Random, 95% CI) | 1.05 [0.88, 1.25] |
| 9 Other adverse events: 4. Cardiovascular | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 9.1 angina pectoris ‐ short term | 1 | 189 | Risk Ratio (M‐H, Random, 95% CI) | 0.57 [0.17, 1.87] |
| 9.2 palpitations ‐ short term | 1 | 189 | Risk Ratio (M‐H, Random, 95% CI) | 0.28 [0.06, 1.33] |
| 10 Other adverse events: 5. Central nervous system | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 10.1 disturbance of consciousness ‐ short term | 1 | 189 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 8.00] |
| 10.2 headache ‐ short term | 1 | 189 | Risk Ratio (M‐H, Random, 95% CI) | 0.90 [0.40, 2.02] |
| 10.3 paraesthesia ‐ short term | 1 | 189 | Risk Ratio (M‐H, Random, 95% CI) | 0.99 [0.25, 3.84] |
| 10.4 vertigo ‐ short term | 1 | 189 | Risk Ratio (M‐H, Random, 95% CI) | 0.82 [0.37, 1.82] |
| 11 Other adverse events: 6. Gastrointestinal | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 11.1 constipation ‐ short term | 1 | 189 | Risk Ratio (M‐H, Random, 95% CI) | 0.84 [0.40, 1.77] |
| 11.2 icterus ‐ short term | 1 | 189 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 11.3 loss of appetite ‐ short term | 1 | 189 | Risk Ratio (M‐H, Random, 95% CI) | 0.75 [0.45, 1.27] |
| 11.4 nausea and vomiting ‐ short term | 1 | 189 | Risk Ratio (M‐H, Random, 95% CI) | 1.62 [0.81, 3.24] |
| 12 Other adverse events: 7. Skin | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 12.1 pruritus ‐ short term | 1 | 189 | Risk Ratio (M‐H, Random, 95% CI) | 0.66 [0.11, 3.86] |
| 12.2 rash ‐ short term | 1 | 189 | Risk Ratio (M‐H, Random, 95% CI) | 1.98 [0.18, 21.46] |
| 13 Other adverse events: 8. Genitourinary | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 13.1 diminished sexual desire | 1 | 189 | Risk Ratio (M‐H, Random, 95% CI) | 0.99 [0.06, 15.59] |
| 13.2 micturition disturbances ‐ short term | 1 | 189 | Risk Ratio (M‐H, Random, 95% CI) | 0.49 [0.09, 2.64] |
| 14 Other adverse events: 9. Others | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 14.1 cramps ‐ short term | 1 | 189 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 8.00] |
| 14.2 sweating ‐ short term | 1 | 189 | Risk Ratio (M‐H, Random, 95% CI) | 0.16 [0.02, 1.34] |
Comparison 15. PERPHENAZINE vs LOXAPINE.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Leaving the study early: 1. Any reason | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 short term | 1 | 47 | Risk Ratio (M‐H, Random, 95% CI) | 0.96 [0.32, 2.88] |
| 2 Leaving the study early: 2. Due to relapse / worsening or no improvement | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 short term | 1 | 47 | Risk Ratio (M‐H, Random, 95% CI) | 0.96 [0.06, 14.43] |
| 3 Global state: 1. Change over time ‐ no better or deterioration (ITT) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 short term | 1 | 47 | Risk Ratio (M‐H, Random, 95% CI) | 0.85 [0.40, 1.82] |
| 4 Adverse events: Movement disorders | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 dystonia ‐ short term | 1 | 47 | Risk Ratio (M‐H, Random, 95% CI) | 0.14 [0.01, 2.52] |
| 4.2 extrapyramidal signs ‐ short term | 1 | 47 | Risk Ratio (M‐H, Random, 95% CI) | 0.90 [0.62, 1.31] |
| 4.3 use of antiparkinsonian drugs ‐ short term | 1 | 47 | Risk Ratio (M‐H, Random, 95% CI) | 1.18 [0.75, 1.86] |
| 5 Other adverse events: 1. Arousal | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 drowsiness ‐ short term | 1 | 47 | Risk Ratio (M‐H, Random, 95% CI) | 1.60 [0.43, 5.93] |
| 6 Other adverse events: 2. At least one | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 6.1 short term | 1 | 47 | Risk Ratio (M‐H, Random, 95% CI) | 1.30 [1.00, 1.68] |
| 7 Other adverse events: 3. lab data | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 7.1 ECG – atrial extrasystole & flattening of T‐wave in V6 ‐ short term | 1 | 47 | Risk Ratio (M‐H, Random, 95% CI) | 6.72 [0.37, 123.33] |
| 7.2 ventricular extrasystole ‐ short term | 1 | 47 | Risk Ratio (M‐H, Random, 95% CI) | 0.32 [0.01, 7.48] |
Comparison 16. PERPHENAZINE vs MEPAZINE.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Leaving the study early: 1. Any reason | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 short term | 2 | 68 | Risk Ratio (M‐H, Random, 95% CI) | 1.18 [0.91, 1.53] |
| 2 Leaving the study early: 2. Due to adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 short term | 1 | 58 | Risk Ratio (M‐H, Random, 95% CI) | 6.97 [0.93, 52.20] |
| 3 Global state: 1. Change over time ‐ no better or deterioration (ITT) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 short term | 1 | 58 | Risk Ratio (M‐H, Random, 95% CI) | 0.40 [0.16, 1.00] |
| 4 Other adverse events: 1. Central nervous system | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 electroencephalogram (increase) ‐ short term | 1 | 10 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.08, 11.93] |
Comparison 17. PERPHENAZINE vs METHYPERIDOL.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Leaving the study early: 1. Any reason | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 short term | 1 | 78 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.35, 2.83] |
| 2 Leaving the study early: 2. Due to adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 short term | 1 | 78 | Risk Ratio (M‐H, Random, 95% CI) | 4.0 [0.47, 34.20] |
| 3 Global state: 1. Change over time ‐ no better or deterioration (ITT) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 short term | 1 | 78 | Risk Ratio (M‐H, Random, 95% CI) | 1.05 [0.68, 1.64] |
| 4 Behaviour: 1. Behaviour subscale of the Psychiatric Rating Scale: no better or worse | 1 | 78 | Risk Ratio (M‐H, Random, 95% CI) | 0.88 [0.61, 1.26] |
| 5 Adverse events: Movement disorders | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 akathisia ‐ short term | 1 | 78 | Risk Ratio (M‐H, Random, 95% CI) | 1.25 [0.36, 4.31] |
| 5.2 ataxia ‐ short term | 1 | 78 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 5.3 dyskinesia ‐ short term | 1 | 78 | Risk Ratio (M‐H, Random, 95% CI) | 0.2 [0.02, 1.63] |
| 5.4 parkinsonism ‐ short term | 1 | 78 | Risk Ratio (M‐H, Random, 95% CI) | 0.8 [0.35, 1.81] |
| 6 Other adverse events: 1. Anticholinergic | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 6.1 blurred vision ‐ short term | 1 | 78 | Risk Ratio (M‐H, Random, 95% CI) | 0.2 [0.01, 4.04] |
| 6.2 dry mouth ‐ short term | 1 | 78 | Risk Ratio (M‐H, Random, 95% CI) | 2.0 [0.19, 21.16] |
| 7 Other adverse events: 2. Arousal | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 7.1 insomnia ‐ short term | 1 | 78 | Risk Ratio (M‐H, Random, 95% CI) | 0.64 [0.28, 1.47] |
| 7.2 sleepiness/sedation ‐ short term | 1 | 78 | Risk Ratio (M‐H, Random, 95% CI) | 0.5 [0.10, 2.57] |
| 8 Other adverse events: 3. At least one | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 8.1 short term | 1 | 78 | Risk Ratio (M‐H, Random, 95% CI) | 0.96 [0.70, 1.33] |
| 9 Other adverse events: 4. Central nervous system | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 9.1 disturbance of consciousness ‐ short term | 1 | 78 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 9.2 headache ‐ short term | 1 | 78 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.06, 15.43] |
| 9.3 paraesthesia ‐ short term | 1 | 78 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 9.4 vertigo ‐ short term | 1 | 78 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 7.94] |
| 10 Other adverse events: 5. Gastrointestinal | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 10.1 constipation ‐ short term | 1 | 78 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 7.94] |
| 10.2 icterus ‐ short term | 1 | 78 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 10.3 loss of appetite ‐ short term | 1 | 78 | Risk Ratio (M‐H, Random, 95% CI) | 0.6 [0.15, 2.34] |
| 10.4 nausea and vomiting ‐ short term | 1 | 78 | Risk Ratio (M‐H, Random, 95% CI) | 1.5 [0.27, 8.49] |
| 11 Other adverse events: 6. Skin | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 11.1 rash ‐ short term | 1 | 78 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.06, 15.43] |
| 12 Other adverse events: 7. Genitourinary | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 12.1 micturition disturbances ‐ short term | 1 | 78 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 7.94] |
| 13 Other adverse events: 8. Others | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 13.1 cramps ‐ short term | 1 | 78 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 13.2 sweating ‐ short term | 1 | 78 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 18. PERPHENAZINE vs OLANZAPINE.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Leaving the study early: 1. Any reason | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 long term | 2 | 657 | Risk Ratio (M‐H, Random, 95% CI) | 1.16 [1.05, 1.29] |
| 2 Leaving the study early: 2. Due to adverse events | 2 | 643 | Risk Ratio (M‐H, Random, 95% CI) | 0.85 [0.59, 1.21] |
| 2.1 long term | 2 | 643 | Risk Ratio (M‐H, Random, 95% CI) | 0.85 [0.59, 1.21] |
| 3 Leaving the study early: 3. Due to relapse / worsening or no improvement | 2 | 643 | Risk Ratio (M‐H, Random, 95% CI) | 1.24 [0.47, 3.29] |
| 3.1 long term | 2 | 643 | Risk Ratio (M‐H, Random, 95% CI) | 1.24 [0.47, 3.29] |
| 4 Mental state: 1a. State ‐ BPRS end score (high = poor, skewed data) | Other data | No numeric data | ||
| 4.1 long term | Other data | No numeric data | ||
| 5 Mental state: 1b. State ‐ PANSS total endpoint score (high = poor, skewed data) | Other data | No numeric data | ||
| 5.1 long term | Other data | No numeric data | ||
| 6 Mental state: 1c. State ‐ PANSS: mean endpoint score from baseline (high = poor) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 6.1 total ‐ short term | 1 | 60 | Mean Difference (IV, Random, 95% CI) | 4.21 [2.17, 6.25] |
| 6.2 positive ‐ short term | 1 | 60 | Mean Difference (IV, Random, 95% CI) | 8.23 [7.26, 9.20] |
| 6.3 negative ‐ short term | 1 | 60 | Mean Difference (IV, Random, 95% CI) | 1.58 [0.64, 2.52] |
| 6.4 general psychopathology ‐ short term | 1 | 60 | Mean Difference (IV, Random, 95% CI) | ‐0.39 [‐0.79, 0.01] |
| 7 Mental state: 1d. State ‐ less than 25% PANSS reduction ('no effect') | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 7.1 short term | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 2.0 [0.40, 10.11] |
| 8 Adverse events: Movement disorders | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 8.1 akathisia ‐ long term | 2 | 591 | Risk Ratio (M‐H, Random, 95% CI) | 1.33 [0.68, 2.60] |
| 8.2 EPS ‐ long term | 1 | 539 | Risk Ratio (M‐H, Random, 95% CI) | 0.79 [0.42, 1.49] |
| 8.3 tardive dyskinesia ‐ long term | 1 | 473 | Risk Ratio (M‐H, Random, 95% CI) | 1.28 [0.83, 1.95] |
| 8.4 use of antiparkinsonian drugs ‐ long term | 1 | 46 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.34, 26.76] |
| 9 Other adverse events: 1. Anticholinergic | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 9.1 increased salivation ‐ long term | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 7.87] |
| 10 Other adverse events: 2. Arousal | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 10.1 agitation ‐ long term | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.33, 27.23] |
| 10.2 anxiety ‐ long term | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 1.25 [0.37, 4.21] |
| 10.3 insomnia ‐ long term | 2 | 657 | Risk Ratio (M‐H, Random, 95% CI) | 2.34 [0.62, 8.89] |
| 10.4 sleepiness/ sedation ‐ long term | 1 | 597 | Risk Ratio (M‐H, Random, 95% CI) | 0.92 [0.71, 1.18] |
| 11 Other adverse events: 3. Cardiovascular | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 11.1 bradycardia ‐ long term | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.13, 70.83] |
| 11.2 faintness, dizziness, weakness ‐ long term | 1 | 597 | Risk Ratio (M‐H, Random, 95% CI) | 1.20 [0.75, 1.95] |
| 11.3 hypotension ‐ long term | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 7.87] |
| 12 Other adverse events: 4. Central nervous system | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 12.1 headache ‐ long term | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 0.71 [0.25, 2.00] |
| 12.2 hypertonia ‐ long term | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.13, 70.83] |
| 13 Other adverse events: 5. Gastrointestinal | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 13.1 gastroenteritis ‐ long term | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.13, 70.83] |
| 13.2 weight gain ‐ long term | 1 | 550 | Risk Ratio (M‐H, Random, 95% CI) | 0.40 [0.27, 0.58] |
| 14 Other adverse events: 6. Skin | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 14.1 cellulitis ‐ long term | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 7.87] |
| 14.2 eczema ‐ long term | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.13, 70.83] |
| 14.3 fungal dermatitis ‐ long term | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 7.87] |
| 14.4 rash ‐ long term | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.07, 15.26] |
| 15 Other adverse events: 7. Genitourinary | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 15.1 diminished sexual desire ‐ long term | 1 | 597 | Risk Ratio (M‐H, Random, 95% CI) | 0.91 [0.69, 1.19] |
| 15.2 galactorrhea/ gynaecomastia ‐ long term | 1 | 597 | Risk Ratio (M‐H, Random, 95% CI) | 0.74 [0.22, 2.49] |
| 16 Other adverse events: 8. Others | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 16.1 allergic reaction ‐ long term | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 7.87] |
| 16.2 back pain ‐ long term | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.13, 70.83] |
| 16.3 incontinence nocturia ‐ long term | 1 | 597 | Risk Ratio (M‐H, Random, 95% CI) | 0.43 [0.17, 1.07] |
| 16.4 suicide attempt ‐ long term | 1 | 597 | Risk Ratio (M‐H, Random, 95% CI) | 0.64 [0.06, 7.06] |
| 16.5 urinary hesitancy, dry mouth, constipation ‐ long term | 1 | 597 | Risk Ratio (M‐H, Random, 95% CI) | 0.93 [0.69, 1.25] |
| 17 Other adverse events: 9. any serious adverse event | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 17.1 long term | 1 | 597 | Risk Ratio (M‐H, Random, 95% CI) | 1.17 [0.72, 1.88] |
| 18 Other adverse events: 10. lab data | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 18.1 anemia ‐ long term | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.07, 15.26] |
| 19 Other adverse events: 11a. lab data | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 19.1 mean weight change (lb) ‐ long term | 1 | 597 | Mean Difference (IV, Random, 95% CI) | ‐11.4 [‐14.19, ‐8.61] |
| 19.2 change in blood glucose ‐ long term | 1 | 597 | Mean Difference (IV, Random, 95% CI) | ‐9.8 [‐16.54, ‐3.06] |
| 19.3 change in glycosylated haemoglobin ‐ long term | 1 | 597 | Mean Difference (IV, Random, 95% CI) | ‐0.31 [‐0.52, ‐0.10] |
| 19.4 change in cholesterol ‐ long term | 1 | 597 | Mean Difference (IV, Random, 95% CI) | ‐9.2 [‐15.30, ‐3.10] |
| 19.5 change in triglycerides ‐ long term | 1 | 597 | Mean Difference (IV, Random, 95% CI) | ‐34.60 [‐62.51, ‐6.69] |
| 19.6 change in prolactin ‐ long term | 1 | 597 | Mean Difference (IV, Random, 95% CI) | 6.5 [2.42, 10.58] |
| 20 Other adverse events: 11b. lab data (skewed) | Other data | No numeric data | ||
| 20.1 weight change | Other data | No numeric data |
Comparison 19. PERPHENAZINE vs PENFLURIDOL.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Leaving the study early: 1. Any reason | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 short term | 1 | 105 | Risk Ratio (M‐H, Random, 95% CI) | 0.49 [0.20, 1.19] |
| 2 Global state: 1. Change over time ‐ no better or deterioration (ITT) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 short term | 1 | 105 | Risk Ratio (M‐H, Random, 95% CI) | 0.92 [0.64, 1.31] |
| 3 Adverse events: Movement disorders | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 akathisia ‐ short term | 1 | 105 | Risk Ratio (M‐H, Random, 95% CI) | 0.66 [0.39, 1.11] |
| 3.2 dyskinesia ‐ short term | 1 | 105 | Risk Ratio (M‐H, Random, 95% CI) | 0.82 [0.33, 2.05] |
| 3.3 parkinsonism ‐ short term | 1 | 105 | Risk Ratio (M‐H, Random, 95% CI) | 1.13 [0.80, 1.60] |
| 4 Other adverse events: 1. Anticholinergic | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 blurred vision ‐ short term | 1 | 105 | Risk Ratio (M‐H, Random, 95% CI) | 1.06 [0.15, 7.24] |
| 4.2 dry mouth ‐ short term | 1 | 105 | Risk Ratio (M‐H, Random, 95% CI) | 1.32 [0.57, 3.09] |
| 5 Other adverse events: 2. Arousal | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 drowsiness ‐ short term | 1 | 105 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.49, 1.94] |
| 5.2 insomnia ‐ short term | 1 | 105 | Risk Ratio (M‐H, Random, 95% CI) | 0.97 [0.62, 1.52] |
| 6 Other adverse events: 3. Cardiovascular | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 6.1 angina pectoris ‐ short term | 1 | 105 | Risk Ratio (M‐H, Random, 95% CI) | 0.53 [0.17, 1.65] |
| 6.2 faintness, dizziness, weakness ‐ short term | 1 | 105 | Risk Ratio (M‐H, Random, 95% CI) | 0.85 [0.50, 1.45] |
| 6.3 palpitations ‐ short term | 1 | 105 | Risk Ratio (M‐H, Random, 95% CI) | 0.12 [0.02, 0.90] |
| 7 Other adverse events: 4. Central nervous system | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 7.1 headache ‐ short term | 1 | 105 | Risk Ratio (M‐H, Random, 95% CI) | 0.79 [0.37, 1.72] |
| 7.2 paraesthesia ‐ short term | 1 | 105 | Risk Ratio (M‐H, Random, 95% CI) | 0.53 [0.05, 5.66] |
| 8 Other adverse events: 5. Gastrointestinal | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 8.1 constipation ‐ short term | 1 | 105 | Risk Ratio (M‐H, Random, 95% CI) | 1.38 [0.66, 2.86] |
| 8.2 loss of appetite ‐ short term | 1 | 105 | Risk Ratio (M‐H, Random, 95% CI) | 0.91 [0.46, 1.77] |
| 8.3 nausea and vomiting ‐ short term | 1 | 105 | Risk Ratio (M‐H, Random, 95% CI) | 0.85 [0.36, 1.98] |
| 9 Other adverse events: 6. Skin | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 9.1 oedema | 1 | 105 | Risk Ratio (M‐H, Random, 95% CI) | 3.17 [0.13, 76.16] |
| 9.2 pruritus ‐ short term | 1 | 105 | Risk Ratio (M‐H, Random, 95% CI) | 0.71 [0.12, 4.05] |
| 9.3 rash ‐ short term | 1 | 105 | Risk Ratio (M‐H, Random, 95% CI) | 1.06 [0.07, 16.48] |
| 10 Other adverse events: 7. Genitourinary | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 10.1 diminished sexual desire | 1 | 105 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 10.2 increased sexual desire ‐ short term | 1 | 105 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 10.3 micturition disturbances ‐ short term | 1 | 105 | Risk Ratio (M‐H, Random, 95% CI) | 1.06 [0.15, 7.24] |
| 11 Other adverse events: 8. Others | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 11.1 sweating ‐ short term | 1 | 105 | Risk Ratio (M‐H, Random, 95% CI) | 0.71 [0.12, 4.05] |
Comparison 20. PERPHENAZINE vs PROCHLORPERAZINE.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Leaving the study early: 1. Any reason | 3 | 175 | Risk Ratio (M‐H, Random, 95% CI) | 0.90 [0.37, 2.19] |
| 1.1 short term | 3 | 175 | Risk Ratio (M‐H, Random, 95% CI) | 0.90 [0.37, 2.19] |
| 2 Leaving the study early: 2. Due to adverse events | 2 | 165 | Risk Ratio (M‐H, Random, 95% CI) | 2.37 [0.80, 7.05] |
| 2.1 short term | 2 | 165 | Risk Ratio (M‐H, Random, 95% CI) | 2.37 [0.80, 7.05] |
| 3 Global state: 1. Change over time ‐ no better or deterioration (ITT) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 short term | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 0.67 [0.24, 1.87] |
| 4 Adverse events: Movement disorders | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 akathisia ‐ short term | 1 | 105 | Risk Ratio (M‐H, Random, 95% CI) | 2.29 [0.63, 8.38] |
| 4.2 akinesia ‐ short term | 1 | 105 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 7.85] |
| 4.3 dyskinesia ‐ short term | 1 | 105 | Risk Ratio (M‐H, Random, 95% CI) | 1.96 [0.18, 20.99] |
| 4.4 parkinsonism ‐ short term | 1 | 105 | Risk Ratio (M‐H, Random, 95% CI) | 1.96 [0.63, 6.12] |
| 5 Other adverse events: 1. Central nervous system | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 electroencephalogram (increase) ‐ short term | 1 | 10 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.05, 2.21] |
Comparison 21. PERPHENAZINE vs QUETIAPINE.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Leaving the study early: 1. Any reason | 2 | 662 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.55, 1.75] |
| 1.1 short term | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.33, 27.33] |
| 1.2 long term | 1 | 598 | Risk Ratio (M‐H, Random, 95% CI) | 0.91 [0.84, 1.00] |
| 2 Leaving the study early: 2. Due to adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 long term | 1 | 598 | Risk Ratio (M‐H, Random, 95% CI) | 1.05 [0.72, 1.55] |
| 3 Leaving the study early: 3. Due to relapse / worsening or no improvement | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 long term | 1 | 598 | Risk Ratio (M‐H, Random, 95% CI) | 0.91 [0.69, 1.20] |
| 4 Mental state: 2a. State ‐ BPRS score reduction ('no effect') | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 less than 25% reduction ‐ short term | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 1.33 [0.32, 5.49] |
| 5 Mental state: 2b. State ‐ BPRS total score (high = worse) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 5.1 short term | 1 | 64 | Mean Difference (IV, Random, 95% CI) | 1.10 [‐1.26, 3.46] |
| 6 Adverse events: Movement disorders | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 6.1 akathisia ‐ short term | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 7.0 [0.91, 53.68] |
| 6.2 akathisia ‐ long term | 1 | 546 | Risk Ratio (M‐H, Random, 95% CI) | 1.27 [0.65, 2.48] |
| 6.3 EPS ‐ long term | 1 | 541 | Risk Ratio (M‐H, Random, 95% CI) | 1.53 [0.73, 3.21] |
| 6.4 rigidity ‐ short term | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 11.00 [0.63, 191.04] |
| 6.5 tardive dyskinesia ‐ long term | 1 | 473 | Risk Ratio (M‐H, Random, 95% CI) | 1.36 [0.88, 2.10] |
| 6.6 torsion dystonia ‐ short term | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 15.0 [0.89, 252.09] |
| 6.7 tremor ‐ short term | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 12.0 [1.66, 86.94] |
| 7 Other adverse events: 1. Anticholinergic | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 7.1 blurred vision ‐ short term | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 4.5 [1.05, 19.22] |
| 7.2 dry mouth ‐ short term | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.65, 13.76] |
| 8 Other adverse events: 2. Arousal | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 8.1 drowsiness ‐ short term | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 0.67 [0.12, 3.73] |
| 8.2 insomnia ‐ short term | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 0.6 [0.16, 2.30] |
| 8.3 insomnia ‐ long term | 1 | 598 | Risk Ratio (M‐H, Random, 95% CI) | 1.37 [1.01, 1.87] |
| 8.4 sleepiness/ sedation ‐ long term | 1 | 598 | Risk Ratio (M‐H, Random, 95% CI) | 0.93 [0.72, 1.19] |
| 9 Other adverse events: 3. Cardiovascular | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 9.1 faintness, dizziness, weakness ‐ long term | 1 | 598 | Risk Ratio (M‐H, Random, 95% CI) | 0.99 [0.62, 1.55] |
| 9.2 orthostatic hypotension ‐ short term | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 0.17 [0.02, 1.31] |
| 9.3 tachycardia ‐ short term | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 2.5 [0.52, 11.96] |
| 10 Other adverse events: 4. Gastrointestinal | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 10.1 constipation ‐ short term | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 2.5 [0.52, 11.96] |
| 10.2 weight gain ‐ short term | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.33, 27.33] |
| 10.3 weight gain ‐ long term | 1 | 548 | Risk Ratio (M‐H, Random, 95% CI) | 0.74 [0.48, 1.14] |
| 11 Other adverse events: 5. Genitourinary | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 11.1 diminished sexual desire ‐ long term | 1 | 598 | Risk Ratio (M‐H, Random, 95% CI) | 1.20 [0.89, 1.62] |
| 11.2 galactorrhoea/ gynaecomastia ‐ long term | 1 | 598 | Risk Ratio (M‐H, Random, 95% CI) | 0.86 [0.25, 3.02] |
| 12 Other adverse events: 6. Others | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 12.1 urinary hesitancy, dry mouth, constipation ‐ long term | 1 | 598 | Risk Ratio (M‐H, Random, 95% CI) | 0.70 [0.53, 0.93] |
| 12.2 suicide attempt ‐ long term | 1 | 598 | Risk Ratio (M‐H, Random, 95% CI) | 1.29 [0.08, 20.55] |
| 12.3 incontinence nocturia ‐ long term | 1 | 598 | Risk Ratio (M‐H, Random, 95% CI) | 0.52 [0.20, 1.31] |
| 13 Other adverse events: 7. any serious adverse event | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 13.1 long term | 1 | 598 | Risk Ratio (M‐H, Random, 95% CI) | 1.17 [0.73, 1.88] |
| 14 Other adverse events: 8a. lab data | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 14.1 abnormal ECG ‐ short term | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 7.0 [0.91, 53.68] |
| 14.2 abnormal liver function ‐ short term | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 4.0 [0.92, 17.40] |
| 15 Other adverse events: 8b. lab data | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 15.1 mean weight change (lb) ‐ long term | 1 | 598 | Mean Difference (IV, Random, 95% CI) | ‐3.1 [‐5.89, ‐0.31] |
| 15.2 change in blood glucose ‐ long term | 1 | 598 | Mean Difference (IV, Random, 95% CI) | ‐1.60 [‐7.50, 4.30] |
| 15.3 change in glycosylated haemoglobin ‐ long term | 1 | 598 | Mean Difference (IV, Random, 95% CI) | 0.05 [‐0.10, 0.20] |
| 15.4 change in cholesterol ‐ long term | 1 | 598 | Mean Difference (IV, Random, 95% CI) | ‐4.8 [‐10.90, 1.30] |
| 15.5 change in triglycerides ‐ long term | 1 | 598 | Mean Difference (IV, Random, 95% CI) | ‐10.90 [‐41.55, 19.75] |
| 15.6 change in prolactin ‐ long term | 1 | 598 | Mean Difference (IV, Random, 95% CI) | 9.70 [5.38, 14.02] |
Comparison 22. PERPHENAZINE vs PROMAZINE.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Leaving the study early: 1. Any reason | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 short term | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 1.20 [0.93, 1.56] |
| 2 Leaving the study early: 2. Due to adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 short term | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 2.49 [0.73, 8.50] |
| 3 Global state: 1. Change over time ‐ no better or deterioration (ITT) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 short term | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 0.39 [0.16, 0.97] |
Comparison 23. PERPHENAZINE vs RISPERIDONE.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Leaving the study early: 1. Any reason | 2 | 709 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.92, 1.11] |
| 1.1 short term | 2 | 709 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.92, 1.11] |
| 2 Leaving the study early: 2. Due to adverse events | 2 | 709 | Risk Ratio (M‐H, Random, 95% CI) | 1.29 [0.73, 2.29] |
| 2.1 short term | 2 | 709 | Risk Ratio (M‐H, Random, 95% CI) | 1.29 [0.73, 2.29] |
| 3 Leaving the study early: 3. Due to relapse / worsening or no improvement | 2 | 709 | Risk Ratio (M‐H, Random, 95% CI) | 0.95 [0.72, 1.24] |
| 3.1 short term | 2 | 709 | Risk Ratio (M‐H, Random, 95% CI) | 0.95 [0.72, 1.24] |
| 4 Global state: 1. Change over time ‐ no better or deterioration (ITT) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 short term | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 1.29 [0.79, 2.12] |
| 5 Mental state: 1a. State ‐ less than 20% BPRS reduction ('no effect') | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 short term | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 0.81 [0.61, 1.08] |
| 6 Mental state: 1b. State ‐ PANSS reduction ('no effect') | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 6.1 less than 20% reduction ‐ short term | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 0.86 [0.64, 1.15] |
| 6.2 less than 40% reduction ‐ short term | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 1.7 [0.30, 9.50] |
| 7 Mental state: 1c. State ‐ PANSS: mean change from baseline to endpoint | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 7.1 short term | 1 | 107 | Mean Difference (IV, Random, 95% CI) | 4.0 [‐5.04, 13.04] |
| 8 Adverse events: Movement disorders | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 8.1 akathisia ‐ long term | 1 | 399 | Risk Ratio (M‐H, Random, 95% CI) | 0.75 [0.38, 1.49] |
| 8.2 dystonia ‐ short term | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 2.47 [0.67, 9.04] |
| 8.3 EPS ‐ short term | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 9.07 [1.20, 68.35] |
| 8.4 EPS ‐ long term | 1 | 446 | Risk Ratio (M‐H, Random, 95% CI) | 2.23 [0.97, 5.11] |
| 8.5 hyperkinesia ‐ short term | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 2.64 [0.88, 7.91] |
| 8.6 tardive dyskinesia ‐ long term | 1 | 363 | Risk Ratio (M‐H, Random, 95% CI) | 1.21 [0.73, 2.02] |
| 8.7 use of antiparkinsonian drugs ‐ short term | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 1.20 [0.67, 2.14] |
| 9 Other adverse events: 1. Anticholinergic | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 9.1 dry mouth ‐ short term | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 7.40 [0.39, 139.81] |
| 10 Other adverse events: 2. Arousal | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 10.1 depression ‐ short term | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 1.34 [0.76, 2.35] |
| 10.2 emotional indifference ‐ short term | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 1.06 [0.58, 1.94] |
| 10.3 increased dream activity ‐ short term | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 1.51 [0.62, 3.67] |
| 10.4 insomnia ‐ short term | 2 | 553 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.52, 1.97] |
| 10.5 sleepiness/sedation ‐ short term | 2 | 553 | Risk Ratio (M‐H, Random, 95% CI) | 1.16 [0.87, 1.54] |
| 10.6 tension ‐ short term | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 0.97 [0.63, 1.50] |
| 11 Other adverse events: 3. At least one | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 11.1 short term | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.85, 1.21] |
| 12 Other adverse events: 4. Cardiovascular | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 12.1 faintness, dizziness, weakness ‐ short term | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 0.93 [0.36, 2.37] |
| 12.2 lassitude ‐ short term | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 0.97 [0.61, 1.52] |
| 12.3 tachycardia ‐ short term | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 0.57 [0.25, 1.32] |
| 13 Other adverse events: 5. Central nervous system | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 13.1 difficulty in concentration ‐ short term | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 1.06 [0.71, 1.58] |
| 13.2 headache ‐ short term | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 0.53 [0.14, 2.01] |
| 13.3 paraesthesia ‐ short term | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 0.71 [0.12, 4.05] |
| 14 Other adverse events: 6. Endocrine | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 14.1 amenorrhoea (women only) ‐ short term | 1 | 30 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.13, 68.26] |
| 14.2 gynaecomastia ‐ short term | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 0.15 [0.01, 2.85] |
| 14.3 lactation ‐ short term | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 0.21 [0.01, 4.30] |
| 14.4 menorrhagia (women only) ‐ short term | 1 | 30 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.13, 68.26] |
| 15 Other adverse events: 7. Gastrointestinal | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 15.1 constipation ‐ short term | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 0.07 [0.00, 1.20] |
| 15.2 diarrhoea ‐ short term | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 0.15 [0.01, 2.85] |
| 15.3 faintness, dizziness, weakness ‐ long term | 1 | 446 | Risk Ratio (M‐H, Random, 95% CI) | 0.86 [0.52, 1.42] |
| 15.4 nausea and vomiting ‐ short term | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 0.76 [0.26, 2.23] |
| 15.5 weight loss ‐ short term | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 2.12 [0.56, 8.02] |
| 15.6 weight gain ‐ short term | 2 | 511 | Risk Ratio (M‐H, Random, 95% CI) | 0.90 [0.29, 2.81] |
| 16 Other adverse events: 8. Skin | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 16.1 pruritus ‐ short term | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 0.79 [0.19, 3.38] |
| 16.2 rash ‐ short term | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 0.71 [0.12, 4.05] |
| 16.3 sweating ‐ short term | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 0.66 [0.23, 1.89] |
| 17 Other adverse events: 9. Genitourinary | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 17.1 diminished sexual desire | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 1.06 [0.22, 5.01] |
| 17.2 ejaculatory dysfunction (men only) ‐ short term | 1 | 77 | Risk Ratio (M‐H, Random, 95% CI) | 0.18 [0.02, 1.43] |
| 17.3 erectile dysfunction (men only) ‐ short term | 1 | 77 | Risk Ratio (M‐H, Random, 95% CI) | 0.90 [0.30, 2.70] |
| 17.4 galactorrhoea ‐ short term | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 0.15 [0.01, 2.85] |
| 17.5 galactorrhoea/ gynaecomastia ‐ long term | 1 | 446 | Risk Ratio (M‐H, Random, 95% CI) | 0.47 [0.14, 1.65] |
| 17.6 increased sexual desire ‐ short term | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 1.06 [0.22, 5.01] |
| 17.7 orgastic dysfunction ‐ short term | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 5.28 [0.26, 107.51] |
| 18 Other adverse events: 10. Others | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 18.1 failing memory ‐ short term | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 1.27 [0.60, 2.68] |
| 18.2 incontinence nocturia ‐ long term | 1 | 446 | Risk Ratio (M‐H, Random, 95% CI) | 0.43 [0.16, 1.15] |
| 18.3 suicide attempt ‐ long term | 1 | 446 | Risk Ratio (M‐H, Random, 95% CI) | 0.71 [0.04, 11.26] |
| 18.4 sweating ‐ short term | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | 0.96 [0.45, 2.07] |
| 18.5 urinary hesitancy, dry mouth, constipation ‐ long term | 1 | 446 | Risk Ratio (M‐H, Random, 95% CI) | 1.09 [0.76, 1.58] |
| 19 Other adverse events: 11. any serious adverse event | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 19.1 long term | 1 | 446 | Risk Ratio (M‐H, Random, 95% CI) | 1.08 [0.63, 1.87] |
| 20 Other adverse events: 12. lab data | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 20.1 mean weight change (lb) ‐ long term | 1 | 602 | Mean Difference (IV, Random, 95% CI) | ‐2.8 [‐5.58, ‐0.02] |
| 20.2 change in blood glucose ‐ long term | 1 | 602 | Mean Difference (IV, Random, 95% CI) | ‐1.5 [‐7.04, 4.04] |
| 20.3 change in glycosylated haemoglobin ‐ long term | 1 | 602 | Mean Difference (IV, Random, 95% CI) | 0.02 [‐0.12, 0.16] |
| 20.4 change in cholesterol ‐ long term | 1 | 602 | Mean Difference (IV, Random, 95% CI) | 2.6 [‐3.25, 8.45] |
| 20.5 change in triglycerides ‐ long term | 1 | 602 | Mean Difference (IV, Random, 95% CI) | 10.9 [‐14.80, 36.60] |
| 20.6 change in prolactin ‐ long term | 1 | 602 | Mean Difference (IV, Random, 95% CI) | ‐15.0 [‐19.44, ‐10.56] |
Comparison 24. PERPHENAZINE vs SULPIRIDE.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Leaving the study early: 1. Any reason | 2 | 111 | Risk Ratio (M‐H, Random, 95% CI) | 1.40 [0.62, 3.20] |
| 1.1 short term | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 0.8 [0.24, 2.71] |
| 1.2 medium term | 1 | 47 | Risk Ratio (M‐H, Random, 95% CI) | 1.92 [0.86, 4.25] |
| 2 Leaving the study early: 2. Due to adverse events | 2 | 111 | Risk Ratio (M‐H, Random, 95% CI) | 2.94 [0.32, 27.41] |
| 2.1 short term | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.13, 71.00] |
| 2.2 medium term | 1 | 47 | Risk Ratio (M‐H, Random, 95% CI) | 2.88 [0.12, 67.29] |
| 3 Leaving the study early: 3. Due to relapse / worsening or no improvement | 2 | 111 | Risk Ratio (M‐H, Random, 95% CI) | 1.26 [0.47, 3.39] |
| 3.1 short term | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.13, 71.00] |
| 3.2 medium term | 1 | 47 | Risk Ratio (M‐H, Random, 95% CI) | 1.15 [0.41, 3.25] |
| 4 Global state: 1. Change over time ‐ no better or deterioration (ITT) | 2 | 111 | Risk Ratio (M‐H, Random, 95% CI) | 0.96 [0.65, 1.43] |
| 4.1 short term | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 0.8 [0.52, 1.24] |
| 4.2 medium term | 1 | 47 | Risk Ratio (M‐H, Random, 95% CI) | 1.20 [0.73, 1.97] |
| 5 Mental state: 1. State ‐ BPRS end score (high = poor, skewed data) | Other data | No numeric data | ||
| 5.1 medium term | Other data | No numeric data | ||
| 6 Adverse events: Movement disorders | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 6.1 akathisia ‐ short term | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 0.75 [0.18, 3.09] |
| 6.2 akathisia ‐ medium term | 1 | 47 | Risk Ratio (M‐H, Random, 95% CI) | 2.88 [0.64, 12.82] |
| 6.3 ataxia ‐ short term | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.07, 15.30] |
| 6.4 dyskinesia ‐ medium term | 1 | 47 | Risk Ratio (M‐H, Random, 95% CI) | 0.48 [0.05, 4.93] |
| 6.5 rigidity ‐ short term | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 0.25 [0.06, 1.09] |
| 6.6 rigidity ‐ medium term | 1 | 47 | Risk Ratio (M‐H, Random, 95% CI) | 0.64 [0.12, 3.48] |
| 6.7 tremor ‐ short term | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 0.64 [0.28, 1.43] |
| 6.8 tremor ‐ medium term | 1 | 47 | Risk Ratio (M‐H, Random, 95% CI) | 1.34 [0.50, 3.63] |
| 6.9 use of antiparkinsonian drugs ‐ medium term | 1 | 47 | Risk Ratio (M‐H, Random, 95% CI) | 0.14 [0.01, 2.52] |
| 7 Other adverse events: 1. Anticholinergic | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 7.1 blurred vision ‐ short term | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 7.89] |
| 7.2 dry mouth ‐ short term | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 5.0 [0.25, 100.20] |
| 7.3 dry mouth ‐ medium term | 1 | 47 | Risk Ratio (M‐H, Random, 95% CI) | 1.92 [0.19, 19.73] |
| 7.4 increased salivation ‐ short term | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 5.0 [0.25, 100.20] |
| 8 Other adverse events: 2. Arousal | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 8.1 excitation ‐ short term | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.07, 1.53] |
| 8.2 excitation ‐ medium term | 1 | 47 | Risk Ratio (M‐H, Random, 95% CI) | 1.28 [0.32, 5.10] |
| 8.3 insomnia ‐ short term | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 1.29 [0.55, 3.03] |
| 8.4 insomnia ‐ medium term | 1 | 47 | Risk Ratio (M‐H, Random, 95% CI) | 0.24 [0.03, 1.99] |
| 8.5 need of additional benzodiazepines ‐ medium term | 1 | 47 | Risk Ratio (M‐H, Random, 95% CI) | 1.60 [0.43, 5.93] |
| 9 Other adverse events: 3. At least one | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 9.1 short term | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 0.76 [0.50, 1.17] |
| 10 Other adverse events: 4. Central nervous system | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 10.1 headache ‐ short term | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 0.5 [0.05, 5.24] |
| 10.2 paraesthesia ‐ short term | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 5.0 [0.25, 100.20] |
| 11 Other adverse events: 5. Endocrine | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 11.1 amenorrhoea ‐ medium term | 1 | 47 | Risk Ratio (M‐H, Random, 95% CI) | 0.32 [0.04, 2.85] |
| 11.2 lactation ‐ medium term | 1 | 47 | Risk Ratio (M‐H, Random, 95% CI) | 0.19 [0.01, 3.80] |
| 12 Other adverse events: 6. Gastrointestinal | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 12.1 constipation ‐ short term | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 0.5 [0.05, 5.24] |
| 12.2 loss of appetite ‐ short term | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 0.8 [0.24, 2.71] |
| 12.3 nausea and vomiting ‐ short term | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 0.4 [0.08, 1.91] |
| 12.4 nausea and vomiting ‐ medium term | 1 | 47 | Risk Ratio (M‐H, Random, 95% CI) | 0.32 [0.04, 2.85] |
| 12.5 weight loss ‐ short term | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 11.00 [0.63, 191.04] |
| 12.6 weight gain ‐ short term | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 5.00 [0.62, 40.44] |
| 13 Other adverse events: 7. Skin | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 13.1 rash ‐ short term | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 7.89] |
| 14 Other adverse events: 8. Others | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 14.1 abdominal pain ‐ medium term | 1 | 47 | Risk Ratio (M‐H, Random, 95% CI) | 0.14 [0.01, 2.52] |
| 14.2 sweating ‐ short term | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 0.2 [0.01, 4.01] |
| 15 Other adverse events: 9. lab data | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 15.1 decrease in white blood cell count and haemoglobin level ‐ short term | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.07, 15.30] |
| 15.2 increase in white blood cell count and haemoglobin level ‐ short term | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 0.5 [0.10, 2.54] |
| 15.3 rise in alkaline phosphate and GOT levels ‐ short term | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.13, 71.00] |
Comparison 25. PERPHENAZINE vs THIOPROPRAZATE.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Leaving the study early: 1. Any reason | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 short term | 1 | 106 | Risk Ratio (M‐H, Random, 95% CI) | 0.73 [0.43, 1.22] |
| 2 Leaving the study early: 2. Due to adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 short term | 1 | 106 | Risk Ratio (M‐H, Random, 95% CI) | 2.0 [0.19, 21.40] |
| 3 Adverse events: Movement disorders | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 akathisia ‐ short term | 1 | 106 | Risk Ratio (M‐H, Random, 95% CI) | 7.0 [0.89, 54.94] |
| 3.2 akinesia ‐ short term | 1 | 106 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.3 dyskinesia ‐ short term | 1 | 106 | Risk Ratio (M‐H, Random, 95% CI) | 5.0 [0.25, 101.73] |
| 3.4 parkinsonism ‐ short term | 1 | 106 | Risk Ratio (M‐H, Random, 95% CI) | 2.67 [0.75, 9.51] |
Comparison 26. PERPHENAZINE vs THIORIDAZINE.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Leaving the study early: 1. Any reason | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 short term | 1 | 106 | Risk Ratio (M‐H, Random, 95% CI) | 0.73 [0.43, 1.22] |
| 2 Leaving the study early: 2. Due to adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 short term | 1 | 106 | Risk Ratio (M‐H, Random, 95% CI) | 2.0 [0.19, 21.40] |
| 3 Adverse events: Movement disorders | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 akathisia ‐ short term | 1 | 106 | Risk Ratio (M‐H, Random, 95% CI) | 15.0 [0.88, 256.16] |
| 3.2 akinesia ‐ short term | 1 | 106 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.3 dyskinesia ‐ short term | 1 | 106 | Risk Ratio (M‐H, Random, 95% CI) | 5.0 [0.25, 101.73] |
| 3.4 parkinsonism ‐ short term | 1 | 106 | Risk Ratio (M‐H, Random, 95% CI) | 2.67 [0.75, 9.51] |
Comparison 27. PERPHENAZINE vs THIOTHIXENE.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Leaving the study early: 1. Any reason | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 short term | 1 | 86 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.06, 15.48] |
| 2 Global state: 1. Change over time ‐ no better or deterioration (ITT) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 short term | 1 | 86 | Risk Ratio (M‐H, Random, 95% CI) | 1.30 [0.93, 1.83] |
| 3 Behaviour: 1. Behaviour subscale of the Psychiatric Rating Scale: no better or worse | 1 | 86 | Risk Ratio (M‐H, Random, 95% CI) | 1.18 [0.90, 1.55] |
| 4 Adverse events: Movement disorders | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 akathisia ‐ short term | 1 | 86 | Risk Ratio (M‐H, Random, 95% CI) | 0.58 [0.25, 1.34] |
| 4.2 ataxia ‐ short term | 1 | 86 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.21, 4.68] |
| 4.3 dyskinesia ‐ short term | 1 | 86 | Risk Ratio (M‐H, Random, 95% CI) | 1.5 [0.26, 8.53] |
| 4.4 parkinsonism ‐ short term | 1 | 86 | Risk Ratio (M‐H, Random, 95% CI) | 0.8 [0.48, 1.32] |
| 4.5 use of antiparkinsonian drugs ‐ short term | 1 | 86 | Risk Ratio (M‐H, Random, 95% CI) | 1.18 [0.81, 1.73] |
| 5 Other adverse events: 1. Anticholinergic | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 blurred vision ‐ short term | 1 | 86 | Risk Ratio (M‐H, Random, 95% CI) | 0.29 [0.06, 1.30] |
| 5.2 dry mouth ‐ short term | 1 | 86 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.38, 2.61] |
| 6 Other adverse events: 2. Arousal | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 6.1 insomnia ‐ short term | 1 | 86 | Risk Ratio (M‐H, Random, 95% CI) | 0.71 [0.39, 1.29] |
| 6.2 need of additional benzodiazepines ‐ short term | 1 | 86 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.21, 4.68] |
| 6.3 sleepiness/sedation ‐ short term | 1 | 86 | Risk Ratio (M‐H, Random, 95% CI) | 0.9 [0.41, 1.99] |
| 7 Other adverse events: 3. At least one | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 7.1 short term | 1 | 86 | Risk Ratio (M‐H, Random, 95% CI) | 0.90 [0.68, 1.20] |
| 8 Other adverse events: 4. Cardiovascular | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 8.1 faintness, dizziness, weakness ‐ short term | 1 | 86 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 9 Other adverse events: 5. Central nervous system | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 9.1 disturbance of consciousness ‐ short term | 1 | 86 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 9.2 headache ‐ short term | 1 | 86 | Risk Ratio (M‐H, Random, 95% CI) | 2.0 [0.19, 21.24] |
| 9.3 paraesthesia ‐ short term | 1 | 86 | Risk Ratio (M‐H, Random, 95% CI) | 5.0 [0.25, 101.18] |
| 10 Other adverse events: 6. Gastrointestinal | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 10.1 constipation ‐ short term | 1 | 86 | Risk Ratio (M‐H, Random, 95% CI) | 1.2 [0.40, 3.64] |
| 10.2 icterus ‐ short term | 1 | 86 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 7.96] |
| 10.3 loss of appetite ‐ short term | 1 | 86 | Risk Ratio (M‐H, Random, 95% CI) | 0.88 [0.35, 2.20] |
| 10.4 nausea and vomiting ‐ short term | 1 | 86 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 11 Other adverse events: 7. Skin | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 11.1 rash ‐ short term | 1 | 86 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.13, 71.65] |
| 12 Other adverse events: 8. Genitourinary | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 12.1 micturition disturbances ‐ short term | 1 | 86 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 13 Other adverse events: 9. Others | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 13.1 cramps ‐ short term | 1 | 86 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 7.96] |
| 13.2 sweating ‐ short term | 1 | 86 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.06, 15.48] |
Comparison 28. PERPHENAZINE vs TIMIPERONE.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Leaving the study early: 1. Any reason | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 short term | 1 | 198 | Risk Ratio (M‐H, Random, 95% CI) | 1.33 [0.67, 2.67] |
| 2 Leaving the study early: 2. Due to adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 short term | 1 | 198 | Risk Ratio (M‐H, Random, 95% CI) | 1.33 [0.67, 2.67] |
| 3 Global state: 1. Change over time ‐ no better or deterioration (ITT) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 short term | 1 | 198 | Risk Ratio (M‐H, Random, 95% CI) | 1.11 [0.79, 1.55] |
| 4 Adverse events: Movement disorders | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 akathisia ‐ short term | 1 | 198 | Risk Ratio (M‐H, Random, 95% CI) | 0.85 [0.56, 1.29] |
| 4.2 dyskinesia ‐ short term | 1 | 198 | Risk Ratio (M‐H, Random, 95% CI) | 0.36 [0.13, 0.95] |
| 4.3 dystonia ‐ short term | 1 | 198 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 8.08] |
| 4.4 parkinsonism ‐ short term | 1 | 198 | Risk Ratio (M‐H, Random, 95% CI) | 1.21 [0.84, 1.73] |
| 5 Other adverse events: 1. Anticholinergic | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 blurred vision ‐ short term | 1 | 198 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.12, 72.76] |
| 6 Other adverse events: 2. Arousal | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 6.1 drowsiness ‐ short term | 1 | 198 | Risk Ratio (M‐H, Random, 95% CI) | 1.43 [0.57, 3.60] |
| 6.2 excitation ‐ short term | 1 | 198 | Risk Ratio (M‐H, Random, 95% CI) | 4.75 [1.68, 13.46] |
| 6.3 insomnia ‐ short term | 1 | 198 | Risk Ratio (M‐H, Random, 95% CI) | 1.31 [0.94, 1.82] |
| 7 Other adverse events: 3. At least one | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 7.1 short term | 1 | 198 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.89, 1.16] |
| 8 Other adverse events: 4. Cardiovascular | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 8.1 faintness, dizziness, weakness ‐ short term | 1 | 198 | Risk Ratio (M‐H, Random, 95% CI) | 1.5 [0.71, 3.18] |
| 8.2 lassitude ‐ short term | 1 | 198 | Risk Ratio (M‐H, Random, 95% CI) | 1.07 [0.55, 2.10] |
| 9 Other adverse events: 5. Central nervous system | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 9.1 confusion ‐ short term | 1 | 198 | Risk Ratio (M‐H, Random, 95% CI) | 0.67 [0.11, 3.90] |
| 9.2 disturbance of consciousness ‐ short term | 1 | 198 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.06, 15.76] |
| 9.3 dysarticulation ‐ short term | 1 | 198 | Risk Ratio (M‐H, Random, 95% CI) | 0.83 [0.38, 1.84] |
| 9.4 headache ‐ short term | 1 | 198 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.09, 1.19] |
| 9.5 seizure ‐ short term | 1 | 198 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 29. PERPHENAZINE vs TRIFLUOPERAZINE.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Leaving the study early: 1. Any reason | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 short term | 1 | 105 | Risk Ratio (M‐H, Random, 95% CI) | 0.63 [0.38, 1.03] |
| 2 Leaving the study early: 2. Due to adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 short term | 1 | 105 | Risk Ratio (M‐H, Random, 95% CI) | 0.65 [0.11, 3.76] |
| 3 Adverse events: Movement disorders | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 akathisia ‐ short term | 1 | 105 | Risk Ratio (M‐H, Random, 95% CI) | 2.29 [0.63, 8.38] |
| 3.2 akinesia ‐ short term | 1 | 105 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 7.85] |
| 3.3 dyskinesia ‐ short term | 1 | 105 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.14, 6.71] |
| 3.4 parkinsonism ‐ short term | 1 | 105 | Risk Ratio (M‐H, Random, 95% CI) | 2.62 [0.73, 9.32] |
Comparison 30. PERPHENAZINE vs TRIFLUOPROMAZINE.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Leaving the study early: 1. Any reason | 3 | 178 | Risk Ratio (M‐H, Random, 95% CI) | 1.14 [0.66, 1.98] |
| 1.1 short term | 3 | 178 | Risk Ratio (M‐H, Random, 95% CI) | 1.14 [0.66, 1.98] |
| 2 Leaving the study early: 2. Due to adverse events | 2 | 168 | Risk Ratio (M‐H, Random, 95% CI) | 2.00 [0.74, 5.39] |
| 2.1 short term | 2 | 168 | Risk Ratio (M‐H, Random, 95% CI) | 2.00 [0.74, 5.39] |
| 3 Global state: 1. Change over time ‐ no better or deterioration (ITT) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 short term | 1 | 62 | Risk Ratio (M‐H, Random, 95% CI) | 0.71 [0.25, 2.01] |
| 4 Adverse events: Movement disorders | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 akathisia ‐ short term | 1 | 106 | Risk Ratio (M‐H, Random, 95% CI) | 7.0 [0.89, 54.94] |
| 4.2 akinesia ‐ short term | 1 | 106 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 4.3 dyskinesia ‐ short term | 1 | 106 | Risk Ratio (M‐H, Random, 95% CI) | 5.0 [0.25, 101.73] |
| 4.4 parkinsonism ‐ short term | 1 | 106 | Risk Ratio (M‐H, Random, 95% CI) | 17.0 [1.01, 287.24] |
| 5 Other adverse events: 1. Central nervous system | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 electroencephalogram (increase) ‐ short term | 1 | 10 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.08, 11.93] |
Comparison 31. PERPHENAZINE vs ZIPRASIDONE.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Leaving the study early: 1. Any reason | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 long term | 1 | 446 | Risk Ratio (M‐H, Random, 95% CI) | 0.95 [0.85, 1.05] |
| 2 Leaving the study early: 2. Due to adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 long term | 1 | 446 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.65, 1.58] |
| 3 Leaving the study early: 3. Due to relapse / worsening or no improvement | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 long term | 1 | 446 | Risk Ratio (M‐H, Random, 95% CI) | 1.05 [0.75, 1.46] |
| 4 Adverse events: Movement disorders (completer‐only data) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 akathisia ‐ long term | 1 | 399 | Risk Ratio (M‐H, Random, 95% CI) | 0.75 [0.38, 1.49] |
| 4.2 EPS ‐ long term | 1 | 395 | Risk Ratio (M‐H, Random, 95% CI) | 1.56 [0.62, 3.94] |
| 4.3 tardive dyskinesia ‐ long term | 1 | 363 | Risk Ratio (M‐H, Random, 95% CI) | 1.21 [0.73, 2.02] |
| 5 Other adverse events: 1. Arousal | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 insomnia ‐ long term | 1 | 446 | Risk Ratio (M‐H, Random, 95% CI) | 0.84 [0.62, 1.13] |
| 5.2 sleepiness/ sedation ‐ long term | 1 | 446 | Risk Ratio (M‐H, Random, 95% CI) | 1.17 [0.85, 1.60] |
| 6 Other adverse events: 2. Cardiovascular | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 6.1 faintness, dizziness, weakness ‐ long term | 1 | 446 | Risk Ratio (M‐H, Random, 95% CI) | 0.86 [0.52, 1.42] |
| 7 Other adverse events: 3. Gastrointestinal | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 7.1 weight gain ‐ long term | 1 | 404 | Risk Ratio (M‐H, Random, 95% CI) | 1.60 [0.84, 3.04] |
| 8 Other adverse events: 4. Genitourinary | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 8.1 diminished sexual desire ‐ long term | 1 | 446 | Risk Ratio (M‐H, Random, 95% CI) | 1.30 [0.90, 1.87] |
| 8.2 galactorrhea/ gynaecomastia ‐ long term | 1 | 446 | Risk Ratio (M‐H, Random, 95% CI) | 0.47 [0.14, 1.65] |
| 9 Other adverse events: 5. Others | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 9.1 urinary hesitancy, dry mouth, constipation ‐ long term | 1 | 446 | Risk Ratio (M‐H, Random, 95% CI) | 1.09 [0.76, 1.58] |
| 9.2 suicide attempt ‐ long term | 1 | 446 | Risk Ratio (M‐H, Random, 95% CI) | 0.71 [0.04, 11.26] |
| 9.3 incontinence nocturia ‐ long term | 1 | 446 | Risk Ratio (M‐H, Random, 95% CI) | 0.43 [0.16, 1.15] |
| 10 Other adverse events: 6. any serious adverse event | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 10.1 long term | 1 | 446 | Risk Ratio (M‐H, Random, 95% CI) | 1.08 [0.63, 1.87] |
| 11 Other adverse events: 7. lab data | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 11.1 mean weight change (lb) ‐ long term | 1 | 446 | Mean Difference (IV, Random, 95% CI) | ‐0.40 [‐3.45, 2.65] |
| 11.2 change in blood glucose ‐ long term | 1 | 446 | Mean Difference (IV, Random, 95% CI) | 2.90 [‐5.69, 11.49] |
| 11.3 change in glycosylated haemoglobin ‐ long term | 1 | 446 | Mean Difference (IV, Random, 95% CI) | 0.20 [‐0.10, 0.50] |
| 11.4 change in cholesterol ‐ long term | 1 | 446 | Mean Difference (IV, Random, 95% CI) | 9.7 [‐1.44, 20.84] |
| 11.5 change in triglycerides ‐ long term | 1 | 446 | Mean Difference (IV, Random, 95% CI) | 26.40 [‐2.71, 55.51] |
| 11.6 change in prolactin ‐ long term | 1 | 446 | Mean Difference (IV, Random, 95% CI) | 4.9 [0.33, 9.47] |
Comparison 32. PERPHENAZINE vs ZOTEPINE.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Leaving the study early: 1. Any reason | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 short term | 1 | 95 | Risk Ratio (M‐H, Random, 95% CI) | 1.48 [0.63, 3.48] |
| 2 Leaving the study early: 2. Due to relapse / worsening or no improvement | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 short term | 1 | 95 | Risk Ratio (M‐H, Random, 95% CI) | 1.34 [0.56, 3.23] |
| 3 Global state: 1. Change over time ‐ no better or deterioration (ITT) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 short term | 1 | 95 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.65, 1.47] |
| 4 Other adverse events: 1. Cardiovascular | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 tachycardia ‐ short term | 1 | 95 | Risk Ratio (M‐H, Random, 95% CI) | 0.31 [0.01, 7.50] |
Comparison 33. PERPHENAZINE vs ZUCLOPENTHIXOL.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Leaving the study early: 1. Any reason | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 short term | 1 | 54 | Risk Ratio (M‐H, Random, 95% CI) | 1.8 [0.69, 4.67] |
| 2 Global state: 1. Change over time ‐ no better or deterioration (ITT) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 short term | 1 | 54 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.07, 15.18] |
| 3 Adverse events: Movement disorders | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 akathisia ‐ short term | 1 | 54 | Risk Ratio (M‐H, Random, 95% CI) | 1.14 [0.48, 2.71] |
| 3.2 akinesia ‐ short term | 1 | 54 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.07, 1.51] |
| 3.3 rigidity ‐ short term | 1 | 54 | Risk Ratio (M‐H, Random, 95% CI) | 2.2 [0.88, 5.48] |
| 3.4 tremor ‐ short term | 1 | 54 | Risk Ratio (M‐H, Random, 95% CI) | 2.2 [0.88, 5.48] |
| 4 Other adverse events: 1. Arousal | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 excitation ‐ short term | 1 | 54 | Risk Ratio (M‐H, Random, 95% CI) | 1.29 [0.56, 2.95] |
| 4.2 insomnia ‐ short term | 1 | 54 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.07, 1.51] |
| 4.3 sleepiness/sedation ‐ short term | 1 | 54 | Risk Ratio (M‐H, Random, 95% CI) | 1.71 [0.80, 3.68] |
| 5 Other adverse events: 2. Cardiovascular | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 faintness, dizziness, weakness ‐ short term | 1 | 54 | Risk Ratio (M‐H, Random, 95% CI) | 1.5 [0.48, 4.72] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Amakusa 1973.
| Methods | Allocation: randomised. Blindness: double. Duration: 6 weeks (preceded by 7 days for decreasing previous drugs and several days wash‐out). Setting: multi‐centre, Japan. | |
| Participants | Diagnosis: chronic schizophrenia (51 hebephrenic, 5 catatonic, 2 delusional type, 6 not otherwise specified).
N = 64 (32 matched‐pairs).
Age: 18‐55 years.
Sex: 20 F, 44 M.
History: chronic, inpatient, ill from 2 months to 30 years. Included: not stated. Excluded: no details. Consent: not stated. |
|
| Interventions | 1. Perphenazine: 4 x 3 mg tablets/day for the first week (12 mg), 6 tablets/day for the second week (18 mg), then increased or decreased according to judgements of each physician in the third week; maximum 10 tablets, N = 32.
2. Sulpiride: 4 x 100 mg tablets/day for the first week (400 mg), 6 tablets/day for the second week (600 mg), then increased or decreased according to judgements of each physician in the third week; maximum 10 tablets, N = 32. Antiparkinsonian and sedative medication was given if needed (including trihexyphenidyl, promethazine, pentobarbital, amobarbital and bromovalerylurea). |
|
| Outcomes | Global state: no better or worse (Global Comparative Judgement (GCJ); General improvement rating (GIR)). Leaving the study early (any reason; adverse effects; relapse/worsening). Adverse events (gastrointestinal; movement disorder; anticholinergic; arousal; CNS; at least one adverse effect; general; lab data). Unable to use ‐ Mental state: psychiatric evaluating scale (PES) (developed by three universities ‐ unpublished scale). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomised – matched‐pair design. Quote: “two patients of the same sex, an age difference of less than 10 years and with similar symptoms were classified as a pair then randomly allocated to either intervention or control (p3). |
| Allocation concealment (selection bias) | Low risk | An independent controller randomly allocated the matched pairs to either Sulpiride or perphenazine. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Double blind – quote: “test and control tablets had similar external appearance… [the tablets] were placed in bags which were packed into cans of identical design and appearance” (p3‐4). An independent controller randomly allocated the matched pairs to either Sulpiride or perphenazine. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 14% (n = 9) excluded from analysis: n = 5 receiving sulpiride (n = 3 refused drugs; n = 1 given electro‐shock therapy; n = 1 given other drugs for marked excitation) and n = 4 receiving perphenazine (n = 1 discarding drugs; n = 1 refusing drugs; n = 1 due to acute colitis; and n = 1 given other drugs) (ITT used). |
| Selective reporting (reporting bias) | Unclear risk | None detected. |
| Other bias | Unclear risk | Funding: no details provided. Raters: not stated to be independent of treatment. |
Bennett 1961.
| Methods | Allocation: randomised. Blindness: unclear. Duration: 12 weeks, preceded by 2 weeks wash‐out. Setting: hospital, single‐centre. | |
| Participants | Diagnosis: all except two had chronic schizophrenia.
N = 30 (n = 25 relevant).
Age: median ˜ 37 years.
Sex: 30 M.
History: chronic; 'ill for more than two years'; median number of years hospitalised 2.6; Multidimensional Scale for Rating Psychiatric Patients (MSRPP) showed patients to be more 'perceptually confused, paranoid and mentally agitated than the usual male patients confined to Veterans Administration hospitals'; n = 22 were previously receiving phenothiazine derivatives without sustained improvement; n = 10 received courses of electric shock therapy; n = 7 had been given insulin coma therapy. Included: not stated. Excluded: history of central nervous system disease, seizures, lobotomy, somatic illnesses, which included duodenal ulcer, liver disease, blood dyscrasia, or those psychotic as a result of infection or toxic factors. Consent: not stated. |
|
| Interventions | 1. Perphenazine: 16 mg/day gradually increased until by the 5th week 48 mg/day, then until the 12th week 96 mg/day, N = 5. 2. Chlorpromazine: 200 mg/day gradually increased until by the 5th week 600 mg/day, then until the 12th week 1200 mg/day, N = 5. 3. Prochlorperazine: 25 mg/day gradually increased until by the 5th week 75 mg/day, then until the 12th week 150 mg/day, N = 5. 4. Triflupromazine: 50 mg/day gradually increased until by the 5th week 150 mg/day, then until the 12th week 300 mg/day, N = 5. 5. Mepazine: 50 mg/day gradually increased until by the 5th week 150 mg/day, then until the 12th week 300 mg/day, N = 5. [6. Phenobarbital: 32 mg/day gradually increased until by the 5th week 100 mg/day, then until the 12th week 200 mg/day, N = 5 ‐ not included in data and analysis, not an antipsychotic]. | |
| Outcomes | Leaving the study early. Adverse events: EEG results. Unable to use ‐ Global state: no better or worse (no data for individual groups). Mental state: MSRPP, CEPS (no data). Adverse events (no data). Laboratory tests (no data). Cognitive functioning: Porteus Maze (no data). |
|
| Notes | Prior to treatment, participants were given inert identical‐appearing placebo for 14 days; "judicious use of conventional short‐acting sedatives was allowed during first 4 weeks to control behaviour/ insomnia". | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised ‐ no further details. |
| Allocation concealment (selection bias) | Unclear risk | No details. |
| Blinding (performance bias and detection bias) All outcomes | High risk | Single‐blind (implied) ‐ all participants were given identical‐looking placebo 14 days prior to treatment; there is no mention of blinding at treatment commencing. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Follow‐up 90% ‐ n = 2 people left early (n = 1 from the chlorpromazine group; n = 1 from the prochlorperazine), no explanation provided; not clear which numbers are completed in full analysis and by which group. |
| Selective reporting (reporting bias) | High risk | Non‐reported data by group include: improvement; behaviour (MSRPP); mental state (CEPS); side effects; laboratory testing. |
| Other bias | Unclear risk | Funding: no details provided. Raters: not stated to be independent of treatment. |
CATIE 2005.
| Methods | Allocation: randomised. Blindness: double. Duration: 18 months. Setting: 57 clinical US sites (16 university clinics, 10 state mental health agencies, 7 Veterans Affairs medical centers, 6 private non‐profit agencies, 4 private‐practice sites, and 14 mixed‐system sites) January 2001 to December 2004. |
|
| Participants | Diagnosis: chronic schizophrenia (DSM‐IV). N = 1,493. Age: 40.6 ± 11.1 years old. Sex: F 380; M 1080. History: exacerbation in previous 3 months n = 402; mean age of first treatment for any behavioural or emotional problem = 24.0 ± 8.9 years old; years since first antipsychotic medication prescribed = 14.4 ± 10.7 years. Included: 18 to 65 years’ old; DSM‐IV schizophrenia diagnosis; adequate capacity to consent; have a condition appropriate for treatment with oral medication. Exclusion: severe adverse reactions/ intolerance/ failure to respond to one of the treatments; diagnosis of schizoaffective disorder or mental retardation, pervasive developmental disorder, delirium, dementia, amnesia or other cognitive disorders; first episode schizophrenia; pregnant women/breastfeeding; past or current treatment with clozapine for treatment resistance; people currently stabilised on haloperidol decanoate or Fluphenazine decanoate who require long‐acting injections to maintain treatment adherence; people with tardive dyskinesia excluded from assignment to conventional treatment arm; people with cardiac conditions (myocardial infarction in past 6 months, QTc prolongation, cardiac arrhythmia, uncompensated congestive heart failure, heart block [PR interval > 0.22 seconds]). |
|
| Interventions | 1. Perphenazine (8 mg oral tablets), mean modal dose 20.8 mg/ day, n = 261. 2. Olanzapine (7.5 mg oral tablets), mean modal dose 20.1 mg/day, n = 336. 3. Quetiapine (200 mg oral tablets), mean modal dose 543.4 mg/day, n = 337. 4. Risperidone (1.5 mg oral tablets), mean modal dose 3.9 mg/day, n = 341. 5. Ziprasidone (40 mg oral tablets), mean modal dose 112.8 mg/day, n = 185. First four weeks after randomisation allowed overlap in administration of antipsychotic medication that participants received prior to study‐entry to allow gradual transition to study medication. Concomitant medication permitted throughout study, except for additional antipsychotic agents. |
|
| Outcomes | Primary outcome: all‐cause treatment discontinuation Secondary outcomes: reasons for discontinuation; weight gain; EPS; sedation; prolactin; (as judged by study doctor). PANSS; CGI; AIMS; Barnes akathisia rating; adverse events. |
|
| Notes | We contacted the authors of this study for more information, which was provided, and we were able to include the above data. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomised: no further details – emailed authors 18/11/13 – Jeffrey Lieberman (jlieberman@columbia.edu). Author reply (unpublished information) “[r]andom assignments were computer‐generated. Site investigators called‐in to an interactive voice response system to get a participant's treatment assignment.” The randomisation scheme prevented people with current tardive dyskinesia to receive perphenazine. |
| Allocation concealment (selection bias) | Unclear risk | As above. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Double‐blind: maintained by prescribing identical‐looking capsules of each medication. Nonequivalent choice of dosing regimen and flexible dose raised question of bias to jeopardise integrity of maintaining blind and influencing results; a further study found no bias or effect on results (Rosenheck 2009). Quote, “[b]ecause of product labelling, quetiapine and ziprasidone are given twice daily and olanzapine, perphenazine, and Risperidone once daily. To protect blinding, half the patients randomly assigned to perphenazine, olanzapine and Risperidone were assigned to twice‐daily dosing and half to once‐daily dosing” (p1211). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Out of N = 1432, n = 371 (26%) completed phase 1 of CATIE. Randomised participants who received at least one dose of study medication were included in ITT analysis (n = 1432). Quote, “[s]eventy‐four percent of patients in the intention‐to‐treat analysis (1,061 of 1,432) discontinued their assigned treatment in phase 1 before 18 months (median, 6).” (p1212). Quote, “all data from one site (33 patients) were excluded before analysis, owing to concern about the integrity of data from that site before the end of the study and before unblinding” (p1212). |
| Selective reporting (reporting bias) | Low risk | None detected. |
| Other bias | Unclear risk | Funding: study sponsored by grant (N01 MH90001) from the NIMH and foundation of Hope and Raleigh, N.C.. Study medications were provided by: AstraZeneca Pharmaceuticals, Bristol‐Myers Squibb, Forest Pharmaceuticals, Janssen Pharmaceutica, Eli Lilly, Otsuka Pharmaceutical, Pfizer, Zenith Goldline Pharmaceuticals, Schering‐Plough, and Novartis. All authors report having received research or consultant/advisory board fees from one or more of the following: Eli Lilly, Janssen, AstraZeneca, Bristol‐Myers Squibb, GlaxoSmith Kline, Pfizer, or Otsuka. Involvement of pharmaceutical companies stated to be limited: quote, “each provided advice on the dose of its own drug; they were otherwise not involved in the design of the study, analysis or interpretation of results” (p1212). Rating scales: raters not stated to be independent of treatment. Other: quote, “Two‐hundred thirty‐one patients with tardive dyskinesia were excluded from random assignment to perphenazine. ziprasidone was added to the trial after approximately 40% of the patients had been enrolled. Consequently, comparisons involving the perphenazine group were limited to patients without tardive dyskinesia, and comparisons involving the ziprasidone group were limited to the cohort of patients who underwent randomisation after ziprasidone was added” (p1211). |
Chouinard 1975.
| Methods | Allocation: randomised. Blindness: double. Duration: 12 weeks, preceded by 2 weeks fixed daily dose of 125 mg chlorpromazine. Setting: multi‐centre, Hôpital St‐Jean‐de‐Dieu, Montréal and Hôpital St‐Charles, Joliette (Canada). | |
| Participants | Diagnosis: chronic (ambulatory) schizophrenia.
N = 96 (n = 48 relevant).
Age: mean ˜ 42 ± 9.3 years.
Sex: 48 F, 48 M.
History: chronic, outpatient, length of previous hospitalisations ranging from 0.5 to 31.3 years. Included: 20 to 60 years old; primary hospital diagnosis of schizophrenia confirmed by research psychiatrist; presence of two or more of the following symptoms/ behaviours – thought or speech disturbance catatonic motor behaviour; paranoid ideation; hallucinations; delusional thinking other than paranoid; blunted or inappropriate affect; disturbance of social behaviour and interpersonal relations. Excluded: physical illness, childhood schizophrenia, chronic or acute brain syndrome, IQ below 70, alcoholism, epilepsy, drug addiction. Consent: all participants gave informed consent after purpose of study and possible side effects were explained. |
|
| Interventions | 1. Perphenazine: 20 mg/day, N = 24.
2. Placebo: 5 tablets a day, N = 24. [3. Amitriptyline‐perphenazine: 20 mg/day, N = 24 ‐ not included in data and analysis, not an antipsychotic]. [4. Amitriptyline hydrochloride: 125 mg/day, N = 24 ‐ not included in data and analysis, not an antipsychotic]. Additional medication: procyclidine hydrochloride for parkinsonism, chlorpromazine (orally in liquid form) for extreme agitation. |
|
| Outcomes | Leaving the study early: any reason.
Mental state: change over time ‐ relapse.
Adverse events: Abnormal laboratory results: SGOT, SGPT, alkaline phosphate, cholesterol, total serum bilirubin, fasting blood glucose. Unable to use ‐ Global state: CGI end score (no SD). Mental state: BPRS, IMPS (no SD). Laboratory tests (no data). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised ‐ quote, "within [groups of] 12 patients, individuals were randomly assigned to one of the four treatment drugs" (p1296). |
| Allocation concealment (selection bias) | Unclear risk | No details. |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind ‐ identical‐appearing tablets used. Each participant received one tablet three times per day and two at bedtime. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Of N = 96 entering the study, n = 16 did not complete the full 12‐week course because of, quote, "poor response to treatment...but all completed at least four weeks" (p1299), i.e. those that relapsed. For participants leaving the study early, they were rated before being excluded from the study, and these evaluations were used in the final analysis. |
| Selective reporting (reporting bias) | High risk | No (useable) data reported for CGI, BPRS, IMPS or laboratory tests (only P values, or df reported). |
| Other bias | Unclear risk | Funding: no details provided. Raters: not stated to be independent of treatment. |
Chouinard 1977.
| Methods | Allocation: randomised. Blindness: double. Duration: 12 weeks, preceded by 2 weeks fixed daily dose of 125 mg chlorpromazine. Setting: single‐centre, Hôpital St‐Jean‐de‐Dieu, Montréal and Hôpital St‐Charles, Joliette (Canada). | |
| Participants | Diagnosis: chronic schizophrenia.
N = 54.
Age: mean ˜ 46 years.
Sex: 27 F, 27 M.
History: all participants previously receiving antipsychotic medication (not specified); mean hospitalisations 13.35. Included: age 20 to 60 years; primary diagnosis of schizophrenia. Excluded: organic heart disease; physical illness/ chronic or acute brain syndrome; IQ lower than 70; alcoholism; epilepsy; drug addiction; electrolyte abnormalities. Consent: all participants gave informed consent after purpose of the study and possible side effects had been explained. |
|
| Interventions | 1. Perphenazine (Trilafan) 20mg/ day, n = 18.
2. Placebo: 5 tablets a day, n = 18. [3. Perphenazine 20 mg/day and amitriptyline 125 mg/day, n = 18 ‐ not included in data and analysis, combined with anti‐depressant]. |
|
| Outcomes | Leaving the study early.
Adverse events: use of antiparkinsonian medication.
Physical tests: ECG abnormalities (primary outcome). "Intensified psychopathological condition" ‐ resulting in hop. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Random ‐ no further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Double: ECG readings (primary outcome) taken by cardiologist unaware “as to factors under study” (p.952). Drugs were administered in identically‐appearing tablets. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Follow‐up ‐ N = 38 (70%), with equal amount of males (n = 19) and females (n = 19) remaining. N = 13 placebo participants were excluded from study because of intensified psychopathological condition resulting in hospitalisation or chlorpromazine use. N = 1 perphenazine and n = 2 perphenazine‐amitriptyline participants were hospitalised and excluded from study. |
| Selective reporting (reporting bias) | Low risk | None detected. |
| Other bias | Unclear risk | Funding: unclear. Raters: not stated to be independent of treatment. |
Collins 1967.
| Methods | Allocation: randomised. Blindness: unclear. Duration: 10 weeks, preceded by 2 weeks wash‐out . Setting: hospital, single‐centre. | |
| Participants | Diagnosis: chronic schizophrenia.
N = 87.
Age: mean ˜ 41 years.
Sex: all male.
History: chronic 'long‐stay' inpatient. Included: no details. Excluded: no details. Consent: not stated. |
|
| Interventions | 1. Perphenazine: 4 mg/day for five weeks, then 8 mg/day for another five weeks, n = 29.
2. Placebo: one tablet daily for the first five weeks, then two tablets daily for another five weeks, n = 29. [3. Perphenazine and amitriptyline: 4 mg/day and 25 mg/day for five weeks, then 8 mg/day and 50 mg/day for another five weeks, n = 29 ‐ not included in data and analysis, combined with anti‐depressant]. |
|
| Outcomes | Leaving the study early.
Behaviour: Wing‐Scale B ‐ social withdrawal, socially embarrassing behaviour (SD imputed from standard error).
Adverse events. Unable to use ‐ Global state: overall improvement; relative improvement (unusable data). Physical: body weight (no SD). Other side effects: nausea; dizziness; skin rash (no data reported). |
|
| Notes | *Night sedatives as needed, but no other medication administered. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Random – “allotted to treatment groups at random”. |
| Allocation concealment (selection bias) | Unclear risk | No details. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Double – quote, “neither nursing staff nor doctors concerned knew which tablets were which” (p1428). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No losses ‐ N = 29 participants in each treatment group completed the trial. |
| Selective reporting (reporting bias) | Low risk | Not all adverse effects reported in full; not all means and SD reported. |
| Other bias | Unclear risk | Funding: Messrs. Allen & Hanburys Ltd supplied matching tablets of Triptafen Forte, Fentazin and placebo. Raters: not stated to be independent of treatment. |
Dehnel 1968.
| Methods | Allocation: randomised. Blindness: double. Duration: 12 weeks. Setting: inpatient, US. |
|
| Participants | Diagnosis: schizophrenia (no further details). N = 50. Age: range 25 to 59 years; mean 42.8 years. Sex: all male. History: length of hospitalisation ranged from 2 months to 23 years; median 42 months. Prior to study, all but two of the 50 participants were taking one or more psychoactive drugs (mainly phenothiazines). Included: under 60 years. Excluded: significant physical illness (such as liver disease/ severe cardiovascular disease). Consent: not described. |
|
| Interventions | 1. Perphenazine: oral, 8 mg tablets, range 8 to 80 mg, n = 25. 2. Clopenthixol: oral, 25 mg tablets, range 25 to 250 mg, n = 25. *Initial dose was one tablet per day to be adjusted for each participant (up to maximum of 10 tablets). |
|
| Outcomes | Adverse events (checklist completed by investigator). Global rating of improvement (investigator judgement). Leaving the study early Unable to use ‐ BPRS (interviewer rating); PIP (nurses rating) (no means or SD reported). |
|
| Notes | *All participants placed on placebo for wash‐out period prior to 12‐week trial. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote, “Randomly divided into two groups” (p170) – no further details. |
| Allocation concealment (selection bias) | Unclear risk | Quote, “the investigator who cared for the patient and the other raters did not know which of the two agents any patient was receiving” (p170). |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Double: all medications, quote, “as well as the placebo were dispensed in tablets identical in appearance. Medications for each patient during the active drug period were supplied by the pharmacy in an individual bottle with only the patient’s name and study number appearing on the bottle label” (p170). Strict double blind conditions adhered to until the end of study. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Four participants left the study early (n = 2 from each treatment group); clopenthixol: n = 1 discontinued after 6 weeks due to side effects (severe hand and arm tremor), n = 1 discontinued due to worsening of psychiatric condition after 4 weeks. Perphenazine: n = 1 lost from the study when released from hospital against medical advice after first week; n = 1 discontinued due to worsening of psychiatric condition after 4 weeks. |
| Selective reporting (reporting bias) | Unclear risk | No means or standard deviation reported for scale data (BPRS or PIP). |
| Other bias | Unclear risk | Funding: Supported in part by grant No. MYP‐5106 from National Institute of Mental Health, US Public Health Service. Clopenthixol supplied by Ayerst Laboratories, NYC. Raters: All outcome measures taken by study investigator or nurse. Unclear whether they were independent of treatment. |
Eckmann 1984.
| Methods | Allocation: randomised. Blindness: unclear. Duration: 30 days. Setting: hospital, single‐centre (Germany). | |
| Participants | Diagnosis: acute paranoid‐hallucinatory schizophrenia (ICD 295.3).
N = 40.
Age: 20 ‐ 45 years, mean ˜ 34 years.
Sex: 16 F, 24 M.
History: acute, inpatient. Included: no details. Excluded: no details. Consent: not stated. |
|
| Interventions | 1. Perphenazine: 12 mg or 24 mg/day, n = 20.
2. Benperidol: 6 mg or 12 mg/day, (2 patients received 9mg/day from 4th‐6th day and one of them also from 19th‐30th day), n = 20. Additional medication: anticholinergics and sedating drugs (low potent neuroleptics, tranquillisers) as needed. |
|
| Outcomes | Leaving the study early.
Global state: no better or worse. Unable to use ‐ Mental state: BPRS end score (no SD), AMDP (no data). Behaviour: NOSIE end score (no SD). Adverse events: SAS (no data). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised ‐ quote: "randomisiert zugeteilt" (English: randomly allocated). No further details. |
| Allocation concealment (selection bias) | Unclear risk | Not described. |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double‐blind: Oral liquid medication. No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No details of people leaving early. |
| Selective reporting (reporting bias) | High risk | No means or standard deviations reported in results section. |
| Other bias | Unclear risk | Funding: no details. Raters: not stated to be independent of treatment. |
Fruensgaard 1978.
| Methods | Allocation: randomised. Blindness: double. Duration: 3 weeks (for acute) and 12 weeks (for chronic). Setting: multi‐centre, Denmark. | |
| Participants | Diagnosis: (group I) acute schizophrenia and “psychogenic (reactive) psychosis”, n = 22; (group II) chronic schizophrenia, n = 25.
N = 47.
Age: mean ˜ 37 years.
Sex: 15 F, 32 M.
History: acute and chronic. Included: no details. Excluded: known hypersensitivity to dibenzoxazepine compounds or butyrophenones, manic‐depressive illness, electro‐convulsive therapy or insulin coma or sub‐coma therapy within the preceding 8 weeks, alcoholism or drug dependence as a significant feature of clinical history, serious impairment of renal or hepatic function, a history of increased intra‐ocular pressure or history of narrow‐angle glaucoma or urinary retention, cardiovascular or metabolic disease (severe cases), pregnancy. Consent: not stated. |
|
| Interventions | 1. Perphenazine: 8 mg per capsule, initially 2 capsules daily, could be increased to a maximum of 15 capsules daily according to the therapeutic efficacy and side effects, n = 24*.
2. Loxapine: 10 mg per capsule, initially 2 capsules daily, could be increased to a maximum of 15 capsules daily according to the therapeutic efficacy and side effects, n = 23**. Additional medication: chloralodol for severe insomnia, antiparkinsonian drugs (orphenadrine and/or biperiden) as necessary. |
|
| Outcomes | Leaving the study early.
Global state: CGI – dichotomised data, no better or deterioration.
Adverse events. Laboratory tests (no data). Unable to use ‐ Mental state: BPRS end score (no mean or SD). Behaviour: NOSIE end score (no mean or SD). Laboratory tests (not all data reported). |
|
| Notes | *Perphenazine mean dose for participants with acute schizophrenia = 34 mg after three weeks of treatment; mean dose for participants with chronic schizophrenia = 90.1 mg after two months of treatment. **Loxapine mean dose for participants with acute schizophrenia = 54.4 mg after three weeks of treatment; mean dose for participants with chronic schizophrenia = 81.1 mg after two months of treatment. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | “Randomly assigned” – no further details. |
| Allocation concealment (selection bias) | Unclear risk | Not stated. |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double‐blind stated – no further details. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 21% (n = 10) dropped‐out of the study and excluded from analysis – n = 5 from perphenazine group and n = 5 for loxapine group (reasons only provided for n = 2 ‘inadequate therapeutic response, and n = 2 aversion to continued treatment). Not fully addressed (ITT used). |
| Selective reporting (reporting bias) | High risk | Not all adverse event data reported; no means or SDs reported for BPRS, CGI, NOSIE. |
| Other bias | Unclear risk | Funding: no details. Raters: not stated to be independent of treatment. |
Hanlon 1964.
| Methods | Allocation: randomised. Blindness: double. Duration: 12 weeks, preceded by 2 weeks wash‐out. Setting: multi‐centre. | |
| Participants | Diagnosis: 115 anergic, withdrawn psychotic patients of which at least 99 with schizophrenia.
N = 115 (n = 58 relevant).
Age: range 21 to 74 years, mean 54±10.5 years.
Sex: 115 F.
History: inpatient. Included: no details. Excluded: over 75 years of age, diagnosed with one of the chronic brain disorders, manifested complicated physical condition. Consent: not stated. |
|
| Interventions | 1. Perphenazine: 4 mg three times daily for two weeks, then 8 mg three times daily for 10 weeks, N = 29. 2. Placebo: one tablet three times daily for two weeks, then two tablets three times daily for 10 weeks, N = 29. [3. Amitriptyline: 25 mg three times daily for two weeks, then 50 mg three times daily for 10 weeks, N = 29.] [4. Amitriptyline ‐ perphenazine: 25 mg and 4 mg three times daily for two weeks, then 50 mg and 8 mg three times daily for 10 weeks, N = 28.] | |
| Outcomes | Leaving the study early – any reason/ adverse events.
Adverse events: hypotension; dry mouth; nausea; body pains; dizziness; drowsiness; insomnia; weakness; oedema; tremors; tenseness; agitation; severe regression. Unable to use ‐ Mental state: IMPS, PRP (no SD/P value). Behaviour: MACC (no SD/P value). Laboratory tests (no data). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised – no further details. |
| Allocation concealment (selection bias) | Unclear risk | Not stated. Quote: “ultimate medical responsibility for the patients remained in the hands of the ward physicians, who were aware of the design and study‐drugs” (p53). |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Stated double blind; quote, “medication, administered in a double blind fashion, was dispensed to the wards from the pharmacy in containers identified only by the patient name and number” (p53). However, see above (allocation concealment). |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 11% (n = 13) dropped‐out of the study. Quote, "n = 5 placebo patients were discontinued and transferred to the infirmary for medical reasons unrelated to treatment; the remaining three patients were described as either overactive, restless or agitated. One perphenazine patient was dropped because of hypotension and elevated temperature, and another because of nausea and weakness" (p55). Another n = 5 participants were lost in the two other treatment arms. |
| Selective reporting (reporting bias) | High risk | No SD or P values reported for scale data. |
| Other bias | High risk | Funding: supported in part by Merck Sharp and Dohme Research Laboratories, West Point, Philadelphia (US). Raters: not independent of treatment. |
Hanlon 1965.
| Methods | Allocation: randomised. Blindness: double. Duration: 30 days. Setting: multi‐centre, inpatient, Baltimore, Maryland (US). | |
| Participants | Diagnosis: n = 270 schizophrenia; n = 52 neurotic or manifesting personality disorders*.
N = 421 (n = 322 remained on the project for at least five days).
Age: range 18 to 60 years, mean ˜ 36 years.
Sex: 162 F, 160 M.
History: acutely disturbed, newly‐admitted state psychiatric hospital inpatients. Included: newly admitted; between 18 to 60 years of age; "considered candidates for tranquilizing drug therapy"**. Excluded: admission under court order; presence of a complicating physical disease or disability; alcoholism; drug addiction; brain syndrome; senility. Consent: not stated. |
|
| Interventions | 1. Perphenazine: oral capsules of 8 mg and 16 mg, at least 24 mg/day, average dosage 38.61 mg/day, at least 65 mg phenobarbital t.i.d. for the first 48 hours, n = 53.
2. Chlorpromazine: oral capsules of 100 mg and 200 mg, at least 300 mg/day, average dosage 395.56 mg/day, at least 65 mg phenobarbital t.i.d. for the first 48 hours, N = 52.
3. Thioridazine: oral capsules of 50 mg and 100 mg, at least 150 mg/day, average dosage 193.46 mg/day, at least 65 mg phenobarbital t.i.d. for the first 48 hours, N = 53.
4. Triflupromazine: oral capsules of 25 mg and 50 mg, at least 75 mg/day, average dosage 95.93 mg/day, at least 65 mg phenobarbital t.i.d. for the first 48 hours, N = 53.
5. Prochlorperazine: oral capsules of 12.5 mg and 25 mg, at least 37.5 mg/day, average dosage 51.22 mg/day, at least 65 mg phenobarbital t.i.d. for the first 48 hours, N = 52.
6.Thiopropazate: oral capsules of 5 mg and 10 mg, at least 15 mg/day, average dosage 20.83 mg/day, at least 65 mg phenobarbital t.i.d. for the first 48 hours, N = 53.
7. Trifluoperazine: oral capsules of 2.5 mg and 5 mg, at least 7.5 mg/day, average dosage 11.49 mg/day, at least 65 mg phenobarbital t.i.d. for the first 48 hours, N = 52.
8. Fluphenazine: oral capsules of 1.25 mg and 2.5 mg, at least 3.75 mg/day, average dosage 5.92 mg/day, at least 65 mg phenobarbital t.i.d. for the first 48 hours, N = 53. All above dosages depended on the clinical judgment of the treating physician. Additional medication: biperiden for parkinsonism, phenobarbital etc. for mild sedation. |
|
| Outcomes | Leaving the study early: any reason; due to adverse events.
Adverse events: skin rash; hypotensive reaction; akathisia; dyskinesia; edema; EPS; Parkinsonism; akinesia. Unable to use ‐ Mental state: MSRPP (no SD), IMPS, MMPI (no data). Behaviour: MACC, PRP (no data). |
|
| Notes | *Those that were in final evaluation. **Quote: "to insure the inclusion of the more disturbed patients, all referrals were administered a minimum parenteral dosage of 65 mg phenobarbital t.i.d for the first 28 hours" (p90). |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised: no further details. |
| Allocation concealment (selection bias) | Unclear risk | Not stated. |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double‐blind: identical pink unmarked capsules. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Follow‐up: 76% in first evaluation at five days; of the n = 421 randomised, n = 322 remained on the project for at least five days. The n = 99 were not included in any of the post‐treatment analyses; the majority of these participants were considered ‘unacceptable on the basis of selection criteria’; n = 5 early side‐effect cases; others refused oral medication or were dropped for administrative treatment ‘unrelated to drug treatment’. Remaining post‐treatment evaluations: 68% in second evaluation at 15th day (n = 287) and 59% in third evaluation at 30th day (n = 248). |
| Selective reporting (reporting bias) | Unclear risk | No SD or P values reported for scale data. |
| Other bias | Unclear risk | Funding: Research Project Grant No. MY‐2152 of the National Advisory Mental Health Council, National Institutes of Health, US Public Health Service, administered by Friends of Psychiatric Research, Inc., Spring Grove State Hospital, Baltimore, Maryland (US). Pharmaceutical companies provided drugs for study (Sandoz; Schering Corp; GD Searle; Smith Kline and French Laboratories; Squibb; Knoll). Raters: not stated to be independent of treatment. |
Hoyberg 1993.
| Methods | Allocation: randomised. Blindness: double. Duration: 8 weeks, preceded by wash‐out ‐ no further details. Setting: multi‐centre. | |
| Participants | Diagnosis: chronic schizophrenia disorder with acute exacerbation (DSM‐III‐R).
N = 107.
Age: 20 to 67 years, mean ˜ 36 years.
Sex: 30 F, 77 M.
History: chronic; outpatient; 93% of participants were receiving drugs of diverse categories pre‐wash‐out (most commonly‐used drugs included phenothiazines and thioxanthenes). Included: aged between 18 to 65; diagnosis of chronic schizophrenia with acute exacerbation (DSM‐III‐R); informed consent. Excluded: mental disorder other than chronic schizophrenic disorder; clinically significant organic disorders; clinically relevant abnormalities in laboratory tests; history of alcohol or drug abuse as defined in DSM‐III‐R within the 12‐month period preceding the study; reception of oral neuroleptic treatment less than 72 hours or depot neuroleptics less than 3 weeks before the start of treatment; commitment to a mental hospital; women of reproductive age without adequate contraception; pregnant or lactating women. Consent: informed consent required from participants or their relatives or legal guardians; study approved by relevant ethics committees in accordance with the Declaration of Helsinki II. |
|
| Interventions | 1. Perphenazine: 8 mg per capsule, initially 2 capsules daily, dose could be changed during the first 4 weeks according to individual needs to a maximum of 48 mg/day, during the last 4 weeks dose was to be kept unchanged, but could be reduced if adverse events occurred*, n = 52. 2. Risperidone: 2.5 mg per capsule, initially 2 capsules daily, dose could be changed during the first 4 weeks according to individual needs to a maximum of 15 mg/day, during the last 4 weeks dose was to be kept unchanged, but could be reduced if adverse effects occurred*, n = 55. | |
| Outcomes | Leaving the study early: any reason; due to adverse events.
Global state: no better or worse (CGI).
Mental state: PANSS (clinical improvement, defined as 'at least 20% reduction in total PANSS score at endpoint).
Adverse events: UKU Side Effect Rating Scale Unable to use ‐ Mental state: BPRS; PANSS (no SD). Global state: CGI (no means or SD). Adverse events: ESRS (no SD). |
|
| Notes | *During the last 4 weeks dose was to be kept unchanged, but could be reduced if adverse effects occurred. **During the treatment period, n = 42 in the Risperidone group and n = 38 in the perphenazine group required concomitant medication (including benzodiazepines and orphenadrine). |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised: no further details. |
| Allocation concealment (selection bias) | Unclear risk | Not stated. |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double‐blind: identical tablets. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Seventy‐eight participants (73%) completed the full 8‐week trial period; n = 29 withdrew prematurely, of these n = 8 receiving risperidone and n = 6 receiving perphenazine were withdrawn due to adverse effects; n = 2 receiving risperidone and n = 3 receiving perphenazine were withdrawn due to lack of therapeutic effect; n = 4 receiving risperidone and n = 6 receiving perphenazine were withdrawn because they stopped attending the control visits. All participants that had attended at least one clinical assessment were included in the ITT analysis or endpoint analysis (N = 101; n = 50 receiving risperidone and n = 51 receiving perphenazine). All prematurely withdrawn participants were included in side effects analysis, whereas other therapeutic measures were analysed ITT. |
| Selective reporting (reporting bias) | High risk | CGI scale reported as an outcome measure, however no data are reported. |
| Other bias | Unclear risk | Funding: not stated; two co‐authors had affiliations with Janssen Pharma, Denmark and Norway. Raters: not stated to be independent of treatment. |
Imai 1980.
| Methods | Allocation: randomised. Blindness: double. Duration: 8 weeks, preceded by 3 to 7 days wash‐out. Setting: multi‐centre, Japan. | |
| Participants | Diagnosis: schizophrenia.
N = 95.
Age: 20 to 55 years.
Sex: 19 F, 76 M.
History: inpatient. Included: no details. Excluded: no details. Consent: no details. |
|
| Interventions | 1. Perphenazine: initially 12 mg daily and inactive placebo, then according to the symptoms and side effects, maximum dosage 24 mg daily, n = 49. 2. Zotepine: initially 75 mg daily and inactive placebo, then according to the symptoms and side effects, maximum dosage 150 mg daily, n = 46. | |
| Outcomes | Leaving the study early: any reason; due to relapse/ worsening.
Global state: no better or worse.
Adverse events: cardiovascular. Unable to use: Mental state: Keio University Psychiatric Rating Scale (unpublished scale). |
|
| Notes | Additional medication: only after 4 days from the beginning of the study; antiparkinsonians, barbiturates, liver protectors, vitamins, haloperidol, levomepromazine, chlorpromazine, diazepam if needed. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised: no further details. |
| Allocation concealment (selection bias) | Unclear risk | Not stated. |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double‐blind: no further details. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No details. |
| Selective reporting (reporting bias) | Unclear risk | No details. |
| Other bias | Unclear risk | Funding: not stated. Raters: not stated to be independent of treatment. |
Itoh 1969.
| Methods | Allocation: randomised. Blindness: double. Duration: 8 weeks, preceded by one week wash‐out. Setting: multi‐centre, Japan. | |
| Participants | Diagnosis: schizophrenia.
N = 80.
Age: mean ˜ 34 years.
Sex: 18 F, 62 M.
History: inpatient; duration of illness ranged from less than 12 months (6%) to over 10 years (33%). Included: showing one or more of the following: state of intense excitement; stuporous state; pronounced hallucinations and/ or delusions; pronounced delusions; markedly devoid of spontaneity and apathetic (fresh or subacute hebephrenia; or chronic, terminal deteriorative state); pronounced neurosis‐like state. Excluded: no details. Consent: not stated. |
|
| Interventions | 1. Perphenazine: (oral, 4 mg) + lactose; initially 45 mg daily, increased to 90 mg within one week, then individual dosage according to symptoms and adverse events, n = 40. 2. Clothiapine: (oral, 15 mg); initially 12mg daily, increased to 24 mg within one week, then individual dosage according to symptoms and adverse events, n = 40. | |
| Outcomes | Leaving the study early.
Global state: no better or worse.
Behaviour: no better or worse.
Adverse events. Unable to use ‐ Mental state: Psychiatric Rating Scale (unpublished scale). |
|
| Notes | Antiparkinsonism drugs and hypnotic agents were prepared and used when deemed necessary by the clinician. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomised: randomisation by numbers, quote: "patients with the same type of disorder, clinical features and sex and with differences in ages of less than ten years were paired among those examined by the same physicians at each institution, and the data obtained were analysed by the sequential trial technique of Armitage upon conclusion of the study period. After the code had been broken, the pairs were untied and the data evaluated for testing for the therapeutic efficacy of the two drugs" (p7). Random number table used (p8). |
| Allocation concealment (selection bias) | Low risk | Quote, "bottles were coded so that neither the investigators nor the patients knew whether clothiapine or placebo was being administered at any given time, [and] every investigator was instructed to retain an allocation schedule in a sealed envelope for each matched pair assigned to him in case of medical emergency of a patient under study" (p8) ‐ no such emergency happened and all envelopes were returned unopened to the central statistical unit upon completion of the study. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Double‐blind: identical shape and colour of tablets. Quote, "medication and analysis of data obtained were performed by the controlled double‐blind method" (p8). |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No details. |
| Selective reporting (reporting bias) | Unclear risk | None detected. |
| Other bias | Unclear risk | Funding: not stated. Raters: not stated to be independent of treatment. |
Itoh 1969 II.
| Methods | Allocation: randomised. Blindness: double. Duration: unclear. Setting: number of participating centres unclear, Japan. | |
| Participants | Diagnosis: schizophrenia.
N = 86.
Age: unclear.
Sex: unclear.
History: no details. Included: no details. Excluded: no details. Consent: no details. |
|
| Interventions | 1. Perphenazine: no further details, n = 43. 2. Thiothixene: no further details, n = 43. | |
| Outcomes | Leaving the study early; any reason.
Global state: no better or worse.
Behaviour: no better or worse.
Adverse events. Unable to use ‐ Mental state: Psychiatric Rating Scale (unpublished scale). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomised ‐ randomisation by numbers. |
| Allocation concealment (selection bias) | Unclear risk | Not clear. |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double‐blind: no further details. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Not clear. |
| Selective reporting (reporting bias) | Unclear risk | Not clear. |
| Other bias | Unclear risk | Funding: not clear. Raters: not stated to be independent of treatment. |
Itoh 1969 III.
| Methods | Allocation: randomised. Blindness: double. Duration: 60 days, preceded by one week wash‐out. Setting: multi‐centre, Japan. | |
| Participants | Diagnosis: schizophrenia.
N = 78.
Age: no details.
Sex: no details.
History: no details. Included: no details. Excluded: no details. Consent: no details. |
|
| Interventions | 1. Perphenazine: initially 9 mg (3 tablets) daily, increased to 18 mg within one week, then individual dosage according to symptoms and adverse events, n = 39. 2. Methylperidol: initially 15 mg (3 tablets) daily, increased to 30 mg within one week, then individual dosage according to symptoms and adverse events, n = 39. | |
| Outcomes | Leaving the study early.
Global state: no better or worse.
Behaviour: no better or worse.
Adverse events. Unable to use ‐ Mental state: Psychiatric Rating Scale (unpublished scale). |
|
| Notes | Additional medication: antiparkinson medication if needed. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomised: randomisation by numbers. |
| Allocation concealment (selection bias) | Unclear risk | Not clear. |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double‐blind: identical shape and colour of tablets. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Not clear. |
| Selective reporting (reporting bias) | Unclear risk | Not clear. |
| Other bias | Unclear risk | Funding: not clear. Raters: not stated to be independent of treatment. |
Itoh 1976.
| Methods | Allocation: randomised. Blindness: double. Duration: 12 weeks, preceded by one week wash‐out. Setting: multi‐centre, Japan. | |
| Participants | Diagnosis: schizophrenia (hebephrenia (n = 74), catatonia (n = 5), paranoid (n = 18) undifferentiated (n = 8)).
N = 105.
Age: range 19 to 50.
Sex: 58 F, 47 M.
History: inpatient; duration of hospitalisation of less than one year (n = 32), one to five years (n = 29), five years plus (n = 44); duration of illness range from less than one year to 10 years. Included: not clear. Excluded: somatic diseases; allergy; history of side effects of antiparkinsonians; glaucoma; prostate‐hypertrophy; children; pregnancy and older patients. Consent: not clear. |
|
| Interventions | 1. Perphenazine: one tablet (a 4 mg) three times daily for the first week, 2 tablets three times daily in the second week, 3 tablets three times daily in the third week, then flexible dose according to the judgement of each physician; maximum 60 mg daily, n = 51. 2. Penfluridol: 1 tablet (a 20 mg) daily for the first week, 6 tablets in the second week, 9 tablets in the third week, then flexible dose according to the judgement of each physician; maximum 100 mg daily, n = 54. | |
| Outcomes | Leaving the study early.
Global state: no better or worse.
Adverse events. Unable to use ‐ Mental state: BPRS (no SD), Keio University Psychiatric Rating Scale (unpublished scale). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised: no further details. |
| Allocation concealment (selection bias) | Unclear risk | Not clear. |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double‐blind: no further details. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Not clear. |
| Selective reporting (reporting bias) | Unclear risk | Not clear. |
| Other bias | Unclear risk | Funding: not clear. Raters: not stated to be independent of treatment. |
Kane 2003.
| Methods | Allocation: randomised. Blindness: double. Duration: 6 weeks (preceded by 2 weeks screening period; 2 days wash‐out; 4‐6 weeks open‐label treatment [olanzapine vs risperidone] to confirm treatment‐resistant; 2‐10 days wash‐out). Setting: multi‐centre, 59 centres in US & Canada (2000‐2002). |
|
| Participants | Diagnosis: schizophrenia (DSM‐IV) classified as ‘treatment resistant*’ (n = 215 paranoid; n = 14 disorganised; n = 71 undifferentiated). N = 300. Age: mean 42.1 (Perphenazine mean 41.6; aripiprazole mean 42.6). Sex: F 92; M 208, (Perphenazine: F 52, M 94; aripiprazole: F 40, M 114). History: mean age at time of first hospitalisation 22.8 (Perphenazine mean 22.9; aripiprazole mean 22.6). Included: > 18 years of age; schizophrenia DSM‐IV; classified as ‘treatment resistant*’; PANSS total score ≥ 75 and a score of ≥ 4 on at least 2 items of conceptual disorganization; suspiciousness, hallucinatory behaviour, or delusions; Clinical Global Impression‐Severity of Illness (CGI‐S) score ≥ 4; treated as outpatient for at least 1 continuous 3‐month period during 2‐years prior to study. Excluded: DSM‐IV schizoaffective disorder, residual schizophrenia, bipolar disorder, clinical presentation or history consistent with delirium, dementia, amnesic or other cognitive disorders; refractory response to prior clozapine treatment administered at therapeutic doses for 6 weeks; previous unsatisfactory response to perphenazine; likelihood to require prohibited concomitant therapy; history of or current drug or alcohol abuse or dependence; history of suicide attempts/ serious suicidal thoughts; known allergy/ hypersensitivity to study drugs; treatment with an investigational drug within 4 weeks of wash‐out phase; previous enrolment in aripiprazole study; acute or unstable medical condition; pregnancy or lactating. Consent: written informed consent required; institutional review board approval obtained. |
|
| Interventions | 1. Perphenazine: range 8 to 64 mg/day, mean 39.1 mg/day; started at 8 mg/day and could be increased to 16 mg/day on day 4 if needed; max 64 mg/day, doses greater than 8 mg/day administered twice daily, n = 146. 2. Aripiprazole: 15 to 30 mg/day, mean 30 mg/day; started at 15 mg/day, dose adjustments made to maximum 30 mg/day after week 1, n = 154. (Dose reductions also permitted provided participants remain in permitted dose range). |
|
| Outcomes |
Primary outcome: PANSS change from baseline. Secondary outcomes: CGI‐I; CGI‐S; BPRS; QoL using QLS (clinically important improvement defined as > 20% improvement in total score from baseline); response to treatment (> 30% decrease in PANSS total score or CGI‐I score of 1 or 2); adverse events; lab data; EPS (SAS, AIMS and BAS). |
|
| Notes | *treatment resistant defined as “failure to experience satisfactory symptom relief despite at least 2 periods of treatment (lasting at least 6 weeks) with adequate doses of antipsychotic agents (one of which had to be a typical antipsychotic) during the 2 years prior to the study. Nor should participants have experienced satisfactory symptom relief with their most recent course of antipsychotic therapy (p214). | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised – no further details; emailed authors 15/11/13. |
| Allocation concealment (selection bias) | Unclear risk | No details. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Double‐blind treatment phase: treatment‐resistant participants, as previously diagnosed, entered a 6‐week, open‐label treatment phase (olanzapine vs risperidone) to confirm treatment‐resistance. If completed, entered 2‐ to 10‐day single‐blind placebo wash‐out phase; afterwards randomised to double‐blind treatment with perphenazine vs aripiprazole. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | N = 512 screened; n = 416 entered open‐label treatment phase with olanzapine and Risperidone to confirm treatment resistant; n = 334 completed; of these, n = 34 discontinued due to reasons including adverse events, withdrawal of consent, lost to follow‐up; lack of efficacy, patient unreliability, and other causes. N = 300 entered double‐blind randomised study phase; of these, n = 225 (75%) completed the full 6‐week study (n = 110 receiving aripiprazole; n = 115 receiving perphenazine); of the n = 75 who discontinued, n = 33 was due to adverse events; n = 18 was due to lack of efficacy; n = 11 was due to consent withdrawal; n = 13 unknown cause. N = 3 participants did not receive study medication (n = 1 aripiprazole; n = 2 perphenazine). A further n = 3 (all aripiprazole) were excluded due to lacking post‐randomisation efficacy rating. QLS analysis included n = 207 participants (n = 104 aripiprazole; n = 103 perphenazine), missing participants were excluded due to lack of baseline/ post‐randomisation data. LOCF used; observed case analyses performed to corroborate data. |
| Selective reporting (reporting bias) | Low risk | None detected from cross‐checking published protocol and review. |
| Other bias | High risk | Funding: supported by Bristol‐Myers Squibb Co. and Otsuka Pharmaceutical Development & Commercialization, Inc. PI is consultant to Abbott, Bristol‐Myers Squibb, Janssen, Eli Lilly, Pfizer, Wyeth and AstraZeneca; received honoraria from Bristol Myers‐Squibb and Janssen. All other co‐authors bar‐one are either consultants, shareholders, have received grant support from or are employees of ACADIA, AstraZeneca, Bristol‐Myers Squibb, Eli Lilly, GlaxoSmithKline, Janssen, Pfizer, Sanofi and Solvay, or Otsuka. Rating scales: raters not stated to be independent of treatment. |
Kurihara 1983.
| Methods | Allocation: randomised. Blindness: double. Duration: 8 weeks. Setting: multi‐centre, Japan. | |
| Participants | Diagnosis: schizophrenia.
N = 286.
Age: no details.
Sex: 124 F, 160 M.
History: inpatient. Included: not clear. Excluded: severe excitement; severe stupor; severely delusional hallucinatory; severely deteriorated; hepatic; cardiac or haematological disturbance; pregnancy or suspected; children; elderly people. Consent: not clear. |
|
| Interventions | 1. Perphenazine: initially 9 mg (3 tablets) daily for one week, 18 mg during the second week, then individual dosage according to symptoms and adverse events, n = 95. 2. Clocapramine: initially 75 mg (3 tablets) daily for one week, 150 mg during the second week, then individual dosage according to symptoms and adverse events, n = 97. 3. Haloperidol: initially 3 mg (3 tablets) daily for one week, 6 mg during the second week, then individual dosage according to symptoms and adverse events, n = 94. | |
| Outcomes | Leaving the study early.
Global state: no better or worse.
Adverse events. Unable to use ‐ Mental state: Psychiatric Rating Scale (unpublished scale). Behaviour: Evaluation of Behaviour (no SD). |
|
| Notes | Additional medication: sedatives such as nitrazepam, amobarbital, bromvalerylurea and antiparkinsonians such aspromethazine, biperiden, trihexyphenidyl were given if needed. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised: no further details. |
| Allocation concealment (selection bias) | Unclear risk | Not clear. |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double‐blind: identical appearance of capsules. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Not clear. |
| Selective reporting (reporting bias) | Unclear risk | Not clear. |
| Other bias | Unclear risk | Funding: not clear. Raters: not stated to be independent of treatment. |
Kurland 1961.
| Methods | Allocation: randomised. Blindness: double. Duration: 6 weeks, preceded by 48 hours wash‐out. Setting: hospital, single‐centre, Baltimore, Maryland (US). | |
| Participants | Diagnosis: "predominantly schizophrenia" ‐ no further details.
N = 238 (n = 209 relevant)
Age: 18‐61 years, mean ˜ 39 years.
Sex: ratio men to women was 1:2 ‐ no further details.
History: inpatient, newly admitted. Included: between 18 to 65 years old; referred on basis of target symptoms of anxiety, agitation and restlessness. Excluded: alcoholism, admission under court order, chronic brain syndrome, major organic diseases, senility. Consent: not stated. |
|
| Interventions | 1. Perphenazine: initially 5 mg three times daily parenterally for 2 days, then 8 or 16 mg or a multitude of 16 mg orally for 40 days, average 30.83 mg/day, n = 31.
2. Prochlorperazine: initially 5 mg three times daily parenterally for 2 days, then 10 or 25 mg or a multitude of 25 mg orally for 40 days, average 45.38 mg/day, n = 29.
3. Triflupromazine: initially 25 mg three times daily parenterally for 2 days, then 25 or 50 mg or a multitude of 50 mg orally for 40 days, average 110.46 mg/day, n = 31.
4. Mepazine: initially 25 mg three times daily parenterally for 2 days, then 25 or 50 mg or a multitude of 50 mg orally for 40 days, average 135.45 mg/day, n = 27.
5. Chlorpromazine: initially 25 mg three times daily parenterally for 2 days, then 100 or 200 mg or a multitude of 200 mg orally for 40 days, average 401.35 mg/day, n = 32.
6. Promazine: initially 50 mg three times daily parenterally for 2 days, then 100 or 200 mg or a multitude of 200 mg orally for 40 days, average 438.92 mg/day, n = 29.
7. Placebo: active (phenobarbital) and inert (saline/ lactose) initially saline three times daily parenterally for 2 days, then lactose orally for 40 days, average 6 capsules/day, n = 30. [8. Phenobarbital: initially 65 mg three times daily parenterally for 2 days, then 32.5 or 65.0 mg or a multitude of 65.0 mg orally for 40 days, average 183.64 mg/day, n = 29] ‐ not included in data and analysis (not an antipsychotic). |
|
| Outcomes | Leaving the study early: any reason; due to adverse events. Response: no better or worse. Unable to use ‐ Mental state: MSRPP (no SD), PRP (no data), Psychiatric Scale of Target Symptoms (unpublished scale). Adverse events (no data). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised: quote, "the choice of the particular drug to be used in any case was based on a predetermined random selection of the compounds" (p4). |
| Allocation concealment (selection bias) | Unclear risk | Quote, "the physician knew a patient had six chances in eight of being given a phenothiazine compound, but did not know which drug was being given, or whether the patient was receiving one of the placebos" (p4). |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double‐blind: identical capsules; quote, "drugs were dispensed in standard unmarked capsules... coloured pink to mask all identifying consistencies and colours of the drugs" (p4). Treatment information was “secured from the pharmacist and recorded in the patient’s chart by a member of the nursing staff” (p5). Quote, "all clinical and research personnel working with patients were bound to the double‐blind stipulation imposed by the research plan" (p6). |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Quote, "those patients on whom data were incomplete, together with those not receiving medication for at least then days, were excluded from the study" (p6). Unclear which participants dropped‐out of which treatment arm. One publication makes reference to N = 277 included in the study and presents data for those. Whereas the subsequently‐published paper refers to N = 238 included after screening people eligible for study. ITT used. |
| Selective reporting (reporting bias) | Unclear risk | No SD or P values reported for scale data. |
| Other bias | Unclear risk | Funding: Research Grant No. MY‐2152 of the National Advisory Mental Health Council, National Institutes of Health, US Public Health Service and administered by Friends of Psychiatric Research Inc, Spring Grove State Hospital, Baltimore, Maryland (US). The study involved "preliminary correspondence with the drug companies"; further, the "ward physician could also drop a patient from the study if her felt he should later the plan of treatment because of the ineffectiveness of the drug" (p5). Raters: not stated to be independent of treatment. |
Lepola 1989.
| Methods | Allocation: randomised. Blindness: double. Duration: 4 months, preceded by 4 weeks wash‐out. Setting: hospital, single‐centre. | |
| Participants | Diagnosis: schizophrenia (n = 17 acute; n = 30 chronic) (RDC).
N = 47.
Age: 18 to 66 years, mean ˜ 39 years.
Sex: 27 F, 20 M.
History: inpatient, Finland. Included: not stated. Excluded: organic cerebral disease, mental retardation apparent from school age, epilepsy, serious physical disability, renal or hepatic insufficiency, haematological disorders, pregnancy. Consent: informed consent required; trial design was accepted by the Ethic Committee of "Harjamāki Mental Hospital". |
|
| Interventions | 1. Perphenazine: 4 mg per tablet, initially 3 tablets daily, could be changed when considered necessary according to response or side effects, maximal daily dose 40 mg, n = 24.
2. Sulpiride: 200 mg per tablet, initially 3 tablets daily, could be changed when considered necessary according to response or side effects, maximal daily dose 2000 mg, n = 23. Additional medication: biperiden for extrapyramidal symptoms, diazepam or nitrazepam for additional sedation. |
|
| Outcomes | Leaving the study early. Global state: no better or worse. Mental state: BPRS. Adverse events. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised: no further details. |
| Allocation concealment (selection bias) | Unclear risk | Not described. |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double‐blind: identical tablets ‐ no further details. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | None detected. |
| Selective reporting (reporting bias) | Unclear risk | None detected. |
| Other bias | Unclear risk | Funding: not stated. Raters: not stated to be independent of treatment. |
Naukkarinen 2000.
| Methods | Allocation: randomised. Blindness: double. Duration: 26 weeks + wash‐out (time not stated). Setting: multi‐centre, Finland. |
|
| Participants | Diagnosis: acute or chronic schizophrenia (DSM‐IV) with acute symptoms (at least ‘moderately ill’ using CGI 4). N = 60. Age: range 18 to 70 years; mean (olanzapine) 37.4 (perphenazine) 37.7 years. Sex: F 18, M 28 (olanzapine: F 10, M 13 – perphenazine: F 8, M 15). History: not stated. Included: acute symptoms present; 18‐70 years of age. Excluded: not described. Consent: not described. |
|
| Interventions | 1. Perphenazine: 8mg to 32 mg/day, n = 30. 2. Olanzapine: 5mg to 29 mg/day, n = 30. |
|
| Outcomes | Mental state: PANSS; BPRS. Global state: CGI; PGI. Adverse events: UKU‐ConSat; UKU side‐effects scale. Leaving study early; adverse events. Additional medication. Change scores in weight increase. Unable to use ‐ CGI, PGI, UKU‐ConSat, UKU side effects (no mean or SD ‐ authors emailed for further information). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomised: quote, “performed on the basis of clusters, or blocks, for each investigator, such that patients within a block for a given investigator were randomised in a 1:1 ratio” (p7). |
| Allocation concealment (selection bias) | Low risk | Randomisation schedule was, quote, “generated on block size, number of treatments, and number of investigative sites included in the study. The study drugs were assigned to medication kits by a computer‐generated random sequence in such a way that there was a 1:1 ratio of olanzapine to perphenazine‐ treated patients” (p7). Participants were assigned on their first visit and those who met inclusion criteria were randomly allocated to double‐blind at the second visit by assignment of a unique kit number. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Medication kit contained double‐blind study medication for each participant; blinding of study was preserved by allowing a “minimum number of Lilly personnel” to see the randomisation table and codes before completion of the study (p7). |
| Incomplete outcome data (attrition bias) All outcomes | High risk | High attrition, with 38% completing the study. Four separate reports provide different numbers of participants included. N = 60 originally randomised, however only n = 46 made reference to follow‐up reports. Of the n = 46, only n = 23 (n = 12 in olanzapine and n = 11 in perphenazine) completed the study. N = 53 included in the safety analysis, and n = 46 included in efficacy analysis. |
| Selective reporting (reporting bias) | High risk | Data not provided for CGI, PGI, UKU‐ConSat, UKU side effects – authors contact 13/11/13 for request of full‐published version, no response. |
| Other bias | High risk | Funding: involvement of Eli Lilly, unclear based on provided study report to what extent. Rating scales: raters not stated to be independent of treatment. |
Remvig 1987.
| Methods | Allocation: randomised. Blindness: double. Duration: 3 to 12 weeks. Setting: inpatient, multi‐centre. | |
| Participants | Diagnosis: acute psychosis or exacerbation of a chronic psychosis.
N = 54.
Age: 20 to 60 years, mean 38 ˜ years.
Sex: 25 F, 15 M (of those having completed at least 3 weeks).
History: recent acute episode; inpatient; more than half of participants had received neuroleptic medication previously, prior to admission. Included: no details. Excluded: suffering from a serious somatic illness (including decreased kidney and liver function); organic brain damage; pregnancy; history of abuse; previous good response to a particular neuroleptic drug. Consent: informed consent from participants had to be obtained. |
|
| Interventions | 1. Perphenazine: initially 16 to 24 mg daily, dose could be increased 2 or 3 times a week if indicated, total average 30 mg daily + zuclopenthixol placebo, n = 27.
2. Zuclopenthixol: initially 20 to 30 mg daily, dose could be increased 2 or 3 times a week if indicated, total average 37 mg daily + perphenazine placebo, n = 27. Additional medication: biperiden or orphenadrine for parkinsonism, benzodiazepines, methotrimeprazine for additional neuroleptic treatment. |
|
| Outcomes | Leaving the study early.
Global state: no better or worse.
Adverse events: UKU‐Scale for side effects. Unable to use ‐ Mental state: CPRS (unpublished scale). CGI score ("slightly modified" version of the scale used). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised: no further details. |
| Allocation concealment (selection bias) | Unclear risk | Not described. |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double‐blind: quote, "double‐blind, double‐dummy" (p147). |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Quote, "all patients treated for 3 weeks (21 days) or more were included in the statistical processing of data" (p148). Fourteen participants (n = 5 on zuclopenthixol; n = 9 on perphenazine) had not been treated for the 3‐week minimum, and were excluded ‐ reasons including insufficient effect, undesired effect, or discharge from hospital. Binary data analysed with 74% (ITT); continuous data using CGI scale presented data for n = 25 (46%), no LOCF used. |
| Selective reporting (reporting bias) | High risk | Quote: "because of the great variation in total scores among the patients, they were stratified post hoc into two subgroups, depending on the total score at baseline" (p149). |
| Other bias | High risk | Funding: not stated ‐ one author was affiliated with Lundbeck (producer of zuclopenthixol for the study drug). Raters: not stated to be independent of treatment. Quote: "as there was no information available about any causal relationship between symptoms and test treatment for may patients... it was assumed, when the side effects were analyzed, that all side effects were drug‐induced" (p148). Quote: "no additional neuroleptic treatment was allowed" (p148), however, it is later stated that "most patients received benzodiazepines... and 12 patients received an additional neuroleptic in the form of methotrimeprazine" (p149). |
Sun 2000.
| Methods | Allocation: randomised. Blindness: not described. Duration: 6 weeks. Setting:no details. |
|
| Participants | Diagnosis: schizophrenia CCMD‐2‐R. N = 103. Age: mean 31 ± 9 years. Sex: male and female, History: length of illness not stated. Included: not stated. Excluded: not stated. Consent: not stated. |
|
| Interventions | 1. Perphenazine: 8‐60 mg/day, n = 51. 2. Clozapine: 50‐600 mg/day, n = 51. 3. Perphenazine + clozapine: 32‐50 mg/day + 100‐300 mg/day, n = 51. |
|
| Outcomes | Curative effect (improvement: cured = psychiatric symptoms disappeared, insight recovered, BPRS reduction >80%; markedly improved = majority of symptoms disappear, some insight, BPRS reduction > 60% to 79%; improvement = some psychiatric symptoms disappeared, no insight, BPRS reduction > 30% to 59%; no effect = no change/ worse symptoms, BPRS reduction < 30%. BPRS, TESS score, adverse events. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised – no further details. |
| Allocation concealment (selection bias) | Unclear risk | No details. |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | No details. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No details. |
| Selective reporting (reporting bias) | Unclear risk | No details. |
| Other bias | Unclear risk | No details as to blinding; two raters involved in administering BPRS and TESS. |
Takahashi 1982.
| Methods | Allocation: randomised. Blindness: double (implied). Duration: 12 weeks. Setting: inpatient, multi‐centre (31 psychiatric institutions), Japan. | |
| Participants | Diagnosis: schizophrenia (n = 127 hebephrenic, n = 24 catatonic, n = 38 paranoid, n = 4 hebephrenic‐catatonic, n = 2 hebephrenic‐paranoid, n = 1 schizoaffective, n = 2 undifferentiated).
N = 205*.
Age: 13 ‐ 60 years.
Sex: no details.
History: inpatient. Included: not described. Excluded: early stages of gestation; severe liver, kidney, cardiovascular and blood diseases; idiosyncrasy to psychotropic drugs; consciousness disturbances or vascular collapse or conditions under strong effects of neuro‐depressants; organic brain disorders; those patients whose conditions in the judgement of the attending physician would make it unlikely that they could tolerate the study treatment. Consent: informed consent obtained from each participant and guardian prior to the trial. |
|
| Interventions | 1. Perphenazine: 4 mg per tablet + placebo tablet matching to the timiperone tablet, initially two or three tablets daily, afterwards individualised according to symptoms and side effects, maximum daily dose was 12 tablets, n = 99.
2. Timiperone: 1 mg per tablet + placebo tablet matching to the perphenazine tablet, initially two or three tablets daily, afterwards individualised according to symptoms and side effects, maximum daily dose was 12 tablets, n = 99. Additional medication: mild hypnotics and anti‐parkinsonian drugs ‐ no further details. |
|
| Outcomes | Leaving the study early.
Global state: no better or worse.
Adverse events. Unable to use ‐ Mental state: Keio University Brief Psychiatric Rating Scale (unpublished scale). |
|
| Notes | *Only 198 participants were evaluated and described. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomised: quote, "participants were assigned randomly" (p258). 'Randomised code' referred to, which was broken only at the end of treatment period. |
| Allocation concealment (selection bias) | Unclear risk | No details. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Double‐blind: implied ‐ decoding of drugs at the end of week 12. Code broken at the end of treatment period. Double‐dummy design. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Of N = 205 randomised, n = 7 were excluded, either because they had been given other neuroleptics or they revealed physical disease during the trial prior to breaking the randomisation code. A total of n = 198 were included in the analysis. |
| Selective reporting (reporting bias) | Unclear risk | No means, SD or P values reported for scale data. |
| Other bias | Unclear risk | Funding: not stated ‐ study drugs were supplied by Daiichi Seiyaku Co, Ltd, Tokyo, Japan. Raters: not stated to be independent of treatment. |
Van Praag 1976.
| Methods | Allocation: randomised. Blindness: double (implied). Duration: 10 days (preceded by 5 to 10 days wash‐out). Setting: inpatient, single‐centre. | |
| Participants | Diagnosis: acute psychosis (n = 3 organic psychosis; n = 13 psychogenic psychosis; and n = 8 acute schizophrenia).
N = 28*.
Age: 19 ‐ 69 years, mean ˜ 35 years.
Sex: 15 F, 9 M.
History: acute, inpatient. Included: according to independent views of attending physician and psychiatrist/ supervisor, the participant showed signs of delusions and/ or hallucinations (Lowe 1973). Excluded: deep melancholia (i.e. deep endogenous depression with delusions of guilt, sin and/or poverty). Consent: not stated. |
|
| Interventions | 1. Perphenazine: 2, 4 and 8 mg per capsule, 3 capsules daily according to constitution, range 0 to 24 mg daily, mean 21 mg daily, n = 15.
2. Clozapine: 50, 100 and 200 mg per capsule, 3 capsules daily according to constitution, range 0 to 600 mg daily, mean 300 mg daily, n = 13. All daily doses determined by a psychiatrist not engaged in scoring; flexible dosing scheme. |
|
| Outcomes | Leaving the study early.
Adverse events. Unable to use ‐ Global state: no better or worse (no data). Mental state: BPRS (only significance tests). Laboratory tests (no data). |
|
| Notes | Only 24 participants were evaluated and described. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised: no further details. |
| Allocation concealment (selection bias) | Unclear risk | No details. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Double‐blind: implied ‐ identical capsules. Quote, "raters and patients were unaware of the type of medication or its dosage; nor were they informed of the biochemical results" (p549). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Four participants (n = 3 receiving perphenazine; n = 1 receiving clozapine) failed to complete the study. All cases were due to presence of severe motor unrest, anxiety or aggression non‐responsive to permitted medication. ITT used. |
| Selective reporting (reporting bias) | Unclear risk | No means or SD reported for scale data. |
| Other bias | Unclear risk | Funding: not stated. Raters: not stated to be independent of treatment. |
Wang 2008a.
| Methods | Allocation: randomised. Blindness: no details. Duration: 8 weeks. Setting: not stated. |
|
| Participants | Diagnosis: schizophrenia (CCMD‐3). N = 64. Age: risperidone: 65±2 years; perphenazine: 64 ± 3 years. Sex: F29, M35 (risperidone: M18, F16; perphenazine: M17, F13) History: length of illness: risperidone: 0.9 ± 1.4; perphenazine: 1.2 ± 1.4 years. Included: schizophrenia; age > 60; no history of taking antipsychotic drugs; PANNS > 60. Excluded: severe physical disease; drug abuse. Consent: not stated. |
|
| Interventions | 1. Perphenazine: start dose 2‐4mg/d, maximum dose 30mg/d, 23.5 ± 1.3mg/d, n = 30. 2. Risperidone: 5mg tablet, start dose 0.25‐0.5mg/d, maximum dose 3.5mg/d, 2.2 ± 1.2mg/d, n = 34. No other combinations permitted. |
|
| Outcomes | PANSS (marked improvement => 60 reduction; improved => 40%; no effect =< 40%). TESS ‐ adverse effects. Unable to use ‐ PANSS score – no means/ SD. TESS scores – no means/ SD. Not all adverse events reported by group. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | ‘Randomised’ – no further details. |
| Allocation concealment (selection bias) | Unclear risk | No details. |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | No description. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Not all outcome measures reported; adverse events not reported fully by group. |
| Selective reporting (reporting bias) | Unclear risk | As above. |
| Other bias | Unclear risk | No details of funding/ who administered rating scales. |
Wang 2008b.
| Methods | Allocation: randomised. Blindness: not reported. Duration: 6 weeks (2007‐2008). Setting: Shandong Center for Mental Health, Jinan, China. |
|
| Participants | Diagnosis: schizophrenia DSM‐IV. N = 60. Age: Olanzapine: 33.1±7.6 years; perphenazine: 31.2±3.2 years. Sex: F26, M34 (Olanzapine F12, M18; perphenazine F14, M16). History: length of illness olanzapine: 78.5 ± 6.8 months; perphenazine: 79.6 ± 9.5 months. Included: schizophrenia combined with positive symptoms; PANSS≥ 70, Positive score ≥ 25. Exclusion: not reported. Consent: not stated. |
|
| Interventions | 1. Perphenazine: start from 8 mg/day, maximum dosage: 60 mg/day, n =30. 2. Olanzapine (Changzhou Watson Pharmacy Ltd.): start from 5 mg/day, maximum dosage: 20 mg/day, n = 30. |
|
| Outcomes | PANSS score. TESS score. Clinical prognosis assessment: decreased rate of PANSS score = (pre‐treatment score‐post‐treatment score)/pre‐treatment score ≥ 75%‐‐recovery; 50%‐ to ‐74% markedly improved; 25% to 49% improved; < 25% no effect. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised – no further details. |
| Allocation concealment (selection bias) | Unclear risk | Not stated. |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Not stated. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No description. |
| Selective reporting (reporting bias) | Low risk | None detected. |
| Other bias | Unclear risk | Funding: not stated. Rating scales: raters not stated to be independent of treatment. |
Wang 2008c.
| Methods | Allocation: randomised. Blindness: not stated. Duration: 8 weeks, 2003‐2007 (one‐week wash‐out) Setting: Baoji rehabilitation hospital, Shanxi, China. |
|
| Participants | Diagnosis: schizophrenia CCMD‐3. N = 64. Age: Quetiapine: 66.08 ± 4.22 years; perphenazine: 65.73 ± 5.53 years. Sex: F27, M37 (Quetiapine (n = 32): M17 F15; perphenazine (n = 32): M20 F12). History: length of illness Quetiapine: 3.6 ± 0.5months; perphenazine: 3.4 ± 0.6months. Included: Schizophrenia CCMD‐3; age ≥ 60 years, first episode; BPRS ≥ 35. Exclusion: drug abuse; organic dysphrenia. Consent: not stated.. |
|
| Interventions | 1. Perphenazine: start from 4 mg/day to treatment dosage: 20‐40 mg/day, average:24.5 ± 5.3mg/day, n = 32. 2. Quetiapine: start from 50 mg/day to the treatment dosage: 200‐400 mg/day, average dosage: 258.92 ± 28.13mg/day, n = 32. |
|
| Outcomes | Lab data: blood test, urine test, liver function, kidney function, ECG, blood pressure. Adverse events. BPRS score. TESS score. Clinical prognosis assessment: decreased rate of BPRS score ≥ 75%‐‐recovery; 50% to 74% markedly improved; 25% to 49% improved; < 25% no effect. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised – no further details. |
| Allocation concealment (selection bias) | Unclear risk | Not described. |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Not described. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | In the quetiapine group, n = 1 left the study due to drowsiness. In the perphenazine group n = 3 left due to EPS. Missing data from participants leaving the study early were not included in analyses. |
| Selective reporting (reporting bias) | Unclear risk | None detected. |
| Other bias | Unclear risk | Funding: not stated. Rating scales: raters not stated to be independent of treatment. |
Woggon 1978.
| Methods | Allocation: randomised. Blindness: double. Duration: 30 days. Setting: inpatient. | |
| Participants | Diagnosis: schizophrenia (n = 5 hebephrenic; n = 29 paranoid, acute schizophrenic episode; n = 2 residual; n = 2 schizo‐affective psychosis; n = 1 other paranoid syndromes).
N = 40.
Age: mean ˜ 39 years.
Sex: 20F, 20M.
History: inpatient; duration of illness 8.7 ± 10.9 years (bromperidol); 10.1 ± 8.0 (perphenazine). Included: no details. Excluded: no details. Consent: not stated. |
|
| Interventions | 1. Perphenazine: capsules of 3 mg; mean 20 mg/day in two administrations; maximum 24 mg daily, n = 20.
2. Bromperidol: capsules of 1 mg; mean 6 mg/day in two administrations; maximum 8 mg daily, n = 20. Additional medication: antiparkinsonian drugs; additional neuroleptics (promazine and levomepromazine); hypnotics (chloralhydrate; nitrazepam; flunitrazepam). |
|
| Outcomes | Leaving the study early.
Global state: no better or worse.
Adverse events. Unable to use ‐ Mental state: AMP (no SD). Lab data: not reported. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised: no further details. |
| Allocation concealment (selection bias) | Unclear risk | No details. |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double‐blind: no further details. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | None detected. |
| Selective reporting (reporting bias) | High risk | No means or SDs reported for continuous outcomes and scale data. |
| Other bias | Unclear risk | Funding: no details. Raters: not stated to be independent of treatment. |
Zhang 2010.
| Methods | Allocation: randomised. Blindness: no further information. Duration: 8 weeks. Setting: not stated. |
|
| Participants | Diagnosis: Schizophrenia CCMD‐3. N = 65 (or 66). Age: aripiprazole 64.6 ± 1.8; perphenazine 62.5 ± 3.1 years. Sex: F34, M32 (aripiprazole M16, F17; perphenazine M16, F17) History: length of illness ‐ aripiprazole 0.5 ± 1.1; perphenazine 0.8 ± 1.2 years. Included: schizophrenia CCMD‐3; age > 60, no history of taking antipsychotic drugs; BPRS > 35. Exclusion: severe physical disease; drug abuse. Consent: not stated. |
|
| Interventions | 1. Perphenazine: start dose 2‐4 mg/day, maximum dose 16 mg/day, average 11.2 ± 3.8mg/day, n = 33. 2. Aripiprazole: 5 mg tablet, start dose 5 mg/day maximum dose 30 mg/ day, average 23.5 ± 1.7mg/day, n = 32. | |
| Outcomes | Mental state: PANSS. Clinical effect: decreased rate of BPRS score ≥ 75%‐‐recovery; ≥ 50% markedly improved; ≥ 25% improved; < 25% no effect (measured at 1, 2 ,4 ,6, 8 weeks before and after treatment) BPRS. TESS (skew). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised – no further details. |
| Allocation concealment (selection bias) | Unclear risk | No details. |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | No further details. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | One participant not included in results nor accounted for. |
| Selective reporting (reporting bias) | High risk | Not all adverse events reported by group – unable to include some missing group data. No data reported using BPRS or TESS scales. |
| Other bias | Unclear risk | No details as to funding or who administered rating scales. |
General abbreviations: CNS – central nervous system ECG ‐ electrocardiogram IQ ‐ intelligence quotient ITT ‐ intention to treat LOCF – Last Observation Carried Forward mg ‐ milligram N = number of participants SD ‐ standard deviation SGOT ‐ serum glutamic oxaloacetic transaminase SGPT ‐ serum glutamic‐pyruvic transaminase t.i.d. ‐ three times daily (Latin: ter in die)
Diagnostic tools: CCMD‐2‐R – Chinese Classification of Mental Disorders, Second Edition, Revised CCMD‐3 – Chinese Classification of Mental Disorders, Third Edition DSM‐III‐R ‐ Diagnostic and Statistical Manual of Mental disorders, Third edition, Revised DSM‐IV – Diagnostic and Statistical Manual, Fourth Edition ICD‐9 ‐ International Classification of Diseases, ninth revision RDC – Research Diagnostic Criteria
Global effect scales: GCJ ‐ Global comparative judgement CGI ‐ Clinical Global Impression CGI‐I – Clinical Global Impression ‐ Improvement CGI‐S –Clinical Global Impression ‐ Severity GIR ‐ General Improvement Rating PGI ‐ Patient Global Impression
Mental state scales: AMDP, AMP ‐ Arbeitsgemeinschaft für Methodik und Dokumentation in der Psychiatrie BPRS ‐ Brief Psychiatric Rating Scale CEPS ‐ Clinical Estimate of Psychiatric Rating Scale CPRS ‐ Comprehensive Psychopathological Rating Scale IMPS ‐ Inpatient Multidimensional Psychiatric Scale MMPI ‐ Minnesota Multiphase Personality Inventory MSRPP ‐ Multidimensional Scale for Rating Psychiatric Patients PANSS ‐ Positive and Negative Syndrome Scale for Schizophrenia PES ‐ Psychiatric Evaluating Scale
Behaviour scales: MACC ‐ Behavioural Adjustment Scale NOSIE ‐ Nurses Obsrevation Scale for Inpatient Evaluation PRP ‐ Psychotic Reaction Profile
Side effect scales: AIMS ‐ Abnormal Involuntary Movement Scale BAS ‐ Barnes Akathisia Scale EPS – Extrapyramidal Symptoms ESRS ‐ Extrapyramidal Symptoms Rating Scale SAS ‐ Simpson and Angus Scale TESS –Treatment Emergent Symptom Severity UKU ‐ Side Effect Rating Scale
Quality of life scales: QoL – Quality of Life Scale QLS ‐ Quality of Life Scale
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Affleck 1969 | Allocation: randomised. Blindness: double. Participants: those with schizophrenia. Interventions: perphenazine vs. penicillamine, perphenazine and a placebo vs. penicillamine, penicillamine vs. placebo. |
| Ahlfors 1980 | Allocation: randomised. Blindness: double. Participants: those with schizophrenia. Interventions: perphenazine enanthate vs. clopenthixol decanoate. |
| Akimoto 1966 | Allocation: 'randomly selected' Participants: schizophrenia Intervention: perphenazine vs chlorpromazine vs levomepromazine vs prochlorperazine (not parallel‐arm study, no control group, all participants received all drugs over one year) |
| Angst 1975 | Allocation: randomised. Blindness: double. Participants: those with schizophrenia. Interventions: perphenazine enanthate vs. fluphenazine decanoate vs. flusipirilene vs. penfluridol vs. pipothiazine palmitate. |
| Anonymous 1966 | Allocation: not randomised. |
| Asada 1976 | Allocation: not randomised ‐ matched pairs. |
| Bakke 1973 | Allocation: randomised. Blindness: double. Participants: those with schizophrenia. Interventions: perphenazine enanthate vs. fluphenazine enanthate. |
| Bandelow 1992 | Allocation: random Participants: people with schizophrenia Interventions: perphenazine enanthate versus perazine versus fluphenazine versus levomepromazine versus thioridazine versus clozapine versus haloperidol decanoate versus fluphenazine decanoate versus flupenthixol decanoate versus fluspirilene |
| Becker 1970 | Allocation: randomised. Blindness: double. Participants: those with chronic schizophrenia. Interventions: perphenazine and amitriptyline vs. perphenazine and placebo. |
| Bjerkelund 1965 | Allocation: randomised. Blindness: double. Participants: those with acute myocardial infarct. |
| Böhlau 1985 | Allocation: unclear, open multi‐centre study. Blindness: double. Participants: those with dementia. |
| Cannistra 1959 | Allocation: unclear, double‐blind prospective study. Participants: obstetric patients. |
| Ceskova 1994 | Allocation: randomised ‐ cross‐over design. Blindness: double. Participants: those with schizophrenia. Interventions: perphenazine vs. risperidone. Data about separate phases not available. |
| Ceskova 1996 | Allocation: randomised ‐ cross‐over design. Blindness: double. Participants: those with schizophrenia. Interventions: perphenazine vs. haloperidol. Data about separate phases not available. |
| Clarke 1981 | Allocation: not randomised, open clinical trial. |
| Cohen 1958 | Allocation: not randomised, open clinical trial. |
| Cooper 1979 | Allocation: randomised. Blindness: unclear. Participants: those with schizophrenia. Interventions: perphenazine vs. amitriptyline vs. perphenazine and amitriptyline. |
| Den Boer 2000 | Allocation: not randomised. |
| Dencker 1994 | Allocation: randomised. Blindness: double. Participants: those with schizophrenia. Interventions: perphenazine decanoate vs. haloperidol decanoate. |
| Eklund 1976 | Allocation: not random |
| Erb 1982 | Allocation: randomised. Blindness: unclear. Participants: healthy normal males. |
| Eufe 1979 | Allocation: randomised. Blindness: double. Participants: those with schizophrenia. Interventions: perphenazine enanthate vs. flupentixol enanthate. |
| Fahn 1964 | Allocation: unclear, double‐blind prospective study. Participants: those with choreic movements. |
| Farzin 2005 | Allocation: random Participants: schizophrenia Interventions: perphenazine + famotidine versus perphenazine + placebo |
| Fitzgerald 1969 | Allocation: randomised. Blindness: double ‐ identical ampoules. Participants: those with acute psychiatric episodes. |
| Galdi 1988 | Allocation: random Participants: people with schizophrenia or depression Intervention: perphenazine vs chlorpromazine vs fluphenazine Outcomes: no usable data |
| Gerlach 1977 | Allocation: not randomised. |
| Gerlach 1988 | Allcation: randomised ‐ cross‐over design. Blindness: double. Participants: those with tardive dyskinesia. |
| Gerson 1964 | Allocation: randomised. Blindness. double. Participants: those with schizophrenia. Interventions: perphenazine or chlorpromazine or trifluoperazine or prochlorperazine or methaminodiazepoxide or fluphenazine or phenelzine or imipramine tranylcypromine vs. 4‐(3, 4‐dimethoxyphenetyl)‐2, 6‐piperazinedione and 2‐methyl‐3‐orthotolyl‐4‐quinazinolone. |
| Gianelli 1990 | Allocation: randomised. Blindness: double. Participants: those with schizophrenia. Interventions: perphenazine enanthate vs. fluphenazine decanoate vs. haloperidol decanoate. |
| Hadlik 1970 | Allocation: randomised ‐ cross‐over design. Blindness: double. Participants: those with schizophrenia. Interventions: perphenazine vs. thiothixem. Data about separate phases no more available. |
| Hanlon 1966 | Allocation: random Participants: 'acutely disturbed psychiatric patients' Intervention: perphenazine vs perphenazine‐benztropine mesylate |
| Hansen 1979 | Allocation: randomised. Blindness: double. Participants: those being on neuroleptic therapy. Interventions: perphenazine vs. perphenazine and biperidine vs. perphenazine and orphenadrine. |
| Haran 1960 | Allocation: not randomised. |
| Holden 1969 | Allocation: not randomised ‐ matched groups. |
| Hollister 1967 II | Allocation: randomised. Blindness: unclear. Participants: those with depression as predominant psychiatric symptom. |
| Hollister 1974 | Allocation: not randomised. |
| Huang 1964 | Allocation: not randomised. |
| Jprn 2011 | Allocation: random Participants: schizophrenia Intervention: algorithm‐guided treatment (different antipsychotic drugs: perphenazine, aripiprazole, olanzapine, blonanserine, perospiron, quetiapine, risperidone, haloperidol) versus treatment as usual |
| Kaim 1972 | Allocation: unclear, double‐blind prospective study. Participants: those with delirium tremens. |
| Kaji 1974 | Allocation: randomised. Blindness: double. Participants: those with schizophrenia. Interventions: perphenazine and levomepromazine vs. clocapramine and levomepromazine. |
| Kawakita 1965 | Allocation: not randomised. |
| Kistrup 1991 | Allocation: randomised. Blindness: double. Participants: those with schizophrenia. Interventions: perphenazine decanoate vs. cis(z)‐flupentixol decanoate. |
| Kline 1968 | Allocation: not randomised. |
| Knudsen 1985 | Allocation: randomised. Blindness: double. Participants: those with schizophrenia. Interventions: perphenazine decanoate vs. perphenazine enanthate. |
| Knudsen 1985a | Allocation: random Participants: schizophrenia Interventions: perphenazine decanoate vs perphenazine enanthate |
| Kothari 1960 | Allocation: not randomised. |
| Lapolla 1967 | Allocation: not randomised. |
| Levine 1997 | Allocation: randomised. Blindness: double. Participants: those with schizophrenia. Interventions: perphenazine vs. chlorpromazine vs. placebo. Contacted on March 12th: data about methods no more available. |
| Lindholm 1978 | Allocation: randomised. Blindness: double. Participants: those with schizophrenia. Interventions: perphenazine enanthate vs. perphenazine enanthate. |
| Linnoila1982 | Allocation: unclear. Blindness: unclear. Participants: those with depression. |
| Loprete 1967 | Allocation: serial placement. Blindness: unclear. Participants: those with schizophrenia and anxiousness and depression. Interventions: perphenazine and amitriptyline (Etrafon) vs. perphenazine and amitriptyline (Etrafon forte) vs. non barbiturate hypnotic sedative (Dormison) vs. phenobarbital vs. placebo. |
| Mason‐Browne 1957 | Allocation: not randomised ‐ matched groups. |
| Mazurek 2003 | Allocation: random Participants: paranoid schizophrenia (ICD‐10‐R) Intervention: typical antipsychotic drugs (haloperidol, levomepromazine, perazine, perphenazine, zuclopenthixol) versus atypical (clozapine, risperidone, olanzapine) ‐ not separated by drug |
| Moore 1958 | Allocation: randomised. Blindness: double. Participants: those with an anaesthetic for operation. |
| Müller 1994 | Allocation: open, randomised, three‐way cross‐over, single‐dose design. Blindness: unclear. Participants: healthy volunteers. |
| Nahunek 1967 | Allocation: open cross‐over design ‐ not randomised. |
| Nahunek 1968 | Allocation: randomised ‐ cross‐over design. Blindness: double. Participants: those with schizophrenia or endogenous depression. Interventions: perphenazine vs. octoclothepin. |
| Nahunek 1969 | Allocation: randomised ‐ cross‐over design. Blindness: double. Participants: those with schizophrenia. Interventions: perphenazine vs. triperidol. Data about separate phases no more available. |
| Nahunek 1970 | Allocation: randomised ‐ cross‐over design. Blindness: double. Participants: those with schizophrenia. Interventions: perphenazine vs. triperidol. Data about separate phases no more available. |
| Nahunek 1970 II | Allocation: randomised ‐ cross‐over design. Blindness: double. Participants: those with schizophrenia. Interventions: perphenazine vs. flupenthixol. Data about separate phases no more available. |
| Nahunek 1975 | Allocation: randomised ‐ cross‐over design. Blindness: double. Participants: those with schizophrenia. Interventions: perphenazine vs. clozapin. Data about separate phases no more available. |
| Nahunek 1976 | Allocation: randomised ‐ cross‐over design. Blindness: double. Participants: those with schizophrenia. Interventions: perphenazine vs. clozapin vs. levopromazin vs. thioridazin vs. chlorpromazin vs. flupenthixol vs. methophenazin vs. oxypertin vs. clothiapin vs. pimozid vs. methylperidol vs. triperidol vs. oxyprothepin vs. octoclothepin vs. thiothixen. Data about separate phases no more available. |
| Nahunek 1980 | Allocation: randomised ‐ cross‐over design. Blindness: double. Participants: those with schizophrenia. Interventions: perphenazine vs. perphenazine. |
| Nordic DS Group 1986 | Allocation: randomised. Blindness: unclear. Participants: those with tardive dyskinesia. |
| O'Reilly 1957 | Allocation: randomised. Blindness: double. Participants: those with schizophrenia and two epileptics. Interventions: perphenazine vs. chlorpromazine or placebo (variable). |
| Oltman 1966 | Allocation: not randomised. |
| Omerov 1989 | Allocation: randomised. Blindness: double. Participants: those with schizophrenia. Interventions: perphenazine vs. perphenazine. |
| Opjordsmoen 2000 | Allocation: not random |
| Pfeiffer 1965 | Allocation: not randomised. |
| Prien 1973 | Allocation: not randomised. |
| Prusoff 1979 | Allocation: randomised. Blindness: unclear. Participants: those with depressive schizophrenia. Interventions: amitriptyline and perphenazine vs. placebo. |
| Rapp 1972 | Allocation: randomised. Blindness: double. Participants: those with schizophrenia. Interventions: perphenazine enanthate vs. fluphenazine decanoate. |
| Rapp 1986 | Allocation: randomised. Blindness: double. Participants: those with schizophrenia. Interventions: perphenazine enanthate vs. haloperidol decanoate. |
| Rappaport 1967 | Allocation: randomised. Blindness: double. Participants: those with schizophrenia. Interventions: perphenazine vs. placebo. Outcomes: intelligibility score. |
| Rappaport 1968 | Allocation: randomised. Blindness: double. Participants: those with schizophrenia. Interventions: perphenazine vs. placebo. Outcomes: intelligibility score. |
| Reznik 2000 | Allocation: randomised. Blindness: double. Participants: those with schizophrenia. Interventions: perphenazine vs. perphenazine and fluvoxamine. |
| Rickels 1982 | Allocation: randomised. Blindness: double. Participants: those with depression. |
| Ritter 1971 | Allocation: randomised. Blindness: double. Participants: those with acute withdrawal syndrome. |
| Rodova 1971 | Allocation: randomised ‐ cross‐over design. Blindness: double. Participants: those with schizophrenia. Interventions: perphenazine vs. clothiapin. Data about separate phases no more available. |
| Rodova 1973 | Allocation: randomised ‐ cross‐over design. Blindness: double. Participants: those with schizophrenia. Interventions: perphenazine vs. clozapine. Data about separate phases no more available. |
| Scurr 1958 | Allocation: unclear. Blindness: unclear. Participants: those with an operation. |
| Shalev 1993 | contacted on March 12th: data about methods no more available. |
| Sharpley 1964 | Allocation: not random (controlled) |
| Shiloh 2002 | Allocation: random Participants: treatment‐resistant schizophrenia Intervention: mianserin versus placebo (in conjunction with haloperidol or perphenazine) |
| Silver 2000 | Allocation: randomised. Blindness: double. Participants: those with schizophrenia. Interventions: antipsychotic drug and fluvoxamine vs. antipsychotic drug and placebo. |
| Simopoulos 1971 | Allocation: randomised. Blindness: unclear. Participants: those with schizophrenia. Interventions: dilantin vs. dilantin and thorazine vs. dilantin and thioridazine vs. dilantin and perphenazine. |
| Simpson 1970 | Allocation: randomised ‐ cross‐over design. Blindness: double. Participants: those with schizophrenia. Interventions: perphenazine vs. perphenazine‐amitriptyline. |
| Smith 1959 | Allocation: not randomised. |
| Spiker 1985 | Allocation: randomised. Blindness: double. Participants: those with delusional depression. |
| Sun 2010 | Allocation: random Participants: schizophrenia Intervention: venlafaxine extended release tablets + perphenazine versus perphenazine + placebo |
| Sun 2010b | Allocation: random Participants: schizophrenia Intervention: aripiprazole + placebo vs aripiprazole + perphenazine |
| Svestka 1970 | Allocation: randomised ‐ cross‐over design. Blindness: double. Participants: those with schizophrenia. Interventions: perphenazine vs. methylperidol. Data about separate phases no more available. |
| Svestka 1972 | Allocation: randomised ‐ cross‐over design. Blindness: double. Participants: those with schizophrenia. Interventions: perphenazine vs. pimozide. Data about separate phases no more available. |
| Svestka 1973 | Allocation: randomised ‐ cross‐over design. Blindness: double. Participants: those with schizophrenia. Interventions: perphenazine vs. oxyprothepine. Data about separate phases no more available. |
| Svestka 1974 | Allocation: randomised ‐ cross‐over design. Blindness: double. Participants: those with schizophrenia. Interventions: perphenazine vs. oxypertine. Data about separate phases no more available. |
| Svestka 1975 | Allocation: randomised ‐ cross‐over design. Blindness: double. Participants: those with schizophrenia. Interventions: perphenazine vs. trifluoperazine. Data about separate phases no more available. |
| Svestka 1989 | Allocation: randomised ‐ cross‐over design. Blindness: double. Participants: those with schizophrenia. Interventions: perphenazine vs. isofloxythepin. |
| Svestka 1989a | Allocation: randomised. Blindness: double. Participants: those with schizophrenia. Interventions: perphenazine vs. carbamazepine. |
| Svestka 1990 | Allocation: randomised ‐ cross‐over design. Blindness: double. Participants: those with schizophrenia. Interventions: perphenazine vs. sulpiride. Data about separate phases no more available. |
| Syvalahti 1997 | Allocation: randomised. Blindness: double. Participants: those with schizophrenia. Interventions: placebo or citalopram vs. perphenazine or haloperidol or chlorpromazine. |
| Tegeler 1979 | Allocation: randomised. Blindness: double. Participants: those with schizophrenia. Interventions: perphenazine enanthate vs. flusipirilene. |
| Wang 2001 | Allocation: random Participants: schizophrenia (ICD‐10 and CCMD‐2‐R) Intervention: perphenazine versus 1‐stepholidine (traditional Chinese medicine) |
| Whittaker 1963 | Allocation: "open cross‐over" ‐ not randomised. |
| Wolf 2007 | Allocation: random Participants: schizophrenia Intervention: aripiprazole versus 'another antipsychotic' ‐ not reported by drug‐group |
| Yagi 1976 | Allocation: randomised. Blindness: double. Participants: those with schizophrenia. Interventions: carpipramine and chlorpromazine vs. perphenazine and chlorpromazine vs. placebo and chlorpromazine. |
| Zhong 2001 | Allocation: random Participants: schizophrenia (ICD‐10 and CCMD‐2‐R) Intervention: perphenazine versus 1‐stepholidine (Chinese herbal medicine) |
| Zhu 2009 | Allocation: random Participants: schizophrenia Intervention: psychological care + perphenazine versus perphenazine alone |
Diagnostic tools: CCMD‐2‐R – Chinese Classification of Mental Disorders, Second Edition, Revised ICD‐10‐R ‐ International Classification of Diseases, tenth revision
Characteristics of studies awaiting assessment [ordered by study ID]
Casey 1960.
| Methods | |
| Participants | |
| Interventions | |
| Outcomes | |
| Notes | No usable data ‐ more information required. |
Covell 2004.
| Methods | |
| Participants | |
| Interventions | |
| Outcomes | |
| Notes | No usable data ‐ more information required (authors contacted October 2013) but no response. |
Ferziger 2010.
| Methods | |
| Participants | |
| Interventions | |
| Outcomes | |
| Notes | No usable data ‐ more information required (authors contacted October 2013) but no response. |
Freedman 1961.
| Methods | |
| Participants | |
| Interventions | |
| Outcomes | |
| Notes | No usable data ‐ more information required. |
Freyhan 1959.
| Methods | |
| Participants | |
| Interventions | |
| Outcomes | |
| Notes | No usable data ‐ more information required. |
Goldstein 1969.
| Methods | |
| Participants | |
| Interventions | |
| Outcomes | |
| Notes | Unclear if randomised, double‐blind stated ‐ more information required. |
Hollister 1967.
| Methods | |
| Participants | |
| Interventions | |
| Outcomes | |
| Notes | Unclear if randomised, double‐blind stated ‐ more information required. |
Kellam 1971.
| Methods | |
| Participants | |
| Interventions | |
| Outcomes | |
| Notes | No usable data ‐ more information required. |
Mahmoud 2004.
| Methods | |
| Participants | |
| Interventions | |
| Outcomes | |
| Notes | No usable data ‐ more information required, study authors contacted but no response. |
NCT 2011.
| Methods | |
| Participants | |
| Interventions | |
| Outcomes | |
| Notes | No usable data ‐ more information required. |
Povlsen 1987.
| Methods | |
| Participants | |
| Interventions | |
| Outcomes | |
| Notes | Requires data extraction (Danish). |
Schulsinger 1958.
| Methods | |
| Participants | |
| Interventions | |
| Outcomes | |
| Notes | Requires data extraction (Danish). |
Szafranski 1999.
| Methods | |
| Participants | |
| Interventions | |
| Outcomes | |
| Notes | No usable data ‐ more information required, authors contacted but no response. |
Vinar 1968.
| Methods | |
| Participants | |
| Interventions | |
| Outcomes | |
| Notes | No usable data ‐ more information required. |
Differences between protocol and review
Minor changes:
1. Additions made to outcomes of interest; economic and patient‐important outcomes added, including:
6. Economic
6.1 Average change in total cost of medical and mental health care 6.2 Total indirect and direct costs 6.3 Direct resource use: 6.3.1 Outpatients ‐ number of contacts (GP consultation, psychiatrist, psychologists, psychiatric nurse, counsellor, social worker) 6.3.2 Hospitalisation (taking battery of tests, patients’ physical, psychiatric and psychological profile and psychological assessment, number of days, relapse) 6.3.3 Medication (different types of antipsychotic drugs to include dose and frequency, treatment of side effects) 6.3.4 Psychological therapies (different types of psychological therapies to include session numbers and frequency) 6.3.5 Other resources (day centres, night shelter) and transportation for medical care visits 6.4 Indirect resource use: 6.4.1 Family, relative and friends resources 6.4.2 Police, criminal justice system 6.4.3 Benefits paid, social security payments 6.4.4 Employment agency workers, absence from work, loss of productivity 6.5 Cost‐effectiveness ratios represented by ICER (incremental cost‐effectiveness ratio) 6.6 Cost‐utilities represented by incremental costs per QALY (quality‐adjusted life year) or DALYs (disability‐adjusted life year) 6.7 Cost benefit represented by net Benefit Ratio, others.
7. Service use and hospitalisation
7.1 Hospital admission 7.2 Medication use 7.3 Engagement with health services
8. Satisfaction
8.1 Satisfaction with treatment 8.2 Satisfaction with care 8.3 Quality of life outcomes 8.4 Employment status"
Contributions of authors
Benno Hartung ‐ protocol development, data extraction, writing of the report. Stephanie Sampson ‐ quality‐assessed; data extracted new studies; wrote update review report Stefan Leucht ‐ protocol development, verification of data extraction, writing of the report.
Sources of support
Internal sources
Freistaat Bayern, Germany.
External sources
-
NIHR, UK.
Cochrane Collaboration Programme Grant 2011, UK
Declarations of interest
Benno Hartung ‐ none known. Stephanie Sampson ‐ none known. Stefan Leucht ‐ S. Leucht has received lecture honoraria from EliLilly, Lundbeck, Pfizer, Janssen, Johnson and Johnson, BristolMyersSquibb, Lundbeck, Roche, SanofiAventis, ICON and Abbvie; honoraria for consulting from Roche, Janssen, Lundbeck, and EliLilly; for the preparation of educational material and publications from the Lundbeck Institute and Roche. EliLilly has provided medication for a clinical trial led by SL as principal investigator.
New search for studies and content updated (no change to conclusions)
References
References to studies included in this review
Amakusa 1973 {published data only}
- Amakusa T, Majima T. The comparison of therapeutic effects of FK‐880 (sulpiride) and perphenazine in schizophrenia by a double‐blind controlled study. Juntendo Igako 1973;19(2):239‐49. [Google Scholar]
Bennett 1961 {published data only}
- Bennett JL, Kooi KA. Five phenothiazine derivates. evaluation and toxicity studies. Archives of General Psychiatry 1961;4:413‐8. [Google Scholar]
CATIE 2005 {published data only}
- CATIE ‐ Schizophrenia Trial. National Institute of Mental Health (NIMH), NCT00014001 Study start: December 2000; Study completion: December 2004.
- Addington D. Impact of second generation antipsychotic drugs and perphenazine on depressive symptoms in a randomized trial of treatment for chronic schizophrenia. Proceedings of the 164th annual general meeting of the American Psychiatric Association; 2011 May 14‐18; Honolulu, Hawaii 2011. [CATIE]
- Addington DE, Mohamed S, Rosenheck RA, Davis SM, Stroup TS, McEvoy JP, et al. Impact of second‐generation antipsychotic drugs and perphenazine on depressive symptoms in a randomized trial of treatment for chronic schizophrenia. Journal of Clinical Psychiatry 2011; Vol. 72, issue 1:75‐80. [CATIE] [DOI] [PMC free article] [PubMed]
- Anon. Antipsychotic medications for schizophrenia on equal footing in improving patients' thinking skills. Science Update 2007:1. [ANON: 2007 h]
- Ascher‐Svanum H, Nyhuis AW, Faries DE, Heiler L, Kinon BJ. Treatment discontinuation following randomization to open‐label olanzapine, risperidone or typical antipsychotic drugs during a one‐year treatment for schizophrenia. Clinical Schizophrenia & Related Psychoses. United States, 2008; Vol. 2, issue 3:226‐34. [ASCHER‐SVANUM: 2008]
- Bick P, Knoesen N, Castle D. Clinical implications of the CATIE schizophrenia trials: day‐to‐day management lessons for Australasian psychiatrists. Australasian Psychiatry 2007; Vol. 15, issue 6:465‐9. [CATIE: 2003] [DOI] [PubMed]
- Essock SM, Covell NH, Davis SM, Stroup TS, Rosenheck RA, Lieberman JA. Effectiveness of switching antipsychotic medications. American Journal of Psychiatry 2006; Vol. 163, issue 12:2090‐5. [ESSOCK: 2006 b] [DOI] [PubMed]
- Gutierrez‐Suela F. Selection of antipsychotic drugs in a psychiatric hospital based on the results of CATIE study. Farmacia Hospitalaria 2008; Vol. 32, issue 2:83‐90. [GUTIERREZ‐SUELA: 2008] [PubMed]
- Haddad PM, Dursun S. Selecting antipsychotic drugs in schizophrenia: lessons from CATIE. Journal of Psychopharmacology (Oxford, England) 2006; Vol. 20, issue 3:332‐4. [CATIE: 2003] [DOI] [PubMed]
- Hermes E, Nasrallah H, Davis V, Meyer J, McEvoy J, Goff D, et al. The association between weight change and symptom reduction in the CATIE schizophrenia trial. Schizophrenia Research 2011; Vol. 128, issue 1‐3:166‐70. [CATIE] [DOI] [PMC free article] [PubMed]
- Hermes E, Rosenheck R. Choice of randomization to clozapine versus other second generation antipsychotic drugs in the CATIE schizophrenia trial. Journal of Psychopharmacology (Oxford, England) 2012; Vol. 26, issue 9:1194‐200. [CATIE] [DOI] [PMC free article] [PubMed]
- Hermes ED, Sokoloff D, Stroup TS, Rosenheck RA. Minimum clinically important difference in the positive and negative syndrome scale with data from the clinical antipsychotic trials of intervention effectiveness (CATIE). Journal of Clinical Psychiatry 2012; Vol. 73, issue 4:526‐32. [CATIE] [DOI] [PMC free article] [PubMed]
- Jin Y, Pollock BG, Coley K, Miller D, Marder SR, Florian J, et al. Population pharmacokinetics of perphenazine in schizophrenia patients from CATIE: impact of race and smoking. Journal of Clinical Pharmacology 2010; Vol. 1, issue 1:73‐80. [CATIE] [DOI] [PMC free article] [PubMed]
- Keefe R. Neurocognitive efficacy in patients with chronic schizophrenia. European Neuropsychopharmacology 2006;16(Suppl 4):S577. [Google Scholar]
- Keefe RSE, Bilder RM, Davis SM, Harvey PD, Palmer BW, Gold JM, et al. CATIE Investigators, Neurocognitive Working Group. Neurocognitive effects of antipsychotic medications in patients with chronic schizophrenia in the CATIE trial. ArchIves of General Psychiatry 2007;64(6):633‐47. [DOI] [PubMed] [Google Scholar]
- Levine SZ, Rabinowitz J, Ascher‐Svanum H, Faries D, Lawson T. Extent of attaining and maintaining symptom remission by antipsychotic medication in the treatment of chronic schizophrenia: Evidence from the CATIE study. Value in Health 2011; Vol. 7, issue 7:14. [CATIE] [DOI] [PubMed]
- Levine SZ, Rabinowitz J, Ascher‐Svanum H, Faries DE, Lawson AH. Extent of attaining and maintaining symptom remission by antipsychotic medication in the treatment of chronic schizophrenia: Evidence from the CATIE study. Schizophrenia Research 2011; Vol. 133, issue 1‐3:42‐6. [CATIE] [DOI] [PubMed]
- Lieberman JA, Stroup TS, McEvoy JP, Swartz MA, Rosenheck RA, Perkins DO, et al. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. New England Journal of Medicine 2005;353(12):1209‐23. [DOI] [PubMed] [Google Scholar]
- Lin LA. Comparing antipsychotic treatments for schizophrenia: A health state approach. Dissertation Abstracts International: Section B: The Sciences and Engineering 2012; Vol. 73, issue 2‐B:863. [CATIE] [DOI] [PubMed]
- McEvoy JP, Lieberman JA, Stroup TS, Davis SM, Meltzer HY, Rosenheck RA, et al. CATIE Investigators. Effectiveness of clozapine versus olanzapine, quetiapine, and risperidone in patients with chronic schizophrenia who did not respond to prior atypical antipsychotic treatment. American Journal of Psychiatry 2006;163(4):600‐10. [DOI: 10.1176/appi.ajp.163.4.600] [DOI] [PubMed] [Google Scholar]
- Meyer JM, McEvoy JP, Davis VG, Goff DC, Nasrallah HA, Davis SM, et al. Inflammatory markers in schizophrenia: comparing antipsychotic effects in phase 1 of the clinical antipsychotic trials of intervention effectiveness study. Biological Psychiatry 2009; Vol. 66, issue 11:1013‐22. [CATIE] [DOI] [PMC free article] [PubMed]
- Miller DD, Caroff SN, Davis SM, Rosenheck RA, McEvoy JP, Saltz BL, et al. Extrapyramidal side‐effects of antipsychotic drugs in a randomised trial. British Journal of Psychiatry 2008; Vol. 193, issue 4:279‐88. [CATIE: 2003] [DOI] [PMC free article] [PubMed]
- Nasrallah HA. Metabolic findings from the CATIE trial and their relation to tolerability. CNS Spectrums 2006; Vol. 11, issue 7 Suppl 7:32‐9. [CATIE] [DOI] [PubMed]
- Olfson M, Ascher‐Svanum H, Faries DE, Marcus SC. Predicting psychiatric hospital admission among adults with schizophrenia. Psychiatric Services (Washington, D.C.) 2011; Vol. 62, issue 10:1138‐45. [CATIE] [DOI] [PubMed]
- Penn DL, Keefe RSE, Davis SM, Meyer PS, Perkins DO, Losardo D, et al. The effects of antipsychotic medications on emotion perception in patients with chronic schizophrenia in the CATIE trial. Schizophrenia Research 2009; Vol. 115, issue 1:17‐23. [CATIE] [DOI] [PMC free article] [PubMed]
- Perlick DA, Rosenheck RA, Kaczynski R, Swartz MS, Canive JM, Lieberman JA. Impact of antipsychotic medication on family burden in schizophrenia: longitudinal results of CATIE trial. Schizophrenia Research 2010; Vol. 116, issue 2‐3:118‐25. [CATIE] [DOI] [PubMed]
- Resnick SG, Rosenheck RA, Canive JM, Souza CD, Stroup TS, McEvoy J, et al. Employment outcomes in a randomized trial of second‐generation antipsychotic drugs and perphenazine in the treatment of individuals with schizophrenia. Journal of Behavioural Health Services and Research 2008;35(2):215‐25. [DOI] [PubMed] [Google Scholar]
- Rosenheck R, Swartz M, McEvoy J, Stroup TS, Davis S, Keefe RS, et al. Second‐generation antipsychotic drugs: Reviewing the cost‐effectiveness component of the CATIE trial. Expert Review of Pharmacoeconomics & Outcomes Research 2007; Vol. 7, issue 2:103‐11. [CATIE] [DOI] [PubMed]
- Rosenheck RA, Davis VG, Davis SM, Stroup S, McEvoy J, Swartz M, et al. Can a nonequivalent choice of dosing regimen bias the results of flexible dose double blind trials? The CATIE schizophrenia trial. Schizophrenia Research 2009; Vol. 113, issue 1:12‐8. [CATIE] [DOI] [PMC free article] [PubMed]
- Rosenheck RA, Leslie DL, Sindelar J, Miller EA, Lin H, Stroup TS, et al. Cost‐effectiveness of second‐generation antipsychotic drugs and perphenazine in a randomized trial of treatment for chronic schizophrenia. American Journal of Psychiatry 2006;163(12):2080‐9. [DOI] [PubMed] [Google Scholar]
- Schulte PFJ, Haan L. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. Tijdschrift voor Psychiatrie 2006;48(3):243‐4. [Google Scholar]
- Stroup S, Appelbaum P, Swartz M, Patel M, Davis S, Jeste D, et al. Decision‐making capacity for research participation among individuals in the CATIE schizophrenia trial. Schizophrenia Research 2005; Vol. 80, issue 1:1‐8. [CATIE] [DOI] [PubMed]
- Stroup TS. Comparison of ziprasidone versus other atypical drugs in prospectively defined, unresponsive patients. Proceedings of the 159th Annual Meeting of the American Psychiatric Association; 2006 May 20‐25, Toronto, Canada. 2006.
- Stroup TS, Leiberman JA, McEvoy JP, Swartz MS, Davis SM. Effectiveness of olanzapine, quetiapine, risperidone, and ziprasidone in patients with chronic schizophrenia following discontinuation of a previous atypical antipsychotic. American Journal of Psychiatry 2006;163:611‐22. [DOI] [PubMed] [Google Scholar]
- Stroup TS, Lieberman JA, McEvoy JP, Davis SM, Swartz MS, Keefe RSE, et al. Results of phase 3 of the CATIE schizophrenia trial. Schizophrenia Research 2009; Vol. 107, issue 1:1‐12. [CATIE: 2003] [DOI] [PMC free article] [PubMed]
- Stroup TS, Lieberman JA, McEvoy JP, Swartz MS, Davis SM, Capuano GA, et al. Effectiveness of olanzapine, quetiapine, and risperidone in patients with chronic schizophrenia after discontinuing perphenazine: a CATIE study. American Journal of Psychiatry 2007;164(3):415‐27. [DOI] [PubMed] [Google Scholar]
- Stroup TS, McEvoy JP, Swartz MS, Byerly MJ, Glick ID, Canive JM, et al. The National Institute of Mental Health Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) project: schizophrenia trial design and protocol developme. Schizophrenia Bulletin 2003;29(1):15‐31. [DOI] [PubMed] [Google Scholar]
- Swartz MS, Perkins DO, Stroup TS, Davis SM, Capuano G, Rosenheck RA, et al. Effects of antipsychotic medications on psychosocial functioning in patients with chronic schizophrenia: findings from the NIMH CATIE study. American Journal of Psychiatry 2007;164(3):428‐36. [DOI] [PubMed] [Google Scholar]
- Swartz MS, Stroup TS, McEvoy JP, Davis SM, Rosenheck RA, Keefe RS, et al. What CATIE found: results from the schizophrenia trial. Psychiatry Online May 2008;59(5):500‐06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swartz MS, Wagner HR, Swanson JW, Stroup TS, McEvoy JP, Reimherr F, et al. The effectiveness of antipsychotic medications in patients who use or avoid illicit substances: Results from the CATIE study. Schizophrenia Research 2008;100(1‐3):39‐52. [DOI] [PubMed] [Google Scholar]
- Tsai HT, Caroff SN, Miller DD, McEvoy J, Lieberman JA, North KE, et al. A candidate gene study of tardive dyskinesia in the CATIE schizophrenia trial. American Journal of Medical Genetics 2010; Vol. 153B, issue 1:336‐40. [CATIE] [DOI] [PMC free article] [PubMed]
- Wessels AM, Bies RR, Pollock BG, Schneider LS, Lieberman JA, Stroup S, et al. Population pharmacokinetic modeling of ziprasidone in patients with schizophrenia from the CATIE study. Journal of Clinical Pharmacology 2011; Vol. 51, issue 11:1587‐91. [CATIE] [DOI] [PubMed]
Chouinard 1975 {published data only}
- Chouinard G, Annable L, Serrano M, Albert JM, Charette R. Amitriptyline‐perphenazine interaction in ambulatory schizophrenic patients. A controlled study of drug interaction. Archives of General Psychiatry 1975;32:1295‐307. [DOI] [PubMed] [Google Scholar]
Chouinard 1977 {published data only}
- Chouinard G, Annable L. Phenothiazine‐induced ECG abnormalities. Effect of a glucose load. Archives of General Psychiatry 1977;34(8):951‐4. [DOI] [PubMed] [Google Scholar]
Collins 1967 {published data only}
- Collins AD, Dundas J. A double‐blind trial of amitriptyline‐perphenazine, perphenazine and placebo in chronic withdrawn inert schizophrenics. British Journal of Psychiatry 1967;113:1425‐9. [DOI] [PubMed] [Google Scholar]
Dehnel 1968 {published data only}
- Dehnel LL, Vestre ND, Schiele BC. A controlled comparison of clopenthixol and perphenazine in a chronic schizophrenic population. Current Therapeutic Research, Clinical and Experimental 1968; Vol. 10, issue 4:169‐76. [DEHNEL: 1968] [PubMed]
Eckmann 1984 {unpublished data only}
- Eckmann F, Weber J. No English version available [Vergleich zwischen Perphenazin und Benperidol. Eine kontrollierte Doppelblind‐Studie]. unpublished manuscript 1984.
Fruensgaard 1978 {published data only}
- Fruensgaard K, Wollenberg J, Hansen KM, Fensbo C, Sihm F. Loxapine versus perphenazine in psychotic patients. A double‐blind, randomized, multicentre trial. Current Medical Research and Opinion 1978;5(8):601‐7. [DOI] [PubMed] [Google Scholar]
Hanlon 1964 {published data only}
- Hanlon TE, Nussbaum K, Wittig B, Hanlon DD, Kurland AA. The comparative effectiveness of amitriptyline, perphenazine, and their combination in the treatment of chronic psychotic female patients. Journal of New Drugs 1964;4:52‐60. [DOI] [PubMed] [Google Scholar]
Hanlon 1965 {published data only}
- Hanlon TE, Michaux MH, Ota KY, Shaffer JW, Kurland AA. The comparative effectiveness of eight phenothiazines. Psychopharmacologia 1965;7(2):89‐106. [DOI] [PubMed] [Google Scholar]
Hoyberg 1993 {published data only}
- Hoyberg OJ, Fensbo C, Remvig J, Lingjaerde O, Sloth Nielsen M, Salvesen I. Risperidone versus perphenazine in the treatment of chronic schizophrenic patients with acute exacerbations. Acta Psychiatrica Scandinavica 1993;88(6):395‐402. [DOI] [PubMed] [Google Scholar]
Imai 1980 {published data only}
- Imai H, Nakamura M, Fukui Y, et al. Comparison of efficacy of zotepine and perphenazine in schizophrenia by double‐blind, controlled study. Shinkei Seishin Yakuri 1980;2(3):285‐99. [Google Scholar]
Itoh 1969 {published data only}
- Itoh H, Okamoto M, Miura S, Suzuki Y, Takemasa K, Shigeta M, et al. A comparison between the clinical effectiveness of a dibenzothiazepine derivative and a phenothiazine derivative in schizophrenia. A controlled double blind study using clotiapine (W 130) and perphenazine. Seishin Igaku 1969;11(6):465‐75. [Google Scholar]
Itoh 1969 II {published data only}
- Itoh H, Miura S, Asai M, et al. Comparison of thiothixene and perphenazine in schizophrenic patients using double‐blind technique. Seishin Igaku 1969;11(4):284‐97. [Google Scholar]
Itoh 1969 III {published data only}
- Itoh H, Okamoto M, Miura S, et al. Comparison of a butyrophenone derivative (methylperidol) and a phenothiazine derivative (perphenazine) in schizophrenic patients using double‐blind technique: statistical analysis by sequential method and wilcoxon test. Seishin Igaku 1969;11(2):131‐42. [Google Scholar]
Itoh 1976 {published data only}
- Itoh H, Miura S, Yagi G, Ogita K, Ohtsuka N, Koga Y, et al. Comparison of clinical effects of penfluridol, a long‐acting oral neuroleptic, and perphenazine in schizophrenia using double‐blind technique. Rinsho Hyoka 1976;4(1):101‐29. [Google Scholar]
Kane 2003 {published data only}
- Bristol‐Myers S. A multicenter, randomized, double‐blind study of flexible doses of aripiprazole versus perphenazine in the treatment of patients with treatment‐resistant schizophrenia. http://www.clinicalstudyresults.org/ 2004. [BRISTOL‐MYERS: 2004]
- Gismondi R, Meltzer H, Kujawa M, Carson W, Stringfellow J, Iwamoto T, et al. Aripiprazole versus perphenazine in treatment‐resistant schizophrenia. Proceedings of the 24th Collegium Internationale Neuro‐Psychopharmacologicum Congress; 2004 June 20–24; Paris, France 2004. [KANE: 2003 a]
- Kane J, Carson W, Kujawa M, Stringfellow J, Marcus R, Sanchez R, et al. Aripiprazole in treatment‐resistant schizophrenia: a 6‐week double‐ blind comparison study versus perphenazine. Schizophrenia Research 2004; Vol. 67, issue 1:155‐6. [KANE: 2003 a]
- Kane J, Carson WH, Kujawa M, Stringfellow J, Marcus R, Sanchez R, et al. Aripiprazole in treatment‐resistant schizophrenia: A 6‐week double‐blind comparison study versus perphenazine. Conference proceedings. Elsevier Science Bv, 2004:155‐6. [KANE: 2004a]
- Kane J, McQuade R, Jody D, Carson W, Kujawa M, Stringfellow J, et al. Aripiprazole in treatment‐resistant schizophrenia: a 6‐week double‐blind comparison study vs perphenazine. Conference proceedings: 12th Biennial Winter Workshop on Schizophrenia, Davos, Switzerland 2004. [KANE: 2003 a]
- Kane JM, Meltzer HY, Carson WH Jr, McQuade RD, Marcus RN, Sanchez R. Aripiprazole Study Group. Aripiprazole for treatment‐resistant schizophrenia: results of a multicenter, randomized, double‐blind, comparison study versus perphenazine. Journal of Clinical Psychiatry 2007; Vol. 68, issue 2:213‐23. [KANE: 2007] [PubMed]
- McQuade R, Jody D, Kane J, Carson W, Kujawa M, Stringfellow J, et al. Efficacy and safety of aripiprazole versus perphenazine in treatment‐resistant schizophrenia. European Neuropsychopharmacology 2003; Vol. 13, issue 4:S326. [KANE: 2003 a]
- Modell S, Jody D, Kujawa M, Carson W, Stringfellow J, Iwamoto T, et al. Efficacy of aripiprazole and perphenazine in severe schizophrenia resistant to treatment with atypical antipsychotic drugs. European Neuropsychopharmacology 2004; Vol. 14, issue Suppl 3:S265. [KANE: 2003 a]
- Sanchez R, Meltzer HY, Marcus RN, Stringfellow J, Carson WH, Kane JM. Aripiprazole versus perphenazine in Treatment ‐ Resistant Schizophrenia. Schizophrenia Bulletin 2005; Vol. 31:502. [KANE: 2003 a]
Kurihara 1983 {published data only}
- Kurihara M, Ito H, Kato N, et al. Clinical evaluation of clocapramine (clofekton) in schizophrenia: a double blind comparison of clocapramine, haloperidol and perphenazine. Rinsho Seishin Igaku 1983;12(4):519‐38. [Google Scholar]
Kurland 1961 {published data only}
- Kurland AA, Hanlon TE, Tatom MH, Ota KY, Simopoulos AM. The comparative effectiveness of six phenothiazine compounds, phenobarbital and inert placebo in the treatment of acutely ill patients: global measures of severity of illness. Journal of Nervous and Mental Disease 1961;133(1):1‐18. [DOI] [PubMed] [Google Scholar]
- Kurland AA, Hanlon TE, Tatom MH, Simopoulos AM. Comparative studies of the phenothiazine tranquilizers: methodological and logistical considerations. Journal of Nervous and Mental Disease 1961;132:61‐74. [PubMed] [Google Scholar]
Lepola 1989 {published data only}
- Lepola U, Koskinen T, Rimon R, Salo H, Gordin A. Sulpiride and perphenazine in schizophrenia. A double‐blind clinical trial. Acta Psychatrica Scandinavica 1989;80(1):92‐6. [DOI] [PubMed] [Google Scholar]
Naukkarinen 2000 {published data only}
- Eli Lilly, Company. HGBJ: olz v perphenazine. Eli Lilly and Company 2001.
- Naukkarinen H, Rimon R, Katila H, Riihikangas R, Heikkilä L. Olanzapine and perphenazine in schizophrenia. Schizophrenia Research 2000;41(1):190. [Google Scholar]
- Rimon RH. Olanzapine versus perphenazine in the treatment of schizophrenia: a double‐blind study. Schizophrenia research 2004;67(1):164‐5. [Google Scholar]
Remvig 1987 {published data only}
- Remvig J, Larsen H, Rask P, Skausig OB, Skov S, Strömgren LS. Zuclopenthixol and perphenazine in patients with acute psychotic states. A double‐blind multicentre study. Pharmacopsychiatry 1987;20:147‐54. [DOI] [PubMed] [Google Scholar]
Sun 2000 {published data only}
- Sun Z, Liu B, Li Z, Zhang D, et al. Clozapine combined with perphenazine in the treatment of schizophrenia [氯氮平联用奋乃静治疗精神分裂症对照研究]. Hebei Mental Health 2000; Vol. 13, issue 2:142‐4. [SUN: 2000 (Perp)]
Takahashi 1982 {published data only}
- Takahashi R, Inanaga K, Samejima K, Sarai K, Asada S, Otsuki S, et al. Comparison of efficacy of a new butyrophenone derivative, timiperone and perphenazine in schizophrenia by a multicentre controlled study. Journal of International Medical Research 1982;10(4):257‐67. [DOI] [PubMed] [Google Scholar]
Van Praag 1976 {published data only}
- Praag HM, Korf J, Dolc LCW. Clozapine versus perphenazine: the value of the biochemical mode of action of neuroleptics in predicting their therapeutic activity. British Journal of Psychiatry 1976;129:547‐55. [DOI] [PubMed] [Google Scholar]
Wang 2008a {published data only}
- Wang XD, Wang YH, Feng TM, Liu ML, Zhang SX. Controlled study of risperidone in elderly treatment of first‐episode schizophrenia [利培酮治疗老年期首发精神分裂症对照研究]. Journal of Psychiatry 2008;21(2):141. [Google Scholar]
Wang 2008b {published data only}
- Wang RF, Yu X, Song ZD. Schizophrenia olanzapine and perphenazine in the treatment of positive symptoms [奥氮平与奋乃静治疗阳性症状为主的精神分裂症对照研究]. Journal of Psychiatry 2008;21(6):423‐4. [Google Scholar]
Wang 2008c {published data only}
- Wang X, Wu J, Chen Z, Zhang JF, et al. Elderly first controlled study of quetiapine in the treatment of schizophrenia [喹硫平治疗首发老年期精神分裂症对照研究]. Journal of Psychiatry 2008;21(6):463‐4. [Google Scholar]
Woggon 1978 {published data only}
- Woggon B. Effects and side‐effects of bromperidol in comparison with other antipsychotic drugs. Acta Psychiatrica Belgica 1978;78(1):155‐72. [PubMed] [Google Scholar]
- Woggon B, Angst J. Double‐blind comparison of bromperidol and perphenazine. International Pharmacopsychiatry 1978;13:165‐76. [DOI] [PubMed] [Google Scholar]
Zhang 2010 {published data only}
- Zhang JL, Zhang J, Shen B. Aripiprazole orally disintegrating tablets in the treatment of schizophrenia in old age were observed [阿立哌唑口腔崩解片治疗老年期精神分裂症疗效的对照观察]. Chinese Journal of Modern Drug Application 2010;4(14):119‐20. [Google Scholar]
References to studies excluded from this review
Affleck 1969 {published data only}
- Affleck JW, Cooper AJ, Forrest AD, Smythies JR, Zealley AK. Penicillamine and schizophrenia ‐ a clinical trial. British Journal of Psychiatry 1969;115(519):173‐6. [DOI] [PubMed] [Google Scholar]
Ahlfors 1980 {published data only}
- Ahlfors UG, Dencker SJ, Gravem A, Remvig J. Clopenthixol decanoate and perphenazine enanthate in schizophrenic patients. A double‐blind Nordic multicentre trial. Acta Psychiatrica Scandinavica Supplementum 1980;279:77‐91. [DOI] [PubMed] [Google Scholar]
Akimoto 1966 {published data only}
- Akimoto H, Shimazaki T, Shibata N, Sato Y, Takahashi R, Naruse H, et al. Current status of pharmacotherapy in schizophrenia. Folia Psychiatrica et Neurologica Japonica 1966;20(1):1‐8. [DOI] [PubMed] [Google Scholar]
Angst 1975 {published data only}
- Angst J, Woggon B. Clinical study on five depot neuroleptics. Comparison of effective profiles of fluphenazine decanoate, flusipirilene, penfluridol, perphenazine enanthate and pipothiazine palmitate. Arzneimittel Forschung 1975;25(2):267‐70. [PubMed] [Google Scholar]
Anonymous 1966 {published data only}
- Anonymous. Schizophrenic and other psychotic reactions. Medical Letter 1966:15‐6.
Asada 1976 {published data only}
- Asada S, Ishimaru T, Kubo S, Kodama H, Masuda K. Study of the clinical effects of sulpiride and perphenazine in 82 schizophrenic patients by the double‐blind method [Etude des effets cliniques du sulpiride et de la perphenazine chez 82 schizophrenes par la methode du double aveugle]. Encephale 1976;2(1):73‐83. [PubMed] [Google Scholar]
Bakke 1973 {published data only}
- Bakke O. Clinical experience with depot neuroleptics. Acta Psychiatrica Scandinavica Supplementum 1973;246:32‐41. [DOI] [PubMed] [Google Scholar]
Bandelow 1992 {published data only}
Becker 1970 {published data only}
- Becker RE. Evaluation of an amitriptyline‐perphenazine combination in chronic schizophrenia. American Journal of Psychiatry 1970;127(6):827‐131. [DOI] [PubMed] [Google Scholar]
Bjerkelund 1965 {published data only}
- Bjerkelund C, Nitter‐Hauge S, Jakobsen E. Perphenazine (trilafon) in the prophylaxis of nausea and vomiting following acute myocardial infarct. Acta Medica Scandinavica 1965;177:729‐37. [DOI] [PubMed] [Google Scholar]
Böhlau 1985 {published data only}
- Böhlau V, Weber J. No English title available [Therapie dementieller Prozesse bei Alterspatienten]. Therapiewoche 1985;35(47):5445‐8. [Google Scholar]
Cannistra 1959 {published data only}
- Cannistra F, Abrams AA. Perphenazine in obstetrics. Evaluation of its use to replace an analgesic. Obstetrics and Gynecology 1959;14(3):337‐41. [PubMed] [Google Scholar]
Ceskova 1994 {published data only}
- Ceskova E, Svestka J. Risperidone vs. perphenazine ‐ a double‐blind comparison and prolactine plasma levels [Risperidone gegen Rispehenazine ‐ ein doppelt‐blinder Vergleich und Prolaktin‐Plasmaspiegel]. Psychiatria Danubina 1994;6(3‐4):151‐5. [Google Scholar]
Ceskova 1996 {published data only}
- Ceskova E, Svestka J. Efficacy and tolerability of risperidone in different dose levels [Ucinnost a snasenlivost risperidonu pri ruznem davkovani (sdeleni z praxe)]. Ceskoslovenska Psychiatrie 1996;92(1):50‐6. [PubMed] [Google Scholar]
Clarke 1981 {published data only}
- Clarke JMC. Amitriptyline and perphenazine (triptafen DA) in chronic pain. Anaesthesia 1981;36:210‐2. [DOI] [PubMed] [Google Scholar]
Cohen 1958 {published data only}
Cooper 1979 {published data only}
- Cooper SF, Dugal R, Elie R, Albert JM. Metabolic interaction between amitriptyline and perphenazine in psychiatric patients. Neuro‐Psychopharmacology 1979;3:369‐76. [DOI] [PubMed] [Google Scholar]
Den Boer 2000 {published data only}
- Boer JA, Vahlne J‐O, Post P, Heck AH, Daubenton F, Olbrich R. Ritanserin as add‐on medication to neuroleptic therapy for patients with chronic or subchronic schizophrenia. Human Psychopharmacology 2000;15:179‐89. [DOI] [PubMed] [Google Scholar]
Dencker 1994 {published data only}
- Dencker SJ, Gios I, Martenson E, Norden T, Nyberg G, Persson R, et al. A long‐term cross‐over pharmacokinetic study comparing perphenazine decanoate and haloperidol decanoate in schizophrenic patients. Psychopharmacology 1994;114(1):24‐30. [DOI] [PubMed] [Google Scholar]
Eklund 1976 {published data only}
- Eklund KL. A double‐blind comparison study between penfluridol and perphenazine in acute schizophrenic patients. Nordisk Psykiatrisk Tidsskrift 1976;30(5):384‐91. [Google Scholar]
Erb 1982 {published data only}
- Erb RJ, Stoltman WP. Serum prolactin level increase in normal subjects following administration of perphenazine oral dosage forms: possible application to bioavailibility testing. Journal of Pharmaceutical Sciences 1982;71(8):883‐8. [DOI] [PubMed] [Google Scholar]
Eufe 1979 {published data only}
- Eufe R, Wegener G. Double‐blind comparison of two depot neuroleptics (perphenazine enanthate and flupentixol enanthate) in chronic schizophrenia. Nervenarzt 1979;50(8):534‐9. [PubMed] [Google Scholar]
Fahn 1964 {published data only}
- Fahn S. Treatment of choreic movements with perphenazine. Diseases of The Nervous System 1972;33:653‐8. [PubMed] [Google Scholar]
Farzin 2005 {published data only}
- Farzin D, Hosseini SH, Shafaat A. A randomized double blind clinical trial in famotidine adjuvant therapy in schizophrenia. Iranian Journal of Medical Sciences 2005; Vol. 30, issue 2:59‐62. [FARZIN: 2005]
Fitzgerald 1969 {published data only}
- Fitzgerald CH. A double‐blind comparison of haloperidol with perphenazine in acute psychiatric patients. Current Therapeutic Research 1969;11(8):515‐9. [PubMed] [Google Scholar]
Galdi 1988 {published data only}
- Galdi J, Bonato RB. Relationship of adverse drug reactions to length of hospital‐stay in genetically subgrouped schizophrenics. Canadian Journal of Psychiatry 1988;33(9):816‐8. [DOI] [PubMed] [Google Scholar]
Gerlach 1977 {published data only}
- Gerlach J, Rasmussen PT, Hansen L, Kristjansen P. Antiparkinsonian agents and long‐term neuroleptic treatment. Effect of G31.406, orphenadrine, and placebo on parkinsonism, schizophrenic symptoms, depression and anxiety. Acta Psychiatrica Scandinavica 1977;55(4):251‐60. [DOI] [PubMed] [Google Scholar]
Gerlach 1988 {published data only}
- Gerlach J. Tardive dyskinesia:Pathophysiological mechanisms and clinical trials. L'Encephale 1988;14:227‐232. [PubMed] [Google Scholar]
Gerson 1964 {published data only}
- Gerson IM, Chat E, Twigger NA. Clinical trial of a potentiated diketopiperazine derivative as a psychopharmacological agent for the treatment of psychotic patients. American Journal of Psychiatry 1964;121:179‐81. [DOI] [PubMed] [Google Scholar]
Gianelli 1990 {published data only}
- Gianelli A, Rabboni M, Zarattini F. No English title available [Profili clinici di azione, indicazioni preferenziali, effeti terapeutici e contraindicazioni di tre neuroletti depot in trial multicentro di confronto]. Rivista di Psichiatria 1990;25(1):7‐24. [Google Scholar]
Hadlik 1970 {published data only}
- Hadlik J, Svestka J, Nahunek K, Rodova A. Controlled studies with thiothixene and perphenzine in schizophrenic psychoses [Kontrolovana studie s thiothixenem a perfenazinem u psychoz schizofrenniho okruhu]. Activitas Nervosa Superior 1970;12(1):60‐1. [PubMed] [Google Scholar]
Hanlon 1966 {published data only}
Hansen 1979 {published data only}
- Hansen BL, Elley J, Christensen RT, Laersen NE, Naestoft J, Hvidberg EF. Plasma levels of perphenazine and its metabolites during simultaneous treatment with anticholinergic drugs. British Journal of clinical Pharmacology 1979;7:75‐80. [DOI] [PMC free article] [PubMed] [Google Scholar]
Haran 1960 {published data only}
- Haran T. Perphenazine (Fentazine) in the management of chronic schizophrenia. Journal of the Irish Medical Association 1960;46:135‐8. [PubMed] [Google Scholar]
Holden 1969 {published data only}
- Holden JM, Itil TM, Keskiner A. Comparison of perphenazine and P‐5227 in chronic schizophrenics: clinical and EEG effects. Journal of Clinical Pharmacology and the Journal of New Drugs 1969;9(3):163‐75. [PubMed] [Google Scholar]
Hollister 1967 II {published data only}
- Hollister LE, Overall JE, Shelton J, Pennington V, Kimbell I, Johnson M. Drug therapy of depression. Archives of General Psychiatry 1967;17:486‐93. [DOI] [PubMed] [Google Scholar]
Hollister 1974 {published data only}
- Hollister LE, Overall JE, Kimbell I Jr, Pokorny A. Specific indications for different classes of phenothiazines. Archives of General Psychiatry 1974;30(1):94‐9. [DOI] [PubMed] [Google Scholar]
Huang 1964 {published data only}
- Huang CL, Kurland AA. Perphenazine (trilafon) metabolism in psychotic patients. Archives of General Psychiatry 1964;10:639‐46. [DOI] [PubMed] [Google Scholar]
Jprn 2011 {published data only}
- Jprn U. An intervention study of optimizing algorism‐based pharmacological treatment for schizophrenia. http://www.umin.ac.jp/ctr/index.htm 2011. [UMIN000004931]
Kaim 1972 {published data only}
- Kaim SC, Klett CJ. Treatment of delirium tremens. A comparative evaluation of four drugs. Quarterly Journal of Studies 1972;33:1065‐72. [PubMed] [Google Scholar]
Kaji 1974 {published data only}
- Kaji S, Tsukada K, Hasegawa S, et al. Comparison of clinical efficacy of clocapramine and perphenazine for chronic schizophrenia by the double‐blind study. Rinsyo Seisin Igaku 1974;3(8):867‐74. [Google Scholar]
Kawakita 1965 {published data only}
- Kawakita Y. Drug therapy of acute schizophrenia. Saishin Igaku 1965;20(9):2440‐8. [PubMed] [Google Scholar]
Kistrup 1991 {published data only}
- Kistrup K, Gerlach J, Aaes Jorgensen T, Larsen NE. Perphenazine decanoate and cis(z)‐flupentixol decanoate in maintenance treatment of schizophrenic outpatients. Serum levels at the minimum effective dose. Psychopharmacology 1991;105(1):42‐8. [DOI] [PubMed] [Google Scholar]
Kline 1968 {published data only}
- Kline NS, Blair J, Cooper TB, Esser AH, Hackett E, Vestergaard P. A controlled seven year study of endocrine and other indices in drug treated chronic schizophrenics. Acta Psychiatrica Scandinavica Supplementum 1968;206:7‐75. [PubMed] [Google Scholar]
Knudsen 1985 {published data only}
- Knudsen P, Hansen LB, Auken G, Waehrens J, Højholdt K, Larsen NE. Perphenazine decanoate vs. Perphenazine enanthate ‐ efficacy and side‐effects in a 6 week double‐blind, comparative study of 50 drug monitored psychotic patients. Acta Psychiatrica Scandinavica Supplementum 1985;322:15‐28. [PubMed] [Google Scholar]
Knudsen 1985a {published data only}
- Knudsen P, Hansen LB, Hojholdt K, Larsen NE. Long‐term depot neuroleptic treatment with perphenazine decanoate. I. Efficacy and side effects in a 12 month study of 42 drug monitored psychotic patients. Acta Psychiatrica Scandinavica. Supplementum 1985; Vol. 322:29‐40. [KNUDSEN: 1985 b] [PubMed]
- Knudsen P, Hansen LB, Hojholdt K, Larsen NE. Long‐term depot neuroleptic treatment with perphenazine decanoate. II. Different depot intervals in the last 6 months of a 12 month study of 42 drug monitored psychotic patients. Acta Psychiatrica Scandinavica. Supplementum 1985; Vol. 322:41‐50. [KNUDSEN: 1985 b] [PubMed]
Kothari 1960 {published data only}
- Kothari NJ, Saunders JC, Kline NS, Griffen JA. A comparison of perphenazine, proketazine, nialamide and MO‐482 in chronic schizophrenics. American Journal of Psychiatry 1960;117:358‐60. [DOI] [PubMed] [Google Scholar]
Lapolla 1967 {published data only}
- Lapolla A. A double‐blind evaluation of chlorpromazine versus a combination of perphenazine and amitriptyline. International Journal of Neuropsychiatry. 1967; Vol. 3, issue 5:403‐5.
Levine 1997 {published data only}
- Levine J, Caspi N, Laufer N. Immediate effects of chlorpromazine and perphenazine following neuroleptic washout on word association of schizophrenic patients. Schizophrenia Research 1997;26:55‐63. [DOI] [PubMed] [Google Scholar]
Lindholm 1978 {published data only}
- Lindholm H, Gullberg B, Ohman A, Sedvall G. Effects of perphenazine injections on prolactin levels in plasma from schizophrenic women and men. Psychopharmacology 1978;57(1):1‐4. [DOI] [PubMed] [Google Scholar]
Linnoila1982 {published data only}
- Linnoila M, George L, Guthrie S. Interaction between antidepressants and perphenazine in psychiatric inpatients. American Journal of Psychiatry 1982;139(10):1329‐31. [DOI] [PubMed] [Google Scholar]
Loprete 1967 {published data only}
- Loprete FP, Palm C. Sleep in psychotic patients: a comparative clinical study. International journal of neuropsychiatry 1967;3(6):497‐500. [PubMed] [Google Scholar]
Mason‐Browne 1957 {published data only}
- Mason‐Browne NL. Perphenazine ‐ a drug modifying consciousness. American Journal of Psychiatry 1957;114:173‐4. [DOI] [PubMed] [Google Scholar]
- Mason‐Browne NL, Borthwick JW. Effect of perphenazine (trilafon) on modification of crude consciousness. Diseases of the Nervous System 1957;18:300‐6. [PubMed] [Google Scholar]
Mazurek 2003 {published data only}
- Mazurek I, Loza B, Lecyk A. Atypical versus typical antipsychotic treatment prognosis based on veps and wcst scores in paranoid schizophrenia. European Neuropsychopharmacology 2003; Vol. 13, issue 4:S347. [LOZA: 2001]
Moore 1958 {published data only}
- Moore DC, Bridenbaugh LD, Ackeren van EG, Cole EV. Control of postoperative vomiting with perphenazine (trilafone): a double‐blind study. Anesthesiology 1958;19:72‐4. [DOI] [PubMed] [Google Scholar]
Müller 1994 {published data only}
- Müller M. Pharmacokinetik/Bioavailibility Evaluation of Three Different perphenazine Formulations (Decentan) in 18 healthy volunteers [Pharmakokinetik/Bioverfügbarkeits‐Studie mit drei verschiedenen Perphenazin‐Zubereitungen (Decentan) mit 18 gesunden Freiwilligen]. E. Merck, Darmstadt 1994.
Nahunek 1967 {published data only}
- Nahunek K, Svestka J, Misurec J. Clinical comparison of methophenazine and perphenazine in schizophrenia. A controlled study. Effects on the photomyoclonic threshold [Klinischer Vergleich von Methophrenazin (Frenolon) und Perphenazin bei Schizophrenie. Kontrollierte Studie. Beeinflussung der photomyoklonischen Schwelle]. Activitas Nervosa Superior 1967;9(4):404‐5. [PubMed] [Google Scholar]
Nahunek 1968 {published data only}
- Nahunek K, Svestka J, Rodova A. Clinical experience with octoclothepin in psychoses. Comparasion with perphenazine. Activitas Nervosa Superior 1968;10(3):339‐40. [PubMed] [Google Scholar]
Nahunek 1969 {published data only}
- Nahunek K, Hadlik J, Svestka J, Rodova A, Vanysek J. A comparison of therapeutic effects of triperidol and perphenazine in schizophrenia [Srovnani lecebneho ucinku triperidolu s perphenazinem u schizofrenie]. Ceskoslovenska Psychiatrie. 1969;65(5):281‐9. [PubMed] [Google Scholar]
Nahunek 1970 {published data only}
- Nahunek K, Hadlik J, Rodova A, Misurec J, Vanysek J. Comparison of triperidol with perphenazine in schizophrenic psychoses. Effect on the photomyoclonic threshold [Srovnani triperidolu s perphenazinem u schizofrennich psychoz vliv na fotomyoklonicky prah]. Activitas Nervosa Superior 1970;12(1):56‐7. [PubMed] [Google Scholar]
Nahunek 1970 II {published data only}
- Nahunek K, Svestka J, Rodova A. Comparison of the therapeutic effect of flupenthixol and perphenazine in schizophrenia. Activitas Nervosa Superior 1970;12(3):247‐8. [PubMed] [Google Scholar]
Nahunek 1975 {published data only}
- Nahunek K, Svestka J, Misurec J, Rodova A. Clinical experience with clozapin [Klinicke zkusenosti s clozapinem]. Ceskoslovenska Psychiatrie 1975;71(1):11‐20. [PubMed] [Google Scholar]
Nahunek 1976 {published data only}
- Nahunek K, Rodova A, Svestka J. Outline classification of neuroleptic drugs based on results of short‐term control crossed studies in schizophrenia [Pokus o klasifikaci neuroleptik na podklade vysledku kradkodobych kontrolovanych zkrizenyich studii u schizofrenie]. Ceskoslovenska Psychiatrie 1976;72(2):104‐14. [Google Scholar]
- Nahunek K, Svestka J, Ceskova E. On the question of differences between therapeutic responses after particular neuroleptics in comparison to perphenazine in schizophrenics [Kotazce rozdilnosti terapeutickych odpovedi po jednotlivych neurolepticich ve srovnani s perphenazinem u schizofrennich nemocnych]. Ceskoslovenska Psychiatrie 1981;77(1):25‐30. [PubMed] [Google Scholar]
Nahunek 1980 {published data only}
- Nahunek K, Svestka J, Ceskova E. Controlled 'comparison' of perphenazine with perphenazine in schizophrenia ‐ a methodical check of a double‐blind cross‐over design. Activitas Nervosa Superior 1980;22(3):154‐5. [Google Scholar]
Nordic DS Group 1986 {published data only}
- Nordic Dyskinesia Study Group. Effect of different neuroleptics in tardive dyskinesia and parkinsonism. A video‐controlled multicentre study with chlorprothixene, perphenazine, haloperidol and haloperidol + biperiden. Psychopharmacology 1986;90(4):423‐9. [PubMed] [Google Scholar]
O'Reilly 1957 {published data only}
- O'Reilly PO, Wojcicki HM, Hrychuk W, Keogh RP. Perphenazine (Trilafon) in the treatment of pschoses. Canadian Medical Association Journal 1957;77:952‐5. [PMC free article] [PubMed] [Google Scholar]
Oltman 1966 {published data only}
- Oltman JE, Friedman S. Perphenazine‐amitriptyline in the treatment of schizophrenia. American Journal of Psychiatry 1966;123(5):607‐9. [DOI] [PubMed] [Google Scholar]
Omerov 1989 {published data only}
- Omerov M, Wistedt B, Bolvig HL, Larsen NE. The relationship between perphenazine plasma levels and clinical response in acute schizophrenia. Progress in Neuropsychopharmacology and Biological Psychiatry 1989;13(1‐2):159‐66. [DOI] [PubMed] [Google Scholar]
Opjordsmoen 2000 {published data only}
- Opjordsmoen S, Brunsvik S, Melle I, Dahl A, Friis S, Haahr U, et al. A comparison between novel and traditional antipsychotic drugs as first‐line medication in early psychosis. Second international conference on early psychosis 2000, 31 March ‐ 2 April. [OPJORDSMOEN: 2000]
Pfeiffer 1965 {published data only}
- Pfeiffer CC, Goldstein L, Murphree HB, Sugerman AA. Time‐series frequency analysis, and electrogenesis of the EEGs of normals and psychotics before and after drugs. American Journal of Psychiatry. 1965;122:1147‐55. [DOI] [PubMed] [Google Scholar]
Prien 1973 {published data only}
- Prien RF, Gillis RD, Caffey EM. Intermittent pharmacotherapy in chronic schizophrenia. Hospital and Community Psychiatry 1973;24(5):317‐322. [DOI] [PubMed] [Google Scholar]
Prusoff 1979 {published data only}
- Prusoff BA, Williams DH, Weissman MM, Astrachan BM. A controlled clinical trial of amitriptyline added to perphenazine in the treatment of depressed schizophrenics. Psychopharmacology Bulletin 1979;15(2):80‐1. [PubMed] [Google Scholar]
Rapp 1972 {published data only}
- Rapp W. Preliminary study of perphenazine enanthate in the treatment of chronic schizophrenia. Pharmakopsychiatrie Neuropsychopharmakologie 1972;5:205‐14. [Google Scholar]
Rapp 1986 {published data only}
- Rapp W, Hellbom E, Norrman O, Palm U, et al. A double blind crossover study comparing haloperidol decanoate and perphenazine enanthate. Current Therapeutic Research 1986;39(5):665‐70. [Google Scholar]
Rappaport 1967 {published data only}
- Rappaport M. Competing voice messages. Effects of a message load and drugs on the ability of acute schizophrenics to attend. Archives of General Psychiatry 1967;17:97‐103. [DOI] [PubMed] [Google Scholar]
Rappaport 1968 {published data only}
- Rappaport M. Attention to competing voice messages by nonacute schizophrenic patients. Effects of message load, drugs, dosage levels and patient background. Journal of Nervous and Mental Disease 1968;146(5):404‐11. [DOI] [PubMed] [Google Scholar]
Reznik 2000 {published data only}
- Reznik I, Sirota P. Treatment of obsessive and compulsive symptoms in schizophrenia with SSRI's and neuroleptics: a randomized controlled trial. 21st Congress of Collegium Internationale Neuro‐psychopharmacologicum, Glasgow, Scotland, UK 1998.
- Reznik I, Sirota P, Psych M. Obsessive and compulsive symptoms in schizophrenia: a randomized controlled trial with fluvoxamine and neuroleptics. Journal of Clinical Psychopharmacology 2000;20(4):410‐6. [DOI] [PubMed] [Google Scholar]
Rickels 1982 {published data only}
- Rickels K, Csanalosi I, Werblowsky J, Weise CC, Weinstock R, Brown AS. Amitriptyline‐perphenazine and doxepin in depressed outpatients: a controlled double‐blind study. Journal of Clinical Psychiatry 1982;43(10):419‐22. [PubMed] [Google Scholar]
Ritter 1971 {published data only}
- Ritter RM, Davidson DE. Haloperidol for acute psychiatric emergencies. A double‐blind comparison with perphenazine in acute alcoholic psychosis. Southern Medical Journal 1971;64(2):249‐50. [PubMed] [Google Scholar]
Rodova 1971 {published data only}
- Rodova A, Nahunek K, Svestka J. Comparison of the therapeutic results of clothiapin and perphenazine in schizophrenia. Activitas Nervosa Superior 1971;13(3):171‐3. [PubMed] [Google Scholar]
Rodova 1973 {published data only}
- Rodova A, Svestka J, Nahunek K, Ceskova E. A blind comparison of clozapine and perphenazine in schizophrenics. Activitas Nervosa Superior 1973;15(2):94‐5. [PubMed] [Google Scholar]
Scurr 1958 {published data only}
- Scurr CF, Robbie DS. Trials of perphenazine in the prevention of postoperative vomiting. British Medical Journal 1958;1:922‐3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Shalev 1993 {published data only}
- Shalev A, Hermesh H, Rothberg J, Munitz H. Poor neuroleptic response in acutely exacerbated schizophrenic patients. Acta Psychiatrica Scandinavica 1993;87(2):86‐91. [DOI] [PubMed] [Google Scholar]
Sharpley 1964 {published data only}
Shiloh 2002 {published data only}
Silver 2000 {published data only}
- Silver H, Barash I, Aharon N, Kaplan A, Poyurovsky M. Fluvoxamine augmentation of antipsychotic drugs improves negative symptoms in psychotic chronic schizophrenic patients: a placebo‐controlled study. International Clinical Psychopharmacology 2000;15(5):257‐61. [DOI] [PubMed] [Google Scholar]
Simopoulos 1971 {published data only}
- Simopoulos AM, Pinto A, Uhlenhuth EH, Mcgee J. Old wine in new bottles: decreased hostility in chronic psychotic inpatients treated with diphenylhydantoin (Dilantin). 5th World Congress of Psychiatry, Ciudad de Mexico. 1971:526.
Simpson 1970 {published data only}
- Simpson GM, Krakov L, Kunz‐Bartholini E. A controlled trial of combined medications on behavioral and extrapyramidal effects. Acta Psychiatrica Scandinavica Supplementum 1970;212:20‐7. [DOI] [PubMed] [Google Scholar]
Smith 1959 {published data only}
- Smith JA, Christian D, Mansfield E, Figaredo A. A graphic comparison of five phenothiazines. American Journal of Psychiatry 1959;116:392‐9. [DOI] [PubMed] [Google Scholar]
Spiker 1985 {published data only}
- Spiker DG, Cofsky Weiss J, Dealy RS, Griffin SJ, Hanin I, Neil JF, et al. The pharmacological treatment of delusional depression. American Journal of Psychiatry 1985;142:430‐6. [DOI] [PubMed] [Google Scholar]
Sun 2010 {published data only}
- Sun S, Liu Y, Long JL, Wang L. Venlafaxine extended release tablets on schizophrenia cognitive function] Google Translate. Chinese Journal of Health Psychology [中国健康心理学杂志] 2010; Vol. 18, issue 4:394‐6. [孙群星//刘勇//龙金亮//王: 2010]
Sun 2010b {published data only}
- Sun J, Xie S, Nisu L, Wang X, Shang X, et al. acute schizophrenic inpatients converted to aripiprazole treatment efficacy and safety of] Google Translate [急性期精神分裂症住院患者转换为阿立哌唑治疗疗效及安全性研究]. Shandong Archives of Psychiatry [山东精神医学] 2010; Vol. 23, issue 3:179‐81. [孙静//谢世平//倪苏琳//王: 2010]
Svestka 1970 {published data only}
- Svestka J, Nahunek K. Controlled comparative study of methylperidol and perphenazine in schizophrenic psychoses [Kontrolovane srovnani metylperidolu s perfenazinem u psychoz schizofrenniho ikruhu]. Activitas Nervosa Superior 1970;12(1):57‐8. [PubMed] [Google Scholar]
Svestka 1972 {published data only}
- Svestka J, Nahunek K. A comparison of pimozide with perphenazine in the treatment of acute schizophrenic psychoses. Activitas Nervositas Superior 1972;14(2):93‐4. [PubMed] [Google Scholar]
Svestka 1973 {published data only}
- Svestka J, Rodova A, Nahunek K. Comparison of the short‐term therapeutic effect of oxyprothepine and perphenazine in schizophrenic patients. A controlled study. Activitas Nervosa Superior 1973;15(2):103‐4. [PubMed] [Google Scholar]
Svestka 1974 {published data only}
- Svestka J, Nahunek K, Rodova A, Ceskova E. A controlled comparison of oxypertine and perphenazine in schizophrenic psychoses. Activitas Nervosa Superior 1974;16(3):165‐6. [PubMed] [Google Scholar]
Svestka 1975 {published data only}
- Svestka J, Nahunek K, Rodova A, Ceskova E. The position of trifluoperazine in the group of neuroleptics (a controlled clinical comparative study). Activitas Nervosa Superior 1975;17(3):208. [PubMed] [Google Scholar]
Svestka 1989 {published data only}
- Svestka J, Nahunek K, Ceskova E, Rysanek R, Balsíková, J. Controlled cross over comparison of isofloxythepin and perphenazine in the treatment of schizophrenic psychoses. Activitas Nervosa Superior 1989;31(1):32‐4. [PubMed] [Google Scholar]
Svestka 1989a {published data only}
- Svestka J, Ceskova E, Rysanek R, Nahunek K. Controlled cross over comparison of carbamazepine with perphenazine in schizophrenic psychoses. 31st Annual Psychopharmacology Meeting (1989, Jesenik, Czechoslovakia). Activitas Nervosa Superior 1989;31(4):276‐7. [PubMed] [Google Scholar]
Svestka 1990 {published data only}
- Svestka J, Rysanek R, Nahunek K, Ceskova E. Therapeutic results in schizophrenic and schizoaffective psychoses with sulpiride (Eglonyl Alkaloid), as compared with perphenazine (Perfenazin Spofa) [Vysledky lecby schizofrennich a schizoafektivnich nemocnych sulpiridem (Eglonyl Alkaloid) ve srovnani s perfenazinem (Perfenazin Spofa)]. Ceskoslovenska Psychiatrie 1990;86(3):145‐56. [PubMed] [Google Scholar]
Syvalahti 1997 {published data only}
- Syvalahti EK, Taiminen T, Saarijarvi S, Lehto H, Niemi H, Ahola V, et al. Citalopram causes no significant alterations in plasma neuroleptic levels in schizophrenic patients. Journal of International Medical Research 1997;25(1):24‐32. [DOI] [PubMed] [Google Scholar]
Tegeler 1979 {published data only}
- Tegeler J, Floru L. A comparative study of the longacting neuroleptics perphenazine enanthate and flusipirilene. Pharmakopsychiatrie Neuropsychopharmakologie 1979;12(5):357‐65. [DOI] [PubMed] [Google Scholar]
Wang 2001 {published data only}
- Wang X, Yin J, Yang L, Li W, Wu G, et al. Double‐blind controlled side reaction of the L‐stepholidine the treatment of schizophrenia and Nursing Strategy. Shandong Archives of Psychiatry [山东精神医学] 2001; Vol. 14, issue 3:204‐6. [王秀英: 2001]
Whittaker 1963 {published data only}
- Whittaker CB, Hoy RM. Withdrawal of perphenazine in chronic schizophrenia. British Journal of Psychiatry 1963;109:422‐7. [DOI] [PubMed] [Google Scholar]
Wolf 2007 {published data only}
- Wolf J, Janssen F, Lublin H, Salokangas RKR, Allain H, Smeraldi E, et al. A prospective, multicentre, open‐label study of aripiprazole in the management of patients with schizophrenia in psychiatric practice in Europe: Broad Effectiveness Trial with Aripiprazole in Europe (EU‐BETA). Current Medical Research and Opinion 2007; Vol. 23, issue 10:2313‐23. [WOLF: 2007] [DOI] [PubMed]
Yagi 1976 {published data only}
- Yagi G. A double‐blind controlled study on the usefulness of carpipramine‐chlorpromazine combination in the pharmacotherapy of chronic schizophrenic patients. Rinsho Hyoka (Clinical Evaluation) 1976;4(3):351‐403. [Google Scholar]
Zhong 2001 {published data only}
- Zhong C, Guozhang J, Duochen W. A double‐blind comparison trial of 1‐stepholidine and perphenazine in treatment of schizophrenia. 7th World Congress of Biological Psychiatry, Berlin, Germany. Suppl 1 2001; Vol. 2. [ZHONG: 2001]
Zhu 2009 {published data only}
- Zhu S, Liu Y, Sun S, Wang L. Psychological care on cognitive function in patients with chronic schizophrenia, the impact of (Google Translate) [心理护理对慢性精神分裂症患者认知功能的影响]. China Practical Medicine (中国实用医药) 2009; Vol. 4, issue 22:190‐2. [朱玉星,: 2009]
References to studies awaiting assessment
Casey 1960 {published data only}
- Casey JF, Lasky JJ, Klett CJ, Hollister LE. Treatment of schizophrenic reactions with phenothiazine derivates. A comparative study of chlorpromazine, triflupromazine, mepazine, prochlorperazine, perphenazine and phenobarbital. American Journal of Psychiatry 1960;117:97‐105. [DOI] [PubMed] [Google Scholar]
Covell 2004 {published data only}
Ferziger 2010 {published data only}
- Ferziger R, Alphs L, Turner N, Mao L, Rodriguez S, Dixon L, et al. A multicenter effectiveness study of paliperidone palmitate vs oral antipsychotic drugs for people with schizophrenia recently released from jail: Study rationale and methodology. Neuropsychopharmacology 2010; Vol. 35:S101‐S2. [FERZIGER: 2010 a]
- Ferziger R, Alphs L, Turner N, Mao L, Rodriguez S, Dixon L, et al. A multicenter effectiveness study of paliperidone palmitate vs oral antipsychotic drugs for people with schizophrenia recently released from jail: Study rationale and methodology. Proceedings of the 49th Annual meeting of the Americam College of Neuropsychopharmacology; 2010 Dec 5‐9; Miami, Florida 2010. [FERZIGER: 2010]
Freedman 1961 {published data only}
- Freedman DX, Jong de J. Thresholds for drug‐induced akathisia. Journal of American Psychiatry 1961;117:930‐1. [DOI] [PubMed] [Google Scholar]
Freyhan 1959 {published data only}
- Freyhan FA. Therapeutic implications of differential effects of new phenothiazine compounds. American Journal of Psychiatry 1959;116:577‐85. [DOI] [PubMed] [Google Scholar]
Goldstein 1969 {published data only}
- Goldstein BJ, Brauzer B, Clyde DJ, Caldwell JM. The differential prediction of response to two anti‐psychotic drugs. Psychosomatics 1969;10(3):193‐7. [DOI] [PubMed] [Google Scholar]
Hollister 1967 {published data only}
- Hollister LE, Overall JE, Bennett JL, Kimbell I, Shelton J. Specific therapeutic actions of acetophenazine, perphenazine, and benzquinamide in newly admitted schizophrenic patients. Clinical Pharmacology and Therapeutics 1967;8(2):249‐55. [DOI] [PubMed] [Google Scholar]
Kellam 1971 {published data only}
- Kellam AMP, Jones KS. A double‐blind controlled trial of thiothixene and perphenazine in chronic schizophrenics shown to require maintenance therapy. Acta Psychiatrica Scandinavica 1971;47(2):174‐85. [DOI] [PubMed] [Google Scholar]
Mahmoud 2004 {published data only}
- Mahmoud RA, Engelhart LM, Janagap CC, Oster G, Ollendorf D. Risperidone versus conventional antipsychotic drugs for schizophrenia and schizoaffective disorder: symptoms, quality of life and resource use under customary clinical care. Clinical drug investigation 2004; Vol. 24, issue 5:275‐86. [MAHMOUD: 2004 a] [DOI] [PubMed]
NCT 2011 {published data only}
- NCT. Decision aid to facilitate shared decision making during treatment in schizophrenia. http://ClinicalTrials.gov/show/NCT01420575 2011. [NCT01420575]
Povlsen 1987 {published data only}
- Povlsen UJ, Noring U, Meidahl B, Korsgaard S, Waehrens J, Gerlach J. The effects of neuroleptics on tardive dyskinesias. A video‐controlled, randomized study of chlorprothixene, perphenazine, haloperidol and haloperidol + biperiden [Neuroleptikas virkning pa tardive dyskinesier. En videokontrolleret, randomiseret undersogelse med klorprotixen, perfenazin, haloperidol og haloperidol+biperiden]. Ugeskrift for Laeger 1987;149(25):1682‐5. [PubMed] [Google Scholar]
Schulsinger 1958 {published data only}
- Schulsinger F, Jensen R. Trilafon (perphenazine) and largactil (chlorpromazine) in chronic schizophrenia. A comparison [Trilafon (perfenazin) og largactil (klorpromazin) hos kronisk sindssyge. En sammenligning]. Ugeskrift for Laeger 1958;120(12):366‐9. [PubMed] [Google Scholar]
Szafranski 1999 {published data only}
- Szafranski T, Jarema M, Olajossy M, Chrzanowski W, Araszkiewicz A, Landowski J, et al. Subjective experiences of schizophrenic patients on atypical antipsychotic olanzapine and typical antipsychotic perphenazine. Journal of the European College of Neuropsychopharmacology 1999;9:273. [Google Scholar]
Vinar 1968 {published data only}
- Vinar O. Differences in therapeutic effects of phenothiazine drugs. Agressolgie 1968;9(2):315‐7. [PubMed] [Google Scholar]
Additional references
Altman 1996
- Altman DG, Bland JM. Detecting skewness from summary information. BMJ 1996;313:1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Begg 1996
- Begg C, Cho M, Eastwood S, Horton R, Moher D, Ollon I, et al. Improving the quality of reporting of randomized controlled trials. The CONSORT statement [comment]. JAMA 1996;276:637‐9. [DOI] [PubMed] [Google Scholar]
Benkert 1996
- Benkert O, Hippius H. Psychiatrische Pharmakotherapie. 6th Edition. Berlin: Springer Verlag, 1996. [Google Scholar]
Bland 1997
- Bland JM, Kerry SM. Trials in clusters. BMJ 1997;315:600. [DOI] [PMC free article] [PubMed] [Google Scholar]
Bleuler 1908
- Bleuler E. [The prognosis of dementia praecox ‐ schizophrenia group ‐ Google translate] [Die Prognose der Dementia praecox ‐ Schizophreniegruppe]. Allgemeine Zeitschrift für Psychiatrie 1908;65:436‐64. [Google Scholar]
BNF 2012
- BMJ Group and Pharmaceautical Press. British National Formulary No. 63. Antipsychotic drugs. BNF 2012.
Boissel 1999
- Boissel JP, Cucherat M, Li W, Chatellier G, Gueyffier F, Buyse M, et al. The problem of therapeutic efficacy indices. 3. Comparison of the indices and their use [Apercu sur la problematique des indices d'efficacite therapeutique, 3: comparaison des indices et utilisation. Groupe d'Etude des Indices D'efficacite]. Therapie 1999;54(4):405‐11. [PUBMED: 10667106] [PubMed] [Google Scholar]
Bundesverband 2001
- Bundesverband der Pharmazeutischen Industrie (editors). No English title given [Fachinformation Decentan]. Fachinformation Decentan 2001:1‐4.
Cannon 1996
- Cannon M, Jones P. Schizophrenia. Journal of Neurology, Neurosurgery and Psychiatry 1996;61:604‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
CEA
- Cost‐Effectiveness Analysis Registry (CEA). https://research.tufts‐nemc.org/cear4/ accessed 11/09/13.
Clarke 2000
- Clarke M, Oxman AD. Cochrane Reviewers' Handbook. The Cochrane Library [database on disk and CDROM]. Oxford: Update Software, 2000. [Google Scholar]
David 2005
- David A, Quraishi SN, Rathbone J. Depot perphenazine decanoate and enanthate for schizophrenia. Cochrane Database of Systematic Reviews 2005, Issue 3. [DOI: 10.1002/14651858.CD001717.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Davies 2007
- Davies LM, Lewis S, Jones PB, Barnes TR, Gaughran F, Hayhurst K, et al. Cost‐effectiveness of first‐ v. second‐generation antipsychotic drugs: results from a randomised controlled trial in schizophrenia responding poorly to previous therapy. British Journal of Psychiatry 2007;191:14‐22. [DOI] [PubMed] [Google Scholar]
Deeks 2000
- Deeks J. Issues in the selection for meta‐analyses of binary data. Abstracts of the 8th International Cochrane Colloquium. Cape Town, South Africa: Cochrane Collaboration, 2000 Oct 25th‐29th.
Divine 1992
- Divine GW, Brown JT, Frazer LM. The unit of analysis error in studies about physicians' patient care behavior. Journal of General Internal Medicine 1992;7:623‐9. [DOI] [PubMed] [Google Scholar]
Donner 2002
- Donner A, Klar N. Issues in the meta‐analysis of cluster randomized trials. Statistics in Medicine 2002;21:2971‐80. [DOI] [PubMed] [Google Scholar]
Egger 1997
- Egger M, Smith GD, Schneider M, Minder C. Bias in meta‐analysis detected by a simple, graphical test. BMJ 1997;315:629‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Elbourne 2002
- Elbourne D, Altman DG, Higgins JPT, Curtina F, Worthingtond HV, Vaile A. Meta‐analyses involving cross‐over trials: methodological issues. International Journal of Epidemiology 2002;31(1):140‐9. [DOI] [PubMed] [Google Scholar]
Furukawa 2006
- Furukawa TA, Barbui C, Cipriani A, Brambilla P, Watanabe N. Imputing missing standard deviations in meta‐analyses can provide accurate results. Journal of Clinical Epidemiology 2006;59(7):7‐10. [DOI] [PubMed] [Google Scholar]
Gulliford 1999
- Gulliford MC, Ukoumunne OC, Chinn S. Components of variance and intraclass correlations for the design of community‐based surveys and intervention studies: data from the Health Survey for England 1994. American Journal of Epidemiology 1999;149:876‐83. [DOI] [PubMed] [Google Scholar]
Guy 1976
- Guy W. ECDEU Assessment Manual for Psychopharmacology. Rockville, MD: National Institute of Mental Health. DHEW Publication NO (ADM), 1976:124‐585. [Google Scholar]
Higgins 2003
- Higgins JPT, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ 2003;327:557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.0.2 [updated September 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Hutton 2009
- Hutton JL. Number needed to treat and number needed to harm are not the best way to report and assess the results of randomised clinical trials. British Journal of Haematology 2009;146(1):27‐30. [DOI] [PubMed] [Google Scholar]
ICH E3 1995
- ICH Expert Working Group. ICH harmonised tripartite guideline: structure and content of clinical study reports. International conference on harmonisation of technical requirements for registration of pharmaceuticals for huma use 30 November 1995.
Jadad 1996
- Jadad AR, Moore RA, Carroll D, Jenkinson C, Reynolds DJM, Gavaghan DJ, et al. Assessing the quality of reports of randomized clinical trials: is blinding necessary?. Controlled Clinical Trials. Vol. 17, New York: Elsevier, 1996:1‐12. [DOI] [PubMed] [Google Scholar]
Kay 1986
- Kay SR, Opler LA, Fiszbein A. Positive and Negative Syndrome Scale (PANSS) Manual. North Tonawanda, NY: Multi‐Health Systems, 1986. [Google Scholar]
Kay 1987
- Kay SR, Fiszbein A, Opler LA. The positive and negative syndrome scale (PANSS) for schizophrenia. Schizophrenia Bulletin 1987;13:261‐76. [DOI] [PubMed] [Google Scholar]
Leucht 2005
- Leucht S, Kane JM, Kissling W, Hamann J, Etschel E, Engel RR. What does the PANSS mean?. Schizophrenia Research 2005;79(2‐3):231‐8. [PUBMED: 15982856] [DOI] [PubMed] [Google Scholar]
Leucht 2005a
- Leucht S, Kane JM, Kissling W, Hamann J, Etschel E, Engel R. Clinical implications of brief psychiatric rating scale scores. British Journal of Psychiatry 2005;187:366‐71. [PUBMED: 16199797] [DOI] [PubMed] [Google Scholar]
Leucht 2007
- Leucht S, Engel RR, Bauml J, Davis JM. Is the superior efficacy of new generation antipsychotic drugs an artifact of LOCF?. Schizophrenia Bulletin 2007;33(1):183‐91. [PUBMED: 16905632] [DOI] [PMC free article] [PubMed] [Google Scholar]
Lowe 1973
- Lowe GR. The phenomenology of hallucinations as an aid to differential diagnosis. British Journal of Psychiatry 1973;123:621‐33. [DOI] [PubMed] [Google Scholar]
Mangalore 2007
- Mangalore R, Knapp M. Cost of schizophrenia in England. Journal of Mental Health Policy and Economics 2007;10(1):23‐41. [PubMed] [Google Scholar]
Marshall 2000
- Marshall M, Lockwood A, Bradley C, Adams C, Joy C, Fenton M. Unpublished rating scales: a major source of bias in randomised controlled trials of treatments for schizophrenia. British Journal of Psychiatry 2000;176:249‐52. [DOI] [PubMed] [Google Scholar]
Moher 2001
- Moher D, Schultz KF, Altman DG. The CONSORT statement: revised recommendations for improving the quality of reports of parallel group randomised trials. Lancet 2001;357:1191‐4. [PubMed] [Google Scholar]
NICE 2010
- National Collaborating Centre for Mental Health. Schizophrenia: core interventions in the treatment and management of schizophrenia in adults in primary and secondary care (updated edition). National Clinical Guideline Number 82 2010.
Overall 1962
- Overall JE, Gorham DR. The Brief Psychiatric Rating Scale. Psychological Reports 1962;10:799‐812. [Google Scholar]
Parfitt 1999
- Parfitt K (editor). Martindale. The Complete Drug Reference. 2nd Edition. London: Pharmaceutical Press, 1999. [Google Scholar]
Rosenheck 2009
- Rosenheck RA, Davis VG, Davis SM, Stroup S, McEvoy J, Swartz M, et al. Can a nonequivalent choice of dosing regimen bias the results of flexible dose double blind trials? The CATIE schizophrenia trial. Schizophrenia Research 2009;113(1):12‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Schünemann 2008
- Schünemann HJ, Oxman AD, Vist GE, Higgins JPT, Deeks JJ, Glasziou P, et al. Chapter 12: Interpreting results and drawing conclusions. In: Higgins JPT, Green S editor(s). Cochrane Handbook for Systematic Reviews of Interventions. The Cochrane Collaboration, 2008:359‐83. [Google Scholar]
Ukoumunne 1999
- Ukoumunne OC, Gulliford MC, Chinn S, Sterne JAC, Burney PGJ. Methods for evaluating area‐wide and organisation‐based interventions in health and health care: a systematic review. Health Technology Assessment 1999;3(5):iii‐92. [MEDLINE: ] [PubMed] [Google Scholar]
Wing 1961
- Wing J. A simple and reliable subclassification of chronic schizophrenia. British Journal of Psychiatry 1967;107:862‐75. [DOI: 10.1192/bjp.107.450.862] [DOI] [PubMed] [Google Scholar]
Xia 2009
- Xia J, Adams CE, Bhagat N, Bhagat V, Bhoopathi P, El‐Sayeh H, et al. Loss to outcomes stakeholder survey: the LOSS study. Psychiatric Bulletin 2009;33(7):254‐7. [Google Scholar]
References to other published versions of this review
Hartune 2005
- Hartung B, Wada M, Laux G, Leucht S. Perphenazine for schizophrenia. Cochrane Database of Systematic Reviews 2005, Issue 1. [DOI: 10.1002/14651858.CD003443.pub2] [DOI] [PubMed] [Google Scholar]