

Abstract
Per- and polyfluoroalkyl substances (PFAS) are an ensemble of persistent organic pollutants of global interest because of their associations with adverse health outcomes. Currently, environmental PFAS pollution is prolific as a result of the widespread manufacturing of these compounds and their chemical persistence. In this work, we demonstrate the advantages of adding ion mobility spectrometry (IMS) separation to existing LC-MS workflows for PFAS analysis. Using a commercially available drift tube IMS-MS, we characterized PFAS species and isomeric content both in analytical standards and wastewater samples. Molecular trendlines based on intrinsic mass and structural relationships were also explored for the PFAS subclasses (e.g. PFSA, PFCA, etc.). Results from rapid IMS-MS analyses provided a link between mass and collision cross sections (CCS) for specific PFAS families and are linked to compositional differences in molecular structure. In addition, CCS values provide additional confidence of annotating prioritized features in untargeted screening studies for potential environmental pollutants. Results from this study show that the IMS separation provides novel information to support traditional LC-MS PFAS analyses and will greatly benefit the evaluation of unknown pollutants in future environmental studies.
Graphical Abstract
Per- and polyfluoroalkyl substances (PFAS) are environmental contaminants characterized by extensive fluorination along extended aliphatic chains, rendering them chemically inert, thermally stable, and hydrophobic. Their physiochemical properties are also uniquely suited for many household and industrial applications, such as the production of stain-resistant carpeting, nonstick cookware, and aqueous film-forming foams (AFFF).1, 2 The production of long chain PFAS, such as perfluorooctane sulfonic acid (PFOS) and perfluorooctanoic acid (PFOA), began in the U.S. during the 1950’s and continued into the early 2000’s. However, towards the end of this time period, manufacturers began to phase out long chain chemistries because of evidence that PFAS exposure is related to various adverse health outcomes including ulcerative colitis,3 thyroid disease,4 cancer,5 elevated cholesterol,6 and decreased immune function,7 and corresponding pressure from regulatory agencies.8, 9 In the U.S., enforceable drinking water standards for PFAS are lacking at the federal level, but the U.S. EPA published a lifetime health advisory level for the sum concentration of PFOA and PFOS at 70 parts-per-trillion (or ng L−1) in May 2016.10 While long chain PFAS production continues in emerging economies (most notably China), many industries have shifted production to favor shorter chain PFAS and fluoroether replacement chemistries because they tend to be less bioaccumulative. Common examples of these molecules include per- and polyfluoroalkyl ether carboxylic acids or PFECAs, such as GenX and ADONA (see Supporting Information Figure S1).11–13 Although toxicological data of emerging PFAS are scarce, the occurrence and persistence of PFECAs in the environment is already being documented.12–17
While the chemical structure and isomeric diversity of legacy PFAS are well-characterized, understanding the evolving structure of emerging fluorinated compounds is a challenge.14 Legacy PFAS isomers result largely from impure chemical syntheses, where the linear form is the desired target (e.g. PFOS, PFOA), but side reactions also produce branched isomers.18 Emerging PFAS possess both linear and branched structures, typically characterized by flexible ether linkages within the CF2 branches (e.g. GenX, see Supporting Information). To date, several analytical strategies have been employed to characterize the isomeric PFAS.19–21 These techniques predominantly consist of gas or liquid chromatography (GC or LC) coupled with mass spectrometry (MS). In isomer characterization studies, spatial annotation of branched isomers is particularly challenging as described by previous studies.22, 23 Highly selective chromatographic methods are often employed in these studies and many take over one hour of analysis time per sample. Analyte derivatization is also regularly utilized, further lengthening the required analysis time.21, 24 Similar fragmentation profiles (MS/MS) for the branched PFAS isomers also complicate the analyses and make the isomer distinction even more difficult (see Supporting Information Figure S2). Additionally, these analyses require reference standards, which are not always available, for replacement PFAS chemistries and manufacturing byproducts.
Ion mobility spectrometry (IMS) is an emerging separation technique that distinguishes ions based on their size, shape, and charge state in the gas phase.25 GC and LC differ from IMS in that they separate analytes based on differences in boiling point and polarity, while IMS separates ions based on differences in gas phase electrophoretic mobility. IMS separations occur post-ionization in a defined mobility separation region filled with an inert gas (also termed as the buffer gas). Ions traverse this region under the influence of an applied electric field, wherein their migration time through the gas (drift time) is directly correlated with ion size. The measured size is commonly described as the ion’s collision cross section (CCS, typically denoted in units of Å2). In the IMS experiments, smaller ions experience fewer collisions with the buffer gas and migrate faster than larger ions whose motion is impeded. Since IMS separations occur in the gas phase, mobility analysis is incredibly rapid and occurs in a typical timescale <100 milliseconds per spectrum. Thus, IMS can be readily interfaced with existing untargeted LC-MS/MS suspect screening workflows,15, 26, 27 enabling an additional separation mechanism and molecular descriptor (CCS) for characterization of analytes without increased analysis time.28 Additionally, previous IMS studies have described intrinsic relationships between CCS and mass for a variety of molecular classes such as lipids, peptides, carbohydrates, and environmental contaminants, such as polyaromatic hydrocarbons (PAHs) and polybrominated diphenyl ethers (PBDEs).29–31 In all cases mentioned, unique trendlines relating CCS to m/z have been observed for the different molecular classes based on their differences in gas phase packing efficiency (compactness). Hines et. al. demonstrated that subclasses of vitamins, antibiotics, and other small biomolecules also possess distinct mass-CCS trendlines, which is useful in providing additional molecular information for unannotated features in untargeted experiments or natural product discovery.32, 33As a consequence of the speed and selectivity afforded by IMS separations, IMS methods are becoming more common and have been developed by various manufacturers to incorporate IMS separations into untargeted MS workflows.34–39 The technique described in this manuscript is drift tube IMS or DTIMS, but for descriptions of other IMS methods we direct the reader to several reviews articles which cover the topics in detail.28, 40, 41 Several of these methods, mainly traveling wave ion mobility spectrometry (TWIMS)42 and differential ion mobility spectrometry (DIMS),23 have been utilized to characterize a few fluorinated isomers previously. However, extensive PFAS analyses have not be performed and relationships between PFAS and CCS have yet to be investigated. In this work we utilized IMS to 1) measure the CCS values of a wide variety of PFAS subclasses and developed mass/CCS correlations for 51 species covering the different groups, 2) explore the potential separation capabilities of IMS for PFAS isomers, and 3) apply LC-IMS-MS as a potential workflow for PFAS analysis in environmental samples.
Experimental Methods
Chemical Standards.
In this study we characterized 51 environmentally relevant PFAS across a diverse range of chemical space to relate molecular structure to CCS. PFAS differed in terms of headgroups, substructures, and isomeric forms, and when possible, we used nomenclature developed by the Interstate Technology Regulatory Council (ITRC).43, 44 Examples subclasses studied include perfluoroalkyl carboxylic acids (PFCA), perfluoroalkyl sulfonic acids (PFSA), fluorotelomer sulfonic acids (FTSA), per- and polyfluoroalkyl ether carboxylic acids (PFECA) and polyfluoroalkyl ether sulfonic acids (PFESA).18, 20 A list of studied PFAS and the corresponding source of procurement is shown in the Supporting Information (Excel Sheet ES1). For both PFOA and PFOS, linear and branched isomers were characterized (see Supporting Information Excel Sheets ES1 and ES2). PFAS not designated as strictly linear from the manufacturer were used to characterize isomeric diversity, such as a PFOS standard from Synquest. Standards were reconstituted and subsequently diluted in 18 MΩ water using an ELGA Purelab Flex purification system (High Wycombe, UK) to provide working concentrations of ca. 5-10 μg/mL for LC-IMS-MS analysis. For eluents, methanol and ammonium acetate were purchased as LC-MS grade from Fischer.
Liquid Chromatography.
To demonstrate the ease of incorporating IMS separations into existing workflows, the HPLC method used here was modified from an application note for quantitative PFAS detection with LC-MS45 and is described in detail in the Supporting Information (Table S1). Briefly, a C18 column (Agilent ZORBAX Eclipse Plus, 2.1 x 50 mm, 1.8 μm) was utilized with the Agilent 1290 Infinity LC system (Agilent Technologies, Santa Clara, CA). Mobile Phase A was comprised of 100% water, while Mobile Phase B consisted of 95% methanol and 5% water. Both Mobile Phase A and B were buffered with 5 mM ammonium acetate. Retention time variation was typically within 0.05 minute variation during a given worklist. For full alignment of features in environmental samples retention times were compared for standards within the same worklist for confident identification.
Ion Mobility and Mass Spectrometry.
The Agilent 6560 IMS-QTOF used in this work has been extensively characterized in previous publications.46–48 A simplified instrument schematic is annotated in Figure 1A. Briefly, the instrument consists of an electrospray ion source, ion funnels for beam focusing and ion trapping, a uniform field drift tube for IMS separation, a quadrupole mass filter, collision induced disassociation (CID) cell, and a high resolution TOF mass analyzer for accurate mass measurement. The ESI source (Agilent Jet Stream) was operated in negative ion mode for all PFAS, monitoring the deprotonated/desalted ion of each molecular form [M-H]− for all analytes and the decarboxylated ion [M-H-CO2]− for some PFCA and PFECA species. Source conditions were modified from the aforementioned application note45 and are referenced in the Supporting Information (Table S1). In all IMS experiments, the ions were pulsed into the drift tube and ion gating was kept short (100 μs) to minimize peak broadening. Each IMS experiment scans all ions in 60 milliseconds, and hence several hundred IMS scans occur in each LC peak. Furthermore, the drift potential was operated at 17.2 V/cm, which provides the optimum resolving power for IMS spectra on this platform.47, 49 The TOF was operated in high sensitivity mode (ca. 20-25,000 mass resolving power) in low mass range (50-1700 m/z). Spectra were acquired using MassHunter Acquisition Software B.09, and data were initially visualized using IM-MS Browser B.08 prior to spreadsheet exportation (Microsoft Excel) for further in depth analyses.
Figure 1.
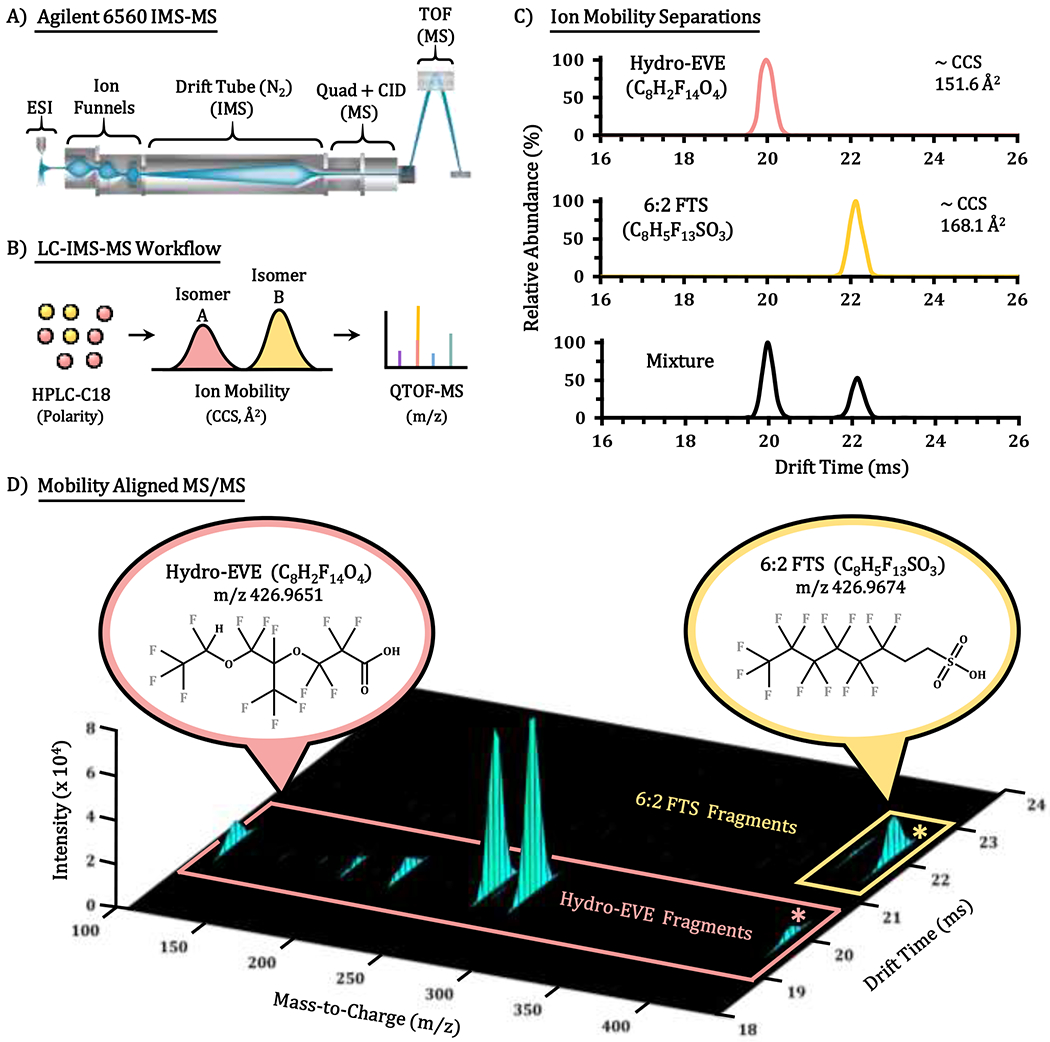
The LC-IMS-MS instrumentation and spectra utilized in this manuscript. (A) The instrumental schematic for the DTIMS-QTOF MS platform used in this work (Agilent 6560). (B) The RPLC-IMS-MS workflow designed to separate and characterize PFAS isomers and subclasses. The RPLC, IMS and MS stages are illustrated separately along with their corresponding mechanisms for structural discrimination. (C) The IMS drift time profiles possible for isobaric and isomeric separations. Deprotonated ions for 6:2 FTS and Hydro-EVE are illustrated along with their resolution in a mixture. (D) The drift time aligned MS/MS fragments of the 6:2 FTS and Hydro-EVE isobars illustrating the different components for each molecule and their distinction with IMS at 5V CID energy and mass selection of precursor ion 426.9 m/z (asterisk).
CCS Values for PFAS.
To assess the relationship between collision cross section (CCS) and mass-to-charge ratio (m/z), CCS values were calculated for the PFAS standards using the previously described single-field method which has been validated across multiple laboratories and is extensively characterized in previous manuscripts.48, 50, 51,52 Briefly, Agilent tune mix ions served as a mobility calibrant for calculating CCS values after measuring analyte drift times. The CCS value for each PFAS standard was measured in separate experiments over three days to ensure method reproducibility and instrument stability. CCS values collected using the single-field method were highly reproducible with RSD < 0.5%, consistent with IMS studies on this platform (see ES1).48 Of the 51 analytes measured in triplicate, 4 compounds did not ionize well in negative ionization mode and 7 did not provide a mobility distribution sufficient to measure a CCS value (see ES1, and Figure S3, S3). In eight cases of PFCA and PFECA, deprotonated ions were not observed or aggregated causing mobility smearing, and the decarboxylated ions [M-H-CO2]− were used to establish trendlines relating CCS to m/z (see ES1). Mobility smearing is currently thought to originate from multimer formation within the ESI, and during the mobility process these multimers disassociate, complicating the measurement of “condensed” drift spectra. For further description of this process, see Supporting Information Figures S3 and S4).
Wastewater Extraction.
Water samples were collected from wastewater outfall of a local chemical plant (Chemours, Fayetteville, NC) and a nearby lake (Marshwood Lake, Grays Creek, NC) to evaluate the robustness of the IMS CCS values for PFAS in complex matrices. Our sampling protocol was modeled after previous studies by Hopkins et. al. using a modified variant of EPA method 537. This method is described in further detail in the Supporting Information (Table S2).13 Briefly, 1 L of water was collected at the sampling site in 1 L polypropylene bottles and five mL of 35% HNO3 was added as a biocide prior to storage. Fifty mL of the collected sample was then filtered through 0.45 μm glass microfiber filters and PFAS were extracted using Oasis Wax Plus Short Cartridges (Waters Corporation, Milford, MA) as recommended by the manufacture protocol.53 After extraction, samples were evaporated to near dryness using a Savant SPD131DDA SpeedVac Concentrator (ThermoFisher Scientific, Waltham, MA) and reconstituted to 0.5 mL in 18 MΩ water prior to LC-IMS-MS analysis.
Results and Discussion
PFAS Initial Isobar Assessment.
To begin our assessment of how well IMS would separate PFAS, we analyzed two PFAS isobars, 6:2 FTS (m/z = 426.9674) and Hydro-EVE (m/z = 426.9651). While these analytes have different chemical formulas, their precursor ions vary in the m/z dimension by less than 10 ppm. Thus, mass separation of these precursors is quite challenging even with high-resolution MS instruments. In the IMS analysis, baseline separation was readily obtained for the isobars as Hydro-EVE elutes from the drift tube several milliseconds prior to 6:2 FTS (Figure 1C). Structural analysis of Hydro-EVE showed that it is highly branched, making it more compact in the gas-phase (smaller CCS) in comparison to 6:2 FTS, which is elongated by the additional CH2-CH2 group in the middle of the structure. MS/MS evaluation was also performed on the isobars for full validation. Since the IMS separation occurs prior to the collision cell, fragment ions of each analyte are drift time aligned to their precursors (Figure 1D) for definitive identification of each species. Thus, the IMS separation significantly enhances distinction of these isobars and their fragments that operates on a timescale (60 millisecond separations) that is easily nested into traditional GC/LC-MS/MS workflows.54
Linear PFAS Mass/CCS Trendlines.
A central aim of this study was to determine CCS values of known PFAS standards and develop correlations between mass and CCS for each PFAS subclass. In our analysis of the PFAS standards, we also observed distinct mass/CCS trendlines for the various subclasses as illustrated in Figure 2 (with data provided in Supporting Information Table S3). In the analyses, the decarboxylated PFCAs have the smallest CCS per mass unit, likely a result of this group possessing many high mass CF2 units with essentially no headgroup. Deprotonated PFSA and PFCA ([M-H]−) had markedly similar trendlines in these analyses, likely a result of shared molecular structure (i.e. perfluoroalkyl backbone terminating in a carboxylic/sulfonic acid headgroup). However, the resolving power of the IMS was still able to show the deprotonated PFCAs were on average larger than the PFSAs. The deprotonated FTSA possess the largest CCS for a given mass, likely a result of the -CH2-CH2- moiety connected to the sulfonic acid headgroup in these structures, which adds less mass than a -CF2-CF2- moiety but increases CCS significantly. A fluorinated replacement for PFOS is F53-B, which possesses two analytes (Supporting Information Figure S1) that form a trendline that falls below those for PFSA and PFCA. Components of F53-B are chlorinated ether sulfonic acids, and our interpretation of their smaller CCS per m/z is that the substitution of a fluorine with chlorine adds a significant amount of mass while only minutely increasing molecular size. For instance, PFOS and 9Cl-PF3ONS have similar CCS values (168Å2, and 170Å2, respectively), yet 9Cl-PF3ONS has significantly more mass (32 m/z units). 9Cl-PF3ONS does possess an ether linkage while PFOS does not, yet PFOS is the closest analogue in this analysis for comparison. The PFECA subgroup also possesses a linear trendline for increasing units of decarboxylated single-ether analytes (PMPA, PEPA, and GenX). As expected, the Nafion byproducts do not possess a linear trend as these molecules do not exhibit a pattern of increasing units with conserved substructure, like FTSA, PFCA or PFSA (for structures see Supporting Information). Instead, the Nafion byproducts share a fluorinated ethanesulfonic acid core, but differ in the number and placement of oxygen and differ widely in their chain termination (for chemical structures, see Supporting Information). The assessment of analytes sharing structural features from multiple groups also made establishing associations between m/z and CCS more challenging. For example, NVHOS possesses a sulfonic acid headgroup (like PFSA), yet it also contains an ether linkage in the middle of the substructure (similar to PFECA). In the IMS-MS analyses, NVHOS was found to possess an m/z and CCS value between the trendlines for the 2 subclasses. Thus, using the information gained from the trendline classification of the subclasses, IMS-MS analyses show great promise for identifying new and emerging PFAS subclasses by plotting their m/z and CCS values onto Figure 2. While many studies have characterized numerical associations for trendlines such as these and utilized their correlations in a predictive manner, a rigorous mathematical evaluation for PFAS trends is beyond the scope of the current work.51, 55
Figure 2.
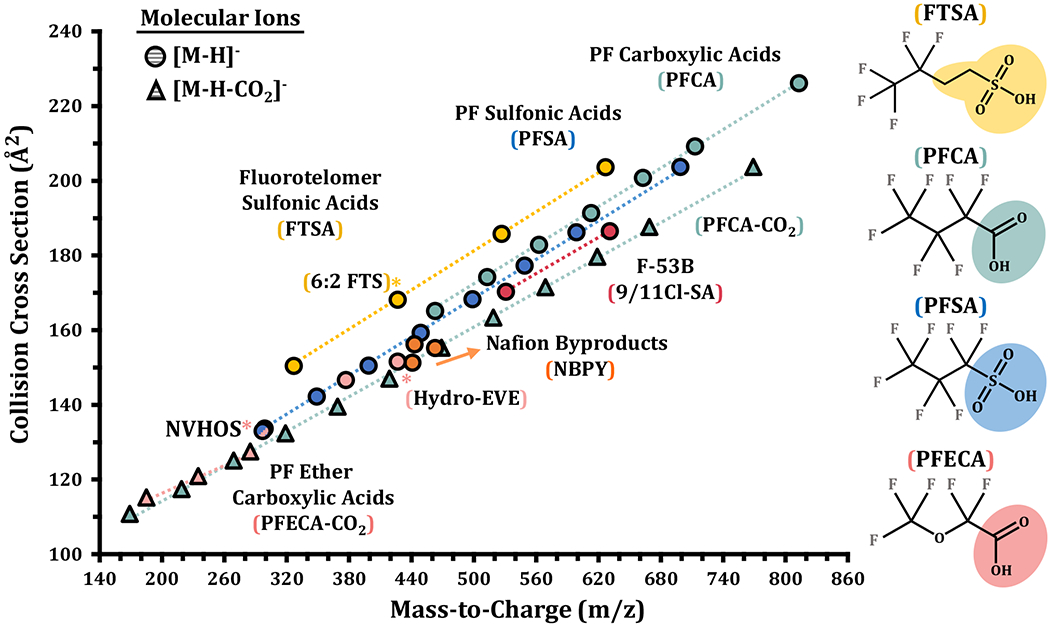
Mass/CCS trendlines for the specific PFAS subclasses analyzed (e.g. FTSA, PFSA, etc.). Specific PFAS headgroups contribute significantly to the observed CCS for a given m/z in each PFAS subclass. The varying chain lengths of CF2 then produce the linear trendlines observed within each subclass. Specific compounds discussed in the main text (e.g. 6:2 FTS and Hydro-EVE) are noted in addition to the PFAS subclasses.
Relationships between Isomeric Branching, CCS, and Retention Time.
As the trendlines for PFCA and PFSA subclasses shown in Figure 2 are based on primarily linear standards, another key focus of this work was to describe how isomeric branching affects CCS and causes deviations from the trendlines. To investigate this question, we obtained standards of specific branched isomers for PFOA and PFOS from Wellington Labs. Linear and branched isomers were characterized using both LC (retention time) and IMS (CCS) to examine the complementary separation of each method (Figure 3). For chemical structures of the singly-methylated and di-methylated isomers, see Supporting Information Figure S5. In all cases, the dimethylated isomers (DMHxA and DMHxS) elute from the LC column first, followed by the singly-methylated isomers (MHpA, MHpS) and finally each respective linear form (PFOA or PFOS, Supporting Information Figure S5). In the IMS analyses, the CCS values of the dimethylated isomers are also smaller than the singly-methylated isomers (with the exception of 5,5 dm-HxS), analogous to the elution pattern noted with reverse-phase liquid chromatography (RPLC). This result makes qualitative sense as the dimethylated and methylated forms should be more compact than the linear forms of PFOA/PFOS and hence have smaller CCS values. In terms of associating specific branch locations to retention time or CCS values for each species, differences were subtle and trends were more difficult to establish. For example, while the 6-m isomer of PFOA and PFOS has the largest CCS (other than the linear forms), CCS differences between the 4-m and 5-m isomers approaches the reproducibility error (ca. 0.3%). However, by combining the information from both approaches, most of the isomers can be resolved cooperatively using both chromatography and ion mobility. It should be noted that the trends put forth here are based solely on isomers for PFOA and PFOS and may not be applicable to all PFAS species.
Figure 3.
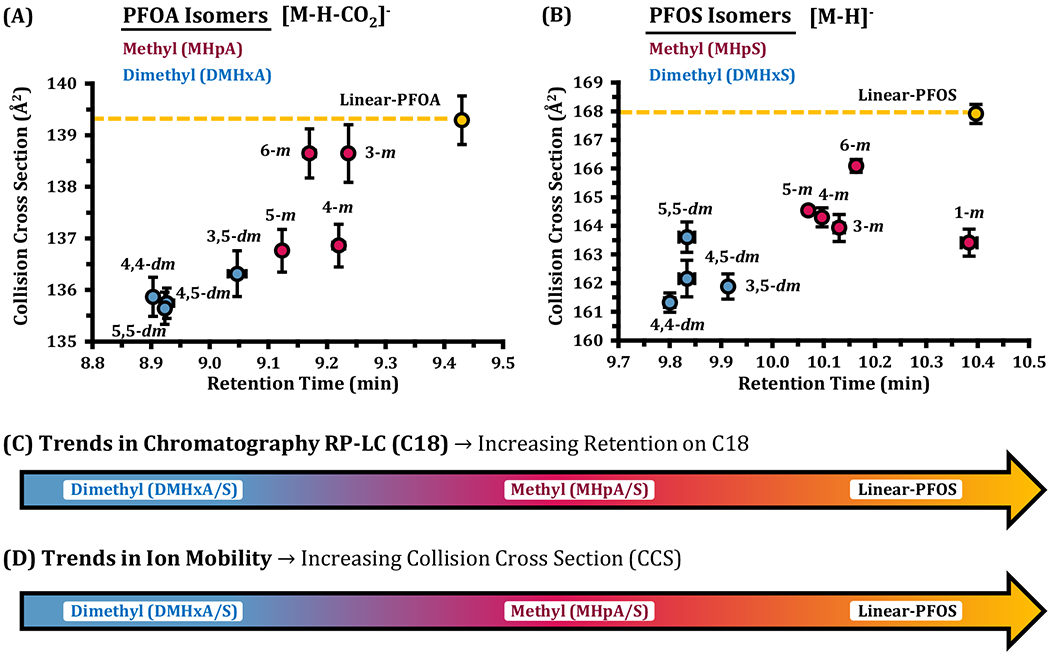
Correlation of RPLC retention time and CCS to specific constitutional isomers for PFOA (A) and PFOS (B). Dimethylated isomers elute earlier in RP-LC and tend to be more compact (smaller CCS) in comparison to singly-methylated isomers or linear forms of PFOA and PFOS (C and D). Error bars for both variation in retention time and CCS are illustrated, though some error bars are within marker size.
Isomeric Analysis in Multi-Component Mixtures.
When both LC and IMS separation are utilized cooperatively, it is possible to begin to characterize PFAS with multiple isomeric contributions, such as the manufacturing sample of PFOS we obtained from Synquest (see Figure 4). Previous characterizations of PFOS have illustrated as many as 16 isomers in a single sample.22, 56 Our analysis of PFOS produced 7 main isomeric contributors by LC-IMS-MS analyses which are labeled A, B, C, D, E, F and G in Figure 4. The observed LC trace is very similar to previous studies which have characterized specific branched PFOS isomers in detail (Supporting information, Figure S6).19, 57 Interestingly the new data added from the IMS dimension shows that the PFOS isomers cover a wide range of CCS space for an individual chemical formula (6.7 Å2, a difference in CCS of 4.1%, Figure 4). Analysis of the LC-IMS data shows that the principal peak (component F) is the linear species, which previous publications cite as contributing ca. 70% of relative abundance.18, 22 Components A and B were annotated as dimethylated PFOS isomers, and components C, D, E, and G are singly-methylated isomers. Complete structural characterization of each component remains challenging, however, as some sites of methylation have extremely similar retention times and CCS values. Therefore, A and C are only tentative assignments.
Figure 4.
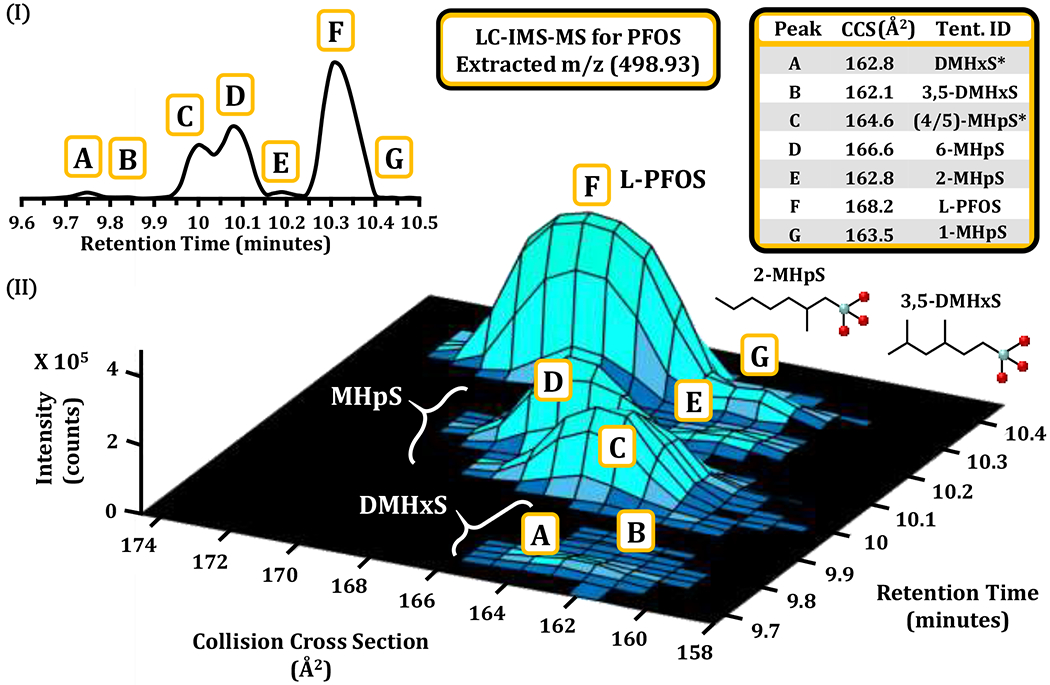
The separation of seven PFOS isomers by LC-IMS-MS. (I) While several publications have noted isomeric diversity for PFOS, this work notes 7 main contributors in our LC-IMS-MS analysis. The complementary separation mechanisms of analyte polarity from LC and molecular size (IMS) provide a more comprehensive snapshot of relative peak volume for each isomer (II). Converting observed drift times to CCS (inset Table) further provides a metric for verification if previously measured standards have been analyzed. The retention time and CCS values from Figure 3B are used to make tentative annotations to the spectra. (*) While some components are easily annotated using both retention time and CCS for characterization, component A is very close to several DMHxS isomers and component C is close to both the 4th and 5th methylation site (4/5)-MHpS isomers.
The LC and IMS separations in Figure 4 also illustrate the resolution afforded by orthogonal separation mechanisms of polarity (LC) and molecular size (IMS). Although not all of the isomeric distributions for PFOS could be resolved by the LC or IMS dimension alone, coupling these two techniques provides a more comprehensive snapshot of relative isomer volume in a 3-dimensional space. For example, Feature G in Figure 3 (1-MHpS) has the same retention time as L-PFOS, but a much lower CCS value; hence both isomers can be observed in the mixture. Conversely, Features A and E are dimethylated and singly-methylated isomers that possess the same nominal CCS, yet are readily resolved by retention time. However, even with the three separation dimensions (LC, IMS and MS) there was still some ambiguity in the identifications as A was very close to several of the DMHxS branched position isomers in retention time and CCS. C is most likely the 4 or 5th methylation site, if not a combination of both. Beyond PFOS and PFOA, other compounds that were noted to possess multiple distributions in our LC-IMS-MS analysis of the provided standards indicate potential isomers or conformers, including PFHpS, N-AP-FHxSA, and Hydro-EVE. For analytes with multiple distributions in the LC dimension CCS values were obtained for each distribution and the extended information is available in Excel Sheet ES1. Most other PFAS however provided well-defined Gaussian distributions in both the LC and IMS dimension, with relatively minor isomer contributions.
PFAS in Wastewater Outfall and Lake Water.
To evaluate the robustness of our LC-IMS-MS method in environmental samples, we analyzed samples collected from the wastewater outfall of a chemical plant (Chemours, Fayetteville, NC, Figure 5A) and nearby Marshwood Lake. This region of the Cape Fear River watershed has been the focus of prior investigations related to PFAS contamination and has been found to contain both legacy and emerging PFAS, such as GenX.12–14 After pre-concentration of 50 mL water using SPE, we detected several PFAS from the sulfonic acid subclass (PFSA) and several emerging PFESAs using LC-IMS-MS (Figure 5A and B). NVHOS and Nafion byproducts have recently been characterized by untargeted LC-MS/MS studies from McCord and Strynar,14 and their presence is not surprising considering our sampling sites are located in the same geographical area. In addition to high mass measurement accuracy and retention time alignment afforded by our instrument setup (see ES1), observed CCS values for detected analytes in the environmental samples are in good agreement with CCS from standards (typically <0.5% difference) as shown in detail in the Supporting Information Table S4. Results such as these illustrate the potential benefits of adding the IMS separation to current workflows.
Figure 5.
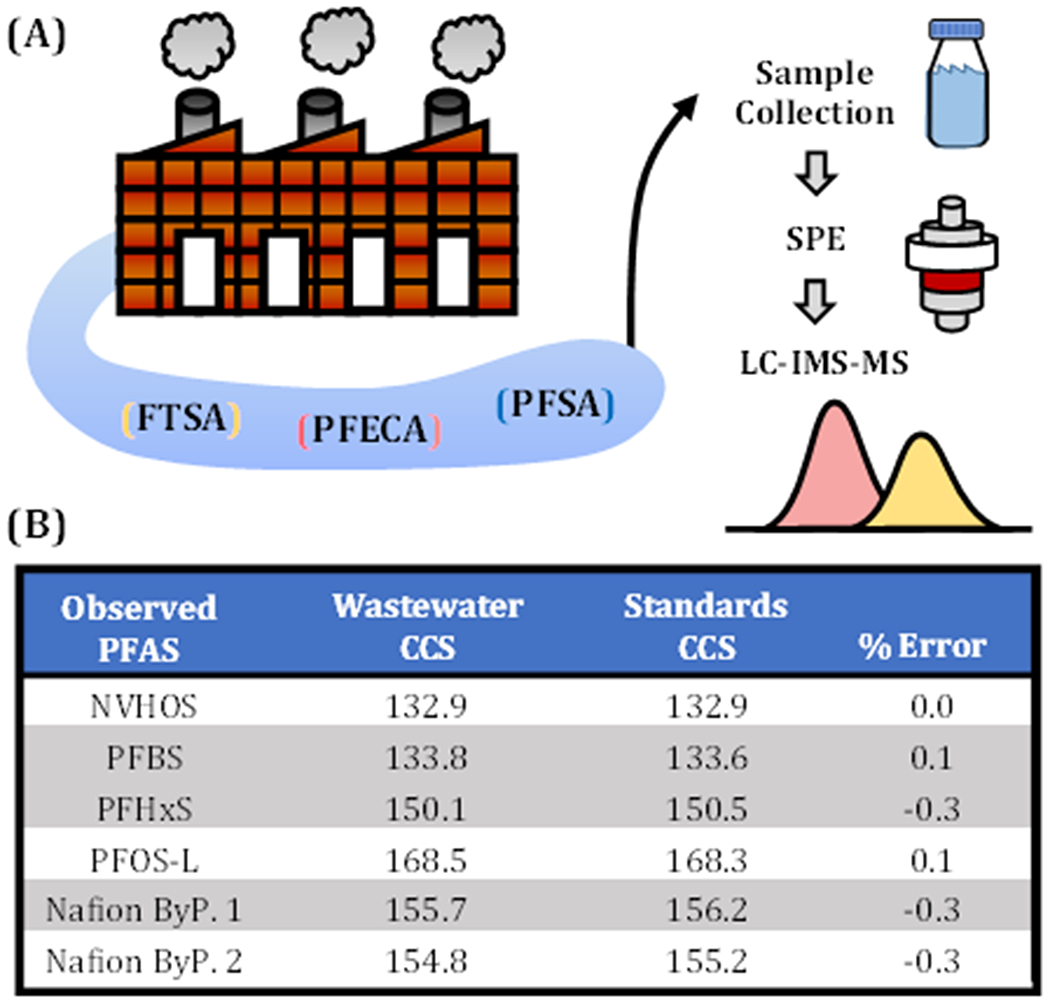
(A) Experimental workflow for PFAS analysis of Marshwood Lake, spatially located near a chemical plant in North Carolina. After pre-concentration with SPE and subsquent LC-IMS-MS analysis, measured CCS of detected PFAS correspond in good agreement to experimental standards (B). Retention time and accurate mass were also used for verification of annotated features (Supporting Information Table S4).
Overall, our results show that the use of IMS as a molecular descriptor in addition to accurate mass and retention time alignment in PFAS subclass and isomer analyses has great potential for rapidly characterizing branching positions and identifying various legacy and emerging PFAS species. In the trendline assessments relating CCS to m/z, each of the different subclasses was separated from the others based on their structural properties. These separations provide additional descriptors for the molecules and can point to new species in environmental and biological samples. It should be noted that the results shown in this manuscript are a proof of concept to demonstrate that measured CCS values in environmental samples can be matched to pre-existing CCS values from standards in order to increase confidence in annotated features. In this workflow we did not evaluate explicit concentration levels for each PFAS. Rather this sampling is strictly a qualitative measure of PFAS detection and CCS reproducibility in complex matrices. The results also highlighted positive characteristics of adding the IMS dimension such as the fact that while LC retention times may shift based on system void volume, variations in solvent composition, or changes in pH, CCS is an intrinsic molecular descriptor of gas phase size, and is not generally thought to be affected by changes in the above variables. Moreover, the CCS values for PFAS measured in this work can be readily incorporated into existing CCS libraries for inclusion into untargeted LC-IMS-MS workflows. As CCS values have been previously shown to be highly reproducible (often less than 1.0% different between laboratories),48 CCS values in this manuscript can be used as reference standards for laboratories that do not have access to analytical standards for PFAS observed in our analyses, especially for manufacturing byproduct chemistries (e.g. NVHOS and Nafion Byproducts). Utilizing database CCS matching for unknown analytes provides additional molecular confidence in annotating unknown features in untargeted studies, especially when combined with other molecular descriptors such as mass defect analysis,14 isotope ratio patterning, and fragmentation methods.51 In addition, as IMS is a chemically independent separation, the methods described in this manuscript can be readily adapted for screening of other environmental pollutants, such as pesticides, PCBs, and PAHs.31
Supplementary Material
ACKNOWLEDGMENT
All LC-IMS-MS measurements were made in the Molecular Education, Technology, and Research Innovation Center (METRIC) at NC State University. Funding for this work was made possible through a grant from the NIH National Institute of Environmental Health Sciences (P42 ES027704) and startup funds from North Carolina State University.
Footnotes
Supporting Information. The Supporting Information is available free of charge on the ACS Publications website.
Tabulated descriptors (retention times, observed m/z’s, drift times, and CCS) for each PFAS are detailed in Supporting Excel Sheets ES1–ES4 along with appropriate CCS calibration from Agilent Tune Mix. IMS smearing for small analytes is also described.
The authors declare no competing financial interest.
REFERENCES
- 1.Rahman MF; Peldszus S; Anderson WB, Behaviour and fate of perfluoroalkyl and polyfluoroalkyl substances (PFASs) in drinking water treatment: A review. Water Research 2014, 50, 318–340. [DOI] [PubMed] [Google Scholar]
- 2.McGuire ME; Schaefer C; Richards T; Backe WJ; Field JA; Houtz E; Sedlak DL; Guelfo JL; Wunsch A; Higgins CP, Evidence of Remediation-Induced Alteration of Subsurface Poly- and Perfluoroalkyl Substance Distribution at a Former Firefighter Training Area. Environmental Science & Technology 2014, 48, (12), 6644–6652. [DOI] [PubMed] [Google Scholar]
- 3.Steenland K; Zhao L; Winquist A; Parks C, Ulcerative Colitis and Perfluorooctanoic Acid (PFOA) in a Highly Exposed Population of Community Residents and Workers in the Mid-Ohio Valley. Environmental Health Perspectives 2013, 121, (8), 900–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lopez-Espinosa M-J; Mondal D; Armstrong B; Bloom Michael S; Fletcher T, Thyroid Function and Perfluoroalkyl Acids in Children Living Near a Chemical Plant. Environmental Health Perspectives 2012, 120, (7), 1036–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barry V; Winquist A; Steenland K, Perfluorooctanoic Acid (PFOA) Exposures and Incident Cancers among Adults Living Near a Chemical Plant. Environmental Health Perspectives 2013, 121, (11–12), 1313–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fletcher T; Galloway TS; Melzer D; Holcroft P; Cipelli R; Pilling LC; Mondal D; Luster M; Harries LW, Associations between PFOA, PFOS and changes in the expression of genes involved in cholesterol metabolism in humans. Environment International 2013, 57-58, 2–10. [DOI] [PubMed] [Google Scholar]
- 7.Chang ET; Adami H-O; Boffetta P; Wedner HJ; Mandel JS, A critical review of perfluorooctanoate and perfluorooctanesulfonate exposure and immunological health conditions in humans. Critical Reviews in Toxicology 2016, 46, (4), 279–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang Y; Starling AP; Haug LS; Eggesbo M; Becher G; Thomsen C; Travlos G; King D; Hoppin JA; Rogan WJ; Longnecker MP, Association between Perfluoroalkyl substances and thyroid stimulating hormone among pregnant women: a cross-sectional study. Environmental Health 2013, 12, (1), 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li M; Zeng X-W; Qian Z; Vaughn MG; Sauvé S; Paul G; Lin S; Lu L; Hu L-W; Yang B-Y; Zhou Y; Qin X-D; Xu S-L; Bao W-W; Zhang Y-Z; Yuan P; Wang J; Zhang C; Tian Y-P; Nian M; Xiao X; Fu C; Dong G-H, Isomers of perfluorooctanesulfonate (PFOS) in cord serum and birth outcomes in China: Guangzhou Birth Cohort Study. Environment International 2017, 102, 1–8. [DOI] [PubMed] [Google Scholar]
- 10.Beauvais J, Lifetime Health Advisories and Health Effects Support Documents for Perfluorooctanoic Acid and Perfluoroctane Sulfonate In Federal Register: Federal Register, 2016; Vol. 81, pp 33250–33251. [Google Scholar]
- 11.Li L; Zhai Z; Liu J; Hu J, Estimating industrial and domestic environmental releases of perfluorooctanoic acid and its salts in China from 2004 to 2012. Chemosphere 2015, 129, 100–109. [DOI] [PubMed] [Google Scholar]
- 12.Sun M; Arevalo E; Strynar M; Lindstrom A; Richardson M; Kearns B; Pickett A; Smith C; Knappe DRU, Legacy and Emerging Perfluoroalkyl Substances Are Important Drinking Water Contaminants in the Cape Fear River Watershed of North Carolina. Environmental Science & Technology Letters 2016, 3, (12), 415–419. [Google Scholar]
- 13.Hopkins ZR; Sun M; DeWitt JC; Knappe DRU, Recently Detected Drinking Water Contaminants: GenX and Other Per- and Polyfluoroalkyl Ether Acids. Journal - American Water Works Association 2018, 110, (7), 13–28. [Google Scholar]
- 14.Au - McCord J; Au - Strynar M, Identifying Per- and Polyfluorinated Chemical Species with a Combined Targeted and Non-Targeted-Screening High-Resolution Mass Spectrometry Workflow. JoVE 2019, (146), e59142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu Y; Qian M; Ma X; Zhu L; Martin JW, Nontarget Mass Spectrometry Reveals New Perfluoroalkyl Substances in Fish from the Yangtze River and Tangxun Lake, China. Environmental Science & Technology 2018, 52, (10), 5830–5840. [DOI] [PubMed] [Google Scholar]
- 16.Appleman TD; Higgins CP; Quiñones O; Vanderford BJ; Kolstad C; Zeigler-Holady JC; Dickenson ERV, Treatment of poly- and perfluoroalkyl substances in U.S. full-scale water treatment systems. Water Research 2014, 51, 246–255. [DOI] [PubMed] [Google Scholar]
- 17.Zhang C; Hopkins ZR; McCord J; Strynar MJ; Knappe DRU, Fate of Per- and Polyfluoroalkyl Ether Acids in the Total Oxidizable Precursor Assay and Implications for the Analysis of Impacted Water. Environmental Science & Technology Letters 2019, 6, (11), 662–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Benskin JP; De Silva AO; Martin JW, Isomer Profiling of Perfluorinated Substances as a Tool for Source Tracking: A Review of Early Findings and Future Applications In Reviews of Environmental Contamination and Toxicology Volume 208: Perfluorinated alkylated substances, De Voogt P, Ed. Springer New York: New York, NY, 2010; pp 111–160. [DOI] [PubMed] [Google Scholar]
- 19.Kärrman A; Langlois I; Bavel B.v .; Lindström G; Oehme M, Identification and pattern of perfluorooctane sulfonate (PFOS) isomers in human serum and plasma. Environment International 2007, 33, (6), 782–788. [DOI] [PubMed] [Google Scholar]
- 20.Langlois I; Oehme M, Structural identification of isomers present in technical perfluorooctane sulfonate by tandem mass spectrometry. Rapid Communications in Mass Spectrometry 2006, 20, (5), 844–850. [DOI] [PubMed] [Google Scholar]
- 21.De Silva AO; Mabury SA, Isolating Isomers of Perfluorocarboxylates in Polar Bears (Ursus maritimus) from Two Geographical Locations. Environmental Science & Technology 2004, 38, (24), 6538–6545. [DOI] [PubMed] [Google Scholar]
- 22.Benskin JP; Yeung LWY; Yamashita N; Taniyasu S; Lam PKS; Martin JW, Perfluorinated Acid Isomer Profiling in Water and Quantitative Assessment of Manufacturing Source. Environmental Science & Technology 2010, 44, (23), 9049–9054. [DOI] [PubMed] [Google Scholar]
- 23.Ahmed E; Mohibul Kabir KM; Wang H; Xiao D; Fletcher J; Donald WA, Rapid separation of isomeric perfluoroalkyl substances by high-resolution differential ion mobility mass spectrometry. Analytica Chimica Acta 2019. [DOI] [PubMed] [Google Scholar]
- 24.De Silva AO; Muir DCG; Mabury SA, Distribution of perfluorocarboxylate isomers in select samples from the north american environment. Environmental Toxicology and Chemistry 2009, 28, (9), 1801–1814. [DOI] [PubMed] [Google Scholar]
- 25.Viehland LA; Mason EA, Gaseous lon mobility in electric fields of arbitrary strength. Annals of Physics 1975, 91, (2), 499–533. [Google Scholar]
- 26.Liu Y; Pereira ADS; Martin JW, Discovery of C5–C17 Poly- and Perfluoroalkyl Substances in Water by In-Line SPE-HPLC-Orbitrap with In-Source Fragmentation Flagging. Analytical Chemistry 2015, 87, (8), 4260–4268. [DOI] [PubMed] [Google Scholar]
- 27.Stephan S; Hippler J; Köhler T; Deeb AA; Schmidt TC; Schmitz OJ, Contaminant screening of wastewater with HPLC-IM-qTOF-MS and LC+LC-IM-qTOF-MS using a CCS database. Analytical and Bioanalytical Chemistry 2016, 408, (24), 6545–6555. [DOI] [PubMed] [Google Scholar]
- 28.May JC; McLean JA, Ion Mobility-Mass Spectrometry: Time-Dispersive Instrumentation. Analytical Chemistry 2015, 87, (3), 1422–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fenn LS; McLean JA, Biomolecular structural separations by ion mobility–mass spectrometry. Analytical and Bioanalytical Chemistry 2008, 391, (3), 905–909. [DOI] [PubMed] [Google Scholar]
- 30.Goodwin CR; Fenn LS; Derewacz DK; Bachmann BO; McLean JA, Structural Mass Spectrometry: Rapid Methods for Separation and Analysis of Peptide Natural Products. Journal of Natural Products 2012, 75, (1), 48–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zheng X; Dupuis KT; Aly NA; Zhou Y; Smith FB; Tang K; Smith RD; Baker ES, Utilizing ion mobility spectrometry and mass spectrometry for the analysis of polycyclic aromatic hydrocarbons, polychlorinated biphenyls, polybrominated diphenyl ethers and their metabolites. Analytica Chimica Acta 2018, 1037, 265–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hines KM; Ross DH; Davidson KL; Bush MF; Xu L, Large-Scale Structural Characterization of Drug and Drug-Like Compounds by High-Throughput Ion Mobility-Mass Spectrometry. Analytical Chemistry 2017, 89, (17), 9023–9030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mairinger T; Causon TJ; Hann S, The potential of ion mobility–mass spectrometry for non-targeted metabolomics. Current Opinion in Chemical Biology 2018, 42, 9–15. [DOI] [PubMed] [Google Scholar]
- 34.Zhang X; Quinn K; Cruickshank-Quinn C; Reisdorph R; Reisdorph N, The application of ion mobility mass spectrometry to metabolomics. Current Opinion in Chemical Biology 2018, 42, 60–66. [DOI] [PubMed] [Google Scholar]
- 35.May JC; Gant-Branum RL; McLean JA, Targeting the untargeted in molecular phenomics with structurally-selective ion mobility-mass spectrometry. Current Opinion in Biotechnology 2016, 39, 192–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Giles K; Williams JP; Campuzano I, Enhancements in travelling wave ion mobility resolution. Rapid Communications in Mass Spectrometry 2011, 25, (11), 1559–1566. [DOI] [PubMed] [Google Scholar]
- 37.Pu Y; Ridgeway ME; Glaskin RS; Park MA; Costello CE; Lin C, Separation and Identification of Isomeric Glycans by Selected Accumulation-Trapped Ion Mobility Spectrometry-Electron Activated Dissociation Tandem Mass Spectrometry. Analytical Chemistry 2016, 88, (7), 3440–3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bijlsma L; Berntssen MHG; Merel S, A Refined Nontarget Workflow for the Investigation of Metabolites through the Prioritization by in Silico Prediction Tools. Analytical Chemistry 2019, 91, (9), 6321–6328. [DOI] [PubMed] [Google Scholar]
- 39.Poland JC; Schrimpe-Rutledge AC; Sherrod SD; Flynn CR; McLean JA, Utilizing untargeted ion mobility-mass spectrometry to profile changes in the gut metabolome following biliary diversion surgery. Analytical Chemistry 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cumeras R; Figueras E; Davis CE; Baumbach JI; Gràcia I, Review on Ion Mobility Spectrometry. Part 1: current instrumentation. Analyst 2015, 140, (5), 1376–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cumeras R; Figueras E; Davis CE; Baumbach JI; Gràcia I, Review on Ion Mobility Spectrometry. Part 2: hyphenated methods and effects of experimental parameters. Analyst 2015, 140, (5), 1391–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McCullagh M; Hodgkinson M; Dillon L; van Bavel B; Burgess JA; Jogsten IE, Determination and Characterization of Perfluoroalkyl and Polyfluoroalkyl Substances (PFAS’s) in Environmental Samples using Ion Mobility MS In Waters Application Note: Online, 2014; pp 1–9. [Google Scholar]
- 43.Buck RC; Franklin J; Berger U; Conder JM; Cousins IT; de Voogt P; Jensen AA; Kannan K; Mabury SA; van Leeuwen SPJ, Perfluoroalkyl and polyfluoroalkyl substances in the environment: Terminology, classification, and origins. Integrated Environmental Assessment and Management 2011, 7, (4), 513–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mueller R; Yingling V, Naming Conventions and Physical and Chemical Properties of Per- and Polyfluoroalkyl Substances (PFAS) In Interstate Technology Regulatory Council: https://pfas-1.itrcweb.org/wp-content/uploads/2017/10/pfas_fact_sheet_naming_conventions_11_13_17.pdf, 2017; pp 1–15.
- 45.Anumol T; Yang D-H; Sosienski T; Batoon P, Analysis of per/polyfluoroalkyl substances (PFASs) in drinking water using the Agilent Ultivo triple quadrupole LC/MS. Agilent Application Note 2018. [Google Scholar]
- 46.May JC; Goodwin CR; Lareau NM; Leaptrot KL; Morris CB; Kurulugama RT; Mordehai A; Klein C; Barry W; Darland E; Overney G; Imatani K; Stafford GC; Fjeldsted JC; McLean JA, Conformational Ordering of Biomolecules in the Gas Phase: Nitrogen Collision Cross Sections Measured on a Prototype High Resolution Drift Tube Ion Mobility-Mass Spectrometer. Analytical Chemistry 2014, 86, (4), 2107–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.May JC; Dodds JN; Kurulugama RT; Stafford GC; Fjeldsted JC; McLean JA, Broadscale resolving power performance of a high precision uniform field ion mobility-mass spectrometer. Analyst 2015, 140, (20), 6824–6833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stow SM; Causon TJ; Zheng X; Kurulugama RT; Mairinger T; May JC; Rennie EE; Baker ES; Smith RD; McLean JA; Hann S; Fjeldsted JC, An Interlaboratory Evaluation of Drift Tube Ion Mobility–Mass Spectrometry Collision Cross Section Measurements. Analytical Chemistry 2017, 89, (17), 9048–9055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dodds JN; May JC; McLean JA, Investigation of the Complete Suite of the Leucine and Isoleucine Isomers: Toward Prediction of Ion Mobility Separation Capabilities. Analytical Chemistry 2017, 89, (1), 952–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nichols CM; Dodds JN; Rose BS; Picache JA; Morris CB; Codreanu SG; May JC; Sherrod SD; McLean JA, Untargeted Molecular Discovery in Primary Metabolism: Collision Cross Section as a Molecular Descriptor in Ion Mobility-Mass Spectrometry. Analytical Chemistry 2018, 90, (24), 14484–14492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Picache JA; Rose BS; Balinski A; Leaptrot, Katrina L.; Sherrod, S. D.; May, J. C.; McLean, J. A., Collision cross section compendium to annotate and predict multi-omic compound identities. Chemical Science 2019, 10, (4), 983–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Revercomb HE; Mason EA, Theory of plasma chromatography/gaseous electrophoresis. Review. Analytical Chemistry 1975, 47, (7), 970–983. [Google Scholar]
- 53.Silcock P; Karrman A; van Bavel B, Advancing Perfluorinated Compound Analysis Using Simultaneous Matrix Monitoring In Waters Application Note: Online, 2014; pp 1–7. [Google Scholar]
- 54.Houel S; Abernathy R; Renganathan K; Meyer-Arendt K; Ahn NG; Old WM, Quantifying the Impact of Chimera MS/MS Spectra on Peptide Identification in Large-Scale Proteomics Studies. Journal of Proteome Research 2010, 9, (8), 4152–4160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Leaptrot KL; May JC; Dodds JN; McLean JA, Ion mobility conformational lipid atlas for high confidence lipidomics. Nature Communications 2019, 10, (1), 985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Langlois I; Berger U; Zencak Z; Oehme M, Mass spectral studies of perfluorooctane sulfonate derivatives separated by high-resolution gas chromatography. Rapid Communications in Mass Spectrometry 2007, 21, (22), 3547–3553. [DOI] [PubMed] [Google Scholar]
- 57.Martin JW; Kannan K; Berger U; Voogt PD; Field J; Franklin J; Giesy JP; Harner T; Muir DCG; Scott B; Kaiser M; Järnberg U; Jones KC; Mabury SA; Schroeder H; Simcik M; Sottani C; Bavel BV; Kärrman A; Lindström G; Leeuwen SV, Peer Reviewed: Analytical Challenges Hamper Perfluoroalkyl Research. Environmental Science & Technology 2004, 38, (13), 248A–255A. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.