Abstract
More than ten years ago, the German Consortium for Hereditary Breast and Ovarian Cancer (GC-HBOC) set up a panel of experts (VUS Task Force) which was tasked with reviewing the classifications of genetic variants reported by individual centres of the GC-HBOC to the central database in Leipzig and reclassifying them, where necessary, based on the most recent data. When it evaluates variants, the VUS Task Force must arrive at a consensus. The resulting classifications are recorded in a central database where they serve as a basis for ensuring the consistent evaluation of previously known and newly identified variants in the different centres of the GC-HBOC. The standardised VUS evaluation by the VUS Task Force is a key element of the recall system which has also been set up by the GC-HBOC. The system will be used to pass on information to families monitored and managed by GC-HBOC centres in the event that previously classified variants are reclassified based on new information. The evaluation algorithm of the VUS Task Force was compiled using internationally established assessment methods (IARC, ACMG, ENIGMA) and is presented here together with the underlying evaluation criteria used to arrive at the classification decision using a flow chart. In addition, the characteristics and special features of specific individual risk genes associated with breast and/or ovarian cancer are discussed in separate subsections. The URLs of relevant databases have also been included together with extensive literature references to provide additional information and cover the scope and dynamism of the current state of knowledge on the evaluation of genetic variants. In future, if criteria are updated based on new information, the update will be published on the website of the GC-HBOC ( https://www.konsortium-familiaerer-brustkrebs.de/ ).
Key words: hereditary breast/ovarian cancer, classification of genetic variants, risk genes
General Principles
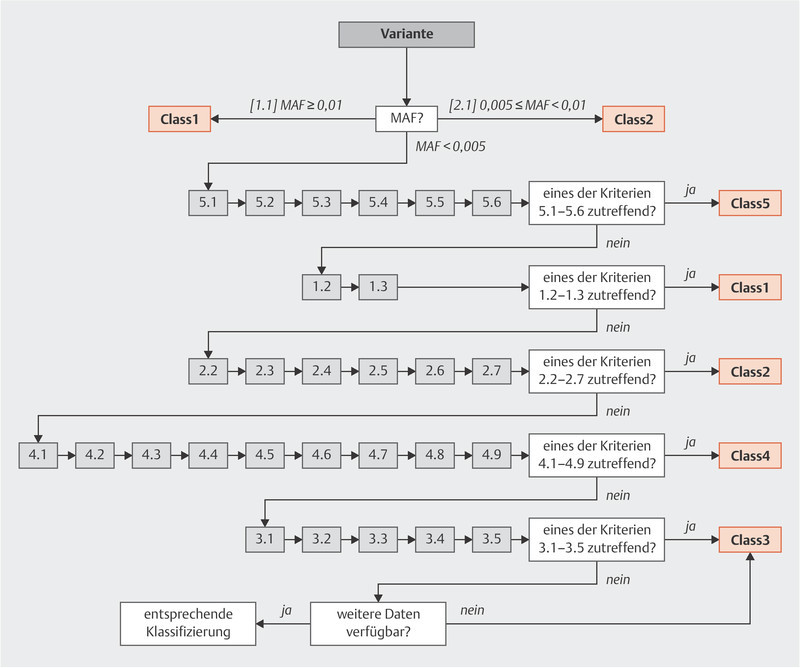
The criteria of the German Consortium for Hereditary Breast and Ovarian Cancer ( http://www.konsortium-familiaerer-brustkrebs.de/ ) for the classification of germline sequence variants in risk genes for hereditary breast and ovarian cancer were developed and compiled by the members of the panel of experts on variant evaluation (VUS Task Force) from the German Consortium for Hereditary Breast and Ovarian Cancer listed above. The task of this panel of experts is to specify binding criteria for the German Consortium for Hereditary Breast and Ovarian Cancer to evaluate variants and verify the classification of variants to ensure that variants are uniformly evaluated by the Consortium. The present criteria are based on the IARC 1 5-class system for high-risk genes 2 which is based on the guidelines issued by the ENIGMA 3 Consortium (ENIGMA BRCA1/2 Classification Criteria, Version 2.5.1, June 2017), as well as the ACMG 4 and ACGS 5 guidelines. Using this 5-class system, germline sequence variants are evaluated in terms of their relevance for a loss of function of the coded protein (Class1: neutral, Class2: likely neutral, Class3: uncertain evidence/no reliable evaluation, Class4: likely relevant loss of function, Class5: relevant loss of function; level of significance, see 4 ). For high-penetrance genes (such as BRCA1, BRCA2 ) for which a clinical correlation (pathogenicity) with loss of function has been described, the functional classification yields a pathogenicity evaluation based on the IARC 5-class system (ranging from Class1: not pathogenic to Class5: pathogenic), as outlined in Appendix A 2, Table 1 . The advantage of such a structured approach is that it starts by checking for defined criteria which can be used for a quick and unambiguous classification, and the extensive data and literature search is only carried out afterwards (see Appendix A 2, Table 2 : Relevant literature and databases, and A 3, Fig. 1 : Evaluation criteria flow chart). As regards the classification of sequence variants of the genes ATM, BRCA1, BRCA2, BRIP1, CDH1, CHEK2, PALB2, RAD51C, RAD51D and TP53 , all of them so-called “core genes” according to the TruRisk panel (version 1/2018, see the homepage of the German Consortium: http://www.konsortium-familiaerer-brustkrebs.de/ ), the specific features of the individual genes listed in Appendix A 5 must be taken into account. Moderate/low-penetrance genes are only evaluated in terms of their functionality (in these cases: loss of function should not be equated with “pathogenicity”). Even variants of high-risk genes may only be associated with an intermediate risk 5 , 6 .
Table 1 IARC 5-tiered classification system with accompanying recommendations for family management a (excerpt from https://enigmaconsortium.org/wp-content/uploads/2018/10/ENIGMA_Rules_2017-06-29-v2.5.1.pdf ).
| Class | Quantitative measure: probability of pathogenicity | Predictive testing of at-risk relatives | Surveillance of at-risk relatives | Research testing of relatives |
|---|---|---|---|---|
|
a
Adapted for clarity from the original published tabular presentation (Plon et al., 2008
4
)
b Continued testing of proband for any additional available testing modalities available for BRCA1/2, e.g. rearrangements, is recommended. | ||||
| 5: Pathogenic | > 0.99 | Yes | Full high-risk guidelines for variant carriers | Not indicated |
| 4: Likely pathogenic | 0.95 – 0.99 | Yes b | Full high-risk guidelines for variant carriers | Yes |
| 3: Uncertain | 0.05 – 0.949 | No b | Based on family history & other risk factors | Yes |
| 2: Likely not pathogenic or of little clinical significance | 0.001 – 0.049 | No b | Based on family history & other risk factors – treat as “no BRCA1/2 pathogenic variant detected” for this disorder | Yes |
| 1: Not pathogenic or of no clinical significance | < 0.001 | No b | Based on family history & other risk factors – treat as “no BRCA1/2 pathogenic variant detected” for this disorder | Not indicated |
Table 2 Relevant literature and databases available for evaluation.
Fig. 1.

VUS evaluation criteria 1.1.
Criteria for the Interpretation of mRNA Analyses
The criteria of the German Consortium for Hereditary Breast and Ovarian Cancer for the evaluation of sequence variants with subsequent mRNA analysis are based on the guidelines of the ENIGMA Consortium (see also 7 ). The respective threshold values of potential splice variants for an empirical predictive prognosis based on three commonly used predictive programmes are given in Appendix A 1. Appendix A 4 ( Fig. 2 ) gives a schematic representation of the areas considered by the VUS Task Force when evaluating the splice variants. mRNA analysis is carried out using fresh blood samples, cultured lymphocytes, cultured lymphoblastoid cell lines, etc. and compared in parallel with at least 5 controls of the same type of material. A sequence variant is described as pathogenic if it has the following effect on mRNA transcription: one or more aberrant transcripts of the variant allele are detected, which lead to a stop codon or an in-frame deletion and result in the destruction of known functional domains. Sequencing of the full-length transcript of the variant allele or the presence of an intronic variant of a cis-acting polymorphism is considered sufficient (evidence of monoallelic expression) to determine the transcript amount (using semi-quantitative or quantitative methods). Variants which show a transcript pattern comparable to the mean value of controls are rated as neutral/not pathogenic due to the lack of aberrant mRNA. As regards the cDNA primer design, the physiological splice variants/naturally occurring isoforms must also be taken into account (see 8 , 9 ).
Fig. 2.

Classification of variants which can affect splicing.
Caution: Certain BRCA1 and BRCA2 variants which are ± 1, 2 bp from the exon border and which are predicted or proven to lead to at least 20 – 30% naturally occurring in-frame RNA isoforms per allele could presumably result in some residual protein activity (see 9 , 10 , 11 , 12 ) and are therefore classified as VUS Class3, unless there is evidence to the contrary (see Overview, Appendix 5, Table 5 )
Table 5 Excerpt from https://enigmaconsortium.org/wp-content/uploads/2018/10/ENIGMA_Rules_2017-06-29-v2.5.1.pdf .
| Table 6: BRCA1 and BRCA2 exon boundary variants predicted or known to lead to naturally occurring in-frame RNA isoforms that may rescue gene functionality. Variants at these positions should be considered Class3 Uncertain unless proven otherwise.* | |||
|---|---|---|---|
| Gene | Alternative splicing event | Variants implicated | Rationale |
| * This summary table does not yet capture the possibility of acceptor site changes leading to small in-frame deletions > 3 bp, e.g. due to NAG (NNN) n NAG sites. It is recommended that bioinformatic prediction analysis is carried out for variation in/near all donor and acceptor sites to assess the likelihood that a variant will or will not cause alternative splicing. | |||
|
Note: It could be argued that nonsense or frameshift variants in
BRCA1
exon 9,
BRCA1
exon 10, or
BRCA2
exon 12 may not be associated with high risk of cancer due to rescue by the expression of in-frame naturally occurring isoforms that bypass the premature termination codon and thus encode a functional protein. A review of multiple clinical and control datasets for the frequency of unique nonsense or frameshift variants –
adjusted for exon size
– does not provide strong support for this hypothesis at present (Spurdle, de la Hoya, unpublished data). Additional research is underway to further investigate the functional/clinical importance of germline nonsense or frameshift variants in these exons.
Moreover, further work is planned within ENIGMA (led by Paolo Radice) to document variants that have undergone splicing assays and are proven to be “leaky” variants, to provide a record of all spliceogenic variants for which additional research is necessary. This resource will identify variants that have already been classified using clinical data, as positive and negative controls for future quantitative mRNA studies. | |||
| BRCA1 | Δ8p | c.442-1 (IVS7-1) c.442-2 (IVS7-2) |
BRCA1 exon 8 acceptor site is an experimentally validated tandem acceptor site (NAGNAG) subject to alternative splicing (Colombo et al., 2014). c.442-1,-2 variants are predicted to inactivate the 5′ acceptor site, but not the 3′ acceptor site, thus producing Δ8p transcripts. |
| Δ9,10 | c.548-1 (IVS8-1) c.548-2 (IVS8-2) c.593 to non-G c.593+1 (IVS9+1) c.593+2 (IVS9+2) c.594-1 (IVS9-1) c.594-2 (IVS9-2) c.670 to non-G c.670+1 (IVS10+1) c.670+2 (IVS10+2) |
Carriers of variants at these positions are predicted to produce normal (or increased) levels of
BRCA1
Δ(9,10), a major in-frame alternative splicing event (Colombo et al., 2014).
The BRCA1 variant c.594-2A>C (shown from ENIGMA research to co-occur in cis with c.641A>G), has been reported to demonstrate clinical characteristics inconsistent with a high risk of cancer expected for a pathogenic BRCA1 variant (Rosenthal et al., 2015). The haplotype of c.[594-2A>C; 641A>G] has been shown from mRNA analysis in human samples to produce high levels of Δ10 transcripts (70% of the overall expression, and has been designated as Class1 Not Pathogenic by the ENIGMA Consortium using multifactorial likelihood analysis that includes genetic (segregation, case-control analysis) and pathology data (de la Hoya et al., 2016). |
|
| Δ11q, Δ11 | c.4096 to non-G c.4096+1 (IVS11+1) c.4097+2 (IVS11+2) |
Data collected by the ENIGMA consortium demonstrates that the BRCA1 c.4096+1G>A variant, proven to result in the production of naturally occurring in-frame transcripts Δ11q (Bonatti et al., 2006) and also Δ11 (Radice, unpublished data), may not exhibit the clinical characteristics of a standard high-risk pathogenic BRCA1 variant (Spurdle, unpublished data). | |
| Δ13p | c.4186-1 (IVS12-1) c.4186-2 (IVS12-2) |
BRCA1 exon 13 acceptor site is an experimentally validated tandem acceptor site (NAGNAG) subject to alternative splicing (Colombo et al., 2014). c.4186-1,-2 variants are predicted to inactivate the 5′ acceptor site, but not the 3′ acceptor site, thus producing Δ13p transcripts. | |
| Δ14p | c.4358-1 (IVS13-1) c.4358-2 (IVS13-2) |
BRCA1 exon 14 acceptor site is an experimentally validated tandem acceptor site (NAGNAG) subject to alternative splicing (Colombo et al., 2014). c.4358-1,-2 variants are predicted to inactivate the 5′ acceptor site, but not the 3′ acceptor site, thus producing Δ14p transcripts. | |
| BRCA2 | Δ12 | c.6842-1 (IVS11-1) c.6842-2 (IVS11-2) c.6937 to non-G c.6937+1 (IVS12+1) c.6937+2 (IVS12+2) |
Carriers of these variants are predicted to produce exon 12 skipping. BRCA2 Δ12 is a naturally occurring in-frame splicing event (Fackenthal et al., 2016). BRCA2 exon 12 is functionally redundant (Li et al., 2009). |
Approach of the VUS Task Force
The Consortium recommends routinely testing for 10 genes which are known (as per 8/19) to be associated with breast and/or ovarian cancer: ATM, BRCA1, BRCA2, BRIP1, CDH1, CHEK2, PALB2, RAD51C, RAD51D and TP53 ( http://www.konsortium-familiaerer-brustkrebs.de/ ).
The Consortiumʼs panel of experts (VUS Task Force) holds monthly telephone conferences and, if necessary, meetings to reach a consensus on the classification of newly reported sequence variants, discuss any new evidence available for the re-evaluation of already known variants, and evaluate variants of unclear significance. In the event of a re-evaluation, the central database of the Consortium will inform all centres about the reclassification (recall system).
It should be expressly noted that new information can lead to changes in the classification of variants and that these classifications are regularly reviewed by the panel of experts. Similarly, new findings can lead to changes in the list of core genes for which the German Consortium for Hereditary Breast and Ovarian Cancer recommends that patients are tested. All such changes along with the inclusion of new findings into the classification are published on the homepage of the German Consortium for Hereditary Breast and Ovarian Cancer ( http://www.konsortium-familiaerer-brustkrebs.de/ ).
Classification of Sequence Variants According to Their Functional Relevance
1. Class1 (functionally irrelevant/no loss of function) if one of the following criteria is met:
1.1 Allele frequency of variants in large population groups (e.g. Caucasians, Africans, or Asians) is ≥ 1% (minor allele frequency [MAF] ≥ 0.01). Caution: An allele frequency of ≥ 1% in subpopulations with a low-diversity gene pool (examples: Finnish population, founder mutations!) is not sufficient.
1.2 Variants with a calculated multifactorial probability of < 0.001 of being pathogenic.
Caution: This currently only applies to the high-risk genes BRCA1/2 (for an exemplary calculation, see 13 ).
1.3 Variants in high-risk genes which occur in at least 10 individuals in appropriate cohorts of persons without disease ( Table 1 ).
2. Class2 (probably no loss of function/functionally irrelevant) if one of the following criteria are met:
2.1 Allele frequency of variants in large population groups (e.g. Caucasians, Africans, or Asians) is 0.5 – 1% (MAF 0.005 – 0.01) Caution: An allele frequency of 0.5 – 1% in subpopulations with a low-diversity gene pool (examples: Finnish population, founder mutations!) is not sufficient.
2.2 Exonic variants (A) which result in substitution of an amino acid (missense variants) or small in-frame insertions/deletions (insertions/deletions of one or fewer amino acid [s]) and whose a priori probability of pathogenicity is ≤ 2% (A-GVGD analysis, http://priors.hci.utah.edu/PRIORS/ ); intronic variants (B) which are more than − 20 bp, + 10 bp from the exon border ; and synonymous variants (C) if these variants (A – C) will, according to bioinformatic prediction programmes (see Appendix A 1), in all probability not change the splicing mechanism. In non- BRCA1/2 genes, the above-mentioned variants must be present in large population groups with an allele frequency of 0.001 ≤ MAF < 0.01.
2.3 Synonymous substitutions or intronic variants which show no mRNA aberrations in the form of exon deletions/duplications or monoallelic expression of the wildtype (wt) transcript in “in-vitro” laboratory tests even if, according to bioinformatic prediction programmes (see Appendix A 1 for programmes and threshold values), in all probability they change the splicing mechanism.
2.4 Variants which occur in the same gene with a clearly pathogenic variant in trans (co-occurrence), if it has been verified that a homozygous or compound heterozygous genotype is associated with a known, clinically unambiguous phenotype.
2.5 Variants with a calculated multifactorial probability of pathogenicity of 0.001 – 0.049.
Caution: This currently only applies to the high-risk genes BRCA1/2 (for an exemplary calculation, see: 13 ).
2.6 Exon variants which code for the same amino acid exchange as a sequence variant which has already been classified as Class1 but are based on a different nucleotide exchange if no aberrant splicing is predicted.
2.7 Missense variants for which information from functional analyses, etc., is available but not sufficient for multifactorial classification and which have been classified as Class2 by a panel of experts (e.g. ENIGMA).
3. Class3 (unclear functional relevance) if one of the following criteria is met: variants which cannot be unambiguously assigned to Class1, Class2, Class4, or Class5, e.g.:
3.1 Special cases which could be assigned to one of the other classes based on the evaluation criteria but are listed in Appendix A 5 among the characteristics of individual core genes or in Table 5, Appendix of BRCA1/2 classification criteria, Version 2.5.1, July 2017 (ENIGMA) ( Table 5 ).
3.2 Variants where the data used for their evaluation is contradictory and for which further studies are still required.
3.3 Variants which are − 20 bp, + 10 bp from the exon border and which, based on bioinformatic predictive programmes (see Appendix A 1), probably change the splicing mechanism as long as no in-vitro mRNA analysis has been done yet ( Fig. 2 , Schematic representation of variants in the vicinity of splice sites).
3.4 Exon duplications which have not been analysed further (e.g. break point analysis, cDNA analysis, etc.).
3.5 Variants with a calculated multifactorial probability of pathogenicity of 0.05 – 0.949.
Caution: This currently only applies to the high-risk genes BRCA1/2 (for an exemplary calculation, see 13 ).
4. Class4 (probable loss of function/functionally relevant) if one of the following criteria is met:
4.1 Variants with a calculated multifactorial probability of pathogenicity of 0.95 – 0.99.
Caution: This currently only applies to the high-risk genes BRCA1/2 (for an exemplary calculation, see Goldgar et al., 2004 13 ).
4.2 Variants which code for a premature termination of protein biosynthesis (nonsense or frameshift variants) and do not necessitate the loss of known clinically relevant functional protein domains as long as the location of the stop codon is not downstream from the Nonsense-mediated decay-(NMD-)relevant site, 50 base pairs before the end of the penultimate exon.
4.3 Intronic variants in position ± 1,2 or G > non-G in the last position of the exon: if there is a positive splicing prediction (see Appendix A 1) and the first 6 bases in the intron are not GTRRGT and an aberrant in-vitro mRNA analysis is not yet available (i.e. has not [yet] been confirmed by a panel of experts or the pathomechanism of loss of function has been confirmed to be exon skipping or allele-specific transcript expression).
Exceptions:
A cryptic splice site (AG/GT) in the vicinity is activated and the (predicted) new exon is spliced in-frame (→ Class3)
The (predicted) skipped exon (or exons) is alternatively spliced in significant quantities (→ Class3) .
The (predicted) skipped exon (or exons) is spliced in-frame and contains no known functional domain (→ Class3)
4.4 Variants which code for the same amino acid exchange as pathogenic missense variants which have already been categorised as Class5 but are caused by another nucleotide exchange and for which there is no positive splicing prediction (see Appendix A 1).
4.5 In-frame deletions (even for just one amino acid) which lead to the loss of a missense variant already categorised as Class5 and which result in the interruption of known, functionally important domains.
4.6 Extensive in-frame deletions which lead to the interruption/loss of known, functionally important domains.
4.7 In-frame insertions verified by in-vitro mRNA analysis which result in the interruption of functionally important domains.
4.8 Variants which lead to mutations of the translation initiation codon (AUG, methionine) and for which there is no evidence (e.g. an alternative start codon in the immediate vicinity) which would support an alternative classification.
4.9 Variants for which information from functional analyses, clinical data, etc., is available but insufficient for a multifactorial classification and which are categorised as Class4 by a panel of experts (e.g. ENIGMA).
5. Class5 (loss of function/functionally relevant) if one of the following criteria are met:
5.1 Variants which code for a premature termination of protein biosynthesis (nonsense or frameshift variants) which prevents the expression of known, clinically relevant, functional protein domains.
5.2 Variants with a calculated multifactorial probability of pathogenicity of > 0.99.
Caution: This currently only applies to the high-risk genes BRCA1/2 (for an exemplary calculation, see Goldgar et al., 2004 13 ).
5.3 Splice variants for which a frameshift effect was established by in-vitro mRNA analysis, which leads to a premature termination of protein biosynthesis and prevents the expression of known, clinically relevant, functional protein domains and for which a wild-type transcript of the mutated allele has not been confirmed (monoallelic expression).
5.4 Splice variants for which in-vitro mRNA analysis detected an in-frame deletion/insertion which leads to the interruption or loss of a known, clinically relevant domain or functional inactivation through changes of the protein structure and for which a wild-type transcript of the mutated allele has not been verified (monoallelic expression).
5.5 Copy-number deletion variants which result in the interruption or loss of one or more exons with known, clinically relevant functional domains or lead to a reading frameshift, which results, according to the prediction, in the inactivation of known, clinically relevant, functional domains.
5.6 Copy-number duplication variants of any size, confirmed by laboratory analysis, which duplicate one or more exons and lead to a reading frameshift, which results, according to the prediction, in the inactivation of known, clinically relevant, functional domains.
Appendix
A 1. Splicing prediction programmes and their threshold values
The splicing prediction programmes MaxEntScan (MES), Splice Site Finder (SSF), and Human Splicing Finder (HSF) are considered relatively reliable and should therefore be used to evaluate possible effects on the splicing process. MaxEntScan results are considered non-normal for a deviation of delta of ≥ 15% 14 , Human Splicing Finder for a delta of ≥ 4.1% 10 and Splice Site Finder for a deviation of delta of ≥ 5% 14 . An mRNA analysis should be done for evaluation in cases with non-normal prediction (at least two of the three programmes mentioned below). The precondition is that the physiological splice site is recognised by the respective prediction software based on the following threshold values.
The threshold values (calculated for BRCA1/2 14 ) are:
MES > 3
SSF > 60
HSF > 80
An approximation of these threshold values can also be for the other genes. Once specific threshold values have been defined, then the defined threshold values must be used.
A 2.
A 3. Evaluation Criteria Flow Chart
Fig. 1 .
A 4. Schematic Representation of Variants in the Vicinity of Splice Sites
Fig. 2 .
The use of predictive programmes to assess possible splicing effects is obligatory for all new mutations including stop mutations as “rescue” effects can occur through alternative transcripts.
A 5. Characteristics of Individual Genes
The above-mentioned general evaluation criteria should apply to all genes. However, some exceptions, variations and special features are present in specific variants and regions of individual genes, which, for the sake of clarity, are listed below.
A 5.1 BRCA1/2
The following should be categorised as Class3: truncating BRCA1 mutations after the amino acid position 1854 and truncating BRCA2 variants after amino acid position 3308 (they are not categorised as Class1, as structural mutations cannot be excluded). Exception: truncating variants after the polymorphic stop codon p.(Lys3326*) are classified as dispensable/neutral (Class1) 17 and ENIGMA: p.(Lys3326*) is a frequently detected polymorphism which is not associated with a higher risk, OR 1.3 – 1.5 depending on breast or ovarian cancer. This means that variants which lead to a stop downstream from p.(Lys3326*) will also not be associated with an increased risk of developing disease.
The following should be categorised as Class5 in BRCA1/2 : all truncating BRCA1 variants up to the last mutation unequivocally identified as pathogenic at amino acid position 1853 18 and all truncating BRCA2 variants up to amino acid position 3308, c.9924C>G 19 . See ENIGMA BRCA1,2 functional domains, Tables 3 and 4 . Caution: Be aware of the potential impact of NMD; the last 50 bp in the penultimate exon and variants in the last exon usually are usually not subject to NMD. The predictive value of RNA analysis of blood may be limited as it does not involve the target tissue.
Table 3 Excerpt from https://enigmaconsortium.org/wp-content/uploads/2018/10/ENIGMA_Rules_2017-06-29-v2.5.1.pdf .
| Table 3: Catalogue of BRCA1 conserved domains/motifs and currently known, clinically important amino acid residues, and relevance for classification of BRCA1 in-frame and terminal exon sequence variants. | |||||
|---|---|---|---|---|---|
| Domain/Motif | AA start | AA end | AA alterations with demonstrated clinical importance a | Classification of in-frame deletions targeting domain/motifs | References and summary interpretation a |
| RING | 1 | 101 | L22S (c.65T>C [p.Leu22Ser]) T37K (c.110C>A [p.Thr37Lys]) C39R (c.115T>C [p.Cys39Arg]) H41R (c.122A>G [p.His41Arg]) C44S (c.130T>A [p.Cys44Ser]) C44Y (c.131G>A [p.Cys44Tyr]) C61G (c.181T>G [p.Cys61Gly]) |
Class5 if at least one clinically relevant residue is removed. Otherwise Class3. | http://www.ncbi.nlm.nih.gov/protein/15988069 ; http://hci-exlovd.hci.utah.edu ; multifactorial analysis for H41R (c.122A>G [p.His41Arg]) (Whiley et al., 2014). |
| NES | 81 | 99 | None reported | Class3 | Domain location description (Rodriguez and Henderson, 2000). |
| NLS1 | 503 | 508 | None reported | Class3 | Domain location description (Chen et al., 1996, Thakur et al., 1997). |
| NLS2 | 607 | 614 | None reported | Class3 | Domain location description (Chen et al., 1996, Thakur et al., 1997). |
| NLS3 | 651 | 656 | None reported | Class3 | Domain location description (Chen et al., 1996). |
| COILED-COIL | 1391 | 1424 | None reported | Class3 | Domain location description (Hu et al., 2000). |
| BRCT DOMAINS | 1650 | 1863 | T1685A (c.5053A>G [p.Thr1685Ala]) T1685I (c.5054C>T [p.Thr1685Ile]) V1688del (c.5062_5064del [p.Val1688del]) R1699W (c.5095C>T [p.Arg1699Trp]) G1706E (c.5117G>A [p.Gly1706Glu]) A1708E (c.5123C>A [p.Ala1708Glu]) S1715R (c.5143A>C [p.Ser1715Arg]) G1738R (c.5212G>A [p.Gly1738Arg]) L1764P (c.5291T>C [p.Leu1764Pro]) I1766S (c.5297T>G [p.Ile1766Ser]) M1775K (c.5324T>A [p.Met1775Lys]) M1775R (c.5324T>G [p.Met1775Arg]) C1787S (c.5359T>A [p.Cys1787Ser]) G1788V (c.5363G>T [p.Gly1788Val]) V1838E (c.5513T>A [p.Val1838Glu]) |
Class5 if at least one clinically relevant residue is removed. Otherwise Class3. |
Domain boundaries derived from X-ray crystallography data are aa1646-1863 (1T15,
http://www.ncbi.nlm.nih.gov/Structure/mmdb/mmdbsrv.cgi?uid=27907
), and ENIGMA functional assay data (Monteiro, unpublished).
Digestion data indicate aa1860-1863 are dispensable based on susceptibility to digestion (Lee et al., 2010), while pathogenic variant data indicate that 1855-1862 are dispensable (Hayes et al., 2000). Position 1854 is implicated as clinically important by the observation that Y1853X (c.5559C>G [p.Tyr1853Ter]) is a recognised high-risk pathogenic variant. These combined data indicate that position 1854 or 1855 is the C-terminal border of the BRCT/BRCA1 relevant for the clinical interpretation of sequence variants in exon 24 of BRCA1. That is, a variant predicted to disrupt expression of protein sequence only downstream* of position 1855 would not be considered clinically important. |
|
a
Missense substitutions in specific functional domains that are designated as Class5 pathogenic based on multifactorial likelihood of the posterior probability of pathogenicity > 0.99 (listed in
http://hci-exlovd.hci.utah.edu
or individual references), and which have no/little effect on the mRNA transcript profile,
unless
the variant results in an aberrant transcript that encodes a discrete in-frame deletion considered informative for the definition of clinically important domains.
* Typo was corrected in version 2.5.1. Note: The following pathogenic exonic variants known to alter mRNA splicing have been excluded from Table 3 above, as justified below: | |||||
| Variant | mRNA Change | Predicted protein change | Reason for exclusion | ||
| BRCA1 R1495M (c.4484G>T [p.Arg1495Met]) | r.[4358_4484del, 4358_4675del] | p.(Ala1453Glyfs Ter10) – predominant transcript | Predominant alternate transcript is out of frame. Loss of function is assumed due to loss of full-length transcript from variant allele (Houdayer et al., 2012, Colombo et al., 2013, Santos et al., 2014). | ||
| BRCA1 E1559K (c.4675G>A [p.Glu1559Lys]) | r.[4665_4675del] | p.(Gln1366Alafs Ter13) | Alternate transcript is out-of-frame. Level of full-length transcript not assessed (Wappenschmidt et al., 2012). | ||
| BRCA1 A1623G (c.4868C>G [p.Ala1623Gly]) | r.[4868_4986del] | p.(Ala1623Aspfs Ter16) | Alternate transcript is out of frame. Variant allele produces some full-length transcripts (Walker et al., 2010). | ||
| BRCA1 D1692N (c.5074G>A [p.Asp1692Asn]) | r.[4987_5074del, 5074_5075ins5074+1_5074+153] | p.(Val1665Serfs Ter8) – predominant transcript | Predominant alternate transcript, based on minigene assay (Ahlborn et al., 2015), is out of frame. | ||
Table 4 Excerpt from https://enigmaconsortium.org/wp-content/uploads/2018/10/ENIGMA_Rules_2017-06-29-v2.5.1.pdf .
| Table 4: Catalogue of BRCA2 conserved domains/motifs and currently known clinically important amino acid residues, and relevance for classification of BRCA2 in-frame and terminal exon sequence variants. | |||||
|---|---|---|---|---|---|
| Domain/Motif | AA start | AA end | AA alterations with demonstrated clinical importance a | Classification of in-frame deletions targeting domain/motifs | References and summary interpretation a |
| PALB2 Binding | 10 | 40 | None reported | Class3 | Domain location description (Oliver et al., 2009, Xia et al., 2006) |
| BRC-1 | 1002 | 1036 | None reported | Class3 | http://www.ncbi.nlm.nih.gov/protein/NP_000050.2 |
| BRC-2 | 1212 | 1246 | None reported | Class3 | http://www.ncbi.nlm.nih.gov/protein/NP_000050.2 |
| BRC-3 | 1422 | 1453 | None reported | Class3 | http://www.ncbi.nlm.nih.gov/protein/NP_000050.2 |
| BRC-4 | 1518 | 1549 | None reported | Class3 | http://www.ncbi.nlm.nih.gov/protein/NP_000050.2 |
| BRC-5 | 1665 | 1696 | None reported | Class3 | http://www.ncbi.nlm.nih.gov/protein/NP_000050.2 |
| BRC-6 | 1837 | 1871 | None reported | Class3 | http://www.ncbi.nlm.nih.gov/protein/NP_000050.2 |
| BRC-7 | 1971 | 2005 | None reported | Class3 | http://www.ncbi.nlm.nih.gov/protein/NP_000050.2 |
| BRC-8 | 2051 | 2085 | None reported | Class3 | http://www.ncbi.nlm.nih.gov/protein/NP_000050.2 |
| DBD (DNA/DSS1 binding domain – helical, OB1, OB2, OB3) | 2481 | 3186 | W2626C (c.7878G>C [p.Trp2626Cys]) I2627F (c.7879A>T [p.Ile2627Phe]) E2663V (c.7988A>T [p.Glu2663Val]) T2722R (c.8165C>G [p.Thr2722Arg]) D2723G (c.8168A>G [p.Asp2723Gly]) D2723H (c.8167G>C [p.Asp2723His]) G2748D (c.8243G>A [p.Gly2748Asp]) I2778_Q2829del (c.8332_8487del [p.Ile2778_Gln2829del]) R3052W (c.9154C>T [p.Arg3052Trp]) |
Class5 if at least one clinically relevant residue (or all of AA2778-2829) is removed. Otherwise Class3. |
http://www.ncbi.nlm.nih.gov/protein/NP_000050.2
;
http://hci-exlovd.hci.utah.edu
.
Pathogenic variant c.8486G>A (also recorded as Gln2829Arg) results in a transcript encoding an in-frame exon 19 deletion only (Houdayer et al., 2012), indicating that genetic variation encompassing loss of this entire exon (AA2778-2829) should be considered clinically important. The clinical impact of alteration/deletion of individual amino acids in exon 19 is not yet established. |
| NLS1 | 3263 | 3269 | None reported | Class3 | Domain local description (Guidugli et al., 2014) |
| BRC-9 or TR2 | 3265 | 3330 | None reported | Class3 | Note: although amino acids 3270-3305 within this fragment are reported to bind RAD51-DNA filaments (Davies and Pellegrini, 2007), there is no sequence conservation with the BRC repeats located between aa1002 and aa2014. Domain boundaries are derived from x-ray crystallography data are aa3265-3330 (Esashi et al., 2005, Esashi et al., 2007). Case-control and frequency data indicate that BRCA2 c.9976A>T (p.Lys3326Ter) does not confer a high risk of cancer (OR 1.3–1.5, dependent on breast or ovarian cancer subtype (Meeks et al., 2016), demonstrating that residues at and downstream of 3327 are likely dispensable. Position 3308 is implicated as clinically important by the observation that a nonsense variant c.9924C>G (p.Tyr3308Ter) is recognized as a high-risk pathogenic variant with known functional relevance ([Vallee et al., 2016]; Bayes score 1122 : 1 from a single large kConFab family, Spurdle unpublished data). There is currently no publicly available clinical information to support pathogenicity of nonsense or frameshift variants located between positions 3309 and 3325. These data combined suggest that the C-terminal border of the BRC-9 relevant to the clinical interpretation of sequence variants in exon 27 of BRCA2 lies between 3309 and 3325. That is, a variant predicted to disrupt expression only of protein sequence downstream of position 3325 would be considered unlikely to be clinically important. Further functional and clinical studies are underway to refine risk, if any, for predicted nonsense or frameshift variants downstream of position 3326. |
| NLS2 | 3381 | 3385 | No | Class3 | Domain location description (Guidugli et al., 2014). This domain is considered unlikely clinically relevant since it lies downstream of position 3326. |
|
a
Missense substitutions in denoted functional domains that are designated as Class5 pathogenic based on multifactorial likelihood posterior probability of pathogenicity > 0.99, and for which there is no/little effect on mRNA transcript profile –
unless
the variant results in an aberrant transcript that encodes a discrete in-frame deletion considered informative to definition of clinically important domains. (Splicing aberrations are reported for
BRCA2
c.7988A>T [p.Glu2663Val] and c.8168A>G [p.Asp2723Gly] (Walker et al., 2010), but these did not lead to complete loss of function of the full length transcript), and missense alterations showed abrogation of functional activity using multiple assays (Walker et al., 2010). An additional conserved region not commonly recognized as a BRCA2 domain/motif is located AA 1110-1183, but no pathogenic missense substitutions have been recorded for this region.
Note – The following pathogenic exonic variants known to alter mRNA splicing have been excluded from Table 4 above, as justified below: | |||||
| Variant | mRNA Change | Predicted protein change | Reason for exclusion | ||
| BRCA2 R2659K (c.7976G>A [p.Arg2659Lys]) | r.[7806_7976del] | p.(Ala2603_ Arg2659del) | Alternate transcript is in-frame but level of full length transcript not assessed (Farrugia et al., 2008) | ||
| BRCA2 R2659T (c.7976G>C [p.Arg2659Thr]) | r.[7806_7976del] | p.(Ala2603_ Arg2659del) | Alternate transcript is in-frame but level of full length transcript not assessed (Farrugia et al., 2008) | ||
| BRCA2 P3039P (c.9117G>A [p.Pro3039Pro]) | r.[8954_9117del] | p.(Val2985 Glyfs*4) | Allele-specific assay shows out-of-frame transcript (Houdayer et al., 2012) | ||
Other special features are listed in the Appendix of the evaluation guidelines of the ENIGMA Consortium, which can be accessed via the following link: https://enigmaconsortium.org/wp-content/uploads/2018/10/ENIGMA_Rules_2017-06-29-v2.5.1.pdf , Tables 3, 4 and 6 ( Tables 3 to 5 ).
A 5.2 ATM
The evaluation criteria for ATM are based on a combination of the following criteria:
The 5-class IARC system for the assessment of the pathogenicity of BRCA1 and BRCA2 variants.
The 3-class system to evaluate the pathogenicity of ATM variants 20 which includes in silico analyses such as Align-GVGD.
The ACMG guidelines on the classification of variants 3 , 21 .
Class1:
If the allele frequency is ≥ 1% (MAF ≥ 0.01) in large population groups (e.g. Caucasians, Africans, or Asians) or there is evidence of homozygous variant carriers in control populations. If this is the case, then the variant is always categorised as Class1. An allele frequency of ≥ 1% in subpopulations with a low-diversity gene pool (examples: Finnish population, founder mutations!) is not sufficient.
Class2:
If the allele frequency is ≥ 0.5–< 1% (MAF ≥ 0.005 – 0.099) in large population groups (e.g. Caucasians, Africans, or Asians), the variant is always categorised as Class2.
Missense variants which, according to in silico analysis (Align-GVGD, SIFT), are very probably neutral and/or outside the functionally critical domain (FATKIN).
Class3:
All variants which cannot be categorised as Class1, 2, 4 or 5.
Class4:
Variants with an in-frame deletion which are within the functionally critical domain (FATKIN).
Missense variants which are within the functionally critical domain (FATKIN) and are, according to in silico analysis (Align-GVGD, SIFT), very probably harmful and described as functionally inactive.
Class5:
Truncating ATM variants up to the FATKIN domain.
Missense variants, in-frame deletion or splice mutations which reduce ATM protein expression to < 20% for the mutated allele 28 , 29 .
Variants associated with classic AT.
Splice variants: see BRCA1 and BRCA2 .
Functional domains: FATKIN with FAT; PI3K-related kinase; FATC
Table 6 .
Table 6 ATM , functional domains and relevance for the interpretation of the clinical importance of sequence variants – catalogue of clinically relevant functional domains and amino acids.
| Region | AA start | AA end | AA alterations with demonstrated clinical importance (AT) characterising known functional domains | References and summary interpretation |
|---|---|---|---|---|
| Substrate binding | 91 | 97 | None reported |
Domain location description
30
Also contains p53- and BRCA1-binding domain |
| NLS | 385 | 388 | None reported | Domain location description 28 , 31 |
| Leucine zipper | 1218 | 1238 | None reported | Domain location description 25 , 28 |
| Proline rich | 1373 | 1382 | None reported | Domain location description 25 , 28 |
| FATKIN | 1893 | 3056 | Yes, e.g. p.(Val2424Gly) p.(2546_2548del), in frame p.(Asp2625Glu) p.(Ala2626Pro) p.(Val2716Ala) p.(Ser2855_Val2856delinsArgIle) |
AA alterations and in-frame deletions
26
,
28
,
29
,
32
,
33
,
34
,
35
Domain location description 25 , 27 , 28 , 36 , 37 Domains: FAT: 1893-2612 KIN: 2612-3056 with ATP-binding: 2716-2730, substrate (nibrin and p53) binding: 2682-3012, FATC with TIP60 binding: 3034-3056 Domain location description |
Additional literature: 22 , 23 , 24 , 25 , 26 , 28 , 33 , 35 , 38 .
A 5.3 PALB2
The p.(Leu939Trp) mutation should be categorised as Class2 39 .
Additional literature: 35 , 38 , 40 , 41 , 42 , 43 , 44 , 45 , 46 .
Table 7 .
Table 7 PALB2 , functional domains and relevance for the interpretation of the clinical importance of sequence variants – catalogue of clinically relevant functional domains and amino acids.
| Region | AA start | AA end | AA alterations with potential clinical importance | References and summary interpretation |
|---|---|---|---|---|
| BRCA1 interaction domain | 9 | 43 | Yes, e.g. p.(Leu35Pro) |
Also covers oligomerisation domain/covers coiled-coiled motif Domain location description 47 , 48 , 49 ; Amino acid alteration in VUS and functional analysis 50 . |
| DNA-binding site | 1 | 200 | None reported | Domain location description 51 |
| RAD51 binding site | 101 | 184 | None reported | Domain location description 51 , 52 |
| DNA-binding site | 372 | 561 | None reported | Covers also chromatin association motif (ChAM, 395-446) Domain location description 51 , 53 |
| MRG15 (MORF4L1) interaction domain | 611 | 764 | None reported | Domain location description 48 |
| WD40 repeat | 853 | 1186 | Yes, e.g. p.(Thr1030Ile) p.(Leu1143Pro) |
BRCA2 (1019-1098), RAD51C, XRCC3 and/or RAD51 complex formation Domain location description 51 , 52 , 54 , 55 , 56 , 57 . Amino acid alterations 48 , 54 , 58 |
A 5.4 CHEK2
In exons 11 – 15, highly homologous, functionally inactive sequences (pseudogenes) on various other chromosomes (2, 7, 10, 13, 15, 16, X, and Y) 59 , 60 which can superimpose the relevant sequences > long-range PCR of exons 11 – 15 and bioinformatic filtering of pseudogene reads, where possible.
Functional domains: SQ/TQ-rich domain*, forkhead-associated (FHA)** domain, kinase domain***, nuclear localisation signal (NLS) 61 , 62 , 63 .
To date, only truncating variants in the SQ/TQ-rich domain have been classified as pathogenic ( Table 8 ).
Table 8 CHEK2 , functional domains and relevance for the interpretation of the clinical importance of sequence variants – catalogue of clinically relevant functional domains and amino acids.
| Region | AA start | AA end | AA alterations with Potential Clinical Importance | References and summary interpretation |
|---|---|---|---|---|
|
* Q/TQ consensus sites are sites phosphorylated by ATM/ATR
116
. e.g. phosphorylation of Thr-68 is important for CHEK2 activation and oligomerisation.
** The phosphorylated Thr-68 site of CHEK2 interacts with the FHA domain of another CHEK2 molecule and thus leads to the formation of CHEK2 oligomers 66 . *** 67 § The CHEK2 c.470T>C p.Ile157Thr variant, although still present with various classifications (VUS/likely pathogenic/pathogenic) in ClinVar and other databases has been reclassified (date: 28.08.2018) as Class2/likely benign by the German Consortium on HBOC on the following basis: It is frequently listed in large unaffected control cohorts (0.5%, gnomAD V. 2.1.1, non-cancer). The population frequency in Finnish Europeans is 2.5% (10 homozygous carriers). In addition, it is present in 47/7325 individuals (0.64%) in the FLOSSIES database (non-cancer female controls of European descent aged > 70 years). Although showing functionally impaired dimerisation and autophosphorylation 61 , 62 , 68 , numerous large case-control studies show results indicating low or no increased breast cancer risk 64 , 69 : breast cancer OR = 1.58 (1.42 – 1.75), colon cancer OR = 1.67 (1.24 – 2.26) 70 , 71 . Additionally, it has been observed with a frequency of 2% in controls 2 . However, it may act as a polygenic risk allele. | ||||
| SQ/TQ-rich | 19 | 69 | e.g. c.85C>T,p.Gln29* | 62 , 65 |
| FHA | 92 [115] | 205 [175] | p.Arg117Gly, p.Arg145Trp, p.Gly167Arg | 61 , 62 , 63 |
| Kinase | 212 | 501 | c.1040A>C, p.Asp347Ala #; c.1100del; c.1164dup; p.Thr476Met, c.1169A>C, p.Tyr390Ser; c.1183G>T, p.Val395Phe#; c.1283C>T, p.Ser428Phe; c.1427C>T, p.Thr476Met | 61 , 62 , #ClinVar |
| NLS | 515 | 538 | e.g. c.1547delC, p.Ser516Leufs#; c.1555C>T, p.Arg519Ter#; | 63 , #ClinVar |
Numerous known missense variants in the FHA domain. Caution: Consult FLOSSIES database during evaluation! (e.g. c.470C>T; p.Ile157Thr: Class2 [see § footnote Table 8 ) or with unclear clinical relevance (e.g. c.434G>A, p.Arg145Gln; c.422A>C, p.Lys141Thr).
Missense variants in the kinase domain with unclear clinical relevance: e.g. c.1216C>T, p.Arg406Cys.
Similarly, in the NLS domain, only truncating variants have been classified as pathogenic to date ( Table 8 ).
The investigation by Ow et al. 63 gives an overview of the identified mutations in the CHEK2 gene region. The publication by Roeb et al. (2012) includes a schematic overview of the functional domains as well as the results of functional analyses of the missense mutations located in these different CHEK2 domains 62 .
A 5.5 TP53
IARC TP53 database; the functional analyses by Kato et al. (2003) and Monti et al. (2007, 2011) are reliable 72 , 73 , 74 , 75 , 76 , 77 .
Functional domains: oligomerisation domain, core domain (DNA-binding).
Possible dominant negative effect of missense variants and stop variants which affect the oligomerisation domain.
Mosaic mutations and clonal haematopoiesis are possible, therefore watch out for the variant allele fraction when carrying out NGS analysis; if necessary, carry out further analysis to confirm germline mutations.
Table 9 .
Table 9 p53 , functional domains and relevance for the interpretation of the clinical importance of sequence variants – catalogue of clinically relevant functional domains and amino acids.
| Region | AA start | AA end | AA alterations with demonstrated clinical importance (including conflicting interpretations of pathogenicity but criteria provided) | References and summary interpretation |
|---|---|---|---|---|
| Transcription activation | 1 | 55 | p.(Val10Ile) p.(Val31Ile) p.(Pro47Ser) |
Amino acid alterations (ClinVar) Domain location description 78 Also binding site for numerous proteins including HDM2 (amino acids 15-29; IARC) |
| Proline-rich domain | 61 | 94 | p.(Pro82Leu) p.(Ala83Val) |
Amino acid alterations (ClinVar) Domain location description 78 |
| DNA-binding region | 102 | 292 | p.(Gly105Asp) p.(Lys120Glu) p.(Thr125Met) p.(Ser127Phe) p.(Asn131Tyr) p.(Cys141Tyr) p.(Pro151Ser) p.(Pro151Thr) p.(Pro152Leu) p.(Arg156His) p.(Arg158Cys) p.(Arg158His) p.(Tyr163Asp) p.(Tyr163Cys) p.(Arg175Leu) p.(Arg175His) p.(Cys176Tyr) p.(His179Tyr) p.(Arg181Cys) p.(Arg181His) p.(Ala189Val) p.(His193Arg) p.(His193Leu) p.(Leu194Phe) p.(Ile195Thr) p.(Arg213Gln) p.(Val.216Met) p.(Tyr220Cys) p.(Tyr220Ser) p.(Ile232Thr) p.(Tyr234Cys) p.(Asn235Ser) p.(Tyr236Asp) p.(Met237Val) p.(Met237Ile) p.(Cys238Tyr) p.(Ser241Phe) p.(Cys242Tyr) p.(Gly245Asp) p.(Gly245Ser) p.(Gly245Cys) p.(Met246Val) p.(Met246Leu) p.(Met246Arg) p.(Arg248Gln) p.(Arg248Trp) p.(Ile251Leu) p.(Ile251Ser) p.(Thr256Ala) p.(Leu257Arg) p.(Glu258Lys) p.(Arg267Trp) p.(Arg267Gln) p.(Val272Leu) p.(Arg273Hisv p.(Arg273Cys) p.(Cys275Tyr) p.(Cys277Tyr) p.(Arg280Thr) p.(Asp281Val) p.(Asp281Gly) p.(Arg282Gly) p.(Arg282Leu) p.(Arg282Trp) p.(Arg283His) p.(Arg283Lys) p.(Glu286Lys) |
Amino acid alterations (ClinVar) Domain location description (IARC) Also binding site for numerous proteins including 53BP1 (IARC) and RAD51 amino acids 94-160 and 264-315 79 |
| Oligomerisation region | 325 | 356 | p.(Gly325Val) p.(Arg337Leu) p.(Arg337Cys) p.(Glu339Lys) p.(Arg342Pro) |
Amino acid alterations (ClinVar) Domain location description (IARC) Also binding site for numerous proteins Covering main nuclear localisation signal (amino acids 316-322) 80 , 81 |
| Basic (repression of DNA-binding region) | 369 | 388 | None reported | Domain location description (IARC) Also binding site for numerous proteins including RAD54 82 |
A 5.6 RAD51D
Functional domains: N-terminal domains and ATP-binding domain with the highly conserved Walker A and B motifs 83 , 84 , 85 .
Table 10 .
Table 10 RAD51D , functional domains and relevance for the interpretation of the clinical importance of sequence variants – catalogue of clinically relevant functional domains and amino acids.
| Region | AA start | AA end | AA alterations with demonstrated clinical importance (including conflicting interpretations of pathogenicity) | References and summary interpretation |
|---|---|---|---|---|
| N-terminal region | 1 | 83 | None reported | N-terminal domain required for ssDNA-specific binding function 86 |
| Linker | 60 | 78 | None reported | Proper interaction with RAD51C and XRCC2 85 |
| ATPase domain and RAD51B, RAD51C, and XRCC2 binding | 99 | 274 |
p.(G112A) (disrupts binding of RAD51D to RAD51C
87
)
p.(S207L) (disrupts RAD51D-XRCC2 interaction 85 ) p.(A210V) (predicted to be potentially pathogenic 88 , 89 ) p.(R266C) 90 , Meindl et al. (unpublished) |
ATPase, AAA+ type Walker A and B motifs crucial for HR. These motifs are also implicated in binding to RAD51C and XRCC2. |
A 5.7 RAD51C
Functional domains: DNA repair/recombination protein RecA-like, ATP-binding domain
Table 11 .
Table 11 RAD51C , functional domains and relevance for the interpretation of the clinical importance of sequence variants – catalogue of clinically relevant functional domains and amino acids.
| Region | AA start | AA end | AA alterations with demonstrated clinical importance (including conflicting interpretations of pathogenicity) | References and summary interpretation |
|---|---|---|---|---|
| N-terminal region | 1 | 66 | None reported | Homology-derived putative DNA-binding domain 91 |
| ATPase domain and RAD51B, XRCC3, and RAD51D binding | 79 | 376 | p.(Gly125Val) p.(Cys135Tyr) p.(Leu138Phe) p.(Gly153Asp) p.(Asp159Asn) p.(Val169Ala) p.(Leu219Ser) p.(Arg258His) p.(Gly264Ser) |
Amino acid alterations and functional consequences 95 , 96 , 97 , 98 Domain location description 91 ATPase domain includes Walker A nucleotide binding motif (amino acids 125-132) and Walker B nucleotide binding motif (amino acids 238-242) 91 , 99 |
| Nuclear localisation signal | 366 | 370 | None reported | Domain location description 99 |
A 5.8 BRIP1
Table 12 .
Table 12 BRIP1 , functional domains and relevance for the interpretation of the clinical importance of sequence variants – catalogue of clinically relevant functional domains and amino acids.
| Region | AA start | AA end | AA alterations with demonstrated clinical importance (including conflicting interpretations of pathogenicity) | References and summary interpretation |
|---|---|---|---|---|
| DEAD/DEAH box helicase domain | 17 | 441 | Domain location description 102 | |
| Helicase superfamily c-terminal domain | 697 | 851 | Domain location description 102 | |
| The BRCA1 interacting region of BRIP1 | 976 | 1006 | Phosphorylation of FANCJ at Ser-990 is important for its interaction with BRCA1 | 103 |
| MLH1 interaction | Lysines 141 and 142 are required for direct interaction of FANCJ with MLH1 | 104 | ||
| Nuclear localisation signal | 158 | 175 | None reported | Domain location description 102 |
Additional literature: 1 , 102 , 105 , 106 , 107 , 108 , 109 , 110
A 5.9 CDH1
References:
Focuses predominantly on molecular genetics: 111
Review of functional analyses: 112
Review of the HDGC Consortium: 113
Review of lobular breast cancer: 114
Distribution of pathogenic variants at the CDH1 locus 111
Known pathogenic CDH1 variants are distributed across the entire locus; it is therefore not possible to define a clinically relevant functional protein domain. The last known truncating pathogenic variant in the last exon is c.2506G>T (p.Glu836*) 115 . All truncating variants upstream must therefore be categorised at least as Class4.
The proposal put forwards by the ClinGen Consortium to categorise variants with a MAF > 0.2% as ACMG Class1, contrary to IARC guidelines, is currently being debated.
CDH1 Rule Specifications for the ACMG/AMP Variant Curation Guidelines ClinGen ( https://www.clinicalgenome.org/site/assets/files/8816/clingen_cdh1_acmg_specifications_v1.pdf ).
Conflict of Interest/Interessenkonflikt The authors declare that they have no conflict of interest./Die Autorinnen/Autoren geben an, dass kein Interessenkonflikt besteht.
International Agency for Research on Cancer.
High-risk genes: at least one sequence variant with an odds ratio for breast and/or ovarian cancer of > 5 (e.g. BRCA1, BRCA2, RAD51C, PALB2, TP53, ATM), see 1 , 2 .
Evidence-based network for the interpretation of germline mutant alleles: http://enigmaconsortium.org/
American College of Medical Genetics and Genomics (ACMG 3 ).
Association for Clinical Genomic Science (ACGS, http://www.acgs.uk.com/ ).
International Agency for Research on Cancer.
Hochrisikogene: mind. eine Sequenzvariante mit Odds Ratio für Brust- und/oder Eierstockkrebs OR > 5 (z.B. BRCA1, BRCA2, RAD51C, PALB2, TP53, ATM), siehe auch 1 , 2 .
Evidence based Network for the Interpretation of Germline Mutant Allels, http://enigmaconsortium.org/
American College of Medical Genetics and Genomics (ACMG 3 ).
Association for Clinical Genomic Science (ACGS, http://www.acgs.uk.com/ ).
References/Literatur
- 1.Couch F J, Shimelis H, Hu C. Associations Between Cancer Predisposition Testing Panel Genes and Breast Cancer. doi:10.1001/jamaoncol.2017.0424. JAMA Oncol. 2017;3:1190–1196. doi: 10.1001/jamaoncol.2017.0424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hauke J, Horvath J, Gross E. Gene panel testing of 5589 BRCA1/2-negative index patients with breast cancer in a routine diagnostic setting: results of the German Consortium for Hereditary Breast and Ovarian Cancer. doi:10.1002/cam4.1376. Cancer Med. 2018;7:1349–1358. doi: 10.1002/cam4.1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Richards S, Aziz N, Bale S. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. doi:10.1038/gim.2015.30. Genet Med. 2015;17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Plon S E, Eccles D M, Easton D. Sequence variant classification and reporting: recommendations for improving the interpretation of cancer susceptibility genetic test results. doi:10.1002/humu.20880. Hum Mutat. 2008;29:1282–1291. doi: 10.1002/humu.20880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moghadasi S, Meeks H D, Vreeswijk M P. The BRCA1 c. 5096G>A p.Arg1699Gln (R1699Q) intermediate risk variant: breast and ovarian cancer risk estimation and recommendations for clinical management from the ENIGMA consortium. doi:10.1136/jmedgenet-2017-104560. J Med Genet. 2018;55:15–20. doi: 10.1136/jmedgenet-2017-104560. [DOI] [PubMed] [Google Scholar]
- 6.Shimelis H, Mesman R LS, Von Nicolai C. BRCA2 Hypomorphic Missense Variants Confer Moderate Risks of Breast Cancer. doi:10.1158/0008-5472.can-16-2568. Cancer Res. 2017;77:2789–2799. doi: 10.1158/0008-5472.CAN-16-2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Walker L C, Whiley P J, Houdayer C. Evaluation of a 5-tier scheme proposed for classification of sequence variants using bioinformatic and splicing assay data: inter-reviewer variability and promotion of minimum reporting guidelines. doi:10.1002/humu.22388. Hum Mutat. 2013;34:1424–1431. doi: 10.1002/humu.22388. [DOI] [PubMed] [Google Scholar]
- 8.Whiley P J, de la Hoya M, Thomassen M. Comparison of mRNA splicing assay protocols across multiple laboratories: recommendations for best practice in standardized clinical testing. doi:10.1373/clinchem.2013.210658. Clin Chem. 2014;60:341–352. doi: 10.1373/clinchem.2013.210658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fackenthal J D, Yoshimatsu T, Zhang B. Naturally occurring BRCA2 alternative mRNA splicing events in clinically relevant samples. doi:10.1136/jmedgenet-2015-103570. J Med Genet. 2016;53:548–558. doi: 10.1136/jmedgenet-2015-103570. [DOI] [PubMed] [Google Scholar]
- 10.Colombo M, Blok M J, Whiley P. Comprehensive annotation of splice junctions supports pervasive alternative splicing at the BRCA1 locus: a report from the ENIGMA consortium. doi:10.1093/hmg/ddu075. Hum Mol Genet. 2014;23:3666–3680. doi: 10.1093/hmg/ddu075. [DOI] [PubMed] [Google Scholar]
- 11.de la Hoya M, Soukarieh O, Lopez-Perolio I. Combined genetic and splicing analysis of BRCA1 c.[594-2A>C; 641A>G] highlights the relevance of naturally occurring in-frame transcripts for developing disease gene variant classification algorithms. doi:10.1093/hmg/ddw094. Hum Mol Genet. 2016;25:2256–2268. doi: 10.1093/hmg/ddw094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li L, Biswas K, Habib L A. Functional redundancy of exon 12 of BRCA2 revealed by a comprehensive analysis of the c.6853A>G (p. I2285 V) variant. doi:10.1002/humu.21101. Hum Mutat. 2009;30:1543–1550. doi: 10.1002/humu.21101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goldgar D E, Easton D F, Deffenbaugh A M. Integrated evaluation of DNA sequence variants of unknown clinical significance: application to BRCA1 and BRCA2. doi:10.1086/424388. Am J Hum Genet. 2004;75:535–544. doi: 10.1086/424388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Houdayer C, Caux-Moncoutier V, Krieger S. Guidelines for splicing analysis in molecular diagnosis derived from a set of 327 combined in silico/in vitro studies on BRCA1 and BRCA2 variants. doi:10.1002/humu.22101. Hum Mutat. 2012;33:1228–1238. doi: 10.1002/humu.22101. [DOI] [PubMed] [Google Scholar]
- 15.Findlay G M, Boyle E A, Hause R J. Saturation editing of genomic regions by multiplex homology-directed repair. doi:10.1038/nature13695. Nature. 2014;513:120–123. doi: 10.1038/nature13695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Starita L M, Young D L, Islam M. Massively Parallel Functional Analysis of BRCA1 RING Domain Variants. doi:10.1534/genetics.115.175802. Genetics. 2015;200:413–422. doi: 10.1534/genetics.115.175802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meeks H D, Song H, Michailidou K. BRCA2 Polymorphic Stop Codon K3326X and the Risk of Breast, Prostate, and Ovarian Cancers. doi:10.1093/jnci/djv315. J Natl Cancer Inst. 2016;108:pii:djv315. doi: 10.1093/jnci/djv315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hayes F, Cayanan C, Barilla D. Functional assay for BRCA1: mutagenesis of the COOH-terminal region reveals critical residues for transcription activation. Cancer Res. 2000;60:2411–2418. [PMC free article] [PubMed] [Google Scholar]
- 19.Kuznetsov S G, Liu P, Sharan S K. Mouse embryonic stem cell-based functional assay to evaluate mutations in BRCA2. doi:10.1038/nm.1719. Nat Med. 2008;14:875–881. doi: 10.1038/nm.1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goldgar D E, Healey S, Dowty J G. Rare variants in the ATM gene and risk of breast cancer. doi:10.1186/bcr2919. Breast Cancer Res. 2011;13:R73. doi: 10.1186/bcr2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maxwell K N, Hart S N, Vijai J. Evaluation of ACMG-Guideline-Based Variant Classification of Cancer Susceptibility and Non-Cancer-Associated Genes in Families Affected by Breast Cancer. doi:10.1016/j.ajhg.2016.02.024. Am J Hum Genet. 2016;98:801–817. doi: 10.1016/j.ajhg.2016.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Teraoka S N, Malone K E, Doody D R. Increased frequency of ATM mutations in breast carcinoma patients with early onset disease and positive family history. Cancer. 2001;92:479–487. doi: 10.1002/1097-0142(20010801)92:3<479::aid-cncr1346>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 23.Fernet M, Moullan N, Lauge A. Cellular responses to ionising radiation of AT heterozygotes: differences between missense and truncating mutation carriers. doi:10.1038/sj.bjc.6601549. Br J Cancer. 2004;90:866–873. doi: 10.1038/sj.bjc.6601549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dörk T, Bendix-Waltes R, Wegner R D. Slow progression of ataxia-telangiectasia with double missense and in frame splice mutations. doi:10.1002/ajmg.a.20601. Am J Med Genet A. 2004;126A:272–277. doi: 10.1002/ajmg.a.20601. [DOI] [PubMed] [Google Scholar]
- 25.Lavin M F, Scott S, Gueven N. Functional consequences of sequence alterations in the ATM gene. doi:10.1016/j.dnarep.2004.03.011. DNA Repair (Amst) 2004;3:1197–1205. doi: 10.1016/j.dnarep.2004.03.011. [DOI] [PubMed] [Google Scholar]
- 26.Renwick A, Thompson D, Seal S. ATM mutations that cause ataxia-telangiectasia are breast cancer susceptibility alleles. doi:10.1038/ng1837. Nat Genet. 2006;38:873–875. doi: 10.1038/ng1837. [DOI] [PubMed] [Google Scholar]
- 27.Tavtigian S V, Oefner P J, Babikyan D. Rare, evolutionarily unlikely missense substitutions in ATM confer increased risk of breast cancer. doi:10.1016/j.ajhg.2009.08.018. Am J Hum Genet. 2009;85:427–446. doi: 10.1016/j.ajhg.2009.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Keimling M, Volcic M, Csernok A. Functional characterization connects individual patient mutations in ataxia telangiectasia mutated (ATM) with dysfunction of specific DNA double-strand break-repair signaling pathways. doi:10.1096/fj.11-185546. FASEB J. 2011;25:3849–3860. doi: 10.1096/fj.11-185546. [DOI] [PubMed] [Google Scholar]
- 29.Gilad S, Chessa L, Khosravi R. Genotype-phenotype relationships in ataxia-telangiectasia and variants. doi:10.1086/301755. Am J Hum Genet. 1998;62:551–561. doi: 10.1086/301755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fernandes N, Sun Y, Chen S. DNA damage-induced association of ATM with its target proteins requires a protein interaction domain in the N terminus of ATM. doi:10.1074/jbc.M412065200. J Biol Chem. 2005;280:15158–15164. doi: 10.1074/jbc.M412065200. [DOI] [PubMed] [Google Scholar]
- 31.Young D B, Jonnalagadda J, Gatei M. Identification of domains of ataxia-telangiectasia mutated required for nuclear localization and chromatin association. doi:10.1074/jbc.M411689200. J Biol Chem. 2005;280:27587–27594. doi: 10.1074/jbc.M411689200. [DOI] [PubMed] [Google Scholar]
- 32.Mitui M, Nahas S A, Du L T. Functional and computational assessment of missense variants in the ataxia-telangiectasia mutated (ATM) gene: mutations with increased cancer risk. doi:10.1002/humu.20805. Hum Mutat. 2009;30:12–21. doi: 10.1002/humu.20805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tavtigian S V, Oefner P J, Babikyan D. Rare, evolutionarily unlikely missense substitutions in ATM confer increased risk of breast cancer. doi:10.1016/j.ajhg.2009.08.018. Am J Hum Genet. 2009;85:427–446. doi: 10.1016/j.ajhg.2009.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barone G, Groom A, Reiman A. Modeling ATM mutant proteins from missense changes confirms retained kinase activity. doi:10.1002/humu.21034. Hum Mutat. 2009;30:1222–1230. doi: 10.1002/humu.21034. [DOI] [PubMed] [Google Scholar]
- 35.Southey M C, Goldgar D E, Winqvist R. PALB2, CHEK2 and ATM rare variants and cancer risk: data from COGS. doi:10.1136/jmedgenet-2016-103839. J Med Genet. 2016;53:800–811. doi: 10.1136/jmedgenet-2016-103839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Khanna K K, Keating K E, Kozlov S. ATM associates with and phosphorylates p 53: mapping the region of interaction. doi:10.1038/3882. Nat Genet. 1998;20:398–400. doi: 10.1038/3882. [DOI] [PubMed] [Google Scholar]
- 37.Gatei M, Scott S P, Filippovitch I. Role for ATM in DNA damage-induced phosphorylation of BRCA1. Cancer Res. 2000;60:3299–3304. [PubMed] [Google Scholar]
- 38.Girard E, Eon-Marchais S, Olaso R. Familial breast cancer and DNA repair genes: Insights into known and novel susceptibility genes from the GENESIS study, and implications for multigene panel testing. Int J Cancer. 2018 doi: 10.1002/ijc.31921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Catucci I, Radice P, Milne R L. The PALB2 p.Leu939Trp mutation is not associated with breast cancer risk. doi:10.1186/s13058-016-0762-9. Breast Cancer Res. 2016;18:111. doi: 10.1186/s13058-016-0762-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Antoniou A C, Casadei S, Heikkinen T. Breast-cancer risk in families with mutations in PALB2. doi:10.1056/NEJMoa1400382. N Engl J Med. 2014;371:497–506. doi: 10.1056/NEJMoa1400382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Antoniou A C, Foulkes W D, Tischkowitz M. Breast-cancer risk in families with mutations in PALB2. doi:10.1056/NEJMc1410673. N Engl J Med. 2014;371:1651–1652. doi: 10.1056/NEJMc1410673. [DOI] [PubMed] [Google Scholar]
- 42.Antoniou A C, Foulkes W D, Tischkowitz M. Breast cancer risk in women with PALB2 mutations in different populations. doi:10.1016/s1470-2045(15)00002-9. Lancet Oncol. 2015;16:e375–e376. doi: 10.1016/S1470-2045(15)00002-9. [DOI] [PubMed] [Google Scholar]
- 43.Southey M C, Teo Z L, Dowty J G. A PALB2 mutation associated with high risk of breast cancer. doi:10.1186/bcr2796. Breast Cancer Res. 2010;12:R109. doi: 10.1186/bcr2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tischkowitz M, Capanu M, Sabbaghian N. Rare germline mutations in PALB2 and breast cancer risk: a population-based study. doi:10.1002/humu.22022. Hum Mutat. 2012;33:674–680. doi: 10.1002/humu.22022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tischkowitz M, Sabbaghian N, Hamel N. Contribution of the PALB2 c.2323C>T [p. Q775X] founder mutation in well-defined breast and/or ovarian cancer families and unselected ovarian cancer cases of French Canadian descent. doi:10.1186/1471-2350-14-5. BMC Med Genet. 2013;14:5. doi: 10.1186/1471-2350-14-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Obermeier K, Sachsenweger J, Friedl T W. Heterozygous PALB2 c.1592delT mutation channels DNA double-strand break repair into error-prone pathways in breast cancer patients. doi:10.1038/onc.2015.448. Oncogene. 2016;35:3796–3806. doi: 10.1038/onc.2015.448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hayakawa T, Zhang F, Hayakawa N. MRG15 binds directly to PALB2 and stimulates homology-directed repair of chromosomal breaks. doi:10.1242/jcs.060178. J Cell Sci. 2010;123:1124–1130. doi: 10.1242/jcs.060178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sy S M, Huen M S, Chen J. MRG15 is a novel PALB2-interacting factor involved in homologous recombination. doi:10.1074/jbc.C109.023937. J Biol Chem. 2009;284:21127–21131. doi: 10.1074/jbc.C109.023937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang F, Ma J, Wu J. PALB2 links BRCA1 and BRCA2 in the DNA-damage response. doi:10.1016/j.cub.2009.02.018. Curr Biol. 2009;19:524–529. doi: 10.1016/j.cub.2009.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Foo T K, Tischkowitz M, Simhadri S. Compromised BRCA1-PALB2 interaction is associated with breast cancer risk. doi:10.1038/onc.2017.46. Oncogene. 2017;36:4161–4170. doi: 10.1038/onc.2017.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Buisson R, Dion-Cote A M, Coulombe Y. Cooperation of breast cancer proteins PALB2 and piccolo BRCA2 in stimulating homologous recombination. doi:10.1038/nsmb.1915. Nat Struct Mol Biol. 2010;17:1247–1254. doi: 10.1038/nsmb.1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dray E, Etchin J, Wiese C. Enhancement of RAD51 recombinase activity by the tumor suppressor PALB2. doi:10.1038/nsmb.1916. Nat Struct Mol Biol. 2010;17:1255–1259. doi: 10.1038/nsmb.1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bleuyard J Y, Buisson R, Masson J Y. ChAM, a novel motif that mediates PALB2 intrinsic chromatin binding and facilitates DNA repair. doi:10.1038/embor.2011.243. EMBO Rep. 2012;13:135–141. doi: 10.1038/embor.2011.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Park J Y, Singh T R, Nassar N. Breast cancer-associated missense mutants of the PALB2 WD40 domain, which directly binds RAD51C, RAD51 and BRCA2, disrupt DNA repair. doi:10.1038/onc.2013.421. Oncogene. 2014;33:4803–4812. doi: 10.1038/onc.2013.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Oliver A W, Swift S, Lord C J. Structural basis for recruitment of BRCA2 by PALB2. doi:10.1038/embor.2009.126. EMBO Rep. 2009;10:990–996. doi: 10.1038/embor.2009.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang F, Fan Q, Ren K. PALB2 functionally connects the breast cancer susceptibility proteins BRCA1 and BRCA2. doi:10.1158/1541-7786.mcr-09-0123. Mol Cancer Res. 2009;7:1110–1118. doi: 10.1158/1541-7786.MCR-09-0123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Caleca L, Catucci I, Figlioli G. Two Missense Variants Detected in Breast Cancer Probands Preventing BRCA2-PALB2 Protein Interaction. doi:10.3389/fonc.2018.00480. Front Oncol. 2018;8:480. doi: 10.3389/fonc.2018.00480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hellebrand H, Sutter C, Honisch E. Germline mutations in the PALB2 gene are population specific and occur with low frequencies in familial breast cancer. doi:10.1002/humu.21478. Hum Mutat. 2011;32:E2176–E2188. doi: 10.1002/humu.21478. [DOI] [PubMed] [Google Scholar]
- 59.Sodha N, Williams R, Mangion J. Screening hCHK2 for mutations. Science. 2000;289:359. doi: 10.1126/science.289.5478.359a. [DOI] [PubMed] [Google Scholar]
- 60.Cybulski C, Gorski B, Huzarski T. CHEK2 is a multiorgan cancer susceptibility gene. doi:10.1086/426403. Am J Hum Genet. 2004;75:1131–1135. doi: 10.1086/426403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cai Z, Chehab N H, Pavletich N P. Structure and activation mechanism of the CHK2 DNA damage checkpoint kinase. doi:10.1016/j.molcel.2009.09.007. Mol Cell. 2009;35:818–829. doi: 10.1016/j.molcel.2009.09.007. [DOI] [PubMed] [Google Scholar]
- 62.Roeb W, Higgins J, King M C. Response to DNA damage of CHEK2 missense mutations in familial breast cancer. doi:10.1093/hmg/dds101. Hum Mol Genet. 2012;21:2738–2744. doi: 10.1093/hmg/dds101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ow G S, Ivshina A V, Fuentes G. Identification of two poorly prognosed ovarian carcinoma subtypes associated with CHEK2 germ-line mutation and non-CHEK2 somatic mutation gene signatures. doi:10.4161/cc.29271. Cell Cycle. 2014;13:2262–2280. doi: 10.4161/cc.29271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Muranen T A, Blomqvist C, Dork T. Patient survival and tumor characteristics associated with CHEK2:p. I157T – findings from the Breast Cancer Association Consortium. doi:10.1186/s13058-016-0758-5. Breast Cancer Res. 2016;18:98. doi: 10.1186/s13058-016-0758-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dong X, Wang L, Taniguchi K. Mutations in CHEK2 associated with prostate cancer risk. doi:10.1086/346094. Am J Hum Genet. 2003;72:270–280. doi: 10.1086/346094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ahn J, Prives C. Checkpoint kinase 2 (Chk2) monomers or dimers phosphorylate Cdc25C after DNA damage regardless of threonine 68 phosphorylation. doi:10.1074/jbc.M208321200. J Biol Chem. 2002;277:48418–48426. doi: 10.1074/jbc.M208321200. [DOI] [PubMed] [Google Scholar]
- 67.Ahn J, Urist M, Prives C. The Chk2 protein kinase. doi:10.1016/j.dnarep.2004.03.033. DNA Repair (Amst) 2004;3:1039–1047. doi: 10.1016/j.dnarep.2004.03.033. [DOI] [PubMed] [Google Scholar]
- 68.Schwarz J K, Lovly C M, Piwnica-Worms H. Regulation of the Chk2 protein kinase by oligomerization-mediated cis- and trans-phosphorylation. Mol Cancer Res. 2003;1:598–609. [PubMed] [Google Scholar]
- 69.Han F F, Guo C L, Liu L H. The effect of CHEK2 variant I157T on cancer susceptibility: evidence from a meta-analysis. doi:10.1089/dna.2013.1970. DNA Cell Biol. 2013;32:329–335. doi: 10.1089/dna.2013.1970. [DOI] [PubMed] [Google Scholar]
- 70.Kleibl Z, Havranek O, Novotny J. Analysis of CHEK2 FHA domain in Czech patients with sporadic breast cancer revealed distinct rare genetic alterations. doi:10.1007/s10549-007-9838-7. Breast Cancer Res Treat. 2008;112:159–164. doi: 10.1007/s10549-007-9838-7. [DOI] [PubMed] [Google Scholar]
- 71.Bak A, Janiszewska H, Junkiert-Czarnecka A. A risk of breast cancer in women – carriers of constitutional CHEK2 gene mutations, originating from the North – Central Poland. doi:10.1186/1897-4287-12-10. Hered Cancer Clin Pract. 2014;12:10. doi: 10.1186/1897-4287-12-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kato S, Han S Y, Liu W. Understanding the function-structure and function-mutation relationships of p 53 tumor suppressor protein by high-resolution missense mutation analysis. doi:10.1073/pnas.1431692100. Proc Natl Acad Sci U S A. 2003;100:8424–8429. doi: 10.1073/pnas.1431692100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mathe E, Olivier M, Kato S. Predicting the transactivation activity of p 53 missense mutants using a four-body potential score derived from Delaunay tessellations. doi:10.1002/humu.20284. Hum Mutat. 2006;27:163–172. doi: 10.1002/humu.20284. [DOI] [PubMed] [Google Scholar]
- 74.Soussi T, Kato S, Levy P P. Reassessment of the TP53 mutation database in human disease by data mining with a library of TP53 missense mutations. doi:10.1002/humu.20114. Hum Mutat. 2005;25:6–17. doi: 10.1002/humu.20114. [DOI] [PubMed] [Google Scholar]
- 75.Leroy B, Fournier J L, Ishioka C. The TP53 website: an integrative resource centre for the TP53 mutation database and TP53 mutant analysis. doi:10.1093/nar/gks1033. Nucleic Acids Res. 2013;41:D962–D969. doi: 10.1093/nar/gks1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Monti P, Ciribilli Y, Jordan J. Transcriptional functionality of germ line p 53 mutants influences cancer phenotype. doi:10.1158/1078-0432.ccr-06-2545. Clin Cancer Res. 2007;13:3789–3795. doi: 10.1158/1078-0432.CCR-06-2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Monti P, Perfumo C, Bisio A. Dominant-negative features of mutant TP53 in germline carriers have limited impact on cancer outcomes. doi:10.1158/1541-7786.mcr-10-0496. Mol Cancer Res. 2011;9:271–279. doi: 10.1158/1541-7786.MCR-10-0496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Saha T, Kar R K, Sa G. Structural and sequential context of p 53: A review of experimental and theoretical evidence. doi:10.1016/j.pbiomolbio.2014.12.002. Prog Biophys Mol Biol. 2015;117:250–263. doi: 10.1016/j.pbiomolbio.2014.12.002. [DOI] [PubMed] [Google Scholar]
- 79.Buchhop S, Gibson M K, Wang X W. Interaction of p 53 with the human Rad51 protein. Nucleic Acids Res. 1997;25:3868–3874. doi: 10.1093/nar/25.19.3868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Liang S H, Clarke M F. The nuclear import of p 53 is determined by the presence of a basic domain and its relative position to the nuclear localization signal. doi:10.1038/sj.onc.1202350. Oncogene. 1999;18:2163–2166. doi: 10.1038/sj.onc.1202350. [DOI] [PubMed] [Google Scholar]
- 81.Shaulsky G, Goldfinger N, Ben-Zeʼev A. Nuclear accumulation of p 53 protein is mediated by several nuclear localization signals and plays a role in tumorigenesis. Mol Cell Biol. 1990;10:6565–6577. doi: 10.1128/mcb.10.12.6565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Linke S P, Sengupta S, Khabie N. p 53 interacts with hRAD51 and hRAD54, and directly modulates homologous recombination. Cancer Res. 2003;63:2596–2605. [PubMed] [Google Scholar]
- 83.Pittman D L, Weinberg L R, Schimenti J C. Identification, characterization, and genetic mapping of Rad51 d, a new mouse and human RAD51/RecA-related gene. doi:10.1006/geno.1998.5226. Genomics. 1998;49:103–111. doi: 10.1006/geno.1998.5226. [DOI] [PubMed] [Google Scholar]
- 84.Cartwright R, Dunn A M, Simpson P J. Isolation of novel human and mouse genes of the recA/RAD51 recombination-repair gene family. Nucleic Acids Res. 1998;26:1653–1659. doi: 10.1093/nar/26.7.1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rivera B, Di Iorio M, Frankum J. Functionally Null RAD51D Missense Mutation Associates Strongly with Ovarian Carcinoma. doi:10.1158/0008-5472.Can-17-0190. Cancer Res. 2017;77:4517–4529. doi: 10.1158/0008-5472.CAN-17-0190. [DOI] [PubMed] [Google Scholar]
- 86.Kim Y M, Choi B S. Structural and functional characterization of the N-terminal domain of human Rad51D. doi:10.1016/j.biocel.2010.11.014. Int J Biochem Cell Biol. 2011;43:416–422. doi: 10.1016/j.biocel.2010.11.014. [DOI] [PubMed] [Google Scholar]
- 87.Gruver A M, Miller K A, Rajesh C. The ATPase motif in RAD51D is required for resistance to DNA interstrand crosslinking agents and interaction with RAD51C. doi:10.1093/mutage/gei059. Mutagenesis. 2005;20:433–440. doi: 10.1093/mutage/gei059. [DOI] [PubMed] [Google Scholar]
- 88.Gutierrez-Enriquez S, Bonache S, de Garibay G R. About 1% of the breast and ovarian Spanish families testing negative for BRCA1 and BRCA2 are carriers of RAD51D pathogenic variants. doi:10.1002/ijc.28540. Int J Cancer. 2014;134:2088–2097. doi: 10.1002/ijc.28540. [DOI] [PubMed] [Google Scholar]
- 89.Janatova M, Soukupova J, Stribrna J. Mutation Analysis of the RAD51C and RAD51D Genes in High-Risk Ovarian Cancer Patients and Families from the Czech Republic. doi:10.1371/journal.pone.0127711. PLoS One. 2015;10:e0127711. doi: 10.1371/journal.pone.0127711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Thompson E R, Rowley S M, Sawyer S. Analysis of RAD51D in ovarian cancer patients and families with a history of ovarian or breast cancer. doi:10.1371/journal.pone.0054772. PLoS One. 2013;8:e54772. doi: 10.1371/journal.pone.0054772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Miller K A, Sawicka D, Barsky D. Domain mapping of the Rad51 paralog protein complexes. doi:10.1093/nar/gkg925. Nucleic Acids Res. 2004;32:169–178. doi: 10.1093/nar/gkg925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Loveday C, Turnbull C, Ramsay E. Germline mutations in RAD51D confer susceptibility to ovarian cancer. doi:10.1038/ng.893. Nat Genet. 2011;43:879–882. doi: 10.1038/ng.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wiese C, Hinz J M, Tebbs R S. Disparate requirements for the Walker A and B ATPase motifs of human RAD51D in homologous recombination. doi:10.1093/nar/gkl366. Nucleic Acids Res. 2006;34:2833–2843. doi: 10.1093/nar/gkl366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Song H, Dicks E, Ramus S J. Contribution of Germline Mutations in the RAD51B, RAD51C, and RAD51D Genes to Ovarian Cancer in the Population. doi:10.1200/jco.2015.61.2408. J Clin Oncol. 2015;33:2901–2907. doi: 10.1200/JCO.2015.61.2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Meindl A, Hellebrand H, Wiek C. Germline mutations in breast and ovarian cancer pedigrees establish RAD51C as a human cancer susceptibility gene. doi:10.1038/ng.569. Nat Genet. 2010;42:410–414. doi: 10.1038/ng.569. [DOI] [PubMed] [Google Scholar]
- 96.Vaz F, Hanenberg H, Schuster B. Mutation of the RAD51C gene in a Fanconi anemia-like disorder. doi:10.1038/ng.570. Nat Genet. 2010;42:406–409. doi: 10.1038/ng.570. [DOI] [PubMed] [Google Scholar]
- 97.Clague J, Wilhoite G, Adamson A. RAD51C germline mutations in breast and ovarian cancer cases from high-risk families. doi:10.1371/journal.pone.0025632. PLoS One. 2011;6:e25632. doi: 10.1371/journal.pone.0025632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Osorio A, Endt D, Fernandez F. Predominance of pathogenic missense variants in the RAD51C gene occurring in breast and ovarian cancer families. doi:10.1093/hmg/dds115. Hum Mol Genet. 2012;21:2889–2898. doi: 10.1093/hmg/dds115. [DOI] [PubMed] [Google Scholar]
- 99.French C A, Tambini C E, Thacker J. Identification of functional domains in the RAD51L2 (RAD51C) protein and its requirement for gene conversion. doi:10.1074/jbc.M308621200. J Biol Chem. 2003;278:45445–45450. doi: 10.1074/jbc.M308621200. [DOI] [PubMed] [Google Scholar]
- 100.Jonson L, Ahlborn L B, Steffensen A Y. Identification of six pathogenic RAD51C mutations via mutational screening of 1228 Danish individuals with increased risk of hereditary breast and/or ovarian cancer. doi:10.1007/s10549-015-3674-y. Breast Cancer Res Treat. 2016;155:215–222. doi: 10.1007/s10549-015-3674-y. [DOI] [PubMed] [Google Scholar]
- 101.Schnurbein G, Hauke J, Wappenschmidt B. RAD51C deletion screening identifies a recurrent gross deletion in breast cancer and ovarian cancer families. doi:10.1186/bcr3589. Breast Cancer Res. 2013;15:R120. doi: 10.1186/bcr3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ali A M, Singh T R, Meetei A R. FANCM-FAAP24 and FANCJ: FA proteins that metabolize DNA. doi:10.1016/j.mrfmmm.2009.04.002. Mutat Res. 2009;668:20–26. doi: 10.1016/j.mrfmmm.2009.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Yu X, Chini C C, He M. The BRCT domain is a phospho-protein binding domain. doi:10.1126/science.1088753. Science. 2003;302:639–642. doi: 10.1126/science.1088753. [DOI] [PubMed] [Google Scholar]
- 104.Peng M, Litman R, Xie J. The FANCJ/MutLalpha interaction is required for correction of the cross-link response in FA-J cells. doi:10.1038/sj.emboj.7601754. EMBO J. 2007;26:3238–3249. doi: 10.1038/sj.emboj.7601754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Weber-Lassalle N, Hauke J, Ramser J. BRIP1 loss-of-function mutations confer high risk for familial ovarian cancer, but not familial breast cancer. doi:10.1186/s13058-018-0935-9. Breast Cancer Res. 2018;20:7. doi: 10.1186/s13058-018-0935-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Castera L, Harter V, Muller E. Landscape of pathogenic variations in a panel of 34 genes and cancer risk estimation from 5131 HBOC families. Genet Med. 2018 doi: 10.1038/s41436-018-0005-9. [DOI] [PubMed] [Google Scholar]
- 107.Li J, Meeks H, Feng B J. Targeted massively parallel sequencing of a panel of putative breast cancer susceptibility genes in a large cohort of multiple-case breast and ovarian cancer families. doi:10.1136/jmedgenet-2015-103452. J Med Genet. 2016;53:34–42. doi: 10.1136/jmedgenet-2015-103452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Norquist B M, Harrell M I, Brady M F. Inherited Mutations in Women With Ovarian Carcinoma. doi:10.1001/jamaoncol.2015.5495. JAMA Oncol. 2016;2:482–490. doi: 10.1001/jamaoncol.2015.5495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ramus S J, Song H, Dicks E. Germline Mutations in the BRIP1, BARD1, PALB2, and NBN Genes in Women With Ovarian Cancer. doi:10.1093/jnci/djv214. J Natl Cancer Inst. 2015;107:pii:djv214. doi: 10.1093/jnci/djv214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lilyquist J, LaDuca H, Polley E. Frequency of mutations in a large series of clinically ascertained ovarian cancer cases tested on multi-gene panels compared to reference controls. doi:10.1016/j.ygyno.2017.08.030. Gynecol Oncol. 2017;147:375–380. doi: 10.1016/j.ygyno.2017.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hansford S, Kaurah P, Li-Chang H. Hereditary Diffuse Gastric Cancer Syndrome: CDH1 Mutations and Beyond. doi:10.1001/jamaoncol.2014.168. JAMA Oncol. 2015;1:23–32. doi: 10.1001/jamaoncol.2014.168. [DOI] [PubMed] [Google Scholar]
- 112.Melo S, Figueiredo J, Fernandes M S. Predicting the Functional Impact of CDH1 Missense Mutations in Hereditary Diffuse Gastric Cancer. doi:10.3390/ijms18122687. Int J Mol Sci. 2017;18:pii:E2687. doi: 10.3390/ijms18122687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Oliveira C, Pinheiro H, Figueiredo J. Familial gastric cancer: genetic susceptibility, pathology, and implications for management. doi:10.1016/S1470-2045(14)71016-2. Lancet Oncol. 2015;16:e60–e70. doi: 10.1016/S1470-2045(14)71016-2. [DOI] [PubMed] [Google Scholar]
- 114.Corso G, Intra M, Trentin C. CDH1 germline mutations and hereditary lobular breast cancer. doi:10.1007/s10689-016-9869-5. Fam Cancer. 2016;15:215–219. doi: 10.1007/s10689-016-9869-5. [DOI] [PubMed] [Google Scholar]
- 115.Krempely K, Karam R. A novel de novo CDH1 germline variant aids in the classification of carboxy-terminal E-cadherin alterations predicted to escape nonsense-mediated mRNA decay . doi:10.1101/mcs.a003012. Cold Spring Harb Mol Case Stud. 2018;4:pii:a003012. doi: 10.1101/mcs.a003012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Matsuoka S, Rotman G, Ogawa A. Ataxia telangiectasia-mutated phosphorylates Chk2 in vivo and in vitro. doi:10.1073/pnas.190030497. Proc Natl Acad Sci U S A. 2000;97:10389–10394. doi: 10.1073/pnas.190030497. [DOI] [PMC free article] [PubMed] [Google Scholar]