

Plasma biomarkers are promising tools for the early diagnosis of dementia and for monitoring its progression. In a population-based study, de Wolf et al. show that low amyloid-β42 and high NfL levels are associated, both independently and in combination, with an increased risk of dementia and Alzheimer’s disease.
Keywords: dementia, Alzheimer’s disease, plasma biomarkers, NfL, Aβ42
Abstract
CSF biomarkers, including total-tau, neurofilament light chain (NfL) and amyloid-β, are increasingly being used to define and stage Alzheimer’s disease. These biomarkers can be measured more quickly and less invasively in plasma and may provide important information for early diagnosis of Alzheimer’s disease. We used stored plasma samples and clinical data obtained from 4444 non-demented participants in the Rotterdam study at baseline (between 2002 and 2005) and during follow-up until January 2016. Plasma concentrations of total-tau, NfL, amyloid-β40 and amyloid-β42 were measured using the Simoa NF-light® and N3PA assays. Associations between biomarker plasma levels and incident all-cause and Alzheimer’s disease dementia during follow-up were assessed using Cox proportional-hazard regression models adjusted for age, sex, education, cardiovascular risk factors and APOE ε4 status. Moreover, biomarker plasma levels and rates of change over time of participants who developed Alzheimer’s disease dementia during follow-up were compared with age and sex-matched dementia-free control subjects. During up to 14 years follow-up, 549 participants developed dementia, including 374 cases with Alzheimer’s disease dementia. A log2 higher baseline amyloid-β42 plasma level was associated with a lower risk of developing all-cause or Alzheimer’s disease dementia, adjusted hazard ratio (HR) 0.61 [95% confidence interval (CI), 0.47–0.78; P < 0.0001] and 0.59 (95% CI, 0.43–0.79; P = 0.0006), respectively. Conversely, a log2 higher baseline plasma NfL level was associated with a higher risk of all-cause dementia [adjusted HR 1.59 (95% CI, 1.38–1.83); P < 0.0001] or Alzheimer’s disease [adjusted HR 1.50 (95% CI, 1.26–1.78); P < 0.0001]. Combining the lowest quartile group of amyloid-β42 with the highest of NfL resulted in a stronger association with all-cause dementia [adjusted HR 9.5 (95% CI, 2.3–40.4); P < 0.002] and with Alzheimer’s disease [adjusted HR 15.7 (95% CI, 2.1–117.4); P < 0.0001], compared to the highest quartile group of amyloid-β42 and lowest of NfL. Total-tau and amyloid-β40 levels were not associated with all-cause or Alzheimer’s disease dementia risk. Trajectory analyses of biomarkers revealed that mean NfL plasma levels increased 3.4 times faster in participants who developed Alzheimer’s disease compared to those who remained dementia-free (P < 0.0001), plasma values for cases diverged from controls 9.6 years before Alzheimer’s disease diagnosis. Amyloid-β42 levels began to decrease in Alzheimer’s disease cases a few years before diagnosis, although the decline did not reach significance compared to dementia-free participants. In conclusion, our study shows that low amyloid-β42 and high NfL plasma levels are each independently and in combination strongly associated with risk of all-cause and Alzheimer’s disease dementia. These data indicate that plasma NfL and amyloid-β42 levels can be used to assess the risk of developing dementia in a non-demented population. Plasma NfL levels, although not specific, may also be useful in monitoring progression of Alzheimer’s disease dementia.
Introduction
Dementia is the most common neurodegenerative disease of the elderly, characterized by behavioural changes and the loss of cognitive and social functioning (Goedert, 2015; Alzheimer’s Association, 2016). It is most frequently diagnosed as the clinical outcome of Alzheimer’s disease (Burns and Iliffe, 2009). Alzheimer’s disease is defined by its underlying pathological process of amyloid-β plaques and neurofibrillary tau tangle formation in the brain, accompanied by progressive neuronal and synaptic loss (Braak et al., 2006; Serrano-Pozo et al., 2011; Nisbet et al., 2015). A preclinical phase of perhaps 20 years or more may occur before the clinical diagnosis of Alzheimer’s disease dementia, in which no or only subtle symptoms of disease are seen (Villemagne et al., 2013; Goudsmit, 2016; Verlinden et al., 2016). This gives the opportunity to postpone the onset of disease or even prevent Alzheimer’s disease dementia by intervening on the preclinical phase of disease. Such early intervention requires timely identification of individuals at high risk of developing Alzheimer’s disease by using quantitative biomarkers that are preferably easily accessible and minimally invasive.
Quantitative measurement of proteins that are associated with the neuropathological characteristics of Alzheimer’s disease, including amyloid-β deposition, tau-tangle formation and neurodegeneration, in CSF have been suggested as part of a biological definition of Alzheimer’s disease (Jack et al., 2018). In this context, CSF levels of amyloid-β42, tau and, more recently, neurofilament light chain (NfL) proteins have been associated with cognition and Alzheimer’s disease (Mattsson et al., 2016a, 2017; Zetterberg et al., 2016; Olsson et al., 2019). Studies in smaller populations have also revealed that the CSF levels of these proteins, each individually and in combination, are associated with hippocampal volume (Hilal et al., 2018) and the clinical onset of Alzheimer’s disease (Mattsson et al., 2016b; Olsson et al., 2016). These proteins can also be measured more quickly and less invasively in serum and plasma. Previous studies have documented the association of plasma amyloid-β42 levels and the amyloid-β42/amyloid-β40 ratio with cognition and risk of all-cause as well as Alzheimer’s disease dementia (van Oijen et al., 2006; Seppala et al., 2010; Fandos et al., 2017; Nakamura et al., 2018). In addition, total-tau plasma levels have been shown to improve the prediction of dementia and are suggested as a potential biomarker for risk stratification in prevention trials (Mielke et al., 2018; Pase et al., 2019). In parallel, elevated plasma levels of NfL were found to be associated with neurodegeneration in Alzheimer’s disease patients (Mattsson et al., 2017, 2019) and in people at genetically determined risk for Alzheimer’s disease compared to control subjects, even before the onset of symptoms (Preische et al., 2019). The repeated measurements of NfL levels over time have also been demonstrated to distinguish mutation carriers of familial Alzheimer’s disease from non-carriers earlier compared to single plasma levels of NfL obtained at baseline (Preische et al., 2019). Although previous studies have confirmed tau, amyloid-β and NfL as potentially valuable plasma biomarkers for Alzheimer’s disease, a head-to-head comparison of the long-term association with all-cause and Alzheimer’s disease dementia in the general population for each individual biomarker and their combinations is currently lacking. Moreover, a longitudinal approach with repeated measurements of the biomarkers in large numbers of healthy individuals at risk from the start of disease incubation and during the long preclinical phase until clinically manifested Alzheimer’s disease is needed to assess the robustness of the biomarkers over time.
In the present study, we first investigated whether plasma concentrations of total-tau, NfL, amyloid-β40, amyloid-β42 and the amyloid-β42/amyloid-β40 ratio are associated with incident all-cause dementia and Alzheimer’s disease dementia in the prospective population-based Rotterdam Study cohort. Second, we quantified the change and course of biomarker plasma levels over time in participants who developed Alzheimer’s disease dementia after baseline and compared the results to the age and sex-matched participants who remained dementia-free during up to 14 years follow-up.
Materials and methods
Study design and participants
This study was embedded in the Rotterdam Study, a large population-based cohort designed to ascertain the occurrence and determinants of age-related diseases in the general population (Ikram et al., 2017). In 1989 and 1990, all inhabitants aged 55 and older from a well-defined suburb (Ommoord district) in the city of Rotterdam, the Netherlands, were invited to participate. This first study wave (RS-I) compromised 7983 participants. In 2000, a second study wave (RS-II) of 3011 participants who had reached 55 years of age or moved into the study district since the start of the study, if aged 55 years and older, were added to the first study wave. In 2006, a third study wave (RS-III) of the cohort was initiated, in which 3932 participants were included, aged 45 years and older. In total, the Rotterdam Study comprises 14 926 participants aged 45 years or over (Ikram et al., 2017). The Rotterdam Study has medical ethics committee approval per the Population Study Act Rotterdam Study, executed by the Ministry of Health, Welfare and Sports of the Netherlands. Written informed consent was obtained from all participants.
The baseline to study associations of individual biomarker levels with risk of all-cause dementia and of Alzheimer’s disease dementia only was defined by the availability of −80°C stored plasma samples obtained from participants during the fourth visit of RS-I between January 2002 and July 2004 and the second visit of RS-II between July 2002 and December 2005. From this selection of 5094 participants, with plasma samples available and informed consent to access medical records during follow-up, we excluded 55 participants without follow-up after this baseline, 77 participants diagnosed with dementia at the baseline visit and 518 participants with one or more missing or invalid test results for plasma levels of biomarkers (for more details see ‘Measurement of plasma concentrations of biomarkers’ section). As a result, the final sample included 4444 participants for incident dementia analyses, of whom demographic and clinical data were recorded at baseline and during follow-up (Supplementary Table 1). For more details regarding the number of included participants, see Supplementary Fig. 1.
For the analyses of biomarker trajectories and risk of Alzheimer’s disease dementia, we compared changes in plasma levels of total-tau, NfL, amyloid-β40 and amyloid-β42 over time of participants who were diagnosed with Alzheimer’s disease (cases) to those participants who remained dementia-free during follow-up (controls). Each Alzheimer’s disease case (with known time to disease onset) was matched with a dementia-free control of the same sex and age (±3 years). The alignment of plasma measurements of each dementia-free control was anchored to the time to Alzheimer’s disease onset of the matched case (for more details, refer to ‘Trajectories of biomarkers’ section), hence it was particularly important to match cases and control subjects in a one-to-one manner based on age, as the best predictor of time to Alzheimer’s disease (Licher et al., 2018).
Measurement of plasma concentrations of total-tau, NfL, amyloid-β40 and amyloid-β42
EDTA plasma was sampled, aliquoted and frozen at −80°C according to standard procedures. Measurements were carried out in two separate batches (5094 samples); the first batch included 2000 samples, obtained from a random selection of 1000 participants from the fourth visit of RS-I and 1000 from the second visit of RS-II. The second batch included 3094 samples from the remaining participants of these two study waves. All measurements were performed at Quanterix on a single molecule array (Simoa) HD-1 analyser platform (Rissin et al., 2010). NfL was measured by using the Simoa NF-light® advantage kit (Gisslen et al., 2016; Kuhle et al., 2016; Rohrer et al., 2016). The Simoa Human Neurology 3-Plex A assay (N3PA) was used to measure the concentration of total-tau, amyloid-β40 and amyloid-β42 (Chang et al., 2017). Samples were tested in duplicate. Two quality control samples were run on each plate for each analyte. When duplicates or single measurements were missing (Supplementary Table 2) or in the case the concentration coefficient of variation exceeded 20% or control samples were out of range (Supplementary Table 3), participant’s data were excluded from the analyses.
Ascertainment of dementia and Alzheimer’s disease
Dementia screening and diagnosis in the Rotterdam Study has been described elsewhere (de Bruijn et al., 2015). In brief, participants were screened for dementia at baseline and subsequent centre visits with the Mini-Mental State Examination (MMSE) and the Geriatric Mental Schedule organic level. Those with a MMSE score <26 or Geriatric Mental Schedule score >0 underwent further investigation and informant interview, including the Cambridge Examination for Mental Disorders of the Elderly. All participants also underwent routine cognitive assessment. In addition, the entire cohort was continuously under surveillance for dementia through electronic linkage of the study database with medical records from general practitioners and the regional institute for outpatient mental healthcare. Available information on cognitive testing and clinical neuroimaging was used when required for diagnosis of dementia subtype. A consensus panel led by a consultant neurologist established the final diagnosis according to standard criteria for dementia (DSM-III-R), Alzheimer’s disease (NINCDS–ADRDA) and vascular dementia (NINDS-AIREN) (Licher et al., 2019). Follow-up until 1 January 2016 was virtually complete (95.7% of potential person-years). Within this period, participants were censored at date of dementia diagnosis, death, loss to follow-up, or 1 January 2016, whichever came first. Detailed information regarding the assessment of dementia subtypes, mild cognitive impairment (MCI) and covariates are described in the Supplementary material.
Statistical analysis
Descriptive statistics were used to summarize baseline characteristics of all included participants. Means (standard deviation, SD), and counts with percentages, were used to report continuous and discrete variables at the baseline visit, respectively. Plasma concentration (pg/ml) of total-tau, NfL, amyloid-β40 and amyloid-β42 were log2 transformed (confirmed by Box-Cox screening, see Supplementary material) for further analysis to comply with the assumption of normality in the distribution of the data. Pearson correlation and linear regression were used to explore the relationship between plasma levels and age.
Associations of baseline levels of each biomarker with risk of all-cause and Alzheimer’s disease dementia
Dementia was considered as the primary study end point. Plasma total-tau, NfL, amyloid-β40 and amyloid-β42 were analysed on a continuous scale and according to quartiles. In each of the two assay batches the plasma concentration for each individual protein was ranked and divided into four equally sized groups. Group Q1 representing the group with the lowest concentration, Q2 the second lowest group, Q3 the second highest group and Q4 the highest group. For each protein the groups for both batches were then combined into one variable (of four equally sized groups). In standard Cox proportional hazard regression models, time was modelled as follow-up time after baseline. The proportional hazard assumption was tested by including time-dependent interactions between time after baseline and total-tau, NfL, amyloid-β40 and amyloid-β42 plasma levels and the amyloid-β40/amyloid-β42 ratio (all log2 transformed) on the risk for all-cause and Alzheimer’s disease dementia. We used a linear interaction between time and plasma proteins. In sensitivity analyses, we repeated the biomarker analyses among cognitively unimpaired participants, by excluding participants with MMSE score <26 or an MCI diagnosis at baseline, to evaluate whether plasma protein levels were still associated with development of all-cause and of Alzheimer’s disease dementia. All analyses were repeated without adjusting for batch number. We used the cause-specific cumulative incidence function to translate results to absolute risks for all-cause and Alzheimer’s disease dementia according to age for each of the marker quartile groups. To overcome issues of competing risks, we analysed the data considering the occurrence of competing events by computing cumulative incidence as left truncated data, with age as the time scale. Observation time started at the age of participants at the date of sampling and ended at the age of diagnosis of all-cause or Alzheimer’s disease dementia, death, loss to follow-up or end of the study, whichever came first (Klein and Moeschberger, 2003; Thiebaut and Benichou, 2004). Death and dementia other than Alzheimer’s disease were treated as competing events for Alzheimer’s disease analysis (Fine and Gray, 1999).
Associations of combined biomarkers with risk of all-cause and Alzheimer’s disease dementia
We investigated the association of combined plasma protein levels with all-cause and Alzheimer’s disease dementia by including an interaction term for the quartile group analysis as well as the analysis on a continuous scale. Significance of multiplicative interaction on a continuous scale and using the four equally sized groups, was tested using the χ2 likelihood test using Wilk’s approximation.
For both the individual plasma proteins as well as for their combinations, we used the following models for adjustment. In a first model (Model I), we adjusted for age (using a linear and a quadratic term), sex, and assay batch number. In a second model (Model II), we further adjusted for baseline educational level, cardiovascular risk factors [smoking status, systolic blood pressure, total and high-density lipoprotein (HDL) cholesterol, body mass index, history of diabetes, and stroke or coronary heart disease] and APOE ε4 status (i.e. presence of 0, 1 or 2 APOE ε4 alleles). Measurements of systolic blood pressure and total and HDL cholesterol were missing in three, three and 12 participants, respectively, in which case we imputed the mean, conditional on age and sex. In a third model (Model III), we additionally adjusted for plasma levels of all other proteins.
Trajectories of biomarkers
Linear mixed models with random intercepts and slopes were used to estimate trajectories of log2 plasma levels of total-tau, NfL, amyloid-β40, amyloid-β42 on a population level for those who developed Alzheimer’s disease dementia (cases) and the age- and sex-matched participants who remained dementia-free (controls) during follow-up. Follow-up times were calculated by subtracting the time of plasma measurement from the date of diagnosis for those who developed Alzheimer’s disease. As a result, negative follow-up times represent the time (in years) before Alzheimer’s disease onset, whereas positive follow-up times represent the time (in years) after the disease onset. For the dementia-free controls (for whom, by definition the date of diagnosis is not observed) an index date identical to the date of diagnosis of the corresponding matching case was used. We included age at baseline for baseline measurements and age at visiting the research centre for follow-up measurements and sex in a primary model, and additionally extended this model with educational level and APOE ε4 carrier status. As a complementary analysis, we assessed the rate of change of Alzheimer’s disease-associated proteins on an individual level to examine non-linearity as a function of disease progression among those with two or more biomarker measurements. A rate (‘slope’) of change for each individual was extracted from a linear mixed effects model, which included an interaction term between time and case/control to allow different rates for cases compared to controls over time. Individual rates of change were then regressed on time from the first measurement taken in cases or controls to conversion or index-date, respectively. In addition, we modelled the trajectories of Alzheimer’s disease-associated plasma protein levels on age as time scale. In this analysis, we adjusted for all of the covariates, and instead of time to diagnosis used age of visiting the centre as timescale.
All analyses were done at the significance level of 0.05 (two-tailed) using SAS version 9.4, SPSS Statistics version 24.0.0.1 (IBM Corp., Armonk, NY) and R version 3.4.3, using the ‘survival’, ‘npsurv’, ‘rstan’, ‘lme4’ and ‘rms’ packages.
Data availability
The data that support the findings of this study are available on request from the corresponding author, subject to local and European regulations.
Results
Baseline characteristics
A total of 4444 participants were included in the analysis for the risk of dementia with an average age at baseline of 71.9 (SD 7.5) years, 2555 (57.5%) participants were female (Table 1). The highest level of attained education varied, with 11.1% having only attained primary education and 43.5% with a lower/intermediate general education or lower vocational education. The median baseline MMSE score was 28 [interquartile range (IQR) 26–30] and 424 participants (9.5%) had a MMSE score <26. At baseline, 419 participants (9.4%) had MCI and 114 (25.7%) were APOE ε4 carriers (homozygous plus heterozygous) (Corder et al., 1993).
Table 1.
Baseline characteristics of the participants
| Variable | Study population (n = 4444) |
|---|---|
| Age | 71.9 (7.5) |
| Female | 2555 (58) |
| MMSEa | 27.8 (2) |
| ≤20 | 30 (1) |
| 21–25 | 394 (9) |
| 26–29 | 3328 (75) |
| 30 | 652 (15) |
| MCIa | 419 (9) |
| Education level | |
| Primary education | 493 (11) |
| Lower-intermediate education level | 1931 (44) |
| Intermediate-higher level | 1342 (30) |
| Higher-university education level | 614 (14) |
| APOE ε4 | |
| 0 alleles | 3083 (69) |
| 1 allele | 1066 (24) |
| 2 alleles | 77 (2) |
| Smoking | |
| Never | 1306 (29) |
| Former | 2379 (54) |
| Current | 673 (15) |
| Body mass index, kg/m2 | 27.6 (4) |
| <18.5 | 23 (1) |
| 18.5–25.0 | 1132 (26) |
| 25.0–30.0 | 2156 (49) |
| 30.0–35.0 | 827 (19) |
| >35.0 | 216 (5) |
| Total cholesterol, mmol/l | 5.6 (1) |
| HDL cholesterol, mmol/l | 1.5 (0.4) |
| Systolic blood pressure, mmHg | 149.1 (21.1) |
| Diastolic blood pressure, mmHg | 79.7 (10.9) |
| Coronary heart disease | 431 (10) |
| Diabetes | 581 (14) |
| Stroke | 197 (4) |
| Biomarker plasma levels, pg/ml, mean (SD), IQR [min, max] | |
| Total-tau | 2.6 (2.3), 1.9–3.0 [0.4, 141.4] |
| NfL | 16.0 (12.9), 10.0–18.3 [3.4, 390.8] |
| Amyloid-β40 | 266.0 (55.0), 230.5–294.1 [52.4, 614.6] |
| Amyloid-β42 | 10.7 (3.0), 8.9–12.1 [0.5, 94.2] |
Non-imputed data presented as frequency (%) for categorical values and mean ± SD for continuous variables, unless indicated otherwise. Data at baseline were virtually complete (<5.0% missing).
For ascertainment of MCI, see Supplementary material.
HDL = high-density lipoprotein; MMSE = Mini-Mental State Examination.
Mean baseline plasma levels of total-tau, NfL, amyloid-β40 and amyloid-β42 measured in the 4444 participants were 2.6 (SD 2.3), 16.0 (SD 12.9), 266.0 (SD 55.0) and 10.7 (SD 3.0) pg/ml, respectively (Table 1). NfL and amyloid-β40 plasma levels increased with age (Pearson coefficient r = 0.59 and r = 0.38, respectively, all P < 0.0001; Fig. 1). Correlations with age were much weaker for total-tau (r = 0.09, P < 0.0001) and amyloid-β42 plasma levels (r = 0.17, P < 0.0001). Plasma levels of amyloid-β40 and amyloid-β42 were correlated (r = 0.59, P < 0.0001, Supplementary Table 4) as was the case for amyloid-β42 plasma levels and the plasma amyloid-β42/amyloid-β40 ratio (r = 0.67, P < 0.0001). Plasma levels of NfL were correlated with those of amyloid-β40 (r = 0.49, P < 0.0001) and amyloid-β42 (r = 0.33, P < 0.0001).
Figure 1.

Correlation of plasma levels of total-tau, NfL, amyloid-β40 and amyloid-β42 with age. The correlation between individual plasma total-tau, NfL, amyloid-β40 (Aβ40), and amyloid-β42 (Aβ42) levels (pg/ml) with age are shown. The black line represents the mean obtained by regressing each biomarker on age. r = Pearson correlation coefficient; P-value for each biomarker <0.0001.
During a total follow-up time of 40 577 person-years, 549 individuals developed dementia (12.4%). The median follow-up time from baseline to dementia diagnosis was 6.7 years (IQR: 3.4–9.1 years). Among those, Alzheimer’s disease dementia was diagnosed in 374 (68%) participants after a median of 6.6 years (IQR: 2.9–9.0 years). Possible Alzheimer’s disease and Alzheimer’s disease plus cardiovascular disease were diagnosed in 75 (13.7%) participants and were not included in the analyses with Alzheimer’s disease as end-point as was the case for the remaining 100 (18.2%) individuals who were diagnosed with vascular dementia, Parkinson dementia, or an undetermined type of dementia (Supplementary Fig. 1). Median follow-up for those who remained alive and dementia-free was 11.2 years (IQR: 10.8–12.1), with a maximum follow-up of 13.9 years.
Association of plasma biomarkers with risk of all-cause and Alzheimer’s disease dementia
A log2 higher concentration of NfL in plasma measured at baseline was associated with an increased risk for developing all-cause dementia {hazard ratio (HR) 1.54 [95% confidence interval (CI), 1.34–1.75]; P < 0.0001}, and for Alzheimer’s disease dementia [HR 1.49 (95% CI, 1.27–1.74); P < 0.0001], while adjusting for age, sex and assay batch number (Model I; Table 2). In the same model, a log2 higher amyloid-β42 concentration in plasma was associated with a decreased risk for all-cause dementia [HR 0.60 (95% CI, 0.49–0.74); P < 0.0001] and for Alzheimer’s disease dementia [HR 0.60 (95% CI, 0.47–0.76); P < 0.0001]. After further adjustment for all other biomarkers, educational level, cardiovascular risk factors and APOE ε4 status (Model III), the association between development of all-cause dementia and a log2 increase of baseline plasma NfL [HR 1.59 (95% CI, 1.38–1.83); P < 0.0001] and amyloid-β42 [HR 0.61 (95% CI, 0.47–0.78); P < 0.0001] remained nearly identical (Table 2). Similar patterns were observed for Alzheimer’s disease dementia (Supplementary Table 5). In addition, baseline log2 amyloid-β42/amyloid-β40 ratios were significantly associated with risk of developing all-cause and Alzheimer’s disease dementia in the first, age, sex and batch adjusted analysis [HR 0.53 (95% CI, 0.42–0.68) and HR 0.52 (95% CI, 0.39–0.70), respectively; P < 0.0001] and after further adjustment in Model III [HR 0.63 (95% CI, 0.48–0.82) and HR 0.62 (95% CI, 0.45–0.85), respectively; P < 0.005]. In contrast, baseline total-tau and amyloid-β40 plasma levels were not significantly associated with risk for all-cause or Alzheimer’s disease dementia (Table 2 and Supplementary Table 5). Repeating the analyses without adjusting for batch number did not meaningfully change effect estimates (Supplementary Table 6). Associations of plasma levels of biomarkers with other types of dementia are presented in Supplementary Tables 7 and 8, showing increase of baseline NfL levels to be associated with vascular dementia [HR 1.95 (95% CI, 1.19–3.20); P = 0.008] and with other subtypes of non-Alzheimer’s disease dementia [HR 1.78 (95% CI, 1.18–2.68); P = 0.006].
Table 2.
Association of plasma levels of total-tau, NfL, amyloid-β40, amyloid-β42 and the amyloid-β42/amyloid-β40 ratio with all-cause dementia
| Biomarker | Association with all-cause dementia | |||||
|---|---|---|---|---|---|---|
| Model I | Model II | Model III | ||||
| HR (95%CI) | P-value (overall) | HR (95%CI) | P-value (overall) | HR (95%CI) | P-value (overall) | |
| Tau (per log2 pg/ml increase) | 1.20 (1.00, 1.45) | 0.049 | 1.16 (0.96, 1.40) | 0.1333 | 1.16 (0.95, 1.41) | 0.13771 |
| NfL (per log2 pg/ml increase) | 1.54 (1.34, 1.75) | <0.0001 | 1.50 (1.31, 1.71) | <0.0001 | 1.59 (1.38, 1.83) | <0.0001 |
| Amyloid-β40 (per log2 pg/ml increase) | 0.87 (0.64, 1.19) | 0.394 | 0.84 (0.62, 1.16) | 0.2995 | 0.86 (0.58, 1.29) | 0.47899 |
| Amyloid-β42 (per log2 pg/ml increase) | 0.60 (0.49, 0.74) | <0.0001 | 0.66 (0.53, 0.83) | 0.0002 | 0.61 (0.47, 0.78) | <0.0001 |
| Amyloid-β42/amyloid-β40 ratioa | 0.53 (0.42–0.68) | <0.0001 | 0.62 (0.47, 0.80) | 0.0004 | 0.63 (0.48-0.82) | 0.00052 |
Model I: Adjusted for age, sex and assay batch number.
Model II: Model I + additional adjustment for highest level of education, smoking status, systolic blood pressure, total and high-density lipoprotein cholesterol, body mass index, history of diabetes, stroke and coronary heart disease, and APOE ε4 status.
Model III: Model II + additional adjustment for plasma levels of other biomarkers.
Model III for amyloid-β42/amyloid-β40 ratio: Model II + additional adjustment only for plasma levels of total-tau and NfL.
To evaluate whether plasma levels of NfL and amyloid-β42 were still associated with development of all-cause and Alzheimer’s disease dementia in cognitively unimpaired participants at baseline, we repeated the analysis excluding 742 (16.7%) individuals who had a baseline MMSE score of <26 or a MCI diagnosis from the analysis. Of the remaining 3702 participants, 357 developed dementia including 239 cases of Alzheimer’s disease dementia. The hazard ratio for all-cause dementia per log2 increase of NfL was 1.70 (95% CI, 1.40–2.06) and for amyloid-β42 was 0.56 (95% CI, 0.40–0.76). Similar results were found for Alzheimer’s disease, with a hazard ratio per log2 increase of NfL of 1.64 (95% CI, 1.29–2.09) and for amyloid-β42 of 0.60 (95% CI, 0.41–0.89), which are very similar to those obtained from the main analyses.
Next, we ranked the baseline plasma levels of total-tau, NfL, amyloid-β40, amyloid-β42 and amyloid-β42/amyloid-β40 ratio and divided them according to quartiles into four equally sized groups (Q1, Q2, Q3, Q4 with Q1 the group with the lowest plasma concentrations) and compared Q2, Q3 and Q4 to Q1 as reference. The risk for all-cause dementia showed a gradual increase per quartile group of plasma NfL (P < 0.0001) with a hazard ratio of 2.70 (95% CI, 1.85–3.85) for Q4 compared to Q1 (Fig. 2A). Similar patterns were found for Alzheimer’s disease [HR 3.28 (95% CI, 2.05–5.24); P < 0.0001] (Supplementary Fig. 2). Moreover, the risk for all-cause and Alzheimer’s disease dementia significantly decreased per quartile group of amyloid-β42 (P < 0.0001 for both). Although there was a significant association between quartile groups of total-tau and the risk of all-cause and Alzheimer’s disease dementia (P = 0.003 and 0.005, respectively), trends were not consistent (Fig. 2A and Supplementary Fig. 2). Quartile groups of increasing amyloid-β40 were not significantly associated with all-cause dementia (P = 0.07) or Alzheimer’s disease dementia (P = 0.12). Repeating the analyses without adjusting for batch number did not meaningfully change the effect estimates (Supplementary Table 9).
Figure 2.

Association of plasma total-tau, NfL, amyloid-β40 and amyloid-β42 levels and amyloid-β42/amyloid-β40 ratio with all-cause dementia. (A) Forest plots showing hazard ratios for all-cause dementia and 95% CI per quartile of plasma levels (pg/ml) of total-tau, NfL, amyloid-β40, amyloid-β42, and amyloid-β42/amyloid-β40 ratio, with the lowest quartile (Q1) as reference group. Hazard ratios were obtained from the Cox proportional hazard models that were adjusted for age, sex, assay batch number, systolic blood pressure, total and HDL cholesterol, smoking status, highest level of education, body mass index, APOE ε4 status, history of diabetes, stroke and coronary heart disease. (B) Cause-specific incidence curves showing the incidence of all-cause dementia with current age for total-tau. P-value for test for equality of the cause-specific cumulative incidence curve between the five groups: total-tau (P = 0.3087), NfL (P = 0.0162), amyloid-β40 (P = 0.0012), amyloid-β42 (P < 0.0001) and amyloid-β42/amyloid-β40 ratio (P < 0.0001) (Klein and Moeschberger, 2003). Plasma levels are categorized into equally sized groups using quartiles (lowest group, Q1: black, Q2: brown, Q3: blue and Q4: green line). In total, 549 individuals had a diagnosis of all-cause dementia and 1229 individuals had death as a competing event. The remaining 2666 individuals were censored. Numbers represent the total number per quartile at risk for all-cause dementia at a given age.
Plasma biomarker levels and cumulative incidence for all-cause and Alzheimer’s disease dementia
Dementia risk profiles according to age for the four quartile groups of baseline plasma levels of total-tau, NfL, amyloid-β40, amyloid-β42 and amyloid-β42/amyloid-β40 ratio are shown in Fig. 2B. For total-tau, as well as for amyloid-β40, curves overlapped indicating that plasma levels were not linearly associated with the development of all-cause dementia. For NfL, the risk for all-cause dementia increased with each quartile group, whereas for amyloid-β42 the risk decreased with each quartile group (Fig. 2B). We repeated the analyses for Alzheimer’s disease dementia and observed similar patterns (Supplementary Fig. 2). We then investigated if the hazard ratio for all-cause and for Alzheimer’s disease dementia at baseline remained constant over the course of follow-up by including a linear interaction with follow-up time and plasma biomarkers in the model (Supplementary Table 10). For NfL, the hazard ratio for Alzheimer’s disease declined significantly (P = 0.003) from 2.29 (95% CI, 1.77–2.95) per log2 NfL higher at baseline to 1.18 (95% CI, 0.88–1.57) 10 years thereafter. The hazard ratio for Alzheimer’s disease per log2 higher amyloid-β42 level did not change significantly over time (P = 0.35), nor did the hazard ratio for total-tau and amyloid-β40 (P = 0.70 and P = 0.86, respectively).
Association of combined plasma levels of NfL and amyloid-β42 with risk of all-cause and Alzheimer’s disease dementia
We observed that 41 (30.4%) of the 135 participants in the highest (Q4) quartile group for NfL combined with the lowest (Q1) quartile group of amyloid-β42 developed all-cause dementia, 28 with Alzheimer’s disease dementia. In contrast, only two (1.9%) incident cases of all-cause dementia were found in the 107 participants in the lowest (Q1) quartile group for NfL in combination with highest (Q4) quartile group for amyloid-β42 (Supplementary Table 11). The hazard ratio for developing all-cause dementia was 9.5 (95% CI, 2.3–40.4; P = 0.0022), and for Alzheimer’s disease dementia was 15.7 (95% CI, 2.1–117.4; P < 0.0001), when the highest quartile group of NfL was combined with the lowest quartile group of amyloid-β42 and compared to the combination of lowest quartile group of NfL and highest quartile group amyloid-β42 (Fig. 3). NfL and amyloid-β42 were each independently associated with the risk of developing all-cause dementia and Alzheimer’s disease dementia as tests for interaction between NfL and amyloid-β42 were not significant (P = 0.15 and P = 0.34, respectively, when NfL and amyloid-β42 were modelled on the continuous log2 scale). However, confidence intervals of hazard ratios for each of the 16 quartile NfL–amyloid-β42 combinations were wide because of the smaller numbers in each combination.
Figure 3.
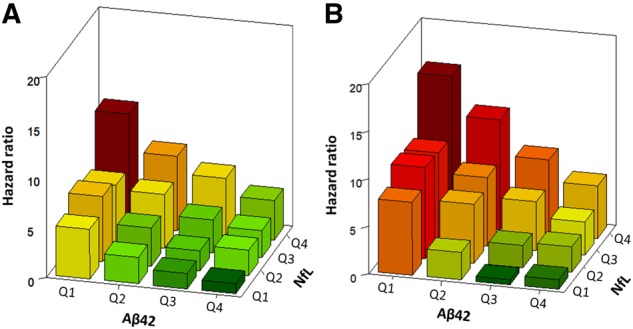
Hazard ratios for all-cause and Alzheimer’s disease dementia per quartile of plasma amyloid-β42 and NfL combined. Bars show the hazard ratios (HRs) for all-cause dementia (A) and for Alzheimer’s disease dementia (B) per quartile of plasma NfL and amyloid-β42, with the combination of the highest quartile group (Q1) of amyloid-β42 and the lowest quartile group (Q4) of NfL as reference group (HR = 1.0). Hazard ratios were obtained with the Cox proportional hazard model adjusted for age, sex, assay batch number, highest level of education, smoking status, systolic blood pressure, total and HDL cholesterol, body mass index, history of diabetes, stroke and coronary heart disease and APOE ε4 status. There were no significant interactions between amyloid-β42 and NfL for all-cause dementia (P = 0.15) and for Alzheimer’s disease dementia (P = 0.32), suggesting that the association between amyloid-β42, NfL and the risk of dementia are independent of each other.
Plasma NfL and amyloid-β42 trajectories during the development of Alzheimer’s disease dementia
As results for all-cause dementia and the largest subgroup of Alzheimer’s disease dementia were shown to be nearly identical, we focused on the latter for our study on changes in plasma concentrations of biomarkers, in particular NfL and amyloid-β42, over time. Mean trajectories and 95% CIs for plasma NfL and amyloid-β42 levels, along with their corresponding individual-level rates of change, were compared between the 374 participants who developed Alzheimer’s disease dementia and the 374 age- and sex-matched controls who remained dementia-free during the follow-up time (Fig. 4). Plots of other biomarkers are shown in Supplementary Fig. 3. Participants contributed one to three plasma measurements over time.
Figure 4.

Longitudinal changes of plasma NfL and amyloid-β42 levels and their rates of change over time before and after Alzheimer’s disease dementia diagnosis. (A) Trajectories of plasma NfL levels on a population-level are shown for Alzheimer’s disease (AD) dementia cases and dementia-free control subjects. (B) Trajectories of plasma amyloid-β42 levels are shown for Alzheimer’s disease dementia cases and dementia-free control subjects. The yellow and blue lines depict estimated means of plasma levels based on repeatedly measured biomarkers for 374 cases with Alzheimer’s disease dementia and their age- and sex-matched controls, respectively. Dotted lines represent 95% CI. Background shaded dots are coloured accordingly and denote single measurements corresponding to data where models were fit upon. (C) Rate of change of plasma NfL at the individual level for cases with Alzheimer’s disease dementia compared to dementia-free control subjects. (D) Rate of change of plasma amyloid-β42 at the individual level for cases with Alzheimer’s disease dementia compared to dementia-free control subjects. For these analyses, a log2-transformed rate of change was estimated for those participants that had two or more biomarker measurements. One datapoint reflects the rate of change for each of these individuals, anchored at the time since disease diagnosis relative to first biomarker measurement. Analyses with linear mixed effect models were adjusted for age at baseline, sex, APOE ε4 status, time between baseline measurement and disease onset and the interaction with case or control status. Rates of change derived from the population level (A and B) are different compared to those derived from the individual level analysis presented in C and D, as population level analyses are based on the entire sample, and individual level analysis required multiple measurements and were therefore only carried out among those participants that contributed two or three measurements.
Plasma NfL increased linearly for both Alzheimer’s disease dementia cases and dementia-free controls (Fig. 4A). The change of NfL per year was 3.4 times faster for participants who developed Alzheimer’s disease dementia [log2 0.04 pg/ml (95% CI, 0.02–0.06)] compared to those who did not [log2 0.01 pg/ml (95% CI, 0.00–0.02), P for interaction < 0.0001]. A significant difference in the rate of change between cases and controls was also found in the individual-level rate of change analysis among those with two or more measurements (P < 0.0001). Absolute plasma NfL levels in participants who developed Alzheimer’s disease dementia were estimated to start at 14.62 pg/ml (95% CI, 13.33–16.03) 13 years before diagnosis compared to 15.93 pg/ml (95% CI, 14.63–17.35) for dementia-free control subjects. A subsequent faster rate of change for cases led to a higher absolute value of NfL at the time of Alzheimer’s disease dementia diagnosis, 20.77 pg/ml (95% CI, 19.21–22.42) compared to 17.66 pg/ml (95% CI, 16.50–18.93) for controls at the time of index date, and 13 years thereafter 29.49 pg/ml (95% CI, 25.22–34.47) for cases compared to 19.60 pg/ml (95% CI, 17.53–21.93) for control subjects.
The mean plasma NfL concentration found over time in the Alzheimer’s disease cases began to deviate from the mean concentration found in the dementia-free controls at 9.6 years prior to Alzheimer’s disease diagnosis or index date (Fig. 4A). On an age scale, absolute plasma NfL levels in the participants who developed Alzheimer’s disease began to deviate from those that remained dementia-free from a mean age of 60.3 years on (Supplementary Fig. 4). Furthermore, the effect of time on the rate of change of plasma NfL did not reach statistical significance (Fig. 4C), indicating that the changes in NfL plasma levels do not accelerate or decelerate over time.
In those who developed Alzheimer’s disease dementia and those who did not, mean plasma amyloid-β42 levels declined and their rates of decline were not significantly different [log2 −0.01 pg/ml per year (95% CI, −0.02, 0.00) for cases and (95% CI, −0.01, 0.00) for controls, P = 0.189] (Fig. 4B). On average, plasma amyloid-β42 levels in participants who developed Alzheimer’s disease dementia declined from 11.03 (95% CI, 10.47–11.62) around 13 years before diagnosis to 10.08 (95% CI, 9.66–10.53) at the time of diagnosis. In control subjects, amyloid-β42 levels declined from 11.10 (95% CI, 10.63–11.58) to 10.61 (95% CI, 10.29–10.96) over the same period. After diagnosis or index date, the levels declined further to 9.22 (95% CI, 8.37–10.15) for cases and 10.15 (95% CI, 9.52–10.81) for control subjects. A significant (P = 0.038) difference was found at the individual-level rate of change in those who developed Alzheimer’s disease dementia and those who did not, however, the effect size was negligible (Fig. 4D). As with plasma NfL, there was no evidence of non-linearity in the changes of amyloid-β42 over time.
In parallel analyses on an age scale, we observed significantly different effects of increasing age on plasma NfL levels between cases with Alzheimer’s disease dementia and dementia-free control subjects for amyloid-β40 and amyloid-β42/amyloid-β40 ratio (P = 0.010 and P = 0.045, respectively), although absolute differences were negligible (Supplementary Fig. 4). The effects of increasing age on plasma total-tau and amyloid-β42 levels were not significant (both P > 0.5) (Supplementary Fig. 4).
Discussion
Within this large prospective population-based cohort study, we investigated whether levels of total-tau, NfL, amyloid-β40 and amyloid-β42 and the amyloid-β42/amyloid-β40 ratio measured in plasma of non-demented participants at baseline were associated with incident all-cause dementia and with Alzheimer’s disease dementia. We demonstrated that independent of age, sex, education, cardiovascular risk factors and APOE ε4 carrier status, high plasma levels of NfL and low plasma levels of amyloid-β42 are each independently associated with an increased risk of developing all-cause and Alzheimer’s disease dementia during 13.9 years of follow-up. When combined, high plasma NfL and low plasma amyloid-β42 levels showed an even stronger association with risk for all-cause as well as Alzheimer’s disease dementia. Our analysis of the rate of changes in plasma levels of biomarkers further showed a stable increase of NfL in those who developed Alzheimer’s disease dementia, where plasma levels begin to deviate from the levels found in those who remained dementia-free from ∼10 years prior to Alzheimer’s disease diagnosis. Plasma levels of total-tau did not show a consistent association with risk for all-cause or Alzheimer’s disease dementia and trajectories found in participants who developed Alzheimer’s disease were also not different from those who did not. Amyloid-β40 levels increased with age but were not associated with all-cause or Alzheimer’s disease dementia risk.
Associations of total-tau, NfL, amyloid-β40 and amyloid-β42 levels in CSF with Alzheimer’s disease have been established in several studies (Mattsson et al., 2016a; Olsson et al., 2016). In their meta-analysis, for example, Olsson and colleagues (2016) showed that high levels of tau and NfL and low levels of amyloid-β42 in CSF are strongly associated with clinically diagnosed Alzheimer’s disease. We reported previously that high amyloid-β40 in combination with low amyloid-β42 plasma levels are associated with risk for Alzheimer’s disease (van Oijen et al., 2006; Hilal et al., 2018). In the present study, we found that higher NfL and lower amyloid-β42 plasma levels are significantly associated with incident all-cause and Alzheimer’s disease dementia. We did not find a clear association between plasma total-tau levels and all-cause or Alzheimer’s disease dementia risk, which is in line with results from a study amongst memory clinic patients recently reported by Verberk et al. (2018). In contrast, Mattsson et al. (2016b) found plasma tau to be significantly associated with worse cognition, atrophy and hypometabolism during follow-up in the ADNI study. Association of plasma total-tau with incident all-cause dementia was also reported from the participants of the Framingham Heart Study (Pase et al., 2019). These contradictory results may signal that plasma total-tau is a non-specific marker (Deters et al., 2017; Jack et al., 2018; Pase et al., 2019) or a relatively late marker for the risk of developing Alzheimer’s disease (Mattsson et al., 2016b). In addition, on the individual level, CSF and plasma tau did not show a strong correlation (Mattsson et al., 2016a), possibly reflecting peripheral degradation of total-tau, rendering tau fragments undetectable in the assay used. This effect may also explain our finding that plasma total-tau is not associated with risk for all-cause and Alzheimer’s disease dementia.
Our findings on the association between plasma levels of NfL in a dementia-free population and development of all-cause and Alzheimer’s disease dementia are in line with cross-sectional studies done in ADNI (Mattsson et al., 2017; Zhou et al., 2017), where significant differences were found between Alzheimer’s disease and cognitively normal control subjects, but also between control subjects and MCI, indicating that the plasma NfL level is altered in a stage of Alzheimer’s disease where early clinical symptoms and cognitive decline become noticeable. We also found that the association between NfL and Alzheimer’s disease dementia is strongest for shorter follow-up and gradually declines as the duration of follow-up increases. The results from our longitudinal analysis that NfL plasma levels start to increase ∼10 years before Alzheimer’s disease diagnosis appear to confirm this. These results may indicate that NfL is a marker of progressive myelinated axonal damage that develops in the mid- to late stage of the preclinical phase of Alzheimer’s disease when subjects are incubating the disease (Ikram et al., 2010; Rohrer et al., 2016). Notably, individuals in this stage of disease development respond better to potential therapies for Alzheimer’s disease than those with symptomatic disease, suggesting NfL as a promising biomarker to detect both degeneration at earlier stages of Alzheimer’s disease and response to treatment (Bacioglu et al., 2016; Mattsson et al., 2019). In our study, amyloid-β42 plasma levels declined over time in the participants who developed Alzheimer’s disease dementia, although the difference was not significant when compared to participants who remained dementia-free during follow-up. This might suggest that amyloid-β42 is a marker earlier in Alzheimer’s disease incubation. In line with this conjecture, previous studies have postulated that the plasma amyloid-β levels are increased before the onset of sporadic Alzheimer’s disease (Seppala et al., 2010; Tijms et al., 2018).
Our results could serve as a basis to investigate further whether baseline plasma NfL levels yield predictive value to identify those asymptomatic individuals that are at significant risk of developing Alzheimer’s disease dementia. However, increased CSF and plasma levels of NfL are also observed in other neurodegenerative diseases than Alzheimer’s disease (Mattsson et al., 2017). In our study higher plasma NfL, but not amyloid-β42, levels were associated with risk of vascular and other dementias, although absolute numbers were small. When we combined plasma levels of NfL and amyloid-β42, we found a strong association with incident Alzheimer’s disease, although the results should be interpreted with caution because of wide confidence intervals due to the small numbers in each of the biomarker combinations. At the same time, this indicates that NfL and amyloid-β42 combined may be even more useful in identifying individuals at high risk of Alzheimer’s disease.
Finally, our results revealed that the average plasma concentration of NfL in participants who develop Alzheimer’s disease dementia rise at a constantly higher rate than in participants who remained dementia-free. On a population level this translates into NfL plasma concentrations in those who develop Alzheimer’s disease starting to deviate from those who remain dementia-free at ∼10 years before diagnosis of disease. These results may allow for further modelling dynamics of plasma marker levels in addition to those in CSF (Ower et al., 2018), establishing NfL as a stable biomarker while taking into account its plasma variability over time.
The findings of this study should be considered in light of some limitations. First, we were unable to compare results of our measurements in plasma with those in CSF of the same participants, as CSF is not systematically collected in the population-based Rotterdam Study. Second, Alzheimer’s disease has been consistently diagnosed according to the clinical NINCDS–ADRDA criteria in the Rotterdam Study, while new frameworks for (preclinical) Alzheimer’s disease diagnoses have been introduced, such as the NIA-AA research framework (Jack et al., 2018). Moreover, amyloid PET or CSF were not available in the Rotterdam Study to further confirm the pathological diagnosis of Alzheimer’s disease. However, final Alzheimer’s disease dementia diagnoses were established via a consensus panel led by a neurologist as is commonly done in large population-based studies (Chibnik et al., 2017). The main strength of our study is that it is based on data obtained from dementia-free participants in a large population-based prospective cohort study with long follow-up. In addition, simultaneous assessment of total-tau, NfL, amyloid-β40 and amyloid-β42 in plasma repeatedly obtained from single individuals facilitated a head-to-head comparison of the biomarkers to quantify their importance in the association with all-cause and Alzheimer’s disease dementia in the general population.
In conclusion, we found that higher levels of NfL and lower levels of amyloid-β42 plasma concentrations are each independently and in combination strongly associated with a higher risk of developing all-cause as well as Alzheimer’s disease dementia. Repeated measurements over time indicated that increasing NfL levels in plasma may occur as early as 10 years prior to Alzheimer’s disease dementia diagnosis. These findings indicate that NfL and amyloid-β42 measured in plasma can potentially contribute to assess the risk for dementia while the disease is still in its preclinical stage. Moreover, plasma NfL levels, although not specific, may be useful in monitoring progression of Alzheimer’s disease dementia.
Supplementary Material
Acknowledgements
The dedication and commitment by which study participants, general practitioners, and pharmacists of the Ommoord district contribute to the Rotterdam Study are gratefully acknowledged. We thank all staff at the Rotterdam Study research center, facilitating assessment of participants throughout the years, and Frank J.A. van Rooij as data manager.
Funding
This study was sponsored by Janssen Pharmaceutical Companies of Johnson & Johnson. Plasma concentrations of total-tau, NfL, amyloid-β40, and amyloid-β42 were assessed through the Janssen Prevention Center in Leiden, the Netherlands, on anonymized plasma samples without knowledge of disease status. Janssen had no role in study design and data collection. The Rotterdam Study is sponsored by the Erasmus Medical Centre and Erasmus University Rotterdam, The Netherlands Organization for Scientific Research (NWO), The Netherlands Organization for Health Research and Development (ZonMW), the Research Institute for Diseases in the Elderly (RIDE), The Netherlands Genomics Initiative, the Ministry of Education, Culture and Science, the Ministry of Health, Welfare and Sports, the European Commission (DG XII), and the Municipality of Rotterdam. None of the funding organizations or sponsors were involved in study design, in collection, analysis, and interpretation of data, in writing of the report, or in the decision to submit the article for publication.
Competing interests
None of the authors has anything to disclose in addition to the sponsorship of the study by Janssen Pharmaceuticals of Johnson and Johnson. G.J.W., P.R.W. and J.M.K. are and F.d.W., L.G., W.K. and J.G. were employed by Janssen Pharmaceuticals of Johnson & Johnson.
Glossary
- MCI =
mild cognitive impairment
- NfL =
neurofilament light chain
References
- Alzheimer’s Association. 2016 Alzheimer’s disease facts and figures. Alzheimer’s Dement 2016; 12: 459–509. [DOI] [PubMed] [Google Scholar]
- Bacioglu M, Maia LF, Preische O, Schelle J, Apel A, Kaeser SA, et al. Neurofilament light chain in blood and CSF as marker of disease progression in mouse models and in neurodegenerative diseases. Neuron 2016; 91: 56–66. [DOI] [PubMed] [Google Scholar]
- Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K.. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol 2006; 112: 389–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns A, Iliffe S.. Dementia. BMJ 2009; 338: b75. [DOI] [PubMed] [Google Scholar]
- Chang L, Shan D, Wickman J, Holdridge M, Raso C, Wilson D. SimoA human neurology 3-plex A (N3PA) immunoassay measures amyloid beta 1-42, amyloid beta 1-40 and tau in blood and CSF samples simultaneously. In: Poster Presented at the Alzheimer’s Association International Conference, Lexington; 2017.
- Chibnik LB, Wolters FJ, Backman K, Beiser A, Berr C, Bis JC, et al. Trends in the incidence of dementia: design and methods in the Alzheimer Cohorts Consortium. Eur J Epidemiol 2017; 32: 931–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993; 261: 921–3. [DOI] [PubMed] [Google Scholar]
- de Bruijn RF, Bos MJ, Portegies ML, Hofman A, Franco OH, Koudstaal PJ, et al. The potential for prevention of dementia across two decades: the prospective, population-based Rotterdam Study. BMC Med 2015; 13: 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deters KD, Risacher SL, Kim S, Nho K, West JD, Blennow K, et al. Plasma tau association with brain atrophy in mild cognitive impairment and Alzheimer’s disease. J Alzheimers Dis 2017; 58: 1245–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fandos N, Perez-Grijalba V, Pesini P, Olmos S, Bossa M, Villemagne VL, et al. Plasma amyloid beta 42/40 ratios as biomarkers for amyloid beta cerebral deposition in cognitively normal individuals. Alzheimers Dement (Amst) 2017; 8: 179–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fine JP, Gray RJ.. A proportional hazards model for the subdistribution of a competing risk. J Am Stat Assoc 1999; 94: 13. [Google Scholar]
- Gisslen M, Price RW, Andreasson U, Norgren N, Nilsson S, Hagberg L, et al. Plasma concentration of the neurofilament light protein (NFL) is a biomarker of CNS injury in HIV infection: a cross-sectional study. EBioMedicine 2016; 3: 135–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M. Parkinson’s diseases: the prion concept in relation to assembled Abeta, tau, and alpha-synuclein. Science 2015; 349: 1255555. [DOI] [PubMed] [Google Scholar]
- Goudsmit J. The incubation period of Alzheimer’s disease and the timing of tau versus amyloid misfolding and spreading within the brain. Eur J Epidemiol 2016; 31: 99–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilal S, Wolters FJ, Verbeek MM, Vanderstichele H, Ikram MK, Stoops E, et al. Plasma amyloid-beta levels, cerebral atrophy and risk of dementia: a population-based study. Alzheimer’s Res Ther 2018; 10: 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikram MA, Brusselle GGO, Murad SD, van Duijn CM, Franco OH, Goedegebure A, et al. The Rotterdam Study: 2018 update on objectives, design and main results. Eur J Epidemiol 2017; 32: 807–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikram MA, Vrooman HA, Vernooij MW, den Heijer T, Hofman A, Niessen WJ, et al. Brain tissue volumes in relation to cognitive function and risk of dementia. Neurobiol Aging 2010; 31: 378–86. [DOI] [PubMed] [Google Scholar]
- Jack CR Jr, Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, et al. NIA-AA Research Framework: toward a biological definition of Alzheimer’s disease. Alzheimer’s Dement 2018; 14: 535–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein JP, Moeschberger ML.. Survivall analysis techniques for censored and truncated data. New York: Springer-Verlag New York; 2003. [Google Scholar]
- Kuhle J, Barro C, Andreasson U, Derfuss T, Lindberg R, Sandelius A, et al. Comparison of three analytical platforms for quantification of the neurofilament light chain in blood samples: eLISA, electrochemiluminescence immunoassay and Simoa. Clin Chem Lab Med 2016; 54: 1655–61. [DOI] [PubMed] [Google Scholar]
- Licher S, Darweesh SKL, Wolters FJ, Fani L, Heshmatollah A, Mutlu U, et al. Lifetime risk of common neurological diseases in the elderly population. J Neurol Neurosurg Psychiatry 2019; 90; 148–56. [DOI] [PubMed] [Google Scholar]
- Licher S, Yilmaz P, Leening MJG, Wolters FJ, Vernooij MW, Stephan BCM, et al. External validation of four dementia prediction models for use in the general community-dwelling population: a comparative analysis from the Rotterdam Study. Eur J Epidemiol 2018; 33: 645–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattsson N, Andreasson U, Zetterberg H, Blennow K.. Alzheimer’s disease neuroimaging i. association of plasma neurofilament light with neurodegeneration in patients with Alzheimer disease. JAMA Neurol 2017; 74: 557–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattsson N, Cullen NC, Andreasson U, Zetterberg H, Blennow K.. Association between longitudinal plasma neurofilament light and neurodegeneration in patients with Alzheimer disease. JAMA Neurol 2019; 76: 791–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattsson N, Insel PS, Palmqvist S, Portelius E, Zetterberg H, Weiner M, et al. Cerebrospinal fluid tau, neurogranin, and neurofilament light in Alzheimer’s disease. EMBO Mol Med 2016a; 8: 1184–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattsson N, Zetterberg H, Janelidze S, Insel PS, Andreasson U, Stomrud E, et al. Plasma tau in Alzheimer disease. Neurology 2016b; 87: 1827–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mielke MM, Hagen CE, Xu J, Chai X, Vemuri P, Lowe VJ, et al. Plasma phospho-tau181 increases with Alzheimer’s disease clinical severity and is associated with tau- and amyloid-positron emission tomography. Alzheimer’s Dement 2018; 14: 989–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura A, Kaneko N, Villemagne VL, Kato T, Doecke J, Dore V, et al. High performance plasma amyloid-beta biomarkers for Alzheimer’s disease. Nature 2018; 554: 249–54. [DOI] [PubMed] [Google Scholar]
- Nisbet RM, Polanco JC, Ittner LM, Gotz J.. Tau aggregation and its interplay with amyloid-beta. Acta Neuropathol 2015; 129: 207–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsson B, Lautner R, Andreasson U, Ohrfelt A, Portelius E, Bjerke M, et al. CSF and blood biomarkers for the diagnosis of Alzheimer’s disease: a systematic review and meta-analysis. Lancet Neurol 2016; 15: 673–84. [DOI] [PubMed] [Google Scholar]
- Olsson B, Portelius E, Cullen NC, Sandelius A, Zetterberg H, Andreasson U, et al. Association of cerebrospinal fluid neurofilament light protein levels with cognition in patients with dementia, motor neuron disease, and movement disorders. JAMA Neurol 2019; 76: 318–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ower AK, Hadjichrysanthou C, Gras L, Goudsmit J, Anderson RM, de Wolf F, et al. Temporal association patterns and dynamics of amyloid-beta and tau in Alzheimer’s disease. Eur J Epidemiol 2018; 33: 657–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pase MP, Beiser AS, Himali JJ, Satizabal CL, Aparicio HJ, DeCarli C, et al. Assessment of plasma total tau level as a predictive biomarker for dementia and related endophenotypes. JAMA Neurol 2019; 76: 598–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preische O, Schultz SA, Apel A, Kuhle J, Kaeser SA, Barro C, et al. Serum neurofilament dynamics predicts neurodegeneration and clinical progression in presymptomatic Alzheimer’s disease. Nat Med 2019; 25: 277–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rissin DM, Kan CW, Campbell TG, Howes SC, Fournier DR, Song L, et al. Single-molecule enzyme-linked immunosorbent assay detects serum proteins at subfemtomolar concentrations. Nat Biotechnol 2010; 28: 595–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrer JD, Woollacott IO, Dick KM, Brotherhood E, Gordon E, Fellows A, et al. Serum neurofilament light chain protein is a measure of disease intensity in frontotemporal dementia. Neurology 2016; 87: 1329–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seppala TT, Herukka SK, Hanninen T, Tervo S, Hallikainen M, Soininen H, et al. Plasma Abeta42 and Abeta40 as markers of cognitive change in follow-up: a prospective, longitudinal, population-based cohort study. J Neurol Neurosurg Psychiatry 2010; 81: 1123–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano-Pozo A, Frosch MP, Masliah E, Hyman BT.. Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med 2011; 1: a006189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiebaut AC, Benichou J.. Choice of time-scale in Cox’s model analysis of epidemiologic cohort data: a simulation study. Stat Med 2004; 23: 3803–20. [DOI] [PubMed] [Google Scholar]
- Tijms BM, Vermunt L, Zwan MD, van Harten AC, van der Flier WM, Teunissen CE, et al. Pre-amyloid stage of Alzheimer’s disease in cognitively normal individuals. Ann Clin Transl Neurol 2018; 5: 1037–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Oijen M, Hofman A, Soares HD, Koudstaal PJ, Breteler M.. Plasma Aβ1–40 and Aβ1–42 and the risk of dementia: a prospective case-cohort study. Lancet Neurol 2006; 5: 655–60. [DOI] [PubMed] [Google Scholar]
- Verberk IMW, Slot RE, Verfaillie SCJ, Heijst H, Prins ND, Bnm V. B, et al. Plasma amyloid as prescreener for the earliest alzheimer pathological changes. Ann Neurol 2018: 84: 648–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verlinden VJA, van der Geest JN, de Bruijn R, Hofman A, Koudstaal PJ, Ikram MA.. Trajectories of decline in cognition and daily functioning in preclinical dementia. Alzheimer’s Dement 2016; 12: 144–53. [DOI] [PubMed] [Google Scholar]
- Villemagne VL, Burnham S, Bourgeat P, Brown B, Ellis KA, Salvado O, et al. Amyloid beta deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: a prospective cohort study. Lancet Neurol 2013; 12: 357–67. [DOI] [PubMed] [Google Scholar]
- Zetterberg H, Skillback T, Mattsson N, Trojanowski JQ, Portelius E, Shaw LM, et al. Association of cerebrospinal fluid neurofilament light concentration with Alzheimer disease progression. JAMA Neurol 2016; 73: 60–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Zhang J, Ye F, Xu G, Su H, Su Y, et al. Plasma neurofilament light chain levels in Alzheimer’s disease. Neurosci Lett 2017; 650: 60–4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author, subject to local and European regulations.