

Abstract
Herein, the synthesis and use of [11C]carbonyl difluoride for labeling heterocycles in [11C]carbonyl groups in high molar activity is described. A very mild single-pass gas-phase conversion of [11C]carbon monoxide into [11C]carbonyl difluoride over silver(II) fluoride provides easy access to this new synthon in robust quantitative yield for labeling a broad range of cyclic substrates e.g., imidazolidin-2-ones, thiazolidin-2-ones, and oxazolidin-2-ones. Labeling reactions may utilize close to stoichiometric precursor quantities and short reaction times at room temperature in a wide range of solvents while also showing high water tolerability. The overall radiosynthesis protocol is both simple and reproducible. The required apparatus can be constructed from widely available parts and is therefore well suited to be automated for PET radiotracer production. We foresee that this straightforward method will gain wide acceptance for PET radiotracer syntheses across the radiochemistry community.
Keywords: carbonyl difluoride, heterocycles, Isotopic labeling, PET radiochemistry, carbon-11
Graphical Abstract

Herein the synthesis and use of [11C]carbonyl difluoride for labeling heterocycles is described. Labeling reactions with [11C]COF2 are very mild and proceed in high yields in short reaction times at room temperature in a wide range of solvents. The radiosynthesis protocol is simple and easily implemented.
Positron emission tomography (PET) is a very important tool in drug development and for medical diagnosis.[1] The two main radionuclides used in PET imaging are the cyclotron-produced positron-emitters carbon-11 (t1/2 = 20.4 min) and fluorine-18 (t1/2 = 110 min).[2] Carbon-11 has an advantage over fluorine-18 in that naturally occurring carbon is present in virtually all pharmaceuticals and theoretically can be replaced with carbon-11 to construct a required radioactive PET probe.
PET radiotracer synthesis with short-lived carbon-11 is challenging. Ideally, radiosyntheses should be fast, utilize nontoxic reagents, and use a low precursor quantity. Moreover, a new radiosynthesis methodology needs to be both reproducible and operationally simple if it is to be widely applied for regular PET radiotracer production.
Although 11C-methylation reactions are the most widely used for introducing carbon-11 into organic molecules, not all molecules have a methyl group amenable to labeling. Radiocarbonylation reactions are an important complement to radiomethylation reactions because they considerably broaden the range of chemotypes that can be labeled as prospective PET radiotracers. It is therefore important to develop reliable and high yielding radiocarbonylation methods.[3–5]
[11C]Phosgene ([11C]COCl2) fulfills a requirement for high reactivity in a labeling synthon for producing [carbonyl-11C]compounds (Scheme 1A). However, [11C]COCl2 synthesis requires hazardous chlorine gas, as well as complicated equipment that may need extensive maintenance and replacement of key components after each use.[6–8] Due to these shortcomings, [11C]COCl2 has only been utilized in a few laboratories worldwide.[4, 9, 10] Significant efforts have therefore been devoted towards developing 11C-carbonylation methods based on alternative reagents.[3, 4]
Scheme 1.
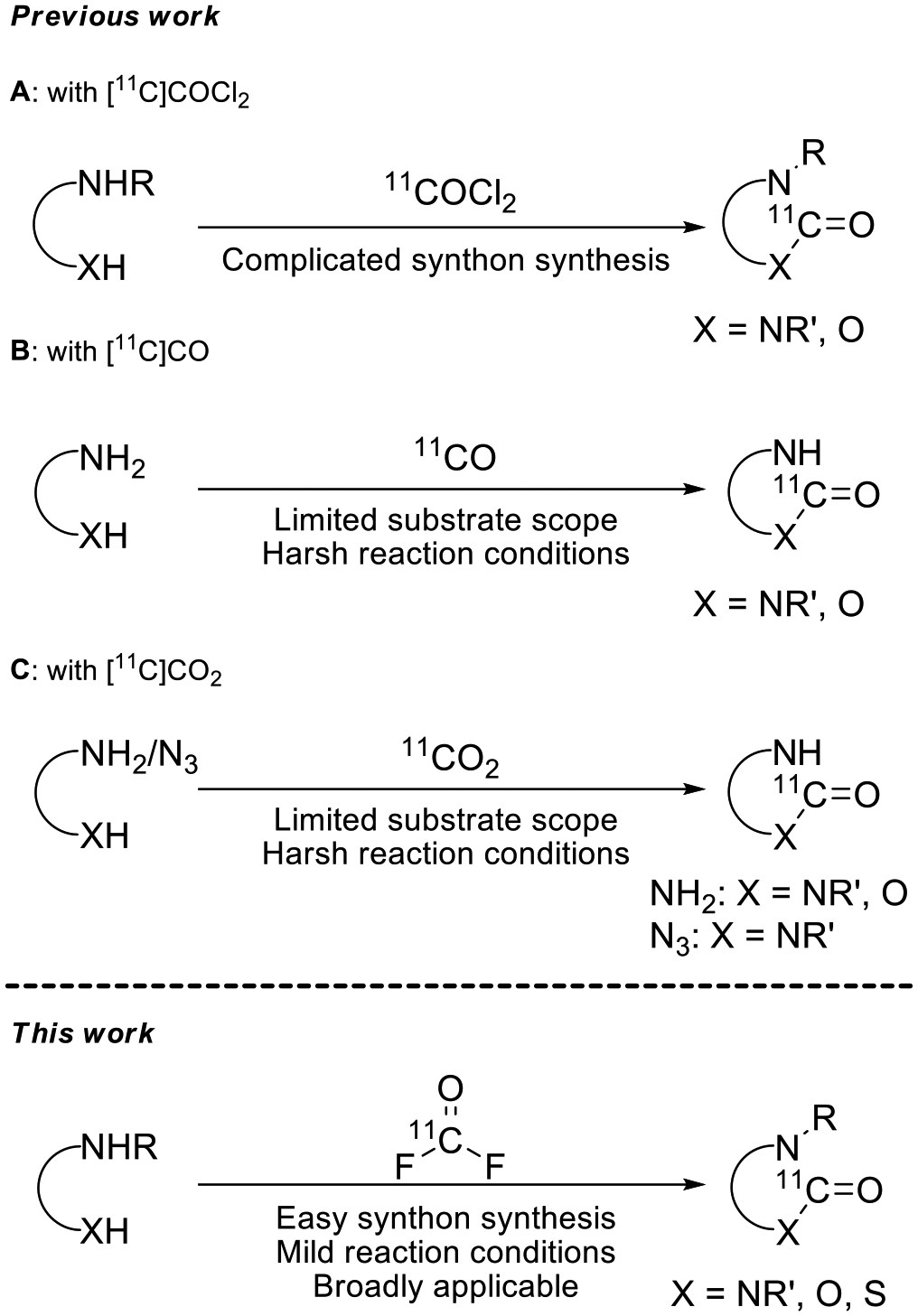
Synthesis of [carbonyl-11C]heterocycles.
Carbonylation reactions with [11C]carbon monoxide have utilized transition metal mediated chemistry, mainly with palladium[11] or selenium activation[12] for the labeling of cyclic ureas and related chemotypes (Scheme 1B).[13] Although [11C]carbon monoxide is nowadays readily accessible from cyclotron-produced [11C]carbon dioxide by various methods,[13] both metal-mediated labelimg methods suffer from the generally low solubility of [11C]CO in organic solvents. Special techniques are needed to utilize [11C]CO efficiently, such as recirculation[14], the use of a micro-autoclave[15–17], or a chemical adjunct for the temporary trapping and release of [11C]CO[11, 18] in solution.
From a perspective of seeking short synthesis time, direct use of cyclotron-produced [11C]CO2 as a labeling synthon is attractive. Some newer methods for labeling cyclic ureas are now based on [11C]CO2 fixation (Scheme 1C).[19, 20] These methods are nonetheless multistep with sequential reagent additions that require cooling and/or heating. This is operationally cumbersome and can be challenging to automate as would be necessary for radiation safety in routine PET radiotracer production. Side reactions are another drawback arising from harsh reaction conditions. Moreover, to maintain high molar activity, stringent precautions are needed to exclude ingress of atmospheric CO2. For example, equipment, reagents, and solutions need to be purged of any residual atmospheric CO2. The substrate scope amenable to this type of 11C-carbonylation chemistry is also quite limited.
Herein we describe the synthesis of [11C]carbonyl difluoride ([11C]COF2) by fluorination of [11C]CO with AgF2 (Scheme 2). Although this reaction has been conducted on a macroscale with non-radioactive carbon monoxide,[21] we needed the fluorination reaction to occur on a no-carrier-added (NCA) scale, typically using [11C]CO with 10–100 nmol of carrier CO. Reactions on such low scale can differ in outcome from the macroscale reaction.[22]
Scheme 2.
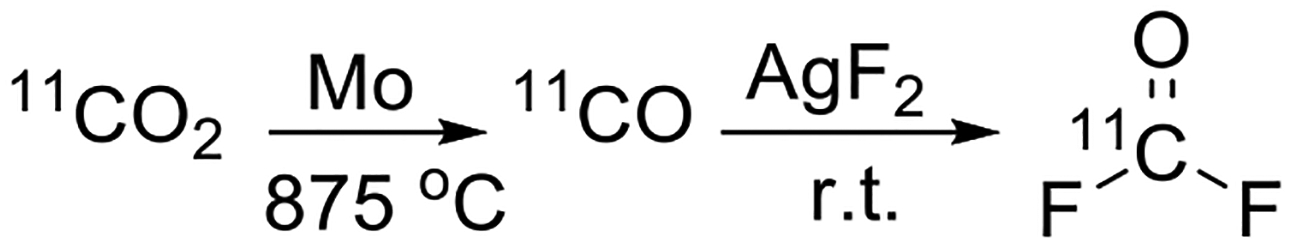
Synthesis of [11C]carbonyl difluoride.
In practice, we synthesised the required [11C]CO from [11C]CO2 through a well-established method operating in our laboratory, namely reduction of [11C]CO2 over molybdenum at 875 °C.[23] The generated [11C]CO was subsequently carried in helium through a stainless steel column of AgF2 at room temperature. As an initial test for the formation of [11C]COF2, the effluent was bubbled into a solution of 1,2-diaminobenzene (1) in acetonitrile with a vent connected to sodium hydroxide on silica to retain any escaping [11C]COF2 or [11C]CO2, followed by a balloon to contain any unreacted species. The cyclic urea ([11C]2) was formed in high decay-corrected radiochemical yields (RCYs; 87 ± 8%), thereby indicating successful syntheses of [11C]COF2. Control reactions omitting the AgF2 column showed no formation of [11C]2 (ESI).
We set out to investigate how much AgF2 was required to achieve optimal conversion of [11C]CO into [11C]COF2 using a helium flow of 5 mL/min. We found that 0.4 g of AgF2 sufficed for almost quantitative (99.7 ± 0.1%) conversion of the incoming [11C]CO into [11C]COF2 (Table 1). A negligible proportion (< 0.1%) of radioactivity was retained on the AgF2 column. No radioactivity passed through the sodium hydroxide solution showing the excellent chemoselectivity of the conversion with no over-oxidation into perfluorinated species. The AgF2 column gave high and reproducible conversion of [11C]CO into [11C]COF2 over ten successive runs.
Table 1.
RCY (mean ± SD; n = 3) of [11C]COF2 on passing [11C]CO in a helium stream over different quantities of AgF2.
 |
|||
|---|---|---|---|
| Entry | AgF2 (g) | Yield (%) | Activity retained on AgF2 (%) |
| 1 | 0.1 | 72 ± 11 | 0.1 ± 0.0 |
| 2 | 0.4 | 99.7 ± 0.1 | 0.1 ± 0.1 |
| 3 | 2.1 | 100 ± 0 | 0.1 ± 0.1 |
Following these highly encouraging results, we set out to investigate the dependence of yield of [11C]2 from [11C]COF2 on quantity of precursor (1). Because this reaction was conducted no-carrier-added (NCA), 4 mg of precursor (37 μmol) would have been in large excess over the amount of carrier COF2 (58 ± 39 nmol; n = 25) that was present in our system in early experiments. We observed that reductions in the amount of precursor from 4 mg to 1 mg and then to 0.25 mg did not change the yield of [11C]2 appreciably; high yields were maintained (i.e., 88 ± 5% and 86 ± 6%, respectively; Figure 1). Use of 0.25 mg of 1 is already a major reduction in the amount of precursor used by Horkka et al.[20] and by Del Vecchio et al.[19] in their methods for producing [11C]2 and is therefore already a considerable improvement. By allowing the use of very low precursor quantities, precursor synthesis demands and separation challenges are reduced which can be important for difficult to access or expensive precursors for PET radiotracers that may need to be produced on a regular basis. We found that acceptable yields of [11C]2 were obtained using even lower precursor quantities. Use of 0.1 mg (930 nmol) of 1 showed a negligibly reduced yield. A ten-fold reduction in precursor quantity to 0.01 mg (93 nmol) gave a clearly lower yield with increased variability (Figure 1). At 93 nmol of 1, the stoichiometry of precursor to [11C]COF2 was close to 1: 1. To the best of our knowledge, such a low reaction stoichiometry and low precursor amount has not been used previously for the successful synthesis of a 11C-labeled compound. At and close to 1: 1 stoichiometry, second order reaction kinetics can be expected instead of pseudo first order reaction kinetics, thus slowing down the radiolabeling reaction.[24] However, regularly performing this reaction with such a low amount of precursor would likely give fluctuating yields because amounts of carrier [11C]CO cannot be tightly controlled from run to run. Therefore, to avoid lower yields for less reactive substrates we decided to set the amount precursor to be used in later experiments to 0.25 mg.
Figure 1.

Average radiochemical yield (mean ± SD) for the conversion of [11C]COF2 into [11C]2. The synthesis time was 5.5 min from release of [11C]CO. Reactions were performed in triplicate except those with 4 mg of 1 which was performed in duplicate. (Error bar is within symbol size where not shown). Note the x-axis is on logarithmic scale.
HPLC purification of [11C]2 from a reaction mixture demonstrated that the yield measured from HPLC analysis corresponded well with isolated yield. Generally, the reaction in acetonitrile proved very reproducible with low RCY deviations in between runs, as would be vital to producing PET radiotracers in a reliable and reproducible manner. In PET radiotracer production, it is advantageous to use water-miscible solvents in order to enable straightforward radiotracer purification with reversed-phase HPLC. We therefore investigated the reactivity of [11C]COF2 towards 1 in other common polar aprotic solvents. We found that the reaction was highly compatible with THF, DMF, and DMSO (Table 2 entries 2–4). We also investigated ethanol as an example of a polar protic solvent. To our delight, [11C]2 was obtained in acceptable yields of 46 ± 7% (Table 2 entry 5). The lower yields were most likely due to the formation of an ethyl carbamate side-product, as indicated by LC-MS analysis of the HPLC purified impurity.
Table 2.
Performance of [11C]COF2 in different solvents.
 | ||
|---|---|---|
| Entry | Solvent | RCY (%) |
| 1 | MeCN | 87 ± 6 |
| 2 | THF | 79 ± 7 |
| 3 | DMF | 56 ± 14 |
| 4 | DMSO | 72 ± 5 |
| 5 | EtOH[a] | 46 ± 17 |
| 6 | H2O/MeCN (1: 99) | 84 ± 3 |
| 7 | H2O/MeCN (10: 90) | 60 ± 7 |
| 8 | H2O | 27 ± 3 |
The synthesis time was 5.5 min from release of [11C]CO. Quantity of 1 was 0.25 mg (2.3 μmol). Entry 1 is reproduced from Figure 1.
RCY for the ethyl carbamate side product was 47 ± 5%. n = 3, except entry 2 (n = 2).
Inspired by the high reproducibility of this reaction in various solvents, we investigated water tolerability and found that 1% water in acetonitrile gave no reduction in yield (84 ± 3%, Table 2, entry 6) and that the yield was only slightly decreased in 10% water in acetonitrile (60 ± 7%, Table 2, entry 7). Reaction also occurred in purely aqueous media but gave much reduced yields of [11C]2 (27 ± 3%, Table 2, entry 8). The tolerability of the reaction towards low water concentration demonstrates that no special precautions are needed to exclude atmospheric moisture from reactions of [11C]COF2.
Having established the optimal labeling conditions for the reaction of [11C]COF2 with 1, we then aimed to investigate the reactivity of [11C]COF2 towards a range of substrates. Although the chemistry of phosgene has been well studied, the chemistry of carbonyl difluoride has been little explored. Some evidence suggests carbonyl difluoride and phosgene have similar chemistries.[25, 26] We therefore explored the use of [11C]COF2 in some types of reaction that have been conducted previously with [11C]COCl2, among some others that have been performed with [11C]CO or [11C]CO2 (Scheme 3).
Scheme 3.

Substrate scope for labeling with [11C]COF2 and examples of labeled PET radiotracers. RCYs are isolated decay-corrected radiochemical yields based on recovered radioactivity from HPLC purification (n = 3). Synthesis time was 5.5 min from release of [11C]CO. [a] Reaction was quenched with water and kept for an additional 5 min; [b] n = 5; [c] 5 mg precursor in water/MeCN (1: 9); [d] 5 mg precursor and KOH (10 mg, 0.18 mmol) in MeCN (3 mL); [e] Quenched with water before helium sparging (10 min) for removing [11C]CO2.
We obtained carbamate [11C]3 in 73 ± 8% yield from o-aminophenol, showing that a change of one heteroatom group from amino to hydroxy was not detrimental (Scheme 3). Thiols are challenging precursors for use in palladium-mediated 11C-carbonylation reactions, presumably because of catalyst poisoning. Gratifyingly, [11C]COF2 was found to react avidly with o-aminothiophenol to give the corresponding cyclized product [11C]4 in 62 ± 4% yield. Furthermore, N,N’-dimethyl o-diaminobenzene gave the cyclization product [11C]5 in 61 ± 5% yield. This labeled compound had been inaccessible through the azide based [11C]CO2 fixation method of Del Vecchio et al.[19] and only accessible in low yields using a dehydration strategy with phosphines and diazacarboxylates by the method of Horkka et al.[20] Ureas [11C]6 and [11C]7, bearing electron-withdrawing substituents, were labeled in 49 ± 12% and 82 ± 4% yields, respectively, thereby further demonstrating the strong reactivity of [11C]COF2. More nucleophilic benzylamines reacted cleanly with [11C]COF2 producing the corresponding ureas [11C]8, [11C]9, and [11C]10 in excellent RCYs (Scheme 3). Compound [11C]9 was also obtained in good yields and in high purity in the cGMP compatible solvents ethanol and water (Scheme 3). Aliphatic amino alcohols proved excellent substrates as they reacted with [11C]COF2 to provide the carbamates [11C]11 and [11C]12 in near quantitative yields, thereby also affirming the high purity of the [11C]COF2 reagent. Sterically hindered electron-deficient urea [11C]13 was furnished in good yields demonstrating compatibility with pyridinyl moieties, a structural feature in many PET radiotracers. The cyclic urea [11C]14 was obtained in 82 ± 17% yield. Full preparative HPLC purification of [11C]14 provided the isolated compound in comparable yield (77%). Compound [11C]14 represents a structural motif found in many drug-like compounds (e.g., Y5 receptor antagonists,[27] μ-opioid antagonists,[28] and the antipsychotics, pimozide[10] and benperidol[29]).
We selected three PET radiotracers to test the utility of [11C]COF2 for labeling more complex and useful structures. The well-known β-adrenoceptor radioligand, [11C](S)-CGP 12177[30, 31], was obtained in 72 ± 15% yield with very little side product formation (ESI), despite the presence of a secondary amino and secondary alcohol group. Carbon-11 produced with a cyclotron by the 14N(p,α)11C reaction with a proton beam (16 MeV, 45 μA) for 20 minutes gave 13.2 ± 2.6 GBq of [11C](S)-CGP 12177 ([11C]15) with a high molar activity of 218 ± 58 GBq/μmol at end of synthesis (EOS) (n = 2). Biologically relevant [11C]5,6-benzouracil ([11C]16) was synthesised in a moderate yield of 30 ± 13%, thereby expanding the substrate scope further to include aminobenzamides. Earlier, this product had been synthesised from [11C]COCl2 only[32] and had been inaccessible through selenium-mediated carbonylation with [11C]CO2.[12] [11C]DMO ([11C]17), a PET radiotracer useful for determining tissue pH in vivo, was synthesised from a hydroxy propionamide precursor. This new method used only 10% of the earlier reported precursor amount[33] and gave [11C]DMO in 41 ± 4% yield. These results further demonstrate the versatility of [11C]COF2 as a new and effective [11C]carbonyl group transfer agent.
In summary, we have successfully synthesized [11C]COF2, a new [11C]carbonyl group transfer agent that provides a new simple means for the 11C-labeling of heterocycles at a carbonyl group between two heteroatoms belonging to amines, alcohols, thiols or amides. The synthesis of [11C]COF2 proceeds quantitatively at room temperature in a single pass over a low quantity of AgF2. Subsequent labeling reactions proceed under very mild conditions using very low precursor quantity at room temperature under ambient pressure to produce labeled products in high yields. Labeling reactions are compatible with a range of solvents, including water, and do not require any additives, thus reaction mixtures that are easily purified with reversed phase HPLC. No special precautions are needed to exclude atmospheric moisture from the reaction mixtures as reactions proceed well in partially aqueous media. We anticipate that this operationally simple 11C-labeling methodology will be implemented for the future synthesis of PET radiotracers for clinical use.
Experimental Section
Supplementary Material
Acknowledgements.
This work was supported by the Intramural Research Program of the National Institutes of Health (NIMH; ZIA MH002793). We thank the NIH PET Department for [11C]CO2 production.
References
- [1].Matthews PM, Rabiner EA, Passchier J, Gunn RN, Br. J. Clin. Pharmacol 2012, 73, 175–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Pike VW, Curr. Med. Chem 2016, 23, 1818–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Miller PW, Long NJ, Vilar R, Gee AD, Angew. Chem. Int. Ed 2008, 47, 8998–9033. [DOI] [PubMed] [Google Scholar]
- [4].Rotstein BH, Liang SH, Placzek MS, Hooker JM, Gee AD, Dolle F, Wilson AA, Vasdev N, Chem. Soc. Rev 2016, 45, 4708–4726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Lu S, Simeon FG, Telu S, Cai L, Pike VW, Adv. Het. Chem.131 In press, DOI: 10.1016/bs.aihch.2019.1011.1005. [DOI] [Google Scholar]
- [6].Landais P, Crouzel C, Appl. Radiat. Isot 1987, 38, 297–300. [Google Scholar]
- [7].Bramoulle Y, Roeda D, Dolle F, Tetrahedron Lett 2010, 51, 313–316. [Google Scholar]
- [8].Ogawa M, Takada Y, Suzuki H, Nemoto K, Fukumura T, Nucl. Med. Biol 2010, 37, 73–76. [DOI] [PubMed] [Google Scholar]
- [9].Wilson AA, Garcia A, Houle S, Sadovski O, Vasdev N, Chem. Eur. J 2011, 17, 259–264. [DOI] [PubMed] [Google Scholar]
- [10].Roeda D, Dolle F, Curr. Top. Med. Chem 2010, 10, 1680–1700. [DOI] [PubMed] [Google Scholar]
- [11].Kealey S, Husbands SM, Bennacef I, Gee AD, Passchier J, J. Label. Compd. Radiopharm 2014, 57, 202–208. [DOI] [PubMed] [Google Scholar]
- [12].Kihlberg T, Karimi F, Langstrom B, J. Org. Chem 2002, 67, 3687–3692. [DOI] [PubMed] [Google Scholar]
- [13].Taddei C, Pike VW, EJNMMI Radiopharm. Chem 2019, 4, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Lidstrom P, Kihlberg T, Langstrom B, J. Chem. Soc., Perkin Trans 1 1997, 2701–2706. [Google Scholar]
- [15].Kihlberg T, Langstrom B, J. Org. Chem 1999, 64, 9201–9205. [Google Scholar]
- [16].Hostetler ED, Burns HD, Nucl. Med. Biol 2002, 29, 845–848. [DOI] [PubMed] [Google Scholar]
- [17].Itsenko O, Kihlberg T, Langstrom B, Nat. Protoc 2006, 1, 798–802. [DOI] [PubMed] [Google Scholar]
- [18].Kealey S, Miller PW, Long NJ, Plisson C, Martarello L, Gee AD, Chem. Commun 2009, 3696–3698. [DOI] [PubMed] [Google Scholar]
- [19].Del Vecchio A, Caille F, Chevalier A, Loreau O, Horkka K, Halldin C, Schou M, Camus N, Kessler P, Kuhnast B, Taran F, Audisio D, Angew. Chem. Int. Ed 2018, 57, 9744–9748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Horkka K, Dahl K, Bergare J, Elmore CS, Halldin C, Schou M, Chemistryselect 2019, 4, 1846–1849. [Google Scholar]
- [21].Ruff OV, Miltschitzky G, Z. Anorg. Allg. Chem 1934, 221, 154–158. [Google Scholar]
- [22].Jakobsson JE, Riss PJ, RSC Advances 2018, 8, 21288–21291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Zeisler SK, Nader M, Theobald A, Oberdorfer F, Appl. Radiat. Isot 1997, 48, 1091–1095. [Google Scholar]
- [24].Holland JP, Chem. Eur. J 2018, 24, 16472–16483. [DOI] [PubMed] [Google Scholar]
- [25].Emeleus HJ, Wood JF, J. Chem. Soc 1948, 2183–2188. [Google Scholar]
- [26].Petzold D, Nitschke P, Brandl F, Scheidler V, Dick B, Gschwind RM, Konig B, Chem. Eur. J 2019, 25, 361–366. [DOI] [PubMed] [Google Scholar]
- [27].Fotsch C, Sonnenberg JD, Chen N, Hale C, Karbon W, Norman MH, J. Med. Chem 2001, 44, 2344–2356. [DOI] [PubMed] [Google Scholar]
- [28].Kennedy NM, Schmid CL, Ross NC, Lovell KM, Yue Z, Chen YT, Cameron MD, Bohn LM, Bannister TD, J. Med. Chem 2018, 61, 8895–8907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Suehiro M, Dannals RF, Scheffel U, Stathis M, Wilson AA, Ravert HT, Villemagne VL, Sanchez-Roa PM, Wagner HN Jr., J. Nucl. Med 1990, 31, 2015–2021. [PubMed] [Google Scholar]
- [30].Aigbirhio F, Pike VW, Francotte E, Waters SL, Banfield B, Jaeggi KA, Drake A, Tetrahedron-Asymmetry 1992, 3, 539–554. [Google Scholar]
- [31].Brady F, Luthra SK, Tochon-Danguy H, Landais PG, Waters SL, Law M, Jaeggi KA, Pike VW, J. Label. Compd. Radiopharm 1991, 30, 251–253. [Google Scholar]
- [32].Brinkman GA, Hasslisewska I, Veenboer JT, Lindner L, Appl. Radiat. Isot 1978, 29, 701–702. [Google Scholar]
- [33].Diksic M, Int. J. Appl. Radiat. Isot 1984, 35, 1035–1037. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.