

Abstract
Rhododendron lapponicum L. is a familiar ornamental plant worldwide with important ornamental and economic value. However, a full-length R. lapponicum transcriptome is still lacking. In the present study, we used the Pacific Biosciences single-molecule real-time sequencing technology to generate the R. lapponicum transcriptome. A total of 346,270 full-length non-chimeric reads were generated, from which we obtained 75,002 high-quality full-length transcripts. We identified 55,255 complete open reading frames, 7,140 alternative splicing events and 2,011 long non-coding RNAs. In gene annotation analyses, 71,155, 33,653, 30,359 and 31,749 transcripts were assigned to the Nr, GO, COG and KEGG databases, respectively. Additionally, 3,150 transcription factors were detected. KEGG pathway analysis showed that 96 transcripts were identified coding for the enzymes associated with anthocyanin synthesis. Furthermore, we identified 64,327 simple sequence repeats from 45,319 sequences, and 150 pairs of primers were randomly selected to develop SSR markers. This study provides a large number of full-length transcripts, which will facilitate the further study of the genetics of R. lapponicum.
Subject terms: RNA sequencing, Transcriptomics
Introduction
Rhododendron is the largest genus in Ericaceae, with more than 1000 species of woody plants, either evergreen or deciduous1. Rhododendron species are widely cultivated around the world ranging from tropical to polar climates, and serve as a potential genetic resource for the development of new plant cultivars adapted to different environmental conditions2. Rhododendron is a familiar ornamental plant worldwide, and is decorative shrub with beautiful flowers that are widespread around the world. There are a remarkably broad range of rhododendron flower colours, including red, white, yellow, and green and so on. Rhododendron lapponicum L., a Rhododendron species found in subarctic regions around the world, is a common ornamental plant worldwide3. Recent advances in sequencing technology have facilitated genome and transcriptome studies in many species. However, genome and transcriptome sequencing in R. lapponicum has lagged behind that in other species, and information about the sequence and structure of its genes is limited. Therefore, the generation of a transcriptome data may establish a very important molecular biology basis for the research of R. lapponicum.
The transcriptome reflects the number and type of genes expressed in different cell types and reveals underlying metabolic pathways and genetic mechanisms4. Transcriptome sequencing is an efficient and feasible approach for generating a large amount of sequence data, and a large number of cDNA sequences provides a useful resource for genomic and genetic research5–9. Thus, third-generation long-read transcriptome sequencing platforms such as the Pacific Biosciences (PacBio), Nanopore and Moleculo platforms were developed. Recently, PacBio single-molecule real-time (SMRT) sequencing technology has served as a better alternative for obtaining full-length transcripts10,11. The major advantage of SMRT is the generation of full-length transcript without the need for fragmentation or post-sequencing assembly12. SMRT sequencing technology has been used in the transcriptome analysis of both model and nonmodel species13–15. Moreover, transcriptome sequencing is a simple and effective strategy for the development of large-scale SSRs at low cost. In recent years, the development of SSR markers by RNA sequencing and it successfully used in genetic improvement has been reported in many nonmodel plants16–18.
In this study, we constructed a full-length cDNA library of R. lapponicum and analysed it using SMRT sequencing technology. More than 15 Gb of sequencing data was produced, and 75,002 high-quality transcripts were obtained. Based on the obtained transcripts, alternative splicing (AS) analysis, long non-coding RNAs (lncRNA) prediction, transcription factor (TF) classification, open reading frame (ORF) prediction, transcript functional annotation and SSR analysis were performed. This is the first systematic report to characterize the full-length transcriptome of R. lapponicum via SMRT sequencing. The transcriptome data generated from this study provide valuable resources for genome annotation that may establish an important basis for future molecular biology research on Rhododendron species.
Materials and Methods
Plant materials
Rhododendron lapponicum L. (Fuli Jinling) was grown at Jiangsu Academy of Agricultural Sciences (Nanjing, China). Samples of the roots, stems, leaves and flowers from three individual plants were collected and frozen in liquid nitrogen, then stored at −70 °C for RNA extraction.
RNA extraction
Total RNA was extracted using TRIzol LS reagent (Invitrogen, USA) following the manufacturer’s instructions. RNA degradation and contamination were monitored using 1% agarose gels. The purity, concentration and absorption peak of RNA were measured using a NanoDrop 2000 spectrophotometer (Thermo Scientific, USA). RNA quality was determined with the RNA Nano 6000 Assay Kit of the Agilent Bioanalyzer 2100 system (Agilent, USA). The total RNA samples from four tissues were mixed together for the following experiments.
Library construction and SMRT sequencing
To construct a full-length transcript sequencing library, 5 μg of mixed total RNA was reverse transcribed into cDNA using the Clontech SMARTer cDNA Synthesis Kit (Takara Clontech Biotech, Dalian, China) following the manufacturer’s protocols. Size fractionation and selection (1–2 kb, 2–3 kb, and 3–6 kb) were performed using the BluePippin Size Selection System (Sage Science, USA). Three SMRT sequencing libraries containing fragments of 1–2, 2–3, and 3–6 kb in length were constructed using the Pacific Biosciences DNA Template Prep Kit 2.0. Finally, 1 (1–2 kb), 1 (2–3 kb) and 1 (3–6 kb) SMRT cells were sequenced on the Pacific Bioscience RS II platform.
Quality filtering and error correction
Raw reads were processed by removing polymerase reads (<50 bp in length). The obtained clean reads were processed into error-corrected ROIs with the following parameters: full passes ≥0 and predicted consensus accuracy >0.75. By identifying the 5′ and 3′ adapters and the poly (A) tail, full-length and non-full-length reads were determined from the ROIs. A full-length read containing both the 5′ and 3′ primer sequences and a poly (A) tail was considered to be a full-length transcript. ROIs with all three elements that did not contain any additional copies of the adapter sequence within the DNA fragment were referred to as full-length non-chimeric (FLNC) reads. Corrected FLNC reads were clustered into transcripts using the ICE algorithm in the PacBio SMRT Analysis (v2.3.0) software. Full-length transcripts with a post-correction accuracy >99% were used for further analysis.
Prediction of ORFs, lncRNAs, TFs and AS events
To predict ORFs in transcripts, the TransDecoder v2.0.1 tool was used to find potential coding sequences. Based on the obtained transcripts with redundancy removed, we predicted AS events with the software AStalavista19. TFs were predicted from the putative protein sequences using the Plant Transcription Factor Database v4.0 tool20. We identified unique transcripts without protein-coding potential as candidate lncRNAs with four analysis tools: the coding-non-coding index (CNCI)21, the coding potential assessment tool (CPAT)22, the coding potential calculator (CPC)23, and Pfam protein structure domain analysis24.
Functional annotation
All transcript sequences were analysed for homology via searches against the non-redundant nucleotide database (Nr)25, Swiss-Prot protein26, protein family (pfam)27, evolutionary genealogy of genes: non-supervised orthologous groups (eggNOG)28, clusters of orthologous groups of proteins (COG)29, eukaryotic ortholog groups (KOG)30, gene ontology (GO)31, kyoto encyclopedia of genes and genomes (KEGG)32 databases with BLAST alignment (E-value ≤ 10−5).
qRT-PCR analysis
Samples of flowers at four flower developmental stages were collected, and RNA was isolated from them using TRizol regent according to the manufacturer’s instructions. The cDNA was synthesized using AMV reverse transcriptase XL for RT-PCR according to the manufacturer’s instructions33. The qRT-PCR was performed under the following conditions: 95 °C for 2 min, followed by 40 cycles of 5 s at 95 °C, 30 s at 55–60 °C, and a final melting curve step. Three biological replicates were performed in a Roche 480 LightCycler. Threshold values (CT) were used to quantify relative gene expression using the comparative 2−ΔΔCt method34. The information of primer used for qRT-PCR analyses is shown in Table S7.
Development of SSR markers
For SSRs analysis, transcripts longer than 500 bp were selected and MISA software was used. The parameters were set for identifcation of mono-, di-, tri-, tetra-, penta-, and hexa-nucleotide motifs with a minimum of ten, six, five, five, five, and five, respectively. SSR primers were designed by Batch Primer 3 tool6. PCR amplifications were performed using the DNA template extracted from R. lapponicum. The PCR products were separated in 8% polyacrylamide gels.
Results
SMRT sequencing data
To obtain a representative full-length transcriptome for R. lapponicum, the total RNA from four tissues (root, stem, leaf and flower) were used for the library construction for SMRT sequencing. In this study, we obtained 957,032 polymerase reads from the sequenced library, and a dataset with 15.37 Gb of clean reads was generated (Supplementary Table S1). A total of 658,338 reads of inserts (ROIs) were generated with full passes ≥0 and a consensus accuracy >0.75. Three SMRT cells (1–2 kb, 2–3 kb, and 3–6 kb) were constructed, and the average length of the ROIs in different libraries was between 1,206 bp and 3,519 bp (Table 1). ROIs were classified into 346,270 FLNC and 274,471 non-full-length reads. According to the clustering algorithm of ICE, we obtained 180,047 consensus isoform sequences, including 105,015 high-quality isoforms and 74,963 low-quality isoforms. After the removal of redundant reads, we obtained 75,002 high-quality full-length transcripts. The length distribution of the ROIs and transcripts is shown in Fig. 1.
Table 1.
Summary of reads of inserts from PacBio single-molecule long-read sequencing.
| Size | Reads of insert | Read bases of insert | Mean read length of insert | Mean read quality of insert | Mean number of passes |
|---|---|---|---|---|---|
| 1–2 K | 239,854 | 289,251,046 | 1,206 | 0.97 | 16 |
| 2–3 K | 265,435 | 626,058,946 | 2,359 | 0.93 | 11 |
| 3–6 K | 153,049 | 538,326,391 | 3,519 | 0.91 | 8 |
Figure 1.

The length distribution of reads of inserts (ROIs) and transcripts. (a) The length distribution of 658,338 ROIs. (b) The length distribution of 75,002 transcripts.
Open reading frame and AS event prediction
Using the software TransDecoder, 72,606 ORFs were predicted. A total of 55,255 complete ORFs were identified, and the length distribution of the complete ORFs was analysed (Fig. 2). Among all transcripts obtained by SMRT sequencing, 7,140 AS events were detected (Supplementary Table S2). Due to the lack of an available R. lapponicum reference genome, further characterization of the types of AS events would be warranted in future studies.
Figure 2.

The length distribution of complete open reading frames (ORFs).
Long non-coding RNA identification
LncRNA are a class of poly-A noncoding RNAs that play roles in the growth and stress responses of plants. In this study, we used four computational approaches to identify lncRNAs, involving the CPC, CNCI, CPAT and Pfam databases. A total of 5,167, 3,380, 11,137 and 12,315 lncRNAs were identified in the CNCI, CPC, CPAT and Pfam databases, respectively (Supplementary Table S3). By filtering transcripts of less than 300 bp, 2,011 transcripts were considered as lncRNAs by all four methods (Fig. 3).
Figure 3.
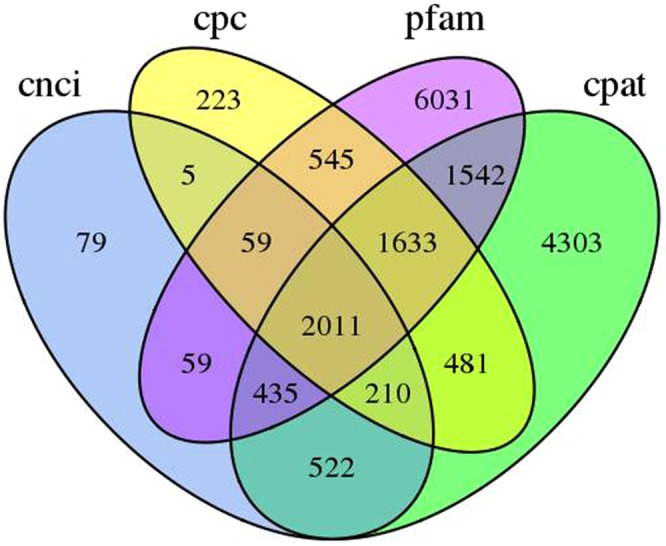
Venn diagram of long non-coding RNAs (lncRNAs) predicted by the Coding-Non-Coding Index (CNCI), Coding Potential Assessment Tool (CPAT), Coding Potential Calculator (CPC) and Pfam protein structure domain analysis methods.
Transcription factor prediction
TFs are key regulators of gene expression and play important roles in plant growth and development. In this study, 3,150 putative TFs were identified and divided into 64 TF families (Supplementary Table S4). The TFs in the R. lapponicum transcriptome mainly belonged to the C3H (231, 7.33%), FAR1 (187, 5.94%), bHLH (182, 5.78%), C2H2 (150, 4.76%), GRAS (136, 4.32%), MYB-related (135, 4.28%), bZIP (130, 4.13%), WRKY (123, 3.90%), RWP-RK (120, 3.81%), and NAC (115, 3.65%) families (Fig. 4).
Figure 4.

The top 15 transcription factor (TF) families in the R. lapponicum transcriptome. The x-axis represents the TFs, and the y-axis indicates the number of transcripts of a specific TF type.
Functional annotation of transcripts
Among the 75,002 transcripts identified, 71,155 (94.87%), 57,837 (77.11%), 33,653 (44.87%), 30,359 (40.48%), 45,925 (61.23%), 69,897 (93.19%), 60,296 (80.39%) and 31,749 (42.33%) transcripts were annotated in the Nr, Swiss-Prot, GO, COG, KOG, eggNOG, Pfam, and KEGG databases (Table 2), respectively. The annotation of the species distribution showed the largest proportion of the transcripts distributed in Vitis vinifera, followed by Quercus suber, Juglans regia and Coffea canephora (Fig. 5).
Table 2.
Summary of the functional annotation of the R. lapponicum transcriptome.
| Annotated database | Number of transcript hits | Percentage (%) |
|---|---|---|
| NR | 71,155 | 94.87 |
| Swiss-Prot | 57,837 | 77.11 |
| GO | 33,653 | 44.87 |
| COG | 30,359 | 40.48 |
| KOG | 45,925 | 61.23 |
| eggNOG | 69,897 | 93.19 |
| Pfam | 60,296 | 80.39 |
| KEGG | 31,749 | 42.33 |
| All annotated | 71,386 | 95.18 |
| All analysed | 75,002 | 100.00 |
Figure 5.
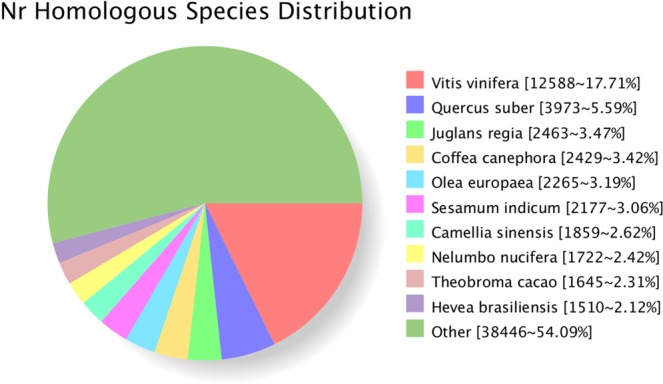
Homologous species distribution of R. lapponicum transcripts annotated in the non-redundant (Nr) database. The numbers and frequencies of the main annotated species are shown.
GO classification
To classify the gene functions of the transcripts, GO analysis was performed. GO analysis showed the enrichment of 33,653 transcripts categorized into 51 functional groups, which could be divided into three major categories: biological process, cellular component and molecular function (Fig. 6). In the biological process group, catalytic activity and binding were the main categories. In the cellular component group, cell part, cell and organelle were the most frequent categories. In the molecular function group, the genes were involved in catalytic activity, binding and other categories.
Figure 6.

Gene Ontology (GO) classification of R. lapponicum transcripts. Green represents biological processes blue represents cellular components, and red represents molecular functions. The x-axis represents GO categories, the right y-axis represents the number of transcripts, and the left y-axis represents the percentage of transcripts.
COG classification
To further study the functional annotation and classification of the R. lapponicum transcripts, all transcripts were subjected to a search against the Clusters of COG database. COG analysis showed that 30,359 transcripts were assigned to 24 categories (Fig. 7). The largest group was general function prediction only (8,967, 19.69%), followed by transcription (4,862, 10.68%) and then replication, recombination and repair (4,722, 10.37%). The percentages of six groups were less than 1.00%, including RNA processing and modification, nuclear structure, and cell motility.
Figure 7.

Clusters of Orthologous Groups of protein (COG) classification of R. lapponicum transcripts. The x-axis represents the subcategories, and the y-axis indicates the number of transcripts in a specific functional cluster.
KEGG pathway analysis
To identify biological pathways in R. lapponicum, transcripts were searched against the KEGG pathway database. A total of 31,749 transcripts were mapped to 128 KEGG functional pathways (Supplementary Table S5). Among these pathways, carbon metabolism (1,378, 4.34%) and protein processing in the endoplasmic reticulum (1290, 4.06%) were the most dominant pathways, followed by the biosynthesis of amino acids (1,196, 3.77%), spliceosomes (946, 2.98%) and ribosomes (909, 2.86%) (Table 3). The KEGG functional classification provided valuable clues for investigating specific processes, functions, and pathways in R. lapponicum.
Table 3.
The top 15 mapped pathways annotated by the KEGG database.
| Number | Name of pathway | Pathway ID | Number of transcripts |
|---|---|---|---|
| 1 | Carbon metabolism | ko01200 | 1,378 |
| 2 | Protein processing in endoplasmic reticulum | ko04141 | 1,290 |
| 3 | Biosynthesis of amino acids | ko01230 | 1,196 |
| 4 | Spliceosome | ko03040 | 1,049 |
| 5 | Ribosome | ko03010 | 946 |
| 6 | RNA transport | ko03013 | 909 |
| 7 | Starch and sucrose metabolism | ko00500 | 839 |
| 8 | Plant hormone signal transduction | ko04075 | 827 |
| 9 | Oxidative phosphorylation | ko00190 | 787 |
| 10 | Glycolysis / Gluconeogenesis | ko00010 | 740 |
| 11 | Plant-pathogen interaction | ko04626 | 701 |
| 12 | mRNA surveillance pathway | ko03015 | 664 |
| 13 | Ubiquitin mediated proteolysis | ko04120 | 616 |
| 14 | Amino sugar and nucleotide sugar metabolism | ko00520 | 581 |
| 15 | Endocytosis | ko04144 | 568 |
Representative genes in the anthocyanin biosynthesis pathway and expression pattern analysis
Anthocyanins are natural bioactive pigments in plants that play important roles in many physiological functions. Through KEGG analysis, a total of 96 transcripts were identified coding for the enzymes associated with anthocyanin synthesis, which included trans-cinnamate 4-monooxygenase (C4H, seven transcripts), chalcone isomerase (CHI, 12 transcripts), chalcone synthase (CHS, 41 transcripts), naringenin 3-dioxygenase (F3H, nine transcripts), flavonoid 3ʹ-hydroxylase (F3ʹH, eight transcripts) and flavonoid 3ʹ,5ʹ-hydroxylase (F3ʹ5ʹH, eight transcripts), anthocyanidin synthase (ANS, five transcripts) and dihydroflavonol 4-reductase (DFR, six transcripts) (Supplementary Table S6). Six genes related to anthocyanin biosynthesis were randomly selected to perform qRT-PCR analysis. The qRT-PCR analysis showed that the highest expression of C4H (F01_cb9736_c8/f1p0/2072) and F3H (F01_cb13925_c0/f2p0/1339) was observed at stage S2, while the expression of F3′5′H (F01_cb7576_c7/f2p0/1921), DFR (F01_cb3655_c0/f2p0/2991) and ANS (F01_cb7563_c31/f1p0/655) was highest at stage S3 (Fig. 8).
Figure 8.

Expression analysis of transcripts involved in flavonoid biosynthesis throughout the flower development in R. lapponicum. (a) Expression levels of C4H, CHS and DFR. (b) Expression levels of F3H, F3′5′H and ANS. Four flower developmental stages were examined in our study: the budding stage (S1), the initial flowering stage (S3), the full-flowering stage (S3), and the end flowering stage (S4). The qRT-PCR validation of six randomly selected transcripts in the three samples. Columns represent the relative expression levels. Error bars represent the standard deviation from three biological replicates.
SSR identification
After screening the 74,031 obtained transcripts, 64,327 potential SSRs were identified from 45,319 transcripts. Among these transcripts, 26,312 contained one SSR, and 19,007 contained two loci or more. Furthermore, 26,312 and 19,007 transcripts contained one SSR and at least two SSRs, respectively. In addition, 11,634 SSRs were considered compound formations. As shown in Table 4, the numbers of mono-, di-, tri-, tetra-, penta- and hexa-nucleotide repeats were 18,064, 27,639, 6,494, 228, 114 and 155, respectively. SSRs with 10 repeat units (7,178, 13.62%) were the most abundant, followed by those with 6 (6,496, 12.33%), 11 (4,969, 9.43%) and 7 (4,549, 8.63%) (Table 5). The most frequent motif type was A/T, followed by AG/CT, GA/TC, CA/TG and GAA/TTC (Fig. 9).
Table 4.
Summary of SSRs identified in the R. lapponicum transcriptome.
| Searching item | Numbers |
|---|---|
| Total number of sequences examined | 74,031 |
| Total size of examined sequences (bp) | 171,584,325 |
| Total number of identified SSRs | 64,327 |
| Number of SSR containing sequences | 45,319 |
| Number of sequences containing more than one SSR | 19,007 |
| Number of SSRs present in compound formation | 11,634 |
| Mono-nucleotide | 18,064 |
| Di-nucleotide | 27,639 |
| Tri-nucleotide | 6,494 |
| Tetra-nucleotide | 228 |
| Penta-nucleotide | 114 |
| Hexa-nucleotide | 155 |
Table 5.
The distribution of SSRs based on the number of repeat units.
| Number of repeat units | Mono | Di | Tri | Tetra | Penta | Hexa | Total | Percentage (%) |
|---|---|---|---|---|---|---|---|---|
| 5 | — | — | 3,853 | 168 | 83 | 96 | 4,200 | 7.97 |
| 6 | — | 4,917 | 1,475 | 46 | 25 | 33 | 6,496 | 12.33 |
| 7 | — | 3,881 | 639 | 12 | 4 | 13 | 4,549 | 8.63 |
| 8 | — | 3,399 | 234 | 1 | 1 | 8 | 3,643 | 6.91 |
| 9 | — | 3,026 | 112 | 1 | — | 1 | 3,140 | 5.96 |
| 10 | 4,635 | 2,461 | 81 | — | 1 | — | 7,178 | 13.62 |
| 11 | 2,964 | 1,968 | 35 | — | — | 2 | 4,969 | 9.43 |
| 12 | 2,062 | 1,627 | 24 | — | — | — | 3,713 | 7.05 |
| 13 | 1,359 | 1,316 | 26 | — | — | — | 2,701 | 5.12 |
| 14 | 1,103 | 1,104 | 8 | — | — | 1 | 2,216 | 4.21 |
| ≥15 | 5,941 | 3,940 | 7 | — | — | 1 | 9,889 | 18.77 |
Figure 9.
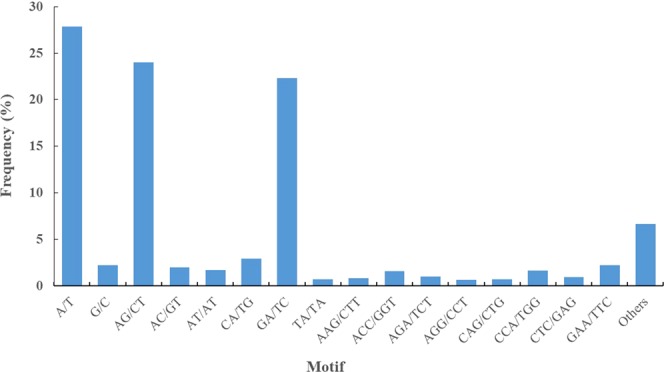
Frequency distribution of SSRs based on motif types. The frequency of the main motif types is shown.
SSR marker development
Using Primer3.0 software, 40,509 primer pairs were designed, and 150 were randomly selected for PCR (Supplementary Table S8). The PCR products of 127 primer pairs were successfully examined, with an amplification efficiency rate of 84.67%. However, the remaining 23 primer pairs failed to achieve amplification at various annealing temperatures.
Discussion
SMRT sequencing technology is an efficient and reliable approach for obtaining the full-length transcripts of certain species35. Recently, long-read SMRT sequencing has been the most reliable and efficient strategy for whole-transcriptome profiling studies, especially for nonmodel plant species without reference genome sequences. In this study, SMRT sequencing technology was applied to investigate the R. lapponicum transcriptome using the PacBio RS II platform. A total of 15.37 Gb of sequencing data were generated, including 658,338 ROIs and 346,270 FLNC reads. The percentage of FLNC reads in all ROIs was 52.59%, and this result was similar findings obtained in alfalfa36 and strawberry37 by SMRT sequencing. After removing redundant sequences, 75,002 full-length transcripts were obtained. SMRT sequencing can capture the very long nucleotide sequences, where one read usually represents a full-length transcript13. The length of the transcripts obtained by SMRT sequencing technology is longer than that of transcripts obtained by next-generation high-throughput sequencing technology. In this study, the average length of the R. lapponicum transcripts was 2,509 bp, which was longer than those obtained in seashore paspalum (970 bp)38, sweet potato (581 bp)39, and sesame (629 bp)40 by Illumina sequencing technology. Furthermore, we found that 58.66% of all transcripts were longer than 2,000 bp in this study, and much higher than that in Rhododendron molle (7.23%)41 and Neottopteris nidus (13.63%)6 using Illumina sequencing technology. These results indicated that PacBio SMRT sequencing technology is an efficient approach to capture the transcript sequences, especially for long transcript sequences.
Alternative splicing is a major cellular mechanism generating transcriptome diversity and proteome complexity in plants42. In this study, 7,140 AS events were detected from the R. lapponicum transcripts. In addition, 3,150 TFs that are key components involved in the transcriptional regulatory system were identified. LncRNAs are a novel class of non-coding transcripts with lengths greater than 200 nucleotides that play important roles in many biological processes43. LncRNAs are largely involved in regulating plant development and growth, secondary metabolism, and the plant stress response44. Recently, an increasing number of studies have focused on the functions of lncRNAs in plants such as in red pineapple45 and hot pepper46. However, no lncRNAs from Rhododendron have been reported. In this study, we identified 2,011 lncRNAs using four methods, and these lncRNAs will be useful for further research in R. lapponicum.
A total of 71,386 transcripts were annotated by sequence alignment in eight databases, suggesting that this study generated a very large number of R. lapponicum genes. The percentage of annotated transcripts was 95.18%, which was consistent with that in alfalfa36 and shrimp47. The remaining 3,616 transcripts presented no BLAST matches and might represent R. lapponicum-specific genes or unknown genes in R. lapponicum. The systematic classification of proteins in the transcriptome is crucial for maximizing the utilization of transcripts for functional and evolutionary studies. The results of GO and COG classification suggested that a large number of transcripts were involved in transcription, replication, recombination and repair, and catalytic activity. There were 31,749 transcripts assigned to specific pathways, such as metabolism, genetic information processing, cellular processes, environmental information processing, and organismal systems pathways. The results of GO, COG and KEGG classification showed that a large number of transcripts had diverse molecular functions and were involved in many biological pathways. Therefore, our data provided abundant genetic information on future molecular survey on the growth and development of R. lapponicum.
Flower colour is one of the most important ornamental characteristics of rhododendrons. The biosynthesis of anthocyanin is critical for a wide range of flower colours. Previous studies have shown that C4H, CHS, F3H, F3’H, F3ʹ5ʹH, DFR and ANS are the key enzymes involved in the biosynthesis of anthocyanin for the determination of different flower colours in plants48. In the present study, a total of 96 transcripts were identified coding for the enzymes associated with anthocyanin synthesis. Gene expression analysis by qRT-PCR showed that the expression levels of C4H, F3H, F3ʹ5ʹH, DFR and ANS genes were low at the early flowering developmental stage and increased as the flowers developed. The increases in the expression of these genes were consistent with the changes in anthocyanin content in the flower petals of R. lapponicum during flower development. In addition, transcription factors such as those of the MYB, bHLH and WD40 families play a key role by regulating the expression of genes in anthocyanin biosynthesis49,50. According to the functional annotation results, 3,150 putative transcription factor genes belonging to 64 TF families were identified. Among these genes, 83 and 182 transcripts belonged to the MYB and bHLH families, respectively (Fig. 4). In conclusion, the identification of key enzymes and related regulatory TF genes involved in anthocyanin biosynthesis and metabolic pathways may contribute to the understanding of colour-regulating mechanisms in rhododendrons.
The rapid development of transcriptome sequencing technology has enabled the massive development of SSR markers51,52. In total, 64,327 SSRs were identified from 45,319 SSR-containing sequences, and the average frequency of SSRs was one in 2.67 kb. Among the six types of repeat motifs, dinucleotide repeats were the most abundant. In the present study, the most frequent mono-, di-, and tri-nucleotide motifs were A/T, AG/CT and GAA/TTC, respectively, which was consistent with the results of studies in non-heading Chinese cabbage53, rubber tree54 and radish55. CT/AG/GA/TC were the most abundant motifs, accounting for 92.11% of the total dinucleotide repeats. CT repeats are typically found in transcribed regions that may be involved in antisense transcription and play a role in gene regulation56,57. Furthermore, the most abundant mononucleotide motif was A/T, which is thought to be frequent in the genomic sequences of plants58. SSR abundance varies among different plant species in different studies. Repeat units of 10, 6, 11, 7, and 5 in SSR sequences accounted for 51.98% of the total SSRs. A total of 150 pairs of PCR primers were designed, and 127 primer pairs successfully amplified PCR products. The failure of 23 primer pairs to achieve amplification might have resulted from the targeting of amplicons with large introns, primers positioned across splice sites or chimeric primers. These results suggested that the development of SSR markers based on R. lapponicum transcripts obtained from PacBio SMRT sequencing is an effective and feasible approach. The newly developed SSR markers from our study will provide a valuable genetic tool that be used in studies on genetic diversity, comparative genomics, gene mapping, and population genetics and other types of genetic studies in rhododendron.
In conclusion, we analysed the full-length transcriptome of R. lapponicum by using the PacBio SMRT sequencing technology. This study represents the first the third-generation long-read transcriptome sequencing of R. lapponicum. Based on the obtained transcriptome data, 7,140 AS events, 2,011 lncRNAs, 55,255 complete ORFs and 3,150 TF members were identified. A total of 96 transcripts were identified coding for the enzymes associated with anthocyanin synthesis. In addition, 64,327 SSRs were detected, and 150 primer pairs were randomly selected to develop SSR markers. The obtained transcriptome data may facilitate further genetic studies on R. lapponicum.
Supplementary information
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant no. 31700627) and the Natural Science Foundation of Jiangsu Province (Grant no. BK20170607).
Author contributions
Xinping Jia and Jiale Su conceived and designed the experiments. Xinping Jia, Ling Tang and Xueying Mei conducted the experiments. Hairong Luo, Yanming Deng and Huazhou Liu analysed the data. Xinping Jia wrote the manuscript with the help of all coauthors. All authors read and approved the manuscript.
Data availability
The following information was supplied regarding data availability: Data are available at the Sequence Read Archive (SRA) (https://www.ncbi.nlm.nih.gov/sra) of NCBI, accession number: PRJNA594084.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
is available for this paper at 10.1038/s41598-020-63814-x.
References
- 1.Huang CC, et al. Genetic population structure of the alpine species Rhododendron pseudochrysanthum sensu lato (Ericaceae) inferred from chloroplast and nuclear DNA. BMC Evolutionary Biology. 2011;11:108. doi: 10.1186/1471-2148-11-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xing W, et al. De novo assembly of transcriptome from Rhododendron latoucheae Franch. using Illumina sequencing and development of new EST-SSR markers for genetic diversity analysis in Rhododendron. Tree Genetics Genomes. 2017;13:53. doi: 10.1007/s11295-017-1135-y. [DOI] [Google Scholar]
- 3.Jonasson S. Resource allocation in relation to leaf retention time of the wintergreen Rhododendron Lapponicum. Ecology. 1995;76:475–485. doi: 10.2307/1941206. [DOI] [Google Scholar]
- 4.Jia D, et al. SMRT sequencing of full-length transcriptome of flea beetle Agasicles hygrophila (Selman and Vogt) Scientific Reports. 2018;8:2197. doi: 10.1038/s41598-018-20181-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zeng J, et al. Application of EST-SSR markers developed from the transcriptome of Torreya grandis (Taxaceae), a threatened nut-yielding conifer tree. Peer J. 2018;6:e5606. doi: 10.7717/peerj.5606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jia XP, et al. De novo assembly of the transcriptome of Neottopteris nidus using Illumina paired-end sequencing and development of EST-SSR markers. Molecular Breeding. 2016;36:94. doi: 10.1007/s11032-016-0519-2. [DOI] [Google Scholar]
- 7.Zhang L, et al. The draft genome assembly of Rhododendron delavayi Franch. var. delavayi. GigaScience. 2017;6:1–11. doi: 10.1093/gigascience/gix076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Soza VL, et al. The Rhododendron genome and chromosomal organization provide insight into shared whole genome duplications across the heath family (Ericaceae) Genome Biology Evolution. 2019;11:3353–3371. doi: 10.1093/gbe/evz245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu XM, et al. De novo assembly and comparative transcriptome analysis: novel insights into terpenoid biosynthesis in Chamaemelum nobile L. Plant Cell Reports. 2019;38:101–116. doi: 10.1007/s00299-018-2352-z. [DOI] [PubMed] [Google Scholar]
- 10.Chaisson MJ, et al. Resolving the complexity of the human genome using single-molecule sequencing. Nature. 2014;517:608–611. doi: 10.1038/nature13907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peng Z, et al. Long read and single molecule DNA sequencing simplifies genome assembly and TAL effector gene analysis of Xanthomonas translucens. BMC Genomics. 2016;17:21. doi: 10.1186/s12864-015-2348-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang JY, et al. A full-length transcriptome of Sepia esculenta using a combination of single-molecule long-read (SMRT) and Illumina sequencing. Marine Genomics. 2019;43:54–57. doi: 10.1016/j.margen.2018.08.008. [DOI] [Google Scholar]
- 13.Sharon D, et al. A single-molecule long-read survey of the human transcriptome. Nature Biotechnology. 2013;31:1009–1014. doi: 10.1038/nbt.2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abdel-Ghany SE, et al. A survey of the sorghum transcriptome using single-molecule long reads. Nature Communications. 2016;7:11706. doi: 10.1038/ncomms11706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu X, et al. Detecting alternatively spliced transcript isoforms from single-molecule long-read sequences without a reference genome. Molecular Ecology Resources. 2017;17(6):1243–1256. doi: 10.1111/1755-0998.12670. [DOI] [PubMed] [Google Scholar]
- 16.Chen JF, et al. Development of EST-SSR markers in flowering Chinese cabbage (Brassica campestris L. ssp. chinensis var. utilis Tsen et Lee) based on de novo transcriptomic assemblies. PLoS One. 2017;12:e0184736. doi: 10.1371/journal.pone.0184736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang S, et al. Transcriptome analysis of the roots at early and late seedling stages using Illumina paired-end sequencing and development of EST-SSR markers in radish. Plant Cell Reports. 2012;31:1437–1447. doi: 10.1007/s00299-012-1259-3. [DOI] [PubMed] [Google Scholar]
- 18.Yagi M, et al. Construction of a reference genetic linkage map for carnation (Dianthus caryophyllus L.) BMC Genomics. 2013;14:734. doi: 10.1186/1471-2164-14-734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Foissac S, Sammeth M. ASTALAVISTA: dynamic and flexible analysis of alternative splicing events in custom gene datasets. Nucleic Acids Research. 2007;35:W297–W299. doi: 10.1093/nar/gkm311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guo AY, et al. PlantTFDB: a comprehensive plant transcription factor database. Nucleic Acids Research. 2007;36:D966–D969. doi: 10.1093/nar/gkm841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Altschul SF, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic acids research. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yangyang D, et al. Integrated nr Database in Protein Annotation System and Its Localization. Computer Engineering. 2006;32:71–72. [Google Scholar]
- 23.Li A, Zhang J, Zhou Z. PLEK: a tool for predicting long non-coding RNAs and messenger RNAs based on an improved k-mer scheme. BMC Bioinformatics. 2014;15:311. doi: 10.1186/1471-2105-15-311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sonnhammer EL, et al. Pfam: a comprehensive database of protein domain families based on seed alignments. Proteins. 1997;28:405–420. doi: 10.1002/(SICI)1097-0134(199707)28:3<405::AID-PROT10>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 25.Deng Y, et al. Integrated nr database in protein annotation system and its localization. Computer Engineering. 2006;32:71–74. [Google Scholar]
- 26.Apweiler R, et al. UniProt: the Universal Protein knowledgebase. Nucleic Acids Research. 2004;32:115–119. doi: 10.1093/nar/gkh131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Finn RD, et al. Pfam: the protein families database. Nucleic Acids Research. 2013;42:D222–D230. doi: 10.1093/nar/gkt1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huerta-Cepas J, et al. eggNOG 4.5: a hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Research. 2016;44:D286–D293. doi: 10.1093/nar/gkv1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tatusov RL, et al. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Research. 2000;28:33–36. doi: 10.1093/nar/28.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Koonin EV, et al. A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. Genome Biology. 2004;5:R7. doi: 10.1186/gb-2004-5-2-r7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Götz S, et al. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Research. 2008;36(10):3420–3435. doi: 10.1093/nar/gkn176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kanehisa M, et al. The KEGG resource for deciphering the genome. Nucleic Acids Research. 2004;32:D277–D280. doi: 10.1093/nar/gkh063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sun YX, et al. The role of wheat jasmonic acid and ethylene pathways in response to Fusarium graminearum infection. Plant Growth Regulation. 2016;80:69–77. doi: 10.1007/s10725-016-0147-1. [DOI] [Google Scholar]
- 34.Livak K, Schmittgen T. Analysis of relative gene expression data using real-time quantitative PCR and the 2−△△Ct method. Methods. 2000;25:4. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 35.Chen SY, et al. A transcriptome atlas of rabbit revealed by PacBio single-molecule long-read sequencing. Scientific Reports. 2017;7:7648. doi: 10.1038/s41598-017-08138-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chao YH, et al. Analysis of transcripts and splice isoforms in Medicago sativa L. by single-molecule long-read sequencing. Plant Molecular Biology. 2019;99:219–235. doi: 10.1007/s11103-018-0813-y. [DOI] [PubMed] [Google Scholar]
- 37.Li Y, et al. Global identification of alternative splicing via comparative analysis of SMRT- and Illumina-based RNA-seq in strawberry. Plant Journal. 2017;90(1):164–176. doi: 10.1111/tpj.13462. [DOI] [PubMed] [Google Scholar]
- 38.Jia XP, et al. Characterization of the global transcriptome using Illumina sequencing and novel microsatellite marker information in seashore paspalum. Genes. Genomics. 2015;37:77–86. [Google Scholar]
- 39.Wang ZY, et al. De novo assembly and characterization of root transcriptome using Illumina paired-end sequencing and development of cSSR markers in sweet potato (Ipomoea batatas) BMC Genomics. 2010;11:726. doi: 10.1186/1471-2164-11-726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wei WL, et al. Characterization of the sesame (Sesamum indicum L.) global transcriptome using Illumina paired-end sequencing and development of EST–SSR markers. BMC Genomics. 2011;12:451. doi: 10.1186/1471-2164-12-451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xiao Z, et al. De novo transcriptome analysis of Rhododendron molle G. Don flowers by Illumina sequencing. Genes. Genomics. 2018;40:591–601. doi: 10.1007/s13258-018-0662-8. [DOI] [PubMed] [Google Scholar]
- 42.Reddy AS. Alternative splicing of pre-messenger RNAs in plants in the genomic era. Annual Review of Plant Biology. 2007;58:267–294. doi: 10.1146/annurev.arplant.58.032806.103754. [DOI] [PubMed] [Google Scholar]
- 43.Kapranov P, et al. RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science. 2007;316:1484–1488. doi: 10.1126/science.1138341. [DOI] [PubMed] [Google Scholar]
- 44.Zhang G, et al. Transcriptomic and functional analyses unveil the role of long non-coding RNAs in anthocyanin biosynthesis during sea buckthorn fruit ripening. DNA Research. 2018;25:465–476. doi: 10.1093/dnares/dsy017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ma J, et al. SMRT sequencing analysis reveals the full-length transcripts and alternative splicing patterns in Ananas comosus var. bracteatus. PeerJ. 2019;7:e7062. doi: 10.7717/peerj.7062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhu, C. et al. Transcriptome profiling using Illumina- and SMRT-based RNA-seq of hot pepper for in-depth understanding of genes involved in CMV infection. Gene S0378111918304815 (2018). [DOI] [PubMed]
- 47.Zeng DG, et al. Single-molecule long-read sequencing facilitates shrimp transcriptome research. Scientific Reports. 2018;8:16920. doi: 10.1038/s41598-018-35066-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Winkel-Shirley B. Flavonoid biosynthesis. A colorful model for genetics, biochemistry, cell biology, and biotechnology. Plant Physiology. 2001;126:485–493. doi: 10.1104/pp.126.2.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gonzalez A, et al. Regulation of the anthocyanin biosynthetic pathway by the TTG1/ bHLH/Myb transcriptional complex in Arabidopsis seedlings. Plant Journal. 2008;53:814–827. doi: 10.1111/j.1365-313X.2007.03373.x. [DOI] [PubMed] [Google Scholar]
- 50.Schaart JG, et al. Identification and characterization of MYB-bHLH-WD40 regulatory complexes controlling proanthocyanidin biosynthesis in strawberry (Fragaria × ananassa) fruits. New Phytologist. 2013;197:454–467. doi: 10.1111/nph.12017. [DOI] [PubMed] [Google Scholar]
- 51.Dutta S, et al. Development of genic-SSR markers by deep transcriptome sequencing in pigeonpea [Cajanus cajan (L.) Millspaugh] BMC Plant Biology. 2011;11:7. doi: 10.1186/1471-2229-11-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhai LL, et al. Novel and useful genic-SSR markers from de novo transcriptome sequencing of radish (Raphanus sativus L.) Molecular Breeding. 2014;33:611–624. doi: 10.1007/s11032-013-9978-x. [DOI] [Google Scholar]
- 53.Song X, et al. Genome-wide identification of SSR and SNP markers from the non-heading Chinese cabbage for comparative genomic analyses. BMC Genomics. 2015;216:328. doi: 10.1186/s12864-015-1534-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li D, et al. De novo assembly and characterization of bark transcriptome using Illumina sequencing and development of EST-SSR markers in rubber tree (Hevea brasiliensis Muell. Arg.) BMC Genomics. 2012;13:192. doi: 10.1186/1471-2164-13-192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lagercrantz U, et al. The abundance of various polymorphic microsatellite motifs differs between plants and vertebrates. Nucleic acids research. 1993;21:1111–1115. doi: 10.1093/nar/21.5.1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Martienssen RA, Colot V. DNA methylation and epigenetic inheritance in plants and filamentous fungi. Science. 2001;293:1070–1074. doi: 10.1126/science.293.5532.1070. [DOI] [PubMed] [Google Scholar]
- 57.Wang HX, et al. Development and cross-species/genera transferability of microsatellite markers discovered using 454 genome sequencing in chokecherry (Prunus virginiana L.) Plant Cell Report. 2012;31:2047–2055. doi: 10.1007/s00299-012-1315-z. [DOI] [PubMed] [Google Scholar]
- 58.Lagercrantz U, et al. The abundance of various polymorphic microsatellite motifs differs between plants and vertebrates. Nucleic Acids Research. 1993;21:1111–1115. doi: 10.1093/nar/21.5.1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The following information was supplied regarding data availability: Data are available at the Sequence Read Archive (SRA) (https://www.ncbi.nlm.nih.gov/sra) of NCBI, accession number: PRJNA594084.