

Abstract
Disorders of Sex Development (DSD) are congenital anomalies in which there is a discordance between chromosomal, genetic, gonadal, and/or internal/external genital sex. In XY individuals, the process of fetal sex differentiation can be disrupted at the stage of gonadal differentiation, resulting in gonadal dysgenesis, a form of early fetal-onset primary hypogonadism characterized by insufficient androgen and anti-Müllerian hormone (AMH) production, which leads to the development of ambiguous or female genitalia. The process of sex differentiation can also be disrupted at the stage of genital differentiation, due to isolated defects in androgen or AMH secretion, but not both. These are forms of fetal-onset hypogonadism with dissociated gonadal dysfunction. In this review, we present a perspective on impaired testicular endocrine function, i.e., fetal-onset male hypogonadism, resulting in incomplete virilization at birth.
Keywords: AMH, DSD, gonadal dysgenesis, steroidogenesis, testosterone
Introduction to Disorders of Sex Development and Hypogonadism
Disorders of Sex Development (DSD) are congenital anomalies in which there is a discordance between chromosomal, genetic, gonadal, and/or internal/external genital sex, as defined by the Chicago consensus published in 2006 (1), more recently revised and endorsed by all major societies of pediatric endocrinology (2). The etiologies and related pathogeneses of DSD can be comprehended once the developmental physiology of fetal sex differentiation is understood, as briefly described below. Most DSD affect the fetal endocrine and/or paracrine hormonal action resulting in the wide spectrum of clinical and hormonal phenotype of these patients.
Hypogonadism is usually defined as the gonadal failure resulting in an impaired steroid hormone production and deficient gamete output. Although steroids and gametes have been recognized as gonadal products for over a century, in the last decades of the 20th century it became evident that the somatic component of the ovaries and testes also secrete protein hormones, like inhibins and anti-Müllerian hormone (AMH), which have a relevant role as biomarkers in reproductive physiology from fetal life through adulthood (3, 4). Therefore, in face of the changes occurring in the physiology of the hypothalamic-pituitary gonadal axis from fetal life to puberty, the definition of hypogonadism should be extended to the impaired function of the ovaries or testes, as compared to what is expected for age, that involves a decreased function of the germ and/or somatic (Sertoli/granulosa, Leydig/theca) cell populations of the gonads, which can result in impaired hormone secretion (estrogens, progestins, androgens, inhibins, and/or AMH) and/or gamete production (4–6).
Physiology of Fetal Sex Differentiation
The process of sexual differentiation in fetal life can be divided into three stages (Figure 1): (i) the formation of the undifferentiated gonads and the anlagen of the genitalia, internal (Müllerian and Wolffian ducts, urogenital sinus) and external (genital tubercle, urethral folds, labioscrotal folds), (ii) gonadal differentiation, and (iii) male or female differentiation of the internal and external genitalia (8).
Figure 1.

The three stages of fetal sex differentiation. During the undifferentiated stage, the mesoderm gives rise to the adreno-gonadal primordia and the Wolffian and Müllerian ducts, whereas the ectoderm and mesoderm contribute to the formation of the genital tubercle and the urethral and labioscrotal folds. During the stage of gonadal differentiation, the testes or ovaries develop from the gonadal ridges. During the stage of genital differentiation, the internal and external genitalia differentiate along the male or the female pathway. Modified with permission from Rey et al. (7) © 2018 McGraw Hill Education.
The initial formation of the undifferentiated gonads, the Müllerian and Wolffian ducts, the urogenital sinus and the undifferentiated external genitalia is identical in the XX and the XY fetuses between the 3rd and the 5th weeks of embryonic life (i.e., 5–7 weeks of amenorrhea). The gonadal ridges, the urogenital sinus, and the external genitalia are bipotential, i.e., they can develop through either the female or the male pathways. The internal ducts are unipotential: Müllerian ducts can give rise only to female functional structures (the Fallopian tubes, uterus, and upper vagina), and Wolffian ducts can differentiate only into male structures (the epididymides, vasa deferentia, and seminal vesicles).
Gonadal differentiation into ovaries or testes, in the 7th week of embryonic life, depends on a complex network of genes: the presence of the SRY gene on the Y chromosome disrupts the balance between pro-testicular and pro-ovarian genes, triggering testis differentiation (9–12). Subsequently, the action of two discrete testicular hormones, AMH and androgens, drive the differentiation of the internal ducts, urogenital sinus, and external genitalia (Figure 2). AMH, secreted by Sertoli cells, provokes the regression of Müllerian ducts in fetal weeks 8 and 9 (14). Androgens produced by Leydig cells induce the stabilization and differentiation of Wolffian ducts as well as the virilization of the urogenital sinus and the external genitalia between fetal weeks 8 and 13. These processes occurring in the first trimester of fetal life are independent of fetal pituitary gonadotropins: basal AMH expression is driven by a set of transcription factors (15), and androgen production is regulated by placental hCG (16). Insulin-like factor 3 (INSL3), produced by Ledyig cells, is involved in testicular descent (17). Ovarian hormones have no influence on fetal sex differentiation (18).
Figure 2.

Male reproductive hormone levels and fetal sex differentiation. (A) Ontogeny of male reproductive hormone levels from fetal to adult periods of life. In the early fetal stage, the initiation of testicular hormones secretion is independent of luteinizing hormone (LH) and follicle-stimulating hormone (FSH). In the second and third trimesters, LH and FSH are the main regulators of Sertoli and Leydig cell hormone production. For ~6 months after birth, LH, FSH, and testicular hormone levels remain at high levels. Afterwards, in infancy and childhood, gonadotropins, testosterone (T), and insulin-like factor 3 (INSL3) levels decrease, while those of anti- Müllerian hormone (AMH) and inhibin B continue to be high. During puberty, gonadotropins, T and INSL3 increase again to attain adult levels. AMH is inhibited by testosterone, and inhibin B is stimulated by FSH. Reprinted, with permission, from Salonia et al. (4). © 2019 Springer Nature Limited. (B) The undifferentiated gonad is exposed to pro-testicular and pro-ovarian factors. In the XY fetus, SRY expression regulated by MAP3K1, GATA4, SF1, and WT1 shifts the balance toward the pro-testicular factors, like SOX9, inducing testis differentiation. Sertoli cells produce anti-Müllerian hormone (AMH), which provokes Müllerian duct regression upon binding to its receptor AMHR2. Leydig cells express the LHCG receptor and steroidogenic proteins involved in androgen synthesis; testosterone acts on Wolffian ducts, the urogenital sinus, and the external genitalia, either directly or after metabolization to the more potent androgen dihydrotestosterone (DHT), to virilize them. In the XX fetus, where SRY is absent, the pro-ovarian factors WNT4, RSPO1, FOXL2, and DAX1 control the differentiation of the ovaries. The latter do not produce androgens or AMH, which results in feminization of the internal and external genitalia. Modified with permission from Rey et al. (13) © 2016 Elsevier Saunders.
In the newborn, testosterone, INSL3, AMH, and inhibin B are significantly higher in the male than in the female. During childhood, testosterone and INSL3 levels are similar between males and females, but AMH and inhibin B are clearly different between sexes, with higher levels in males (19–21).
DSD: Abnormal Fetal Sex Differentiation
The process of fetal sex differentiation can be disrupted at any of the three stages. The early morphogenesis of the reproductive tract may fail independently of gonadal differentiation: these are malformative, non-endocrine DSD (22) and will not be dealt with in this review. Gonadal differentiation failure is known as gonadal dysgenesis and results in primary hypogonadism. Testicular dysgenesis drives to incomplete virilization, and the condition is known as dysgenetic DSD (11, 14, 23). According to the gene defect and probably the timing of its abnormal expression, variable degrees of dysgenesis will result, thus eliciting variable compromise of endocrine and paracrine secretion of androgens and AMH. This will be reflected in different degrees of virilization of the external genitalia and Wolffian ducts as well as in the magnitude of regression of Müllerian structures. Conversely, ovarian dysgenesis does not affect the development of female genitalia. Finally, the third stage of fetal sex differentiation can be disrupted either by a deficient production of testicular hormones (24, 25) or an impaired action due to receptor defects (26, 27) in the XY fetus, or to an excess androgen exposure in the XX fetus (28–30). In this review, we will address impaired testicular endocrine function during fetal life resulting in incomplete virilization.
Hypogonadism: Classifications
Male hypogonadism, i.e., the impaired function of the testes as compared to what is expected for age, may involve a decreased hormone secretion (AMH, inhibins, and/or androgens) and/or an impaired sperm production. It can be due to a primary disorder or secondary to a defect in the hypothalamic-pituitary axis, respectively resulting in primary (testicular or hypergonadotropic) or secondary (central or hypogonadotropic) hypogonadism (Table 1). The terms hypergonadotropic and hypogonadotropic, extensively used in adult medicine (4), should be applied with caution in pediatric patients (6). Indeed, although elevated gonadotropins, especially FSH, are a typical feature of primary hypogonadism in newborns and infants (31), normal gonadotropin levels can occur in up to 30–70% of anorchid boys later in childhood (32). This highlights that, throughout childhood, the normal development of the intrinsic central inhibitory tone exerted on GnRH pulse generator can overcome the rise of FSH due to low serum inhibin B in circulation.
Table 1.
Classification of fetal-onset male hypogonadism.
| Primary Hypogonadism | ||
|---|---|---|
| Whole gonadal dysfunction | Dissociated gonadal dysfunction | |
| First trimester | Gonadal dysgenesis | Leydig cells: LHCG-R mutation, steroidogenic defects |
| Sertoli cells: AMH mutation | ||
| Central Hypogonadism | ||
| Whole gonadal dysfunction | Cell-specific gonadal dysfunction | |
| Second–third trimesters | Multiple pituitary hormone deficiency | Leydig cells: LHβ-subunit gene mutation |
| Isolated hypogonadotropic hypogonadism (IHH) | Sertoli cells: FSHβ-subunit gene mutation | |
As regards to the testicular compartment that is primarily affected, hypogonadism may present with whole gonadal dysfunction or a dissociated, cell-specific gonadal dysfunction. In the latter, one of the cell populations (Sertoli, Leydig, or germ cells) is primarily disrupted while the function of the others remains intact, at least for some time (5). Gonadal dysgenesis is an example of primary hypogonadism with whole gonadal dysfunction. Mutations of the FSH receptor and AMH genes are examples of Sertoli cell-specific hypogonadism. Mutations of the LH receptor and of steroidogenic proteins provoke Leydig cell-specific hypogonadism. Finally, deletions of the Y chromosome leading to azoospermia are examples of germ cell-specific hypogonadism.
Finally, hypogonadism may be congenital or acquired during postnatal life. The clinical presentation varies according to the age at which hypogonadism is established (4, 33, 34); particularly, fetal-onset hypogonadism results in DSD only when it is established in the first trimester of gestation, as it can be deduced from the knowledge of the physiology of fetal sex differentiation discussed above.
Hypogonadism and Endocrine-Related DSD
Usually, the initial diagnostic approach of patients with DSD is based on the karyotype (35). In 46,XY individuals, the etiology of DSD is classified into: disorders of gonadal development (or gonadal dysgenesis), disorders of androgen synthesis (in non-dysgenetic gonads), disorders of androgen action, and disorders of AMH synthesis or action (resulting in the Persistent Müllerian Duct Syndrome, PMDS) (36). Gonadal dysgenesis and isolated disorders of androgen or AMH synthesis represent early fetal-onset forms of primary hypogonadism resulting in 46,XY DSD (31). Disorders of androgen or AMH action do not initially affect testicular hormone production and, together with non-endocrine DSD (or unclassified disorders), will not be further discussed in this review.
Disorders of gonadal development (or gonadal dysgenesis) also occur in sex-chromosomal DSD (35). The most frequent forms of sex-chromosome aneuploidies, Turner (45,X) and Klinefelter (47,XXY) syndromes, generally do not present at birth with genital anomalies. Therefore, they do not appear as a differential diagnosis in the newborn. Conversely, patients with 45,X/46,XY or 46,XX/46,XY (or other mosaicisms/chimerisms) frequently bear diverse forms of gonadal dysgenesis (partial testicular dysgenesis, asymmetric gonadal differentiation, ovotestes, etc.) which lead to the existence of ambiguous genitalia. Furthermore, ovotestes and dysgenetic testes can develop in 46,XX fetuses, both in the presence or absence of SRY (37).
Disorders of Gonadal Development: Early Fetal-Onset Primary Hypogonadism With Whole Gonadal Dysfunction
Regardless of the karyotype and the pathogenesis, gonadal dysgenesis represents a typical form of primary hypogonadism established in the first trimester of fetal life. Because this is the period when internal and external genitalia are masculinized in the presence of testicular hormones or feminized in their absence, primary hypogonadism results in deficient masculinization of the fetus carrying a Y chromosome (46,XY or chimeras or mosaicisms with a Y chromosome) or in XX fetuses carrying a translocated SRY or gene imbalances provoking ovotesticular or dysgenetic testicular development, as previously explained.
The impaired gonadal development is reflected in a dysfunction of all testicular cell populations: Sertoli cells are less numerous, thus resulting in an insufficient AMH output leading to the existence of Müllerian remnants (Figure 3). Germ cells are also scarce and less differentiated, with a consequently increased risk of infertility and testicular neoplasia (38, 39). Leydig cell number and function is also impaired, resulting in a hypoandrogenism that leads to undervirilization of the internal and external genitalia.
Figure 3.
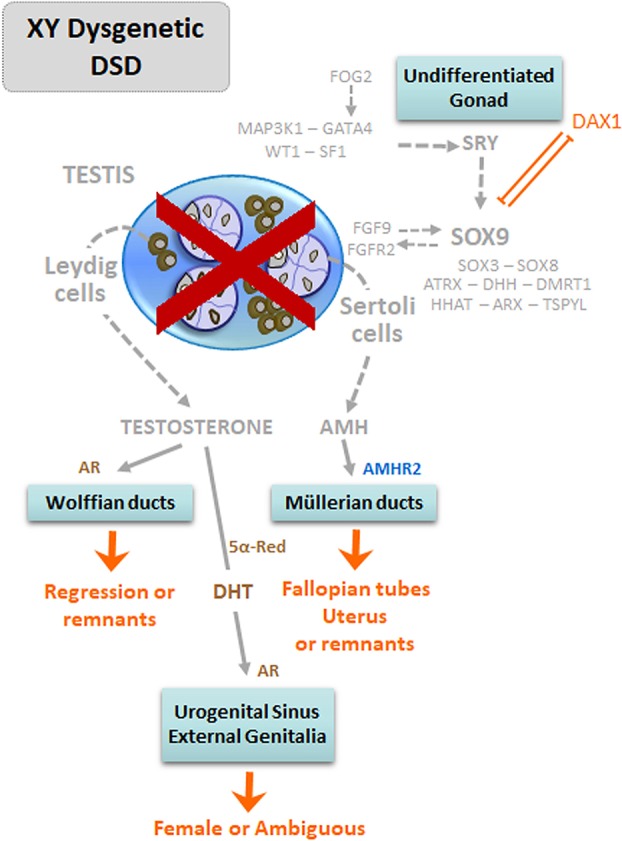
Dysgenetic DSD in 46,XY patients: early fetal-onset primary hypogonadism with whole gonadal dysfunction. The lack of normal testis differentiation, due to mutations of pro-testicular genes or to overexpression of DAX1 results in gonadal dysgenesis. Consequently, the insufficient production of androgens and anti-Müllerian hormone (AMH) results in undervirilization or complete feminization of the internal and external genitalia. Modified with permission from Rey et al. (13) © 2016 Elsevier Saunders.
The most severe forms of dysgenesis—pure or complete gonadal dysgenesis—present with a complete development of the uterus and Fallopian tubes, reflecting AMH deficiency, and absence of Wolffian remnants together with female external genitalia, as a consequence of androgen deficiency (Figure 4). In partial forms of gonadal dysgenesis, the degree of virilization is variable depending on the mass of functional testicular tissue: from micropenis, hypospadias, and cryptorchidism with variable development of Wolffian and Müllerian remnants to an almost female phenotype in severe cases (40). When the condition is asymmetric—known as mixed gonadal dysgenesis (41) or asymmetric gonadal differentiation (42)—, a hemi-uterus and a fallopian tube exist on the side of the streak gonad, and external genitalia may also show asymmetric development.
Figure 4.

Development of the internal genitalia according to fetal testicular function.
Gonadal Dysgenesis in 46,XY patients
Gonadal dysgenesis may be isolated or associated with dysmorphic features of non-reproductive organs (Table 2). Isolated gonadal dysgenesis is caused by mutations in pro-testicular genes. Inactivating mutations or deletions of SRY are present in ~15% of 46,XY females with pure gonadal dysgenesis (Swyer syndrome) (43, 44). Non-reproductive organs are not affected. Loss-of-function variants of MAP3K1 lead to an increased expression of pro-ovarian factors, like β-catenin, and reduced expression of pro-testicular factors, like SOX9/FGF9 (45). A similar pathogenesis has been proposed for mutations in ZNRF3 (46). Dysgenetic DSD can also be attributed to mutations in DHX37 (47, 48) particularly those individuals exhibiting the embryonic testicular regression syndrome and rearrangements involving SOX8 (49). Finally, isolated gonadal dysgenesis was described in 46,XY females with partial duplications of Xp21.3–p21.2, known as the DSS (dosage sensitive sex-reversal) locus encompassing NR0B1 that encodes DAX1; the increased expression of DAX1 is believed to impair testis differentiation (50, 51).
Table 2.
Disorders of Sex Development (DSD) due to 46,XY gonadal dysgenesis.
| Associated syndrome | Implicated gene | Locus |
|---|---|---|
| Swyer syndrome or partial testicular dysgenesis | SRY | Yp11.2 |
| MAP3K1 | 5q11.2 | |
| ZNRF3 | 22q12.1 | |
| DSS | Xp21.2 | |
| DHX37 | 12q24.31 | |
| Campomelic dysplasia: bowing of long bones, hip dislocation, scapula hypoplasia, small thoracic cage, micrognathia, macrocephaly, low-set ears, flat nasal bridge, and cardiac and renal defects | SOX9 | 17q24.3 |
| Denys-Drash syndrome: nephropathy and Wilms tumor | WT1 | 11p13 |
| Frasier syndrome: nephropathy and gonadoblastoma | ||
| Adrenal insufficiency | NR5A1 | 9q33.3 |
| ATRX syndrome: α-thalassemia and mental retardation | ATRX | Xq21.1 |
| Minifascicular neuropathy | DHH | 12q13.12 |
| Clinodactyly, hydrocephaly, and autistic spectrum disorder | FOG2/ZFPM2 | 8q23.1 |
| Craniosynostosis | FGFR2 | 10q26.13 |
| Microcephaly, mental retardation, short stature, and/or bronchial or digestive malformations | DMRT1/DMRT2 | 9p24.3 |
| Chondrodysplasia, cerebellar hypoplasia, and coloboma of the optic disks | HHAT | 1q32.2 |
| Lissencephaly and epilepsy | ARX | Xp21.3 |
| Cardiac malformations | MYRF | 11q12.2 |
| Sudden death | TSPYL | 6q22.1 |
Gonadal dysgenesis associated with dysmorphic features of non-reproductive organs (Table 2) has been seen in patients with mutations or deletions of SOX9 (52), WT1 (53), NR5A1 encoding SF1 (54), ATRX (55), DHH (56), FOG2/ZFPM2 (57), FGFR2 (58), 9p24 encompassing DMRT1 and DMRT2 (59), HHAT (60), ARX (61), MYRF (62), and TSPYL (63). Mutations in other genes, like CBX2 (64), ESR2 (65), MALD1 (66, 67), STARD8 (68), and WWOX (69), have been described in patients with gonadal dysgenesis but their pathogenic relationship still needs to be clearly established.
In a newborn with complete (or pure) gonadal dysgenesis, the external genitalia are typically female. There is also a normal development of the Fallopian tubes and uterus. The phenotype is due to a complete lack of androgen and AMH secretion. Because of the typical female aspect, the diagnosis is not suspected until pubertal age. Then, the absence of pubertal signs is the main complaint. Gonadal hormones are undetectable (14) and gonadotropins are very high in newborns. During childhood, gonadotropin levels decrease to almost normal values by the age of 6–9 years (70), but they increase again to extremely high levels in pubertal age. Replacement therapy with estrogens and progestins results in normal development of secondary sex characteristics, and gestations have been successful after oocyte donation (71). Gonads are characterized by the existence of bilateral fibrous streaks, with no gonadal tissue, although in rare cases abnormal germ cells can be observed embedded in cord-like structures. The risk of gonadoblastoma after puberty is increased.
In patients with partial dysgenesis, the degree of virilization of the internal and external genitalia correlates with the amount of functional testicular tissue present during the first trimester of intrauterine development exerting endocrine and paracrine hormonal action. The mildest forms may present as males with infertility. Serum levels of AMH and androgens are low for males but above the female range, while gonadotropins are elevated, though less than in pure gonadal dysgenesis. Although low INSL3 production might be involved in failure of testicular descent, no information is available on serum INSL3 in patients with dysgenetic DSD. Testes are small, have a thin albuginea with scarce seminiferous tubules separated by wide spaces of fibrous connective tissue. Germ cell number is significantly decreased, and gonadal tumor risk is increased (38, 72, 73). The trend for male assignment in these cases has increased over the last decades (74), and the surgical repair of hypospadias and management of tumor risk have become the main challenges in the management (36). At the age of puberty, replacement with testosterone leads to satisfactory development of secondary sex characteristics and growth, but infertility is the rule in adulthood (71).
Gonadal Dysgenesis in 46,XX Patients
As already mentioned, ovarian dysgenesis has no impact on fetal differentiation of the female genitalia; these patients do not seek medical assistance until the age of puberty, when the lack of ovarian steroids results in delayed puberty. These disorders will not be discussed amongst the DSD due to fetal hypogonadism addressed in this review. Congenital adrenal hyperplasia, aromatase deficiency, and other conditions characterized by fetal virilization not due to fetal-onset hypogonadism are also out of the scope of this review.
Disorders of gonadal differentiation leading to the development of testicular tissue and ambiguous or male genitalia may occur in 46,XX individuals. In the originally described “XX male” (75), testes differentiate almost normally—only minor signs of dysgenesis are present, characterized by lower numbers of germ cells (76)—and the patients are fully virilized. These patients usually seek medical attention in adulthood owing to infertility (34). This is a typical case of a cell-specific dissociated hypogonadism, where only the germ cell population is initially affected: spermatogenesis is disturbed by the presence of two X chromosomes and the lack of Y-chromosome genes that are essential for germ cell development. Approximately 90% of XX males are SRY-positive, i.e., they bear part of the short arm of the Y chromosome translocated onto the X chromosome or an autosome (77).
SRY-negative cases have been intriguing for decades (78), until the importance of a balance between pro-testicular and pro-ovarian genes was apprehended. The development of testicular tissue has been attributed to: (i) overexpression of pro-testicular genes, (ii) insufficient expression of pro-ovarian genes, and (iii) mixed or unknown pathogenic mechanisms (37).
Overexpression of testicular factors was initially described in patients bearing duplications of the SOX9 gene or duplications/triplications of its regulatory sequences (79–85), and subsequently found in individuals with duplications or rearrangements of the SOX3 gene (84, 86–88) or its regulatory sequences (89, 90), and duplications of SOX10 (91–95) or FGF9 (96). Insufficient expression of pro-ovarian genes has been proposed as the underlying pathogenesis in patients with loss-of-function mutations or deletions of RSPO1 (97–100) or WNT4 (101). More recently, 46,XX testicular or ovotesticular DSD has been attributed to mutations in NR5A1, encoding SF1 (102–107), WT1 (108) and NR2F2, encoding COUP-TF2 (109, 110).
The resulting phenotype can be either testicular or ovotesticular DSD; the testicular tissue usually shows signs of typical dysgenesis, justifying a primary hypogonadism with whole gonadal dysfunction. External genitalia are ambiguous, reflecting insufficient androgen levels to fully virilize the fetus, and Müllerian remnants may be present, indicating deficient AMH production.
Sex assignment in these cases follows the general suggestions for DSD (2). In patients raised as boys, there is a need for ovarian tissue removal, if present, before the age of puberty to prevent potential complications of cystic follicle development and to avoid gynecomastia due to estrogen secretion (111). The risk of tumor development in the testicular tissue seems to be low, likely due to the absence of Y-chromosome peri-centromeric sequences (72). In patients with ambiguous genitalia and a diagnosis of ovotesticular DSD, attention should be driven to the fact that the ovarian tissue may produce oocytes after pubertal development, which raises the possibility of fertility (112). In these cases, assignment to the female gender may be preferred, and special care should be taken during testicular tissue removal to preserve the ovarian tissue.
Gonadal Dysgenesis in Sex-Chromosome DSD
The lack of the short arm of the Y chromosome, where SRY maps, is responsible for gonadal dysgenesis in patients with sex-chromosome mosaicisms or chimeras. It is noteworthy that the frequent sex-chromosome aneuploidies observed in patients with Klinefelter syndrome (47,XXY and variants) and in triple X syndrome (47,XXX and variants) most usually present typical genitalia at birth and do not represent a differential diagnosis of the conditions discussed in this review. Several different karyotypes have been described in patients with sex-chromosome DSD presenting with ambiguous genitalia. The most prevalent is 45,X/46,XY, which is usually—though not always—associated with asymmetric gonadal differentiation (41, 42, 73). 46,XX/46,XY chimeras usually present with ovotesticular DSD.
Disorders of Testicular Hormone Synthesis: Early Fetal-Onset Primary Hypogonadism With Dissociated Dysfunction
Congenital specific disorders of androgen or AMH synthesis are examples of non-dysgenetic, early-onset, primary hypogonadism characterized by a cell-specific testicular dysfunction in 46,XY patients. The main feature is that only Leydig cell function is impaired in one case and exclusively Sertoli cell secretion is affected in the other, leading to specific clinical and biochemical presentations.
46,XY DSD Due to Disorders of Androgen Synthesis
The main sources of androgens are the gonads and the adrenals. There are steps of steroidogenesis that are common to the testis and the adrenal cortex, and others are specific to the gonads (Figure 5) (24, 29). Disorders of androgen synthesis represent a “dissociated” or “cell-specific” fetal-onset primary hypogonadism, characterized by impaired Leydig cell, but normal Sertoli cell, function. Testosterone levels are low, but serum AMH is within the normal male range or elevated (113), and Müllerian derivatives are absent (Figure 6). Steroidogenic defects, like that of insufficient testosterone conversion to dihydrotestosterone (DHT) occurring locally in the end-organ due to mutations in the gene encoding 5α-reductase type 2, will not be discussed in this review, since they do not represent a form of impaired gonadal function.
Figure 5.

Steroidogenesis. The steroidogenic pathway occurring in testicular Leydig cells is schematically shown in the box. Steroidogenic steps taking place in the adrenals, placenta, and ovaries are outside the box. Modified with permission from Rey et al. (14) © 2010 Elsevier Ltd.
Figure 6.
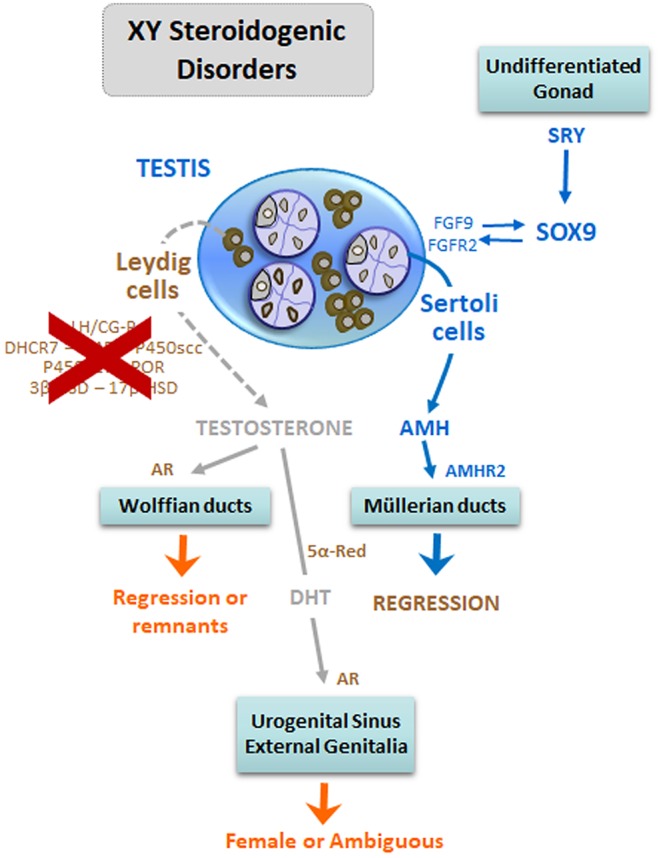
DSD due to steroidogenic defects in 46,XY patients: early fetal-onset primary hypogonadism with Leydig cell-specific dysfunction. The lack of Leydig cell differentiation, due to mutations in the LHCGR gene, or in steroidogenic proteins leads to insufficient androgen production by the testes resulting in undervirilization or complete feminization of the external genitalia. In these patients, there is no uterus or Fallopian tubes because anti-Müllerian hormone (AMH) is produced by Sertoli cells. Modified with permission from Rey et al. (13) © 2016 Elsevier Saunders.
Disorders of androgen synthesis affecting the testes and the adrenals
Cholesterol is the initial substrate for steroid synthesis. Smith-Lemli-Opitz Syndrome (SLOS) results from a defect in dehydrocholesterol reductase (DHCR), involved in the last step of cholesterol synthesis. Cholesterol also plays a major role in the formation of the nervous system, face, and limbs; therefore, patients with SLOS present with DSD associated with polymalformations including microcephaly, facial malformations, growth retardation and congenital heart defects (114). Clinical diagnosis of SLOS is suspected in patients with elevated 7–dehydrocholesterol and low cholesterol.
The first step of gonadal and adrenal steroidogenesis is cholesterol transfer from the cytoplasm into the inner mitochondrial membrane (Figure 5), under control of the steroidogenic acute regulatory protein (StAR). Defects in the STAR gene lead to lipoid congenital adrenal hyperplasia. Most 46,XY newborns present with female external genitalia and severe glucocorticoid and mineralocorticoid deficiency. Milder forms have been reported with ambiguous or male genitalia. All steroids are low and no response is observed after hCG and ACTH stimulation. There is massive adrenal enlargement, due to accumulation of cholesterol esters in the adrenal cortex (115).
Subsequently, cytochrome P450 cholesterol side-chain cleavage enzyme cytochrome P450 (P450scc) catalyzes the synthesis of pregnenolone. P450scc deficiency, due to mutations in CYP11A1, also results in very low steroid levels but without adrenal enlargement. Most 46,XY patients are born with female external genitalia, but partial deficiencies present with ambiguous genitalia (115, 116).
Cytochrome P450c17 has two enzymatic activities (Figure 5): 17α-hydroxylase, which hydroxylates pregnenolone or progesterone into 17α-hydroxypregnenolone or 17α-hydroxyprogesterone (17-OHP), and 17,20-lyase, required for the synthesis of dehydroepiandrosterone (DHEA) and Δ4-androstenedione. Mutations in CYP17A1, encoding P450c17, may induce a combined deficiency of 17α-hydroxylase and 17,20-lyase, but isolated 17,20-lyase deficiency has also been described. The 46,XY newborns are undervirilized. Progesterone and 17-OHP are high in serum whereas DHEA, Δ4-androstenedione, and testosterone are low. Combined 17α-hydroxylase/17,20-lyase deficiency leads to hypertension and hypokalemia due to the accumulation of deoxycorticosterone and corticosterone. In isolated 17,20-lyase deficiency, testicular, but not adrenal, function is impaired (116, 117).
Cytochrome b5 enhances 17,20-lyase activity of P450c17 but does not modify 17-hydroxylase activity. It also reduces methemoglobin (ferric hemoglobin) to normal hemoglobin (ferrous hemoglobin). Mutations in CYB5A result in 46,XY DSD with impaired 17,20-lyase activity and methemoglobinemia (116).
P450 oxidoreductase (POR) is essential for the catalytic activity of P450c17 (preferentially its 17,20-lyase activity), 21-hydroxylase and aromatase (116, 118). XY newborns with POR mutations are undervirilized but present mild signs of congenital adrenal hyperplasia (e.g., elevated 17-OHP), and their mothers virilize due to impaired placental aromatase. Severe mutations lead to the Antley-Bixler syndrome, characterized by craniosynostosis, fusion of long bones, midface hypoplasia, and choanal stenosis. Laboratory findings reflect the combined deficiencies of 17α-hydroxylase and 21-hydroxylase: elevated serum progesterone and 17-OHP but low DHEA and androstenedione.
3β-hydroxysteroid dehydrogenase (HSD) type 2 catalyzes the conversion of Δ5- to Δ4-steroids (pregnenolone to progesterone, 17α-hydroxypregnenolone to 17-OHP, DHEA to androstenedione, and androstenediol to testosterone; Figure 5). Mutations in HSD3B2 leads to 3β-HSD type 2 deficiency, characterized by ambiguous genitalia and variable adrenal impairment (116). Laboratory findings are characterized by an elevated Δ5/Δ4 steroids ratio, particularly after ACTH or hCG stimulation.
Disorders of androgen synthesis specifically affecting the testes
LH and hCG bind to the same receptor on the Leydig cell membrane to induce their typical steroidogenic phenotype. Inactivating mutations in the LHCGR gene lead to Leydig cell aplasia or hypoplasia resulting in various degrees of undervirilization: from micropenis, hypospadias, and cryptorchidism to female external genitalia. All androgen levels are low in basal conditions and after hCG stimulation, but adrenal steroid response to ACTH is normal (119).
The enzyme 17β-HSD type 3 converts Δ4-androstenedione to testosterone in the testes (Figure 5) (24, 116). HSD17B3 gene mutations lead to 46,XY DSD characterized by a female or ambiguous external genital phenotype and unaffected adrenal function. Virilization may occur at pubertal age owing to androgen synthesis by other isoforms, like that encoded by HSD17B5 (also called AKR1C3) involved in the “backdoor” steroidogenic pathway (116). Laboratory findings are characterized by elevated Δ4-androstenedione and low testosterone after hCG stimulation (24).
46,XY DSD Due to Disorders of AMH Synthesis
Defects in Sertoli cell AMH secretion, due to mutations in the AMH gene, are a typical form of “dissociated” or “cell-specific” fetal-onset primary hypogonadism, characterized by impaired Sertoli cell, but normal Leydig cell, function (Figure 7). The resulting disorder is known as the persistent Müllerian duct syndrome (PMDS), which is characterized by the presence of the uterus and Fallopian tubes in an otherwise normally virilized newborn (27). The existence of ambiguous external genitalia, which reflects a concomitant defect in androgen synthesis, rules out the diagnosis of PMDS. Clinical manifestations are cryptorchidism with or without inguinal hernia, and the diagnosis is not suspected until Müllerian derivatives are unexpectedly found at surgery. Mutations in the AMH gene explain ~45% of PMDS patients. Serum testosterone and gonadotropins are in the male range. AMH is undetectable in patients with AMH mutations. In patients with PMDS due to AMH receptor defects, testicular function is not affected and AMH levels are within the male range (25). Malignant tumors of the testes occur in ~1/3 of adults with the disorder, while malignancies of the Müllerian derivatives are less frequent (27).
Figure 7.
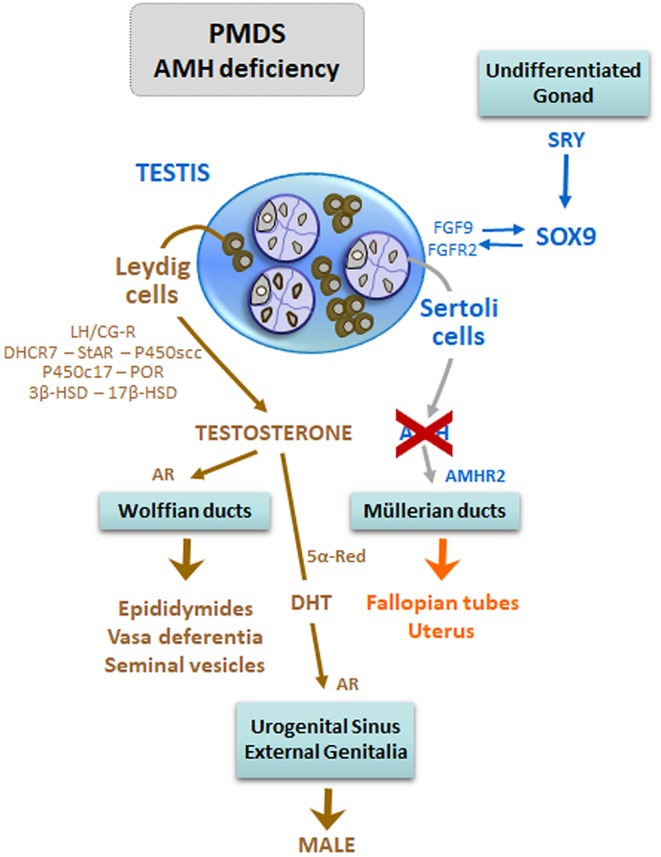
DSD due to anti-Müllerian hormone (AMH) deficiency in 46,XY patients: early fetal-onset primary hypogonadism with Sertoli cell-specific dysfunction. Mutations of the AMH gene are responsible for the persistence of the uterus and Fallopian tubes, a condition known as Persistent Müllerian Duct Syndrome (PMDS), in otherwise normally virilized newborns. Modified with permission from Rey et al. (13) © 2016 Elsevier Saunders.
Early Fetal-Onset Central Hypogonadism Does Not Lead to DSD
The GnRH neurons derive from cells present in the nasal placode (120), which migrate together with olfactory axons through the cribriform plate and arrive in the developing forebrain in the 9th−10th weeks. The pituitary develops from the Rathke's pouch, and functional gonadotropes secrete LH from fetal week 12 (121), when sex differentiation is almost complete. As mentioned, Leydig cells secrete androgens driven by hCG rather by pituitary LH during the period of sex differentiation. This explains why 46,XY fetuses virilize in spite of complete absence of gonadotropin secretion in congenital central (hypogonadotropic) hypogonadism. Because testicular androgen synthesis is dependent on fetal LH during the second and third trimesters, these patients present with micropenis and cryptorchidism at birth (31).
Concluding Remarks
In patients with a Y chromosome (46,XY, 45,X/46,XY, or variants), DSD usually results from testicular dysgenesis, an early fetal-onset hypogonadism characterized by whole gonadal dysfunction presenting with serum levels of testosterone and AMH that are below the male range (Figure 8). In 46,XY individuals, DSD may result from isolated Leydig cell dysfunction; this dissociated form of fetal-onset hypogonadism is characterized by androgen levels below the male range and AMH within the male range. When there is an isolated Sertoli cell dysfunction, AMH is low while testosterone is within the male range. If both testosterone and AMH are within the male range, DSD is not due to fetal-onset hypogonadism, but rather to a defect in the target organ, e.g., androgen insensitivity or 5α-reductase deficiency, or to an anatomical malformation. In 46,XX newborns with ambiguous or male genitalia, testicular or ovotesticular DSD may be the underlying cause; Sertoli and Leydig cell populations may be affected, resulting in undervirilization but they may be functionally normal, like in the XX male presenting with testosterone and AMH levels within the male range. In newborns with androgen levels above but AMH within the female range, virilization is not related to hypogonadal states but rather to adrenal or placental dysfunction. In chimeric forms of DSD, e.g., 46,XX/46,XY or variants, ovotesticular differentiation usually occurs, characterized by androgen and AMH levels that are above the female but below the male range, indicating that there is a whole testicular tissue dysfunction. All these are primary forms of fetal hypogonadism. Fetal-onset central hypogonadism usually leads to micropenis or cryptorchidism, reflecting insufficient androgen production in the second half of intrauterine life, but does not result in ambiguous genitalia.
Figure 8.

DSD and fetal-onset male hypogonadism. 46,XY DSD may result from various forms of primary fetal-onset male hypogonadism, or from defects in hormone action in target organs (AIS: androgen insensitivity syndrome, or 5α-reductase deficiency), without hypogonadism. In 46,XX individuals with testicular differentiation, partial virilization results from impaired testicular function; in completely virilized newborns, Leydig and Sertoli cell function is preserved and only germ cell development is impaired; however, the most frequent situation of XX virilization results from dysfunction of the adrenals (e.g., congenital adrenal hyperplasia, CAH) or the placenta (aromatase deficiency). Chromosomal DSD with 45,X/46,XY or 46,XX/46,XY karyotypes, testicular tissue is characterized by impaired function of all cell populations, as shown by the existence of testosterone and AMH levels that are below the male range. AIS, androgen insensitivity syndrome; CAH, congenital adrenal hyperplasia; Def., deficiency; Malformative, anatomical malformation of the genitalia not due to hormonal disorders; PMDS, persistent Müllerian duct syndrome, due to specific AMH defects, < or > female or male: below or above normal female or male range.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
Funding. Partially supported by grants PIP 11220130100687 of Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET) and PID-C 2017-0032 and PICT 2014-2490 of Fondo para la Investigación Científica y Tecnológica (FONCYT).
References
- 1.Hughes IA, Houk C, Ahmed SF, Lee PA, LWPES/ESPE consensus group . Consensus statement on management of intersex disorders. Arch Dis Child. (2006) 91:554–63. 10.1136/adc.2006.098319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee PA, Nordenstrom A, Houk CP, Ahmed SF, Auchus R, Baratz A, et al. global disorders of sex development update since 2006: perceptions, approach and care. Horm Res Paediatr. (2016) 85:158–80. 10.1159/000442975 [DOI] [PubMed] [Google Scholar]
- 3.Freire AV, Grinspon RP, Rey RA. Importance of serum testicular protein hormone measurement in the assessment of disorders of sex development. Sex Dev. (2018) 12:30–40. 10.1159/000479572 [DOI] [PubMed] [Google Scholar]
- 4.Salonia A, Rastrelli G, Hackett G, Seminara SB, Huhtaniemi IT, Rey RA, et al. Paediatric and adult-onset male hypogonadism. Nat Rev Dis Primers. (2019) 5:38. 10.1038/s41572-019-0087-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rey RA, Grinspon RP, Gottlieb S, Pasqualini T, Knoblovits P, Aszpis S, et al. Male hypogonadism: an extended classification based on a developmental, endocrine physiology-based approach. Andrology. (2013) 1:3–16. 10.1111/j.2047-2927.2012.00008.x [DOI] [PubMed] [Google Scholar]
- 6.Grinspon RP, Freire AV, Rey RA. Hypogonadism in pediatric health: adult medicine concepts fail. Trends Endocrinol Metab. (2019) 30:879–90. 10.1016/j.tem.2019.08.002 [DOI] [PubMed] [Google Scholar]
- 7.Rey RA, Houk C, Witchel S, Lee PA. Disorders of sex development. In: Gardner DG, Shoback D, editors. Greenspan's Basic and Clinical Endocrinology, 10th ed New York, NY: McGraw-Hill Education; (2018). p. 501–46. [Google Scholar]
- 8.Rey R, Picard JY. Embryology and endocrinology of genital development. Baillieres Clin Endocrinol Metab. (1998) 12:17–33. 10.1016/S0950-351X(98)80427-8 [DOI] [PubMed] [Google Scholar]
- 9.Lin YT, Capel B. Cell fate commitment during mammalian sex determination. Curr Opin Genet Dev. (2015) 32:144–52. 10.1016/j.gde.2015.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grinspon RP, Rey RA. Disorders of sex development with testicular differentiation in SRY-negative 46,XX individuals: clinical and genetic aspects. Sex Dev. (2016) 10:1–1. 10.1159/000445088 [DOI] [PubMed] [Google Scholar]
- 11.Baetens D, Verdin H, De Baere E, Cools M. Update on the genetics of differences of sex development (DSD). Best Pract Res Clin Endocrinol Metab. (2019) 33:101271. 10.1016/j.beem.2019.04.005 [DOI] [PubMed] [Google Scholar]
- 12.Makela JA, Koskenniemi JJ, Virtanen HE, Toppari J. Testis development. Endocr Rev. (2019) 40:857–905. 10.1210/er.2018-00140 [DOI] [PubMed] [Google Scholar]
- 13.Rey RA, Josso N. Diagnosis and treatment of disorders of sexual development. In: Jameson JL, De Groot LC, editors. Endocrinology: Adult and Pediatric, 7th ed Philadelphia, PA: Elsevier Saunders; (2016). p. 2086–118. [Google Scholar]
- 14.Rey RA, Grinspon RP. Normal male sexual differentiation and aetiology of disorders of sex development. Best Pract Res Clin Endocrinol Metab. (2011) 25:221–38. 10.1016/j.beem.2010.08.013 [DOI] [PubMed] [Google Scholar]
- 15.Josso N, Rey RA, Picard JY. Anti-müllerian hormone: a valuable addition to the toolbox of the pediatric endocrinologist. Int J Endocrinol. (2013) 2013:674105. 10.1155/2013/674105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klonisch T, Fowler PA, Hombach-Klonisch S. Molecular and genetic regulation of testis descent and external genitalia development. Dev Biol. (2004) 270:1–18. 10.1016/j.ydbio.2004.02.018 [DOI] [PubMed] [Google Scholar]
- 17.Ivell R, Anand-Ivell R. Biological role and clinical significance of insulin-like peptide 3. Curr Opin Endocrinol Diabetes Obes. (2011) 18:210–6. 10.1097/MED.0b013e3283453fe6 [DOI] [PubMed] [Google Scholar]
- 18.Jost A. Problems of fetal endocrinology: the gonadal and hypophyseal hormones. Recent Prog Horm Res. (1953) 8:379–418. [Google Scholar]
- 19.Grinspon RP, Bedecarrás P, Ballerini MG, Iñíguez G, Rocha A, Mantovani Rodrigues Resende EA, et al. Early onset of primary hypogonadism revealed by serum anti-Müllerian hormone determination during infancy and childhood in trisomy 21. Int J Androl. (2011) 34:e487–e98. 10.1111/j.1365-2605.2011.01210.x [DOI] [PubMed] [Google Scholar]
- 20.Bergadá I, Milani C, Bedecarrás P, Andreone L, Ropelato MG, Gottlieb S, et al. Time course of the serum gonadotropin surge, inhibins, and anti-Mullerian hormone in normal newborn males during the first month of life. J Clin Endocrinol Metab. (2006) 91:4092–8. 10.1210/jc.2006-1079 [DOI] [PubMed] [Google Scholar]
- 21.Bay K, Virtanen HE, Hartung S, Ivell R, Main KM, Skakkebæk NE, et al. Insulin-like factor 3 levels in cord blood and serum from children: effects of age, postnatal hypothalamic-pituitary-gonadal axis activation, and cryptorchidism. J Clin Endocrinol Metab. (2007) 92:4020–7. 10.1210/jc.2007-0974 [DOI] [PubMed] [Google Scholar]
- 22.Grinspon RP, Rey RA. When hormone defects cannot explain it: Malformative disorders of sex development. Birth Defects Res C Embryo Today. (2014) 102:359–73. 10.1002/bdrc.21086 [DOI] [PubMed] [Google Scholar]
- 23.Ladjouze A, Donaldson M. Primary gonadal failure. Best Pract Res Clin Endocrinol Metab. (2019) 33:101295. 10.1016/j.beem.2019.101295 [DOI] [PubMed] [Google Scholar]
- 24.Mendonça BB, Costa EM, Belgorosky A, Rivarola MA, Domenice S. 46,XY DSD due to impaired androgen production. Best Pract Res Clin Endocrinol Metab. (2010) 24:243–62. 10.1016/j.beem.2009.11.003 [DOI] [PubMed] [Google Scholar]
- 25.Josso N, Rey R, Picard JY. Testicular anti-Mullerian hormone: clinical applications in DSD. Semin Reprod Med. (2012) 30:364–73. 10.1055/s-0032-1324719 [DOI] [PubMed] [Google Scholar]
- 26.Hughes IA, Deeb A. Androgen resistance. Best Pract Res Clin Endocrinol Metab. (2006) 20:577–98. 10.1016/j.beem.2006.11.003 [DOI] [PubMed] [Google Scholar]
- 27.Picard JY, Cate RL, Racine C, Josso N. The persistent mullerian duct syndrome: an update based upon a personal experience of 157 cases. Sex Dev. (2017) 11:109–25. 10.1159/000475516 [DOI] [PubMed] [Google Scholar]
- 28.Belgorosky A, Guercio G, Pepe C, Saraco N, Rivarola MA. Genetic and clinical spectrum of aromatase deficiency in infancy, childhood and adolescence. Horm Res. (2009) 72:321–30. 10.1159/000249159 [DOI] [PubMed] [Google Scholar]
- 29.Turcu AF, Auchus RJ. Adrenal steroidogenesis and congenital adrenal hyperplasia. Endocrinol Metab Clin North Am. (2015) 44:275–96. 10.1016/j.ecl.2015.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hakim C, Padmanabhan V, Vyas AK. Gestational hyperandrogenism in developmental programming. Endocrinology. (2017) 158:199–212. 10.1210/en.2016-1801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grinspon RP, Loreti N, Braslavsky D, Valeri C, Schteingart H, Ballerini MG, et al. Spreading the clinical window for diagnosing fetal-onset hypogonadism in boys. Front Endocrinol (Lausanne). (2014) 5:51. 10.3389/fendo.2014.00051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grinspon RP, Ropelato MG, Bedecarrás P, Loreti N, Ballerini MG, Gottlieb S, et al. Gonadotrophin secretion pattern in anorchid boys from birth to pubertal age: pathophysiological aspects and diagnostic usefulness. Clin Endocrinol (Oxf). (2012) 76:698–705. 10.1111/j.1365-2265.2011.04297.x [DOI] [PubMed] [Google Scholar]
- 33.Basaria S. Male hypogonadism. Lancet. (2014) 383:1250–63. 10.1016/S0140-6736(13)61126-5 [DOI] [PubMed] [Google Scholar]
- 34.Juul A, Almstrup K, Andersson AM, Jensen TK, Jorgensen N, Main KM, et al. Possible fetal determinants of male infertility. Nat Rev Endocrinol. (2014) 10:553–62. 10.1038/nrendo.2014.97 [DOI] [PubMed] [Google Scholar]
- 35.Cools M, Nordenström A, Robeva R, Hall J, Westerveld P, Flück C, et al. Caring for individuals with a difference of sex development (DSD): a consensus statement. Nat Rev Endocrinol. (2018) 14:415–29. 10.1038/s41574-018-0010-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wisniewski AB, Batista RL, Costa EMF, Finlayson C, Sircili MHP, Denes FT, et al. Management of 46,XY differences/disorders of sex development (DSD) throughout life. Endocr Rev. (2019) 40:1547–72. 10.1210/er.2019-00049 [DOI] [PubMed] [Google Scholar]
- 37.Grinspon RP, Rey RA. Molecular characterization of XX maleness. Int J Mol Sci. (2019) 20:6089. 10.3390/ijms20236089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chemes HE, Venara M, Del Rey G, Arcari AJ, Musse MP, Papazian R, et al. Is a CIS phenotype apparent in children with disorders of sex development? Milder testicular dysgenesis is associated with a higher risk of malignancy. Andrology. (2015) 3:59–69. 10.1111/andr.301 [DOI] [PubMed] [Google Scholar]
- 39.Berney DM, Looijenga L, Idrees M, Oosterhuis JW, Rajpert-De Meyts E, Ulbright TM, et al. Germ cell neoplasia in situ (GCNIS). Evolution of the current nomenclature for testicular pre-invasive germ cell malignancy. Histopathology. (2016) 69:7–10. 10.1111/his.12958 [DOI] [PubMed] [Google Scholar]
- 40.Ahmed SF, Achermann JC, Arlt W, Balen A, Conway G, Edwards Z, et al. Society for endocrinology UK guidance on the initial evaluation of an infant or an adolescent with a suspected disorder of sex development (Revised 2015). Clin Endocrinol (Oxf). (2016) 84:771–88. 10.1111/cen.12857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sohval AR. “Mixed” gonadal dysgenesis: a variety of hermaphroditism. Am J Hum Genet. (1963) 15:155–8. [PMC free article] [PubMed] [Google Scholar]
- 42.Bergadá C, Cleveland WW, Jones HW, Jr, Wilkins L. Gonadal histology in patients with male pseudohermaphroditism and atypical gonadal dysgenesis: relation to theories of sex differentiation. Acta Endocrinol (Copenh). (1962) 40:493–520. 10.1530/acta.0.0400493 [DOI] [PubMed] [Google Scholar]
- 43.Fan W, Wang B, He S, Zhang T, Yin C, Chen Y, et al. A novel missense mutation 224G>T (R75M) in SRY coding region interferes with nuclear import and results in 46, XY complete gonadal dysgenesis. PLoS One. (2016) 11:e0168484. 10.1371/journal.pone.0168484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bashamboo A, Eozenou C, Rojo S, McElreavey K. Anomalies in human sex determination provide unique insights into the complex genetic interactions of early gonad development. Clin Genet. (2017) 91:143–56. 10.1111/cge.12932 [DOI] [PubMed] [Google Scholar]
- 45.Pearlman A, Loke J, Le CC, White S, Chin L, Friedman A, et al. Mutations in MAP3K1 cause 46,XY disorders of sex development and implicate a common signal transduction pathway in human testis determination. Am J Hum Genet. (2010) 87:898–904. 10.1016/j.ajhg.2010.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Harris A, Siggers P, Corrochano S, Warr N, Sagar D, Grimes DT, et al. ZNRF3 functions in mammalian sex determination by inhibiting canonical WNT signaling. Proc Natl Acad Sci U S A. (2018) 115:5474–9. 10.1073/pnas.1801223115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.da Silva TE, Gomes NL, Lerario AM, Keegan CE, Nishi MY, Carvalho FM, et al. Genetic evidence of the association of DEAH-Box helicase 37 defects with 46,XY gonadal dysgenesis spectrum. J Clin Endocrinol Metab. (2019) 104:5923–34. 10.1210/jc.2019-00984 [DOI] [PubMed] [Google Scholar]
- 48.McElreavey K, Jorgensen A, Eozenou C, Merel T, Bignon-Topalovic J, Tan DS, et al. Pathogenic variants in the DEAH-box RNA helicase DHX37 are a frequent cause of 46,XY gonadal dysgenesis and 46,XY testicular regression syndrome. Genet Med. (2019) 22:150–9. 10.1038/s41436-019-0606-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Portnoi MF, Dumargne MC, Rojo S, Witchel SF, Duncan AJ, Eozenou C, et al. Mutations involving the SRY-related gene SOX8 are associated with a spectrum of human reproductive anomalies. Hum Mol Genet. (2018) 27:1228–40. 10.1093/hmg/ddy037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bardoni B, Zanaria E, Guioli S, Floridia G, Worley KC, Tonini G, et al. A dosage sensitive locus at chromosome Xp21 is involved in male to female sex reversal. Nat Genet. (1994) 7:497–501. 10.1038/ng0894-497 [DOI] [PubMed] [Google Scholar]
- 51.Barbaro M, Oscarson M, Schoumans J, Staaf J, Ivarsson SA, Wedell A. Isolated 46,XY gonadal dysgenesis in two sisters caused by a Xp21.2 interstitial duplication containing the DAX1 gene. J Clin Endocrinol Metab. (2007) 92:3305–13. 10.1210/jc.2007-0505 [DOI] [PubMed] [Google Scholar]
- 52.Schafer AJ, Foster JW, Kwok C, Weller PA, Guioli S, Goodfellow PN. Campomelic dysplasia with XY sex reversal: diverse phenotypes resulting from mutations in a single gene. Ann N Y Acad Sci. (1996) 785:137–49. 10.1111/j.1749-6632.1996.tb56252.x [DOI] [PubMed] [Google Scholar]
- 53.Niaudet P, Gubler MC. WT1 and glomerular diseases. Pediatr Nephrol. (2006) 21:1653–60. 10.1007/s00467-006-0208-1 [DOI] [PubMed] [Google Scholar]
- 54.Suntharalingham JP, Buonocore F, Duncan AJ, Achermann JC. DAX-1 (NR0B1) and steroidogenic factor-1 (SF-1, NR5A1) in human disease. Best Pract Res Clin Endocrinol Metab. (2015) 29:607–19. 10.1016/j.beem.2015.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gibbons R. Alpha thalassaemia-mental retardation, X linked. Orphanet J Rare Dis. (2006) 1:15. 10.1186/1750-1172-1-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Werner R, Merz H, Birnbaum W, Marshall L, Schroder T, Reiz B, et al. 46,XY Gonadal dysgenesis due to a homozygous mutation in desert hedgehog (DHH) identified by exome sequencing. J Clin Endocrinol Metab. (2015) 100:E1022–9. 10.1210/jc.2015-1314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bashamboo A, Brauner R, Bignon-Topalovic J, Lortat-Jacob S, Karageorgou V, Lourenco D, et al. Mutations in the FOG2/ZFPM2 gene are associated with anomalies of human testis determination. Hum Mol Genet. (2014) 23:3657–65. 10.1093/hmg/ddu074 [DOI] [PubMed] [Google Scholar]
- 58.Bagheri-Fam S, Bird AD, Zhao L, Ryan JM, Yong M, Wilhelm D, et al. Testis determination requires a specific FGFR2 isoform to repress FOXL2. Endocrinology. (2017) 158:3832–43. 10.1210/en.2017-00674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Quinonez SC, Park JM, Rabah R, Owens KM, Yashar BM, Glover TW, Keegan CE. 9p partial monosomy and disorders of sex development: review and postulation of a pathogenetic mechanism. Am J Med Genet A. (2013) 161A:1882–96. 10.1002/ajmg.a.36018 [DOI] [PubMed] [Google Scholar]
- 60.Callier P, Calvel P, Matevossian A, Makrythanasis P, Bernard P, Kurosaka H, et al. Loss of function mutation in the palmitoyl-transferase HHAT leads to syndromic 46,XY disorder of sex development by impeding Hedgehog protein palmitoylation and signaling. PLoS Genet. (2014) 10:e1004340. 10.1371/journal.pgen.1004340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kitamura K, Yanazawa M, Sugiyama N, Miura H, Iizuka-Kogo A, Kusaka M, et al. Mutation of ARX causes abnormal development of forebrain and testes in mice and X-linked lissencephaly with abnormal genitalia in humans. Nat Genet. (2002) 32:359–69. 10.1038/ng1009 [DOI] [PubMed] [Google Scholar]
- 62.Rossetti LZ, Glinton K, Yuan B, Liu P, Pillai N, Mizerik E, et al. Review of the phenotypic spectrum associated with haploinsufficiency of MYRF. Am J Med Genet A. (2019) 179:1376–82. 10.1002/ajmg.a.61182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Puffenberger EG, Hu-Lince D, Parod JM, Craig DW, Dobrin SE, Conway AR, et al. Mapping of sudden infant death with dysgenesis of the testes syndrome (SIDDT) by a SNP genome scan and identification of TSPYL loss of function. Proc Natl Acad Sci U S A. (2004) 101:11689–94. 10.1073/pnas.0401194101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Biason-Lauber A, Konrad D, Meyer M, deBeaufort C, Schoenle EJ. Ovaries and female phenotype in a girl with 46,XY karyotype and mutations in the CBX2 gene. Am J Hum Genet. (2009) 84:658–63. 10.1016/j.ajhg.2009.03.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Baetens D, Guran T, Mendonca BB, Gomes NL, De Cauwer L, Peelman F, et al. Biallelic and monoallelic ESR2 variants associated with 46,XY disorders of sex development. Genet Med. (2018) 20:717–27. 10.1038/gim.2017.163 [DOI] [PubMed] [Google Scholar]
- 66.Ogata T, Sano S, Nagata E, Kato F, Fukami M. MAMLD1 and 46,XY disorders of sex development. Semin Reprod Med. (2012) 30:410–6. 10.1055/s-0032-1324725 [DOI] [PubMed] [Google Scholar]
- 67.Flück CE, Audí L, Fernández-Cancio M, Sauter KS, Martínez de LaPiscina I, Castaño L, et al. Broad phenotypes of disorders/differences of sex development in MAMLD1 patients through oligogenic disease. Front Genet. (2019) 10:746. 10.3389/fgene.2019.00746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ilaslan E, Calvel P, Nowak D, Szarras-Czapnik M, Slowikowska-Hilczer J, Spik A, et al. A case of two sisters suffering from 46,XY gonadal dysgenesis and carrying a mutation of a novel candidate sex-determining gene STARD8 on the X chromosome. Sex Dev. (2018) 12:191–5. 10.1159/000489692 [DOI] [PubMed] [Google Scholar]
- 69.White S, Hewitt J, Turbitt E, van der Zwan Y, Hersmus R, Drop S, et al. A multi-exon deletion within WWOX is associated with a 46,XY disorder of sex development. Eur J Hum Genet. (2012) 20:348–51. 10.1038/ejhg.2011.204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Conte FA, Grumbach MM, Kaplan SL. A diphasic pattern of gonadotropin secretion in patients with the syndrome of gonadal dysgenesis. J Clin Endocrinol Metab. (1975) 40:670–4. 10.1210/jcem-40-4-670 [DOI] [PubMed] [Google Scholar]
- 71.Guercio G, Costanzo M, Grinspon RP, Rey RA. Fertility issues in disorders of sex development. Endocrinol Metab Clin North Am. (2015) 44:867–81. 10.1016/j.ecl.2015.07.012 [DOI] [PubMed] [Google Scholar]
- 72.Cools M, Looijenga LH, Wolffenbuttel KP, T'Sjoen G. Managing the risk of germ cell tumourigenesis in disorders of sex development patients. Endocr Dev. (2014) 27:185–96. 10.1159/000363642 [DOI] [PubMed] [Google Scholar]
- 73.Andrade JGR, Fabbri-Scallet H, Dos Santos AP, Cools M, Werner R, Hiort O, et al. Clinical findings and follow-up of 46,XY and 45,X/46,XY testicular dysgenesis. Sex Dev. (2019). 10.1159/000504239. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 74.Kolesinska Z, Ahmed SF, Niedziela M, Bryce J, Molinska-Glura M, Rodie M, et al. Changes over time in sex assignment for disorders of sex development. Pediatrics. (2014) 134:e710–5. 10.1542/peds.2014-1088 [DOI] [PubMed] [Google Scholar]
- 75.de la Chapelle A. Analytic review: nature and origin of males with XX sex chromosomes. Am J Hum Genet. (1972) 24:71–105. [PMC free article] [PubMed] [Google Scholar]
- 76.Boucekkine C, Toublanc JE, Abbas N, Chaabouni S, Ouahid S, Semrouni M, et al. Clinical and anatomical spectrum in XX sex reversed patients. Relationship to the presence of Y specific DNA-sequences. Clin Endocrinol (Oxf). (1994) 40:733–42. 10.1111/j.1365-2265.1994.tb02506.x [DOI] [PubMed] [Google Scholar]
- 77.Vorona E, Zitzmann M, Gromoll J, Schuring AN, Nieschlag E. Clinical, endocrinological, and epigenetic features of the 46,XX male syndrome, compared with 47,XXY Klinefelter patients. J Clin Endocrinol Metab. (2007) 92:3458–65. 10.1210/jc.2007-0447 [DOI] [PubMed] [Google Scholar]
- 78.McElreavey K, Vilain E, Abbas N, Herskowitz I, Fellous M. A regulatory cascade hypothesis for mammalian sex determination: SRY represses a negative regulator of male development. Proc Natl Acad Sci U S A. (1993) 90:3368–72. 10.1073/pnas.90.8.3368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cox JJ, Willatt L, Homfray T, Woods CG. A SOX9 duplication and familial 46,XX developmental testicular disorder. N Engl J Med. (2011) 364:91–3. 10.1056/NEJMc1010311 [DOI] [PubMed] [Google Scholar]
- 80.Benko S, Gordon CT, Mallet D, Sreenivasan R, Thauvin-Robinet C, Brendehaug A, et al. Disruption of a long distance regulatory region upstream of SOX9 in isolated disorders of sex development. J Med Genet. (2011) 48:825–30. 10.1136/jmedgenet-2011-100255 [DOI] [PubMed] [Google Scholar]
- 81.Xiao B, Ji X, Xing Y, Chen YW, Tao J. A rare case of 46, XX SRY-negative male with approximately 74-kb duplication in a region upstream of SOX9. Eur J Med Genet. (2013) 56:695–8. 10.1016/j.ejmg.2013.10.001 [DOI] [PubMed] [Google Scholar]
- 82.Hyon C, Chantot-Bastaraud S, Harbuz R, Bhouri R, Perrot N, Peycelon M, et al. Refining the regulatory region upstream of SOX9 associated with 46,XX testicular disorders of sex development (DSD). Am J Med Genet A. (2015) 167A:1851–8. 10.1002/ajmg.a.37101 [DOI] [PubMed] [Google Scholar]
- 83.Kim GJ, Sock E, Buchberger A, Just W, Denzer F, Hoepffner W, et al. Copy number variation of two separate regulatory regions upstream of SOX9 causes isolated 46,XY or 46,XX disorder of sex development. J Med Genet. (2015) 52:240–7. 10.1136/jmedgenet-2014-102864 [DOI] [PubMed] [Google Scholar]
- 84.Vetro A, Dehghani MR, Kraoua L, Giorda R, Beri S, Cardarelli L, et al. Testis development in the absence of SRY: chromosomal rearrangements at SOX9 and SOX3. Eur J Hum Genet. (2015) 23:1025–32. 10.1038/ejhg.2014.237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ohnesorg T, van den Bergen JA, Belluoccio D, Shankara-Narayana N, Kean AM, Vasilaras A, et al. A duplication in a patient with 46,XX ovo-testicular disorder of sex development refines the SOX9 testis-specific regulatory region to 24 kb. Clin Genet. (2017) 92:347–9. 10.1111/cge.12976 [DOI] [PubMed] [Google Scholar]
- 86.Sutton E, Hughes J, White S, Sekido R, Tan J, Arboleda V, et al. Identification of SOX3 as an XX male sex reversal gene in mice and humans. J Clin Invest. (2011) 121:328–41. 10.1172/JCI42580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Moalem S, Babul-Hirji R, Stavropolous DJ, Wherrett D, Bagli DJ, Thomas P, et al. XX male sex reversal with genital abnormalities associated with a de novo SOX3 gene duplication. Am J Med Genet A. (2012) 158A:1759–64. 10.1002/ajmg.a.35390 [DOI] [PubMed] [Google Scholar]
- 88.Grinspon RP, Nevado J, Mori Alvarez ML, del Rey G, Castera R, Venara M, et al. 46,XX ovotesticular DSD associated with a SOX3 gene duplication in a SRY-negative boy. Clin Endocrinol (Oxf). (2016) 85:669–75. 10.1111/cen.13126 [DOI] [PubMed] [Google Scholar]
- 89.Mizuno K, Kojima Y, Kamisawa H, Moritoki Y, Nishio H, Nakane A, et al. Elucidation of distinctive genomic DNA structures in patients with 46,XX testicular disorders of sex development using genome wide analyses. J Urol. (2014) 192:535–41. 10.1016/j.juro.2014.02.044 [DOI] [PubMed] [Google Scholar]
- 90.Haines B, Hughes J, Corbett M, Shaw M, Innes J, Patel L, et al. Interchromosomal insertional translocation at Xq26.3 alters SOX3 expression in an individual with XX male sex reversal. J Clin Endocrinol Metab. (2015) 100:E815–20. 10.1210/jc.2014-4383 [DOI] [PubMed] [Google Scholar]
- 91.Nicholl RM, Grimsley L, Butler L, Palmer RW, Rees HC, Savage MO, et al. Trisomy 22 and intersex. Arch Dis Child Fetal Neonatal Ed. (1994) 71:F57–8. 10.1136/fn.71.1.F57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Aleck KA, Argueso L, Stone J, Hackel JG, Erickson RP. True hermaphroditism with partial duplication of chromosome 22 and without SRY. Am J Med Genet. (1999) 85:2–4. [DOI] [PubMed] [Google Scholar]
- 93.Seeherunvong T, Perera EM, Bao Y, Benke PJ, Benigno A, Donahue RP, et al. 46,XX sex reversal with partial duplication of chromosome arm 22q. Am J Med Genet. (2004) 127A:149–51. 10.1002/ajmg.a.20630 [DOI] [PubMed] [Google Scholar]
- 94.Polanco JC, Wilhelm D, Davidson TL, Knight D, Koopman P. Sox10 gain-of-function causes XX sex reversal in mice: implications for human 22q-linked disorders of sex development. Hum Mol Genet. (2010) 19:506–16. 10.1093/hmg/ddp520 [DOI] [PubMed] [Google Scholar]
- 95.Falah N, Posey JE, Thorson W, Benke P, Tekin M, Tarshish B, et al. 22q11.2q13 duplication including SOX10 causes sex-reversal and peripheral demyelinating neuropathy, central dysmyelinating leukodystrophy, waardenburg syndrome, and hirschsprung disease. Am J Med Genet A. (2017) 173:1066–70. 10.1002/ajmg.a.38109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chiang HS, Wu YN, Wu CC, Hwang JL. Cytogenic and molecular analyses of 46,XX male syndrome with clinical comparison to other groups with testicular azoospermia of genetic origin. J Formos Med Assoc. (2013) 112:72–8. 10.1016/j.jfma.2012.02.009 [DOI] [PubMed] [Google Scholar]
- 97.Parma P, Radi O, Vidal V, Chaboissier MC, Dellambra E, Valentini S, et al. R-spondin1 is essential in sex determination, skin differentiation and malignancy. Nat Genet. (2006) 38:1304–9. 10.1038/ng1907 [DOI] [PubMed] [Google Scholar]
- 98.Tomaselli S, Megiorni F, De Bernardo C, Felici A, Marrocco G, Maggiulli G, et al. Syndromic true hermaphroditism due to an R-spondin1 (RSPO1) homozygous mutation. Hum Mutat. (2008) 29:220–6. 10.1002/humu.20665 [DOI] [PubMed] [Google Scholar]
- 99.Naasse Y, Bakhchane A, Charoute H, Jennane F, Bignon-Topalovic J, Malki A, et al. A novel homozygous missense mutation in the FU-CRD2 domain of the R-spondin1 gene associated with familial 46,XX DSD. Sex Dev. (2017) 11:269–74. 10.1159/000485393 [DOI] [PubMed] [Google Scholar]
- 100.Tallapaka K, Venugopal V, Dalal A, Aggarwal S. Novel RSPO1 mutation causing 46,XX testicular disorder of sex development with palmoplantar keratoderma: a review of literature and expansion of clinical phenotype. Am J Med Genet A. (2018) 176:1006–10. 10.1002/ajmg.a.38646 [DOI] [PubMed] [Google Scholar]
- 101.Mandel H, Shemer R, Borochowitz ZU, Okopnik M, Knopf C, Indelman M, et al. SERKAL syndrome: an autosomal-recessive disorder caused by a loss-of-function mutation in WNT4. Am J Hum Genet. (2008) 82:39–47. 10.1016/j.ajhg.2007.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bashamboo A, Donohoue PA, Vilain E, Rojo S, Calvel P, Seneviratne SN, et al. A recurrent p.Arg92Trp variant in steroidogenic factor-1 (NR5A1) can act as a molecular switch in human sex development. Hum Mol Genet. (2016) 25:3446–53. 10.1093/hmg/ddw186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Domenice S, Machado AZ, Ferreira FM, Ferraz-de-Souza B, Lerario AM, Lin L, et al. Wide spectrum of NR5A1-related phenotypes in 46,XY and 46,XX individuals. Birth Defects Res C Embryo Today. (2016) 108:309–20. 10.1002/bdrc.21145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Miyado M, Inui M, Igarashi M, Katoh-Fukui Y, Takasawa K, Hakoda A, et al. The p.R92W variant of NR5A1/Nr5a1 induces testicular development of 46,XX gonads in humans, but not in mice: phenotypic comparison of human patients and mutation-induced mice. Biol Sex Differ. (2016) 7:56 10.1186/s13293-016-0114-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Baetens D, Stoop H, Peelman F, Todeschini AL, Rosseel T, Coppieters F, et al. NR5A1 is a novel disease gene for 46,XX testicular and ovotesticular disorders of sex development. Genet Med. (2017) 19:367–76. 10.1038/gim.2016.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Knarston IM, Robevska G, van den Bergen JA, Eggers S, Croft B, Yates J, et al. NR5A1 gene variants repress the ovarian-specific WNT signaling pathway in 46,XX disorders of sex development patients. Hum Mutat. (2019) 40:207–16. 10.1002/humu.23672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Saito-Hakoda A, Kanno J, Suzuki D, Kawashima S, Kamimura M, Hirano K, et al. A follow-up from infancy to puberty in a Japanese male with SRY-negative 46,XX testicular disorder of sex development carrying a p.Arg92Trp mutation in NR5A1. Sex Dev. (2019) 13:60–6. 10.1159/000496777 [DOI] [PubMed] [Google Scholar]
- 108.Gomes NL, de Paula LCP, Silva JM, Silva TE, Lerario AM, Nishi MY, et al. A 46,XX testicular disorder of sex development caused by a Wilms' tumour Factor-1 (WT1) pathogenic variant. Clin Genet. (2019) 95:172–6. 10.1111/cge.13459 [DOI] [PubMed] [Google Scholar]
- 109.Bashamboo A, Eozenou C, Jorgensen A, Bignon-Topalovic J, Siffroi JP, Hyon C, et al. Loss of function of the nuclear receptor NR2F2, encoding COUP-TF2, causes testis development and cardiac defects in 46,XX children. Am J Hum Genet. (2018) 102:487–93. 10.1016/j.ajhg.2018.01.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Carvalheira G, Malinverni AM, Moyses-Oliveira M, Ueta R, Cardili L, Monteagudo P, et al. The natural history of a man with ovotesticular 46,XX DSD caused by a novel 3-Mb 15q26.2 deletion containing NR2F2 gene. J Endocr Soc. (2019) 3:2107–13. 10.1210/js.2019-00241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Dutta D, Shivaprasad KS, Das RN, Ghosh S, Chatterjee U, Chowdhury S, et al. Ovotesticular disorder of sexual development due to 47,XYY/46,XY/45,X mixed gonadal dysgenesis in a phenotypic male presenting as cyclical haematuria: clinical presentation and assessment of long-term outcomes. Andrologia. (2014) 46:191–3. 10.1111/and.12048 [DOI] [PubMed] [Google Scholar]
- 112.Guercio G, Rey RA. Fertility issues in the management of patients with disorders of sex development. Endocr Dev. (2014) 27:87–98. 10.1159/000363633 [DOI] [PubMed] [Google Scholar]
- 113.Rey RA, Belville C, Nihoul-Fékété C, Michel-Calemard L, Forest MG, Lahlou N, et al. Evaluation of gonadal function in 107 intersex patients by means of serum antimüllerian hormone measurement. J Clin Endocrinol Metab. (1999) 84:627–31. 10.1210/jc.84.2.627 [DOI] [PubMed] [Google Scholar]
- 114.Porter FD. Smith-Lemli-Opitz syndrome: pathogenesis, diagnosis and management. Eur J Hum Genet. (2008) 16:535–41. 10.1038/ejhg.2008.10 [DOI] [PubMed] [Google Scholar]
- 115.Miller WL. Disorders in the initial steps of steroid hormone synthesis. J Steroid Biochem Mol Biol. (2017) 165:18–37. 10.1016/j.jsbmb.2016.03.009 [DOI] [PubMed] [Google Scholar]
- 116.Auchus RJ, Miller WL. Defects in androgen biosynthesis causing 46,XY disorders of sexual development. Semin Reprod Med. (2012) 30:417–26. 10.1055/s-0032-1324726 [DOI] [PubMed] [Google Scholar]
- 117.Miller WL. The syndrome of 17,20 lyase deficiency. J Clin Endocrinol Metab. (2012) 97:59–67. 10.1210/jc.2011-2161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Miller WL. P450 oxidoreductase deficiency: a disorder of steroidogenesis with multiple clinical manifestations. Sci Signal. (2012) 5:pt11. 10.1126/scisignal.2003318 [DOI] [PubMed] [Google Scholar]
- 119.Latronico AC, Arnhold IJ. Inactivating mutations of the human luteinizing hormone receptor in both sexes. Semin Reprod Med. (2012) 30:382–6. 10.1055/s-0032-1324721 [DOI] [PubMed] [Google Scholar]
- 120.Schwanzel-Fukuda M, Pfaff DW. Origin of luteinizing hormone-releasing hormone neurons. Nature. (1989) 338:161–4. 10.1038/338161a0 [DOI] [PubMed] [Google Scholar]
- 121.Asa SL, Kovacs K, Singer W. Human fetal adenohypophysis: morphologic and functional analysis in vitro. Neuroendocrinology. (1991) 53:562–72. 10.1159/000125775 [DOI] [PubMed] [Google Scholar]