

ABSTRACT
The Drosophila male germline stem cell (GSC) lineage provides a great model to understand stem cell maintenance, proliferation, differentiation and dedifferentiation. Here, we use the Drosophila GSC lineage to systematically analyze the transcriptome of discrete but continuously differentiating germline cysts. We first isolated single cysts at each recognizable stage from wild-type testes, which were subsequently applied for RNA-seq analyses. Our data delineate a high-resolution transcriptome atlas in the entire male GSC lineage: the most dramatic switch occurs from early to late spermatocyte, followed by the change from the mitotic spermatogonia to early meiotic spermatocyte. By contrast, the transit-amplifying spermatogonia cysts display similar transcriptomes, suggesting common molecular features among these stages, which may underlie their similar behavior during both differentiation and dedifferentiation processes. Finally, distinct differentiating germ cell cyst samples do not exhibit obvious dosage compensation of X-chromosomal genes, even considering the paucity of X-chromosomal gene expression during meiosis, which is different from somatic cells. Together, our single cyst-resolution, genome-wide transcriptional profile analyses provide an unprecedented resource to understand many questions in both germ cell biology and stem cell biology fields.
KEY WORDS: Transcription, RNA-seq, Germ cells, Proliferation, Differentiation, Spermatogenesis, Drosophila
Summary: Single cyst-resolution, genome-wide transcriptional profile analysis of Drosophila testes has offered an unprecedented data resource at each crucial stage of differentiation in an adult stem cell system under physiological conditions.
INTRODUCTION
Maintenance of a multicellular organism during homeostasis and tissue repair requires active replenishment of depleted or injured cells during aging and regeneration. Adult stem cells fulfill this requirement owing to their unique ability to both self-renew and give rise to differentiated special cell types (Betschinger and Knoblich, 2004; Clevers, 2005; Inaba and Yamashita, 2012; Morrison and Kimble, 2006). In many adult stem cell lineages, progenitor cells often undergo a proliferative stage to expand their population before commitment for terminal differentiation. The switch from proliferation to differentiation must be tightly regulated, because mis-regulation of this transition might lead to tumorigenesis or tissue dystrophy (Clarke and Fuller, 2006). On the other hand, progenitor cells remain plastic and can dedifferentiate to become stem cell-like cells in multiple stem cell lineages (Barroca et al., 2009; Boyle et al., 2007; Brawley and Matunis, 2004; Cheng et al., 2008; Friedmann-Morvinski et al., 2012; Kai and Spradling, 2004; Lim et al., 2015; Nakagawa et al., 2010; Schwitalla et al., 2013; Sheng et al., 2009; Talchai et al., 2012; Wallenfang et al., 2006; Wong and Jones, 2012). In order to properly differentiate adult stem cells or progenitor cells in vitro and/or to promote dedifferentiation in vivo for regenerative medicine, we need to fully understand the molecular changes underlying the normal differentiation program of adult stem cells in vivo.
Drosophila spermatogenesis provides a great model system to study mechanisms that regulate the maintenance, proliferation and proper differentiation of adult stem cells (Fig. 1A) (Brawley and Matunis, 2004; Kiger et al., 2001; Tulina and Matunis, 2001; Yamashita et al., 2003, 2007). In adult testes of Drosophila melanogaster, germline stem cells (GSCs) can be precisely located by their proximity and attachment to a group of post-mitotic somatic cells called hub cells at the apical tip of testis (Fig. 1A). A GSC typically divides asymmetrically to self-renew and to give rise to a gonialblast (GB), the daughter cell that initiates differentiation. GBs first go through a transit-amplifying stage as spermatogonial cells, for which they undergo exactly four rounds of mitosis in D. melanogaster. Once this stage is complete, germ cells enter the spermatocyte stage, which is an elongated G2 phase of meiosis I. During this stage, each spermatocyte grows ∼25-fold in volume, which involves a robust gene expression program allowing for meiotic divisions and spermatid terminal differentiation program (reviewed by Davies and Fuller, 2009; Fuller, 1998; Gleason et al., 2018; Greenspan et al., 2015; Lim et al., 2012; White-Cooper and Caporilli, 2013; Yamashita, 2018). In parallel with GSC asymmetric cell division, the cyst stem cell (CySC), two of them surrounding each GSC, also divides asymmetrically, resulting in one daughter cell retaining CySC identity and the other daughter cell becoming a differentiated cyst cell (Cheng et al., 2011). Two cyst cells encapsulate the differentiating germ cells throughout the entire spermatogenesis and they never divide again. It has been demonstrated that CySCs and cyst cells communicate with their accompanying germ cells via multiple signaling pathways for maintaining germ cell fate and regulating proper germline proliferation and differentiation throughout spermatogenesis (Amoyel et al., 2016a,b, 2013; Chen et al., 2013; Court et al., 2017; Eun et al., 2014; Feng et al., 2017; Hasan et al., 2015; Herrera and Bach, 2019; Kiger et al., 2000; Leatherman and Dinardo, 2008, 2010; Li et al., 2014; Lim and Fuller, 2012; Parrott et al., 2012; Sarkar et al., 2007; Schulz et al., 2004; Stine et al., 2014; Tarayrah et al., 2013; Tran et al., 2000). During this process, dynamic changes in the gene expression program are orchestrated by both extrinsic cues, such as paracrine factors that trigger signaling pathways, and intrinsic factors, such as chromatin regulators (reviewed by Davies and Fuller, 2008; de Cuevas and Matunis, 2011; Feng and Chen, 2015; Lim et al., 2012).
Fig. 1.

Summary of the TASC method and overview of this dataset. (A) Schematic illustration of spermatogenesis in Drosophila melanogaster and experimental setup. Germline stem cells (GSC; green) and somatic cyst cells (CySC; gray) are connected to the hub cells (H; blue). GSC asymmetrically divide to self-renew and generate gonialblasts (GB; orange). GBs undergo four rounds of mitosis to generate a cyst of 2, 4, 8 and 16 spermatogonia (S2, S4, S8 and S16; yellow). Spermatocytes initiate meiotic and terminal differentiation (early spermatocyte: EC16 and late spermatocyte: LC16; pink) and undergo meiotic divisions to generate 64 haploid round spermatids (RS), which later elongate to become sperm (elongated sperm: ES). (B) Multidimensional scaling plots showing distribution of the dataset. (C) Heat map showing unsupervised clustering using pair-wise Spearman's correlation coefficient. The color scale represents a correlation coefficient: the hotter the color, the higher the coefficient. (D) Analysis of transcriptome change between each two consecutive differentiation stages along the spermatogenesis. Significantly differentially expressed genes are displayed from one stage to the next stage, using differential expression analysis with Benjamini multiple test correction. Red represents significantly activated genes and black represents significantly repressed genes.
Previous studies have attempted to parse the transcriptional networks underlying GSC differentiation by comparing gene expression profiles of mutant testes that accumulate germ cells at distinct cellular differentiation stages with wild-type testes (Chen et al., 2011; Gan et al., 2010a; Terry et al., 2006). Although these approaches have led to many interesting functional studies using a candidate gene approach, the information gleaned from intact tissues is limited by the inherently mixed population of cells, and the difficulty in extrapolating results obtained from mutant backgrounds to normal situation in wild-type tissue. In this report, we systematically study the gene expression profile of the Drosophila male GSC lineage at every recognizable and isolatable stage. Using this dataset, we are interested in the following questions: Do GSCs and GBs, the two daughter cells derived from GSC asymmetric division, have similar or distinct transcriptional profiles? How does the transcriptome change in continuously proliferating spermatogonial cells? Is the switch from mitosis to meiosis accompanied by a transcriptome change that leads to another transcriptome change during spermatocyte maturation? Does dosage compensation occur in germ cells? Here, we address these issues. In summary, our single-cyst transcriptome profiles provide a comprehensive dataset at a resolution that has not been achieved before, which yields much-needed information on transcriptional status at each crucial stage from an endogenous stem cell system. Researchers from both germ cell biology and stem cell biology fields should benefit from using this resource to screen for genes with a particular expression pattern or examine genes from specific pathway(s), before designing detailed functional analyses.
RESULTS
Development of the transcriptome analysis using single germline cyst (TASC) technique
To elucidate endogenous gene expression profiles in germ cells at discrete but continuous differentiation stages, we developed a new strategy that we named transcriptome analysis using single germline cyst (TASC). As germ cells at various differentiation stages can be recognized by their distinct anatomical positions and morphological characteristics in wild-type testes (Fuller, 1998), it is feasible to isolate single germ cell cysts at a particular stage during differentiation. We combined these features with cell type- and stage-specific green fluorescent protein (GFP) markers (Fig. S1A,B, Materials and Methods) (Boyle et al., 2007; Chen and McKearin, 2003; Chen et al., 2005; Santel et al., 1997). Specifically, a hub-specific GFP marker [unpaired1 (upd)-Gal4>UAS-GFP (Boyle et al., 2007)] was used to isolate the niche sample including hub cells, GSCs and CySCs. In addition, spermatogonial cysts from different stages were identified and isolated with the spermatogonia-specific Bag of marbles (Bam)-GFP marker (Chen and McKearin, 2003). Finally, the spermatocyte-specific Spermatocyte arrest (Sa)-GFP marker (Chen et al., 2005) was used to isolate the spermatocyte cysts. Together, single germline cyst samples from seven distinct stages were identified and isolated from wild-type testes: niche, GB, four-cell spermatogonia (S4), eight-cell spermatogonia (S8), 16-cell spermatogonia (S16), early spermatocyte (EC) and late spermatocyte (LC). Furthermore, arrested spermatocyte cyst samples were isolated from always early (aly) (White-Cooper et al., 2000) mutant testes. The aly mutations arrest germline differentiation at the early spermatocyte stage, resulting in testes enriched with early spermatocytes without entry into meiotic division or spermatid differentiation (White-Cooper et al., 2000). Molecular characterization of the aly gene reveals that it encodes a component of the testis-specific meiotic arrest complex (tMAC), a tissue-specific version of the mammalian MIP/dREAM complex and the Caenorhabditis elegans SynMuv complexes (Ayyar et al., 2003; Beall et al., 2007; Jiang et al., 2007; Jiang and White-Cooper, 2003; Perezgasga et al., 2004; White-Cooper et al., 2000, 1998). In the aly mutant, no further germline differentiation could occur beyond the early spermatocyte stage because of this genetic mutation, so we used this sample as another source for early-stage spermatocytes. Meanwhile, we recognized the early-stage wild-type spermatocyte cyst based on its morphology (i.e. relatively smaller size), a criterion that could vary. Therefore, the aly mutant spermatocyte cyst sample could represent the earliest spermatocyte stage. We then performed RNA-seq using isolated germline cysts as the starting material to systematically profile their transcriptomes. We used an adapted amplification method for single cell transcriptome analysis (Fig. S1C, Materials and Methods) (Kurimoto et al., 2007; Tang et al., 2009). The purity of our samples offered an unprecedented opportunity to study transcriptional dynamics in continuously differentiating germline cysts at a genome-wide level.
The final reads from 20 sample libraries (Fig. S2 and Table S1) were assigned to annotated genes based on their merged transcript regions (Gan et al., 2010a). The 20 samples include independent biological replicates for: niche (two samples), GB (two samples), S4 (two samples), S8 (two samples), S16 (one sample), aly mutants (two samples), EC (three samples), LC1 (relatively smaller LC, three samples) and LC2 (the largest LC, three samples). The original RPKM (sequencing reads per kilobase merged exonic region, per million mapped reads) value (Gan et al., 2010a) was normalized by total read counts across different libraries and length of various transcripts. Here, we combined RPKM computation with the trimmed mean of M-values (TMM) and upper-quantile normalization methods to better control variations among different libraries. We then used post-normalization read counts (pseudo.alt generated by EdgeR) followed by normalization with merged transcript length to get corrected RPKM (cRPKM) values (Materials and Methods).
Overview of the TASC data: dynamic transcriptional changes during Drosophila spermatogenesis
We first set up a threshold for a gene to be called ‘expressed’. We compared the cRPKM value with published microarray data (Chen et al., 2011; Terry et al., 2006), which showed that genes with a cRPKM value >20 mostly have ‘present’ calls. If we used cRPKM=20 as a cutoff for expressed genes, which likely represents a stringent cutoff because of the higher sensitivity of RNA-seq compared with microarray (Wang et al., 2009), 51.4% (9256 out of 18,000) annotated Drosophila genes were expressed in at least one staged germ cell cyst during Drosophila spermatogenesis (Table S2).
In order to gain a global picture of the transcriptome change during spermatogenesis, we performed the multidimensional scaling assay (Fig. 1B), as well as unsupervised clustering using Spearman's correlation coefficient between two samples among all samples (Fig. 1C). Among the 20 samples, biological replicates showed the highest degree of clustering (Fig. 1B) and correlation coefficient (Fig. 1C), demonstrating reproducibility of the TASC method. Furthermore, both analyses revealed three discrete clusters (Fig. 1B,C): the first cluster includes the niche samples and all transit-amplifying staged samples from GB to S16; the second one represents the early spermatocyte stage including the aly spermatocyte and three EC samples; and the third one is for late spermatocyte samples (i.e. three LC1 and three LC2 samples). As the first cluster is for mitotic cells and the remaining two represent meiotic stages, changes from one cluster to the next represent the major transcriptional switches during spermatogenesis, which coincide with morphological changes at the cellular level (Fuller, 1998). We then analyzed the transcriptome change between every two consecutive stages along the differentiation pathway (Fig. 1D, Fig. S3, Tables S3-S10), the switch with the largest number of genes changing expression is from early to late spermatocyte stage (Tables S9 and S10), followed by the switch from S16 to early spermatocyte stage (Tables S7 and S8). These findings are consistent with previous studies comparing testes enriched with spermatogonial cells, testes enriched with arrested early spermatocytes, and wild-type testes containing all stages of germ cells (Chen et al., 2011; Gan et al., 2010a; Terry et al., 2006). Interestingly, these analyses also indicate that the changes from niche to GB (Fig. 1D, Fig. S3, Tables S3 and S4) and from GB to S4 (Fig. 1D, Fig. S3, Tables S5 and S6) are accompanied by hundreds of gene expression changes, including both upregulated and downregulated expression patterns.
Validation of the TASC data
To validate the TASC data, we collected genes with either well-characterized expression patterns or known stage-specific biological functions for Drosophila spermatogenesis from the published literature. We then checked their transcription profiles in our TASC dataset (Fig. 2). Most of these gene expression patterns in the TASC dataset are consistent with previous publications, indicating that these data are of high quality and could be used to identify genes with interesting expression patterns. For example, the JAK-STAT signaling pathway ligand-encoding unpaired1 (upd; also known as os) gene has been reported to have high expression levels in the niche (Kiger et al., 2001; Toledano et al., 2012; Tulina and Matunis, 2001) (Fig. S4: upd in ‘niche’ class). Here, in the TASC dataset, upd has a distinctive high expression in the niche sample (Fig. S5A). The detectable level of upd in late spermatocytes could be for a yet-to-be defined role of the JAK-STAT pathway during late spermatogenesis. Furthermore, Piwi, an essential RNA-binding protein required for GSC and CySC function (Gonzalez et al., 2015), is highly transcribed in the niche sample (Fig. S5A), consistent with the previously reported expression pattern at the apical tip of adult testes (Gonzalez et al., 2015; Kalmykova et al., 2005) (Fig. S4: piwi in niche class).
Fig. 2.
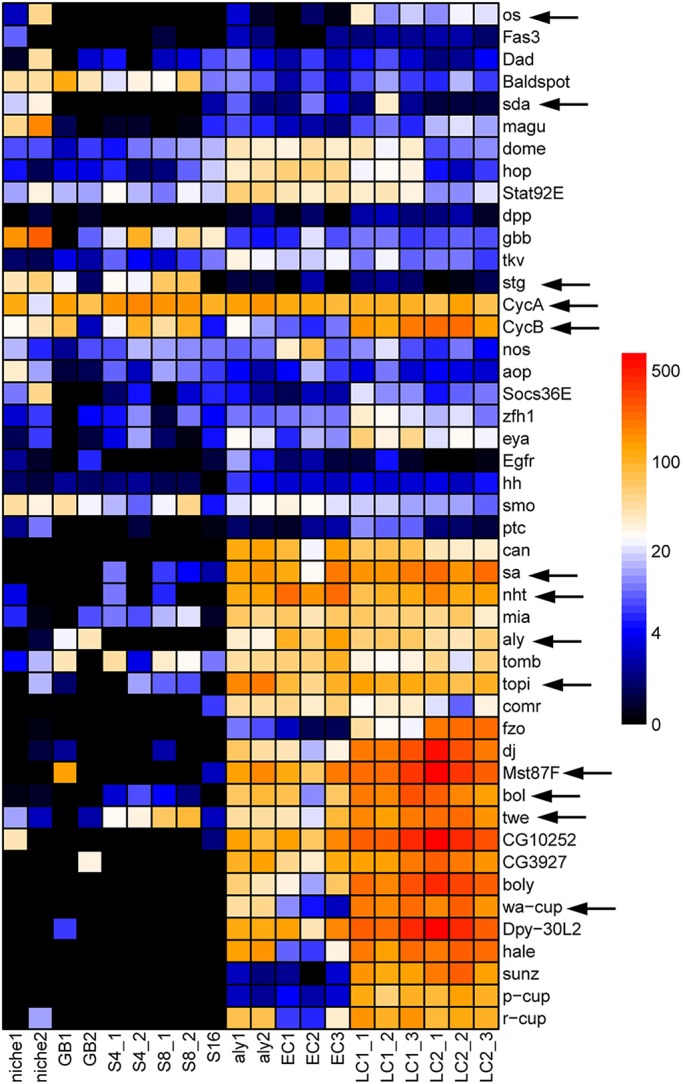
Validation of TASC data. A list of genes whose expression patterns have been demonstrated in previous publications. Arrows indicate those genes that are discussed in the main text. The heat map represents their cRPKM values. See main text for detailed examples and cross comparison of the TASC data with the published results.
During the transit-amplification stage, the mitotic Cdc25-encoding string (stg) gene showed a dramatically increased expression in the mitotic spermatogonial cyst samples (Figs S5B and S4: stg-GFP in TA class), in contrast to the meiotic Cdc25-encoding twine (twe) gene, which displayed enhanced expression in late spermatocyte cyst samples (Figs S5D and S4: twine in LC class), consistent with previous reports (Courtot et al., 1992; Inaba et al., 2011; Maines and Wasserman, 1999). In addition, two groups of meiotic arrest genes for both the tMAC (Ayyar et al., 2003; Beall et al., 2007; Jiang et al., 2007; Jiang and White-Cooper, 2003; Perezgasga et al., 2004; White-Cooper et al., 2000, 1998) and the testis-specific TBP-associated factor (tTAF) (Chen et al., 2005, 2011; Hiller et al., 2004, 2001; Lin et al., 1996) components had a stereotypic early-stage spermatocyte-enriched expression pattern, consistent with a previous assay using in situ hybridization [Figs S5C and S4: cookie monster (comr), no hitter (nht, Taf4L), rye (Taf12L), spermatocyte arrest (sa, Taf8L), meiosis I arrest (mia, Taf6L) in EC class]. By contrast, genes required for meiotic divisions such as boule (bol) (Maines and Wasserman, 1999) and twe (Courtot et al., 1992; Inaba et al., 2011; Maines and Wasserman, 1999), as well as terminal differentiation genes such as mst87F (Barckmann et al., 2013; Hiller et al., 2001) and mst77F (Barckmann et al., 2013), were all detected at the highest level in late spermatocyte cyst sample (Figs S5D and S4: boule, twine, mst77F, mst87F in LC class). Finally, CycB is known to be required in both early mitotic and meiotic dividing germ cells (Baker and Fuller, 2007; Baker et al., 2015; Feng et al., 2018; White-Cooper et al., 1998). Consistently, the cycB transcript was detectable in both spermatogonial cyst and late spermatocyte cyst samples (Fig. S5E), whereas a constitutively expressed cycA gene (Chen et al., 2011) had consistently high expression level throughout spermatogenesis (Fig. S5F).
Distinct clusters of genes showing similar expression pattern during spermatogenesis
Next, we examined the overall dynamics of the 9256 expressed genes along the cellular differentiation program of spermatogenesis by performing clustering analysis. Among the 10 clusters identified using the K-means algorithm (Fig. S6), six clusters show differential gene expression. Cluster #4, containing 548 genes, shows overall downregulation, with enriched expression in early-stage germ cells including the GSC-containing niche sample (Fig. 3A). Cluster #2 consists of 287 genes, which display spermatogonia-specific expression patterns, with enriched expression from GB to S16 samples but reduced expression in both niche and spermatocyte samples (Fig. 3B). Cluster #3, with 1148 genes, shows slightly elevated expression in early-stage spermatocytes but robust expression in late stage spermatocytes, suggesting their functions in terminal differentiation (Fig. 3C). In addition, three other clusters – #1, #7 and #8 – have 2536 genes in total, which show spermatocyte-specific expression starting from early-stage spermatocyte samples (Fig. 3D). The remaining four clusters (#5, #6, #9 and #10) together have 4737 genes, which show overall stable gene expression (Fig. S7), likely representing constitutively expressed housekeeping genes.
Fig. 3.

Distinct clusters of genes that share similar differential gene expression patterns during spermatogenesis with respective GO analyses. (A) A cluster of 548 genes with an overall downregulation pattern and with enriched expression in early-stage germ cells, and the corresponding GO terms. (B) A cluster of 287 genes with a spermatogonia-specific expression pattern with enriched expression from GB to S16 samples but declined expression elsewhere, and the corresponding GO terms. (C) A cluster of 1148 genes with spermatocyte-specific expression starting from early-stage spermatocyte samples, and the corresponding GO terms. (D) Three clusters of a total of 2536 genes with overall stable gene expression throughout all samples, and the corresponding GO terms. The box plots are generated using the R Boxplot default setting: the box represents the 25th and 75th percentiles, with the 50th percentile shown as a black bar. The upper and lower whiskers are defined using the default setting of the R Boxplot function. The y-axis represents the log2cRPKM value.
Based on these patterns (Fig. 3), it appears that gene activation is likely to be the major mode of differential gene expression during Drosophila spermatogenesis. On the other hand, genes with stable levels of transcripts (Fig. S7) could still be regulated post-transcriptionally, which is extensively used in the germline (Cinalli et al., 2008; Rangan et al., 2008; Seydoux and Braun, 2006).
The transcriptome changes among the mitotic stage germline cyst samples
In Drosophila testes, a GSC normally divides asymmetrically: the self-renewed stem daughter cell remains physically attached to the hub cells via adherens junctions, whereas the other daughter cell (GB) is displaced away from the hub and initiates transit-amplification followed by meiosis and terminal differentiation (Yamashita et al., 2003). It remained unclear to what extent the GSC and GB have different molecular characteristics. Previously, we have identified distinct chromatin states between GSC and GB (Tran et al., 2012), suggesting their potential distinct gene expression profiles. Here, we sought to gain insight into this by comparing the TASC data between niche and GB samples. However, a caveat in this comparison is the heterogeneity of the niche sample, which contains hub cells and CySCs in addition to GSCs. Nevertheless, we reason that the upregulated genes in the GB sample should still represent newly transcribed genes, whereas enriched transcripts in the niche sample would still shed light on putative regulators required for stem cell maintenance or niche functionality. Both lines of information should enhance our knowledge regarding how stem cells maintain their unique properties as well as how their immediate daughter cells initiate the proper differentiation program.
First, we compared transcription level of genes in niche versus GB samples (Fig. 4A, Tables S3 and S4). Among the 792 differentially expressed genes (P<0.05, based on negative binomial model with Benjamini multiple testing correction, Fig. S3B), 511 genes are more enriched in the niche sample and 281 have higher expression in the GB sample. A gene ontology (GO) analysis on these differentially expressed genes revealed three top categories with biological significance (Fig. 4A): cell-cell adhesion molecules (21 genes), genes that regulate cell fate commitment (10 genes) and signaling pathway components (71 genes). Some genes are present in more than one category, such as niche-enriched Socs36E gene, which is a known JAK-STAT target (Amoyel et al., 2016a; Issigonis et al., 2009; Tarayrah et al., 2013).
Fig. 4.

Transcriptome changes among discrete stages of germline cyst samples. (A) Analysis of differential gene expression in niche versus GB samples. (B) Analysis of differential gene expression in GB versus S4 samples. (C) Analysis of differential gene expression in S16 spermatogonia versus early spermatocytes (EC). Heatmaps of differentially expressed genes in multiple GO function terms during stage transition. The statistically significant enrichment of genes in the GO term of interest is calculated based on negative binomial model with Benjamini multiple testing correction (FDR). Color scale represents the Z value of normalized expressions for individual genes. See main text for a detailed discussion of each category.
Next, we compared the transcription level of genes in GB versus S4 samples (Fig. 4B, Tables S5 and S6). Among the 1488 differentially expressed genes (P<0.05, based on negative binomial model with Benjamini multiple testing correction, Fig. S3B), 667 are GB-enriched and 821 genes have higher expression in the S4 sample. A GO analysis on these differentially expressed genes revealed three top categories with biological significance (Fig. 4B): genes that regulate aminoglycan metabolic process (26 genes), genes that regulate monosaccharide metabolic process (17 genes) and signaling transduction (119 genes). Therefore, from both niche to GB and GB to S4 along the GSC differentiation process, genes regulating signaling pathways change their expression most dramatically. However, the progression from niche to GB is accompanied by decreased expression of cell adhesion molecules, whereas from GB to S4, genes involved in metabolic processes change their levels, indicating a potentially different metabolism when germ cells leave the niche and prepare for the differentiation program.
Furthermore, the transit-amplification stages from S4 and S8 to S16 showed >0.9 correlation coefficient by pair-wise comparison. A similar transcriptome among transit-amplifying cells may provide a molecular basis to explain their similar behavior during both dedifferentiation and the differentiation processes. Dedifferentiation of spermatogonial cells has been identified as a mechanism to replenish lost stem cells during genetic ablation or aging (Brawley and Matunis, 2004; Cheng et al., 2008; Eun et al., 2017; Herrera and Bach, 2018; Lim et al., 2015; Sheng et al., 2009; Sheng and Matunis, 2009; Wong and Jones, 2012). On the other hand, transition from spermatogonia to spermatocyte prematurely or at a later point does not appear to affect meiosis and terminal differentiation, suggesting their similar differentiation potential (Eun et al., 2013; Insco et al., 2012, 2009; Parrott et al., 2011).
The transcriptome changes from the mitotic to the meiotic staged germline cyst samples
To date, the best understood transcriptional switch during Drosophila spermatogenesis is from the S16 to spermatocyte, representing the change from the mitotic to the meiotic program (Lim et al., 2012). Previous studies on key regulators, such as the tTAFs and the tMAC complex, demonstrate an orchestrated mode of transcriptional activation (Ayyar et al., 2003; Beall et al., 2007; Chen et al., 2011; Hiller et al., 2004, 2001; Jiang et al., 2007; Jiang and White-Cooper, 2003; Lin et al., 1996; Perezgasga et al., 2004; White-Cooper et al., 2000, 1998). In addition, spermatocyte-specific tTAF and tMAC proteins antagonize the Polycomb group (PcG) transcriptional repressive complex to derepress differentiation genes that are silenced in spermatogonial cells (Chen et al., 2005, 2011; Gan et al., 2010b). Based on these results, it has been hypothesized that there are at least two transcriptional waves in spermatocytes – one at the spermatogonia-to-spermatocyte transition, which turns on transcriptional and chromatin regulators to set up the chromatin landscape, in preparation for the next one, from early to late spermatocytes, when terminal differentiation genes for spermiogenesis are turned on (Chen et al., 2011). It is very likely that genes turned on during the second wave are target genes controlled by genes that have been turned on at the first wave.
Analysis of the transition from the S16 spermatogonia to early spermatocyte stage demonstrates more than 1266 differentially expressed genes: only 125 genes are downregulated whereas 1141 are upregulated (Fig. S3B, Tables S7 and S8). Therefore, transcriptional activation is the major mode at the mitosis-to-meiosis switch. A GO analysis on these upregulated genes revealed three top categories with biological significance (Fig. 4C): genes that regulate male gamete generation (34 genes), including both tTAF [e.g. nht, mia, cannonball (can, Taf5L), sa] and tMAC [e.g. comr, aly, matotopetli (topi)] genes, microtubule-based movement (29 genes), as well as protein ubiquitination (40 genes).
This mitosis-to-meiosis switch needs to be tightly regulated, as any mis-regulation of this transition may lead to infertility due to insufficient germ cells or testicular tumor due to spermatogonial overproliferation. Analysis of the TASC data has revealed that many more key transcriptional regulators (Fig. S8A), alternative splicing factors (Fig. S8B) and enzymes such as kinases (Fig. S8C) all show a dynamic bi-modal pattern (i.e. low-to-high or high-to-low) at the mitosis-to-meiosis switch. Interestingly, histone-modifying enzymes, including both histone methyltransferase (Fig. S8D) and histone demethylase (Fig. S8E), display similar patterns at this switch. These results are consistent with the previous results using bam or benign gonial cell neoplasm (bgcn) mutant testes enriched with mitotic spermatogonial cells for comparison with wild-type testes (Chen et al., 2011; Gan et al., 2010a; Terry et al., 2006). Functional analysis of these candidate genes will shed more light on the molecular mechanisms underlying exit of mitosis and onset of meiosis. On the other hand, the few transcription factors (group #1 in Fig. S8A) that are downregulated from S16 to early spermatocyte stage could be responsible for shutting down mitosis-specific genes to avoid overproliferation and tumorigenesis. To our current knowledge, this decision makes an irreversible commitment in this lineage, as spermatocytes lose the dedifferentiating potential (Sheng et al., 2009).
The transcriptome change from early to late spermatocyte during spermatocyte maturation
The maturation of spermatocytes takes the longest time during the entire process of spermatogenesis, lasting for 3-4 days and involving a 25-fold increase in volume. Here, the TASC data provide a high-resolution transcriptome profile during spermatocyte maturation, with aly and EC samples representing early spermatocyte and LC1 representing the late and mature spermatocyte stage (Fig. 1B-D, Fig. S3B, Tables S9 and S10; LC1 samples share a very similar expression profile with LC2 so we used LC1 to represent late spermatocytes). Because, to our knowledge, aly mutations arrest spermatocyte differentiation at the earliest stage (White-Cooper et al., 2000), the progression of spermatocyte maturation is very likely as the following: aly mutant→wild-type EC spermatocyte→wild-type LC1 spermatocyte.
Next, we analyzed differential gene expression at these spermatocyte stages and compared them with the 16-cell spermatogonial cyst sample (Fig. 5A). The number of genes that are moderately or highly expressed (i.e. RPKM≥20) in the wild-type S16 sample, but are absent or expressed at low levels in the aly mutant spermatocyte sample (S16+aly−), is 654, whereas the number of genes that are moderately or highly expressed in the aly mutant spermatocyte sample, but are absent or are expressed at low levels in the S16 sample (S16−aly+) is 2307. Likewise, the number of genes that are moderately or highly expressed in the S16 sample, but are absent or expressed at low levels in the wild-type early spermatocyte EC sample (S16+EC−) is 401 whereas the number of genes that are moderately or highly expressed in the wild-type early spermatocyte EC sample, but are absent or expressed at low levels in the S16 sample (S16−EC+) genes is 1850. Therefore, the major change of gene expression from S16 to early-stage spermatocytes is activation. Between the 2307 S16−aly+ genes and 1850 S16−EC+ genes, 1521 are common. In addition, 289 of the 654 S16+aly− genes and 401 S16+EC− genes are shared. These data are consistent with previous analyses that show the transition from mitosis to meiosis is accompanied by a dynamic transcriptome change, and that most changes are transcriptional activation.
Fig. 5.

Transcriptome changes among different early and late spermatocyte samples during spermatocyte maturation. (A) Venn diagrams display the overlapping number of expressed genes in three different spermatocyte staged samples (i.e. aly mutant, EC and LC samples) versus the S16 cyst sample. (B) Venn diagram among different groups of stage-enriched genes, see main text. (C) Three classes of GO analysis results (BP, biological process; MF, molecular function; CC, cellular component) of late spermatocyte-enriched genes, immediately before the onset of the meiotic division and terminal differentiation process known as spermiogenesis. Here, based on a negative binomial model: N, total gene number of the study; B, number of N in each category of interest; n, the total number of differentially expressed genes; b, the number of n in each category.
Next, analyzing differentially expressed genes between the S16 sample and LC1 sample revealed an even bigger change: there are 1026 differentially expressed S16+LC1− genes that are moderately or highly expressed in the S16 sample, but are absent or are expressed at low levels in the wild-type late spermatocyte LC1 sample, and 3184 differentially expressed S16−LC1+ genes that are moderately or highly expressed in the wild-type late spermatocyte LC1 sample, but are absent or are expressed at low levels in the S16 sample (Fig. 5A). Among the 3184 S16−LC1+ genes, 1724 are shared with the S16−aly+ group and 1427 are common with the S16−EC+ genes, with the majority of 1298 common genes among all three groups, likely representing the wave of transcriptional activation from spermatogonia to spermatocytes. On the other hand, the 1354 genes that are more enriched in the LC1 sample, but in neither aly nor EC sample, represent the transcriptional activation from early to late spermatocytes. To further understand this sequential change among spermatocyte samples in more detail, we compared four categories of genes (Fig. 5B): high in LC1 but low in aly sample (i.e. LC1>aly), high in LC1 but low in EC sample (LC1>EC), high in EC but low in aly sample (EC>aly), and high in aly but low in EC sample (aly>EC). Among these four categories, 2144 LC1>aly genes and 3176 LC1>EC genes share 1854 common genes, consistent with a transcriptional activation mode during spermatocyte maturation. GO analyses of these late spermatocyte-enriched genes indicate that translational regulators, spindle elongation and microtubule-based process factors are among the top three enriched genes based on biological process GO analysis. Alternatively, with molecular function GO analysis, receptor activity, especially G-protein coupled receptors (GPCR), and ion transmembrane transporters, are among the top three categories, whereas using cellular component GO analysis, dynein complex and microtubule-associated complex are the top two categories (Fig. 5C). These gene categories, such as translational regulators and microtubule-associated factors, are consistent with the known developmental program and cellular processes required for meiotic divisions and terminal differentiation (Fabian and Brill, 2012; Fuller, 2016). In addition, these findings may shed light on new regulators for the sperm differentiation program. For example, bioelectric patterning through ion transporters is known to play a role during Drosophila oogenesis (Weiss and Bohrmann, 2019); however, little is known about their potential functions in spermiogenesis. On the other hand, although the function of GPCRs has been reported for stem cell niche structure and function in Drosophila (Papagiannouli and Lohmann, 2015) and planarians (Saberi et al., 2016), their potential roles in spermiogenesis have not been well studied at all.
Lack of evidence for dosage compensation in germline cysts at individual stages throughout Drosophila spermatogenesis
In somatic cells of male Drosophila, dosage compensation equilibrates X chromosomal and autosomal gene expression by hyperactivation of X-chromosomal gene transcription (Deng et al., 2011; Gupta et al., 2006; Parisi et al., 2003). The dosage compensation complex (DCC) generates an H4K16ac active histone modification and promotes RNA polymerase II elongation on X-linked genes (Gelbart et al., 2009). Previous RNA-seq using gonadectomized flies (adult flies with testes or ovaries removed, labeled as ‘male_carcassed’ and ‘female_carcassed’; Fig. S9) showed little difference between X-linked genes and autosomal genes in their median expression level, confirming dosage compensation in somatic cells. However, despite extensive knowledge about dosage compensation in somatic cells, whether and how X-chromosomal genes are compensated in male germ cells is not fully understood. Analyses of RNA-seq data using isolated wild-type testes (Gan et al., 2010a) showed a significantly lower expression level of X-linked genes compared with autosomal genes (‘wt_testis’; Fig. S9), suggesting potential lack of dosage compensation in male germ cells. By contrast, analysis of bam mutant testes showed comparable level between X-linked genes and autosomal genes, suggesting possible existence of dosage compensation. Because wild-type testes are enriched with meiotic germ cells, whereas bam testes mainly contain mitotic germ cells, analysis of RNA-seq data for these tissues suggests dosage compensation may exist in the mitotic but not meiotic male germ cells in Drosophila. However, using the entire testes unavoidably includes other cell types, such as somatic gonadal cells in addition to germ cells, which may obscure the results.
Here, using the TASC RNA-seq data, we revisited dosage compensation in the male germline. In order to avoid any potential bias introduced by the unique and dynamic gene expression program during spermatogenesis, we restricted our analysis to non-sex-biased and non-spermatogenesis stage-biased genes. Even though our isolated cyst samples contain somatic cells, the cyst samples are enriched with germ cells. For example, in the S16 and all spermatocyte samples, the somatic cyst cell to germ cell ratio is 2:16; and in the S8 sample, the somatic cyst cell to germ cell ratio is 2:8, etc. In all staged samples containing mitotic germ cells (i.e. GB, S4, S8 and S16; Fig. 6A), X-linked genes are expressed at a significantly lower level than autosomal genes, different from the previous results obtained using bam testes (Gan et al., 2010a). There are two possibilities that make the TASC data more directly address the issue of dosage compensation in germ cells: first, bam testes are heterogeneous and contain many other somatic cell types such as cyst cells, muscle sheathe cells, and pigment cells; second, bam mutant germ cells are different from wild-type spermatogonia cells, as shown by the distinct transcriptome of bam testes compared with all spermatogonial cyst samples (Fig. S10). We also confirmed that all meiotic cyst samples showed no obvious evidence of dosage compensation (i.e. all EC and LC samples; Fig. 6A).
Fig. 6.

Lack of evidence for dosage compensation in germline cysts at individual stages throughout Drosophila spermatogenesis. (A) Analysis of non-sex-biased and non-stage-biased genes in all of the staged samples containing mitotic germ cells (N, GB, S4, S8 and S16) and meiotic cyst (EC and LC). The expression levels of genes located on each chromosome arm (2L/2R/3L/3R/4/X) is as follows. For the x-axis: 2L and 2R denote the left and the right arm of the second chromosome, respectively. 3L and 3R denote the left and the right arm of the third chromosome, respectively. 4 denotes the fourth chromosome, while X denotes the X chromosome. The y-axis represents the log2cRPKM value. The box plots are generated using the R Boxplot default setting: box represents the 25th and 75th percentiles, with the 50th percentile is shown as a black bar. The upper and lower whiskers are defined using the default setting of the R Boxplot function. Green bars mark the median level of X chromosomal gene expression. (B) Compensation index (CI) with the average expression level from X chromosome as well as other autosomes showing a higher degree of dosage compensation for the wild-type niche sample compared with other germline cyst samples, including wild-type GB, S4, S8, S16 spermatogonial cysts, aly and can mutant spermatocyte cysts, as well as wild-type EC16, LC1 and LC2 cysts. The wild-type testis (wtT) sample also shows low dosage compensation, whereas bam mutant testis (bamT) sample and wild-type male carcassed (carM) show more dosage compensation. Here, wild-type ovary (wtO), bam mutant ovary (bamO) and wild-type female carcassed (carF) samples are shown as controls. CT=1 indicates full compensation, whereas CT=0 indicates complete loss of dosage compensation.
Next, we summarized the dosage compensation index defined by (2−A:X): here 0 indicates no compensation; 1 indicates twofold upregulation of X-linked gene expression, therefore full compensation (Fig. 6B). We then compared the single germline cyst samples from TASC dataset with the previous RNA-seq data using tissue samples (Gan et al., 2010a). Although most of the previous tissue samples showed compensation, among all TASC data samples only the niche sample showed a higher degree of compensation, probably owing to the presence of somatic hub cells and CySCs in this sample. Overall, these analyses suggest that male germ cells likely lack dosage compensation, when pure starting material containing fewer somatic cells are used. Here, even if a couple of somatic cells are present in the TASC sample, low or no dosage compensation strongly suggests the lack of dosage compensation in germ cells.
DISCUSSION
Our single cyst-resolution, genome-wide transcriptional profile analyses provide a data resource at each crucial stage of differentiation in an adult stem cell system under their wild-type situation, which will help in understanding many interesting issues in stem cell and germ cell biology fields. Previously, RNA-seq analyses were carried out using the entire testes with genetic mutations that arrest cellular differentiation pathways at distinct stages (Gan et al., 2010a) or dissected different regions of wild-type testes enriched with distinct stages of germ cells (Vedelek et al., 2018). However, caveats arise from mixed cell types within either intact or dissected tissues, as well as complications using genetic mutations. Furthermore, although single cell RNA-seq could be currently carried out using dissociated single cells, the relatively small number of early-stage cells, including germline stem cells, make it very challenging to obtain their transcriptomes. Single post-meiotic cysts have been isolated and applied for quantitative RT-PCR, which has revealed two dozen transcribed genes in spermatids (Barreau et al., 2008). Here, for the TASC experiments, we isolated single germline cysts from niche to late spermatocyte stages using cell type- and stage-specific markers (Fig. S1A), which allowed us to precisely reconstruct the single-cyst transcriptome atlas during the entire spermatogenesis process. Therefore, both the original technologies we have developed and the new knowledge we have gained in this study represent technical and conceptual advances, which will have a broad and long-term impact on studies in the stem cell and germ cell biology fields.
As a proof of principle, we have already made several new discoveries using the TASC dataset. For example, we have identified a candidate gene called slamdance (sda) which has enriched transcription in the niche sample (Fig. S5A). The sda gene encodes an aminopeptidase, the function of which had never been reported in any adult stem cell systems. We have carried out extensive functional analyses and demonstrated that the Sda protein indeed acts as an aminopeptidase, the in vivo function of which depends on its enzymatic domain. Sda plays important roles in regulating GSC maintenance and progenitor germline dedifferentiation (Lim et al., 2015). In addition, we found that the known germline differentiation factor bam has prolonged transcription in early-stage spermatocytes (aly and EC stages of the bam profile in Fig. S5B), even though Bam protein is downregulated. This difference between mRNA expression pattern and protein enrichment indicated a potential post-transcriptional regulation of the bam transcript, which was revealed later through recognition of the 3′ untranslated region of bam by microRNAs (Eun et al., 2013). Therefore, the TASC dataset can be used to identify genes with interesting expression patterns for in-depth functional analysis, or combined with other data such as protein expression patterns for studying post-transcriptional regulation in the germline, etc. Together, these studies will contribute to better understanding of gene regulation during spermatogenesis, which will also significantly enhance our knowledge for reproductive biology.
In summary, our single cyst-resolution, genome-wide transcriptional profile analysis provides a supreme and unprecedented data resource at each crucial stage of differentiation in an adult stem cell system under its physiological conditions, which will help in understanding many issues in germ cell biology and stem cell biology fields. The new technologies that we have developed and the knowledge we have gained in this study will have broad and high impact on basic research as well as on regenerative medicine.
MATERIALS AND METHODS
Fly strains and single cyst preparation
All flies were grown using standard Bloomington medium at 25°C: upd-gal4 (a gift from T. Xie, Stowers Institute, Kansas City, MO, USA), Bam-GFP (Chen and McKearin, 2003; a gift from Dennis McKearin, UT Southwestern Medical Center, Dallas, TX, USA) and Sa-GFP (Chen et al., 2005). Testes were dissected in S2 insect media (Invitrogen, 11720-034) with 10% fetal bovine serum (FBS; Invitrogen, 16000-044). Two tungsten micro-needles were used to tear the muscle sheath layer, resulting in germ cell cysts spilling into the media. The cysts were viewed using an inverted microscope (Zeiss Axiovert 40 CFL) and were picked using a glass needle (Drummond, 1-000-0250) pulled by a Sutter instrument P97 using the following parameters: P=200; heat=669; pull=30; velocity=120; time=200. The picked cyst was transferred into PCR tubes by breaking the fine tip at the side of tube.
The upd-Gal4; UAS-GFP labels the niche sample (i.e. hub cells with both GSCs and CySCs) unambiguously (Fig. S1B). However, some ambiguity exists in other sample isolation due to technical limitation, and here we discuss our strategy to reduce it. For example, spermatogonial samples can be distinguished by the expression of Bam-GFP marker (Fig. S1B). However, any apparent S4 cyst could come from a broken S8 cyst or S16 cyst. Although it is unlikely for an S8 cyst to precisely break into two 4-cell cysts or a S16 cyst into four 4-cell cysts, we tried to avoid this caveat by selecting cysts that have ‘smooth’ sphere appearance indicating intact cysts (Insco et al., 2009). We also isolated at least two biological replicates for each of these ‘ambiguous’ stage cysts, namely: GB, S4 and S8, from different flies in totally independent experiments. Our results showed very high Pearson's correlation coefficient (>0.9) between two biological replicates for GB, S4 and S8 samples. This high correlation between biological replicates also suggested the high reproducibility of our single cyst isolation and sequencing procedures. In addition, even though spermatocyte samples can be precisely labeled using the Sa-GFP marker (Fig. S1B), we recognized early-stage spermatocytes (i.e. relatively smaller size) versus late-stage spermatocytes (i.e. relatively larger size) based on their morphology from wild-type testis. However, as this criterion could vary, we also included an aly mutant spermatocyte cyst sample. Because in the aly mutant, no further differentiation could occur beyond the very early spermatocyte stage due to the requirement of this gene product to proceed beyond the early spermatocyte stage, we used this sample as another source for early-stage spermatocytes and compared with the early-stage spermatocyte samples isolated from wild-type testis.
Library preparation for RNA-seq
The mRNA libraries were prepared according to previously described methods (Kurimoto et al., 2007) with modifications (Tang et al., 2009). Immediately after isolation, we treated the cysts with mild detergent to permeabilize the plasma membrane while keeping the nucleus intact. Permeabilization allowed mRNA to be reverse transcribed using poly-dT attached to one universal primer sequence. Poly A was added to the single strand DNA 3′ end using terminal transferase. Second strand DNA was synthesized using poly-dT attached to another universal primer sequence. Finally, the resulting library was amplified using both universal primers. To generate libraries for sequencing, ∼300 ng dsDNA of each sample was fragmented by sonication using Bioruptor (Diagenode, UCD-200-TM-EX) under medium power output for 30 min in ice water. The resulting DNA fragments were analyzed using agarose gel to verify a ∼100-300 bp size range. Sequencing libraries were prepared as follows: end-repair (DNA end-repair kit, Epicenter, ER0720); A-tailing [Poly(A) Tailing Kit, Thermo Fisher, AM1350]; Solexa adaptor ligation (300 ng dsDNA, 4 μl DNA ligase buffer, 1 μl Solexa adaptor mixture, 3 μl DNA ligase, at 70°C overnight); PCR amplification (98°C 10 s, 65°C 30 s, 72°C 30 s for 16 cycles; then additional 72°C for 5 min) with adaptor primers; and size selection (200-400 bp). Lastly, dsDNA library for each sample was used on the Solexa 1G sequencer at a concentration of 10 ng per lane.
Alignment of short reads onto Drosophila genome and assignment into annotated gene regions
The sequencing was performed in the National Institutes of Health sequencing facility or in the Johns Hopkins University Bayview campus core facility. We collected 36- or 50-nucleotide-long single-end short reads from the Illumina GAIII or Hiseq2500 sequencer in FASTQ format. The Fastqc package was utilized for quality control on these short read libraries. There are 20 libraries of short reads from eight distinct spermatogenesis stages, all but one (S16) with at least two biological duplicates. Bowtie software (ref, version 0.12.7) aligned these short reads to the Drosophila genome sequence (Flybase dmel_r5.43, as of Jan 2012, ftp://ftp.flybase.net/releases/FB2012_01/dmel_r5.43/). Only those short reads that are uniquely mapped, with at most two mismatches, were retained for later analysis (later referred to as ‘aligned’). The detailed parameters for running BOWTIE are ‘-a --phred33-quals -n 2 -e 70 -l 28 -m 1 --best –strata’. Those filtered short reads were further searched for possible mapping onto exon junction regions because of splicing of pre-mRNA using Tophat software (version 1.3.3). The same criteria were performed as when running Bowtie. The detailed running parameters were ‘-g1 --butterfly-search -I 50000 --segment-length 15 --max-segment-intron 50000 –G’. The annotation of exon structures are described in the following paragraph. Approximately only 1% of aligned reads are from splice junction regions. The reads aligned to the genome sequence and the junction reads are combined and sifted for non-redundancy. Finally, we concentrated on ∼2 million non-redundant reads per stage sample (Fig. S1). The bam mutant and wild-type testis data were retrieved from a previous RNA-seq study (Gan et al., 2010a).
We then assigned each read into gene regions. The annotation for protein coding genes, rRNAs, tRNAs, snoRNA, snRNAss, pre_miRNAs and other non-coding RNAs was retrieved from Flybase (ftp://ftp.flybase.net/releases/FB2012_01/dmel_r5.43/fasta/). The exons from different alternative splicing isoforms were merged to find the maximum genome coverage regions per gene. An in-house perl script assigned each aligned short read to these merged transcription regions, and read counts per gene region are the output. When a read is mapped to a region with more than one gene, i.e. one merged exon region overlapping with a non-coding gene, the count is split as equal possibilities into these two genes, half count for each. On average, only 2% of aligned reads are involved in gene overlapping regions.
Gene annotation and gene expression calculation
We used EdgeR package (Robinson et al., 2010) to quantitatively measure the level of transcripts normalized across different samples (cRPKM). cRPKM generated by EdgeR algorithm compensates for the difference in the sequence reads among samples, as well as the different number of samples for each individual staged cyst. Thus, cRPKM values are comparable among different samples.
Read counts from biological replicates are fitted into a negative binomial distribution model to obtain a dispersion estimate for each gene (‘tagwise dispersion’ in EdgeR), followed by differential expression inference. One single estimate of transcript concentration is evaluated by the maximum likelihood method (qCML in EdgeR) for biological replicates. This single estimate is called the ‘estimated transcription level’.
Estimation of transcription level per gene per stage and differential expression analysis
RPKM was used to estimate the transcription level for short read data by normalizing transcript length and library size. In this study, we used the edgeR software package in R to find the normalization factors for multiple libraries with various sizes using the TMM method (Robinson et al., 2010). The term RPKM in this study differs in that it uses the normalized library size after the TMM normalization. Then, the EdgeR method models short reads into a negative binomial distribution and estimates the biological replicate variance (dispersion). Tagwise dispersion estimation was performed in different groupings of libraries using the ‘estimateGLMTagwiseDisp’ function in EdgeR. Initially, the 20 libraries came from eight sample stages: niche, GB, S4, S8, S16, aly, EC (early spermatocyte) and LC (late spermatocyte). The six LC samples appeared to reflect a gradual transition (usually taking two days) from EC to LC. They were further grouped into LC1 and LC2 according to the correlation coefficient clustering result. Between any pair of conditions/samples, differential expression (DE) can be determined using the generalized linear model (GLM) likelihood ratio test by the glmFit and glmLRT functions. The DE genes were decided upon using Benjamini–Hochberg multiple testing correction and adjusted P-value <0.05.
Other bioinformatics tools
Multidimension scaling, K-means clustering methods are from R package. Heatmaps were generated using pheatmap and RcolorBrewer. Batch effect was examined using single vector decomposition and sva package (Leek et al., 2012). There is no obvious batch effect from time of procedure and a switch from 36 bp to 50 bp sequencing runs. KEGG pathways were downloaded from www.genome.jp/kegg/pathway.html. The gene set enrichment test was performed using flymine, Gorilla (http://cbl-gorilla.cs.technion.ac.il/) and topGO R package.
Supplementary Material
Acknowledgements
We thank Chen lab members for their critical comments on this paper. We thank Nan Yu for assistance with the RNA-seq data storage, deposit and analyses.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: C.L., X.C.; Methodology: Z.S., C.L., V.T., K.C., X.C.; Validation: C.L., V.T.; Formal analysis: Z.S.; Investigation: C.L., V.T., K.C., K.Z., X.C.; Resources: C.L., X.C.; Data curation: Z.S.; Writing - original draft: Z.S., C.L., X.C.; Supervision: K.Z., X.C.; Project administration: X.C.; Funding acquisition: K.Z., X.C.
Funding
This work has been supported by the Pujiang Talent Program (jointly funded by the Shanghai Municipal Human Resources and Social Security Bureau and the Science and Technology Commission of Shanghai Municipality; 17PJ1432000 to Z.S., 2017-present); the Division of Intramural Research at the National Heart, Lung, and Blood Institute (K.Z.); the David and Lucile Packard Foundation; the Howard Hughes Medical Institute; the 49th Mallinckrodt Scholar Award from the Edward Mallinckrodt, Jr. Foundation; the Eunice Kennedy Shriver National Institute of Child Health and Human Development (R01HD065816); the National Institute of General Medical Sciences (R35GM127075); and Johns Hopkins University startup funds (X.C.). Deposited in PMC for release after 12 months.
Data availability
RNA-seq data have been deposited in GEO under accession number GSE142717.
Supplementary information
Supplementary information available online at http://dev.biologists.org/lookup/doi/10.1242/dev.184259.supplemental
Peer review history
The peer review history is available online at https://dev.biologists.org/lookup/doi/10.1242/dev.184259.reviewer-comments.pdf
References
- Amoyel M., Sanny J., Burel M. and Bach E. A. (2013). Hedgehog is required for CySC self-renewal but does not contribute to the GSC niche in the Drosophila testis. Development 140, 56-65. 10.1242/dev.086413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amoyel M., Anderson J., Suisse A., Glasner J. and Bach E. A. (2016a). Socs36E controls niche competition by repressing MAPK signaling in the Drosophila testis. PLoS Genet. 12, e1005815 10.1371/journal.pgen.1005815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amoyel M., Hillion K.-H., Margolis S. R. and Bach E. A. (2016b). Somatic stem cell differentiation is regulated by PI3K/Tor signaling in response to local cues. Development 143, 3914-3925. 10.1242/dev.139782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayyar S., Jiang J., Collu A., White-Cooper H. and White R. A. H. (2003). Drosophila TGIF is essential for developmentally regulated transcription in spermatogenesis. Development 130, 2841-2852. 10.1242/dev.00513 [DOI] [PubMed] [Google Scholar]
- Baker C. C. and Fuller M. T. (2007). Translational control of meiotic cell cycle progression and spermatid differentiation in male germ cells by a novel eIF4G homolog. Development 134, 2863-2869. 10.1242/dev.003764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker C. C., Gim B. S. and Fuller M. T. (2015). Cell type-specific translational repression of Cyclin B during meiosis in males. Development 142, 3394-3402. 10.1242/dev.122341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barckmann B., Chen X., Kaiser S., Jayaramaiah-Raja S., Rathke C., Dottermusch-Heidel C., Fuller M. T. and Renkawitz-Pohl R. (2013). Three levels of regulation lead to protamine and Mst77F expression in Drosophila. Dev. Biol. 377, 33-45. 10.1016/j.ydbio.2013.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barreau C., Benson E., Gudmannsdottir E., Newton F. and White-Cooper H. (2008). Post-meiotic transcription in Drosophila testes. Development 135, 1897-1902. 10.1242/dev.021949 [DOI] [PubMed] [Google Scholar]
- Barroca V., Lassalle B., Coureuil M., Louis J. P., Le Page F., Testart J., Allemand I., Riou L. and Fouchet P. (2009). Mouse differentiating spermatogonia can generate germinal stem cells in vivo. Nat. Cell Biol. 11, 190-196. 10.1038/ncb1826 [DOI] [PubMed] [Google Scholar]
- Beall E. L., Lewis P. W., Bell M., Rocha M., Jones D. L. and Botchan M. R. (2007). Discovery of tMAC: a Drosophila testis-specific meiotic arrest complex paralogous to Myb-Muv B. Genes Dev. 21, 904-919. 10.1101/gad.1516607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betschinger J. and Knoblich J. A. (2004). Dare to be different: asymmetric cell division in Drosophila, C. elegans and vertebrates. Curr. Biol. 14, R674-R685. 10.1016/j.cub.2004.08.017 [DOI] [PubMed] [Google Scholar]
- Boyle M., Wong C., Rocha M. and Jones D. L. (2007). Decline in self-renewal factors contributes to aging of the stem cell niche in the Drosophila testis. Cell Stem Cell 1, 470-478. 10.1016/j.stem.2007.08.002 [DOI] [PubMed] [Google Scholar]
- Brawley C. and Matunis E. (2004). Regeneration of male germline stem cells by spermatogonial dedifferentiation in vivo. Science 304, 1331-1334. 10.1126/science.1097676 [DOI] [PubMed] [Google Scholar]
- Chen D. and McKearin D. M. (2003). A discrete transcriptional silencer in the bam gene determines asymmetric division of the Drosophila germline stem cell. Development 130, 1159-1170. 10.1242/dev.00325 [DOI] [PubMed] [Google Scholar]
- Chen X., Hiller M., Sancak Y. and Fuller M. T. (2005). Tissue-specific TAFs counteract Polycomb to turn on terminal differentiation. Science 310, 869-872. 10.1126/science.1118101 [DOI] [PubMed] [Google Scholar]
- Chen X., Lu C., Prado J. R. M., Eun S. H. and Fuller M. T. (2011). Sequential changes at differentiation gene promoters as they become active in a stem cell lineage. Development 138, 2441-2450. 10.1242/dev.056572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H., Chen X. and Zheng Y. (2013). The nuclear lamina regulates germline stem cell niche organization via modulation of EGFR signaling. Cell Stem Cell 13, 73-86. 10.1016/j.stem.2013.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J., Türkel N., Hemati N., Fuller M. T., Hunt A. J. and Yamashita Y. M. (2008). Centrosome misorientation reduces stem cell division during ageing. Nature 456, 599-604. 10.1038/nature07386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J., Tiyaboonchai A., Yamashita Y. M. and Hunt A. J. (2011). Asymmetric division of cyst stem cells in Drosophila testis is ensured by anaphase spindle repositioning. Development 138, 831-837. 10.1242/dev.057901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cinalli R. M., Rangan P. and Lehmann R. (2008). Germ cells are forever. Cell 132, 559-562. 10.1016/j.cell.2008.02.003 [DOI] [PubMed] [Google Scholar]
- Clarke M. F. and Fuller M. (2006). Stem cells and cancer: two faces of eve. Cell 124, 1111-1115. 10.1016/j.cell.2006.03.011 [DOI] [PubMed] [Google Scholar]
- Clevers H. (2005). Stem cells, asymmetric division and cancer. Nat. Genet. 37, 1027-1028. 10.1038/ng1005-1027 [DOI] [PubMed] [Google Scholar]
- Court H., Ahearn I. M., Amoyel M., Bach E. A. and Philips M. R. (2017). Regulation of NOTCH signaling by RAB7 and RAB8 requires carboxyl methylation by ICMT. J. Cell Biol. 216, 4165-4182. 10.1083/jcb.201701053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtot C., Fankhauser C., Simanis V. and Lehner C. F. (1992). The Drosophila cdc25 homolog twine is required for meiosis. Development 116, 405-416. [DOI] [PubMed] [Google Scholar]
- Davies E. L. and Fuller M. T. (2008). Regulation of self-renewal and differentiation in adult stem cell lineages: lessons from the Drosophila male germ line. Cold Spring Harb. Symp. Quant. Biol. 73, 137-145. 10.1101/sqb.2008.73.063 [DOI] [PubMed] [Google Scholar]
- de Cuevas M. and Matunis E. L. (2011). The stem cell niche: lessons from the Drosophila testis. Development 138, 2861-2869. 10.1242/dev.056242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng X., Hiatt J. B., Nguyen D. K., Ercan S., Sturgill D., Hillier L. W., Schlesinger F., Davis C. A., Reinke V. J., Gingeras T. R. et al. (2011). Evidence for compensatory upregulation of expressed X-linked genes in mammals, Caenorhabditis elegans and Drosophila melanogaster. Nat. Genet. 43, 1179-1185. 10.1038/ng.948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eun S. H., Stoiber P. M., Wright H. J., McMurdie K. E., Choi C. H., Gan Q., Lim C. and Chen X. (2013). MicroRNAs downregulate Bag of marbles to ensure proper terminal differentiation in the Drosophila male germline. Development 140, 23-30. 10.1242/dev.086397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eun S. H., Shi Z., Cui K., Zhao K. and Chen X. (2014). A non-cell autonomous role of E(z) to prevent germ cells from turning on a somatic cell marker. Science 343, 1513-1516. 10.1126/science.1246514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eun S. H., Feng L., Cedeno-Rosario L., Gan Q., Wei G., Cui K., Zhao K. and Chen X. (2017). Polycomb group gene E(z) is required for spermatogonial dedifferentiation in drosophila adult testis. J. Mol. Biol. 429, 2030-2041. 10.1016/j.jmb.2017.04.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabian L. and Brill J. A. (2012). Drosophila spermiogenesis: big things come from little packages. Spermatogenesis 2, 197-212. 10.4161/spmg.21798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng L. and Chen X. (2015). Epigenetic regulation of germ cells-remember or forget? Curr. Opin. Genet. Dev. 31, 20-27. 10.1016/j.gde.2015.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng L., Shi Z. and Chen X. (2017). Enhancer of polycomb coordinates multiple signaling pathways to promote both cyst and germline stem cell differentiation in the Drosophila adult testis. PLoS Genet. 13, e1006571 10.1371/journal.pgen.1006571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng L., Shi Z., Xie J., Ma B. and Chen X. (2018). Enhancer of polycomb maintains germline activity and genome integrity in Drosophila testis. Cell Death Differ. 25, 1486-1502. 10.1038/s41418-017-0056-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedmann-Morvinski D., Bushong E. A., Ke E., Soda Y., Marumoto T., Singer O., Ellisman M. H. and Verma I. M. (2012). Dedifferentiation of neurons and astrocytes by oncogenes can induce gliomas in mice. Science 338, 1080-1084. 10.1126/science.1226929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller M. T. (1998). Genetic control of cell proliferation and differentiation in Drosophila spermatogenesis. Semin. Cell Dev. Biol. 9, 433-444. 10.1006/scdb.1998.0227 [DOI] [PubMed] [Google Scholar]
- Fuller M. T. (2016). Differentiation in stem cell lineages and in life: explorations in the male germ line stem cell lineage. Curr. Top. Dev. Biol. 116, 375-390. 10.1016/bs.ctdb.2015.11.041 [DOI] [PubMed] [Google Scholar]
- Gan Q., Chepelev I., Wei G., Tarayrah L., Cui K., Zhao K. and Chen X. (2010a). Dynamic regulation of alternative splicing and chromatin structure in Drosophila gonads revealed by RNA-seq. Cell Res. 20, 763-783. 10.1038/cr.2010.64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan Q., Schones D. E., Ho Eun S., Wei G., Cui K., Zhao K. and Chen X. (2010b). Monovalent and unpoised status of most genes in undifferentiated cell-enriched Drosophila testis. Genome Biol. 11, R42 10.1186/gb-2010-11-4-r42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelbart M. E., Larschan E., Peng S., Park P. J. and Kuroda M. I. (2009). Drosophila MSL complex globally acetylates H4K16 on the male X chromosome for dosage compensation. Nat. Struct. Mol. Biol. 16, 825-832. 10.1038/nsmb.1644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleason R. J., Anand A., Kai T. and Chen X. (2018). Protecting and diversifying the germline. Genetics 208, 435-471. 10.1534/genetics.117.300208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez J., Qi H., Liu N. and Lin H. (2015). Piwi is a key regulator of both somatic and germline stem cells in the Drosophila testis. Cell Rep. 12, 150-161. 10.1016/j.celrep.2015.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenspan L. J., de Cuevas M. and Matunis E. (2015). Genetics of gonadal stem cell renewal. Annu. Rev. Cell Dev. Biol. 31, 291-315. 10.1146/annurev-cellbio-100913-013344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta V., Parisi M., Sturgill D., Nuttall R., Doctolero M., Dudko O. K., Malley J. D., Eastman P. S. and Oliver B. (2006). Global analysis of X-chromosome dosage compensation. J. Biol. 5, 3 10.1186/jbiol30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasan S., Hétié P. and Matunis E. L. (2015). Niche signaling promotes stem cell survival in the Drosophila testis via the JAK-STAT target DIAP1. Dev. Biol. 404, 27-39. 10.1016/j.ydbio.2015.04.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrera S. C. and Bach E. A. (2018). JNK signaling triggers spermatogonial dedifferentiation during chronic stress to maintain the germline stem cell pool in the Drosophila testis. eLife 7, e36095 10.7554/eLife.36095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrera S. C. and Bach E. A. (2019). JAK/STAT signaling in stem cells and regeneration: from Drosophila to vertebrates. Development 146, dev167643 10.1242/dev.167643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiller M. A., Lin T. Y., Wood C. and Fuller M. T. (2001). Developmental regulation of transcription by a tissue-specific TAF homolog. Genes Dev. 15, 1021-1030. 10.1101/gad.869101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiller M., Chen X., Pringle M. J., Suchorolski M., Sancak Y., Viswanathan S., Bolival B., Lin T.-Y., Marino S. and Fuller M. T. (2004). Testis-specific TAF homologs collaborate to control a tissue-specific transcription program. Development 131, 5297-5308. 10.1242/dev.01314 [DOI] [PubMed] [Google Scholar]
- Inaba M. and Yamashita Y. M. (2012). Asymmetric stem cell division: precision for robustness. Cell Stem Cell 11, 461-469. 10.1016/j.stem.2012.09.003 [DOI] [PubMed] [Google Scholar]
- Inaba M., Yuan H. and Yamashita Y. M. (2011). String (Cdc25) regulates stem cell maintenance, proliferation and aging in Drosophila testis. Development 138, 5079-5086. 10.1242/dev.072579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Insco M. L., Leon A., Tam C. H., McKearin D. M. and Fuller M. T. (2009). Accumulation of a differentiation regulator specifies transit amplifying division number in an adult stem cell lineage. Proc. Natl. Acad. Sci. USA 106, 22311-22316. 10.1073/pnas.0912454106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Insco M. L., Bailey A. S., Kim J., Olivares G. H., Wapinski O. L., Tam C. H. and Fuller M. T. (2012). A self-limiting switch based on translational control regulates the transition from proliferation to differentiation in an adult stem cell lineage. Cell Stem Cell 11, 689-700. 10.1016/j.stem.2012.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Issigonis M., Tulina N., de Cuevas M., Brawley C., Sandler L. and Matunis E. (2009). JAK-STAT signal inhibition regulates competition in the Drosophila testis stem cell niche. Science 326, 153-156. 10.1126/science.1176817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J. and White-Cooper H. (2003). Transcriptional activation in Drosophila spermatogenesis involves the mutually dependent function of aly and a novel meiotic arrest gene cookie monster. Development 130, 563-573. 10.1242/dev.00246 [DOI] [PubMed] [Google Scholar]
- Jiang J., Benson E., Bausek N., Doggett K. and White-Cooper H. (2007). Tombola, a tesmin/TSO1-family protein, regulates transcriptional activation in the Drosophila male germline and physically interacts with always early. Development 134, 1549-1559. 10.1242/dev.000521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kai T. and Spradling A. (2004). Differentiating germ cells can revert into functional stem cells in Drosophila melanogaster ovaries. Nature 428, 564-569. 10.1038/nature02436 [DOI] [PubMed] [Google Scholar]
- Kalmykova A. I., Klenov M. S. and Gvozdev V. A. (2005). Argonaute protein PIWI controls mobilization of retrotransposons in the Drosophila male germline. Nucleic Acids Res. 33, 2052-2059. 10.1093/nar/gki323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiger A. A., White-Cooper H. and Fuller M. T. (2000). Somatic support cells restrict germline stem cell self-renewal and promote differentiation. Nature 407, 750-754. 10.1038/35037606 [DOI] [PubMed] [Google Scholar]
- Kiger A. A., Jones D. L., Schulz C., Rogers M. B. and Fuller M. T. (2001). Stem cell self-renewal specified by JAK-STAT activation in response to a support cell cue. Science 294, 2542-2545. 10.1126/science.1066707 [DOI] [PubMed] [Google Scholar]
- Kurimoto K., Yabuta Y., Ohinata Y. and Saitou M. (2007). Global single-cell cDNA amplification to provide a template for representative high-density oligonucleotide microarray analysis. Nat. Protoc. 2, 739-752. 10.1038/nprot.2007.79 [DOI] [PubMed] [Google Scholar]
- Leatherman J. L. and Dinardo S. (2008). Zfh-1 controls somatic stem cell self-renewal in the Drosophila testis and nonautonomously influences germline stem cell self-renewal. Cell Stem Cell 3, 44-54. 10.1016/j.stem.2008.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leatherman J. L. and Dinardo S. (2010). Germline self-renewal requires cyst stem cells and stat regulates niche adhesion in Drosophila testes. Nat. Cell Biol. 12, 806-811. 10.1038/ncb2086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leek J. T., Johnson W. E., Parker H. S., Jaffe A. E., and Storey J. D. (2012). The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 28, 882-883. 10.1093/bioinformatics/bts034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Ma Q., Cherry C. M. and Matunis E. L. (2014). Steroid signaling promotes stem cell maintenance in the Drosophila testis. Dev. Biol. 394, 129-141. 10.1016/j.ydbio.2014.07.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim J. G. Y. and Fuller M. T. (2012). Somatic cell lineage is required for differentiation and not maintenance of germline stem cells in Drosophila testes. Proc. Natl. Acad. Sci. USA 109, 18477-18481. 10.1073/pnas.1215516109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim C., Tarayrah L. and Chen X. (2012). Transcriptional regulation during Drosophila spermatogenesis. Spermatogenesis 2, 158-166. 10.4161/spmg.21775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim C., Gandhi S., Biniossek M. L., Feng L., Schilling O., Urban S. and Chen X. (2015). An aminopeptidase in the Drosophila testicular niche acts in germline stem cell maintenance and spermatogonial dedifferentiation. Cell Rep. 13, 315-325. 10.1016/j.celrep.2015.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin T. Y., Viswanathan S., Wood C., Wilson P. G., Wolf N. and Fuller M. T. (1996). Coordinate developmental control of the meiotic cell cycle and spermatid differentiation in Drosophila males. Development 122, 1331-1341. [DOI] [PubMed] [Google Scholar]
- Maines J. Z. and Wasserman S. A. (1999). Post-transcriptional regulation of the meiotic Cdc25 protein Twine by the Dazl orthologue Boule. Nat. Cell Biol. 1, 171-174. 10.1038/11091 [DOI] [PubMed] [Google Scholar]
- Morrison S. J. and Kimble J. (2006). Asymmetric and symmetric stem-cell divisions in development and cancer. Nature 441, 1068-1074. 10.1038/nature04956 [DOI] [PubMed] [Google Scholar]
- Nakagawa T., Sharma M., Nabeshima Y., Braun R. E. and Yoshida S. (2010). Functional hierarchy and reversibility within the murine spermatogenic stem cell compartment. Science 328, 62-67. 10.1126/science.1182868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papagiannouli F. and Lohmann I. (2015). Stage-specific control of stem cell niche architecture in the Drosophila testis by the posterior Hox gene Abd-B. Comput. Struct. Biotechnol. J. 13, 122-130. 10.1016/j.csbj.2015.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisi M., Nuttall R., Naiman D., Bouffard G., Malley J., Andrews J., Eastman S. and Oliver B. (2003). Paucity of genes on the Drosophila X chromosome showing male-biased expression. Science 299, 697-700. 10.1126/science.1079190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrott B. B., Chiang Y., Hudson A., Sarkar A., Guichet A. and Schulz C. (2011). Nucleoporin98-96 function is required for transit amplification divisions in the germ line of Drosophila melanogaster. PLoS ONE 6, e25087 10.1371/journal.pone.0025087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrott B. B., Hudson A., Brady R. and Schulz C. (2012). Control of germline stem cell division frequency--a novel, developmentally regulated role for epidermal growth factor signaling. PLoS ONE 7, e36460 10.1371/journal.pone.0036460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perezgasga L., Jiang J., Bolival B. Jr, Hiller M., Benson E., Fuller M. T. and White-Cooper H. (2004). Regulation of transcription of meiotic cell cycle and terminal differentiation genes by the testis-specific Zn-finger protein matotopetli. Development 131, 1691-1702. 10.1242/dev.01032 [DOI] [PubMed] [Google Scholar]
- Rangan P., DeGennaro M. and Lehmann R. (2008). Regulating gene expression in the Drosophila germ line. Cold Spring Harb. Symp. Quant. Biol. 73, 1-8. 10.1101/sqb.2008.73.057 [DOI] [PubMed] [Google Scholar]
- Robinson M. D., McCarthy D. J. and Smyth G. K. (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139-140. 10.1093/bioinformatics/btp616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saberi A., Jamal A., Beets I., Schoofs L. and Newmark P. A. (2016). GPCRs direct germline development and somatic gonad function in planarians. PLoS Biol. 14, e1002457 10.1371/journal.pbio.1002457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santel A., Winhauer T., Blümer N. and Renkawitz-Pohl R. (1997). The Drosophila don juan (dj) gene encodes a novel sperm specific protein component characterized by an unusual domain of a repetitive amino acid motif. Mech. Dev. 64, 19-30. 10.1016/S0925-4773(97)00031-2 [DOI] [PubMed] [Google Scholar]
- Sarkar A., Parikh N., Hearn S. A., Fuller M. T., Tazuke S. I. and Schulz C. (2007). Antagonistic roles of Rac and Rho in organizing the germ cell microenvironment. Curr. Biol. 17, 1253-1258. 10.1016/j.cub.2007.06.048 [DOI] [PubMed] [Google Scholar]
- Schulz C., Kiger A. A., Tazuke S. I., Yamashita Y. M., Pantalena-Filho L. C., Jones D. L., Wood C. G. and Fuller M. T. (2004). A misexpression screen reveals effects of bag-of-marbles and TGFβ class signaling on the Drosophila male germ-line stem cell lineage. Genetics 167, 707-723. 10.1534/genetics.103.023184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwitalla S., Fingerle A. A., Cammareri P., Nebelsiek T., Göktuna S. I., Ziegler P. K., Canli O., Heijmans J., Huels D. J., Moreaux G. et al. (2013). Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like properties. Cell 152, 25-38. 10.1016/j.cell.2012.12.012 [DOI] [PubMed] [Google Scholar]
- Seydoux G. and Braun R. E. (2006). Pathway to totipotency: lessons from germ cells. Cell 127, 891-904. 10.1016/j.cell.2006.11.016 [DOI] [PubMed] [Google Scholar]
- Sheng X. R. and Matunis E. L. (2009). Make room for dedifferentiation. Fly (Austin) 3, 283-285. 10.4161/fly.10413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng X. R., Brawley C. M. and Matunis E. L. (2009). Dedifferentiating spermatogonia outcompete somatic stem cells for niche occupancy in the Drosophila testis. Cell Stem Cell 5, 191-203. 10.1016/j.stem.2009.05.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stine R. R., Greenspan L. J., Ramachandran K. V. and Matunis E. L. (2014). Coordinate regulation of stem cell competition by Slit-Robo and JAK-STAT signaling in the Drosophila testis. PLoS Genet. 10, e1004713 10.1371/journal.pgen.1004713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talchai C., Xuan S., Lin H. V., Sussel L. and Accili D. (2012). Pancreatic β cell dedifferentiation as a mechanism of diabetic β cell failure. Cell 150, 1223-1234. 10.1016/j.cell.2012.07.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang F., Barbacioru C., Wang Y., Nordman E., Lee C., Xu N., Wang X., Bodeau J., Tuch B. B., Siddiqui A. et al. (2009). mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Methods 6, 377-382. 10.1038/nmeth.1315 [DOI] [PubMed] [Google Scholar]
- Tarayrah L., Herz H.-M., Shilatifard A. and Chen X. (2013). Histone demethylase dUTX antagonizes JAK-STAT signaling to maintain proper gene expression and architecture of the Drosophila testis niche. Development 140, 1014-1023. 10.1242/dev.089433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry N. A., Tulina N., Matunis E. and DiNardo S. (2006). Novel regulators revealed by profiling Drosophila testis stem cells within their niche. Dev. Biol. 294, 246-257. 10.1016/j.ydbio.2006.02.048 [DOI] [PubMed] [Google Scholar]
- Toledano H., D'Alterio C., Czech B., Levine E. and Jones D. L. (2012). The let-7-Imp axis regulates ageing of the Drosophila testis stem-cell niche. Nature 485, 605-610. 10.1038/nature11061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran J., Brenner T. J. and DiNardo S. (2000). Somatic control over the germline stem cell lineage during Drosophila spermatogenesis. Nature 407, 754-757. 10.1038/35037613 [DOI] [PubMed] [Google Scholar]
- Tran V., Lim C., Xie J. and Chen X. (2012). Asymmetric division of Drosophila male germline stem cell shows asymmetric histone distribution. Science 338, 679-682. 10.1126/science.1226028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tulina N. and Matunis E. (2001). Control of stem cell self-renewal in Drosophila spermatogenesis by JAK-STAT signaling. Science 294, 2546-2549. 10.1126/science.1066700 [DOI] [PubMed] [Google Scholar]
- Vedelek V., Bodai L., Grezal G., Kovács B., Boros I. M., Laurinyecz B. and Sinka R. (2018). Analysis of Drosophila melanogaster testis transcriptome. BMC Genomics 19, 697 10.1186/s12864-018-5085-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallenfang M. R., Nayak R. and DiNardo S. (2006). Dynamics of the male germline stem cell population during aging of Drosophila melanogaster. Aging Cell 5, 297-304. 10.1111/j.1474-9726.2006.00221.x [DOI] [PubMed] [Google Scholar]
- Wang Z., Gerstein M. and Snyder M. (2009). RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 10, 57-63. 10.1038/nrg2484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss I. and Bohrmann J. (2019). Electrochemical patterns during Drosophila oogenesis: ion-transport mechanisms generate stage-specific gradients of pH and membrane potential in the follicle-cell epithelium. BMC Dev. Biol. 19, 12 10.1186/s12861-019-0192-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- White-Cooper H. and Caporilli S. (2013). Transcriptional and post-transcriptional regulation of Drosophila germline stem cells and their differentiating progeny. Adv. Exp. Med. Biol. 786, 47-61. 10.1007/978-94-007-6621-1_4 [DOI] [PubMed] [Google Scholar]
- White-Cooper H., Schafer M. A., Alphey L. S. and Fuller M. T. (1998). Transcriptional and post-transcriptional control mechanisms coordinate the onset of spermatid differentiation with meiosis I in Drosophila. Development 125, 125-134. [DOI] [PubMed] [Google Scholar]
- White-Cooper H., Leroy D., MacQueen A. and Fuller M. T. (2000). Transcription of meiotic cell cycle and terminal differentiation genes depends on a conserved chromatin associated protein, whose nuclear localisation is regulated. Development 127, 5463-5473. [DOI] [PubMed] [Google Scholar]
- Wong C. and Jones D. L. (2012). Efficiency of spermatogonial dedifferentiation during aging. PLoS ONE 7, e33635 10.1371/journal.pone.0033635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita Y. M. (2018). Subcellular specialization and organelle behavior in germ cells. Genetics 208, 19-51. 10.1534/genetics.117.300184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita Y. M., Jones D. L. and Fuller M. T. (2003). Orientation of asymmetric stem cell division by the APC tumor suppressor and centrosome. Science 301, 1547-1550. 10.1126/science.1087795 [DOI] [PubMed] [Google Scholar]
- Yamashita Y. M., Mahowald A. P., Perlin J. R. and Fuller M. T. (2007). Asymmetric inheritance of mother versus daughter centrosome in stem cell division. Science 315, 518-521. 10.1126/science.1134910 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.