

Abstract
Background and Purpose
Respiratory viral infections play central roles in the initiation, exacerbation and progression of asthma in humans. An acute paramyxoviral infection in mice can cause a chronic lung disease that resembles human asthma. We sought to determine whether reduction of Sendai virus lung burden in mice by stimulating innate immunity with aerosolized Toll‐like receptor (TLR) agonists could attenuate the severity of chronic asthma‐like lung disease.
Experimental Approach
Mice were treated by aerosol with 1‐μM oligodeoxynucleotide (ODN) M362, an agonist of the TLR9 homodimer, and 4‐μM Pam2CSK4 (Pam2), an agonist of the TLR2/6 heterodimer, within a few days before or after Sendai virus challenge.
Key Results
Treatment with ODN/Pam2 caused ~75% reduction in lung Sendai virus burden 5 days after challenge. The reduction in acute lung virus burden was associated with marked reductions 49 days after viral challenge in eosinophilic and lymphocytic lung inflammation, airway mucous metaplasia, lumenal mucus occlusion and hyperresponsiveness to methacholine. Mechanistically, ODN/Pam2 treatment attenuated the chronic asthma phenotype by suppressing IL‐33 production by type 2 pneumocytes, both by reducing the severity of acute infection and by down‐regulating Type 2 (allergic) inflammation.
Conclusion and Implications
These data suggest that treatment of susceptible human hosts with aerosolized ODN and Pam2 at the time of a respiratory viral infection might attenuate the severity of the acute infection and reduce initiation, exacerbation and progression of asthma.
What is already known
Respiratory viral infections play a major role in the pathogenesis of asthma.
We have previously identified a pharmacological means to stimulate innate immunity of lung epithelial cells.
What this study adds
Activation of innate immunity in lung epithelial cells prevents chronic viral asthma‐like disease in mice.
This activation reduces acute viral burden and modulates long‐term immune responses.
What is the clinical significance
Aerosolized toll‐like receptor agonists have been demonstrated as safe in human clinical trials.
This study provides proof‐of‐principle that aerosolized toll‐like agonists could have clinical efficacy against virus‐induced asthma.
1. INTRODUCTION
Respiratory viral infections play a major role in the pathogenesis of asthma at every stage of disease—initiation, exacerbation and progression (Busse, Lemanske, & Gern, 2010; Farne & Johnston, 2017; Holgate, 2012; Holt & Sly, 2012; Peebles & Aronica, 2019). Regarding asthma initiation, children who experience a respiratory infection with rhinovirus or respiratory syncytial virus have a substantially increased chance of subsequently developing asthma. Regarding asthma exacerbation, new molecular techniques have allowed the identification of viral infections in more than 80% of exacerbation episodes in both children and adults. Last, the progression of underlying asthma severity, defined as poorly reversible airflow obstruction and persistent symptoms, is associated with frequent respiratory viral infections. Rhinoviruses are the most common cause of asthma exacerbations, but paramyxoviruses such as respiratory syncytial virus and parainfluenza virus often cause severe disease and have a prominent role in disease initiation (Bønnelykke, Vissing, Sevelsted, Johnston, & Bisgaard, 2015; Busse et al., 2010; Liu et al., 2013; Peebles & Aronica, 2019).
Although the epidemiologic association between respiratory virus infection and asthma is strong, until recently a mechanistic explanation of the interaction has been lacking. It now appears that the interaction operates in both directions. The presence of asthma results in an increased susceptibility to lower respiratory tract infection by viruses, possibly due at least in part to deficient interferon (IFN) responses resulting from Type 2 immune deviation (Farne & Johnston, 2017; Holgate, 2012; Holt & Sly, 2012; Holtzman, Patel, Zhang, & Patel, 2011; Lambrecht & Hammad, 2015). Conversely, respiratory viral infection can result in long‐term reprogramming of the lung immune environment towards a Type 2 polarity that results in parenchymal changes characteristic of asthma and chronic obstructive pulmonary disease (COPD; Farne & Johnston, 2017; Holgate, 2012; Holt & Sly, 2012; Holtzman, 2012; Lambrecht & Hammad, 2015). Mechanistic insight in this regard has come from studies of a mouse paramyxoviral lung infection with Sendai virus (SeV) by Holtzman and colleagues (Patel et al., 2006; Walter, Morton, Kajiwara, Agapov, & Holtzman, 2002). Analysis of this model has revealed that an acute SeV infection, in which virus is cleared within 12 days, can lead to a chronic, possibly lifetime, asthmatic phenotype. In susceptible mouse strains, the lungs 49 days after infectious challenge are characterized by Type 2 inflammation, epithelial mucous metaplasia and hyper‐responsiveness to bronchoconstrictor stimuli such as https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7438 (Holtzman, 2012; Holtzman et al., 2011; Patel et al., 2006; Walter et al., 2002). In recent work, this group has shown that the acute SeV infection leads to long‐term elevated expression of https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5880 by lung epithelial cells that drives the persistent elaboration of high levels of https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4980 by immune cells within the lungs, which in turn induces the lung parenchymal changes of epithelial mucous metaplasia and smooth muscle hyperresponsiveness (Byers et al., 2013).
In view of the high incidence and substantial morbidity resulting from virus‐induced asthma, treatments to mitigate the severity of respiratory virus infection are greatly needed. We have previously identified a pharmacological means to stimulate Type 3, extracellular pathogen‐directed (Annunziato, Romagnani, & Romagnani, 2015) innate immunity of lung epithelial cells. This treatment has shown activity against multiple bacterial, fungal and viral pathogens in mice (Alfaro et al., 2014; Cleaver et al., 2014; Duggan et al., 2011; Leiva‐Juarez et al., 2018; Leiva‐Juárez et al., 2016; Tuvim, Gilbert, Dickey, & Evans, 2012; Ware et al., 2019) and guinea pigs (Drake, Evans, Dickey, Fryer, & Jacoby, 2013). The stimulus consists of a class C oligodeoxynucleotide (ODN M362), which is an agonist of the https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1759 (TLR9) homodimer, and https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=8560, which is an agonist of the https://www.guidetoimmunopharmacology.org/GRAC/ObjectDisplayForward?objectId=1752/https://www.guidetoimmunopharmacology.org/GRAC/ObjectDisplayForward?objectId=1756 heterodimer. Administration of these two agonists (hereafter called O/P) in a fixed molar ratio shows synergistic activity against microbial pathogens that is dependent upon myeloid differentiation primary response 88 (MyD88) expression in lung epithelial cells and the generation of microbicidal reactive oxygen species (ROS; Cleaver et al., 2014; Duggan et al., 2011; Evans et al., 2011; Kirkpatrick et al., 2018). We hypothesized that treatment with O/P at the time of respiratory infection with SeV would reduce lung virus burden during the acute infection and thereby attenuate the development of a late asthma phenotype.
2. METHODS
2.1. Mice and chemicals
Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010) and with the recommendations made by the British Journal of Pharmacology (McGrath & Lilley, 2015). Seven‐week‐old female C57BL/6J mice were obtained from the Jackson Laboratory (Sacramento, CA; Stock# 000664; RRID:IMSR_JAX:000664) and housed in specific pathogen‐free conditions on a 12‐hr light/dark cycle with free access to food and water. For euthanasia, mice were injected intraperitoneally with 2,2,2‐tribromoethanol (250 mg·kg−1) and exsanguinated by transection of the abdominal aorta. All procedures were performed in accordance with the Institutional Animal Care and Use Committee of MD Anderson Cancer Center and the Texas A&M Institute for Biosciences and Technology. Chemicals were obtained from MilliporeSigma (St. Louis, MO) unless otherwise specified.
2.2. Viral challenge
Sendai virus (parainfluenza type 1) was obtained from the ATCC (cat# VR‐105, RRID:SCR_001672) and expanded in primary rhesus monkey kidney cells obtained from Cell Pro Labs (Golden Valley, MN; cat#103–175). In most experiments, mice were infected with 2.4 × 106 plaque forming units (pfu) in 40 μl PBS instilled into the oropharynx and aspirated into the lungs, with mice suspended by the upper incisors on a board at 60° from horizontal under isoflurane inhalation anaesthesia. The dose of virus was chosen to result in severe bronchopneumonia, but with death in <20% of mice. Lower and higher concentrations of SeV were given in pilot experiments to establish an optimal dose for chronic studies (Figure 1c) and in a high‐dose challenge study (1.2 × 108 pfu) to examine interactive effects of O/P and https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4967 (Figure 8d). Lung virus burden was measured by reverse transcription quantitative PCR (RT‐qPCR) of the Sendai matrix (M) protein normalized to mouse 18S rRNA as described (Moreno‐Vinasco et al., 2009), except for substitution of the primers to ACTGGGACCCTATCTAAGACAT and TAGTAGCGGAAATCACGAGG and analysed by the comparative CT method (Schmittgen & Livak, 2008).
Figure 1.
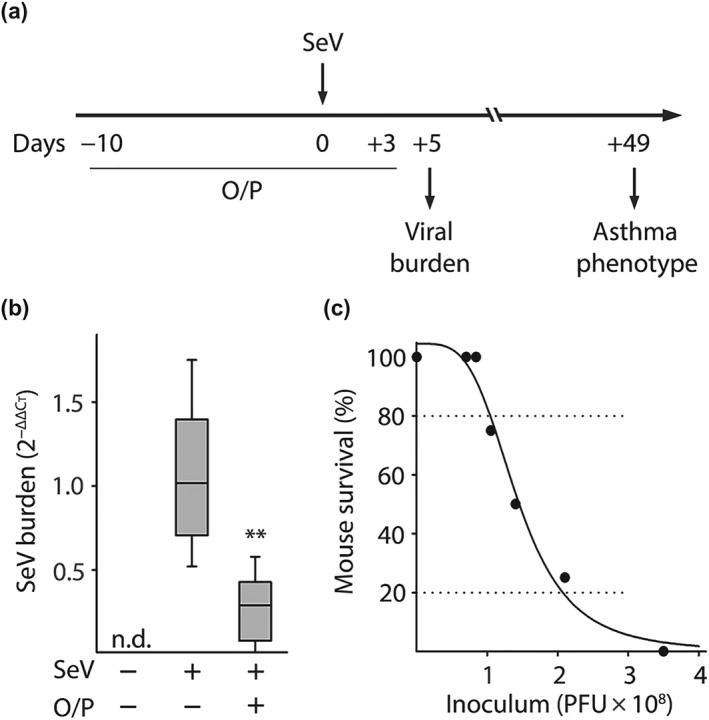
Experimental paradigm, effect of O/P treatment on lung SeV burden and correlation between SeV burden and mouse survival. (a) Illustration of the experimental paradigm with O/P administered by aerosol a few days before or after SeV challenge, lung SeV burden assessed 5 days after challenge and development of an asthma phenotype assessed 49 days after challenge. (b) Lung SeV burden measured by qRT‐PCR 5 days after mice were challenged with SeV, with or without treatment 1 day earlier with O/P (boxes show median and interquartile range, whiskers show 10th and 90th percentiles; **P < .01 for [SeV+, O/P+] versus [SeV+, O/P−] by Mann–Whitney U test; N = 24 mice per group pooled from four experiments). (c) Various SeV inocula were administered to determine the correlation between lung SeV burden and mouse survival (N = 4 mice per group in a representative experiment performed five times)
Figure 8.
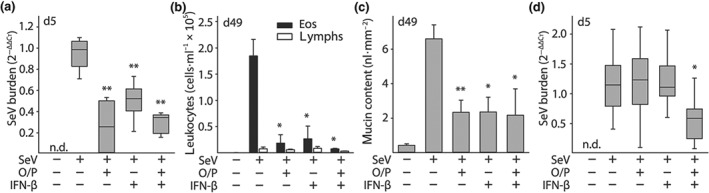
Comparison of the effects of O/P and IFN‐β on acute lung SeV burden and late asthma phenotypes. (a) Lung SeV burden by qRT‐PCR 5 days after mice were challenged with SeV, with or without pretreatment 1 day earlier with O/P, IFN‐β, or both (boxes show median and interquartile range, whiskers show 10th and 90th percentiles; *P < .05 and **P < .01 for [SeV+, drug+] versus [SeV+, drug−], with P > .05 for all other possible pairwise comparisons by one‐way ANOVA with Holm–Sidak's test for multiple comparisons; N = 4–6 mice per group in a single experiment; n.d. = not detectable). (b) Eosinophils and lymphocytes in lung lavage fluid were enumerated 49 days after SeV challenge as in (a) (bars show mean ± SEM; P values as in (a); N = 4–6 mice per group in a single experiment). (c) Quantification of intracellular mucin content of airway epithelium 49 days after SeV challenge as in (a) (bars and P values are as in (a); N = 3–4 in a single experiment). (d) Lung SeV burden by RT‐qPCR 5 days after mice were challenged with a high dose of SeV (2.5 × LD50), with or without pretreatment 1 day earlier with O/P, IFN‐β, or both (boxes show median and interquartile range, whiskers show 10th and 90th percentiles, n.d. = not detectable; *P < .05 for both drugs together versus either drug alone by one‐way ANOVA with Holm–Sidak's test for multiple comparisons; N = 3 for the uninfected group, 10 for all other groups in a single experiment)
2.3. Treatment with aerosolized O/P
This was performed as described (Alfaro et al., 2014). Briefly, ODN 5′ TCG TCG TCG TTC GAA CGA CGT TGA T 3′ as the sodium salt on a phosphorothioate backbone (ODN M362) was purchased from TriLink BioTechnologies (San Diego, CA) and 2,3‐bis (palmitoyloxy)‐2‐propyl‐Cys‐Ser‐Lys‐Lys‐Lys‐Lys‐OH (Pam2CSK4) as the trifluoroacetic acid salt was purchased from Peptides International (Louisville, KY). A solution of ODN (1 μM) and Pam2CSK4 (4 μM) in endotoxin‐free sterile water (8 ml) was placed in an Aerotech II nebulizer (Biodex Medical Systems, Shirley, NY) driven by 10 L·min−1 of 5% CO2 in air to promote deep breathing. The nebulizer was connected by polyethylene tubing (30 cm × 22 mm) to a 10‐L polyethylene chamber vented to a biosafety hood. Mice were exposed to the aerosol for 20 min, resulting in the nebulization of ~4 ml of O/P solution. In one set of experiments determining dose–response relationships, the concentration of O/P solution was varied as indicated.
2.4. Lung lavage
This was performed by instilling and collecting two 1‐ml aliquots of PBS through a 20‐gauge cannula inserted through rings of the exposed trachea of killed animals, then combining the aliquots as described (Alfaro et al., 2014). Total leukocyte count was determined using a haemocytometer and differential counts by cytocentrifugation of 200 μl of lavage fluid at 300 g for 5 min followed by Wright–Giemsa staining.
2.5. Epithelial mucin content and lumenal mucus occlusion
Epithelial mucin content was measured as described (Evans et al., 2004; Piccotti, Dickey, & Evans, 2012; Tuvim et al., 2009). Briefly, lungs were fixed by intratracheal inflation with 10% formalin to 20 cm H2O pressure for 12 hr and then embedded in paraffin. Tissue blocks were cut into 5‐μm sections, mounted on frosted glass slides (Medline, Northfield, IL), deparaffinized with xylene, washed with ethanol and then rehydrated and stained with periodic acid fluorescent Schiff reagent (PAFS). Images were acquired by investigators blinded to mouse treatment and morphometric analysis of the images for quantitation of intracellular mucin was performed using ImagePro (Media Cybernetics, Bethesda, MD). Data are presented as epithelial mucin volume density, signifying the measured volume of mucin overlying a unit area of epithelial basal lamina. Airway mucus plugging was measured by modifications, as follows, of a method we have described previously (Evans et al., 2015). Lungs were fixed by immersion, to avoid displacement of lumenal mucus by inflation, in methanol‐based Carnoy's solution (methacarn), to minimize changes in mucus volume, for 48 hr at 4°C. Lungs were then excised and the left lung was placed in a precision cutting tool to generate 1‐mm transverse slices (Figure S3D). Four slices were embedded in paraffin, with two slices taken below the hilum and two above, each separated by a 1‐mm slice that was discarded. One 5‐μm section was taken from each slice, deparaffinized, rehydrated and then stained with PAFS. The total area of lumenal mucus in all four slices was measured for each lung using ImageJ software (Schindelin et al., 2012).
2.6. Lung mechanics
These were analysed using a flexiVent (Scireq, Montreal, Canada) forced oscillation ventilator system as described (Ren et al., 2015). Briefly, mice were anaesthetized with urethane (2 mg·g−1 i.p.) and placed under neuromuscular blockade with https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4004 chloride (500 μg i.p. followed by continuous i.p. infusion at 10 μg·g−1·min). This anesthesia protocol had been previously determined to suppress blink reflexes for at least one hour without neuromuscular blockage, which was sufficient to cover the total experiment time which lasted ~20 min. They were then tracheotomised and ventilated at 150 breaths·min−1, 10 μl g−1, against 2–3 cm H2O positive end‐expiratory pressure. Respiratory resistance was assessed at baseline and in response to three incremental doses of aerosolized https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7438 (MCh) (3, 10 and 30 mg·ml−1) administered by an in‐line ultrasonic nebulizer (4–6 μm, Aerogen, Ireland). Total respiratory resistance was calculated by averaging five values measured for each dose of methacholine for each mouse. Secondary euthanasia via cervical dislocation was performed immediately after data acquisition from the 30 mg·ml−1 dose.
2.7. Histochemistry
Cellular elements and tissue morphology were examined with haematoxylin and eosin (H&E) staining, and collagen and lipids in nodules were visualized with picrosirius red (PSR) and Sudan black B staining, respectively. For H&E and PSR staining, lungs were fixed and sectioned as above. For H&E staining, slides were incubated in Harris' haematoxylin, eosin, and graded ethanol. For PSR staining, slides were incubated in direct red 80 with picric acid for 1 hr and then rinsed with acidified water. To image lipids, fixed lungs were cut into lobes and immersed in 10% Optimal Cutting Temperature compound (Tissue‐Tek OCT; Sakura, Torrance, CA) and 20% sucrose in PBS on a rocker at 4°C overnight. Tissue was then frozen in OCT, cut into 10‐μm sections and mounted on Superfrost Plus glass slides (Fisher Scientific, Hampton, NH) for this and all other studies using frozen tissue. Sections were stained with Sudan black B (Rowley Biochemical, Danvers, MA) for 1 hr and counterstained with Kernechtrot's nuclear fast red (Rowley) for 20 min, followed by mounting with AquaMount medium (Polysciences, Warrington, PA). All images were acquired with an Olympus BX 60 microscope at 10× or 40× objectives unless otherwise specified.
2.8. Immunohistochemistry
The immuno‐related procedures used comply with the recommendations made by the British Journal of Pharmacology. Mouse lungs were fixed, embedded, sectioned and deparaffinized as above, then exposed for 10 min to 3% H2O2 in 90% methanol and then heated for 10 min in 10‐mM sodium citrate, pH 6.0, for antigen retrieval. Slides were rinsed in water, blocked in horse serum (Vector Laboratories, Cat# S‐2012, RRID:AB_2336618) or goat serum (Jackson ImmunoResearch Labs, Cat# 005‐000‐001, RRID:AB_2336983) for 1 hr and then rinsed again and incubated with primary antibodies diluted in blocking solution at 4°C overnight. Primary antibodies used were goat anti‐mouse IL‐33 (R and D Systems, Cat# AF3626, RRID:AB_884269; 1:1000), rabbit anti‐Muc5ac (gift from Dr. Camille Ehre, 1:500; Ehre et al., 2012) and mouse monoclonal antibody MDA‐3E1 (Millipore, Cat# MABT899;1:500) to detect Muc5b (Ren et al., 2015). After incubation, secondary antibodies—biotinylated horse anti‐goat IgG (Vector Laboratories, Cat# AP‐9500, RRID:AB_2336122), HRP‐labelled goat anti‐rabbit (Millipore, Cat# AP132P, RRID:AB_90264) or HRP‐labelled goat anti‐mouse antibody (Thermo Fisher Scientific, Cat# 31430, RRID:AB_228307)—were added for 2 hr at room temperature. Tissue sections were then washed with PBS, counterstained with H&E and mounted with VectaMount (Vector).
2.9. Immunofluorescence
Lungs were frozen in OCT and cut into 10‐μm sections as above. Sections mounted on glass slides were thawed, washed with water, blocked with donkey serum (Jackson ImmunoResearch), and then incubated with primary antibodies overnight at 4°C. Primary antibodies used were rabbit anti‐prosurfactant protein C (Millipore, Cat# AB3786, RRID:AB_91588; 1:1000), rabbit anti‐keratin 14 (Thermo Fisher Scientific, Cat# PA5–29608, RRID:AB_2547084; 1:500), rat anti‐mouse CD68 (BioLegend, Cat# 137001, RRID:AB_2044003; 1:200) and anti‐Muc5ac and anti‐Muc5b as above. This was followed by the addition of secondary antibodies conjugated to Alexa 555 (Thermo Fisher Scientific, Cat# A32732, RRID:AB_2633281), Alexa 488 (Jackson ImmunoResearch Labs, Cat# 715‐545‐151, RRID:AB_2341099) and DAPI (Jackson ImmunoResearch) for 2 hr. A confocal microscope (A1plus, Nikon) was used to acquire all images. For quantitative studies, random images were acquired by investigators blinded to subject identity. Percentages of IL‐33 positive cells were counted using ImageJ (Figure 6c–e). To measure IL‐33 fluorescence intensity (Figure 6b), the left lung was sectioned at the axial bronchus between lateral branches 1 and 2 using a precision cutting tool (Figure S4) and then imaged using an upright microscope (Olympus BX 60) with a 40× lens objective and identical parameters of exposure time, colour intensity, contrast, and magnification. Images were uploaded to ImageJ and a grey scale image was extracted from the red channel. Pixel intensity was summated across the entire image to determine fluorescence intensity. Each data point represents the mean of three images from a single mouse, plotted as arbitrary fluorescence intensity units (Figure 6b).
Figure 6.
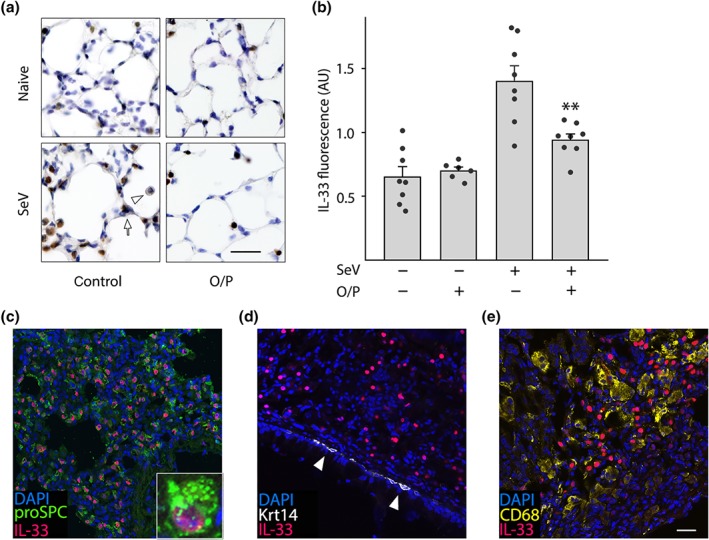
Persistent IL‐33 expression after SeV challenge. (a) Images of the lungs of mice treated or not with O/P, then challenged or not with SeV 1 day later and then killed 49 days later. Brown colour is immunohistochemical staining for IL‐33. Arrow points to an intensely stained Type 2 pneumocyte, and arrowhead points to a faintly stained macrophage. Scale bar = 100 μm. (b) Same groups as in (a), but showing quantification of total IL‐33 immunofluorescence intensity (bars indicate mean ± SEM; **P < .01 for [SeV+, O/P+] versus [SeV+, O/P−] by unpaired Student's t‐test; N = 6–8 mice, with three fields examined per mouse). (c–e) Images of the lungs of mice challenged with SeV and then killed 49 days later. (c) Fluorescence staining for pro‐surfactant protein C (proSPC) to identify type 2 pneumocytes (green), IL‐33 (red) and DAPI to identify nuclei (blue). Inset shows IL‐33 expression in a type 2 pneumocyte. (d) Fluorescence staining for cytokeratin 14 (Krt14) to identify airway basal cells (white, white arrowhead), IL‐33 and DAPI as in (c), shows no apparent expression of IL‐33 in basal cells. (e) Fluorescence staining for CD68 to identify macrophages (yellow), and IL‐33 and DAPI as in (c), shows no apparent expression of IL‐33 in macrophages (scale bar for (c–e) = 200 μm, and for inset in (c) = 30 μm; N = 3 mice/antibody pair)
2.10. Whole mount immunofluorescence
Fixed lung lobes were embedded in OCT as above and then frozen sections of 100‐ to 200‐μm thickness were cut. Sections were thawed and blocked in 2 ml Eppendorf tubes containing PBS with 0.3% TritonX‐100 and 2.5% donkey serum (017‐000‐121, Jackson ImmunoResearch) on a rocker at room temperature for 2 hr. After blocking, lung tissues were incubated with primary antibodies diluted in PBS with 0.3% TritonX‐100 on a rocker at 4°C overnight. Primary antibodies used (Figure 5) were rat anti‐E‐cadherin (Thermo Fisher Scientific, Cat# 13‐1900, RRID:AB_2533005; 1:1000), goat anti‐CCSP (1:2,500, a gift from Dr. Barry Stripp) and mouse anti‐acetylated tubulin (Sigma‐Aldrich, Cat# T6793, RRID:AB_477585; 1:2,000). On the second day, lung tissues were washed in PBS with 1% TritonX‐100 and 1% Tween‐20 for 3 hr and then incubated with secondary antibodies diluted in PBS with 0.3% Triton X‐100 on a rocker at 4°C overnight. Fluorescent secondary antibodies were obtained from Jackson ImmunoResearch and used at 1:1,000. On the third day, lung tissues were again washed in PBS with 1% TritonX‐100 and 1% Tween‐20 for 3 hr, washed with PBS twice for 5 min, fixed with 2% PFA for 2 hr on a rocker, and washed with PBS for 1 hr. After the PBS wash, lung tissues were mounted with AquaMount and airways of the left main bronchi were imaged on a Nikon A1plus confocal microscope. For each lung, six image fields—each consisting of several hundred epithelial cells—were counted.
Figure 5.
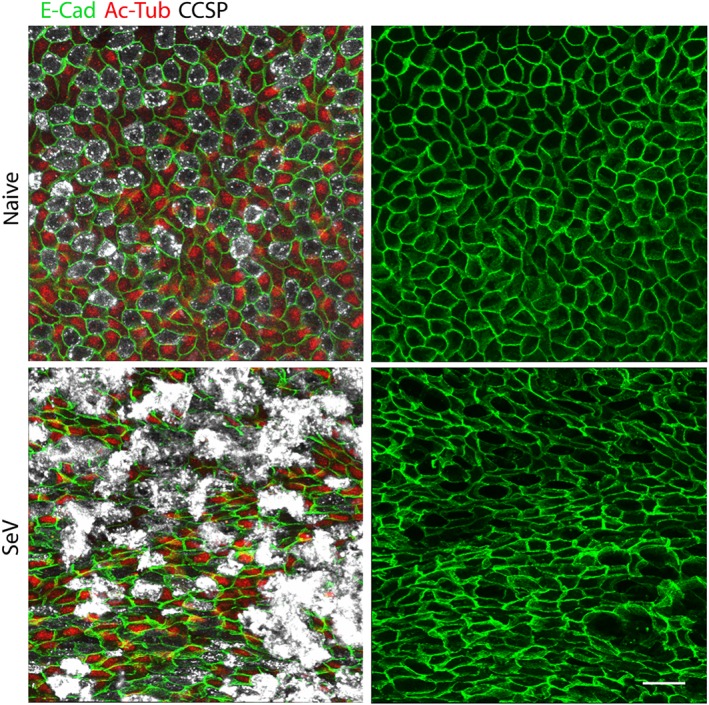
SeV infection alters the normal airway epithelial mosaic. Whole mount immunofluorescence images of proximal portions of the axial bronchi of mice infected or not with SeV 5 weeks earlier (N = 5 mice). Images on the left show staining for E‐cadherin (E‐Cad, green) to outline cell borders, acetylated tubulin (Ac‐Tub, red) to identify ciliated cells and club cell secretory protein (CCSP, white) to identify secretory cells. Images on the right only show staining for E‐cadherin to better illustrate changes in cell shape and pattern. Scale bar = 20 μm
2.11. Treatment with aerosolized interferon (INF)‐β
In time‐course and dose‐finding pilot studies, mice were treated with varying concentrations of aerosolized IFN‐β (R&D Systems, Minneapolis, MN) with 0.05% BSA in water (8 ml) as described above for O/P, except that the aerosolization was continued until all IFN‐β was delivered (approximately 40 min). In efficacy studies, mice were treated with 400,000 units of aerosolized IFN‐β and then challenged the next day with Sendai virus by oropharyngeal instillation and killed 5 days later for measurement of lung virus burden.
2.12. Measurement of cytokines in lung lavage fluid
At varying times after treatment, mice were killed and underwent bronchoalveolar lavage. Lavage fluid supernatant was collected by centrifugation at 500 g for 5 min. Cytokines were quantified using Milliplex MAP mouse cytokine bead panels (Millipore‐Sigma) and analysed by the Quantitative Proteomics core laboratory at Baylor College of Medicine.
2.13. Statistical analyses
The data and statistical analysis comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology (Curtis et al., 2018). All studies were designed to randomly generate groups of equal size; however, in some cases, mice reached euthanasia criteria during the acute infection so were not able to be included in analyses other than survival. Group sizes were selected to detect between‐group differences with a power of 90% and two‐tailed significance of 5%, with calculations based on effect sizes from our prior studies or pilot studies. Statistical analyses were only carried out when N was greater than or equal to 5. Analyses were blinded to group identity. Declared group sizes are the number of individual mice. Units of Y axes are direct linear measures of physical values except for RT‐qPCR. Data normalization was only conducted for RT‐qPCR analysis, as described (Schmittgen & Livak, 2008), and no outliers were excluded. All data sets were first analysed for normality and for variance inhomogeneity using the Shapiro–Wilk and Levene's tests, respectively.For analyses where there was only a single comparison between two groups, data was analyzed by an unpaired Student's t‐test or Mann‐Whitney U test for normally and non‐normally distributed data, respectively. For analyses comparing more than two groups, data were first analysed by one‐way ANOVA or ANOVA on ranks for normally and non‐normally distributed data, respectively. If ANOVA revealed a significant difference among groups, the data were further subjected to post hoc analysis and statistical significance determined from adjusted P values. For analyses where multiple experimental groups were compared against a single control, data were further analysed using Dunn's test. For analyses where each experimental group was compared to every other group, data were further analysed using Holm–Sidak's test for one‐way ANOVA or Dunn's test for ANOVA on ranks. For studies comparing O/P and IFN‐β, an interaction score from linear regression analysis was used to determine whether there was a synergistic effect of the combination compared to either drug alone. All data were analysed using SigmaPlot (version 12.5, Systat Software). P < .05 was considered statistically significant.
2.14. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY(Harding et al., 2018) and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20 (Alexander et al., 2019).
3. RESULTS
3.1. O/P treatment before SeV challenge reduces lung virus burden
Our central hypothesis was that treatment with O/P at the time of acute SeV infection would reduce lung virus burden and thereby attenuate the developement of a late asthma phenotype. The experimental paradigm is illustrated in Figure 1a. To test the first component of our hypothesis, mice were treated with aerosolized O/P 1 day before challenge with SeV because previous studies showed that this is the interval of maximal benefit of a single O/P treatment for challenge with influenza virus or any of several bacterial pathogens (Cleaver et al., 2014; Duggan et al., 2011; Tuvim et al., 2012). Lung SeV load was measured by RT‐qPCR 5 days after challenge based upon this being the time of maximal viral burden in prior studies by others (Hines et al., 2014; Massion et al., 1993; Patel et al., 2006; Walter et al., 2002) and our confirmatory study (Figure S1). Treatment with O/P resulted in a 70–80% reduction in SeV burden (Figures 1b and S1). Pilot studies titrating SeV challenge to mouse survival showed a steep dose–response relationship, with a 1.6‐fold increase in virus load resulting in a decrease in mouse survival from 80% to 20% (Figure 1c). Based on this tight relationship between viral burden and outcome, we anticipated a substantial change in late asthma phenotype with treatment, so we proceeded to test the second component of the central hypothesis.
3.2. O/P treatment before SeV challenge attenuates the late asthma phenotype
Mice were treated with aerosolized O/P 1 day before challenge with SeV and the severity of an asthma phenotype 49 days after viral challenge was measured. Treatment resulted in an 88% reduction in lung lavage eosinophil number, indicating the attenuation of persistent Type 2 lung inflammation and an 86% reduction in lymphocyte number, suggesting a possible reduction in Th2 cells that sustain Type 2 lung inflammation (Figure 2a,b). We next examined whether the reduction in Type 2 inflammation resulted in reduced effects on lung parenchymal cells. There was a 52% reduction in mucous metaplasia measured as airway epithelial mucin content (Figure 2c,d) and a 53% reduction in airway hyperresponsiveness (AHR) measured as lung resistance to inflation after 30 mg·ml−1 methacholine (Figure 2e,f). These results suggest a causal relationship between the reduction in virus load from O/P treatment and the chronic asthma phenotype, so further evidence was sought by examining dose–response and time–response relationships.
Figure 2.
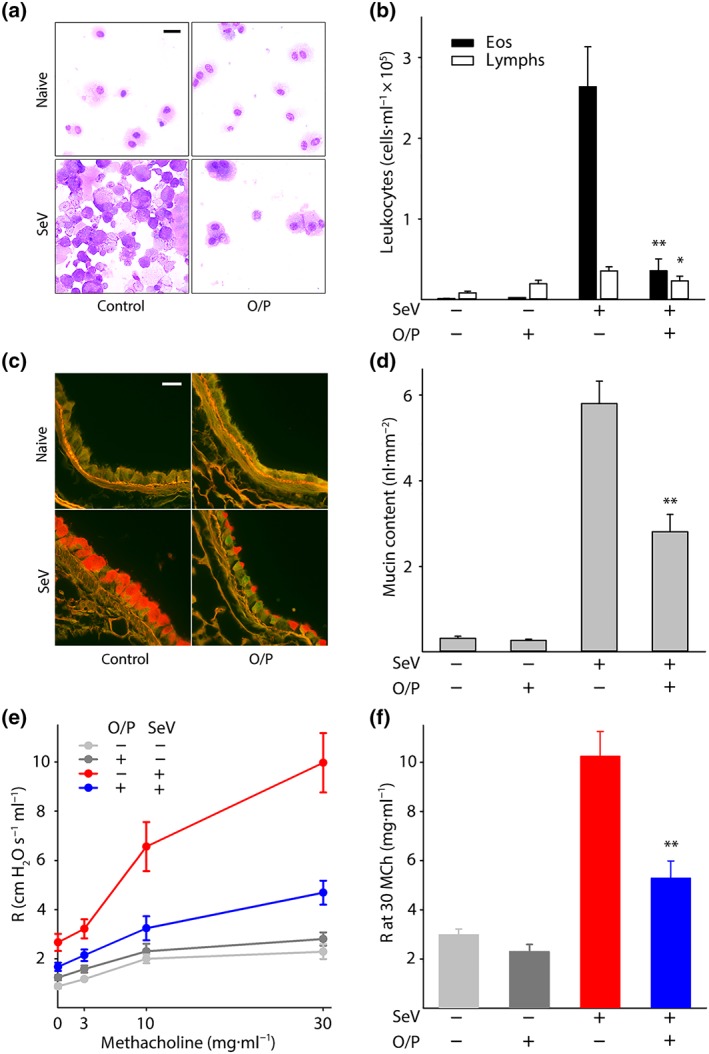
Effects of O/P treatment on asthma phenotype 49 days after SeV challenge. (a) Leukocytes obtained by lung lavage were pelleted onto glass slides by centrifugation and stained with Wright‐Giemsa. Scale bar = 20 μm. (b) Eosinophils and lymphocytes in lung lavage fluid were enumerated (bars show mean ± SEM; *P < .05 and **P < .01 for [SeV+, O/P+] versus [SeV+, O/P−] by unpaired Student's t‐test; N = 21 mice per group pooled from three experiments). (c) Airway epithelium stained with PAFS to demonstrate intracellular mucin in red. Scale bar = 20 μm. (d) Quantification of intracellular mucin content by image analysis of airway as in (c) (bars and P values as in (b); N = 22–25 mice per group pooled from five experiments). (e) Dose–response relationship between the concentration of aerosolized methacholine and total respiratory resistance measured by forced oscillation (points show mean ± SEM; N = 9–14 mice per group pooled from three experiments). (f) Plot of total respiratory resistance at 30 mg·ml−1 methacholine (MCh) using data from (e) (bars and P values as in (b))
3.3. Parallel dose–response relationships between acute SeV burden and late asthma phenotype
Our previous studies had shown that the combination of 1‐μM ODN and 4‐μM Pam2CSK4 in 4 ml (1× O/P) is on the inflection of the upper plateau of the dose–response curves for airway epithelial activation, as measured by cytokine release (Alfaro et al., 2014), and for antimicrobial resistance, as measured by the survival of mice from a microbial challenge (Alfaro et al., 2014; Evans et al., 2011). Now, we evaluated the responses to two halving doses of O/P below the 1× dose to interrogate the steep portion of the dose–response curve and to two doubling doses above the 1× dose to confirm that responses were saturated. Mice treated 1 day before SeV challenge showed dose‐dependent reductions in lung virus burden 5 days after challenge (Figure 3a) and in lung lavage eosinophil and lymphocyte numbers (Figure 3b) and mucous metaplasia (Figure 3c) 49 days after challenge.
Figure 3.
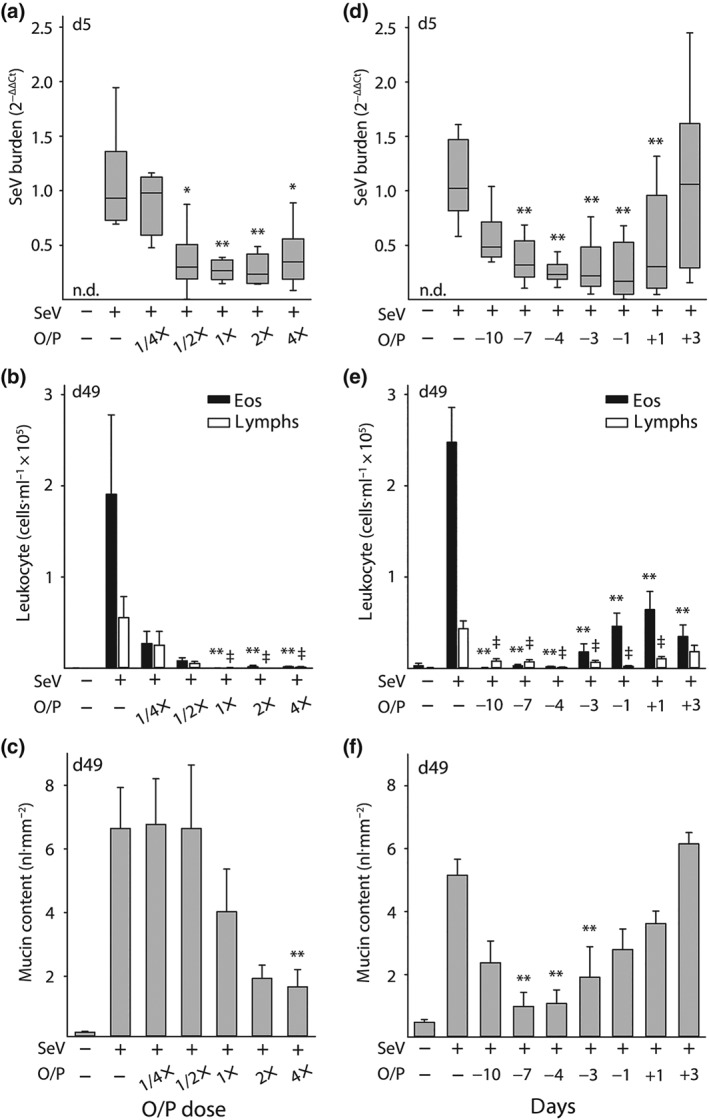
Dose and temporal response relationships between O/P treatment and acute lung SeV burden and late asthma phenotypes. (a) Relationship between O/P dose and lung SeV burden 5 days after SeV challenge (boxes show median and interquartile range, whiskers show 10th and 90th percentiles; *P < .05 and **P < .01 for ANOVA on ranks with Dunn's test for multiple comparisons against [SeV+, O/P−] control; N = 5–6 mice per group in a single experiment; n.d. = not detectable). (b) Relationship between O/P dose and lung lavage eosinophil and lymphocyte numbers 49 days after SeV challenge (bars show mean ± SEM; **P < .01 as in (a), ǂP < .01 for comparisons on lymphocytes; N = 5 mice/group in a single experiment). (c) Relationship between O/P dose and intracellular mucin content 49 days after SeV challenge (bars show mean ± SEM; **P < .01 as in (a); N = 6–7 in a single experiment). (d) Relationship of the interval in days between O/P treatment and SeV challenge in SeV lung burden 5 days after challenge (box plot and P values as in (a); N = 25–39 mice per group pooled from seven experiments). (e) Relationship of the interval between O/P treatment and SeV challenge in lung lavage eosinophil and lymphocyte numbers 49 days after challenge (bars and P values as in (b); N = 25–29 mice per group pooled from six experiments). (f) Relationship of the interval between O/P treatment and SeV challenge in intracellular mucin content 49 days after challenge (bars and P values as in (c); N = 6–12 mice per group pooled from three experiments)
The correlation among these dose–response relationships supports a mechanistic interaction between the reduction in acute viral lung burden by O/P treatment and the attenuation of a late asthma phenotype. However, it is notable that the maximal effect of O/P on lung virus burden appeared to be at the 1/2× dose, on lung lavage eosinophil and lymphocyte levels at the 1× dose, and on mucous metaplasia at the 2× dose. These small concentration‐dependent differences among outcomes were consistent across multiple experiments and suggest that small unmeasurable differences in lung virus burden at higher O/P concentrations (Figure 3a) are sufficient to cause measurable differences in persistent lung inflammation (Figure 3b), leading in turn to downstream effects on lung parenchymal phenotype (Figure 3c). This interpretation is consistent with the steep dose–response relationship between lung virus burden and mouse mortality (Figure 1c).
3.4. Time–response relationships between acute SeV burden and late asthma phenotype
To test temporal relationships, mice were treated with a 1× dose of O/P at varying intervals before and after challenge with SeV. Our previous studies showed variable durations of protection depending upon the intensity of the infectious challenge, with benefit observed with a pretreatment interval as long as 8 days before a bacterial challenge of mild intensity (Duggan et al., 2011) but as short as 3 days before a challenge of severe intensity (Clement et al., 2008). For treatments given after challenge, we have found a great difference between bacterial and viral pathogens, with a therapeutic window of less than 1 day for bacterial pathogens (Clement et al., 2008; Duggan et al., 2011) but of several days for influenza virus (Kirkpatrick et al., 2018; Tuvim et al., 2012). Based on these prior observations, responses to O/P from 10 days before to 3 days after SeV challenge were evaluated. A significant reduction in virus burden was seen from 7 days before challenge to 1 day after challenge (Figure 3d), with a trend towards reduction when treatment was administered 10 days before challenge (P = .08). Reductions in eosinophils and lymphocytes were observed at all time points (Figure 3e) and for mucous metaplasia at all time points except 3 days after challenge (Figure 3f). Overall, there was a trend towards greater effects with shorter intervals between treatment and challenge. Similar to the dose–response relationship, the time‐dependent effects of O/P treatment on leukocyte numbers were greater than on SeV burden (Figure 3e,f).
Whereas the efficacy of O/P in reducing late leukocytes was seen up to 10 days before challenge, the reduction in SeV burden waned at times greater than 7 days. This prompted us to test whether O/P might also be influencing the development of a chronic asthma phenotype by exerting an immunomodulatory effect, so O/P was administered at time points substantially outside its known window of antimicrobial efficacy. Treatment with O/P 20 days before SeV challenge resulted in a reduction 49 days after challenge in eosinophils by 85% and lymphocytes by 75%; similarly, treatment 20 days after SeV challenge resulted in a reduction 49 days after challenge in eosinophils by 73% and lymphocytes by 74% (Figure S2). These findings indicate that O/P treatment attenuates the late asthma phenotype in SeV‐challenged mice by a combination of a reduction in acute lung virus burden and long‐term immunomodulation away from a T2 lung inflammatory milieu.
3.5. O/P treatment reduces airway mucus occlusion and acidophilic pneumonitis
We observed multiple yellow nodules on the surface of the lungs of untreated mice challenged with SeV 49 days earlier (Figure 4a). Transillumination allowed us to enumerate nodules within the lungs (Figure 4b) and to measure their mean diameter at 1.1 mm (SEM = 45 μm, N = 37). Mice treated with O/P 1 day before SeV challenge had a 92% reduction in nodule number (Figure 4c). Histopathologic examination of the nodules showed alveolar accumulation of foamy macrophages, eosinophils and eosinophilic crystals (Figure 4d,e) characteristic of “acidophilic pneumonitis” (Guo, Johnson, & Schuh, 2000; Murray & Luz, 1990). The identity of the macrophages was confirmed by immunofluorescence staining for CD68 (Figure S3D) and Sudan black staining showed the macrophages to be heavily laden with lipids (Figure 4f), even more so than the normal staining of lamellar bodies in alveolar T2 cells (Figure S3E).
Figure 4.
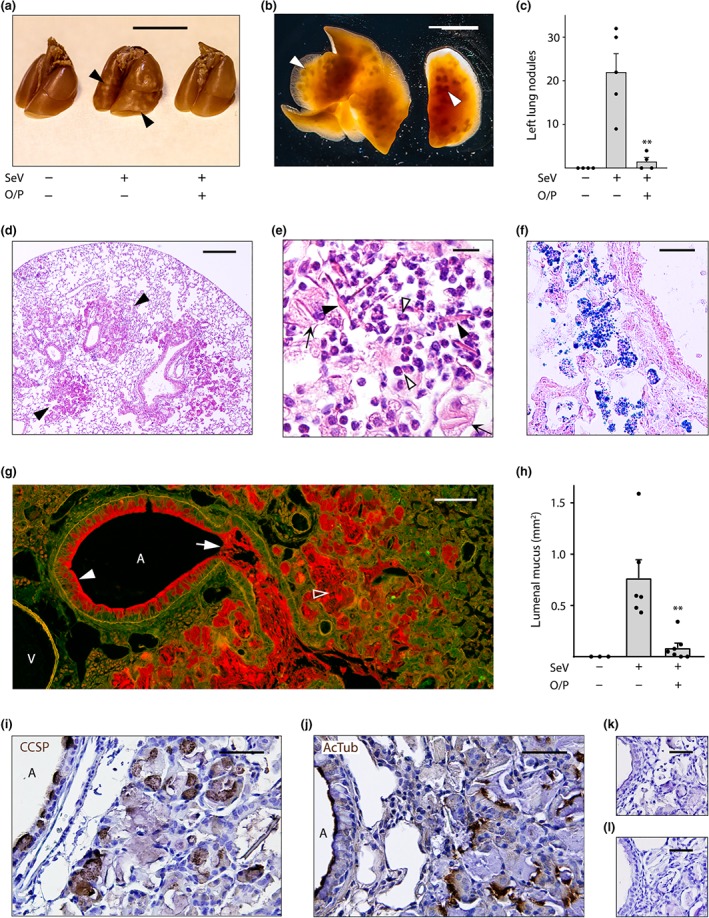
Airway mucus occlusion, acidophilic pneumonitis and alveolar bronchiolization 49 days after SeV infection. (a) The lungs of mice treated or not with O/P, then challenged or not with SeV 1 day later, and then killed 49 days later with lungs inflated with 10% formalin to 20 cm H2O pressure. Arrowheads point to nodules on the lung surface. Scale bar = 1 cm. (b) Transilluminated lungs from a mouse challenged with SeV but not treated with O/P, as in (a). Arrowheads point to nodules in the lung interior. Scale bar = 1 cm. (c) Enumeration of nodules in the left lungs of mice treated with O/P and challenged with SeV as in (a) (bars indicate mean ± SEM; **P < .01 for [SeV+, O/P+] versus [SeV+, O/P−] by Student's t test; N = 4–5 in a single experiment). (d) Section of the lung of a mouse challenged with SeV but not treated with O/P, stained with H&E. Arrowheads point to nodules. Scale bar = 0.5 mm (N = 4 mice, d–f). (e) High magnification image of a nodule from a mouse as in (d), with closed arrowheads pointing to eosinophilic crystals, open arrowheads pointing to eosinophils and arrows pointing to foamy macrophages. Scale bar = 30 μm. (f) Image of a nodule from a mouse as in (d), but with lungs frozen and stained with Sudan black to show lipids within macrophages. Scale bar = 50 μm. (g) Section of the lung of a mouse challenged with SeV but not treated with O/P, fixed by immersion in methacarn and stained with PAFS. Closed arrowhead points to abundant intracellular mucin, arrow points to lumenal mucus occlusion, and open arrowhead points to mucus in the alveolar region. A = airway, V = vessel. Scale bar = 100 μm. (h) The area of lumenal mucus of mice treated or not with O/P, then challenged or not with SeV and then fixed and stained as in (g), with left lungs sectioned at fixed intervals using a precision cutting tool (bars indicate mean ± SEM; **P < .01 for [SeV+, O/P+] versus [SeV+, O/P−] by Mann–Whitney U test; N = 13–15 mice per group in a single experiment). (i–l) Alveolar region of a mouse challenged with SeV but not treated with O/P, stained with antibodies against (i) club cell specific protein (CCSP) or (j) acetylated tubulin (both brown) and counterstained with H&E (blue). A = airway lumen. Primary antibodies against (k) club cell specific protein or (l) acetylated tubulin were omitted to assess the specificity of antibody staining. Scale bar in all four images = 50 μm (N = 4 mice)
We hypothesized that mucus plugs caused airway obstruction leading to lipid‐laden macrophage accumulation in alveoli. To evaluate this, uninflated lungs were fixed with methacarn to minimize changes in mucus volume and position and then stained with PAFS to visualize mucins (Johansson & Hansson, 2012). Supporting this hypothesis, widespread mucus plugs were visible in the airways of mice challenged 49 days earlier with SeV (Figure 4g, arrow) and were greatly reduced in the airways of mice treated with O/P 1 day before challenge (Figure 4h). The mucus plugs were contiguous with areas of intense airway epithelial mucous metaplasia (Figure 4g, filled arrowhead) and the plugs contained both Muc5ac and Muc5b (Figure S3A‐B). In the process of measuring airway lumenal mucus using a precision lung‐slicing tool (Figure S4), we observed that lungs from untreated SeV‐challenged mice were larger (25–30% greater rostral‐caudal length) than those from unchallenged or treated and challenged mice, consistent with the obstructive hyperinflation observed at autopsy in the lungs of human subjects who die from asthma (Hogg, 1997).
Red PAFS staining also extended into alveolar regions distal to mucus plugs (Figure 4h, open arrowhead), and these areas were also positive for Muc5ac and Muc5b immunohistochemical staining (Figure S3A), with relatively more Muc5ac staining in airways and relatively more Muc5b staining in alveolar regions. To assess whether mucus in alveolar regions might be due to local mucin production resulting from bronchiolization of alveolar epithelium, we stained for club cell specific protein and acetylated tubulin, markers of airway secretory and ciliated cells, respectively. This showed normal staining in conducting airways as well as abnormal staining in alveolar regions (Figure 4i,j).
3.6. O/P treatment attenuates the disruption of epithelial mosaicism
We considered that a possible additional contributor to airway lumenal mucus accumulation besides mucin hyperproduction might be impaired ciliary clearance because we and others have observed extensive epithelial cell death in the first 2 weeks after SeV challenge (Jakab & Green, 1972; Kirkpatrick et al., 2018; Look et al., 2001; Massion et al., 1993; Walter, Kajiwara, Karanja, Castro, & Holtzman, 2001). The airway epithelium was examined 2–7 weeks after SeV challenge by whole mount immunofluorescence microscopy and the normal mosaic of secretory and ciliated cells was altered in multiple ways. Secretory cells, marked by club cell specific protein staining, became more prominent than ciliated cells, marked by acetylated tubulin staining (Figures 5 and S5). This prominence was due to a modest increase in secretory cell number (Figure S5B) and a more substantial increase in secretory cell size and intensity of club cell specific protein staining (Figures 5 and S5A). The increase in secretory cell size is likely due to distension by increased intracellular mucin content (Figures 2c, 4g, S3A‐B, and S5C), as we have shown previously during mucous metaplasia (Evans et al., 2004; Nguyen et al., 2008). The increase in intensity of club cell specific protein staining (Figures 5 and S5A) likely reflects a combination of increased expression and redistribution from small apical secretory granules in naïve cells into large mucin‐containing granules during Type 2 inflammation (Evans et al., 2004; Nguyen et al., 2008). Besides these changes in cell proportions and size, an alteration of the normal epithelial polygonal mosaic was revealed by staining cell boundaries using antibodies to E‐cadherin (Figure 5), with cells taking on more elongated shapes and occurring in abnormally large clusters of secretory cells. We observed these changes as early as 2 weeks after SeV challenge (Figure S5), found they partially persisted through 7 weeks (not shown) and that there was a trend towards improvement by pretreatment with O/P (Figure S5B).
3.7. O/P treatment before SeV challenge reduces lung IL‐33 expression 49 days later
Holtzman and colleagues have shown that the effects of acute SeV infection are translated into a chronic asthma phenotype by persistently increased IL‐33 expression in the lungs (Byers et al., 2013). Therefore, we measured the effect of O/P treatment during acute SeV infection on IL‐33 expression 49 days later. Naïve mice that were neither challenged with SeV nor treated with O/P showed scattered faint immunohistochemical staining for IL‐33 in cells with the localization (corners of polygonal alveoli) and morphology (cuboidal and protruding into the alveolar lumen) of type 2 pneumocytes (Figure 6a, isotype controls Figure S6). This finding is consistent with reports of substantial expression of IL‐33 in type 2 pneumocytes of healthy mice (Oczypok, Perkins, & Oury, 2017; Treutlein et al., 2014). There was no apparent change in IL‐33 expression in the lungs of unchallenged mice treated with O/P, but mice challenged with SeV and not treated with O/P showed a dramatic increase in the intensity and frequency of IL‐33 staining of type 2 pneumocytes and faint staining of macrophages (Figure 6a). Using the same antibody against IL‐33, this was quantified by immunofluorescence staining (Figure 6b), and mice treated with O/P and then challenged 1 day later with SeV showed a marked reduction in IL‐33 staining (Figure 6a,b). As shown in Figure 6c, immunofluorescence co‐localization of IL‐33 staining with lineage markers showed that high‐level IL‐33 expression in SeV‐challenged mice occurred exclusively in type 2 pneumocytes, with 97% of IL‐33‐positive cells also positive for pro‐SP‐C (pro‐surfactant protein C) and no measurable IL‐33 staining in Krt14‐positive basal cells of the conducting airways (Figure 6d) or in CD68‐positive alveolar macrophages (Figure 6e) (N = 100 cells in 3 mice for each lineage marker). In SeV‐challenged mice, 71% of pro‐SP‐C‐positive cells were positive for IL‐33 staining (N = 100 cells in three mice).
3.8. Multiple doses of O/P neither induce tachyphylaxis nor augment efficacy
We next sought to determine whether multiple O/P doses induce tachyphylaxis or improve outcomes. Repetitive treatment with two to four doses of O/P given daily or separated by 2 or 3 days between doses, with the final dose for each regimen given 1 day before SeV challenge (Figure 7a), did not result in any measurable differences in the reduction of lung viral burden by O/P compared to a single treatment (Figure 7b). Therefore, we concluded that there is no tachyphylaxis of the antiviral effect of O/P treatment, similar to what we have previously observed with influenza virus and bacterial pathogens (Evans et al., 2010; Tuvim, Evans, Clement, Dickey, & Gilbert, 2009), so we proceeded to test whether multiple dosing improved outcomes.
Figure 7.
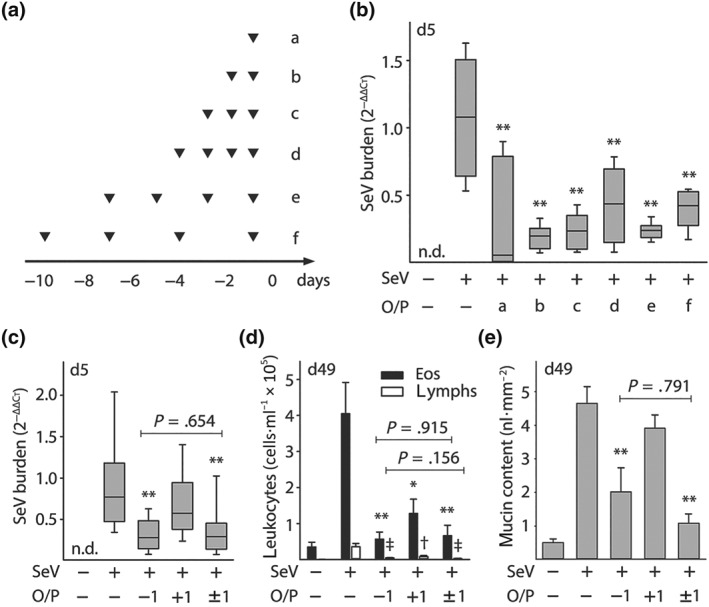
Effects of multiple O/P doses on tachyphylaxis and efficacy. (a) Experimental paradigm illustrating the timing of O/P doses for each experimental group in black triangles in relation to the SeV challenge given on Day 0. (b) Lung SeV burden by qRT‐PCR 5 days after mice were challenged with SeV, with or without pretreatment with O/P as outlined in (a) (boxes show median and interquartile range, whiskers show 10th and 90th percentiles; **P < .01 for analysis by ANOVA on ranks with Dunn's test for multiple comparisons against [SeV+, O/P−] control; N = 4–7 mice per group in a single experiment; n.d. = not detectable). (c) Lung SeV burden by RT‐qPCR 5 days after mice were challenged with SeV, with or without treatment with O/P 1 day before challenge (−1), 1 day after challenge (+1), or both (±1) (box plot and P values as in (b); bar and labelled P values for single comparison between [SeV+, O/P−1] versus [SeV+, O/P ± 1] by unpaired Student's t‐test; N = 14–25 mice per group pooled from three experiments). (d) Eosinophils and lymphocytes in lung lavage fluid were enumerated 49 days after SeV challenge, with or without treatment with O/P as in (c) (bars show mean ± SEM; *P < .05 and **P < .01 as in (b), † P < .05 and ǂ P < .01 for comparisons on lymphocytes; bar and labelled P values as in (c); N = 8–11 mice per group pooled from two experiments). (e) Quantification of intracellular mucin content of airway epithelium 49 days after SeV challenge, with or without treatment with O/P as in (c) (bars and P values as in (c); N = 8–11 mice per group pooled from two experiments)
Treatment with O/P both 1 day before and 1 day after SeV challenge did not reduce lung virus burden more than a single treatment before challenge (Figure 7c). Similarly, when late asthma outcomes were examined, treatment both before and after challenge was no more effective than a single treatment before challenge in reducing lung lavage eosinophils and lymphocytes (Figure 7d) or epithelial mucin content (Figure 7e). In each case, treatment 1 day after challenge was less effective than treatment 1 day before challenge (Figure 7c–e).
3.9. Comparison of O/P to interferon‐β effects on lung SeV burden and asthma phenotype
Interferon‐β (IFN‐β) has been shown to be a key signalling molecule in defence against respiratory viral infections, and aerosolized IFN‐β has progressed to clinical trials for the treatment of virus‐induced asthma exacerbations (Djukanović et al., 2014). To compare treatment with O/P to IFN‐β, we first developed an IFN‐β treatment paradigm in mice. Serum https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4430 levels were used as a biomarker of anti‐viral activity in the published clinical trial (Djukanović et al., 2014), so we measured CXCL10 levels in BALF in mouse pilot studies. The time of peak CXCL10 level in BALF was 2 hr after aerosolized IFN‐β treatment (Figure S7A) and a dose of 400,000 IFN‐β units resulted in a CXCL10 level that lay on the edge of the upper plateau of the dose–response curve (Figure S7B), so this treatment timing and dose was used in efficacy studies. Reductions in lung SeV burden acutely (Figure 8a), and in lavage eosinophils and lymphocytes (Figure 8b) and in epithelial mucin content (Figure 8c) 49 days after SeV challenge, were similar between O/P and IFN‐β treatments.
O/P treatment does not cause a rise in Type I IFNs (Cleaver et al., 2014; Duggan et al., 2011; Kirkpatrick et al., 2018; Tuvim et al., 2012), so we wondered whether O/P and IFN‐β might have an additive effect. Supporting separate mechanisms of action, pilot studies showed that treatment with O/P or IFN‐β elicited different cytokine profiles, with O/P causing a substantially greater rise in CXCL2 and TNF and IFN‐β causing a greater rise in https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4430 (Figure S7C). In addition, both treatments together caused a synergistic rise in https://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=307 (Figure S7C). Despite these promising preliminary data, there was no apparent benefit to treating mice with both drugs simultaneously on lung SeV burden, lavage eosinophils and lymphocytes, or epithelial mucin content (Figure 8a–c). To further test the possibility of additive activity of the two drugs, we gave a high‐intensity SeV challenge (2.5 × LD50), for which there was no apparent reduction in lung SeV burden by either drug alone (Figure 8d). However, the combination of both drugs reduced lung SeV burden significantly compared to the absence of treatment or to treatment with either drug along (Figure 8d).
4. DISCUSSION
Recent advances in molecular diagnostics have deepened knowledge of the tight association between viral infection and asthma initiation, exacerbation and progression (Busse et al., 2010; Farne & Johnston, 2017; Holgate, 2012; Holt & Sly, 2012). Complementing this data, recent work with animal models has supported a causal role of respiratory viral infections in promoting long‐term asthma‐like disease and enabled the identification of critical pathways in translating acute infection into chronic disease (Byers et al., 2013; Drake et al., 2013; Holtzman et al., 2011; Look et al., 2001; Moreno‐Vinasco, Verbout, Fryer, & Jacoby, 2009; Peebles & Aronica, 2019). Together, these developments highlight the importance of attenuating the severity of respiratory viral infections to prevent chronic airway diseases. We have previously identified a means of effectively stimulating the airway epithelium to induce innate immune resistance to respiratory viral infection (Alfaro et al., 2014; Cleaver et al., 2014; Drake et al., 2013; Duggan et al., 2011; Evans et al., 2011; Kirkpatrick et al., 2018; Leiva‐Juarez et al., 2018; Leiva‐Juárez et al., 2016; Tuvim et al., 2012; Ware et al., 2019). Here, we show that treatment of mice with this efficacious combination of aerosolized TLR ligands around the time of SeV infection attenuates chronic lung disease, providing proof‐of‐principle that this intervention might have clinical efficacy.
Based upon our prior studies showing that treatment with O/P results in rapid pathogen killing within the lungs, we hypothesized that the major mechanism of action of O/P in ameliorating chronic SeV‐induced lung disease would be by reducing lung virus burden. The close dose–response relationship between acute SeV lung burden and chronic asthma phenotypes supports such a causal relationship (Figure 3a–c). The relationship between SeV lung burden and acute disease is steep, with a 1.6‐fold decrease in viral burden resulting in a fourfold decrease in mouse death (Figure 1c). Similarly, reduction of SeV lung burden by 70–80% has a dramatic effect on chronic disease (Figures 1b, 3a,d, 7b,c, and 8a). This contrasts with the need for multiple log reductions in pathogen burden in chronic mycobacterial lung infection and acute bacterial blood infections to achieve comparable benefit, reflecting the capability of small numbers of residual organisms to perpetuate disease or develop drug resistance in those infections.
The major mechanism of pathogen killing with O/P treatment is the activation of reactive oxygen generators in airway epithelial cells, which persists at a high level for several days and then slowly declines to its baseline level (Cleaver et al., 2014; Kirkpatrick et al., 2018; Ware et al., 2019). This is consistent with the reduction in SeV lung burden when mice are treated in the few days before or after challenge (Figure 1d). However, the substantial reduction in lung inflammation 49 days after SeV challenge observed in mice treated 10 days before challenge with O/P (Figure 3e), when there is no significant effect on lung viral burden in this (Figure 3d) or our previous studies (Kirkpatrick et al., 2018; Tuvim et al., 2012), suggested an additional effect besides viral killing. To further explore this, we treated mice with O/P either 20 days before or 20 days after SeV challenge and observed substantial effects on lung inflammation 49 days after SeV challenge (Figure S2). This suggests that O/P also exerts an immunomodulatory effect on lung epithelium that additionally attenuates the chronic inflammatory lung phenotype independent of its antiviral effect. Holtzman and colleagues have shown that persistent expression of high levels of IL‐33 in the lungs of SeV‐challenged mice connects the acute infection to the chronic disease (Byers et al., 2013). Consistent with this, O/P treatment markedly reduced IL‐33 expression in type 2 pneumocytes (Figure 6), which are thought to signal to distal airways in an “outside‐in” manner (Klaßen et al., 2017; Oczypok et al., 2017; Pichery et al., 2012; Treutlein et al., 2014). The degree to which the reduction in IL‐33 expression by O/P treatment reflects decreased epithelial cell injury and death and the degree to which it reflects immune modulation will require further study.
A prominent feature of chronic lung disease in SeV‐challenged mice was the presence of multiple millimetre‐sized nodules (Figure 4a–c). By microscopic analysis, these were comprised of areas of acidophilic pneumonitis (Guo et al., 2000; Murray & Luz, 1990), characterized by lipid‐laden foamy macrophages, eosinophilic and lymphocytic inflammation, and eosinophilic (Charcot–Leyden‐like) crystals (Figure 4d–f), as well as areas of bronchiolization characterized by alveolar regions replaced by nests of club and ciliated cells (Figure 4i,j). Abundant mucus was observed occluding airway lumens and also in alveolar regions (Figures 4g and S3). Airway lumenal mucus occlusion was likely due primarily to high levels of mucin production and secretion (Figures 2c,d, 3c,f, 4g, and S3A‐B), but epithelial destruction during the first 2 weeks after infection, as observed previously (Jakab & Green, 1972; Look et al., 2001; Massion et al., 1993), with a failure to restore a normal mucociliary epithelial mosaic during the following 5 weeks (Figures 5 and S5) probably contributed as well. Other possible mechanisms contributing to airway mucus occlusion might include airway surface liquid depletion due to inflammation during viral infection that has been observed by others but was not examined here (Boucher, 2019). The presence of mucus in alveolar regions (Figures 4g and S3A) was more likely due to mucin synthesis and secretion by bronchiolized epithelium than by reflux from the conducting airways because of the tapering anatomy of airways and inability of terminal bronchioles to produce mucin (Evans et al., 2004; Zhu et al., 2008). The presence of intraepithelial mucin in bronchiolized alveolar epithelium, indicating local production (Figure S3A), and the greater abundance of Muc5b relative to Muc5ac in alveolar regions compared to conducting airways suggests different sources of mucus (Figure S3A). Both the number of macroscopic lung nodules (Figure 4b) and the extent of airway mucus occlusion (Figure 4h) were markedly reduced by treatment with O/P.
Our study has several limitations in predicting whether O/P will be an effective therapeutic agent in humans. These include the inherent differences between mice and humans in anatomy, molecular biology and interactions with pathogens. Offsetting this limitation, SeV is a paramyxovirus closely related to respiratory syncytial virus and parainfluenza viruses that cause considerable respiratory disease in humans (Bønnelykke et al., 2015; Busse et al., 2010; Liu et al., 2013; Peebles & Aronica, 2019) and SeV is a natural pathogen of rodents that replicates in their lungs and causes injury resembling that of human paramyxoviral infections (Byers et al., 2013; Holtzman, 2012; Holtzman et al., 2011; Jakab & Green, 1972; Look et al., 2001; Massion et al., 1993). In addition, the efficacy of O/P in inducing epithelial resistance to viral infection in human airway cells has been demonstrated in vitro (Kirkpatrick et al., 2018). Another limitation is that SeV infection of the lungs was induced in mice by bolus aspiration, whereas human infection most commonly proceeds by inhalation of small infected droplets from the upper respiratory tract. However, this should mostly affect the timing of treatment, which can be determined empirically in human subjects, rather than the underlying therapeutic principle.
Treatment of susceptible human subjects with O/P has the potential to prevent the substantial morbidity associated with respiratory viruses in the pathogenesis of airway diseases including asthma, COPD and cystic fibrosis. We have treated 49 normal human subjects in a single‐dose, ascending, dose‐strength trial (NCT02124278) and a multiple‐dose safety and tolerability trial (NCT02566252). Treatment was well tolerated and we believe we reached the upper plateau of the dose–response curve based upon small rises in blood neutrophils and C‐reactive protein levels resembling the small sigmoidal‐shaped plots of rises in blood IL‐6 levels in mice (Alfaro et al., 2014). Next steps in development of the drug include viral challenge studies in subjects with airway disease (Adura et al., 2014; Jackson et al., 2014; Mallia et al., 2011), followed by natural infection prevention and pre‐emption studies in subjects with airway disease or immune compromise (Chemaly, Shah, & Boeckh, 2014; Djukanović et al., 2014; Evans et al., 2011; Sheshadri et al., 2018). O/P showed reductions in acute SeV lung burden and chronic asthma phenotypes comparable to those seen with aerosolized IFN‐β in mice (Figure 8). This was congruent with a clinical trial where IFN‐β administered within 24 hr of the onset of cold symptoms reduced asthma symptoms and improved recovery of peak expiratory flow in the subset of patients with difficult‐to‐treat asthma (Djukanović et al., 2014). Together, O/P and IFN‐β show additive or synergistic activity in mice (Figures 8d and S6C), which is not surprising in view of their differing mechanisms of action with O/P causing no induction of type I IFNs (Cleaver et al., 2014; Duggan et al., 2011; Kirkpatrick et al., 2018; Tuvim et al., 2012). Whether either or both of these drugs, or some other stimulant of innate epithelial resistance to infection, can ameliorate the burden of virus‐caused airway disease will be determined in future clinical trials of efficacy and safety.
AUTHOR CONTRIBUTIONS
B.L.S., S.E.E., M.J.T., and B.F.D. conceived the study; D.L.G., J.R.F., G.V., A.M.J., S.T., S.W., B.H. and J.C. performed the studies; D.E.O. oversaw the statistical analyses; M.J.T. made the figures; D.L.G. and B.F.D. wrote the manuscript.
CONFLICT OF INTEREST
S.E.E., M.J.T. and B.F.D. are inventors on US patent 8,883,174 “Compositions for Stimulation of Mammalian Innate Immune Resistance to Pathogens,” which has been licensed by their employer, the University of Texas MD Anderson Cancer Center, to Pulmotect, Inc., which is developing O/P as a therapeutic for respiratory infections. In addition, B.L.S., S.E.E., M.J.T. and B.F.D. hold equity in Pulmotect, Inc. and B.L.S. is employed by Pulmotect, Inc.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for https://bpspubs.onlinelibrary.wiley.com/doi/abs/10.1111/bph.14207, https://bpspubs.onlinelibrary.wiley.com/doi/abs/10.1111/bph.14208, https://bpspubs.onlinelibrary.wiley.com/doi/abs/10.1111/bph.14206 and as recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Fig. S1. Lung SeV burden days 3 through 7 after infection.
Figure S2. Effect of O/P treatment 20 days before or after SeV infection.
Figure S3. Staining for Muc5ac and Muc5b, collagen, and macrophages in lung nodules.
Figure S4. Location of left axial bronchus sections for mucin content, and image of the precision cutting instrument.
Figure S5. Whole mount and cross‐sectional immunofluorescence staining of airway epithelium 14 days after SeV infection.
Figure S6. Immunohistochemical staining for IL‐33 done 49 days after SeV infection.
Figure S7. Lung lavage fluid cytokine responses to aerosolized O/P and IFN‐β.
ACKNOWLEDGEMENTS
The authors thank Mark McArthur, DVM, for helpful discussions of pathological findings. Funding from NIH, NHLBI, R01 HL129795 to B.F.D., R44 HL115903 to B.L.S., and R01 HL117976, DP2 HL123229 and R35 HL144805 to S.E.E. is acknowledged; Cystic Fibrosis Foundation grants Dickey15PO and Dickey18GO to B.F.D. D.L.G. was a Howard Hughes Medical Institute Medical Research Fellow. S.W. was a graduate student at the MD Anderson‐UTHealth Graduate School of Biomedical Sciences.
Goldblatt DL, Flores JR, Valverde Ha G, et al. Inducible epithelial resistance against acute Sendai virus infection prevents chronic asthma‐like lung disease in mice. Br J Pharmacol. 2020;177:2256–2273. 10.1111/bph.14977
David L. Goldblatt, Jose R. Flores and Gabriella Valverde Ha contributed equally to this work.
Contributor Information
David L. Goldblatt, Email: dgoldblatt@mdanderson.org.
Burton F. Dickey, Email: bdickey@mdanderson.org.
REFERENCES
- Adura, P. T. , Reed, E. , Macintyre, J. , del Rosario, A. , Roberts, J. , Pestridge, R. , … Djukanović, R. (2014). Experimental rhinovirus 16 infection in moderate asthmatics on inhaled corticosteroids. The European Respiratory Journal, 43, 1186–1189. 10.1183/09031936.00141713 [DOI] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Mathie, A. , Peters, J. A. , Veale, E. L. , … CGTP Collaborators (2019). The concise guide to pharmacology 2019/20: Catalytic receptors. British Journal of Pharmacology, 176: S247‐S296. 10.1111/bph.14751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfaro, V. Y. , Goldblatt, D. L. , Valverde, G. R. , Munsell, M. , Quinton, L. J. , Walker, A. K. , … Dickey, B. F. (2014). Safety, tolerability, and biomarkers of the treatment of mice with aerosolized Toll‐like receptor ligands. Frontiers in Pharmacology, 5(8). 10.3389/fphar.2014.00008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annunziato, F. , Romagnani, C. , & Romagnani, S. (2015). The 3 major types of innate and adaptive cell‐mediated effector immunity. The Journal of Allergy and Clinical Immunology, 135, 626–635. 10.1016/j.jaci.2014.11.001 [DOI] [PubMed] [Google Scholar]
- Bønnelykke, K. , Vissing, N. H. , Sevelsted, A. , Johnston, S. L. , & Bisgaard, H. (2015). Association between respiratory infections in early life and later asthma is independent of virus type. The Journal of Allergy and Clinical Immunology, 136, 81–86.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucher, R. C. (2019). Muco‐obstructive lung diseases. The New England Journal of Medicine, 380, 1941–1953. 10.1056/NEJMra1813799 [DOI] [PubMed] [Google Scholar]
- Busse, W. W. , Lemanske, R. F. , & Gern, J. E. (2010). Role of viral respiratory infections in asthma and asthma exacerbations. Lancet, 376, 826–834. 10.1016/S0140-6736(10)61380-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byers, D. E. , Alexander‐Brett, J. , Patel, A. C. , Agapov, E. , Dang‐Vu, G. , Jin, X. , … Holtzman, M. J. (2013). Long‐term IL‐33–producing epithelial progenitor cells in chronic obstructive lung disease. The Journal of Clinical Investigation, 123, 3967–3982. 10.1172/JCI65570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chemaly, R. F. , Shah, D. P. , & Boeckh, M. J. (2014). Management of respiratory viral infections in hematopoietic cell transplant recipients and patients with hematologic malignancies. Clinical Infectious Diseases, 59, S344–S351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleaver, J. O. , You, D. , Michaud, D. R. , Pruneda, F. A. , Juarez, M. M. , Zhang, J. , … Evans, S. E. (2014). Lung epithelial cells are essential effectors of inducible resistance to pneumonia. Mucosal Immunology, 7, 78–88. 10.1038/mi.2013.26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement, C. G. , Evans, S. E. , Evans, C. M. , Hawke, D. , Kobayashi, R. , Reynolds, P. R. , … Tuvim, M. J. (2008). Stimulation of lung innate immunity protects against lethal pneumococcal pneumonia in mice. American Journal of Respiratory and Critical Care Medicine, 177, 1322–1330. 10.1164/rccm.200607-1038OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis, M. J. , Alexander, S. , Cirino, G. , Docherty, J. R. , George, C. H. , Giembycz, M. A. , … Ahluwalia, A. (2018). Experimental design and analysis and their reporting II: Updated and simplified guidance for authors and peer reviewers. British Journal of Pharmacology, 175, 987–993. 10.1111/bph.14153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djukanović, R. , Harrison, T. , Johnston, S. L. , Gabbay, F. , Wark, P. , Thomson, N. C. , … INTERCIA Study Group (2014). The effect of inhaled IFN‐β on worsening of asthma symptoms caused by viral infections. A Randomized Trial. American Journal of Respiratory and Critical Care Medicine, 190, 145–154. 10.1164/rccm.201312-2235OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake, M. G. , Evans, S. E. , Dickey, B. F. , Fryer, A. D. , & Jacoby, D. B. (2013). Toll‐like receptor–2/6 and Toll‐like receptor–9 agonists suppress viral replication but not airway hyperreactivity in guinea pigs. American Journal of Respiratory Cell and Molecular Biology, 48, 790–796. 10.1165/rcmb.2012-0498OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duggan, J. M. , You, D. , Cleaver, J. O. , Larson, D. T. , Garza, R. J. , Guzmán Pruneda, F. A. , … Evans, S. E. (2011). Synergistic interactions of TLR2/6 and TLR9 induce a high level of resistance to lung infection in mice. Journal of Immunology, 186, 5916–5926. 10.4049/jimmunol.1002122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehre, C. , Worthington, E. N. , Liesman, R. M. , Grubb, B. R. , Barbier, D. , O'Neal, W. K. , … Boucher, R. C. (2012). Overexpressing mouse model demonstrates the protective role of Muc5ac in the lungs. Proceedings of the National Academy of Sciences, 109, 16528–16533. 10.1073/pnas.1206552109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans, C. M. , Raclawska, D. S. , Ttofali, F. , Liptzin, D. R. , Fletcher, A. A. , Harper, D. N. , … Dickey, B. F. (2015). The polymeric mucin Muc5ac is required for allergic airway hyperreactivity. Nature Communications, 6, 6281 10.1038/ncomms7281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans, C. M. , Williams, O. W. , Tuvim, M. J. , Nigam, R. , Mixides, G. P. , Blackburn, M. R. , … Dickey, B. F. (2004). Mucin is produced by Clara cells in the proximal airways of antigen‐challenged mice. American Journal of Respiratory Cell and Molecular Biology, 31, 382–394. 10.1165/rcmb.2004-0060OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans, S. E. , Scott, B. L. , Clement, C. G. , Larson, D. T. , Kontoyiannis, D. , Lewis, R. E. , … Dickey, B. F. (2010). Stimulated innate resistance of lung epithelium protects mice broadly against bacteria and fungi. American Journal of Respiratory Cell and Molecular Biology, 42, 40–50. 10.1165/rcmb.2008-0260OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans, S. E. , Tuvim, M. J. , Fox, C. J. , Sachdev, N. , Gibiansky, L. , & Dickey, B. F. (2011). Inhaled innate immune ligands to prevent pneumonia. British Journal of Pharmacology, 163, 195–206. 10.1111/j.1476-5381.2011.01237.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farne, H. A. , & Johnston, S. L. (2017). Immune mechanisms of respiratory viral infections in asthma. Current Opinion in Immunology, 48, 31–37. 10.1016/j.coi.2017.07.017 [DOI] [PubMed] [Google Scholar]
- Guo, L. , Johnson, R. S. , & Schuh, J. C. L. (2000). Biochemical characterization of endogenously formed eosinophilic crystals in the lungs of mice. The Journal of Biological Chemistry, 275, 8032–8037. 10.1074/jbc.275.11.8032 [DOI] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR (2018). The IUPHAR/BPS guide to pharmacology in 2018: Updates and expansion to encompass the new guide to immunopharmacology. Nucleic Acids Research, 46, D1091–D1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hines, E. A. , Szakaly, R. J. , Leng, N. , Webster, A. T. , Verheyden, J. M. , Lashua, A. J. , … Lemanske, R. F. (2014). Comparison of temporal transcriptomic profiles from immature lungs of two rat strains reveals a viral response signature associated with chronic lung dysfunction. PLoS ONE, 9(12), e112997 10.1371/journal.pone.0112997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogg, J. C. (1997). The pathology of asthma. APMIS, 105, 735–745. 10.1111/j.1699-0463.1997.tb05079.x [DOI] [PubMed] [Google Scholar]
- Holgate, S. T. (2012). Innate and adaptive immune responses in asthma. Nature Medicine, 18, 673–683. 10.1038/nm.2731 [DOI] [PubMed] [Google Scholar]
- Holt, P. G. , & Sly, P. D. (2012). Viral infections and atopy in asthma pathogenesis: New rationales for asthma prevention and treatment. Nature Medicine, 18, 726–735. [DOI] [PubMed] [Google Scholar]
- Holtzman, M. J. (2012). Asthma as a chronic disease of the innate and adaptive immune systems responding to viruses and allergens. The Journal of Clinical Investigation, 122, 2741–2748. 10.1172/JCI60325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtzman, M. J. , Patel, D. A. , Zhang, Y. , & Patel, A. C. (2011). Host epithelial–viral interactions as cause and cure for asthma. Current Opinion in Immunology, 23, 487–494. 10.1016/j.coi.2011.05.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson, D. J. , Makrinioti, H. , Rana, B. M. , Shamji, B. W. , Trujillo‐Torralbo, M. B. , Footitt, J. , … Johnston, S. L. (2014). IL‐33–dependent Type 2 inflammation during rhinovirus‐induced asthma exacerbations in vivo. American Journal of Respiratory and Critical Care Medicine, 190, 1373–1382. 10.1164/rccm.201406-1039OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakab, G. J. , & Green, G. M. (1972). The effect of Sendai virus infection on bactericidal and transport mechanisms of the murine lung. The Journal of Clinical Investigation, 51, 1989–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson, M. E. V. , & Hansson, G. C. (2012). Preservation of mucus in histological sections, immunostaining of mucins in fixed tissue, and localization of bacteria with FISH. Methods in Molecular Biology, 229–235 Humana Press. 10.1007/978-1-61779-513-8_13 [DOI] [PubMed] [Google Scholar]
- Kilkenny, C. , Browne, W. , Cuthill, I. C. , Emerson, M. , & Altman, D. G. (2010). Animal research: Reporting in vivo experiments: The ARRIVE guidelines. British Journal of Pharmacology, 160, 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkpatrick, C. T. , Wang, Y. , Leiva Juarez, M. M. , Shivshankar, P. , Pantaleón García, J. , Plumer, A. K. , … Evans, S. E. (2018). Inducible lung epithelial resistance requires multisource reactive oxygen species generation to protect against viral infections. MBio, 9(3). 10.1128/mBio.00696-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaßen, C. , Karabinskaya, A. , Dejager, L. , Vettorazzi, S. , Van Moorleghem, J. , Lühder, F. , … Reichardt, H. M. (2017). Airway epithelial cells are crucial targets of glucocorticoids in a mouse model of allergic asthma. Journal of Immunology, 199, 48 LP–61. [DOI] [PubMed] [Google Scholar]
- Lambrecht, B. N. , & Hammad, H. (2015). The immunology of asthma. Nature Immunology, 16, 45–56. 10.1038/ni.3049 [DOI] [PubMed] [Google Scholar]
- Leiva‐Juarez, M. M. , Kirkpatrick, C. T. , Gilbert, B. E. , Scott, B. , Tuvim, M. J. , Dickey, B. F. , … Markesich, D. (2018). Combined aerosolized Toll‐like receptor ligands are an effective therapeutic agent against influenza pneumonia when co‐administered with oseltamivir. European Journal of Pharmacology, 818, 191–197. 10.1016/j.ejphar.2017.10.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leiva‐Juárez, M. M. , Ware, H. H. , Kulkarni, V. V. , Zweidler‐McKay, P. , Tuvim, M. J. , & Evans, S. E. (2016). Inducible epithelial resistance protects mice against leukemia‐associated pneumonia. Blood, 128, 982–992. 10.1182/blood-2016-03-708511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, W.‐K. , Liu, Q. , Chen, D. H. , Liang, H. X. , Chen, X. K. , Huang, W. B. , … Zhou, R. (2013). Epidemiology and clinical presentation of the four human parainfluenza virus types. BMC Infect Dis, 13, 28 10.1186/1471-2334-13-28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Look, D. C. , Walter, M. J. , Williamson, M. R. , Pang, L. , You, Y. , Sreshta, J. N. , … Brody, S. L. (2001). Effects of paramyxoviral infection on airway epithelial cell Foxj1 expression, ciliogenesis, and mucociliary function. The American Journal of Pathology, 159, 2055–2069. 10.1016/S0002-9440(10)63057-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallia, P. , Message, S. D. , Gielen, V. , Contoli, M. , Gray, K. , Kebadze, T. , … Johnston, S. L. (2011). Experimental rhinovirus infection as a human model of chronic obstructive pulmonary disease exacerbation. American Journal of Respiratory and Critical Care Medicine, 183, 734–742. 10.1164/rccm.201006-0833OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massion, P. P. , Funari, C. C. , Ueki, I. , Ikeda, S. , McDonald, D. , & Nadel, J. A. (1993). Parainfluenza (Sendai) virus infects ciliated cells and secretory cells but not basal cells of rat tracheal epithelium. American Journal of Respiratory Cell and Molecular Biology, 9, 361–370. 10.1165/ajrcmb/9.4.361 [DOI] [PubMed] [Google Scholar]
- McGrath, J. C. , & Lilley, E . (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.). British Journal of Pharmacology, 172, 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno‐Vinasco, L. , Verbout, N. G. , Fryer, A. D. , & Jacoby, D. B. (2009). Retinoic acid prevents virus‐induced airway hyperreactivity and M2 receptor dysfunction via anti‐inflammatory and antiviral effects. Am. J. Physiol. Cell. Mol. Physiol., 297, L340–L346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray, A. B. , & Luz, A. (1990). Acidophilic macrophage pneumonia in laboratory mice. Veterinary Pathology, 27, 274–281. 10.1177/030098589002700409 [DOI] [PubMed] [Google Scholar]
- Nguyen, L. P. , Omoluabi, O. , Parra, S. , Frieske, J. M. , Clement, C. , Ammar‐Aouchiche, Z. , … Bond, R. A. (2008). Chronic exposure to β‐blockers attenuates inflammation and mucin content in a murine asthma model. American Journal of Respiratory Cell and Molecular Biology, 38, 256–262. 10.1165/rcmb.2007-0279RC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oczypok, E. A. , Perkins, T. N. , & Oury, T. D. (2017). Alveolar epithelial cell–derived mediators: Potential direct regulators of large airway and vascular responses. American Journal of Respiratory Cell and Molecular Biology, 56, 694–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel, A. C. , Morton, J. D. , Kim, E. Y. , Alevy, Y. , Swanson, S. , Tucker, J. , … Holtzman, M. J. (2006). Genetic segregation of airway disease traits despite redundancy of calcium‐activated chloride channel family members. Physiological Genomics, 25, 502–513. 10.1152/physiolgenomics.00321.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peebles, R. S. , & Aronica, M. A. (2019). Proinflammatory pathways in the pathogenesis of asthma. Clinics in Chest Medicine, 40, 29–50. 10.1016/j.ccm.2018.10.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccotti, L. , Dickey, B. F. , & Evans, C. M. (2012). Assessment of intracellular mucin content in vivo. Methods in Molecular Biology, 279–295Humana Press. 10.1007/978-1-61779-513-8_17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichery, M. , Mirey, E. , Mercier, P. , Lefrancais, E. , Dujardin, A. , Ortega, N. , & Girard, J. P. (2012). Endogenous IL‐33 is highly expressed in mouse epithelial barrier tissues, lymphoid organs, brain, embryos, and inflamed tissues: in situ analysis using a novel Il‐33‐LacZ gene trap reporter strain. Journal of Immunology, 188, 3488–3495. 10.4049/jimmunol.1101977 [DOI] [PubMed] [Google Scholar]
- Ren, B. , Azzegagh, Z. , Jaramillo, A. M. , Zhu, Y. , Pardo‐Saganta, A. , Bagirzadeh, R. , … Dickey, B. F. (2015). SNAP23 is selectively expressed in airway secretory cells and mediates baseline and stimulated mucin secretion. Bioscience Reports, 35(3). 10.1042/BSR20150004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin, J. , Arganda‐Carreras, I. , Frise, E. , Kaynig, V. , Longair, M. , Pietzsch, T. , … Cardona, A. (2012). Fiji: An open‐source platform for biological‐image analysis. Nature Methods, 9, 676–682. 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmittgen, T. D. , & Livak, K. J. (2008). Analyzing real‐time PCR data by the comparative CT method. Nature Protocols, 3, 1101–1108. 10.1038/nprot.2008.73 [DOI] [PubMed] [Google Scholar]
- Sheshadri, A. , Shah, D. P. , Godoy, M. , Erasmus, J. J. , Song, J. , Li, L. , … Ost, D. E. (2018). Progression of the Radiologic Severity Index predicts mortality in patients with parainfluenza virus‐associated lower respiratory infections. PLoS ONE, 13(5), e0197418 10.1371/journal.pone.0197418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treutlein, B. , Brownfield, D. G. , Wu, A. R. , Neff, N. F. , Mantalas, G. L. , Espinoza, F. H. , … Quake, S. R. (2014). Reconstructing lineage hierarchies of the distal lung epithelium using single‐cell RNA‐seq. Nature, 509, 371–375. 10.1038/nature13173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuvim, M. J. , Evans, S. E. , Clement, C. G. , Dickey, B. F. , & Gilbert, B. E. (2009). Augmented lung inflammation protects against influenza A pneumonia. PLoS ONE, 4(1), e4176 10.1371/journal.pone.0004176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuvim, M. J. , Gilbert, B. E. , Dickey, B. F. , & Evans, S. E. (2012). Synergistic TLR2/6 and TLR9 Activation Protects Mice against Lethal Influenza Pneumonia. PLoS ONE, 7(1), e30596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuvim, M. J. , Mospan, A. R. , Burns, K. A. , Chua, M. , Mohler, P. J. , Melicoff, E. , … Dickey, B. F. (2009). Synaptotagmin 2 couples mucin granule exocytosis to Ca2+ signaling from endoplasmic reticulum. The Journal of Biological Chemistry, 284, 9781–9787. 10.1074/jbc.M807849200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter, M. J. , Kajiwara, N. , Karanja, P. , Castro, M. , & Holtzman, M. J. (2001). Interleukin 12 p40 production by barrier epithelial cells during airway inflammation. The Journal of Experimental Medicine, 193, 339–351. 10.1084/jem.193.3.339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter, M. J. , Morton, J. D. , Kajiwara, N. , Agapov, E. , & Holtzman, M. J. (2002). Viral induction of a chronic asthma phenotype and genetic segregation from the acute response. The Journal of Clinical Investigation, 110, 165–175. 10.1172/JCI14345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ware, H. H. , Kulkarni, V. V. , Wang, Y. , Pantaleón García, J. , Leiva Juarez, M. , Kirkpatrick, C. T. , … Evans, S. E. (2019). Inducible lung epithelial resistance requires multisource reactive oxygen species generation to protect against bacterial infections. PLoS ONE, 14(2), e0208216 10.1371/journal.pone.0208216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, Y. , Ehre, C. , Abdullah, L. H. , Sheehan, J. K. , Roy, M. , Evans, C. M. , … Davis, C. W. (2008). Munc13‐2 −/− baseline secretion defect reveals source of oligomeric mucins in mouse airways. The Journal of Physiology, 586, 1977–1992. 10.1113/jphysiol.2007.149310 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Lung SeV burden days 3 through 7 after infection.
Figure S2. Effect of O/P treatment 20 days before or after SeV infection.
Figure S3. Staining for Muc5ac and Muc5b, collagen, and macrophages in lung nodules.
Figure S4. Location of left axial bronchus sections for mucin content, and image of the precision cutting instrument.
Figure S5. Whole mount and cross‐sectional immunofluorescence staining of airway epithelium 14 days after SeV infection.
Figure S6. Immunohistochemical staining for IL‐33 done 49 days after SeV infection.
Figure S7. Lung lavage fluid cytokine responses to aerosolized O/P and IFN‐β.