

Abstract
Background and Purpose
The midbrain periaqueductal grey (PAG) plays a central role in modulating pain through a descending pathway that projects indirectly to the spinal cord via the rostroventral medial medulla (RVM). While opioids are potent analgesics that target the PAG, their cellular actions on descending projection neurons are unclear.
Experimental Approach
Patch clamp recordings in voltage‐ and current‐clamp mode were made from acutely prepared PAG slices from animals that received retrograde tracer injections into the RVM.
Key Results
The μ‐agonist DAMGO reduced GABAergic evoked inhibitory postsynaptic currents (IPSCs) in retro‐labelled, RVM‐projecting neurons to a greater extent than in unlabelled neurons. The κ‐opioid agonist U69593 reduced evoked IPSCs to a similar extent in both neuronal groups, while the δ‐opioid agonist deltorphin‐II was without effect. DAMGO and U69593 both produced a reduction in the rate, but not amplitude of spontaneous miniature IPSCs and asynchronous evoked IPSCs in retro‐labelled neurons. DAMGO and U69593 also suppressed glutamatergic EPSCs in retro‐labelled and unlabelled neurons. The DAMGO inhibition of evoked EPSCs, however, was less than that for evoked IPSCs in retro‐labelled, but not unlabelled neurons. In current clamp, DAMGO produced a depolarizing increase in evoked postsynaptic potentials in retro‐labelled neurons, but directly inhibited unlabelled neurons.
Conclusion and Implications
These findings suggest that μ‐opioids activate the descending analgesic pathway from the midbrain PAG by a combination of presynaptic disinhibition of RVM‐projecting neurons and postsynaptic inhibition of presumptive interneurons.
Abbreviations
- AP5
2‐amino‐5‐phosphovaleric acid
- CNQX
6‐cyano‐7‐nitroquinoxaline‐2,3‐dione
- CTAP
D‐Ala2, N‐MePhe4, Gly‐ol]‐enkephalin
- DAMGO
D‐Ala2, N‐MePhe4, Gly‐ol]‐enkephalin
- deltorphin‐II
Tyr‐d‐Ala‐Phe‐Glu‐Val‐Val‐Gly‐NH2
- gabazine
6‐Imino‐3‐(4‐methoxyphenyl)‐1(6H)‐pyridazinebutanoic acid hydrobromide
- ICI174864
α‐aminoisobutyric acid
- IPSC
inhibitory postsynaptic current
- Nor‐BNI
17,17′‐(Dicyclopropylmethyl)‐6,6′,7,7′‐6,6′‐imino‐ 7,7′‐binorphinan‐3,4′,14,14′‐tetrol dihydrochloride
- PAG
periaqueductal grey
- PSP
postsynaptic potential
- RVM
rostroventral medial medulla
- TTX
tetrodotoxin
- U69593
(+)‐(5α,7α,8β)‐N‐Methyl‐N‐[7‐(1‐pyrrolidinyl)‐1‐oxaspiro[4.5]dec‐8‐yl]‐benzeneacetamide
What is already known
Opioids produce analgesia partly by activating a midbrain descending pathway.
Within these pathways, opioids directly inhibit neuronal subpopulations and reduce inhibitory and excitatory transmitter release.
What this study adds
μ‐Opioids presynaptically disinhibited midbrain descending projection neurons by targeting their inhibitory inputs.
Non‐projection midbrain neurons were directly inhibited by μ‐opioids and did not display differential presynaptic inhibition.
What is the clinical significance
An understanding of both presynaptic and postsynaptic coupling mechanisms is needed to improve opioid analgesics.
1. INTRODUCTION
To improve opioid analgesics, we need to understand how they engage analgesic systems within the brain. The midbrain periaqueductal grey (PAG) contains relatively high levels of inhibitory Gi/o‐coupled μ‐https://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=50 and serves as a major site of action of opioid analgesics (Commons, Aicher, Kow, & Pfaff, 2000; Gutstein, Mansour, Watson, Akil, & Fields, 1998; Lane, Patel, & Morgan, 2005; Mansour, Khachaturian, Lewis, Akil, & Watson, 1987; Wang & Wessendorf, 2002). While the PAG also expresses δ‐ and κ‐opioid receptors (Gutstein et al., 1998; Wang & Wessendorf, 2002), their role in modulating PAG activity is less well defined (Ossipov, Kovelowski, Nichols, Hruby, & Porreca, 1995; Rossi, Pasternak, & Bodnar, 1994; Smith et al., 1988).
The ventrolateral column of the PAG forms part of a descending analgesic pathway that projects via the rostroventromedial medulla (RVM) to the spinal cord dorsal horn, where it inhibits ascending nociceptive transmission to the brain (Fields & Basbaum, 1978). A disinhibition hypothesis originally proposed that μ‐opioids indirectly activate the descending pathway by inhibiting https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1067ergic interneurons, thereby releasing projection neurons from intrinsic inhibition (Depaulis, Morgan, & Liebeskind, 1987; Moreau & Fields, 1986). In accordance with this hypothesis, anatomical studies have shown that the PAG contains a substantial population of GABAergic neurons and nerve terminals (Barbaresi & Manfrini, 1988; Park et al., 2010; Reichling & Basbaum, 1990a, 1990b). Furthermore, GABAergic neurons within the PAG express postsynaptic μ‐opioid receptors (Commons et al., 2000; Kalyuzhny & Wessendorf, 1998). While the majority of PAG neurons are directly inhibited by μ‐opioids, ventrolateral PAG neurons that project to the RVM are largely μ‐opioid insensitive (Chieng & Christie, 1994a; Chiou & Huang, 1999; Osborne, Vaughan, Wilson, & Christie, 1996). This direct postsynaptic opioid inhibition is consistent with the disinhibition hypothesis.
Opioids, however, also have presynaptic actions within the PAG and other brain regions. Indeed, μ‐opioids presynaptically inhibit GABAergic synaptic transmission onto unidentified neurons within the ventrolateral PAG (Chieng & Christie, 1994b; Chiou & Huang, 1999; Vaughan & Christie, 1997). While it is unknown whether this occurs in RVM‐projecting PAG neurons, GABAergic terminals that express μ‐opioid receptors oppose the cell bodies of these projection neurons (Commons et al., 2000; Kalyuzhny & Wessendorf, 1998). This suggests that presynaptic mechanisms might also be involved in opioid disinhibition. However, there are inconsistencies that challenge the disinhibition hypothesis (Lau & Vaughan, 2014). For example, opioids also inhibit glutamatergic synaptic transmission within the PAG (Chieng & Christie, 1994b; Vaughan & Christie, 1997), and this can functionally oppose GABAergic modulation of pain (Samineni et al., 2017). This has led to the alternative hypothesis that opioid analgesia is mediated by distinct and functionally opposing GABAergic and glutamatergic descending pathways (Cleary, Neubert, & Heinricher, 2008). Furthermore, the presynaptic actions of δ‐ and κ‐opioids within the PAG are less well understood. In the present study, we used a combination of electrophysiology and tract tracing to identify the opioid receptors and circuitry underlying opioid modulation of the descending analgesic pathway within the PAG.
2. METHODS
Experiments were carried out on male and female Sprague Dawley rats in accordance with guidelines set by the National Health and Medical Research Council “Australian code of practice for the care and use of animals for scientific purposes.” All experiments were approved by the Royal North Shore Hospital Animal Ethics Committee (protocols 0912‐021A and 1311‐013A). Pregnant female rats were obtained from the Animal Resources Centre (Canning Vale, Australia) and were housed in the Kolling Institute Facility. After weaning, animals of the same gender were housed in groups of two to four in individually ventilated cages under controlled light (12‐hr light–dark cycles) and temperature (23 ± 1°C, 70% humidity) with ad libitum access to water and food pellets. Cages were enriched with a house igloo, tissues for nesting, and straws or paddle pop sticks on alternate weeks. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny, Browne, Cuthill, Emerson, & Altman, 2010) and with the recommendations made by the British Journal of Pharmacology.
2.1. Retrograde tracer microinjections
For brain microinjections, 22‐ to 35‐day‐old rats were anaesthetized (1–2% isoflurane in O2), placed in a stereotaxic frame and the dura exposed by trephination over the RVM. Red‐orange fluorescent microspheres (Molecular Probes) were injected into the RVM (10–12 × 10 nl injections, total = 100–120 nl) via a glass micropipette (tip diameter 30 μm, Drummond Nanoject), as previously described (Drew, Lau, & Vaughan, 2009). The hole was then filled with bone wax, the incision was closed and animals received a single dose of an antibiotic (amoxicillin 15 mg·kg−1, s.c.) and an analgesic (https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1670 0.05 mg·kg−1, s.c.). Animals were recovered from anaesthesia in a clean cage warmed by a heating pad before being returned to their holding room. Intra‐RVM tracer‐injected animals were used for in vitro slice experiments at 1–2 weeks post‐surgery. The core of the tracer injections was examined post hoc to verify correct placement within the RVM (see Figure 1a inset), which was defined as a triangular region bordered rostrocaudally by the trapezoid body and inferior olive, ventrally by the pyramidal tract and laterally by the extent of the pyramidal tract (Gu & Wessendorf, 2007).
Figure 1.

Met‐enkephalin inhibits GABAergic synaptic transmission onto PAG projection neurons. (a[i]) Low power (20×) 3x3 tiled and z‐stacked confocal images (z = 62 μm) of a PAG slice showing retrogradely labelled (Retro) neurons from the RVM in animals that received an intra‐RVM injection of fluorescent rhodamine microspheres (red). (a[ii,iii]) High power (63×) z‐stacked images (z = 46 μm) respectively showing retro‐labelled (red) and unlabelled PAG neurons recorded and filled with biocytin (blue). Scale bars: (a[i]) 200 μm, (a[ii–iii]) 25 μm. Inset in (a[i]) shows a retrograde tracer injection site within the RVM. (b) Averaged traces of (i) evoked IPSCs and (ii) normalized paired evoked IPSCs in a retro‐labelled PAG neuron before, during and after washout of met‐enkephalin (ME, 10 μM). (c) Time plot of evoked IPSC amplitude in the retro‐labelled PAG neuron from (b) during superfusion of met‐enkephalin (ME). Scatter plots of the effect of ME on the (d) amplitude and (e) paired‐pulse ratio (PPR) of evoked IPSCs in retro‐labelled (Retro) and unlabelled PAG neurons from animals that received an intra‐RVM injection of retrograde tracer, and in blind (Control) PAG neurons from animals that did not receive a tracer injection. In (d–e), data are expressed as a percentage of the pre‐agonist value; *P < .05 (Student's t‐tests, pre‐ vs. post‐drug). In (b), stimulus artefacts have been blanked for clarity [Colour figure can be viewed at http://wileyonlinelibrary.com]
2.2. Electrophysiology
For slice experiments, animals were deeply anaesthetized with isoflurane and decapitated, and coronal midbrain slices (300 μm) containing the PAG were cut using a vibratome (VT1200S, Leica Microsystems) in ice‐cold artificial CSF (ACSF) of composition (in mM): 126 NaCl, 2.5 KCl, 1.4 NaH2PO4, 1.2 MgCl2, 2.4 CaCl2, 11 glucose, and 25 NaHCO3, equilibrated with 95% O2 and 5% CO2. PAG slices were maintained at 34°C in a submerged chamber containing ACSF for 1 hr before being transferred to room temperature ACSF prior to recording. For recording, slices were individually transferred to a chamber on an upright fluorescence microscope (Olympus BX51) and superfused continuously with ACSF (34°C, flow rate 2.5 ml·min−1). PAG neurons were visualized with a 40× water‐immersion objective using Dodt gradient contrast optics. In animals that received retrograde tracer, the presence or absence of red‐orange microspheres was ascertained under concurrent fluorescent/light illumination prior to recording from retro‐labelled or unlabelled neurons. In animals that did not receive retrograde tracer, recordings were made from blind, unidentified neurons.
Whole‐cell voltage‐clamp recordings (Axopatch 200B, or 700B, Molecular Devices) of synaptic currents (holding potential, −65 mV) were made from PAG neurons using a CsCl‐based internal solution containing (in mM): 140 CsCl, 0.2 EGTA, 10 HEPES, 1 MgCl2, 2 MgATP, 0.3 NaGTP, and 0.1% biocytin (pH 7.3; osmolarity, 280–285 mOsm·L−1). Whole‐cell current‐clamp recordings of synaptic potentials were made using a K‐gluconate‐based internal solution containing (in mM): 135 K‐gluconate, 4 NaCl, 0.5 EGTA, 10 HEPES, 1 MgCl2, 2 MgATP, 0.3 NaGTP, and 0.1% biocytin (pH 7.3; osmolarity, 280–285 mOsm·L−1). Series resistance (<25 MΩ) was compensated by 65% and continuously monitored during experiments.
Electrically evoked postsynaptic currents or potentials (PSCs and PSPs) were elicited every 6–12 s using either monopolar glass stimulating electrodes (tip width 20 μm) or nickel‐chromium bipolar electrodes. In voltage‐clamp experiments, paired electrically evoked synaptic currents were elicited at an inter‐stimulus interval of 70 ms. Inhibitory postsynaptic currents (IPSCs) were pharmacologically isolated in the presence of the AMPA/kainate receptor antagonist, 6‐cyano‐7‐nitroquinoxaline‐2,3‐dione (https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5475, 5–10 μM), the glycine receptor antagonist, https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=347 (3 μM) and the NMDA receptor antagonist, dl‐2‐Amino‐5‐phosphonopentanoic acid (https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4168, 25 μM). EPSCs were isolated in the presence of the GABAA receptor antagonist, https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4197 (3 μM), AP5 and strychnine. Spontaneous miniature IPSCs were recorded in the additional presence of the voltage‐gated sodium channel blocker, https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2616 (TTX, 1 μM), while evoked miniature IPSCs were recorded by adding strontium (8 mM) to the external ACSF. In current‐clamp experiments, single electrically evoked PSPs were elicited every 12 s in either blocker‐free ACSF or in the presence of gabazine.
All recordings of synaptic currents or potentials were filtered (4‐ to 10‐kHz low‐pass filter) and sampled (10–20 kHz) for on‐line and later off‐line analysis using AxographX (Axograph Scientific Software, RRID:SCR_014284). The peak amplitude of electrically evoked PSCs and PSPs were measured relative to a 2‐ms baseline preceding the stimulus artefact. Miniature IPSCs above a preset threshold (3–4 SDs above baseline noise) were automatically detected by a sliding template algorithm, then manually checked off‐line.
2.3. Immunohistochemistry and confocal imaging
Post hoc staining was carried out according the journal's guidelines (Alexander et al., 2018). PAG and RVM slices were fixed overnight at 4°C in 4% PFA in 0.1‐M phosphate buffer (PB) and then washed three times for 10 min with 0.1‐M PB and stored (<2 weeks) at 4°C prior to staining. Slices were incubated for 1 hr at room temperature in 5% horse serum, 1% BSA, and 0.3% Triton X‐100 in PB. For staining biocytin‐filled neurons in PAG slices, Streptavidin‐647 (1:1000, ThermoFisher) was diluted in 1% BSA/0.1% Triton X‐100 in PB, and slices were then incubated for 2 hr at room temperature or overnight at 4°C (light protected). In all cases, the nuclear stain, DAPI (1:2000, Sigma), was added for the last 30 min of the final incubation period. Slices were then washed three to four times (10 min) with PB and mounted onto slides using Fluoromount‐G (SouthernBiotech). All sections were visualized using a Leica TCS SP5 confocal microscope (lasers: 405, 488, 561, and 633 nm), and images were captured with LAS AF (Leica) software. Images were taken sequentially with different lasers using either a 20× (NA 0.7) or a 63× (NA 1.4) oil immersion objective using LAS AF software. Z‐stacks and tiled scans were collected where indicated, and images were processed using ImageJ software (NIH). The immuno‐related procedures used comply with the recommendations made by the British Journal of Pharmacology.
2.4. Experimental design and statistical analysis
The data and statistical analysis comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology (Curtis et al., 2018). We have previously observed opioid effect sizes of 2.0 and 1.5 for voltage‐ and current‐clamp experiments respectively (Drew et al., 2009; Osborne et al., 1996); this requires a minimum of six and eight successful recordings from individual neurons (for two‐tailed tests, paired t tests with α and β errors of .05). In order to ensure this minimum sample size, we prepared eight animals per experimental group because we have previously had a 90% success rate with RVM tracer injections and obtain an average of 1.5 successful neuron recordings per animal (only one neuron recording was obtained per slice and between one and three PAG slices were obtained per animal). The number of neuron recordings obtained per experimental group reflected the variability in successful neuron recordings from eight animals per group. Neuron recordings were only excluded from analysis if (a) the RVM injection site was incorrect, (b) series resistance varied by more than 25%, or (c) evoked synaptic currents/potentials during the pre‐agonist, baseline period changed by more than 20%. Data outliers were not excluded.
The researchers performing and analysing electrophysiology experiments were not blinded to the PAG neuron type (retro‐labelled, unlabelled and control blind) or agonist applied. After obtaining a stable baseline recording for at least 5 min, opioid agonists and antagonists were applied for periods of 5–6 min. To avoid bias, the order in which opioid agonists were applied was sequentially rotated between neuron recordings for each experimenter. Drug effects on electrophysiological parameters were made by measuring at fixed time points: over the last 2 min prior to agonist application, during agonist application and then during antagonist application. In voltage‐clamp experiments, the effects of opioid agonists on PSC parameters were calculated as a percentage agonist value divided by the average of the pre‐agonist and antagonist values. In current‐clamp experiments, the effects of opioid agonists on membrane current and synaptic potentials were calculated as a difference to the average of the pre‐agonist and antagonist values; neurons were considered to have a postsynaptic response to DAMGO if the change in membrane potential was greater than two times the SD of the pre‐DAMGO baseline.
All numerical data are expressed as mean ± SEM and two‐sided statistical analyses were performed (Prism v8, GraphPad Software, La Jolla, USA). Statistical comparisons of individual drug effects were made using one‐sample paired Student's t‐tests. Comparisons of opioid agonist effects between multiple groups were made using one‐ or two‐way ANOVAs. One‐ and two‐way ANOVAs satisfied homogeneity of variance (Brown–Forsythe test) and sphericity (Mauchly's test) respectively. When one‐way main effects or two‐way interactions were significant, post hoc comparisons were performed using the Tukey or Bonferroni adjustments. Comparisons of proportions were made using Fisher's exact test. Differences were considered significant when P < .05.
2.5. Drugs and reagents
All reagents were obtained from Sigma‐Aldrich, Abcam, and Tocris Bioscience. Stock solutions of neurochemicals were made in distilled water, or DMSO, aliquoted, and then frozen. Individual aliquots of opioids were then transferred to a fridge every 2–3 weeks and used for experiments. During each neuron recording, stock drugs were diluted to working concentrations in ACSF (≤1:3,000 drug solvent: ACSF) immediately before use and applied by bath superfusion.
2.6. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20 (Alexander et al., 2019; Alexander et al., 2019).
3. RESULTS
3.1. The endogenous opioid met‐enkephalin inhibits GABAergic synaptic transmission onto PAG‐RVM projection neurons
While prior studies have established that opioids inhibit GABAergic synaptic transmission onto unidentified ventrolateral PAG neurons, their actions on neurons that project via the descending analgesic pathway are unknown. To address this, whole‐cell patch clamp electrophysiology and anatomical tract tracing was used to examine the actions of opioids on PAG neurons identified as projecting to the RVM. Following intra‐RVM injection of fluorescent rhodamine microspheres, descending RVM‐projecting neurons were identified bilaterally throughout the ventrolateral PAG by the presence of retrogradely transported tracer within their cell bodies (Figure 1a(i) and inset).
In slices from tracer‐injected animals, recordings were made from neurons that were identified as containing retrograde tracer and confirmed post hoc by staining for biocytin (Figure 1a(ii), retro‐labelled neurons). In the presence of CNQX, AP5 and strychnine, local electrical stimulation evoked IPSCs in retro‐labelled PAG neurons that were abolished by addition of gabazine (n = 12). In these neurons, application of the endogenous opioid https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1614 (10 μM) produced a reversible reduction in the amplitude of evoked IPSCs (Figure 1b–d; n = 10). As a comparison, recordings were also made from neurons that were identified as not containing retrograde tracer and confirmed post hoc by staining for biocytin (Figure 1a(iii), unlabelled neurons). Like retro‐labelled PAG neurons, met‐enkephalin produced a reduction in the amplitude of evoked IPSCs in unlabelled neurons (Figure 1d; n = 8). As a control for the tracer injection surgery, we also examined the effect of met‐enkephalin in blind, unidentified PAG neurons (control) in slices from animals that did not receive intra‐RVM tracer injections. Met‐enkephalin also produced a reduction in the amplitude of evoked IPSCs in these control neurons (Figure 1d; n = 8). The effect of met‐enkephalin on evoked IPSCs did not differ significantly between retro‐labelled, unlabelled and control PAG neurons (P > .05, one‐way ANOVA).
3.2. μ‐ and κ‐opioids inhibit GABAergic synaptic transmission onto PAG‐RVM projection neurons
As met‐enkephalin is a mixed μ/δ‐opioid receptor agonist, we next determined the specific opioid receptor subtypes that modulate inhibitory GABAergic synaptic transmission onto PAG‐RVM projection neurons. The selective μ‐opioid agonist DAMGO (3 μM) produced a reduction in the amplitude of evoked IPSCs in retro‐labelled neurons (Figure 2a–c; n = 8). This inhibition was reversed by addition of the μ‐opioid antagonist, https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1635 (1 μM; Figure 2b, n = 7). In these neurons, the δ‐opioid receptor agonist, deltorphin‐II (300 nM) and the δ‐opioid receptor antagonist ICI174864 (300 nM) had no effect on the amplitude of evoked IPSCs (Figure 2a–c; n = 9,). The κ‐opioid receptor agonist, https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1655, (300 nM) reduced the amplitude of evoked IPSCs and this was reversed by the κ‐opioid antagonist https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1642 (300 nM; Figure 2a–c; n = 10).
Figure 2.
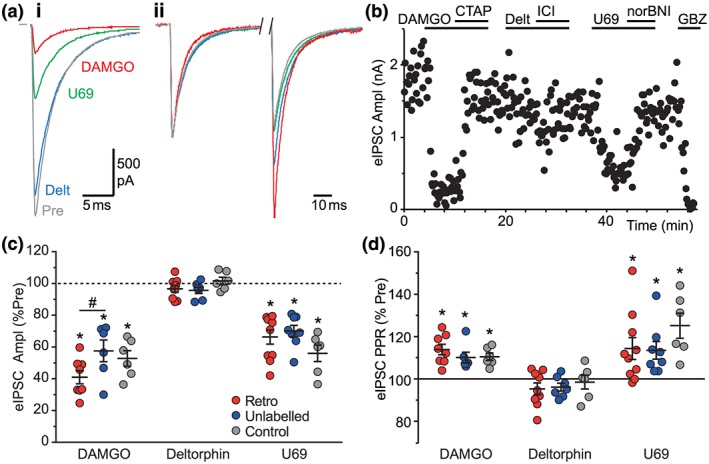
Opioid inhibition of GABAergic synaptic transmission onto PAG projection neurons is μ‐ and κ‐receptor mediated. (a) Averaged raw traces of (i) evoked IPSCs and (ii) normalized paired evoked IPSCs in a retro‐labelled PAG neuron before and during the opioid agonists DAMGO (3 μM), deltorphin‐II (300 nM), and U69593 (300 nM). (b) Time plot of evoked IPSC amplitude in the retro‐labelled PAG neuron from (a) during DAMGO, deltorphin II, and U69393, and the opioid antagonists CTAP (1 μM), ICI174863 (300 nM) and nor‐BNI (300 nM). Scatter plots of the effect of DAMGO, deltorphin‐II, and U69393 on the (c) amplitude and (d) paired‐pulse ratio (PPR) of evoked IPSCs in retro‐labelled and unlabelled PAG neurons from animals that received an intra‐RVM injection of retrograde tracer and in blind (Control) PAG neurons from animals that did not receive a tracer injection. In (c–d), data are expressed as a percentage of the pre‐agonist value; *P < .05 (paired Student's t‐tests, pre‐ vs. post‐drug); # P < .05, post hoc comparisons. In (a), stimulus artefacts have been blanked for clarity [Colour figure can be viewed at http://wileyonlinelibrary.com]
Like retro‐labelled PAG neurons, DAMGO and U69593, but not deltorphin‐II, produced a significant reduction in the amplitude of evoked IPSCs in unlabelled neurons (Figure 2c). Similarly, DAMGO and U69593, but not deltorphin‐II, produced a significant reduction in the amplitude of evoked IPSCs in control blind neurons (Figure 2c). The effect of the opioid agonists differed significantly between retro‐labelled and unlabelled neurons from RVM tracer‐injected animals and control blind neurons from animals that did not receive a tracer. While DAMGO produced a greater reduction in evoked IPSCs in retro‐labelled neurons compared with unlabelled neurons, there were no significant differences in the effect of U69593 or deltorphin‐II between the three neuronal groups (Figure 2c). Together, these observations indicate that μ‐ and κ‐opioid receptor activation inhibits GABAergic synaptic transmission onto all PAG neurons, including those that project along the descending RVM pathway.
3.3. The μ‐ and κ‐opioid inhibition of GABAergic transmission is presynaptically mediated
To examine the locus of opioid action on GABAergic transmission onto RVM projection neurons, we first assessed the effect of agonists on the paired‐pulse ratio of evoked IPSCs. In retro‐labelled neurons, met‐enkephalin, DAMGO and U69593 all produced an increase in the ratio of the amplitude of the second IPSC compared to the first IPSC (Figures 1b(ii),e and 2a(ii),d;). By contrast, deltorphin‐II had no effect on the paired‐pulse ratio of evoked IPSCs in these neurons (Figure 2a(ii),d). Furthermore, met‐enkephalin, DAMGO and U69593, but not deltorphin‐II, produced an increase in evoked IPSC paired‐pulse ratio in unlabelled and control blind neurons (Figures 1e and 2d).
To further examine the locus of action of μ‐ and κ‐opioid agonists specifically in retro‐labelled neurons, we assessed the effect of DAMGO and U69593 on spontaneous, quantal miniature IPSCs in the additional presence of TTX (1 μM), to block action potential‐dependent neurotransmitter release. In retro‐labelled neurons, we observed spontaneous TTX‐resistant miniature IPSCs that were abolished by gabazine (3 μM, n = 6). In these neurons, DAMGO produced a reduction in the mean rate, but not the amplitude of miniature IPSCs (Figure 3a,g). The effect of DAMGO was associated with a rightward shift in the cumulative frequency distribution of miniature IPSC inter‐event intervals and no change in their cumulative amplitude distributions (Figure 3b,c). Similarly, U69593 produced a reduction in the mean rate, but not the amplitude of miniature IPSCs (Figure 3d,g, n = 9). U69593 produced a rightward shift in the cumulative frequency distribution of miniature IPSC inter‐event intervals and no change in their cumulative amplitude distributions (Figure 3e,f). The effect of DAMGO and U69593 on miniature IPSCs was reversed by addition of CTAP and nor‐BNI respectively (Figure 3a–f, n = 11, 9).
Figure 3.

μ‐ and κ‐opioids inhibit GABAergic spontaneous miniature IPSCs in PAG projection neurons. Raw traces of spontaneous miniature IPSCs in retro‐labelled PAG neurons, before (Pre) and during superfusion of (a) DAMGO and CTAP, and (d) U69593 and nor‐BNI. Cumulative frequency distribution plots of miniature IPSC inter‐event interval and amplitude, before and during (b) DAMGO/CTAP and (e) U69593/nor‐BNI. Cumulative amplitude distribution plots of miniature IPSC inter‐event interval and amplitude, before and during (c) DAMGO/CTAP and (f) U69593/nor‐BNI. (g) Scatter plots of the effect of DAMGO and U69593 on the rate and amplitude of spontaneous miniature IPSCs (mIPSC) in retro‐labelled PAG neurons, shown as a percentage of the pre‐agonist value. In (g), *P < .05 (paired Student's t‐tests, pre‐ vs. post‐drug) [Colour figure can be viewed at http://wileyonlinelibrary.com]
Given that spontaneous miniature IPSCs are not action potential dependent and may reflect transmitter release from a different vesicle pool or synapse, we also examined electrically evoked quantal release in the presence of Sr2+. Being a less effective replacement for Ca2+ in modulating transmitter release, Sr2+ results in a desynchronization of an evoked PSC, which leads to delayed asynchronous release of individual transmitter. In retro‐labelled neurons, substitution of Ca2+ (2.4 mM) with Sr2+ (8 mM) in the external solution led to a reduction in the amplitude of synchronous evoked IPSCs and the emergence of smaller asynchronous IPSCs that were defined as quantal during the 50‐ to 500‐ms period following the stimulus artefact (Figure 4a, n = 12). In these neurons, DAMGO produced a reduction in the rate, but not amplitude of the asynchronous miniature IPSCs (Figure 4b,c). Similarly, U69593 produced a reduction in the rate, but not amplitude of the asynchronous miniature IPSCs (Figure 4c; n = 8). Together, these observations indicate that μ‐ and κ‐opioids specifically act via a presynaptic mechanism to decrease the probability of evoked GABA release onto all PAG neurons, including descending projection neurons.
Figure 4.
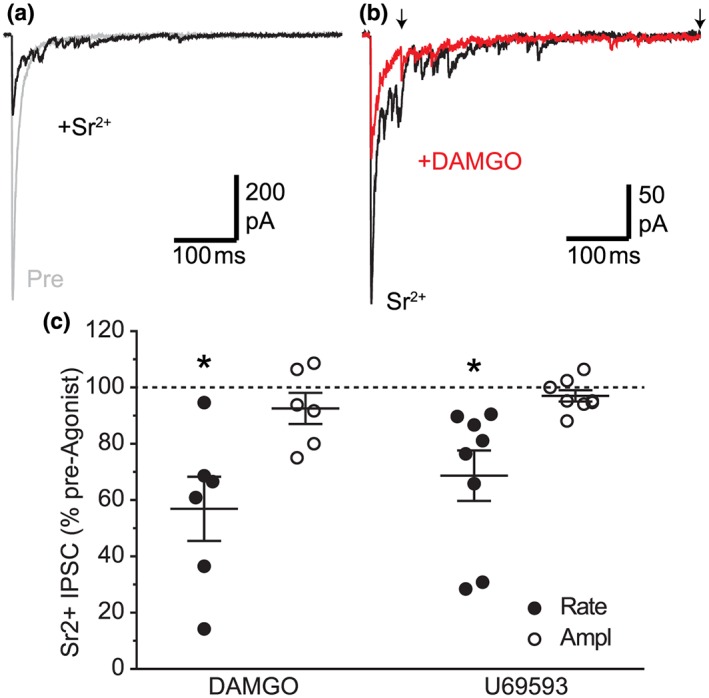
μ‐ and κ‐opioids inhibit GABAergic Sr2+‐evoked IPSCs in PAG projection neurons. (a) Raw traces of electrically evoked IPSCs in a retro‐labelled PAG neuron before (Pre) and after substitution of Ca2+ (2.4 mM) with Sr2+ (8 mM) in the external ACSF solution. (b) Representative traces of Sr2+‐evoked IPSCs from the same neuron in (a), before and during addition of DAMGO. (c) Scatter of the effect of DAMGO on the rate and amplitude of asynchronous Sr2+‐evoked miniature IPSCs (Sr2+‐eIPSC) in retro‐labelled PAG neurons, shown as a percentage of the pre‐DAMGO and U69593 value; *P < .05 (pre‐ vs. post‐drug). In (b), the arrows indicate the start and end of period over which synchronous IPSCs were analysed. In (a, b), stimulus artefacts have been blanked for clarity [Colour figure can be viewed at http://wileyonlinelibrary.com]
3.4. μ‐ and κ‐opioids also inhibit glutamatergic synaptic transmission onto PAG‐RVM projection neurons
We next sought to determine whether opioids affect excitatory glutamatergic transmission onto descending RVM‐projecting neurons. In retro‐labelled neurons from RVM tracer‐injected animals, local electrical stimulation also evoked EPSCs in the presence of gabazine, AP5 and strychnine that were abolished by addition of CNQX (n = 8). As observed for evoked IPSCs, both DAMGO and U69593 produced a reduction in the amplitude of evoked EPSCs in retro‐labelled neurons (Figure 5a,b, n = 10, 11). By contrast, deltorphin‐II did not have a significant effect on the amplitude of evoked EPSCs in these neurons (Figure 5a,b, n = 11). The DAMGO‐ and U69593‐induced inhibition of evoked EPSCs in these neurons was reversed by CTAP and nor‐BNI respectively (n = 9, 11).
Figure 5.

μ‐ and κ‐opioids also inhibit glutamatergic synaptic transmission onto PAG descending projection neurons. (a) Averaged traces of evoked EPSCs before (Pre) and during superfusion of DAMGO, U69593, and deltorphin‐II (Delt) in a retro‐labelled PAG‐RVM neuron. (b) Scatter plots of the effect of DAMGO, U69593, and deltorphin‐II on the amplitude of evoked EPSCs in retro‐labelled and unlabelled PAG neurons from animals that received an intra‐RVM tracer injection and in blind (Control) PAG neurons from animals that did not receive a tracer injection; data expressed as a percentage of the pre‐agonist value. (c) Averaged traces of paired evoked EPSCs from the neuron in (a). (d) Scatter plots of the effect of DAMGO, U69593, and deltorphin‐II on the paired‐pulse ratio of evoked EPSCs in retro‐labelled, unlabelled, and control neurons, as in (b). (e) Summary bar charts comparing the effect of DAMGO and U69 on evoked IPSCs and EPSCs in retro‐labelled and unlabelled neurons. In (b,d,e), *P < .05 (paired Student's t‐tests, pre‐ vs. post‐drug); # P < .05, post hoc comparisons. In (a,c), stimulus artefacts have been blanked for clarity [Colour figure can be viewed at http://wileyonlinelibrary.com]
We also examined the effect of the opioid agonists on evoked EPSCs in unlabelled neurons from tracer‐injected animals and control blind neurons from animals that did not receive tracer injections. The effect of DAMGO on evoked EPSCs differed between retro‐labelled, unlabelled, and control blind neurons (Figure 5b, n = 9, 10 for unlabelled and control blind neurons). Thus, the DAMGO‐induced inhibition of evoked EPSCs was less in retro‐labelled neurons compared to either unlabelled or control blind neurons (Figure 5b). The effect of deltorphin‐II and U69593 on evoked EPSCs did not differ between the three neuronal groups (Figure 5b, n = 7, 8 for deltorphin‐II and n = 9, 9 for U69593 in unlabelled and control blind neurons). This indicates that while both μ‐ and κ‐opioids inhibit glutamatergic transmission onto all PAG neuronal types, these inputs onto retro‐labelled PAG neurons are less sensitive to μ‐opioids than those onto unlabelled and control blind neurons.
DAMGO and U69593, but not deltorphin, produced a significant increase in the paired‐pulse ratio of evoked EPSCs in retro‐labelled PAG neurons (Figure 5c,d). Similarly, DAMGO and U69593, but not deltorphin‐II, produced a significant increase in the paired‐pulse ratio of evoked EPSCs in unlabelled and blind control neurons (Figure 5c,d). The effect of the three agonists on the paired‐pulse ratio did not differ between retro‐labelled, unlabelled and control blind PAG neurons. This indicates that both μ‐ and κ‐opioids inhibit glutamatergic inputs onto all PAG neurons via a presynaptic mechanism.
We finally made a comparison of the effect of μ‐ and κ‐opioids on evoked IPSCs and EPSCs in neurons from tracer‐injected animals. The DAMGO‐induced significant inhibition of evoked IPSCs and EPSCs significantly differed between retro‐labelled and unlabelled PAG neurons. The effect of DAMGO on evoked IPSCs was significantly greater than that for evoked EPSCs in retro‐labelled, but not unlabelled neurons (Figure 5e,). By contrast, the effect of U69593 on evoked IPSCs and EPSCs did not differ between retro‐labelled and unlabelled neurons (Figure 5e).
3.5. The net functional effects of μ‐opioids differ between PAG‐RVM projection neurons and random PAG neurons
In addition to the above presynaptic inhibitory actions, it has previously been shown that μ‐opioids but not δ‐ and κ‐opioids postsynaptically inhibit specific subpopulations of PAG neurons (Chieng & Christie, 1994a; Osborne et al., 1996; Vaughan et al., 2003). To determine the overall consequences of the differential presynaptic and postsynaptic μ‐opioid actions, we compared the net effect of DAMGO on retro‐labelled and unlabelled PAG neurons from tracer‐injected animals, under current‐clamp conditions and with blocker‐free ACSF. We first examined the effect of DAMGO on action potential firing by holding neurons within 5 mV of the threshold for their generation. In retro‐labelled neurons, electrical stimulation rarely elicited actions potentials under basal conditions (Figure 6a, n = 8). In most of these neurons, DAMGO had no effect on the basal membrane potential but led to the generation of action potential firing in response to electrical stimulation (Figure 6a). By contrast, in unlabelled neurons, electrical stimulation often elicited action potential firing under basal conditions (Figure 6b, n = 9). In these neurons, DAMGO produced a hyperpolarization and the cessation of stimulation evoked action potential firing (Figure 6b). These effects of DAMGO were reversed by https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1638, or CTAP (Figure 6a,b).
Figure 6.

μ‐Opioids disinhibit retrogradely labelled neurons but directly inhibit unlabelled neurons. Representative continuous traces of action potential firing in (a) retro‐labelled and (b) unlabelled PAG neuron during superfusion of DAMGO (3 μM) and naloxone (1 μM); current‐clamp recordings were performed in blocker‐free ACSF, and electrical stimulation was applied every 12 s. In (a) and (b), insets show representative traces during electrical stimulation (arrows), before and during DAMGO. PSPs were evoked every 12 s; action potentials have been clipped to show details of the membrane potential. (c–d) Single traces of evoked PSPs in retro‐labelled and unlabelled PAG neurons before (Pre) and during superfusion of DAMGO (3 μM). (e–f) Similar traces of evoked PSPs, but in the presence of gabazine (GBZ, 3 μM). (g) Scatter plots of the baseline evoked PSP amplitude (ePSP) in retro‐labelled and unlabelled neurons, in the absence and presence of gabazine. (h) Scatter plots of the effect of DAMGO on the membrane potential (Vm), the evoked PSP amplitude (ePSP), and the combined effect on Vm + ePSP in retro‐labelled and unlabelled PAG neurons. In (c–f), arrows denote the combined shift in Vm plus ePSP amplitude with DAMGO. In (g,h), *P < .05 (paired Student's t‐test, pre‐ vs. post‐drug); # P < .05 (post hoc comparisons). In (a,b,c,f), stimulus artefacts have been blanked for clarity [Colour figure can be viewed at http://wileyonlinelibrary.com]
We further characterized the effect of DAMGO on evoked PSPs and membrane potential by holding neurons 15 mV below the threshold for action potential firing to avoid their interference with the measurement of these parameters. These experiments were performed in blocker‐free ACSF (n = 15, 14 for retro‐labelled and unlabelled neurons respectively) and then in gabazine to abolish the influence of presynaptic GABAA‐mediated inputs (n = 11 for retro‐labelled and unlabelled neurons). The effect of DAMGO on membrane potential significantly differed between retro‐labelled and unlabelled PAG neurons, although this did not vary between control untreated and gabazine treated slices. In untreated slices, DAMGO produced a membrane hyperpolarization in a greater proportion of unlabelled neurons compared to retro‐labelled neurons (Figure 6c,d). Thus, DAMGO produced a significant membrane hyperpolarization in unlabelled but not retro‐labelled neurons (Figure 6h). Similarly, in the presence of gabazine, DAMGO produced a significant membrane hyperpolarization in unlabelled but not in retro‐labelled neurons (Figure 6e,f,h). This suggests that DAMGO produces a membrane hyperpolarization mainly in unlabelled neurons that is independent of presynaptic GABAergic inputs.
Prior to application of DAMGO, the amplitude of evoked PSPs was significantly more hyperpolarizing in retro‐labelled compared to unlabelled neurons in control untreated slices (Figure 6c,d,g, see also inset Figure 6a). Furthermore, the pre‐DAMGO amplitude of evoked PSPs in retro‐labelled neurons was more depolarizing in gabazine compared to control untreated slices (Figure 6c,e,g). The effect of DAMGO on evoked PSPs differed between retro‐labelled and unlabelled PAG neurons, and this varied significantly between control untreated and gabazine treated slices. In untreated slices, DAMGO produced a depolarizing shift in evoked PSPs in retro‐labelled neurons, but a hyperpolarizing shift in unlabelled neurons (Figure 6c,d,h). By contrast, DAMGO produced a hyperpolarizing shift in evoked PSPs in both retro‐labelled and unlabelled neurons from slices that were pretreated with gabazine (Figure 6e,f,h). This suggests that DAMGO produces a depolarizing shift in evoked PSPs in retro‐labelled neurons that is mediated by suppression of GABAergic synaptic inputs.
Finally, the effect of DAMGO on the combination of membrane potential and evoked PSP amplitude differed between retro‐labelled and unlabelled neurons, and this varied significantly between control untreated and gabazine treated slices. In untreated slices, DAMGO produced a depolarizing shift in the combined membrane potential and evoked PSP in retro‐labelled neurons, but a hyperpolarizing shift in unlabelled neurons (Figure 6c,d,h) By contrast, DAMGO produced a hyperpolarizing shift in the combined membrane potential and evoked PSP in both retro‐labelled and unlabelled neurons from gabazine pretreated slices (Figure 6e,f,h). Taken together, the above results indicate that while DAMGO produces a net depolarization in retro‐labelled neurons and this is dependent upon GABAergic inputs, it directly hyperpolarizes unlabelled neurons.
4. DISCUSSION
Here, we have demonstrated that opioids control a descending analgesic pathway from the midbrain PAG by acting on intrinsic systems that indirectly control its output neurons, which project to the RVM. Specifically, opioids acted via μ‐ and κ‐opioid, but not δ‐opioid, receptors to presynaptically inhibit GABAergic and glutamatergic synaptic inputs onto all PAG neurons, including those that projected to the RVM. However, μ‐opioids inhibited GABAergic synaptic inputs onto PAG projection neurons to a greater extent than their glutamatergic inputs. This differential control of synaptic transmission was not observed for non‐projection neurons, nor was it observed for κ‐opioids in projection and non‐projection neurons. Functionally, the net effect of μ‐opioids was presynaptic disinhibition of evoked synaptic potentials in RVM‐projecting neurons and direct postsynaptic hyperpolarization of putative interneurons within the PAG.
In the present study, it was demonstrated that μ‐ and κ‐opioid, but not δ‐opioid receptor activation inhibited GABAergic synaptic transmission onto PAG neurons identified as projecting to the RVM. In these experiments, evoked GABAA‐mediated IPSCs were readily observed in PAG neurons retrogradely labelled from the RVM, indicating these projection neurons are under inhibitory GABAergic control. GABAergic IPSCs on RVM‐projecting neurons were inhibited by the μ‐ and κ‐opioid agonists, DAMGO and U69593, and this inhibition was reversed by the selective antagonists, CTAP and nor‐BNI. This provides direct evidence that μ‐opioid receptor activation disinhibits PAG projection neurons and complements prior studies on descending projection neurons in other components of the descending analgesic pathway, including the amygdala and RVM (Finnegan, Chen, & Pan, 2005; Finnegan, Li, Chen, & Pan, 2004). Interestingly, however, the observed κ‐opioid disinhibition of PAG projection neurons differed to amygdala and RVM projections where κ‐opioid disinhibition has not been observed (Finnegan et al., 2004, 2005). While the observed κ‐opioid inhibition was similar to that previously observed in unidentified mouse PAG neurons (Vaughan et al., 2003), it differed to prior rat PAG studies where κ‐opioid receptor mediated inhibition of evoked IPSCs has not been observed (Chieng & Christie, 1994b; Vaughan & Christie, 1997). The discrepancy to prior rat studies was unlikely to be due to tracer injection surgery because a similar level of κ‐opioid inhibition was also observed in unidentified PAG neurons from operated and unoperated animals. The difference may therefore have been due to the use of a less selective κ‐opioid agonist U50488H and/or the use of younger pre‐weaned animals in prior studies.
The μ‐ and κ‐opioid inhibition of GABAergic transmission onto RVM‐projecting neurons was likely to be mediated by a presynaptic mechanism. Firstly, the DAMGO‐ and U69593‐induced inhibition of evoked IPSCs was associated with an increase in their paired‐pulse ratio. Secondly, DAMGO and U69593 both produced a reduction in the rate, but not the amplitude of spontaneous TTX‐resistant miniature IPSCs, as observed previously for unidentified PAG neurons (Vaughan & Christie, 1997; Vaughan, Ingram, Connor, & Christie, 1997). We also examined asynchronous evoked miniature IPSCs, using Sr2+ as a less efficient substitute for Ca2+ (Goda & Stevens, 1994). The latter observation was important because the actions of opioids on spontaneous miniature IPSCs and evoked IPSCs in PAG might be mediated by different mechanisms (Vaughan et al., 1997; Vaughan & Christie, 1997). In these experiments, both μ‐ and κ‐opioids produced a reduction in the rate, but not the amplitude of asynchronous evoked miniature IPSCs in projection neurons. Overall, these findings indicate that μ‐ and κ‐opioid disinhibition of descending PAG projection neurons involves presynaptic inhibition of transmitter release from GABAergic nerve terminals. It should be noted that the source of GABAergic synaptic inputs could not be resolved in the current electrical stimulation experiments because of the heterogeneous structure of the PAG. While the midbrain PAG contains a substantial population of GABAergic neurons (Barbaresi & Manfrini, 1988; Park et al., 2010; Reichling & Basbaum, 1990a, 1990b), it also receives extrinsic GABAergic inputs from other regions such as the amygdala and ventral tegmental area (Day, Curran, Watson, & Akil, 1999; Gray & Magnuson, 1992; Kirouac, Li, & Mabrouk, 2004; Tovote et al., 2016). Thus, the relative role of opioid presynaptic modulation of intrinsic and extrinsic GABAergic inputs remains to be resolved.
While μ‐opioid inhibition of GABAergic transmission onto projection neurons would be expected to disinhibit PAG neurons that project to the RVM, there were other complicating presynaptic actions. It was observed that μ‐opioids also inhibited evoked IPSCs in unlabelled PAG neurons from tracer‐injected animals. However, the DAMGO inhibition of evoked IPSCs was less in these unlabelled neurons than in retro‐labelled neurons. This suggests that μ‐opioids will have a lesser disinhibitory effect on unlabelled PAG neurons compared to RVM‐projecting neurons. It might be postulated that a substantial proportion of the unlabelled neurons observed in the present study were GABAergic interneurons. This is because the vast majority of PAG interneurons are GABAergic (Barbaresi & Manfrini, 1988; Reichling & Basbaum, 1990a). Furthermore, only a small percentage (<2%) of PAG neurons project to other brain regions such as the RVM (Reichling & Basbaum, 1990a), although an absence of cell body tracer cannot rule out projections to large structures such as the RVM, or other PAG targets not examined in the present study. Finally, it might be noted that a recent optogenetic study demonstrated that medullary magnocellular‐projection neurons, which are involved in motor control, receive monosynaptic GABAergic inputs from PAG interneurons but not extrinsic descending GABAergic inputs from the amygdala (Tovote et al., 2016). It is therefore possible that μ‐opioid disinhibition of RVM‐projecting neurons is mediated specifically by intrinsic inputs from GABAergic interneurons, although this would have to be confirmed using a similar optogenetic approach.
The other complication was that μ‐opioids also inhibited evoked glutamatergic EPSCs in RVM‐projecting PAG neurons, as observed in prior studies on unidentified neurons (Chieng & Christie, 1994b; Chiou & Huang, 1999; Vaughan & Christie, 1997). This μ‐opioid inhibition of excitatory glutamatergic inputs onto RVM‐projecting neurons would be expected to functionally oppose GABAergic disinhibition (Samineni et al., 2017). However, it was observed that μ‐opioids inhibited glutamatergic synaptic inputs onto RVM‐projecting neurons to a lesser extent than their GABAergic inputs. This has some parallels to the amygdala and RVM, where glutamatergic inputs onto descending projection neurons display only partial, or no μ‐opioid, presynaptic sensitivity (Finnegan et al., 2004, 2005). Importantly, the differential inhibition of evoked IPSCs and EPSCs observed in retro‐labelled neurons did not extend to unlabelled neurons. These observations suggest that μ‐opioid disinhibition predominates in RVM‐projecting neurons, leading to activation of the descending analgesic pathway. It might be noted that there was no difference between the presynaptic κ‐opioid modulation of inhibitory and excitatory synaptic inputs onto retro‐labelled and unlabelled neurons, suggesting that they would be unlikely to have a net presynaptic effect on either type of PAG neuron.
While the voltage‐clamp experiments provide a cellular basis for μ‐opioid presynaptic disinhibition of PAG output neurons, μ‐opioids also have postsynaptic actions within this brain region (Chieng & Christie, 1994a; Chiou & Huang, 1999; Vaughan et al., 2003). We therefore examined the net effect of DAMGO in current‐clamp mode, using internal and external solutions that sustained postsynaptic G‐protein coupling, and enabled the simultaneous detection of inhibitory GABAergic and excitatory glutamatergic PSPs (Chiou & Huang, 1999). In these experiments, it was observed that DAMGO had no direct postsynaptic effect on most projection neurons within the ventrolateral PAG but directly hyperpolarized the majority of unlabelled neurons which is consistent with a prior study (Osborne et al., 1996). In addition, these postsynaptic effects were consistent with the original GABA disinhibition model, which proposed that opioids activate descending analgesic systems by directly inhibiting GABAergic interneurons, thereby suppressing the release of GABA onto descending projection neurons (Fields & Basbaum, 1978; Lau & Vaughan, 2014). Interestingly, it was also observed that DAMGO produced an excitatory depolarizing shift in evoked PSPs in RVM‐projecting neurons, but not in unlabelled neurons. This selective presynaptic disinhibition of RVM‐projecting neurons was likely to be due to a number of factors. Firstly, there was a greater inhibitory GABAergic drive onto RVM‐projecting neurons compared to unlabelled neurons. Secondly, GABAergic inputs onto these projection neurons displayed the greatest μ‐opioid sensitivity, as described above for the voltage‐clamp experiments. Thus, the current findings provide a complementary presynaptic mechanism of disinhibition. Together, the differential presynaptic and postsynaptic μ‐opioid actions led to an increase in action potential firing in PAG descending projection neurons and a cessation of action potential firing in unlabelled, putative PAG interneurons.
The present findings suggest that opioid activation of the descending analgesic pathway from the midbrain PAG is indirectly mediated by intrinsic GABAergic systems. In doing so, opioids act to shift the balance of PAG neuronal activity, such that there is net inhibition of the substantial population of intrinsic neurons and activation of the relatively small population of descending projection neurons. This has implications for global recording and imaging of brain regions involved in pain modulation (Knudsen et al., 2018), since the current findings suggest that opioid inhibition of the PAG at a gross level leads to activation of its descending analgesic output. Furthermore, the opioid disinhibition was pleiotropic, involving both postsynaptic inhibition of intrinsic PAG neurons and presynaptic inhibition of their outputs onto descending projection neurons. In this regard, presynaptic and postsynaptic opioid receptors couple via different intracellular pathways and effectors (Williams, Christie, & Manzoni, 2001). This is likely to have implications for the chronic use of opioids for pain management because presynaptic opioid receptors display different levels and mechanisms of desensitization compared to postsynaptic receptors (Pennock, Dicken, & Hentges, 2012; Pennock & Hentges, 2016; Wilson‐Poe, Jeong, & Vaughan, 2017). Thus, a better understanding of the interactions between presynaptic and postsynaptic opioid signalling in brain analgesic systems is required to improve opioid analgesics.
AUTHOR CONTRIBUTIONS
B.K.L. and C.W.V. devised the project; all authors performed and analysed the experiments; all authors wrote and approved the manuscript.
CONFLICT OF INTEREST
The authors have no conflicts of interest to declare.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for https://bpspubs.onlinelibrary.wiley.com/doi/full/10.1111/bph.14207, https://bpspubs.onlinelibrary.wiley.com/doi/full/10.1111/bph.14208, and https://bpspubs.onlinelibrary.wiley.com/doi/full/10.1111/bph.14206, and as recommended by funding agencies, publishers, and other organisations engaged with supporting research.
ACKNOWLEDGEMENTS
The authors thank Ms. Vanessa Mitchell for assistance with brain microinjections. This study was funded by the NHMRC Grant 1083569. B. Lau received an Australian Postgraduate Award.
Lau BK, Winters BL, Vaughan CW. Opioid presynaptic disinhibition of the midbrain periaqueductal grey descending analgesic pathway. Br J Pharmacol. 2020;177:2320–2332. 10.1111/bph.14982
REFERENCES
- Alexander, S. P. H. , Christopoulos, A. , Davenport, A. P. , Kelly, E. , Mathie, A. , Peters, J. A. , … Pawson, A. J. (2019). The Concise Guide to PHARMACOLOGY 2019/20: G protein‐coupled receptors. British Journal of Pharmacology, 176(Suppl 1), S21–S141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Mathie, A. , Peters, J. A. , Veale, E. L. , Striessnig, J. , Kelly, E. , … Sharman, J. L. (2019). The Concise Guide to PHARMACOLOGY 2019/20: Ion channels. British Journal of Pharmacology, 176(Suppl 1), S142–S228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Roberts, R. E. , Broughton, B. R. S. , Sobey, C. G. , George, C. H. , Stanford, S. C. , … Ahluwalia, A. (2018). Goals and practicalities of immunoblotting and immunohistochemistry: A guide for submission to the British Journal of Pharmacology. British Journal of Pharmacology, 175, 407–411. 10.1111/bph.14112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbaresi, P. , & Manfrini, E. (1988). Glutamate decarboxylase‐immunoreactive neurons and terminals in the periaqueductal gray of the rat. Neuroscience, 27, 183–191. 10.1016/0306-4522(88)90229-1 [DOI] [PubMed] [Google Scholar]
- Chieng, B. , & Christie, M. J. (1994a). Hyperpolarization by opioids acting on μ‐receptors of a sub‐population of rat periaqueductal gray neurones in vitro. British Journal of Pharmacology, 113, 121–128. 10.1111/j.1476-5381.1994.tb16183.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chieng, B. , & Christie, M. J. (1994b). Inhibition by opioids acting on μ‐receptors of GABAergic and glutamatergic postsynaptic potentials in single rat periaqueductal gray neurones in vitro. British Journal of Pharmacology, 113, 303–309. 10.1111/j.1476-5381.1994.tb16209.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiou, L. C. , & Huang, L. Y. (1999). Mechanism underlying increased neuronal activity in the rat ventrolateral periaqueductal grey by a μ‐opioid. Journal of Physiology (London), 518, 551–559. 10.1111/j.1469-7793.1999.0551p.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleary, D. R. , Neubert, M. J. , & Heinricher, M. M. (2008). Are opioid‐sensitive neurons in the rostral ventromedial medulla inhibitory interneurons? Neuroscience, 151, 564–571. 10.1016/j.neuroscience.2007.10.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Commons, K. G. , Aicher, S. A. , Kow, L. M. , & Pfaff, D. W. (2000). Presynaptic and postsynaptic relations of μ‐opioid receptors to γ‐aminobutyric acid‐immunoreactive and medullary‐projecting periaqueductal gray neurons. The Journal of Comparative Neurology, 419, 532–542. [DOI] [PubMed] [Google Scholar]
- Curtis, M. J. , Alexander, S. , Cirino, G. , Docherty, J. R. , George, C. H. , Giembycz, M. A. , … Ahluwalia, A. (2018). Experimental design and analysis and their reporting II: updated and simplified guidance for authors and peer reviewers. British Journal of Pharmacology, 175, 987–993. 10.1111/bph.14153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day, H. E. , Curran, E. J. , Watson, S. J. Jr. , & Akil, H. (1999). Distinct neurochemical populations in the rat central nucleus of the amygdala and bed nucleus of the stria terminalis: evidence for their selective activation by interleukin‐1β. The Journal of Comparative Neurology, 413, 113–128. [DOI] [PubMed] [Google Scholar]
- Depaulis, A. , Morgan, M. M. , & Liebeskind, J. C. (1987). GABAergic modulation of the analgesic effects of morphine microinjected in the ventral periaqueductal gray matter of the rat. Brain Research, 436, 223–228. 10.1016/0006-8993(87)91665-9 [DOI] [PubMed] [Google Scholar]
- Drew, G. M. , Lau, B. K. , & Vaughan, C. W. (2009). Substance P drives endocannabinoid‐mediated disinhibition in a midbrain descending analgesic pathway. The Journal of Neuroscience, 29, 7220–7229. 10.1523/JNEUROSCI.4362-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields, H. L. , & Basbaum, A. I. (1978). Brainstem control of spinal pain‐transmission neurons. Annual Review of Physiology, 40, 217–248. 10.1146/annurev.ph.40.030178.001245 [DOI] [PubMed] [Google Scholar]
- Finnegan, T. F. , Chen, S. R. , & Pan, H. L. (2005). Effect of the μ‐opioid on excitatory and inhibitory synaptic inputs to periaqueductal gray‐projecting neurons in the amygdala. The Journal of Pharmacology and Experimental Therapeutics, 312, 441–448. 10.1124/jpet.104.074633 [DOI] [PubMed] [Google Scholar]
- Finnegan, T. F. , Li, D. P. , Chen, S. R. , & Pan, H. L. (2004). Activation of μ‐opioid receptors inhibits synaptic inputs to spinally projecting rostral ventromedial medulla neurons. The Journal of Pharmacology and Experimental Therapeutics, 309, 476–483. 10.1124/jpet.103.064808 [DOI] [PubMed] [Google Scholar]
- Goda, Y. , & Stevens, C. F. (1994). Two components of transmitter release at a central synapse. Proceedings of the National Academy of Sciences of the United States of America, 91, 12942–12946. 10.1073/pnas.91.26.12942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray, T. S. , & Magnuson, D. J. (1992). Peptide immunoreactive neurons in the amygdala and the bed nucleus of the stria terminalis project to the midbrain central gray in the rat. Peptides, 13, 451–460. 10.1016/0196-9781(92)90074-d [DOI] [PubMed] [Google Scholar]
- Gu, M. , & Wessendorf, M. (2007). Endomorphin‐2‐immunoreactive fibers selectively appose serotonergic neuronal somata in the rostral ventral medial medulla. The Journal of Comparative Neurology, 502, 701–713. 10.1002/cne.21343 [DOI] [PubMed] [Google Scholar]
- Gutstein, H. B. , Mansour, A. , Watson, S. J. , Akil, H. , & Fields, H. L. (1998). Mu and kappa opioid receptors in periaqueductal gray and rostral ventromedial medulla. Neuroreport, 9, 1777–1781. 10.1097/00001756-199806010-00019 [DOI] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Research, 46, D1091–D1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalyuzhny, A. E. , & Wessendorf, M. W. (1998). Relationship of μ‐ and δ‐opioid receptors to GABAergic neurons in the central nervous system, including antinociceptive brainstem circuits. The Journal of Comparative Neurology, 392, 528–547. [PubMed] [Google Scholar]
- Kilkenny, C. , Browne, W. , Cuthill, I. C. , Emerson, M. , & Altman, D. G. (2010). Animal research: Reporting in vivo experiments: The ARRIVE guidelines. British Journal of Pharmacology, 160, 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirouac, G. J. , Li, S. , & Mabrouk, G. (2004). GABAergic projection from the ventral tegmental area and substantia nigra to the periaqueductal gray region and the dorsal raphe nucleus. The Journal of Comparative Neurology, 469, 170–184. 10.1002/cne.11005 [DOI] [PubMed] [Google Scholar]
- Knudsen, L. , Petersen, G. L. , Norskov, K. N. , Vase, L. , Finnerup, N. , Jensen, T. S. , & Svensson, P. (2018). Review of neuroimaging studies related to pain modulation. Scandinavian Journal of Pain, 2, 108–120. 10.1016/j.sjpain.2011.05.005 [DOI] [PubMed] [Google Scholar]
- Lane, D. A. , Patel, P. A. , & Morgan, M. M. (2005). Evidence for an intrinsic mechanism of antinociceptive tolerance within the ventrolateral periaqueductal gray of rats. Neuroscience, 135, 227–234. 10.1016/j.neuroscience.2005.06.014 [DOI] [PubMed] [Google Scholar]
- Lau, B. K. , & Vaughan, C. W. (2014). Descending modulation of pain: the GABA disinhibition hypothesis of analgesia. Current Opinion in Neurobiology, 29, 159–164. 10.1016/j.conb.2014.07.010 [DOI] [PubMed] [Google Scholar]
- Mansour, A. , Khachaturian, H. , Lewis, M. E. , Akil, H. , & Watson, S. J. (1987). Autoradiographic differentiation of μ, δ, and κ opioid receptors in the rat forebrain and midbrain. The Journal of Neuroscience, 7, 2445–2464. [PMC free article] [PubMed] [Google Scholar]
- Moreau, J. L. , & Fields, H. L. (1986). Evidence for GABA involvement in midbrain control of medullary neurons that modulate nociceptive transmission. Brain Research, 397, 37–46. 10.1016/0006-8993(86)91367-3 [DOI] [PubMed] [Google Scholar]
- Osborne, P. B. , Vaughan, C. W. , Wilson, H. I. , & Christie, M. J. (1996). Opioid inhibition of rat periaqueductal grey neurones with identified projections to rostral ventromedial medulla in vitro. Journal of Physiology (London), 490, 383–389. 10.1113/jphysiol.1996.sp021152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossipov, M. H. , Kovelowski, C. J. , Nichols, M. L. , Hruby, V. J. , & Porreca, F. (1995). Characterization of supraspinal antinociceptive actions of opioid δ agonists in the rat. Pain, 62, 287–293. 10.1016/0304-3959(94)00231-3 [DOI] [PubMed] [Google Scholar]
- Park, C. , Kim, J. H. , Yoon, B. E. , Choi, E. J. , Lee, C. J. , & Shin, H. S. (2010). T‐type channels control the opioidergic descending analgesia at the low threshold‐spiking GABAergic neurons in the periaqueductal gray. Proceedings of the National Academy of Sciences of the United States of America, 107, 14857–14862. 10.1073/pnas.1009532107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennock, R. L. , Dicken, M. S. , & Hentges, S. T. (2012). Multiple inhibitory G‐protein‐coupled receptors resist acute desensitization in the presynaptic but not postsynaptic compartments of neurons. The Journal of Neuroscience, 32, 10192–10200. 10.1523/JNEUROSCI.1227-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennock, R. L. , & Hentges, S. T. (2016). Desensitization‐resistant and ‐sensitive GPCR‐mediated inhibition of GABA release occurs by Ca2+‐dependent and ‐independent mechanisms at a hypothalamic synapse. Journal of Neurophysiology, 115, 2376–2388. 10.1152/jn.00535.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichling, D. B. , & Basbaum, A. I. (1990a). Contribution of brainstem GABAergic circuitry to descending antinociceptive controls: I. GABA‐immunoreactive projection neurons in the periaqueductal gray and nucleus raphe magnus. The Journal of Comparative Neurology, 302, 370–377. 10.1002/cne.903020213 [DOI] [PubMed] [Google Scholar]
- Reichling, D. B. , & Basbaum, A. I. (1990b). Contribution of brainstem GABAergic circuitry to descending antinociceptive controls: II. Electron microscopic immunocytochemical evidence of GABAergic control over the projection from the periaqueductal gray to the nucleus raphe magnus in the rat. The Journal of Comparative Neurology, 302, 378–393. 10.1002/cne.903020214 [DOI] [PubMed] [Google Scholar]
- Rossi, G. C. , Pasternak, G. W. , & Bodnar, R. J. (1994). μ and δ opioid synergy between the periaqueductal gray and the rostro‐ventral medulla. Brain Research, 665, 85–93. 10.1016/0006-8993(94)91155-X [DOI] [PubMed] [Google Scholar]
- Samineni, V. K. , Grajales‐Reyes, J. G. , Copits, B. A. , O'Brien, D. E. , Trigg, S. L. , Gomez, A. M. , … Gereau, R. W. IV (2017). Divergent modulation of nociception by glutamatergic and GABAergic neuronal subpopulations in the periaqueductal gray. eNeuro, 4(2). 1‐13 10.1523/ENEURO.0129-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, D. J. , Perrotti, J. M. , Crisp, T. , Cabral, M. E. , Long, J. T. , & Scalzitti, J. M. (1988). The mu opiate receptor is responsible for descending pain inhibition originating in the periaqueductal gray region of the rat brain. European Journal of Pharmacology, 156, 47–54. 10.1016/0014-2999(88)90145-8 [DOI] [PubMed] [Google Scholar]
- Tovote, P. , Esposito, M. S. , Botta, P. , Chaudun, F. , Fadok, J. P. , Markovic, M. , … Lüthi, A. (2016). Midbrain circuits for defensive behaviour. Nature, 534, 206–212. 10.1038/nature17996 [DOI] [PubMed] [Google Scholar]
- Vaughan, C. W. , Bagley, E. E. , Drew, G. M. , Schuller, A. , Pintar, J. E. , Hack, S. P. , & Christie, M. D. J. (2003). Cellular actions of opioids on periaqueductal grey neurons from C57B16/J mice and mutant mice lacking MOR‐1. British Journal of Pharmacology, 139, 362–367. 10.1038/sj.bjp.0705261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan, C. W. , & Christie, M. J. (1997). Presynaptic inhibitory action of opioids on synaptic transmission in the rat periaqueductal grey in vitro. Journal of Physiology (London), 498, 463–472. 10.1113/jphysiol.1997.sp021872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan, C. W. , Ingram, S. L. , Connor, M. , & Christie, M. J. (1997). How opioids inhibit GABA mediated neurotransmission. Nature, 390, 611–615. 10.1038/37610 [DOI] [PubMed] [Google Scholar]
- Wang, H. , & Wessendorf, M. W. (2002). μ‐ and δ‐opioid receptor mRNAs are expressed in periaqueductal gray neurons projecting to the rostral ventromedial medulla. Neuroscience, 109, 619–634. 10.1016/s0306-4522(01)00328-1 [DOI] [PubMed] [Google Scholar]
- Williams, J. T. , Christie, M. J. , & Manzoni, O. (2001). Cellular and synaptic adaptations mediating opioid dependence. Physiological Reviews, 81, 299–343. 10.1152/physrev.2001.81.1.299 [DOI] [PubMed] [Google Scholar]
- Wilson‐Poe, A. R. , Jeong, H. J. , & Vaughan, C. W. (2017). Chronic morphine reduces the readily releasable pool of GABA, a presynaptic mechanism of opioid tolerance. Journal of Physiology (London), 595, 6541–6555. 10.1113/JP274157 [DOI] [PMC free article] [PubMed] [Google Scholar]