

Abstract
Detailed mapping of genetic and environmental influences on the functional connectome is a crucial step toward developing intermediate phenotypes between genes and clinical diagnoses or cognitive abilities. We analyzed resting-state functional magnetic resonance imaging data from two adult twin samples (Nos = 446 and 371) to quantify genetic and environmental influence on all pairwise functional connections between 264 brain regions (~35 000 functional connections). Nonshared environmental influence was high across the whole connectome. Approximately 14–22% of connections had nominally significant genetic influence in each sample, 4.6% were significant in both samples, and 1–2% had heritability estimates greater than 30%. Evidence of shared environmental influence was weak. Genetic influences on connections were distinct from genetic influences on a global summary measure of the connectome, network-based estimates of connectivity, and movement during the resting-state scan, as revealed by a novel connectome-wide bivariate genetic modeling procedure. The brain’s genetic organization is diverse and not as one would expect based solely on structure evident in nongenetically informative data or lower resolution data. As follow-up, we make novel classifications of functional connections and examine highly localized connections with particularly strong genetic influence. This high-resolution genetic taxonomy of brain connectivity will be useful in understanding genetic influences on brain disorders.
Keywords: fMRI, functional connectivity, genetic neuroimaging, resting-state connectivity, twin study
Introduction
The functional connectome refers to intrinsically correlated activity between brain regions when individuals are not engaged in a particular task (i.e., measured during the “resting state”; Fox and Raichle 2007). Patterns within the functional connectome are associated with clinical diagnoses (for reviews, see Greicius 2008; Zhang and Raichle 2010) and individual differences in cognitive abilities (for a broad review of 125 studies, see Vaidya and Gordon 2013). Recent work has showcased reliable and generalizable predictive models of individual differences in behavior that utilize many measurements of the connectome as features (Finn et al. 2015; Rosenberg et al. 2016). These patterns of connectivity may be candidate intermediate phenotypes between genes and traits (i.e., endophenotypes; Hall and Smoller 2010; Kendler and Neale 2010), if genetic influences exist. However, prior work has only quantified genetic and environmental influences on the connectome at the level of large regions/networks of interest. In the current study, we conduct a highly detailed analysis of the etiology of functional brain connections and find a high degree of diversity in genetic influence across the connectome.
Genetic analyses of functional connections could span several units of analysis: from connections between a small number of large networks with correlated activity (Yeo et al. 2011) and related function (Smith et al. 2009) to connections between nearly a million individual voxels. Due to the computational power needed to perform classic twin models at the level of voxels or small regions, current efforts have focused on quantifying genetic and environmental influence on global summary measures of functional connectivity, resting-state networks (i.e., large and spatially separated groups of regions that are all moderately correlated at rest and thus appropriate to model as a single unit), or large regions of interest (ROIs). At the coarsest level of detail, several studies have revealed moderate heritability (i.e., “h2” or the proportion of phenotypic variance explained by genetic variance; h2 = 0.43–0.64) of the degree to which an individual’s connectome is globally efficient (i.e., maximizes information transfer while reducing long path lengths and unnecessary connections; Fornito et al. 2011; van den Heuvel et al. 2013; Sinclair et al. 2015). However, while global efficiency may be an informative phenotype, it may not be a thorough summary of the connectome, possibly summarizing only connections amongst the brains’ densely connected and metabolically costly hub regions (Heuvel et al. 2012).
At the level of networks, there was a substantial early interest in the genetics of the default network, a set of regions implicated in internal mentation functions (Andrews-Hanna 2012), perhaps due to its involvement in a variety of clinical disorders including schizophrenia, depression, and attention-deficit/hyperactivity disorder (Whitfield-Gabrieli et al. 2009; Anticevic et al. 2012; Whitfield-Gabrieli and Ford 2012; Mattfeld et al. 2014; Kaiser et al. 2016). The default network is moderately heritable as a whole (h2 = 0.42), while connectivity of its subcomponents are weakly-to-moderately heritable (h2 = 0.10–0.42; Glahn et al. 2010). Other work has reported moderate heritability of a precuneus-dorsal posterior cingulate network, visual network, default network, frontoparietal (FP) network, and dorsal attention (DA) network (h2 = 0.23–0.65), nonsignificant heritability for the salience and sensory-somatomotor networks, and evidence of environmental effects on functional connectivity between all networks (Yang et al. 2016). Finally, a recent study investigated the genetic etiology of functional connections among seven networks and pairwise connections between 51 brain areas, finding moderate-to-strong heritability of seven networks (h2 = ~0.60 to ~0.75) using a linear-mixed-effects-model approach to account for unreliability across multiple resting-state scans (Ge et al. 2017). At the level of the 51-region parcellation, the authors found that heritability estimates for components of some networks, such as the default network, were consistent, but found evidence of heterogeneity for regions of other networks, such as the limbic and cognitive control networks. In summary, existing studies have provided heritability estimates for functional connectivity at global, network, or large ROI levels of analysis.
Although coarser levels of analysis are undoubtedly informative, they are not without caveats. First, large networks have only vague overarching functional labels (such as “vision”) as opposed to distinct functional labels ascribed to regions of finely detailed parcellations (e.g., those that contain 200–500 regions). Second, the anatomical literature indicates heritability may be overestimated for larger versus smaller pieces of cortex (Eyler et al. 2012). Third, examining heritability at the network level assumes that areas within the networks are homogeneous in terms of their genetic connections to areas in other networks. Fourth, individual differences in within-network connectivity cannot be examined, and these individual differences may have important implications for behavior (i.e., as contributors to connectivity-based predictive models or “fingerprints” of cognitive processes (Rosenberg et al. 2016) or psychopathology Elliott et al. (2018)). Finally, a recent attempt to quantify the utility of parcellations of varying granularity for connectivity-based predictive modeling has shown that fine-grained parcellations lead to higher predictive accuracy Li and Atluri (2018).
Important questions that can be examined at a finer level of analysis are (1) Do within- and between-network connections show similar levels of genetic and environmental influences? (2) Are regions of a particular resting-state network homogeneous in terms of their pattern of genetic and environmental influences across all connections? (3) What are useful applications of high-resolution genetic mapping? For example, can differences between regions’ patterns of genetic connectivity be used to generate hypotheses about or explain differences in their functions? Answering these questions speaks to recent efforts to “carve nature at its joints” and thus has important implications for how we conceptualize resting-state connectivity as a biomarker or candidate endophenotype for behaviors of interest.
To answer these questions, we analyzed resting-state data from two comparably aged adult twin samples: the Colorado Longitudinal Twin Study (LTS; N = 446, including 94 complete monozygotic [MZ], 84 complete same-sex dizygotic [DZ] pairs, and 79 singletons) and the Human Connectome Project (HCP; N = 371, including 89 complete MZ and 79 complete same-sex and opposite-sex DZ pairs). The inclusion of both samples allowed us to examine replicability of general patterns as the main criterion for significance rather than relying on a classic correction for multiple tests in a single, modestly sized (for genetic analysis) sample, which may lead to many false negatives given the limited power in each sample to detect significance at a threshold stringent enough to correct for over 34 000 tests. We decomposed the functional connectome of each individual into pairwise correlations between 264 individual regions (referred to as connections) from a widely used and independently derived brain parcellation (34 716 connections, Power et al. 2011). In comparison with coarser parcellations, this parcellation was developed to reflect functional distinctions between small parts of cortex (Wig et al. 2011), is accompanied by metadata assigning each region to one of 14 function-specific resting-state communities (e.g., visual network, default network, etc.), and is within a window of optimum dimensionality that maximizes reproducibility (Thirion et al. 2014).
We addressed our primary questions by applying a classic univariate twin model to each connection (see section Materials and Methods—Genetic Models) to estimate the proportion of variance in connection with strength explained by additive genetic influence (A or heritability; the sum of a large number of genetic variants that additively influence a trait), shared environmental influence (C; influences that increase sibling similarity), and nonshared environmental influence (E; influences that decrease sibling similarity; including measurement error). The resulting high-resolution genetic and environmental maps allowed us to investigate differences between within-network and between-network connections (question 1) and also investigate the distribution and patterns of genetic influence for regions of a priori resting-state networks (question 2).
Overall, we expect to find a distribution of connection heritability, such that some connections are not heritable while others have moderate heritability estimates, as is found in the network-based and large regional literature (i.e., Glahn et al. 2010; Ge et al. 2017). These results should provide a new degree of spatial specificity that builds on this important historical genetic imaging research and would provide a means, in future work, to discover novel classifications of brain areas beyond existing network parcellations or to generate hypothesis about discriminant functions of brain areas. We investigate these questions in the context of within- versus between-network connectivity but do not make specific predictions regarding the nature of those findings. Another hypothesis is based on a very large functional neuroimagaing literature linking specific brain areas to specific functions (e.g., discriminant functions of medial prefrontal and posterior cingulate regions within the default network). We expect local connections to have genetic influence that is separable from genetic influence on superordinate measures of connectivity (the default network as a whole). We expect the current study to elucidate differences in the genetic and environmental etiology of connections of different types/functions and demonstrate possible applications in a variety of domains.
Materials and Methods
The current study is a parallel analysis of resting-state data from a sample of adults recruited from the Colorado LTS and adults from a publicly available data set from the HCP.
Participant Details
Participants from the LTS sample were of 446 individuals (189 male; Mage = 28.7 years, SDage = 0.63 years, range = 28–32 years) after 31 participants were removed due to incidental anatomical findings, excessive movement during the scanning session based on the criteria of greater than 3-mm translation (motion in x, y, or z plane) or 3° rotation (roll, pitch, or yaw motion), or failure of the presentation on computer to display a fixation cross during the resting scan. Of the 446 individuals, there were 94 pairs of MZ twins, 84 pairs of DZ twins, 41 MZ twin singletons, and 49 DZ twin singletons. Singletons are members of twin pairs whose cotwins either did not participate or were excluded from analysis. Singletons were only informative as to connectivity means and variances. LTS twins were recruited from the Colorado Twin Registry based on birth records (see Rhea et al. 2006; Rhea et al. 2013 for additional information). Comparisons with normative data on several measures suggest that the sample is cognitively, academically, and demographically representative of the state of Colorado. Based on self-report, the entire LTS sample is 92.6% White, 5.0% more than one race, <1% American Indian/Alaskan Native, <1% Pacific Islander, and 1.2% unknown/not reported. Hispanic individuals composed 9.1% of the sample. Participants were paid $150 for participation in the study or $25 per half an hour for those who did not finish the entire 3-h session. The study session involved the administration of behavioral tasks that measured cognitive abilities as well as acquisition of anatomical and functional brain data via magnetic resonance imaging.
HCP participants were 371 individuals (157 male; Mage = 29.1 years, SDage = 3.47 years, range = 22–36 years) selected from the most recent HCP data release because they were part of complete pairs of twins who completed the anatomical and functional imaging components of the study (more information about the HCP sample and project can be found in Van Essen et al. 2013). This subset of HCP participants were 89 MZ pairs and 79 DZ pairs with race reported as 82.7% White, 11.3% Black/African American, 4.5% Asian/Native Hawaiian/other Pacific Islander, <1% American Indian/Alaskan Native, <1% more than one race, and <1% unknown/not reported.
Method Details
Procedure
For the LTS sample, testing took place in a single 3-h session. Following review and obtainment of informed consent, participants were familiarized with the imaging procedures. If both twins of a pair participated on the same day, the twins completed the protocol sequentially (twin order randomized) with the same ordering of behavioral testing and imaging acquisition. The resting-state scan always occurred first in the imaging protocol, before tasks. All study procedures were fully approved by the Institutional Review Board of the University of Colorado Boulder. Testing procedures for participants in the HCP sample has been described in prior work (Van Essen et al. 2013).
Brain Imaging
Participants from the LTS sample were scanned in a Siemens Tim Trio 3T (n = 250) or Prisma 3T (n = 215) scanner (Scanner was included as a nuisance regressor in all analyses involving the LTS dataset.) Neuroanatomical data were acquired with T1-weighted magnetization prepared using rapid gradient echomagnetization prepared rapid gradient echo sequence (acquisition parameters: repetition time (TR) = 2400 ms, echo time (TE) = 2.07, matrix size = 320 × 320 × 224, voxel size = 0.80 × 0.80 × 0.80 mm, flip angle (FA) = 8.00°, slice thickness = 0.80 mm). Resting-state data were acquired with a 6.25-min T2*-weighted echo-planar functional scan (acquisition parameters: number of volumes = 816, TR = 460 ms, TE = 27.2 ms, matrix size = 82 × 82 × 56, voxel size = 3.02 × 3.02 × 3.00 mm, FA = 44.0°, slice thickness = 3.00 mm, field of view (FOV) = 248 mm). During the resting-state scan, participants were instructed to relax and stare at a fixation cross while blinking as they normally would.
Resting-state acquisition in the HCP sample is described in detail elsewhere (Smith et al. 2013), but briefly, each participant completed an anatomical and four 15-min resting-state scans (eyes fixated) in the context of a large imaging and behavioral testing battery. In the current study, we used the first two 15-min resting-state scans.
Preprocessing and Connectome Extraction
All processing of LTS brain data was performed in a standard install of FMRIB Software Library (FSL) build 509 (Jenkinson et al. 2012). To account for signal stabilization, the first 10 volumes of each individual functional scan were removed, yielding 806 volumes per subject for additional analysis. The functional scans were corrected for head motion using MCFLIRT (FMRIB’s linear image registration tool for motion correction). Brain extraction was used to remove signal associated with nonbrain material (e.g., skull, sinuses, etc.). FMRIB’s linear image registration tool (FLIRT) was used to perform a boundary-based registration of each participant’s functional scan to his or her anatomical volume and a 6-degree-of-freedom affine registration to MNI152 standard space. To account for motion and other noise signals known to pollute resting-state analyses, LTS scans were subjected to AROMA (ICA-based Automatic Removal Of Motion Artifacts), an automated independent components analysis-based, single-subject denoising procedure (Pruim et al. 2014). Signal extracted from masks of the lateral ventricles, white matter, and whole brain volume was regressed out, along with a set of six motion regressors and associated first and second derivatives. Finally, the scans were band-pass filtered (0.001–0.080 Hz band).
Preprocessing for HCP data is described elsewhere (Glasser et al. 2013). Briefly, HCP scans were subjected to minimal preprocessing and FIX, a semiautomated single-subject denoising procedure (Salimi-Khorshidi et al. 2014). Additionally, we regressed out the mean grey ordinate time series from each scan as a proxy for the global signal (as suggested by Burgess et al. 2016). HCP scans were band-pass filtered (0.001–0.080-Hz band).
For each participant, we extracted the blood-oxygen-level-dependent time series from each of 264 1-cm spherical ROIs, drawn from Power et al. (2011), which serve as the nodes for the present analysis. This analysis was performed in volume space to maximize similarity between the two samples. We used these nodes, as they are drawn from a meta-analysis of functional activations and have a community structure that agrees with task-based functional networks (i.e., are organized into networks such as default mode network and FP task control network). We chose 1-cm spherical ROIs because they provide the largest possible size for a given ROI but preclude overlap with neighboring ROIs. Within each participant, all pairwise Pearson’s correlations were calculated, yielding a 264 × 264 correlation matrix. All correlations were subjected to the Fisher’s z-transformation to normalize the variance in correlation values. All genetic analyses used the per-participant z-correlations after regressing out several nuisance variables: scanner, gender, and summary measures of movement during the resting-state scan (average motion in the x, y, and z planes and average of roll, pitch, and yaw) for the LTS sample and gender and summary measures of movement during the resting-state scan (average motion in the x, y, and z planes and average of roll, pitch, and yaw) for the HCP sample. The gender regressor is particularly important for the HCP sample given the presence of opposite-sex DZ pairs. Bivariate analyses utilized a global summary measure of each participant’s connectivity matrix, which was calculated as the reciprocal of the average shortest path length between all 264 regions as calculated on a proportionally thresholded (15%) connectivity matrix using the Python package Networkx (Hagberg et al. 2008). Calculation and manipulation of connectivity matrices as well as plotting was also done in Python using the Pandas (McKinney 2010), Seaborn (http://seaborn.pydata.org/), and Matplotlib packages (Hunter 2007).
Statistical Analysis
Genetic Models
All genetic analyses conducted were run as structural equation models in R through the OpenMx (Boker et al. 2011) and UMX packages Bates, Maes, and Neale (2019). As all measures were continuous, these models utilized maximum likelihood estimation (Bentler and Weeks 1980). Univariate genetic models were run on each connection. A univariate model decomposes total phenotypic variation in a connection into additive genetic (A), shared environmental (C), and nonshared environmental (E) components. MZ twins share all of their genes, whereas DZ twins share on average 50% of their genes by descent, and both types are reared together. Genetic influences (A) are indicated when the MZ twin correlation is higher than the DZ correlation; shared environmental influences (C) are indicated when the DZ correlation is greater than half the MZ correlation; and nonshared environmental influences (E), which include measurement error, are indicated when the MZ correlation is less than unity.
Statistical analysis of differences between within- and between-network connectivity heritability estimates was performed using the statsmodels package for Python. For tests of network-wide differences in within- and between- network connectivity heritability, we report two statistical significance criteria: Bonferroni correction for 14 network comparisons and nominal significance (P < 0.05) in both samples. Only tests of regional differences between within- and between-network connectivity heritability that were significant in both samples (P < 0.05) are reported. We additionally report which results withstood a correction for 264 regional tests (P < 0.00018).
For the association between connections and either network connectivity, global efficiency, or movement, we utilized bivariate correlated ACE models (Fig. 1a) derived from bivariate Cholesky decompositions (Fig. 1b). The Cholesky decomposition is a common form of bivariate twin analysis and can be used to calculate the genetic correlation and correlation predicted from A, C, and E overlaps (Neale and Cardon 1992). In this Cholesky decomposition, the first set of A, C, and E latent variables predicting network connectivity are allowed to predict the local connection (via paths x2, y2, and z2), and the local connection also has residual A, C, and E variances (obtained by squaring paths x3, y3, and z3). Given our interest in genetic variance of connections not shared with superordinate measures of connectivity or motion, the residual A values are of particular interest. The matrix of residual A variances (i.e., squared x3 paths) enables us to ascertain whether the finely detailed genetic map of the connectome is simply a redescription of network connectivity (or global efficiency and movement, in those models). That is, it depicts where there are genetic influences on local connections that are independent of the genetic influences on network connectivity. If residual genetic influence is present across the connectome, this analysis supports high-resolution analysis approaches as independent and complementary to analyses that utilize network-derived connectivity measures.
Figure 1.
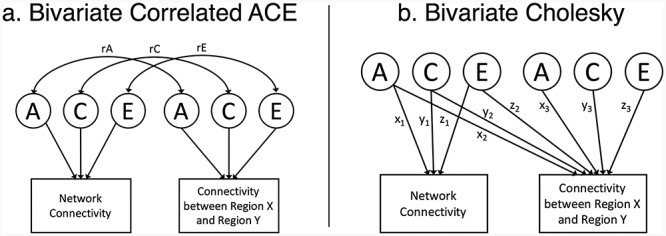
Bivariate models. (a) Bivariate correlated ACE model. rA, genetic correlation; rC, shared environmental correlation; rE, nonshared environmental correlation. (b) Bivariate Cholesky decomposition. Additive genetic (A), shared environmental (C), and nonshared environmental (E) latent variables (left side) predicting network connectivity (via paths x1, y1, and z1) and functional connectivity (via paths x2, y2, and z2). Functional connectivity has residual A, C, and E influences (right side). The variance explained by each influence is obtained by squaring the paths (e.g., x3, y3, and z3). The univariate model of network connectivity is equivalent to left side of the figure (i.e., removing the local connectivity measure). The models include variables for both twins, but for simplicity, only one twin is shown.
Clustering Analysis
We clustered patterns of heritability estimates (rows/columns of Fig. 2a). Ward clustering was implemented in Python using the Fastcluster package (Müllner 2013). Ward clustering is commonly applied in brain-imaging contexts and is known to be accurate and reliable (Thirion et al. 2014). We applied clustering to the 264 × 264 matrix of A estimates to find 2–20 clusters of regions. To estimate the stability of each clustering solution, we calculated the silhouette score for each sample and averaged all scores for each clustering solution (Fig. 3a). The silhouette score compares the distance between a region and other members of its cluster to the distance between that region and the nearest neighboring cluster in similarity space. In the current analysis, similarity was defined as the Euclidean distance between two regions’ vectors of heritability estimates. The silhouette analysis revealed several “stable” solutions in which the average silhouette score reached a local maximum, as seen in the peaks of Fig. 2a at solutions of k = 3, 7, and 18. We describe the clustering results at the coarsest levels in the main body because they are a demonstration of novel genetic communities without the added complexity of describing many clusters.
Figure 2.
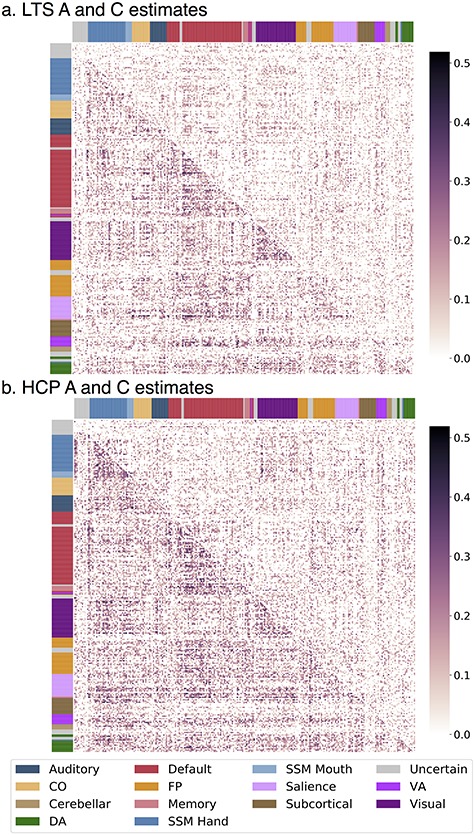
Connection-wise estimates of additive genetic (A) and shared environmental (C) influences. Matrices contain estimates from univariate twin models with the spatial location of each cell (estimate) corresponding to the functional connection between two regions. Assignment to a priori networks is represented by colored bars along x- and y-axes. Different estimates are displayed in upper and lower triangles. (a) LTS sample A and C estimates are in the lower and upper triangles, respectively. (b) HCP sample A and C estimates are in the lower and upper triangles, respectively. CO, cingulo-opercular; DA, dorsal attention; FP, frontoparietal; SSM, sensory/somatomotor; VA, ventral attention.
Figure 3.
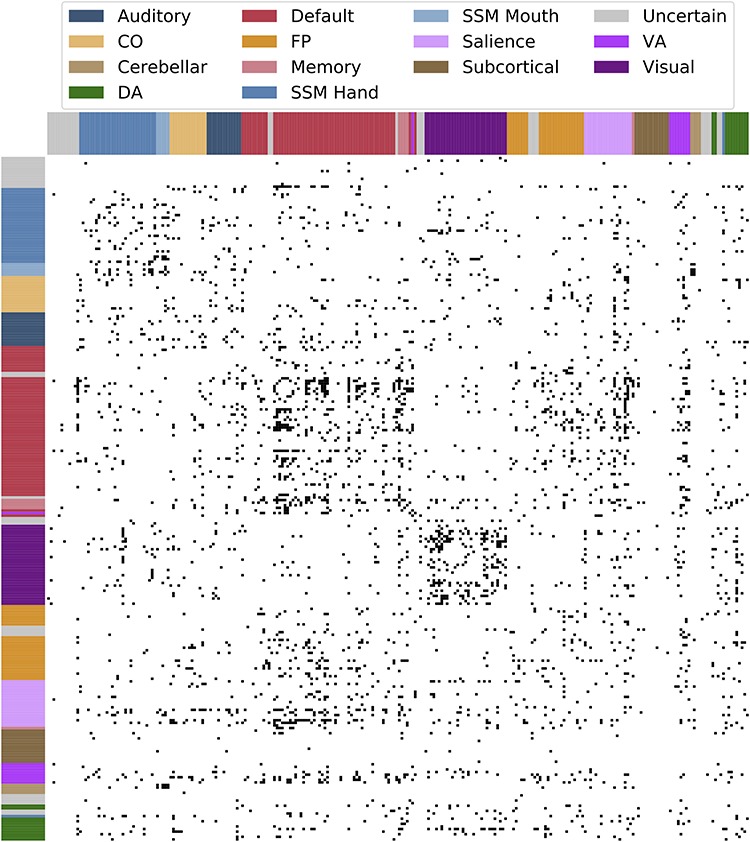
Connections with significant heritability estimate in both LTS and HCP samples. Black cells indicate functional connections between two regions with nominally significant (P < 0.05) heritability in both samples. Assignment to a priori networks is represented by colored bars along x- and y-axes.
Data and Software Availability
Sample genetic model and A, C, and E estimates for all pairwise connections are available for download at: https://github.com/AReineberg/genetic_connectome.
Results
Group Average Connectomes
Visual comparison of mean phenotypic connectivity matrices for each sample to one another and to matrices reported in prior work using independent samples (e.g., Fig. 3 of Cole et al. 2014; Fig. 2 of Reineberg and Banich 2016) reveals striking similarity, especially in the prominence of resting-state networks along the diagonals (Supplementary Fig. S1 (LTS) and Supplementary Fig. S2 (HCP)).
Univariate Twin Models
Connection-wise estimates of additive genetic influence for the LTS and HCP samples are shown in Figure 2a,b (lower triangles). In the LTS sample, additive genetic influence was moderate and bimodally distributed across the connectome such that 16 350 of 34 716 unique connections were estimated as having zero heritability while a separate, positively skewed distribution described the heritability of 18 366 connections (M = 0.114, SD = 0.079, skew = 0.769, min = 0.001, max = 0.485; distribution presented in Supplementary Fig. S3): 5021 connections (14.5% of all connections) had significant (P < 0.05) heritability based on a 1 degree-of-freedom chi-square difference test comparing an AE model to an E model, and 408 (1.2% of total connections) had heritability greater than 30% (30% threshold chosen arbitrarily to represent cutoff to be considered a moderately heritable trait). Similarly, for the HCP sample, 14 302 of 34 716 unique connections were estimated as having zero heritability while a separate, positively skewed distribution described the heritability of 20 414 connections (M = 0.131, SD = 0.085, skew = 0.631, min = 0.001, max = 0.519, distribution presented in Supplementary Fig. S3): 7626 connections (22.0% of all connections) had significant (P < 0.05) heritability based on a 1-degree-of-freedom chi-square difference test comparing an AE model to an E model, and 809 (2.3% of total connections) had heritability greater than 30%. Of 5021 (LTS) and 7626 (HCP) connections with statistically significant heritability, 1612 connection (4.6% of all connections) were overlapping (i.e., the same connections had significant heritability in both samples). Connections with significant heritability in both samples are visualized in Figure 3.
Shared environmental influences generally explained less variance than genetic influences, as shown in Figure 2a,b (upper triangles). In the LTS sample, shared environmental influence was weak to moderate and bimodally distributed across the connectome such that 21.023 of 34 716 unique connections were estimated as having zero shared environmental influence while a separate, positively skewed distribution described the shared environmental influence of 13 693 connections (M = 0.083, SD = 0.058, skew = 0.785, min = 0.001, max = 0.339, distribution presented in Supplementary Fig. S3). Similarly, for the HCP sample, 20 655 of 34 716 unique connections were estimated as having zero shared environmental influence while a separate, positively skewed distribution described the shared environmental influence of 14 061 connections (M = 0.099, SD = 0.067, skew = 0.726, min = 0.001, max = 0.439, distribution presented in Supplementary Fig. S3). Although C estimates were low, there is a suggestion of moderate shared environmental influence in several pieces of the connectome (e.g., within visual connections and visual-to-sensory/somatomotor (SSM) connections) as can be seen in Figure 2a,b (upper triangles). In the current study, no significance testing was done to formalize areas of enriched shared environmental influence.
In both samples, nonshared environmental influences were high across the entire connectome (MLTS = 0.907, SDLTS = 0.082; MHCP = 0.883, SDHCP = 0.090) and negatively skewed (SkewLTS = −0.757, SkewHCP = −0.546). Connection-wise estimates of nonshared environmental influences are shown in the lower and upper triangles of Supplementary Figure S4 for the HCP and LTS samples, respectively. Note that E estimates include measurement error. Reliability of connections was tested for the HCP sample and found to be high (M = 0.825, SD = 0.059; see Supporting Information—Reliability), suggesting that the high E estimates across the connectome are unlikely to solely reflect random measurement error.
In general, both samples had very similar patterns of heritability. To be sure any differences in estimates were due to true differences between the two samples, we compared 6 minutes to 30 minutes of data within the HCP sample. This analysis leads us to believe the differences between the LTS and HCP samples are related to sample differences rather than to data quantity. When comparing 6 to 30 min of HCP data, heritability estimates changed in magnitude (mean h26 min = 0.090, mean h230 min = 0.131) but remained similar in pattern (i.e., were correlated with one another), see Supporting Information—6 versus 30 min of Resting Data for more information about this analysis.
Within- and Between-Network Connections
To examine whether high-resolution mapping of genetic influence reveals differences in within- versus between-network connections, (question 1), we investigated heritability estimates for connections of those types. As stated previously, 1612 of 34 716 connections had significant (P < 0.05) additive genetic influence in both samples. Of those 1612 connection, 364 were within-network connections (from a total of 3748 within-network connections; i.e., 9.71%) and 1248 were between-network connections (from a total of 30 968 between-network connections; i.e., 4.03%). This difference in proportion of significantly heritable connections was statistically significant (χ2(1) = 243.77, P < 0.001), suggesting that within-network connections are over-represented in the heritable connections that replicated across samples.
First, we considered average heritability across all connections considered to be within the same a priori network versus all between-network connections. In both samples, within-network connectivity was more heritable than between network connectivity (Supplementary Table S1a; whole connectome results). This effect was present even when controlling for the estimated test–retest reliability of each connection in the HCP sample (see Supporting Information—Reliability).
We also quantified differences in heritability for within- and between-network connections for each a priori network individually. In both samples, within-network connections tended to be more heritable on average than between-network connections. In both samples, the default, sensory-somatomotor hand, and visual networks had significantly higher heritability for within- than between-network connections (Supplementary Table S1b) after accounting for 14 tests (one per a priori network). In the HCP sample, the uncategorized network had significantly higher heritability for between- than within-network connections. Although within-network connections tended to be more heritable on average than between-network connections, the distributions of between-network connections tended to be more positively skewed, perhaps suggesting there are a minimal number of highly heritable between-network connections. We also tested for differences in shared environmental and nonshared environmental influence for within- versus between-network connectivity. There was significantly higher shared environmental influence on within- versus between-network connections for the visual network in both samples (Supplementary Table S1b). In the LTS sample, there was significantly higher shared environmental influence on within- versus between-network connections for the sensory-somatomotor hand network. In the HCP sample, there was significantly higher shared environmental influence on within- versus between-network connections for the default and sensory-somatomotor networks and significantly higher shared environmental influence on between- versus within-network connections for the ventral attention (VA) network. There was significantly lower nonshared environmental influence on within- versus between-network connections for the FP, default, sensory-somatomotor hand, sensory-somatomotor mouth, and visual networks in both samples and significantly higher nonshared environmental influence on within- versus between-network connections for the uncategorized network of regions in both sample (Supplementary Table S1c). In the LTS sample, there was significantly lower nonshared environmental influence on within- versus between-network connections for the DA network.
Finally, we quantified differences in heritability for within- and between-network connections at the level of regions (each of the 264 regions of the parcellation). Of regions that had significantly different heritability for within- than between-network connections in the LTS (n = 83) and HCP (n = 85) samples, 39 regions showed the effect in both samples, in the same direction (see Supplementary Table S2). Regions from the sensory-somatomotor (4), default (20), visual (11), and FP (1) networks had significantly higher average heritability for within- versus between-network connections. Three uncategorized regions had significantly higher average heritability for between- versus within-network connections.
Clustering Reveals Large Genetic Communities
Given the heterogeneity in genetic influence described above, we explored whether the best way to describe genetic communities in the connectome was in terms of a priori functional networks. Regions of any given resting-state community have a variety of different patterns of heritable connectivity across the connectome. Variation in the patterns of genetic influence for the different regions of each network can be explored with clustering analysis, which groups regions based on similar patterns of heritable connectivity. Ward clustering is a hierarchical procedure that groups together rows of the additive genetic influence matrix (Fig. 2) that have similar patterns of heritable connectivity with all other regions. This analysis could reveal that the 264 regions cluster together in a manner similar to a priori networks, or in a novel way (e.g., a cluster of regions with highly heritable connectivity to some default and FP network regions, but minimally heritable connectivity to other regions). We analyzed average silhouette scores for clustering solutions (i.e., k-values) from 2 to 20, as shown in Figure 4a for the LTS sample (see Supplementary Fig. S5a for HCP sample version), and discovered stable solutions at k-values of 3, 7, and 18 in the LTS sample and k-values of 3, 7, and 18 in the HCP sample.
Figure 4.
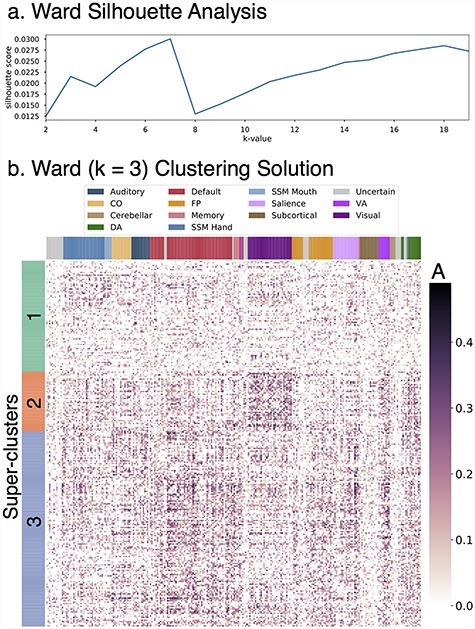
Ward three-cluster solution. Row-wise clustering of LTS additive genetic (A) estimates reveals several stable clustering solutions of regions with similar patterns of connectivity heritability. Superclusters (k = 3) are described in detail. (a) Silhouette analysis reveals stable clustering solutions at k-values of 3, 7, and 18. (b) Clustered version of LTS A estimates for k = 3 solution.
We explored the k = 3 clustering solution first. This level provides the highest level overview of patterns of genetic influence across the connectome. The three clusters from the k = 3 stable solution will be referred to as superclusters throughout the remainder of the manuscript. The three-cluster solution for the LTS sample is shown in Figure 4b (see Supplementary Fig. S5b for HCP version). Figure 5 provides an overview of both the spatial location of the regions in each LTS supercluster (a–c) and also the composition of those superclusters in terms of the region assignments to a priori networks (rightmost column). Supplementary Figure S6 provides the same information for the HCP sample.
Figure 5.
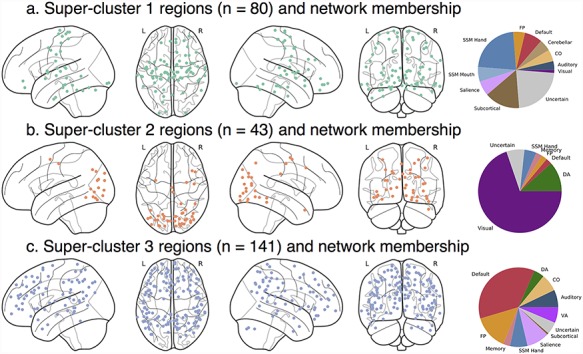
Ward three-cluster summary. Spatial location of regions from LTS superclusters 1–3 of k = 3 solution. (a) Supercluster 1 regions were widely distributed across the brain. (b) Supercluster 3 regions were located primarily in sensory and somatomotor areas. (c) Supercluster 2 regions were located across lateral prefrontal, lateral parietal, mid and anterior temporal, midline frontal, and cingulate areas. n, number of regions.
Overall, regions clustered at a level superordinate to a priori notions of resting-state community structure. Supercluster 1 was composed of 80 regions from all a priori networks with no distinct pattern of heritable connectivity. Generally, heritability was low for all connections in these 80 regions. Supercluster 2 regions had especially heritable connectivity to visual regions as well as moderately heritable connectivity to other regions. Supercluster 2 was composed of 43 regions from a variety of pure sensory (e.g., visual) and DA networks. Supercluster 3 regions had especially heritable connectivity to default, FP, salience, DA, and VA regions. Supercluster 3 was composed of 141 regions that can best be summarized as the majority of the default network as well many frontoparietal regions, among others. The k = 3 solution of the LTS sample maps closely on to the k = 3 solution of the HCP sample (see Supporting Information—Clustering) with only one notable difference: supercluster 2 in the HCP sample contained many SSM regions that were part of supercluster 1 in the LTS sample.
Higher order clustering solutions from the LTS sample give insight into how these large genetic communities break down into more specific patterns of genetic influence. Supplementary Figure S7 shows the composition of subclusters from the 18-cluster solution. Some of the subclusters of the 18-cluster solution remain large and highly heterogeneous (i.e., composed of regions from many different a priori resting-state communities). Others are smaller and relatively pure, such as subclusters 2, 3, 6, 7, 8, 14, 15, 17, and 18, which contain one to three types of regions. Of note is that regions from most a priori resting-state communities split into several different clusters, supporting the conclusion that a priori networks contain several sets of regions that have unique patterns of heritable connectivity across the connectome. For example, default network regions can be found in 10 of the 18 subclusters. Future work could explore this and other higher dimensionality clustering solutions (as we limited our clustering analysis to between 2 and 20 clusters) to possibly reveal novel communities of brain regions.
Applications
The goal of the following sections is to demonstrate the utility of high-resolution genetic estimates while simultaneously validating our approach. First, we explore the difference between high- and low-resolution genetic estimates in magnitude and spatial specificity. Second, we conduct several bivariate analyses that demonstrate genetic separability of local connectivity and more superordinate measures derived from the connectome—a network-level connection and a global summary measure of the connectome. Future bivariate models could be applied to clinical and cognitive measures, but here we chose superordinate connectivity measures to validate the high-resolution approach.
Revealing Particularly Heritable Connections between Two Resting-State Networks
We performed a series of follow-up analyses in both samples on an example set of between-network connections to explore heterogeneity of genetic influence in a more focused system. Prior work has found that the most heritable default network connection is the connection to the SSM network (Yang et al. 2016). We extracted many connections between default network regions and SSM regions for a more detailed investigation (n = 2030, all pairwise connections between 35 SSM and 58 default network regions). Figure 6a shows distribution of nonzero heritability estimates for LTS default-to-SSM connections (h2max LTS = 0.385, h2max HCP = 0.396; see Supplementary Fig. S8a for HCP version).
Figure 6.
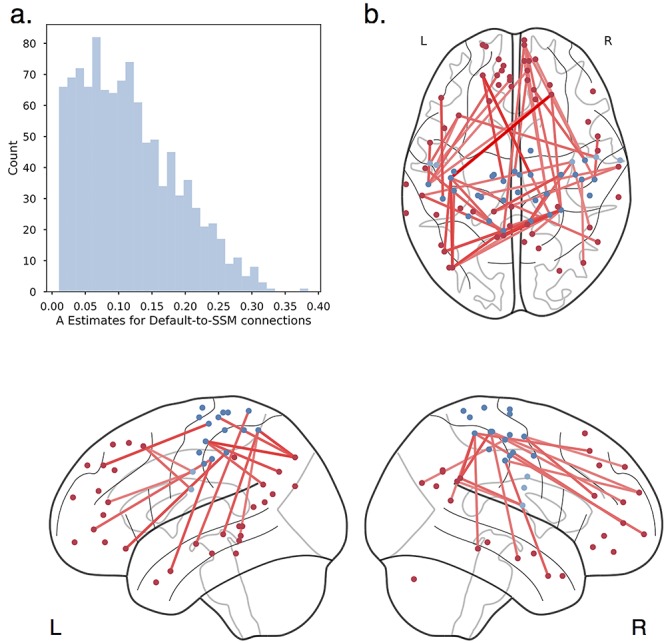
Default-to-SSM connections and heritability. (a) Distribution of nonzero heritability estimates for default-to-SSM connections. (b) Top 10% heritable connections between default (red) and SSM (blue) regions projected onto the brain. The most heritable connections between the default and SSM network are between the sensory/motor strips and superior, medial frontal, posterior cingulate, precuneus, and middle temporal regions of the default network.
The fine-grained analysis revealed that a subset of default to SSM connections had moderate genetic influence, whereas many had minimal to no genetic influence. Figure 6b shows the most heritable of the default to SSM connections in the LTS sample (see Supplementary Fig. S8b for HCP version). We found the most heritable connections are between superior, medial frontal cortex and the sensory/motor strips; hub regions/precuneus of the default network and the sensory/motor strips; as well as connections between the middle temporal lobes and the sensory/motor strips. A recent meta-analysis of thousands of functional MRI studies revealed that the function of many of these superior, medial regions is related to “conflict,” “working memory,” and “inhibition” (de le Vega et al. 2016). The hubs of the default network have an important role in the valuation of motivationally salient and personally significant information (Andrews-Hanna 2012): Precuneus is a highly sensory integrated component of the default network, and the middle temporal gyrus is part of a default network subsystem responsible for introspection about mental states. Here, we have identified particularly strong heritability for connectivity between these places and SSM regions. Future work could explore other network connections of interest in the same way to reveal other novel characterizations of between-network connections.
In summary, across 2000+ connections between the default and SSM, we found many connections are not heritable, but some show moderate heritability in line with prior work. Studies that utilize network-derived estimates should note they may be averaging many connections with heterogeneous genetic influence, which could result in a network-derived heritability estimate that is an underestimate of the maximum heritability between smaller functional units or an overestimate biased heavily by a small number of heritable connections.
ROI-based Local Connectivity Estimates are Genetically Separable from Network-Derived Estimates
Variation in heritability estimates does not imply separate genetic influences (i.e., sets of genes responsible for the difference in heritability). For example, the network estimates quantified in historical work could be driven by the same or different genetic variants as the connections in the current report. Here, we performed a bivariate genetic analysis (models pictured in Fig. 1 and described in section Methods—Genetic Models) between the many default-to-SSM connections described above and the network-derived connectivity estimate for these two networks as a whole. For this analysis, we quantified connectivity for the default-to-SSM network connection as a whole using network templates from a popular network parcellation (Yeo et al. 2011). While the univariate models described above quantify genetic influences on local connections alone, these bivariate analyses quantify the degree to which local functional connectivity is genetically separable from the network-derived estimate of connectivity between the default and SSM networks.
First, we found connectivity between default network and SSM network as a whole was moderately-to-strongly heritable (h2LTS = 0.324; h2HCP = 0.476). Second, local and network connectivity does have strong genetic correlation (“rA”; pictured in Fig. 7a for LTS and Supplementary Fig. S9a for HCP; M|rA|LTS = 0.790, SD|rA|LTS = 0.324, minLTS = −1.000, maxLTS = 1.000; M|rA|HCP = 0.790, SD|rA|HCP = 0.324, minHCP = −1.000, maxHCP = 1.000), as would be expected given the network-derived connectivity estimate includes the same time series data as the more focused 2030 connections. However, we find residual genetic influence not accounted for by the network-derived estimate, with an overall pattern very similar to the univariate A estimates. For the LTS sample, 659 of 2030 connection had nonzero residual additive genetic influence (pictured in Fig. 7b; MLTS = 0.090, SDLTS = 0.066, maxLTS = 0.326). For the HCP sample, 19 085 connections had approximately zero residual additive genetic influence while a separate distribution of 15 631 connections had nonzero residual additive genetic variance (MHCP = 0.104, SDHCP = 0.071, maxHCP = 0.360). Together, these analyses demonstrate that the local connections have unique genetic influences, although there is certainly a substantial, common genetic component captured when utilizing network-derived connectivity estimates.
Figure 7.

Default-to-SSM connections—bivariate analysis. (a) Genetic correlation between network-derived estimate of default to SSM connectivity and many default to SSM connections. (b) Residual additive genetic influence on many default to SSM connections after accounting for genetic variance shared with a network-derived estimate of default to SSM connectivity.
Genetic correlations between individual connections and higher level measures of individual differences in connectivity may be of interest to those interested in graph theoretic analysis of the brain. Graph theory analysis offers many possible summary measures of connectivity and seeks to summarize the brain in the context of complex network dynamics—for example, the degree to information can be shared amongst distributed brain systems (i.e., integration as measured by global efficiency; for review, see Rubinov and Sporns 2010). In Supporting Information—Bivariate Analysis, we describe an analysis in which we quantify the degree to which connections are genetically separable from global efficiency. We find local connections have residual genetic influence not accounted for by genetic influences on global efficiency (Supplementary Fig. S10).
Investigating Genetic Correlation of In-scanner Movement and Connectivity
Prior work has found in-scanner movement, as measured by mean frame displacement, is heritable (h2 = 0.313–0.427; Hodgson et al. 2017). In all analyses reported above, we controlled for movement via single subject denoising and summary movement covariates, so the results do not reflect covariance with movement (i.e., are equivalent to a “specific heritability” estimate as described in the bivariate analyses presented previously). However, we wondered whether or not some connections (before regressing out summary movement covariates) might be genetically related to movement. We utilized bivariate genetic models (Fig. 1 and Methods—Genetic Models) to quantify where in the connectome there is an overlapping genetic influence between connectivity (without summary movement covariates) and in-scanner movement (i.e., genetic correlations or “rA”). In an initial univariate analysis, we found translation (average motion in the x, y, and z planes) and rotation (average roll, pitch, and yaw movements) were weakly-to-moderately heritable (h2translation LTS = 0.368, h2rotation LTS = 0.118; h2translation HCP = 0.221, h2rotation HCP = 0.212), suggesting the analysis was viable to pursue.
Genetic correlations between rotation movement and connections were strong (M|rA| = 0.722, SD|rA| = 0.355, min = −1.000, max = 1.000). Genetic correlations between translation movement and connections were also strong (M|rA| = 0.648, SD|rA| = 0.388, min = −1.000, max = 1.000). The matrix of genetic correlations is presented in Supplementary Figure S11 for the LTS sample and Supplementary Figure S12 for the HCP sample. Interestingly, there are strong positive genetic correlations, either indicating that there may be common genes that are associated with increased level of movement and higher connectivity strengths or vice-versa, or reflecting that movement may induce artifactual connectivity differences in these places. Within-network connections seem to have the weakest absolute genetic correlations between movement and connectivity strength, with the exception of within-SSM network connections. Within-sensory somatomotor connections are also notable in that the direction of the genetic correlations is mostly positive for the LTS sample but mostly negative for the HCP sample. Future work could investigate possible causes of this and other results that go in opposite direction between the two samples analyzed here. Negative genetic correlations were also present, indicating there may be common genes that are associated with decreased level of movement and higher connectivity strengths or vice-versa. Visual network connections may be enriched for negative genetic correlations; however, the pattern is complex and best suited for detailed analysis in future work. Future work could map movement-connectivity genetic correlations onto existing models of movement-related susceptibility for connections of different type and distance.
In summary, had we not residualized connectivity with regard to movement in the main analyses of the current study, we would have obtained heritability estimates across the entire connectome that were driven partially by genetic influence on rotation movement. However, because we controlled for movement prior to genetic analysis, our results described above are equivalent to the residual genetic influence on connections after accounting for genetic influence on motion in the bivariate analyses.
Discussion
Across all analyses, we found converging evidence of etiological heterogeneity in the functional connectome. High-resolution mapping reveals a distribution of genetic and environmental influence that may be missed by approaches that summarize functional connectivity at the level of larger ROIs, networks, and global summary measures of the connectome. More specifically, we found differences in genetic influences for connections of different type (i.e., higher heritability of connections between regions of the same functional network versus between regions of different functional networks). This pattern was present across the whole connectome and especially for the default, SSM, and visual networks. This result provides preliminary evidence that the organization of the brain into networks based on function may be driven by genetic influences on connections between regions involved in the same processes. Prior work has established specific patterns of gene expression within functional networks (Richiardi and Altmann 2015), a possible mechanism linking these observations of genetic influence to specific functions. Importantly, we showed how high-resolution heritability estimates might be used to define novel communities of regions based on their pattern of genetic influences, as well as how to isolate pieces of the connectome with particularly high genetic influence (i.e., which may be candidate endophenotypes). Finally, we showed that genetic influences on connections are separable from genetic influences on network connectivity, a global summary measure of the connectome, and in-scanner movement during the resting-state scan (a frequently discussed source of nuisance signals in functional imaging studies).
Although a number of our analyses were summarized by a priori functional networks, the broad range of genetic estimates across the connectome led us to question whether alternative groupings could better describe patterns of heritability in the connectome. A clustering procedure revealed a novel finding: Regions grouped together based on patterns of heritable connectivity at a level that was superordinate to that of classic resting-state communities. In both samples, we found stable superclusters of regions. Most notably, a set of “cognitive” and “visual” or “SSM” regions had characteristic patterns of highly heritable connections to regions of higher-level (e.g., default network) and lower-level (e.g., visual) functions, for the cognitive and visual or SSM clusters respectively. Although the description of these superclusters as higher and lower-level is likely an oversimplification, it is a worthwhile descriptive tool until future work dissects the role of these sets of brain regions. In both samples, we found evidence of a supercluster of regions with very consistent low-to-moderate heritability to all regions (i.e., no distinct features) and high nonshared environmental influence. Analyses of connection-wise reliability (very high in all connections in the HCP sample) suggest that these nonshared environmental influence estimates do not simply reflect random measurement error. Thus, future work should seek a more thorough understanding of the environmental factors influencing this nondescript set of regions.
Stable clustering solutions were also found at levels of granularity similar to classic resting-state communities, but, interestingly, these genetic clusters were quite dissimilar to the a priori networks. Notably, in our example 18-cluster analysis, regions from the default network broke into several subclusters, which were differentiated on heritability of connectivity to other default network regions and to regions of other networks such as the FP network. Future work should dissect these finer-grained parcels in more detail, especially the degree to which they represent novel or previously described communities. Finer-grained, stable clustering solutions could be explored in more detail too as those may reveal small communities with highly characteristic patterns of heritable connectivity that may not align with known clusters of regions based on community detection performed on phenotypic functional connectivity.
Our use of bivariate genetic models represents a substantial development in neuroimaging genetics. We found that local connections showed genetic influences independent from genetic influences on network connectivity, in-scanner movement, and a global summary measure of the brain. Residual genetic influence justifies analysis at the level of small regions and is an important commentary on an ongoing debate about the proper level of analysis of connectivity, suggesting all levels may be complementary. A practical application of this evidence of residual genetic influence would be to the study of multivariate functional connectivity signals as a predictor of individual differences in some cognitive ability or clinical variation (i.e., connectivity-based predictive modeling,“fingerprinting,” or “connectotyping”). Our results suggest influence on a whole-connectome signal will be diverse and not accurately represented in network-based or global summary measures of the connectome. Specifically, our results show network-derived estimates mostly over-estimate heritability of different pieces of a predictive model by assigning the network-derived measure to all connections of the same type, which we have shown here to have a distribution of genetic influence and to be genetically separable from the network-derived measure of connectivity.
Regarding movement, we showed evidence of both overlapping and distinct genetic influences for in-scanner movement and connections in an analysis using connectivity estimates that did not control for individual variation in movement. This analysis supports the strong emphasis in the resting-state literature on adequately controlling for nuisance signals associated with in-scanner movement.
Although we opted to utilize bivariate genetic models for the purpose of clarifying the specificity of genetic effects with regard to nuisance signal and broader measures of connectivity, future work could apply the same bivariate analysis to the relationship between local connections and clinical symptomatology and/or cognitive abilities. Such an application could identify novel brain-based candidate endophenotypes or focus intervention studies to novel locations. A similar approach has been used in the neuroanatomical/clinical endophenotyping literature in which bivariate genetic models have been used to identify the genetically influenced neurobiological underpinning of disorders such as major depressive disorder (Glahn et al. 2012) and the genetically influenced neurobiological underpinnings of schizophrenia that are shared with other psychiatric disorders (Lee et al. 2016).
Our approach is not without caveats. There may be concerns regarding the magnitude of effects both when comparing the LTS sample to the HCP sample and when comparing our results to previous genetic neuroimaging research. Notably, there were more nonzero estimates and more estimates over 30% heritability in the HCP sample. Sample differences could reflect reliability differences in the measurement of resting-state functional connectivity. Specifically, a scan time of 30 min [HCP] versus 6 min [LTS] is known to produce more reliable results (Gordon et al. 2017), although we did find that 6 min of HCP resting data produced patterns of heritability estimates very similar to those produced using 30 min of data. Our results support the common recommendation to collect as much data as possible within the constraints of a scanning session.
In addition, these sample sizes, while large for phenotypic analysis, are relatively small for heritability analysis, leading to uncertainty in the estimates and a decrease in replicability. The issue of power is increasingly solved in the genetics literature with consortium analyses in which data from many independent studies are harmonized and combined in a single analysis (particularly for analysis involving genomic data). Resting-state consortia are a desirable step for the future of the genetic neuroimaging literature to obtain the most accurate estimates, but future consortia-level analyses should be aware that decisions to increase sample size come at the cost of accepting inferior quality data that could be shorter in duration than desired. Moreover, future consortia should carefully consider at what levels of granularity resting-state data should be examined. Though broader levels may increase heritability, our results show that they do not capture all of the genetic effects within networks. Future work investigating how these local and global genetic effects relate to behaviors of interest (e.g., cognitive abilities or psychopathology symptoms) could inform decisions about the optimal levels for analysis.
Regarding differences between imaging modalities, our results are in line with other resting-state studies in providing estimates that are lower than estimates typically provided by studies of anatomical genetics. We do not feel this difference precludes resting-state phenotypes from being considered as potential endophenotypes. Additionally, we do not wish to leave the impression that local connectivity is not a viable phenotype for genetic analysis. In the current study, we found 400–800 connections with greater than 30% heritability and 1612 connections with significant heritability in 2 separate samples. When a brain phenotype has genetic influence, it may be shared with a condition or ability of interest. Future work will likely be able to directly compare the utility of resting versus anatomical phenotypes for the purposes of endophenotyping, and an important question to investigate is whether the same genetic influences operate on anatomical and resting measures. If not, then considering both simultaneously may increase genetic insights and/or prediction.
There are several changes to experimenter degrees of freedom in the imaging analysis context that could reveal additional heritability for high-resolution estimates when compared to anatomical estimates: (1) A different parcellation than the one used in the current study may more closely map on to the genetic structure of the connectome. (2) utilizing connectivity analysis techniques that boost reliability by quantify only stable variation in connectivity after accounting for transient components (as in Ge et al. 2017), and (3) increases in sample size from consortia-level analysis.
We observed other sample-specific results. Notably, shared environmental influence was nominally lower across the entire connectome in the LTS sample than in the HCP sample, and particularly in connections such as those bridging sensory and visual areas. Although it is not unusual to find a lack of shared environmental influence (e.g., in the anatomical MRI (Eyler et al. 2011) and cognitive literatures (Friedman et al. 2008)), sample differences could be due to demographic differences in the two samples (e.g., the LTS sample is less racially diverse and sampled from higher socioeconomic status communities than the HCP sample). Socioeconomic status differences could certainly explain sample differences in the current study given prior work showing elevated shared environmental influence on variation in IQ for individuals near or below the poverty line (Turkheimer et al. 2003). Effects of SES in a subset of the HCP sample have been partially explored previously and shown to influence brain connectivity (Smith et al. 2015).
As a final caveat regarding modeling of genetic influences, a small literature suggests classic twin modeling procedures may bias estimates (upward in the case of A and downward in the case of E) when compared to models that do not impose boundary constraints on parameters (Carey 2005). Future work should compare these approaches and report notable differences, if any, in the genetic profile of affected connections.
Overall, we demonstrate the utility of fine-grained A, C, and E estimates by showing that the genetic organization of the brain is diverse and not as one would expect based solely off the classic functional organization of the phenotypic connectome. Our analysis sits on a continuum of dimensionality reductions that spans multiple levels of brain organization (i.e., from global summary measures to voxels), so, obviously one must ask if genetic neuroimaging studies should continue to assess the etiology of finer grained parcellations in the future. Our demonstration of residual genetic variance for local connections in the bivariate analyses certainly demonstrates the added value of a fine-grained approach in addition to a single summary measure of the connectome. But, our results also suggest a trade-off between reliability and interpretability/application: large networks maximize heritability estimates (Ge et al. 2017) but are of imprecise function and cannot be used to dissect the etiology of highly dimensional signals that are most useful for predictive modeling. Parcellations in the range of 200–500 might be recommended for region-based approaches in the future because there are numerous well-vetted atlases (Power et al. 2011; Craddock et al. 2012; Gordon et al. 2016) designed to differentiate homogeneous functional brain units while maximizing reliability (which could become an issue in voxel-based approaches). There is still room for determining the best functional parcellation scheme among these possible alternatives, with genetic etiology as one possible mechanism for evaluating the quality/usefulness of the parcellations. In conclusion, our approach has important implications for investigations of neuroimaging-based biomarkers by (1) quantifying which pieces of the connectome are heritable and thus can be investigated as a potential endophenotype or marker of genetic risk, (2) serving as a model for future studies seeking a greater understanding of a broad literature of traits, and (3) establishing the foundation of a taxonomy of functional connections based on genetic influence.
Supplementary Material
Funding
National Institutes of Health (grants MH063207 and MH016880).
Notes
Conflict of Interest: None declared.
References
- Andrews-Hanna JR. 2012. The Brain’s Default Network and its Adaptive Role in Internal Mentation. Neuroscientist. 18(3):251–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anticevic A, Cole MW, Murray JD, Corlett PR, Wang X-J, Krystal JH. 2012. The role of default network deactivation in cognition and disease. Trends Cogn Sci. 16(12):584–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates TC, Maes H, Neale MC.. 2019. umx: Twin and Path-BasedStructural Equation Modeling in R. Twin Research and Human Genetics. 22:27–41, 10.1017/thg.2019.2. [DOI] [PubMed] [Google Scholar]
- Bentler PM, Weeks DG. 1980. Linear structural equations with latent variables. Psychometrika. 45(3):289–308. [Google Scholar]
- Boker S, Neale MC, Maes H, Wilde M, Spiegel M, Brick T, Spies J, Estabrook R, Kenny S, Bates TC, et al. . 2011. OpenMx: an open source extended structural equation modeling framework. Psychometrika. 76(2):306–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess GC, Kandala S, Nolan D, Laumann TO, Power J, Adeyemo B, Harms MP, Petersen SE, Barch DM. 2016. Evaluation of Denoising strategies to addressmotion-correlated Artifact in resting state fMRI data from the human Connectome project. Brain Connect. 6(9), 669–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey G. 2005. Cholesky problems. Behav Genet. 35(5):653–665. [DOI] [PubMed] [Google Scholar]
- Cole MW, Bassett DS, Power JD, Braver TS, Petersen SE. 2014. Intrinsic and task-evoked network architectures of the human brain. Neuron. 83(1):238–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craddock RC, James GA, Holtzheimer PE, Hu XP, Mayberg HS. 2012. A whole brain fMRI atlas generated via spatially constrained spectral clustering. Human Brain Mapp. 33(8):1914–1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de le Vega A, Chang LJ, Banich MT, Wager TD, Yarkoni T. 2016. Large-scale meta-analysis of human medial frontal cortex reveals tripartite functional organization. J Neurosci. 36(24):6553–6562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott ML, Romer A, Knodt AR, Hariri AR. 2017. A connectome-wide functional signature of transdiagnostic risk for mental illness. Biol Psychiatry, 84(6):452–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyler LT, Chen C-H, Panizzon MS, Fennema-notestine C, Neale MC, Jak A, Jernigan TL, Fischl B, Franz CE, Lyons MJ, et al. . 2012. A comparison of heritability maps of cortical surface area and thickness and the influence of adjustment for whole brain measures: a magnetic resonance imaging twin study. Twin Res Hum Genet. 15(3):304–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyler LT, Prom-Wormley E, Panizzon MS, Kaup AR, Fennema-Notestine C, Neale MC, Jernigan TL, Fischl B, Franz CE, Lyons MJ, et al. . 2011. Genetic and environmental contributions to regional cortical surface area in humans: a magnetic resonance imaging twin study. Cerebral Cortex. 21(10):2313–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn ES, Shen X, Scheinost D, Rosenberg MD, Huang J, Chun MM, Papademetris X, Constable RT. 2015. Functional connectome fingerprinting: identifying individuals using patterns of brain connectivity. Nature Neurosci. 18(11):1664–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fornito A, Zalesky A, Bassett DS, Meunier D, Ellison-Wright I, Yucel M, Wood SJ, Shaw K, O’Connor J, Nertney D, et al. . 2011. Genetic influences on cost-efficient organization of human cortical functional networks. J Neurosci. 31(9):3261–3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox MD, Raichle ME. 2007. Spontaneous fluctuations in brain activity observed with functional magnetic resonance imaging. Nature Rev Neurosci. 8(9):700–711. [DOI] [PubMed] [Google Scholar]
- Friedman NP, Miyake A, Young SE, Defries JC, Corley RP, Hewitt JK. 2008. Individual differences in executive functions are almost entirely genetic in origin. J Exp Psychol Gen. 137(2):201–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge T, Holmes AJ, Buckner RL, Smoller JW, Sabuncu MR. 2017. Heritability analysis with repeat measurements and its application to resting-state functional connectivity. Proc Nat Acad Sci. 114(21):201700765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glahn DC, Curran JE, Winkler AM, Carless MA, Kent JW, Charlesworth JC, Johnson MP, Göring HHH, Cole SA, Dyer TD, et al. . 2012. High dimensional endophenotype ranking in the search for major depression risk genes. Biol Psychiatry. 71(1):6–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glahn DC, Winkler AM, Kochunov P, Almasy L, Duggirala R, Carless MA, Curran JC, Olvera RL, Laird AR, Smith SM, et al. . 2010. Genetic control over the resting brain. Proc Nat Acad Sci USA. 107(3):1223–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasser MF, Sotiropoulos SN, Wilson JA, Coalson TS, Fischl B, Andersson JL, Xu J, Jbabdi S, Webster MA, Polimeni JR, et al. . 2013. The minimal preprocessing pipelines for the human Connectome project. NeuroImage. 80:105–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon EM, Laumann TO, Adeyemo B, Huckins JF, Kelley WM, Petersen SE. 2016. Generation and evaluation of a cortical area Parcellation from resting-state correlations. Cerebral Cortex. 26(1):288–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon EM, Laumann TO, Gilmore AW, Newbold DJ, Greene DJ, Berg JJ, Ortega M, Hoyt-Drazen C, Gratton C, Sun H, et al. . 2017. Precision functional mapping of individual human NeuroResource precision functional mapping of individual human brains. Neuron. 95(4):791–807.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greicius M. 2008. Resting-state functional connectivity in neuropsychiatric disorders. Current opinion in neurology. 21(4):424–430. [DOI] [PubMed] [Google Scholar]
- Hagberg AA, Schult DA, Swart PJ. 2008. Exploring network structure, dynamics, and function using NetworkX In: Proceedings of the 7th Python in Science Conference (SciPy 2008), (SciPy), pp. 11–15. This work was self-published by the SciPy 2008 conference.
- Hall M-H, Smoller JW. 2010. A new role for endophenotypes in the GWAS era: functional characterization of risk variants. Harvard Rev Psychiatry. 18(1):67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuvel MPVD, Kahn RS, Goñi J, Sporns O. 2012. High-cost, high-capacity backbone for global brain communication. Proc Nat Acad Sci. 109(28):11372–11377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgson K, Poldrack RA, Curran JE, Knowles EE, Mathias S, Göring HH, Yao N, Olvera RL, Fox PT, Almasy L, et al. . 2017. Shared genetic factors influence head motion during MRI and body mass index. Cerebral Cortex. 27(12): 5539–5546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter JD. 2007. Matplotlib: a 2D graphics environment. Comput Sci Eng. 9(3):90–95. [Google Scholar]
- Jenkinson M, Beckmann CF, Behrens TEJ, Woolrich MW, Smith SM. 2012. FSL. NeuroImage. 62(2):782–790. [DOI] [PubMed] [Google Scholar]
- Kaiser RH, Andrews-hanna JR, Wager TD, Pizzagalli DA. 2016. Large-scale network dysfunction in major depressive disorder: meta-analysis of resting-state functional connectivity. JAMA Psychiatry. 72(6):603–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendler KS, Neale MC. 2010. Endophenotype: a conceptual analysis. Mol Psychiatry. 15(8):789–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee PH, Baker JT, Holmes AJ, Jahanshad N, Ge T, Jung J-Y, Cruz Y, Manoach DS, Hibar DP, Faskowitz J, et al. . 2016. Partitioning heritability analysis reveals a shared genetic basis of brain anatomy and schizophrenia. Mol Psychiatry. 21(12):1680–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li K, Atluri G A, Gowtham A. 2018. Connectomics in neuroimaging In: Wu G, Rekik I, Schirmer M, Chung A, Munsell B, editors. Connectomics in NeuroImaging. Vol 11083 Granada: Spain, Springer International Publishing, pp. 107–116. [Google Scholar]
- Mattfeld AT, Gabrieli JDE, Biederman J, Spencer T, Brown A, Kotte A, Kagan E, Whitfield-Gabrieli S. 2014. Brain differences between persistent and remitted attention deficit hyperactivity disorder. Brain. 137(9):2423–2428. [DOI] [PubMed] [Google Scholar]
- McKinney W. 2010. Data structures for statistical computing in python In: Proceedings of the 9th Python in Science Conference, pp. 51–56. This work was self-published by the Python in Science Conference.
- Müllner D. 2013. Fastcluster: fast hierarchical, agglomerative clustering routines for R and python. J Stat Software. 53(9):1–18. http://www.jstatsoft.org/v53/i09/. [Google Scholar]
- Neale MC, Cardon LR. 1992. Methodology for Genetic Studies of Twins and Families. Dordrecht, The Netherlands: Springer.
- Power JD, Cohen AL, Nelson SM, Wig GS, Barnes KA, Church JA, Vogel AC, Laumann TO, Miezin FM, Schlaggar BL, et al. . 2011. Functional network organization of the human brain. Neuron. 72(4):665–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruim RH, Mennes M, van Rooij D, Llera Arenas A, Buitelaar JK, Beckmann CF.. 2014. ICA-AROMA: a robust ICA-based strategy for removing motion artifact from fMRI data NeuroImage, 112:267–277. [DOI] [PubMed] [Google Scholar]
- Reineberg AE, Banich MT. 2016. Functional connectivity at rest is sensitive to individual differences in executive function: a network analysis. Human Brain Mapp, 37(8):2959–2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhea S-A, Gross AA, Haberstick BC, Corley RP. 2006. Colorado twin registry. Twin Res Human Gen. 9(6):941–949. [DOI] [PubMed] [Google Scholar]
- Rhea S-A, Gross AA, Haberstick BC, Corley RP. 2013. Colorado twin registry - an update. Twin Res Human Gen. 16(1):1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richiardi J, Altmann A. 2015. Correlated gene expression supports synchronous activity in brain networks. Science. 348(6240):11–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg MD, Finn ES, Scheinost D, Papademetris X, Shen X, Constable RT, Chun MM. 2016. A neuromarker of sustained attention from whole-brain functional connectivity. Nature Neurosci. 19(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinov M, Sporns O. 2010. Complex network measures of brain connectivity: uses and interpretations. NeuroImage. 52(3):1059–1069. [DOI] [PubMed] [Google Scholar]
- Salimi-Khorshidi G, Douaud G, Beckmann CF, Glasser MF, Griffanti L, Smith SM. 2014. Automatic denoising of functional MRI data: combining independent component analysis and hierarchical fusion of classifiers. NeuroImage. 90(0):449–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinclair B, Hansell NK, Blokland GA, Martin NG, Thompson PM, Breakspear M, de Zubicaray GI, Wright MJ, McMahon KL. 2015. Heritability of the network architecture of intrinsic brain functional connectivity. NeuroImage. 121:243–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SM, Andersson J, Auerbach EJ, Beckmann CF, Bijsterbosch J, Douaud G, Duff E, Feinberg DA, Griffanti L, Harms MP, et al. . 2013. Resting-state fMRI in the human connectome project. NeuroImage. 80:144–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SM, Fox PT, Miller KL, Glahn DC, Fox PM, Mackay CE, Filippini N, Watkins KE, Toro R, Laird AR, et al. . 2009. Correspondence of the brain’ s functional architecture during activation and rest. Proc Nat Acad Sci USA. 106(31):13040–13045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SM, Nichols TE, Vidaurre D, Winkler AM, Behrens TEJ, Glasser MF, Ugurbil K, Barch DM, Essen DCV, Miller KL. 2015. A positive-negative mode of population covariation links brain connectivity, demographics and behavior. Nature Neurosci. 18:1565–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thirion B, Varoquaux G, Dohmatob E, Poline JB. 2014. Which fMRI clustering gives good brain parcellations? Front Neurosci. 8(8 Jul):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turkheimer E, Haley A, Waldron M, D'Onofrio B, Gottesman II. 2003. Socioeconomic status modified heritability of IQ in young children. Psychol Sci. 14(6):623–628. [DOI] [PubMed] [Google Scholar]
- Vaidya CJ, Gordon EM. 2013. Phenotypic variability in resting-state functional connectivity: current status. Brain Connect. 3(2):99–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Heuvel MP, van Soelen ILC, Stam CJ, Kahn RS, Boomsma DI, Hulshoff Pol HE. 2013. Genetic control of functional brain network efficiency in children. Eur Neuropsychopharmacol. 23(1):19–23. [DOI] [PubMed] [Google Scholar]
- Van Essen DC, Smith SM, Barch DM, Behrens TEJ, Yacoub E, Ugurbil K, Consortium W-MH. 2013. The WU-Minn human connectome project: an overview. NeuroImage. 15(18):62–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitfield-Gabrieli S, Ford JM. 2012. Default mode network activity and connectivity in psychopathology. Ann Rev Clin Psychol. 8:49–76. [DOI] [PubMed] [Google Scholar]
- Whitfield-Gabrieli S, Thermenos HW, Milanovic S, Tsuang MT, Faraone SV, McCarley RW, Shenton ME, Green AI, Nieto-Castanon A, LaViolette P, et al. . 2009. Hyperactivity and hyperconnectivity of the default network in schizophrenia and in first-degree relatives of persons with schizophrenia. Proc Nat Acad Sci USA. 106(4):1279–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wig GS, Schlaggar BL, Petersen SE. 2011. Concepts and principles in the analysis of brain networks. Ann NY Acad Sci. 1224:126–146. [DOI] [PubMed] [Google Scholar]
- Yang Z, Zuo X-N, McMahon KL, Craddock RC, Kelly C, de Zubicaray GI, Hickie I, Bandettini PA, Castellanos FX, Milham MP, et al. . 2016. Genetic and environmental contributions to functional connectivity architecture of the human brain. Cerebral cortex (New York, NY: 1991). 26(5):2341–2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo BTT, Krienen FM, Sepulcre J, Sabuncu MR, Lashkari D, Hollinshead M, Roffman JL, Smoller JW, Zöllei L, Polimeni JR, et al. . 2011. The organization of the human cerebral cortex estimated by intrinsic functional connectivity. J Neurophysiol. 106(3):1125–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Raichle ME. 2010. Disease and the brain’s dark energy. Nature reviews. Neurology. 6(1):15–28. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sample genetic model and A, C, and E estimates for all pairwise connections are available for download at: https://github.com/AReineberg/genetic_connectome.