

Abstract
DNA‐templated self‐assembly represents a rich and growing subset of supramolecular chemistry where functional self‐assemblies are programmed in a versatile manner using nucleic acids as readily‐available and readily‐tunable templates. In this review, we summarize the different DNA recognition modes and the basic supramolecular interactions at play in this context. We discuss the recent results that report the DNA‐templated self‐assembly of small molecules into complex yet precise nanoarrays, going from 1D to 3D architectures. Finally, we show their emerging functions as photonic/electronic nanowires, sensors, gene delivery vectors, and supramolecular catalysts, and their growing applications in a wide range of area from materials to biological sciences.
Keywords: DNA-templating, DNA recognition, supramolecular chemistry, nucleic acids, nanowires, delivery
Inspired by DNA: By making use of DNA as templates, supramolecular interactions can be exploited for generating complex yet precise nano‐arrays of small‐molecules. In this review, we summarize how this growing approach has been explored so far, present the emerging functions, and the wide applications of such self‐assemblies in materials and biological sciences.

1. Introduction
1.1. Why Organizing Molecules in Precise Nanoarrays?
Supramolecular assemblies can display distinct and unique properties compared to the individual molecules they are made of. For instance, light‐harvesting complexes serve as a beautiful example showing how the organization of chromophores is instrumental for directing the cascade of energy and electron transfer processes.1 Viruses constitute another striking example, for which a precise number of proteins forming a capsid are arranged with a high level of organization around the genomic nucleic acid, which is essential for its delivery, and assembly/disassembly processes.2 Recently, researchers have reported many examples in the fields of electronics, sensing, delivery, and catalysis that illustrate emerging collective properties arising from supramolecular organizations occurring at different levels.3 Such a change in the properties when going from molecules to organized supramolecular assemblies is due to the intermolecular communication that is made possible by the close proximity of molecules held together through non‐covalent interactions. There is therefore a strong interest in developing non‐covalent methodologies that enable the robust, precise, and predictable arrangement of molecules into functional supramolecular systems.4
1.2. Why Using Nucleic Acids as Templates?
Besides their prominent biological functions as encoding matter, nucleic acids such as DNA and RNA have been recently considered as building blocks for the bottom‐up construction of 1D, 2D or even 3D nanomaterials. For instance, the field of DNA nanotechnology,5 based in part on DNA origami,6 where the sequence of nucleic acids can be harnessed and computed to program the self‐fabrication of complex multi‐component nanostructures, is blooming. The interest in nucleic acids as biomolecular templates originates from their water‐solubility, their ease of production through chemical synthesis or molecular biology techniques, and the diverse yet highly programmable primary structure (sequence), secondary structure (folding, shape, chirality) and tertiary structures (e. g. origami, tiles, cubes) they can adopt.7 In addition, DNA contains a huge information density that can serve to encode information for storage (up to 2 bits per nucleotide) or, for instance, to program sequence‐controlled polymers that are structurally unrelated to nucleic acids.8
While the covalent modification and functionalization of nucleic acids require delicate synthetic methodologies, supramolecular approaches interestingly use non‐modified and readily‐available nucleic acids.7b, 9 A supramolecular approach also offers room for self‐correction during self‐assembly, erasing intermediates formed under kinetic control and leading to the formation of the thermodynamic product. However, achieving the controlled organization of small molecules onto nucleic acid requires a deep understanding of the binding mechanisms and non‐covalent interactions at play with nucleic acids. Consequently, this knowledge guides the adequate molecular design of nucleic acid ligands. Finally, by characterizing the resulting templated self‐assembly one may evidences emerging functions arising from the collective supramolecular organization of the system.
In this review, we aim at providing an overview of this field, which could help in guiding the reader toward the design of functional DNA‐templated supramolecular assemblies. It is specifically oriented towards organic molecules appropriately engineered to bind DNA and to be organized via a template effect. For reports on DNA recognition by metal complexes and DNA‐templated assembly of inorganic nanostructures, the reader is referred to as recent accounts (see, for example, references10 and,11 respectively).
We start this review by providing a short overview of the possible recognition modes between single molecules and DNA (or RNA). Then, we describe the recent advances of nucleic acids templating of small organic molecules, partitioned by the main type of recognition mode at play. Various types of supramolecular organizations driven by the template effect are considered, and we particularly show the main trends towards sophisticated supramolecular systems. We then shortly review the recent developments on DNA‐templated polymerization, i. e. the polymerization of (supra)molecular guests pre‐assembled onto the nucleic acid template, in particular for dynamic polymerization processes. Finally, we focus this account on the unique functions that may be achieved using DNA‐templated supramolecular self‐assemblies, with potential applications in photonic wires, conducting nanowires, (bio)sensors, delivery systems, and catalysts.
2. Binding to DNA
Nucleic acids are like necklaces of nucleobases (Adenine A, Cytosine C, Guanine G, Thymine T or possibly uracil U) covalently tethered to a ribose sugar, assembled through phosphodiester bridges. Nucleic acids are poly‐anions in which nucleobases are engaged in π‐stacking interactions with one another. Base pairing, through complementary Watson‐Crick‐type or Hoogsteen‐type hydrogen bonds, results in the folding of single‐stranded DNA (ssDNA), the formation of helicoidal double‐stranded DNA (dsDNA), or more complex structures like triplex, i‐motif and G‐quadruplexes. While single‐stranded DNA can be seen as a flexible helicoidal polymer (persistence length from 0.8 nm to 4.0 nm, depending on the ionic strength12), B‐type double‐stranded DNA is a rigid right‐handed helix having a diameter ∼2.4 nm, a pitch of 10 base pairs (ca. 3.4 nm), and a persistence length on the order of 50 nm which amounts to about 150 base pairs,13 thus providing local stiffness which can be useful to program linear 1D nanostructures. Double‐stranded DNA features a major and a minor groove – differing in size and hydration – that can accommodate ligands. In summary, DNA can offer four handles for supramolecular interactions with ligands: base pairing, recognition of phosphodiesters, recognition by groove binding, and recognition by intercalation (Figure 1).14
Figure 1.

Possible recognition modes of organic molecules to single‐stranded DNA (top) and double‐stranded DNA (bottom). Examples of minor groove‐binding and intercalation modes were extracted from the Protein Data Bank.
2.1. Recognition of Nucleobases
Base‐pairing is a straightforward way to bind DNA in a selective manner and would make possible the binding of different molecules in a precise array based on the DNA sequence. Indeed, the Watson‐Crick base‐pairing approach has been undertaken by several researchers, who covalently attached a nucleobase to a non‐natural molecule, in view of pairing the synthetic molecule to complementary nucleobases of a single‐strand DNA (Figure 1 top left). The G⋅C pairing offers a much stronger interaction than that of A⋅T pairing, not only on the number of primary H‐bonds (3 for G⋅C, 2 for A⋅T), but also on the secondary H‐bonding interactions (overall attractive for G⋅C, while repulsive in the case of A⋅T). It has been experimentally observed that the binding free energy, determined in CDCl3, is +190 % higher (more stable) for G⋅C than for A⋅T in the same conditions (ΔG=−24.5 kJ/mol for G⋅C and ΔG=−8.5 kJ/mol for A⋅T).15 Therefore, G⋅C pairing has been thoroughly used to achieve a wide range of self‐assembled structures,16 but to a lower extent in DNA‐templating approaches.
The main reason for not relying on G⋅C pairing when considering DNA templates made of a single type of nucleobase arises from the tendency of G‐rich structures to form G‐quartets via Hoogsteen hydrogen bonds. Indeed, G‐rich oligonucleotides are prone to form intra‐ or inter‐molecular G‐quadruplexes,17 which is detrimental for achieving DNA‐templated assembly. In the case of the guests, guanine‐capped molecules have a tendency self‐assemble into G‐quartets or in ribbon‐like structures.18 which is also unfavorable when considering the binding to an oligoC template. Another practical aspect relates to synthesis since the commercial availability of readily functionalized pyrimidine nucleosides such as dU‐I (5‐iodo‐2’‐deoxyuridine) facilitates the access to nucleobase‐modified oligo‐pyrimidine templates. Consequently, oligo(adenine) or oligo(thymine) templates were most often considered for achieving DNA‐templated assemblies (see examples Scheme 1), as discussed in Section 3.1. Alternatively, non‐natural moieties that pair to nucleobases were also envisioned, which permits the tuning of the H‐bonding interactions with nucleobases. For instance, a diaminotriazine unit pairs thymine via 3 primary H‐bonds (Scheme 1 bottom), which allowed us to form stable monodisperse DNA‐templated assemblies that extend over tens (up to forty) bases, and can also bind very long DNA templates, made of a few thousand bases.19 Other units possessing a large number of H‐bonding donors and acceptors can yield sophisticated templated structures, as for instance a cyanuric acid (possessing 3 ‘faces’ like thymine) can in principle interact with 3 adenines via H‐bonds. When cyanuric acid is mixed with an oligoadenine template, supramolecular self‐assembly yields rosette‐like structures forming elongated fibers, as reported by Sleiman et al. (see Section 3.1).20 The use of non‐natural bases (often referred to as ‘xenobases’) is appealing to expand the possibilities of templating, by increasing the number of template sites to attach molecular guests, and to permit orthogonal binding in complex mixtures made of different guest molecules.
Scheme 1.

Examples of base pairing approaches utilized in the context of DNA‐templated supramolecular self‐assembly. Pairing between: A) a thymine nucleotide‐based guest and oligoadenine; B) an uracil nucleoside‐based guest and oligoadenine; C) a diaminotriazine‐based guest and oligothymidine
2.2. Recognition of Phosphodiesters
In natural nucleic acids, nucleotides are linked through a phosphodiester group that bears a negative charge, particularly important for endowing water solubility and contributing to the well know helical structure of DNA. On top of that, phosphodiester groups can also engage in electrostatic interactions with cationic entities such as inorganic salts and organic cations (see example Figure 1 top right), which can be of particular interest for generating supramolecular DNA‐templated self‐assemblies.
The phosphodiester groups of DNA are deprotonated at pH >2.5 and roughly 50–75 % of them interact with cations.21 This means that interaction with the phosphodiester backbone must be thought of as a competition with condensed cations. While the interaction with a single compound of interest has thus to compete with the salts (e. g. Na+) naturally present along DNA, the use of multivalent ligands usually solves this issue by enabling stronger interactions to be established with DNA. For instance, the Farrell group has developed polynuclear platinum compounds that interact with DNA by recognition of phosphodiester backbone (Figure 2).22 This new mode of interaction was coined the “phosphate clamp”.23
Figure 2.

Chemical structure of octacationic trinuclear platinum complex (top) and X‐ray crystal structures (views perpendicular and along the helical axis, respectively on bottom left and bottom right) of its complex with DNA duplex [d(CGCGAATTCGCG)]2). The phosphodiester backbone of the DNA is represented by a tube. Reproduced with permission from reference.23b Copyright 2006 American Chemical Society.
Cationic organic compounds can also be the basis of phosphodiester ligands. For instance, among organic cations, the guanidinium group clearly stands out since, similarly to diaminoplatinum complexes, it can establish hydrogen bonds in addition to attractive electrostatic interactions (Scheme 2). Such so‐called salt‐bridge interaction makes the guanidinium group a solid choice for designing polycationic systems that recognize polyanionic biomolecules (for reviews, see for instance references24).
Scheme 2.

Structural representation of salt bridge interactions with phosphodiesters by cationic platinum complex (left), and guanidiniums (right).
2.3. Recognition by Groove Binding
Given the DNA double‐helix structure, shape‐selective molecular recognition processes may occur with its minor‐groove or its major‐groove. Particularly, small cationic molecules that adopt a convex shape are known to bind the minor‐groove by shape complementarity, electrostatic interactions with the phosphodiester backbone and hydrogen bonding with nucleobases. For instance, small molecules containing pyrrole rings or phenylindole moieties, with cations such as amidinium groups, are well‐known binders to A−T rich regions of dsDNA.25 This is the case for the well‐known DAPI (4’,6‐diamidino‐2‐phenylindole), a vastly used fluorescent DNA marker for staining cell nuclei.
DNA sequence‐selectivity of minor‐groove binders has been achieved by the synthesis of oligomers containing defined heterocycles containing H‐bonding donors/acceptors, as for instance pyrrole and imidazole rings. These pyrrole‐imidazole polyamides were pioneered by Dervan and coworkers, who designed a series of oligomers that bind in pairs or in hairpin, with DNA sequence‐selectivity,26 to the DNA minor‐groove (see examples Figure 3).27 These selective DNA binding ligands have been successfully applied for modulating gene expression28 but have received less attention as a supramolecular tool to generate DNA‐templated self‐assemblies.
Figure 3.

Sequence‐selective DNA minor‐groove recognition by pyrrole‐imidazole polyamides. a) Schematic representation of the binding of a hairpin polyamide (Py: Pyrrole; Im: Imidazole; Hp: Hydroxypyrrole, adapted from ref. [27]. b) Model of the crystal structure of a polyamide dimer into the DNA minor‐groove (sequence CCAGATCTGG), as extracted from the Protein Data Bank (PDB ID: 1CVY).
Recently, Wilson et al. proposed that the thiophene ring can also bring sequence‐selectivity, through the thiophene “sigma‐hole” (σ‐hole), i. e. the interaction between low‐lying σ* orbitals and positive electrostatic potential and electron donors, such as a nitrogen atom of a benzimidazole next to the thiophene ring. This interaction restricts conformationally the molecule to interact with the guanine amino group within the DNA minor‐groove via hydrogen bonding, and yield specificity towards G⋅C base pairs centered in a track of A ⋅ T base pairs29 This study was then extended to a wide range of compounds able to bind via σ‐hole, and the effect of the shape of the molecule on the specificity towards mixed sequences were studied.30
2.4. Recognition by Intercalation
Watson‐Crick base pairing in double‐stranded DNA leads to the side‐by‐side arrangement of the π‐aromatic surfaces of nucleobases (total surface are ca. 280 Å2).31 As a consequence, π–π stacking interactions between neighboring base pairs play an important role in the stability of dsDNA. Interestingly, it also gives a way to design synthetic compounds that recognize DNA by intercalation within those adjacent base pairs, like a “coin inserted into a stack of base pairs” (see example Figure 1 bottom right, for an acridine‐based intercalator), as initially proposed by Lerman.32 DNA intercalation of planar molecules results in an expansion along the DNA helix main axis which increases intrinsic viscosity upon ligand intercalation. Binding typically occurs with association binding constants in the range 103–107 m −1. Multiple binding sites are offered by dsDNA but the neighbor‐exclusion principle makes that every second (next‐neighbor) intercalation site along DNA main axis remains unoccupied.33 Poly‐intercalation of multiple ligands onto DNA is a multi‐step process that usually occurs non‐cooperatively. Bisintercalation into dsDNA by natural34 or synthetic35 molecular tweezers36 displaying two planar ligands linked through an appropriate spacer37 is a common mode of recognition of natural DNA, as well as DNA containing abasic or mismatch sites.38 Iverson and co‐workers even reported a tetra‐intercalator compound able to bind DNA with exceptional kinetic stability (half‐life of dissociation: 16 days at 100 mM NaCl).39 The design was optimized to install most‐appropriate linkers spanning four base pairs through both the major and the minor grooves of DNA.
3. DNA‐Templated Self‐Assembly of Small Molecules
One critical aspect in generating functional systems by DNA‐templated supramolecular self‐assemblies is to be able to control the precise assembly of multiple ligands in dense arrays where communication and interaction between those ligands are possible so that emerging properties appears. In order to do so, the most effective strategy is to design a cooperative system where binding of the first ligand favors the binding of the following one, and so on until the DNA template is populated at its maximum density. Cooperativity in DNA‐templated self‐assembly can be achieved under circumstances where there is an appropriate balance of ligand—ligand and ligand‐DNA free energy of interaction.19b, 40 Another important aspect to consider is the chirality transfer from the nucleic acid template to the assembly of small achiral molecules, possibly yielding well‐defined helical stacks, as recently reviewed in reference.41
3.1. Assemblies Driven by Interactions with Nucleobases
To the best of our knowledge, one of the first examples of DNA‐templated supramolecular self‐assembly of synthetic molecules was reported by Shimizu et al., who designed a bolaamphiphile made of an alkyl chain (20‐mer) end‐capped on both extremities with thymidine nucleotides. When mixed with an oligoadenine (oligoA) in water, supramolecular self‐assembly of the bolaamphiphile through A⋅T base pairing yields nanofibers, whose lengths greatly surpasses the length of the DNA template.42 This is due to a “double‐zipper” self‐assembly process, where the templates bound to each extremity of the molecule are in a staggered arrangement. The same approach of double‐zipper assembly was then used to achieve nanofibers of π‐conjugated molecules such as oligo(para‐phenylene vinylene)s.43
The “single‐zipper” approach, where the molecular guest to be templated contains one moiety able to pair nucleobases via hydrogen bonding to the DNA template, was explored by several groups, in particular the groups of Balaz, Meijer and Schenning, Stulz, Wagenknecht, and us.19, 44 In such approach, different types of mechanisms of templated self‐assembly were observed, depending on template‐guest interactions and guest‐guest interactions. This has been rationalized by theoretical models of templated supramolecular polymerization developed by Jabbari‐Farouji and van der Schoot, who provided a coarse‐grained model based on the free energies involved between the different partners and showed the important factors that contribute to cooperativity.40
Notable developments of this approach are expected by harnessing the power of DNA‐templating to program a well‐defined sequence of multiple chromophores based on the DNA mother sequence. A significant step in this direction has been recently reported by Wagenknecht et al., who synthesized a pyrene derivative bound to an A‐type nucleoside, and another compound made of a nile red derivative bound to U‐type nucleoside (Figure 4).45 The former pairs to a thymine base, while the latter pairs to an adenine base. They mixed these two chromophores with various types of DNA templates made of either pure T, pure A, and mixed A−T sequences. In the mixed sequences, optical spectroscopy clearly indicates the formation of arrays based on a sequence‐selective assembly of the two chromophores along the DNA template.45 Furthermore, the same group reported the design of identical chromophores (ethynylpyrene and nile red derivatives), each attached to the same uracil base, but with either to d‐ or l‐sugars (2’‐deoxyribofuranosides). They studied the DNA‐templated assemblies of each chromophore using either d‐DNA or l‐DNA templates. Remarkably, they observed that the supramolecular chirality of the chromophore/DNA assemblies is not simply controlled by the configuration of the DNA template, but depends on the nature of the chromophore. The helicity of the pyrene‐based DNA‐templated assemblies are controlled by that of the DNA. In contrast, the nile red already form chiral stacks in the absence of template (whose helicity depends on the configuration of the sugar) and, upon addition of the template, overrule the chirality of the whole DNA‐templated assembly.46 The different behaviors between these two types of chromophores is ascribed to the strong π‐π interactions in the case of nile red, which influences the helical organization of the DNA/chromophores assembly (as the helical pitch), as also observed by us in the case of other types of π‐conjugated molecules.19a
Figure 4.

Top: structure of the pyrene‐adenine and nile red‐uracil derivatives prepared for DNA‐templated self‐assembly of bichromophore arrays. Bottom: schematic representation of their assembly along two different templates. Reproduced with permission from reference,45 Copyright 2018 Wiley‐VCH.
Besides these so‐called “zipper‐like” approaches via base pairing, yielding cable‐like plain structures, another manner to achieve complex supramolecular systems is to harness multiple patterns of H‐bonding donors and acceptors, yielding for instance “rosette‐like” or hexaplex structures. This has been proposed by Sleiman and colleagues, who carried out an in‐depth study of the supramolecular self‐assembly of cyanuric acid with relatively short oligoadenines (oligonucleotides of 15 adenine units).20 They observed that hexameric rosette‐like structures were formed, which yielded long fiber‐like structures that extend over micrometers, very long compared to the size of the oligoadenine (approximately 5 nm fully extended). The fiber growth was found to occur through a cooperative self‐assembly mechanism involving a staggered arrangement of the DNAs around the cyanuric acids, as shown in Figure 5.
Figure 5.

Top: Model of templated assembly between cyanuric acid (red) and oligodeoxyadenine dAn (blue) into rosette‐like structures. Bottom: AFM images in solution, showing long fibers by staggered supramolecular self‐assembly of dA15 with cyanuric acid. Adapted from reference.20 Copyright 2016 Nature Publishing Group.
Another recent example of such rosette‐like structure obtained with non‐natural bases was reported by Asanuma et al., who designed an artificial nucleic acid based on a d‐threoninol scaffold bearing amino‐pyrimidine or cyanuric acid. As these two derivatives have complementary H‐bonding patterns, the chains self‐assemble to form hexaplex structures upon mixing the two types of artificial nucleic acids.47 Remarkably, this hexaplex structure has a pore at its center, which could be further extended to achieve specific ion channels.
3.2. Assemblies Driven by Interactions with Phosphodiesters
The resort to multivalent binding is a powerful solution to the issue that the weak interaction of a single monocationic organic ligand has to compete with condensed inorganic cations. In this line, Vázquez, Mascareñas and co‐workers reported a DNA‐recognition hybrid system that combines a DNA‐binding peptide with an oligoguanidinium fragment interacting with the neighboring phosphodiester backbone (Figure 6).48
Figure 6.

Hybrid DNA recognition system combines DNA‐binding α‐helix peptide with oligoguanidiniums that interacting with the neighboring phosphodiester backbone. a) chemical structures of oligoguanidiniums; b) structural proposition for the interaction of the oligoguanidiniums with the phosphodiester backbone (view along DNA axis); c) cartoon representation of DNA recognition by the hybrid system. Reproduced with permission from reference.48 Copyright 2015 The Royal Society of Chemistry.
Very recently, Lynn and co‐workers have also reported a study showing how such salt‐bridge interactions could explain the role of nucleic acids in facilitating the growth of amyloid assemblies.49 In a different topic, Herrmann and co‐workers have shown how such salt‐bridge interactions can be used for preparing PEGylated DNA supramolecular complexes that are able to hybridize in salt‐free water.50 These examples show that the phosphodiester backbone binding mode has now been recognized as a common way of interacting with nucleic acids. However, the main caveat to note here is that the recognition of the phosphodiester backbone by polycationic ligands, usually at valency ≥3,21 leads to a strong local compensation of charges which in turns may strongly alter the overall structure of nucleic acids if it propagates through long distances. For instance, the octacationic trinuclear platinum complex (Figure 2) has recently been found to effectively condense DNA and RNA oligonucleotides as short as 20 base pairs.51 Therefore, in the context of DNA‐templated self‐assembly, caution must be taken as to the impact of the self‐assembly, by multivalent cations via phosphodiester recognition, on the structure of the DNA template within the final nanostructure, even though both extreme cases – DNA unaffected vs. DNA condensed21 – can be interesting.
Oligoguanidiniums such as those described above (e. g. in Figure 6) usually require tedious synthesis, and their purification and isolation can be tricky as well. An alternative is to design monocationic organic ligands capable of self‐assembly, through non‐covalent interactions, which result in multivalent binding and stable DNA‐templated self‐assembly. In this line, Ulrich and co‐workers developed a dynamic combinatorial chemistry approach that led to the identification of cationic and aromatic side‐groups as stabilizing ssDNA‐templated self‐assemblies of guanidinium compounds (Figure 7).52 The fact that cationic side‐groups have a stabilizing effect was expected since it would promote multivalent binding. The role of aromatic groups was more surprising and attracted our attention. Thus, our groups studied the contribution of π‐stacking interactions to the stabilization of such DNA‐templated self‐assemblies based on guanidinium‐phosphodiester recognition.
Figure 7.

Principle of the dynamic combinatorial chemistry approach implemented for identifying side‐groups that stabilize ssDNA‐templated self‐assemblies of bisfunctionalized guanidiniums.
Three bisfunctionalized guanidinium compounds were prepared that bears aromatic side‐groups varying in size (benzene in GuaBiPhe, naphthalene in GuaBiNaph, pyrene in GuaBiPy (Figure 8).53 Binding of GuaBiPy to dT40 has been evidenced by fluorescence and circular dichroism (CD) spectroscopies. While the former showed a decrease in the monomer emission band concomitant with the appearance of an excimer emission band upon ssDNA addition, the latter revealed an induced CD band centered on the pyrene absorption band at 340 nm. These results showed that multiple ligands were interacting with the ssDNA template and were arranged in close proximity. Binding was found to be fully thermally‐reversible, dissociation occurring when increasing temperature. Titration experiments indicated a loading of up to 30 ligands per dT40. In addition, the presence of phosphate buffer was shown to destabilize the resulting self‐assembly, thereby indicating that salt‐bridge interactions between guanidinium groups and phosphodiesters backbone are the driving force for self‐assembly. Comparison of the three different ligands, assisted by MALDI‐ToF mass spectrometry analysis of competition experiments, revealed that the larger aromatic side‐groups, the more stable the resulting ssDNA‐templated self‐assembly. Altogether, these results show that these bisfunctionalized ligands are able to self‐assemble onto ssDNA through salt‐bridge interactions with the phosphodiester backbone, assisted by stabilizing secondary π‐stacking interactions between the ligands (Figure 8). Further exploiting this prime role of π‐ stacking interactions, we have recently inserted a photoswitchable azobenzene‐based molecule, which was observed to bind to the DNA minor‐groove and was expected to influence these interactions depending on its twisted Z or coplanar E configuration. Remarkably, when interacting with ssDNA, the photoswitching of the azobenzene‐based compound allowed us to control the DNA binding of the pyrene‐based compound GuaBiPy, which is detected by its unique fluorescence signals.54 These results demonstrated that it is possible to achieve a photocontrol of DNA binding in heteromolecular templated assemblies, using photoswitchable ligands.
Figure 8.

Chemical structure of bisfunctionalized guanidinium compounds bearing aromatic side‐groups of varying sizes (left), and model for their templated self‐assembly onto single‐stranded dT DNA (right). The black spheres represent the aromatic side‐groups.
George, Lazzaroni, Surin and co‐workers reported a perylene diimide end‐capped with two dipicolyl‐ethylenediamine‐ZnII receptor motif, which undergoes a ssDNA‐templated self‐assembly mediated by electrostatic interactions between the metal center and DNA phosphodiesters of single‐stranded dA20 and dA40 (Figure 9).55 It was observed that the larger π‐conjugated system PDPA gives rise to the most stable DNA‐templated self‐assembly when compared with NDPA. Interestingly, a cooperative self‐assembly mechanism took place in this case, yielding left‐handed supramolecular stack of PDPA.
Figure 9.

Left: chemical structures of PDPA and NDPA compounds for DNA‐templated self‐assembly in the presence of single‐stranded dA20 and dA40. Counter‐anions are, respectively, perchlorate and nitrate. Right: model of supramolecular organization of ssDNA‐templated stacks of PDPA in a M‐helix (left‐handed helical stack), obtained via molecular modeling simulations. The Zn atoms are represented in balls and the DNA phosphodiester backbones in bold sticks. Adapted with permission from reference.55 Copyright 2016 The Royal Society of Chemistry.
3.3. Assemblies Driven by DNA Groove Binding
Armitage and co‐workers studied the DNA‐templated self‐assembly of tricationic cyanine dyes (Figure 10).56 The authors found precise conditions where J‐type aggregates form spontaneously in the presence of double‐stranded DNA with a high degree of cooperativity. Monomeric minor groove binding is inferred from weak positive induced circular dichroism (CD) signals while further aggregation of the dye along the DNA minor groove gave intense split CD signals. Those split CD signals occurred at surprisingly low dye/DNA ratios, pointing to a cooperative mechanism of binding. The first binding widens the minor groove which in turns promotes binding of subsequent ligands, thus explaining the origin of the cooperativity observed. A model was proposed where π‐stacked dimers of dyes bind to the DNA minor groove and propagate along the floor of the groove in a chain‐polymerization mechanism, until reaching changes in sequence or saturation of the DNA template (Figure 10).56, 57
Figure 10.

Chemical structure of tricationic cyanine dyes (top), and proposed cooperative mechanism of binding onto dsDNA through lateral aggregation within the DNA minor groove (bottom).
Very recently, Peng, Ding and co‐workers reported that achiral cyanine molecules made of a carbazole core self‐assemble in the presence of dsDNA through minor‐groove recognition.58 Interestingly, the effective transfer of chiral information from the DNA template to the bounds ligands resulted in a strong circularly polarized luminescence (CPL), with a dependence of DNA composition and chirality onto the amplitude and sign of the CPL output.
Exploiting multivalent binding with polytopic ligands, Vázquez, Mascareñas, and co‐workers reported the design of peptidic DNA binders that combine two modules of ‘zinc finger’ transcription factor (GAGA) and one peptide referred to as “AT‐hook” (Figure 11).59 This trimeric molecule binds DNA via “major‐minor‐major” groove interaction, which was shown to yield high affinity towards DNA and excellent sequence‐selectivity. This ‘multi‐groove’ binding approach is very appealing to build sophisticated DNA‐templated assemblies, permitting the positioning of different units along a relatively large section of DNA.
Figure 11.

a) Design approach for achieving DNA‐binding molecules with major‐minor‐major groove recognition with different DNA binding modules (GAGA and A−T hook); b) Sketch of the recognition molecule by this trimeric molecule. The sequence of the peptide linkers connecting the modules is shown in red, and the DNA sequence is written below. Reproduced with permission from reference.59 Copyright 2018 The Royal Society of Chemistry.
3.4. Assemblies Driven by Intercalation Through Base Pairs
A single intercalating agent can be used to assemble hybrid systems by DNA templating through intercalation within base pairs. For instance, O'Reilly, Stulz and co‐workers tethered an acridine group – well known for its propensity to intercalate within double‐stranded DNA (see Figure 1 bottom right) – at the end of synthetic polymers obtained by Reversible addition‐fragmentation chain transfer (RAFT) polymerization process.60 Using a short 63 base pairs DNA sequence as template, discrete (10 nm diameter, as estimated by DLS) and well‐defined DNA–polymer hybrids were obtained and observed by AFM (Figure 12).
Figure 12.
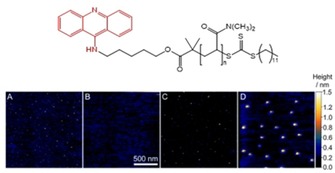
Top: chemical structure of acridine‐terminated polymer; bottom: DNA‐templated self‐assembly of DNA‐polymer hybrid systems driven by intercalation. AFM images of A) DNA alone, B) polymer with the acridine group alone, C) DNA in the presence of a polymer which does not bear an acridine group, and D) DNA in the presence of the polymer bearing the acridine group. Reproduced with permission from reference.60 Copyright 2014 The Royal Society of Chemistry.
Tuite, Pike and co‐workers reported the DNA‐templated self‐assembly of multiple proflavine‐derived compounds (Figure 13). Binding to DNA was shown to occur following the nearest‐neighbor intercalation, as evidenced by negative induced circular dichroism signals as well as linear dichroism, with association constants in the range 104–106 m −1 and little sequence selectivity.61 λ‐DNA was used as a template onto which up to 24251 intercalators can bind per helix. Conditions for full loading were described, even though binding did not occur in a cooperative manner. Interestingly, azide‐modified proflavines behave similarly and enabled further functionalization by cycloaddition with corresponding alkynes. The authors demonstrated in situ functionalization of such 1D DNA‐templated self‐assembly (Figure 13).62 For instance, the click reaction with an alkyne‐bearing green fluorescent dye gave DNA‐templated nanostructures, which was investigated by fluorescence microscopy (Figure 13).
Figure 13.

Chemical structures of proflavine‐derived intercalator and alkyne‐bearing green fluorescent dye (top), mechanism of poly‐intercalation into dsDNA followed by in situ functionalization by cycloaddition (bottom), and AFM and fluorescence images (bottom right, reproduced with permission from reference,62 Copyright 2015 Wiley‐VCH.
4. DNA‐Templated Polymerization
Nature utilizes nucleic acids‐templated polymerization at the very heart of information transfer in cells, using an enzymatic machinery. For eukaryotes, this occurs both in transcription (DNA‐templated polymerization of RNA) and translation (RNA‐templated polymerization of proteins). These biological processes are inspiring to achieve a very high level of precision both on the number and on the sequence of monomers along the polymer backbone. Different approaches were undertaken to polymerize monomer units pre‐assembled on a DNA template for achieving precision polymers, which is the subject of a minireview in 2016 by one of us.63 Here we report only the latest developments in this field, driven by using either 1D nucleic acids to scaffold dynamic polymers or supramolecular polymers, or using 2D or 3D DNA scaffolds to pattern polymers into more complex structures.
4.1. Dynamic Polymerization with 1D DNA Templates
The field of DNA‐templated synthetic polymers has been pioneered by the group of Lynn, who used dynamic covalent chemistry (DCC) to polymerize thymidine derivatives along an oligodeoxyadenine template, using dynamic imine bonds.64 In 2012, Schenning et al. reported the use of DCC to polymerize hydrazine units (bearing naphthalene chromophores) along a ssDNA template made of 40 thymine bases.65 Upon cooling a mixture of hydrazine monomers and template, the units assembled on the template via hydrogen bonding interactions, and the subsequent addition of a dialdehyde (glyoxal) yielded polymerization into a hydrazone polymer, as confirmed by comparing experimental and theoretical CD spectra. Heating the solution permitted the disassembly the DNA/polymer hybrid and a successive cooling led to the precipitation of the polymer. Remarkably, this offered the possibility to re‐use the template for a new cycle of templated polymerization. More recently, Aida et al. reported the design of molecules containing several guanidinium groups substituted with ethylene oxide chains and terminated on both extremities by thiol groups (Figure 14a).66 Note that these molecules contain guanidinum groups, which are particularly effective to bind phosphates of nucleic acids (see above). In presence of siRNA, templated self‐assembly occurs, and the proximity of thiol groups along the template allowed the oxidative polymerization into disulfide polymers upon addition of the oxidant KI/I2 (Figure 14b). This templated polymerization yields nanoobjects (made of siRNA and disulfide polymers) that are uniform in size and have a diameter of around 7 nm, called nanocaplets. The latter were utilized to release the siRNA into cells (see further details in Section 5.4 related to delivery applications).
Figure 14.

Nucleic acid‐templated polymerization of a water‐soluble tetraguanidininium derivative containing thiol groups at both termini. (a) Molecular structure of the building block TEGGu4; (b) Principle of siRNA‐templated oxidative polymerization and reductive depolymerization. Reproduced with permission from reference.67 Copyright 2017 The Royal Society of Chemistry.
Recently, Sollogoub, Bouteiller and colleagues reported the supramolecular polymerization of β‐cyclodextrin (β‐CD) derivatives along a DNA template.68 The β‐CD is designed in such a way that it is bridged on the lower rim, and an adamantane is attached with a linker to one amino group at the edge of the bridge (Figure 15). This geometry prevents self‐inclusion and dimerization processes (Figure 15, left). In aqueous solutions in the mM range, this β‐CD self‐assembles into supramolecular polymers via a host‐guest recognition, in a non‐cooperative manner (Figure 15, top right). Another important feature of this compound is that it possesses two positive charges located on the amino groups of the bridge. The researchers explored the assembly of such compound with a dsDNA (T4‐DNA, 166 kbp) as template for supramolecular polymerization. Even at very low concentration in β‐CD (down to 3 μM), the researchers observed the complexation of DNA (0.1 μM), and the supramolecular polymerization of β‐CD templated by DNA occurred in a cooperative manner (Figure 15, bottom right). The complexation also yielded the compaction of DNA, as evidenced by fluorescence microscopy. Remarkably, the DNA compaction was promoted at very low concentration (3 μM) with only two positive charges per β‐CD molecule, whereas for a well‐known compacting agent such as spermine (with four cationic charges), DNA compaction occurs at 5 μM in the same conditions.
Figure 15.

Structure of the bridged β‐CD−adamantane molecule able to form supramolecular polymers by host/guest complexation. Isodesmic supramolecular polymerization occurs by self‐assembly in the mM range, whereas cooperative supramolecular polymerization occurs in the μM range presence of a DNA template. Adapted with permission from reference.68 Copyright 2018 Wiley‐VCH.
Ultimately, the control over the sequence of the DNA/RNA template would permit the design of a monodisperse, sequence‐controlled polymers which are different in nature than the template, as it is the case in cells at the level of translation. A notable and encouraging advance in this perspective has been achieved by the group of Liu, who developed an enzyme‐free approach based on the designing of a cleavable macrocycle containing a peptide nucleic acid (PNA) that hybridizes on the DNA template.8a This allowed the production of various types of sequence‐controlled polymers (with no structural relationship with DNA) with a molecular weight up to the range of 26 kDa.
4.2. Polymerization with 2D and 3D DNA Templates
In 2014, Wang and Ding introduced the utilization of DNA origami as 2D templates to polymerize aniline, through the use of DNAzymes (catalyzing the oxidation of aniline by H2O2) that were incorporated at preprogrammed locations within the origami.69 Very recent examples show the extension of this approach to better controlled polymerizations, as reported by Weil et al. for the polymerization by Atom‐Transfer Radical Polymerization (ATRP) on pre‐programmed locations of a DNA origami template.70 They selected poly(ethylene glycol) methyl ether methacrylate (PEGMEMA) as a biocompatible polymer. To achieve this goal, Weil, Wu, et al. immobilized the initiator (bound to a DNA) at precise locations on the surface of DNA origami. The ATRP process was achieved on different types of origami, yielding nanopatterned polymers of various sizes. Remarkably, by changing the ratio monomer/initiator, the degree of polymerization could be tuned. This concept has then been extended to the polymerization of dopamine through a different polymerization process.71 The polymerization of dopamine only occurs at pre‐programmed locations on the DNA origami, at the exact position of nanodomains of G‐quadruplex/hemin (Figure 16). This DNAzyme oxidized the dopamine into dopaminochrome, which oligomerized and eventually led to polydopamine nanostructures precisely located on G4/hemin nanodomains. This is illustrated in Figure 15, showing that the pre‐programmed domains of the DNAzyme on the origami are coated after the reaction with dopamine, the lines or crosses on the DNA origami being identified as polydopamine nanopatterns, as shown by AFM. Furthermore, the same group recently reported the photo‐polymerization of dopamine on DNA origami tubes, by using a protoporphyrin photosensitizer.72 Altogether, these studies open the way to the controlled fabrication of polymer nanopatterns on DNA, with many possibilities of 2D and 3D shapes and sizes owing to the addressability offered by DNA origami templates.
Figure 16.

Top: proposed mechanism of polydopamine formation on DNA origami containing G‐quaduplex (G4)/hemin nanodomains. Bottom: sketch of DNA origami with different shapes of G4/hemin nanodomains (orange), and the corresponding AFM images. Adapted from reference.71 Copyright 2018 Wiley‐VCH.
5. Towards Functional Self‐Assemblies
5.1. DNA‐Templated Photonic Nanowires
In the search for light‐harvesting antenna and nanophotonic devices, DNA‐based photonic wires have attracted strong interest, since the rigidity of DNA together with the possibility to precisely position the dyes with a sub‐nm resolution via DNA‐DNA hybridization makes DNA an ideal template to direct the energy transfer flow between dyes. An important result has been reported by García‐Parajò, van Hulst, and colleagues, who reported the hybridization of various ssDNA‐dye molecules to complementary parts of a DNA template at well‐defined positions. Their approach led to photonic wires made of five chromophores, regularly spaced of 10 base pairs, the distance between the dyes being controlled by the hybridization process. The dyes were selected for unidirectional energy transfer, starting from the excitation of a Rhodamine Green (excited at 470 nm), and energy transfer occurred along this multichromophoric array up to the dye emitting at the lower energy (Atto 680, emitting at 680 nm). The pre‐programmed position of the 5 different dyes is such that the distance between two consecutive dyes is 10 base pairs, i. e. the DNA pitch (3.4 nm). In this photonic wire, FRET occurred over a distance of 13.6 nm with a spectral range of around 200 nm. Single‐molecule spectroscopy revealed collective effects that are characteristic of coupled multichromophoric systems. Importantly, about 10 % of the photonic wires exhibited FRET efficiencies up to 90 %.73
Ruiz‐Carretero, Schenning, and co‐workers carried out another approach consisting in the DNA templated self‐assembly of naphthalene‐based donor chromophores assembled along a ssDNA template via hydrogen bonds (Figure 17).74 On its 5’‐end extremity, the ssDNA template is terminated with a cyanine dye (called Cy 3.5), which absorbs in the region where the donor guest emits. This system is designed to harvest light by the stack of donors (up to 40 molecules, as dictated by the DNA template) and direct energy transfer to the acceptor dye. By varying the size of the template from 10 to 40 thymine units, the researchers observed an optimum in the acceptor emission for a stack of 30 donor chromophores (Figure 17, bottom). The combination between time‐resolved spectroscopy and molecular modelling simulations showed that, for longer templates, efficient energy transfer occurs in excess of that expected with simple Förster Resonance Energy Transfer calculations.75
Figure 17.

Directing energy transfer through DNA‐templated self‐assembly of donor guest molecules. Top: sketch of the self‐assembly and FRET process. Bottom: fluorescence spectra (excitation at 400 nm) for DNA templates of different lengths (from 10 to 40 thymine units). Adapted with permissions from references75 and.76 Copyrights 2011 American Chemical Society and 2011 The Royal Society of Chemistry.
Other approaches were undertaken to direct energy transfer, as for instance the use of an oxazole yellow attached to a sequence‐specific polyamide minor‐groove binder dye, which binds a dsDNA terminated by two different dyes (a pacific blue dye and a cyanine dye).77 The oxazole yellow dye located at the center of the DNA via the polyamide groove binding permitted the energy transfer in between the two dyes at the DNA extremities, distant from 21 base pairs. Recently, Wagenknecht and colleagues reported the DNA‐templated self‐assembly of two chromophores (nile red and pyrene), which has been achieved by base paring (A⋅U recognition) using a ssDNA template (Figure 18).78 Indeed, energy transfer between the pyrene and nile red was observed and could be tuned by varying the mixing ratio between the two dyes.79
Figure 18.

Sketch of ssDNA‐templated self‐assembly of two chromophores by base pairing. Adapted with permission from reference.79 Copyrights 2015 American Chemical Society.
Furthermore, the same group studied these mixed assemblies of nile red and pyrene derivative templated by an oligodeoxyadenine (ssDNA) template, which was covalently linked to a fullerene derivative, as the prototype acceptor in organic solar cells. The supramolecular assembly was integrated in the active layer of a solar cell, and the results demonstrated charge‐carrier generation in the spectral range where the three π‐conjugated molecules absorb.80 This is a promising result that demonstrates the potential of DNA‐templated self‐assembly for organic optoelectronic devices.
5.2. DNA‐Templated Electronic Nanowires
The DNA‐templated self‐assembly of conducting polymers has been investigated as a way to construct well‐defined nanowires. A common strategy explored hitherto was to graft cationic groups in the side‐chains of π‐conjugated monomers, thus giving rise to electrostatic interactions with DNA. The polymerization of the monomers bound the DNA template is usually carried out by chemical oxidation, for instance using FeCl3. Using this approach, researchers reported the DNA‐templated synthesis of polyaniline,81 polyindole,82 and polypyrrole,83 as well as polymers containing different conjugated units such as poly‐2,5‐bis(2‐thienyl)pyrrole.84 The electronic properties of single DNA/polymer nanowires were explored, both by depositing DNA/polymer wires onto arrays of microelectrodes and more locally by Conductive AFM, permitting the measurements of I‐V characteristics along the nanowires (Figure 19).82, 85
Figure 19.

Examples of electrical characterization of single DNA/polymer nanowires. a) Tapping‐Mode AFM image showing a DNA/poly(dithienylpyrrole) sandwiched between two Au electrodes (SiO2 substrate). The gap between electrodes is 2.5 μm. b) I‐V measurements on a single of polyindole/DNA nanowire by Conductive AFM. The inset is an AFM image of the nanowire before electrical characterization (scale bar is 1.1 μm). Adapted with permission from references82 and.85 Copyrights 2010 American Chemical Society and 2014 The Royal Society of Chemistry.
The mechanism of templated polymerization was explored by Houlton and colleagues, for the case of DNA‐templated formation of poly‐2,5‐bis(2‐thienyl)pyrrole.86 AFM and coarse‐grained simulations supported a model where fragments reversibly associate onto DNA at low density, like beads‐on‐a‐string, and then react with each other to form the polymer in a DNA‐templated fashion. This approach nicely allows the full expression of a template effect, directing the outcome of the polymerization process to produce precise and regular objects which structures are dictated by their interaction with DNA. In turn, this process was shown to reduce defects and improve electrical conductivity.
On the other hand, DNA can also serve as a template to assemble, rather through non‐covalent interactions, π‐conjugated molecules into 1D stacks for fabricating conducting organic nanowires of defined length by using, for instance, triarylamines.87 However, this approach remains overlooked, as the mechanisms of charge transport by DNA itself remain elusive, which renders difficult to discriminate the charge transport along the stack of molecular guests assembled and that of the stack of nucleobases, together with possible coupling effects.
5.3. Sensing
DNA‐templated reactions,7a, 7e, 88 covalent DNA‐scaffolded assemblies of chromophores,79, 89 as well as hybrid inorganic DNA nanostructures90 have already attracted strong interest for (bio)sensing applications. For instance, the covalent DNA‐scaffolded assemblies of chromophores can adopt preferential chiral folds, which can be affected by different conditions or by the presence of a binding ligand, potentially an analyte of interest. It is precisely the structural dynamic of these nanostructures which makes them appealing for sensing applications. When going toward DNA‐templated supramolecular assemblies, one adds a new dynamic and responsive dimension which is the reversible ligand association/dissociation onto the DNA template. Therefore, ligands can be recruited onto or expelled from the DNA templates, depending on the conditions, and this complex (i. e. multicomponent and dynamic) self‐assembly triggers a functional output that can be exploited for sensing applications. Indeed, the chiral assembly of chromophores on a DNA template can be influenced by DNA sequence or by changes in the microenvironment, which can be exploited for (chir)optical biosensing (Figure 20). Conceptually, such approach is similar to the supramolecular DNA‐templated polymerization of proteins which is part of the innate immune system that sense foreign dsDNA/RNA.91
Figure 20.

Sketch of the concept of nucleic acids optical sensing by chromophore/fluorophore.
One of us studied the self‐assembly of π‐conjugated polymers such as cationic polythiophenes (Figure 21A) with DNA.92 Chiral induction from the DNA to the achiral conjugated polymer was observed by CD, as frequently detected for other DNA‐templated self‐assemblies. CD signals typical of right‐ or left‐handed helical assemblies of the polymer were shown to depend on the DNA sequence, polymer/DNA ratios, and the temperature. These signals were utilized notably to monitor the enzymatic cleavage of DNA. This supramolecular approach allowed the researchers to detect the enzymatic activity in a continuous, label‐free manner.93 Others reported the use of fluorescence signals of conjugated polymers to detect single‐nucleotide polymorphisms and DNA damages, using dye‐labeled DNA.94
Figure 21.

Chemical structures of aromatic compounds (A) polythiophene, B) triarylamines, C) perylene, D) tetraphenylethene) that undergo templated assembly with DNA, cationic polymer, guanine‐quadruplexes, and which have been used for potential sensing applications.
Besides, the DNA‐templated self‐assembly of small molecules can also be used in sensing applications. For instance, Barboiu and co‐workers described simple tricationic compounds, made of triarylamine aromatic core (Figure 21B), which undergoes a templated self‐assembly in the presence of single‐stranded DNA through phosphodiester backbone recognition. This DNA‐small molecule interaction results in different CD outputs depending on the length and composition of the single‐stranded DNA template, thereby opening an opportunity for sensing applications by CD detection.87b, 95
Yu and co‐workers reported an example of a supramolecular displacement assay for nucleic acid sensing.96 The experiment uses a bisanionic peylene probe (Figure 21C) which undergoes self‐assembly in the presence of cationic polymers, resulting in a strong fluorescence quenching. Upon addition of ssDNA nucleic acid, competitive binding took place and polymer‐nucleic acid prevailed because of multivalent interactions. As a result, the pyrene probe was released, which leads to a turn‐on sensing of ssDNA with a remarkable detection limit of 2 pM. This approach has also been implemented for the detection of enzymatic activity of alkaline phosphatase.
In a collaboration between the group of Clément and ours, we have recently reported a water‐soluble tetraimidazolium tetraphenylethene (Figure 21D) acting as a light‐up probe for sensing DNA guanine‐quadruplexes (G4s) by fluorescence spectroscopy.97 The compound displays fluorescence turn‐on and large emission wavelengths shifts triggered by G4 binding, which are attractive features for future sensing applications.
5.4. Delivery
The templated formation of nucleic acid‐protein self‐assemblies is at the basis of the self‐organization of viruses. In this context, Schmuck and co‐workers have for instance shown that tethering a non‐natural arginine analogue that promote salt‐bridge interactions with phosphodiesters can trigger DNA‐templated self‐assembly of cyclic peptides into nanofibers that acts as efficient gene transfection vectors.98 Alternatively, secondary interactions stabilizing DNA‐templated self‐assemblies can also be brought up by lipids, thus making cationic‐ as well as nucleo‐lipids of interest in the context of gene delivery.99 Lipid composition has been shown to affect the dynamic stability of the corresponding lipoplexes which is an essential feature to control in order to achieve high efficiency in gene delivery (meaning combining successfully stable lipoplex formation, effective cell entry, then full release). Stupp and co‐workers reported a beautiful example of an artificial self‐assembly system that mimics the capsid formation of the famous Tobacco Mosaic virus through a subtle combination of non‐covalent interactions to enable the hierarchical formation of dsDNA‐templated self‐assembly (Figure 22).100
Figure 22.

Engineered amphiphilic peptides that undergo a hierarchical self‐assembly to produce virus‐like nanoparticles upon interaction with DNA. Reprinted with permission from reference.100 Copyright 2013 American Chemical Society.
DNA‐templated polymerization is also attracting growing interest en route toward the self‐fabrication of synthetic vectors that are self‐fitted to the nucleic acid cargo. In this context, Aida and co‐workers described the siRNA‐templated polymerization of a multicationic vector that, in turns, promotes cell delivery.66, 67 The tetraguanidinylated dithiol molecule described above (see section 4.1 and Figure 14), forming nanocaplets by siRNA‐templated self‐assembly and polymerization, were incubated with live (Human hepatocellular carcinoma Hep3B) cells, and cellular uptake was observed. Remarkably, the nanocaplets underwent de‐polymerization in the reductive cytosolic environment, by breaking the disulfide polymer into dithiol monomer units, releasing the siRNA that acted as template. The researchers observed gene silencing, via the monitoring of luciferase activity, and showed that gene expression was suppressed in Hep3B cells (Figure 23).
Figure 23.
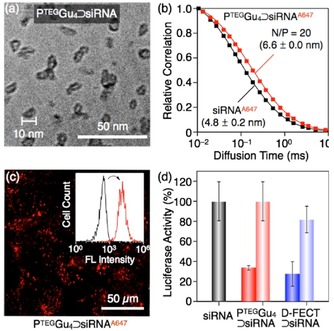
(a) Cryo‐TEM image of the nanocaplets obtained by siRNA‐templated self‐assembly and polymerization of a tetraguanidinium derivative (see Figure 13). (b) Relative autocorrelation profiles in FCS, with the estimated hydrodynamic diameters; (c) Confocal laser scanning microscopy image (λext=638 nm) of Hep3B cells after a 24 h incubation. Inset: flow cytometry histograms before (black and after (red) incucation; (d) Normalized luciferase activities of Hep3B‐luc cells using a luciferase assay. Reprinted with permission from reference.66 Copyright 2015 American Chemical Society.
Gokel and co‐workers have reported a very illustrative example of the potential applications, in delivery, enabled by the DNA‐templated supramolecular self‐assembly of small molecules. They studied small‐molecule neutral anion binders based on isophthalamide and dipicolinamide derivatives and found that they are effective vectors for transporting plasmid DNA into Escherichia coli.[101] The proposed mechanism of action is that binding of multiple ligands occurred through recognition of phosphodiester backbone, which then rendered the whole DNA‐templated self‐assembly more hydrophobic and thus able to penetrate bacteria cell walls (Figure 24).
Figure 24.

Chemical structures of isophthalamide and dipicolinamide derivatives used to deliver plasmid DNA in bacteria (left), and proposed mode of DNA binding that combine salt‐bridge and π‐π stacking interactions (right, reprinted with permission from reference,101 copyright 2012 American Chemical Society). X=H, Cl, CN, OCH3; Y=CH, N.
Exploiting such a supramolecular self‐assembly approach, one of us has recently reported the DNA‐ and siRNA‐templated self‐assembly of guanidylated porphyrins, partly mediated by interactions with phosphodiesters assisted by π–π stacking interactions, which enable dual therapy through siRNA delivery and photodynamic therapy.102
Remarkably, the cationic bridged β‐CD system discussed above (section 4.1, Figure 14), developed by Bouteiller, Sollogoub and co‐workers, was shown to efficiently complex siRNA by gel electrophoresis experiments.68 The researchers tested the ability of the supramolecular self‐assemblies to transfect cells, using a GL3 siRNA (directed against the expression of luciferase) and HEK‐293 cells, expressing the firefly luciferase GL3. In contrast to similar systems that cannot form supramolecular polymers, the β‐CD system that underwent cooperative self‐assembly with siRNA was shown to transfect cells at concentration above 2 mM, and no toxicity on cells was observed. This study demonstrates the importance of cooperative supramolecular self‐assembly using multivalent systems for achieving efficient cell transfection.
5.5. Catalysis
DNA‐templated synthesis is nowadays a rich field that finds wide applications in drug discovery, material sciences, biosensing, and bioimaging.88, 103 This approach capitalizes on proximity effects, enabled by multicomponent DNA‐templated self‐assembly through hybridization, which leads to catalyzed ligation reactions or facilitated group transfers. Such catalytic effects can be exploited in DNA‐accelerated multi‐step organic synthesis for achieving the preparation of sequence‐controlled macrocycles104 or oligomers, which is a topic beyond the scope of this review.8a, 105
Another approach used in asymmetric catalysis, pioneered by Feringa, Roelfes and co‐workers, exploits the DNA‐templated assembly of catalysts.106 Here it is the chiral transfer from the DNA template to the catalyst that is of interest for asymmetric catalysis, as sketched in Figure 25 (for reviews of the field, see for instance references107). Supramolecular anchoring of the catalysts within the DNA template can be achieved by using specific ligands which will direct DNA binding through intercalation or groove binding, possibly with sequence selectivity.108 Precise positioning is key to optimize transfer of chiral information from the template to the catalyst.109
Figure 25.

Sketch of the asymmetric catalysis using a DNA chiral template.
Interestingly, one can also play with the chirality of the nucleic acid template in order to tune the absolute configuration of the products,110 and even extend the approach to RNA templates.111 Sign that the field is reaching maturity is the recent successful application in the total synthesis of a natural product through a key transformation based on template‐accelerated [2+2] photoinduced cycloaddition.112
6. Conclusion and Perspectives
Nucleic acids are information‐rich biomolecules that are readily‐available and readily‐tunable by custom‐design. Besides, our understanding of the supramolecular interactions that can be established with nucleic acids has considerably developed along the last decades. For those reasons, it is nowadays timely to consider DNA‐templated self‐assembly approaches where custom‐made nucleic acids are used to trigger the self‐production of well‐defined and organized hybrid systems that feature emerging applications due to the organization of multiple compounds in precise nanoarrays. However, more efforts remain necessary in order to control DNA sequence recognition, overall assembly, with all‐or‐nothing process being a target of choice to avoid intermediate species. This will require designing cooperative pathways for the self‐assembly of multiple ligands onto nucleic acids. By generating organized and finite arrays of small molecules with precision in terms of length and three dimensional arrangement (density, chirality), one will expand a growing number of applications from materials sciences to biomedical applications. On the materials side, developments are expected on DNA‐templated multichromophoric assemblies for photonics and on the utilization of molecular photoswitches to control the DNA recognition/templating by light. Another important aspect is the chirality induction by the DNA, notably with the recent findings on the chiral‐induced spin selectivity effect with DNA and other biohybrid systems.113 The precise control over the organization in DNA‐templated assemblies and polymers could possibly offer ways to amplify and direct this effect. Besides, the combination of supramolecular polymers and DNA‐based assemblies will be important for achieving sophisticated materials for light‐harvesting systems, switches, accelerated computing, and delivery systems.114 On the biomedical side, the research on gene delivery would certainly benefit to further developments on RNA‐templated self‐assembled structures, in particular through the use of dynamic approaches to possibly release functional RNA together with other bioactive compounds in cells.115
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Mathieu Surin carried out his PhD with Prof. Roberto Lazzaroni (University of Mons) and a post‐doc with Prof. Paolo Samorì (ISIS, Université de Strasbourg). In 2009, he was appointed as research associate at the Fund for Scientific Research (FNRS, Belgium) and he developed a research line on DNA‐based supramolecular assemblies. In 2015, he was invited professor at the University of Montpellier. Since 2017, he is associate professor within the University of Mons. His main research interests are supramolecular chemistry and bio‐inspired (nano)materials.

Biographical Information
Sébastien Ulrich carried out his PhD with Prof. Jean‐Marie Lehn (Université de Strasbourg, France), and post‐docs with Prof. Harry L. Anderson (Oxford University, UK) and Prof. Eric T. Kool (Stanford University, CA, USA). In 2011 he joined the group of Prof. Pascal Dumy, first in Grenoble, then in Montpellier, France where he was recruited by the CNRS in 2012 to develop his current research interests in the field of supramolecular bioorganic chemistry. In 2017, he was awarded the CNRS Bronze Medal.

Acknowledgements
Research in Mons was supported by the Fund for Scientific Research (F.R.S.‐FNRS) under the grants MIS No. F.4532.16 (SHERPA) and EOS No. 30650939 (PRECISION). SU thanks the ANR (ANR‐17‐CE07‐0042‐01) for funding. The authors thank their close collaborators at UMONS, CNRS, and Université de Montpellier. Molecular graphics of a few figures were performed with UCSF Chimera, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco.
M. Surin, S. Ulrich, ChemistryOpen 2020, 9, 480.
Dedicated to Professor Jean‐Marie Lehn on the occasion of his 80th birthday.
Contributor Information
Dr. Mathieu Surin, Email: mathieu.surin@umons.ac.be.
Dr. Sébastien Ulrich, Email: sebastien.ulrich@enscm.fr.
References
- 1. Roszak A. W., Howard T. D., Southall J., Gardiner A. T., Law C. J., Isaacs N. W., Cogdell R. J., Science 2003, 302, 1969–1972. [DOI] [PubMed] [Google Scholar]
- 2. Ni R., Zhou J. L., Hossain N., Chau Y., Adv. Drug Delivery Rev. 2016, 106, 3–26. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Samorì P., Biscarini F., Chem. Soc. Rev. 2018, 47, 4675–4676; [DOI] [PubMed] [Google Scholar]
- 3b. Moulin E., Cid J. J., Giuseppone N., Adv. Mater. 2013, 25, 477–487; [DOI] [PubMed] [Google Scholar]
- 3c. Klosterman J. K., Yamauchi Y., Fujita M., Chem. Soc. Rev. 2009, 38, 1714–1725. [DOI] [PubMed] [Google Scholar]
- 4. Vantomme G., Meijer E. W., Science 2019, 363, 1396–1397. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Seeman N. C., Sleiman H. F., Nat. Rev. Mater. 2017, 3, 17068; [Google Scholar]
- 5b. Zhang F., Nangreave J., Liu Y., Yan H., J. Am. Chem. Soc. 2014, 136, 11198–11211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.
- 6a. Zheng J. P., Birktoft J. J., Chen Y., Wang T., Sha R. J., Constantinou P. E., Ginell S. L., Mao C. D., Seeman N. C., Nature 2009, 461, 74–77; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6b. Rothemund P. W. K., Nature 2006, 440, 297–302. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Sadhu K. K., Rothlingshofer M., Winssinger N., Isr. J. Chem. 2013, 53, 75–86; [Google Scholar]
- 7b. McLaughlin C. K., Hamblin G. D., Sleiman H. F., Chem. Soc. Rev. 2011, 40, 5647–5656; [DOI] [PubMed] [Google Scholar]
- 7c. Becerril H. A., Woolley A. T., Chem. Soc. Rev. 2009, 38, 329–337; [DOI] [PubMed] [Google Scholar]
- 7d. Aldaye F. A., Palmer A. L., Sleiman H. F., Science 2008, 321, 1795–1799; [DOI] [PubMed] [Google Scholar]
- 7e. Pianowski Z. L., Winssinger N., Chem. Soc. Rev. 2008, 37, 1330–1336. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Niu J., Hili R., Liu D. R., Nat. Chem. 2013, 5, 282–292; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8b. Rutten M. G. T. A., Vaandrager F. W., Elemans J. A. A. W., Nolte R. J. M., Nat. Rev. Chem. 2018, 2, 365–381. [Google Scholar]
- 9. Bandy T. J., Brewer A., Burns J. R., Marth G., Nguyen T., Stulz E., Chem. Soc. Rev. 2011, 40, 138–148. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Komor A. C., Barton J. K., Chem. Commun. 2013, 49, 3617–3630; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10b. Marcelis L., Ghesquiere J., Garnir K., Kirsch-De Mesmaeker A., Moucheron C., Coord. Chem. Rev. 2012, 256, 1569–1582; [Google Scholar]
- 10c. Song H., Kaiser J. T., Barton J. K., Nat. Chem. 2012, 4, 615–620; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10d. Niyazi H., Hall J. P., O′Sullivan K., Winter G., Sorensen T., Kelly J. M., Cardin C. J., Nat. Chem. 2012, 4, 621–628; [DOI] [PubMed] [Google Scholar]
- 10e. Zeglis B. M., Pierre V. C., Barton J. K., Chem. Commun. 2007, 4565–4579; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10f. Erkkila K. E., Odom D. T., Barton J. K., Chem. Rev. 1999, 99, 2777–2795. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Li N., Shang Y. X., Han Z. H., Wang T., Wang Z. G., Ding B. Q., ACS Appl. Mater. Interfaces 2019, 11, 13835–13852; [DOI] [PubMed] [Google Scholar]
- 11b. Kumar A., Hwang J. H., Kumar S., Nam J. M., Chem. Commun. 2013, 49, 2597–2609; [DOI] [PubMed] [Google Scholar]
- 11c. Ongaro A., Griffin F., Nagle L., Iacopino D., Eritja R., Fitzmaurice D., Adv. Mater. 2004, 16, 1799; [Google Scholar]
- 11d. Nakao H., Shiigi H., Yamamoto Y., Tokonami S., Nagaoka T., Sugiyama S., Ohtani T., Nano Lett. 2003, 3, 1391–1394; [Google Scholar]
- 11e. Braun E., Eichen Y., Sivan U., Ben-Yoseph G., Nature 1998, 391, 775–778. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Chen H., Meisburger S. P., Pabit S. A., Sutton J. L., Webb W. W., Pollack L., Proc. Natl. Acad. Sci. USA 2012, 109, 799–804; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12b. Tinland B., Pluen A., Sturm J., Weill G., Macromolecules 1997, 30, 5763–5765. [Google Scholar]
- 13.
- 13a. Zheng G. H., Czapla L., Srinivasan A. R., Olson W. K., Phys. Chem. Chem. Phys. 2010, 12, 1399–1406; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13b. Hagerman P. J., Ann. Rev. Biophys. Biophys. Chem. 1988, 17, 265–286. [DOI] [PubMed] [Google Scholar]
- 14. Hannon M. J., Chem. Soc. Rev. 2007, 36, 280–295. [DOI] [PubMed] [Google Scholar]
- 15. Biedermann F., Schneider H. J., Chem. Rev. 2016, 116, 5216–5300. [DOI] [PubMed] [Google Scholar]
- 16. Sessler J. L., Lawrence C. M., Jayawickramarajah J., Chem. Soc. Rev. 2007, 36, 314–325. [DOI] [PubMed] [Google Scholar]
- 17. Doluca O., Withers J. M., Filichev V. V., Chem. Rev. 2013, 113, 3044–3083. [DOI] [PubMed] [Google Scholar]
- 18. Spada G. P., Lena S., Masiero S., Pieraccini S., Surin M., Samorì P., Adv. Mater. 2008, 20, 2433–2438. [Google Scholar]
- 19.
- 19a. Surin M., Janssen P. G. A., Lazzaroni R., Leclère P., Meijer E. W., Schenning A. P. H. J., Adv. Mater. 2009, 21, 1126–1130; [Google Scholar]
- 19b. Janssen P. G. A., Jabbari-Farouji S., Surin M., Vila X., Gielen J. C., de Greef T. F., Vos M. R., Bomans P. H., Sommerdijk N. A., Christianen P. C., Leclère P., Lazzaroni R., van der Schoot P., Meijer E. W., Schenning A. P. H. J., J. Am. Chem. Soc. 2009, 131, 1222–1231. [DOI] [PubMed] [Google Scholar]
- 20. Avakyan N., Greschner A. A., Aldaye F., Serpell C. J., Toader V., Petitjean A., Sleiman H. F., Nat. Chem. 2016, 8, 368–376. [DOI] [PubMed] [Google Scholar]
- 21. Estévez-Torres A., Baigl D., Soft Matter 2011, 7, 6746–6756. [Google Scholar]
- 22. Mangrum J. B., Farrell N. P., Chem. Commun. 2010, 46, 6640–6650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.
- 23a. Komeda S., Moulaei T., Chikuma M., Odani A., Kipping R., Farrell N. P., Williams L. D., Nucleic Acids Res. 2011, 39, 325–336; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23b. Komeda S., Moulaei T., Woods K. K., Chikuma M., Farrell N. P., Williams L. D., J. Am. Chem. Soc. 2006, 128, 16092–16103. [DOI] [PubMed] [Google Scholar]
- 24.
- 24a. Bartolami E., Bouillon C., Dumy P., Ulrich S., Chem. Commun. 2016, 52, 4257–4273; [DOI] [PubMed] [Google Scholar]
- 24b. Hargrove A. E., Nieto S., Zhang T., Sessler J. L., Anslyn E. V., Chem. Rev. 2011, 111, 6603–6782; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24c. Rehm T. H., Schmuck C., Chem. Soc. Rev. 2010, 39, 3597–3611; [DOI] [PubMed] [Google Scholar]
- 24d. Theodossiou T. A., Pantos A., Tsogas I., Paleos C. M., ChemMedChem 2008, 3, 1635–1643; [DOI] [PubMed] [Google Scholar]
- 24e. Pantos A., Tsogas I., Paleos C. A., Biochim. Biophys. Acta Biomembr. 2008, 1778, 811–823; [DOI] [PubMed] [Google Scholar]
- 24f. Wender P. A., Galliher W. C., Goun E. A., Jones L. R., Pillow T. H., Adv. Drug Delivery Rev. 2008, 60, 452–472; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24g. Tamaru S., Hamachi I., in Structure and Bonding, Recognition of Anions, Vol. 129 (Ed.: R. Vilar), Springer, 2008, pp. 95–125; [Google Scholar]
- 24h. Blondeau P., Segura M., Perez-Fernandez R., de Mendoza J., Chem. Soc. Rev. 2007, 36, 198–210; [DOI] [PubMed] [Google Scholar]
- 24i. Sessler J. L., Gale P. A., Cho W.-S., Anion-Receptor Chemistry, The Royal Society of Chemistry, Cambridge, 2006; [Google Scholar]
- 24j. Schmidtchen F. P., Coord. Chem. Rev. 2006, 250, 2918–2928; [Google Scholar]
- 24k. Schug K. A., Lindner W., Chem. Rev. 2005, 105, 67–113; [DOI] [PubMed] [Google Scholar]
- 24l. Kubik S., Reyheller C., Stuwe S., J. Inclusion Phenom. Macrocyclic Chem. 2005, 52, 137–187; [Google Scholar]
- 24m. Best M. D., Tobey S. L., Anslyn E. V., Coord. Chem. Rev. 2003, 240, 3–15. [Google Scholar]
- 25. Hannon M. J., Chem. Soc. Rev. 2007, 36, 280–295. [DOI] [PubMed] [Google Scholar]
- 26.
- 26a. Chenoweth D. M., Meier J. L., Dervan P. B., Angew. Chem. Int. Ed. 2013, 52, 415–418; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 433–436; [Google Scholar]
- 26b. Warren C. L., Kratochvil N. C. S., Hauschild K. E., Foister S., Brezinski M. L., Dervan P. B., Phillips G. N., Ansari A. Z., Proc. Natl. Acad. Sci. USA 2006, 103, 867–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.
- 27a. Doss R. M., Marques M. A., Foister S., Chenoweth D. M., Dervan P. B., J. Am. Chem. Soc. 2006, 128, 9074–9079; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27b. Dervan P. B., Edelson B. S., Curr. Opin. Struct. Biol. 2003, 13, 284–299; [DOI] [PubMed] [Google Scholar]
- 27c. Dervan P. B., Bioorg. Med. Chem. 2001, 9, 2215–2235; [DOI] [PubMed] [Google Scholar]
- 27d. Dervan P. B., Science 1986, 232, 464–471. [DOI] [PubMed] [Google Scholar]
- 28.
- 28a. Kang J. S., Meier J. L., Dervan P. B., J. Am. Chem. Soc. 2014, 136, 3687–3694; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28b. Raskatov J. A., Nickols N. G., Hargrove A. E., Marinov G. K., Wold B., Dervan P. B., Proc. Natl. Acad. Sci. USA 2012, 109, 16041–16045; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28c. Chenoweth D. M., Dervan P. B., Proc. Natl. Acad. Sci. USA 2009, 106, 13175–13179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Guo P., Paul A., Kumar A., Farahat A. A., Kumar D., Wang S., Boykin D. W., Wilson W. D., Chem. Eur. J. 2016, 22, 15404–15412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Guo P., Farahat A. A., Paul A., Harika N. K., Boykin D. W., Wilson W. D., J. Am. Chem. Soc. 2018, 140, 14761–14769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Guckian K. M., Schweitzer B. A., Ren R. X. F., Sheils C. J., Tahmassebi D. C., Kool E. T., J. Am. Chem. Soc. 2000, 122, 2213–2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.
- 32a. Lerman L. S., J. Cell. Comp. Physiol. 1964, 64, 1–18; [PubMed] [Google Scholar]
- 32b. Lerman L. S., J. Mol. Biol. 1961, 3, 18–30. [DOI] [PubMed] [Google Scholar]
- 33. Rao S. N., Kollman P. A., Proc. Natl. Acad. Sci. USA 1987, 84, 5735–5739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.
- 34a. Dawson S., Malkinson J. P., Paumier D., Searcey M., Nat. Prod. Rep. 2007, 24, 109–126; [DOI] [PubMed] [Google Scholar]
- 34b. Wang A. H.-J., Ughetto G., Quigley G. J., Hakoshima T., v. d. Marel G. A., v. Boom J. H., Rich A., Science 1984, 225, 1115–1121; [DOI] [PubMed] [Google Scholar]
- 34c. Waring M. J., Wakelin L. P. G., Nature 1974, 252, 653–657. [DOI] [PubMed] [Google Scholar]
- 35. Mbarek A., Moussa G., Chain J. L., Molecules 2019, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.
- 36a. Zimmerman S. C., Beilstein J. Org. Chem. 2016, 12, 125–138; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36b. Leblond J., Petitjean A., ChemPhysChem 2011, 12, 1043–1051. [DOI] [PubMed] [Google Scholar]
- 37. Fechter E. J., Olenyuk B., Dervan P. B., Angew. Chem. Int. Ed. 2004, 43, 3591–3594; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004, 116, 3675–3678. [Google Scholar]
- 38.
- 38a. Granzhan A., Kotera N., Teulade-Fichou M. P., Chem. Soc. Rev. 2014, 43, 3630–3665; [DOI] [PubMed] [Google Scholar]
- 38b. Granzhan A., Largy E., Saettel N., Teulade-Fichou M. P., Chem. Eur. J. 2010, 16, 878–889. [DOI] [PubMed] [Google Scholar]
- 39. Holman G. G., Zewail-Foote M., Smith A. R., Johnson K. A., Iverson B. L., Nat. Chem. 2011, 3, 875–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jabbari-Farouji S., van der Schoot P., Macromolecules 2010, 43, 5833–5844. [Google Scholar]
- 41.
- 41a. Balaz M., Tannir S., Varga K., Coord. Chem. Rev. 2017, 349, 66–83; [Google Scholar]
- 41b. Liu M., Zhang L., Wang T., Chem. Rev. 2015, 115, 7304–7397. [DOI] [PubMed] [Google Scholar]
- 42. Iwaura R., Yoshida K., Masuda M., Ohnishi-Kameyama M., Yoshida M., Shimizu T., Angew. Chem. Int. Ed. 2003, 42, 1009–1012; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2003, 115, 1039–1042. [Google Scholar]
- 43. Iwaura R., Hoeben F. J. M., Masuda M., Schenning A. P. H. J., Meijer E. W., Shimizu T., J. Am. Chem. Soc. 2006, 128, 13298–13304. [DOI] [PubMed] [Google Scholar]
- 44.
- 44a. Ishutkina M. V., Berry A. R., Hussain R., Khelevina O. G., Siligardi G., Stulz E., Eur. J. Org. Chem. 2018, 2018, 5054–5059; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44b. Sargsyan G., Schatz A. A., Kubelka J., Balaz M., Chem. Commun. 2013, 49, 1020–1022; [DOI] [PubMed] [Google Scholar]
- 44c. Fendt L. A., Bouamaied I., Thöni S., Amiot N., Stulz E., J. Am. Chem. Soc. 2007, 129, 15319–15329. [DOI] [PubMed] [Google Scholar]
- 45. Hofsäb R., Sinn S., Biedermann F., Wagenknecht H. A., Chem. Eur. J. 2018, 24, 16257–16261. [DOI] [PubMed] [Google Scholar]
- 46. Hofsäb R., Ensslen P., Wagenknecht H. A., Chem. Commun. 2019, 55, 1330–1333. [DOI] [PubMed] [Google Scholar]
- 47. Kashida H., Hattori Y., Tazoe K., Inoue T., Nishikawa K., Ishii K., Uchiyama S., Yamashita H., Abe M., Kamiya Y., Asanuma H., J. Am. Chem. Soc. 2018, 140, 8456–8462. [DOI] [PubMed] [Google Scholar]
- 48. Mosquera J., Sanchez M. I., Valero J., de Mendoza J., Vazquez M. E., Mascarenas J. L., Chem. Commun. 2015, 51, 4811–4814. [DOI] [PubMed] [Google Scholar]
- 49. Rha A. K., Das D., Taran O., Ke Y., Mehta A. K., Lynn D. G., Angew. Chem. Int. Ed. 2019, in press, DOI 10.1002/anie.201907661. [Google Scholar]
- 50. Chakraborty G., Balinin K., Portale G., Loznik M., Polushkin E., Weil T., Herrmann A., Chem. Sci. 2019, 10, 10097–10105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.
- 51a. Malina J., Farrell N. P., Brabec V., Chem. Eur. J. 2019, 25, 2995–2999; [DOI] [PubMed] [Google Scholar]
- 51b. Malina J., Cechova K., Farrell N. P., Brabec V., Inorg. Chem. 2019, 58, 6804–6810. [DOI] [PubMed] [Google Scholar]
- 52. Paolantoni D., Cantel S., Dumy P., Ulrich S., Int. J. Mol. Sci. 2015, 16, 3609–3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Paolantoni D., Rubio-Magnieto J., Cantel S., Martinez J., Dumy P., Surin M., Ulrich S., Chem. Commun. 2014, 50, 14257–14260. [DOI] [PubMed] [Google Scholar]
- 54. Rubio-Magnieto J., Phan T. A., Fossepre M., Matot V., Knoops J., Jarrosson T., Dumy P., Serein-Spirau F., Niebel C., Ulrich S., Surin M., Chem. Eur. J. 2018, 24, 706–714. [DOI] [PubMed] [Google Scholar]
- 55. Rubio-Magnieto J., Kumar M., Brocorens P., Idé J., George S. J., Lazzaroni R., Surin M., Chem. Commun. 2016, 52, 13873–13876. [DOI] [PubMed] [Google Scholar]
- 56. Wang M. M., Silva G. L., Armitage B. A., J. Am. Chem. Soc. 2000, 122, 9977–9986. [Google Scholar]
- 57. Hannah K. C., Armitage B. A., Acc. Chem. Res. 2004, 37, 845–853. [DOI] [PubMed] [Google Scholar]
- 58. Jiang Q., Xu X. H., Yin P. A., Ma K., Zhen Y. G., Duan P. F., Peng Q., Chen W. Q., Ding B. Q., J. Am. Chem. Soc. 2019, 141, 9490–9494. [DOI] [PubMed] [Google Scholar]
- 59. Rodriguez J., Learte-Aymami S., Mosquera J., Celaya G., Rodriguez-Larrea D., Vazquez M. E., Mascarenas J. L., Chem. Sci. 2018, 9, 4118–4123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wilks T. R., Pitto-Barry A., Kirby N., Stulz E., O′Reilly R. K., Chem. Commun. 2014, 50, 1338–1340. [DOI] [PubMed] [Google Scholar]
- 61. MoradpourHafshejani S., Hedley J. H., Haigh A. O., Pike A. R., Tuite E. M., RSC Adv. 2013, 3, 18164–18172. [Google Scholar]
- 62. Hafshejani S. M., Watson S. M. D., Tuite E. M., Pike A. R., Chem. Eur. J. 2015, 21, 12611–12615. [DOI] [PubMed] [Google Scholar]
- 63. Surin M., Polym. Chem. 2016, 7, 4137–4150. [Google Scholar]
- 64. Li X., Zhan Z. Y. J., Knipe R., Lynn D. G., J. Am. Chem. Soc. 2002, 124, 746–747. [DOI] [PubMed] [Google Scholar]
- 65. Lin J., Surin M., Beljonne D., Lou X., van Dongen J. L. J., Schenning A. P. H. J., Chem. Sci. 2012, 3, 2732–2736. [Google Scholar]
- 66. Hashim P. K., Okuro K., Sasaki S., Hoashi Y., Aida T., J. Am. Chem. Soc. 2015, 137, 15608–15611. [DOI] [PubMed] [Google Scholar]
- 67. Mogaki R., Hashim P. K., Okuro K., Aida T., Chem. Soc. Rev. 2017, 46, 6480–6491. [DOI] [PubMed] [Google Scholar]
- 68. Evenou P., Rossignol J., Pembouong G., Gothland A., Colesnic D., Barbeyron R., Rudiuk S., Marcelin A. G., Menand M., Baigl D., Calvez V., Bouteiller L., Sollogoub M., Angew. Chem. Int. Ed. 2018, 57, 7753–7758; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 7879–7884. [Google Scholar]
- 69. Wang Z.-G., Liu Q., Ding B., Chem. Mater. 2014, 26, 3364–3367. [Google Scholar]
- 70. Tokura Y., Jiang Y., Welle A., Stenzel M. H., Krzemien K. M., Michaelis J., Berger R., Barner-Kowollik C., Wu Y., Weil T., Angew. Chem. Int. Ed. 2016, 55, 5692–5697; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 5786–5791. [Google Scholar]
- 71. Tokura Y., Harvey S., Chen C., Wu Y., Ng D. Y. W., Weil T., Angew. Chem. Int. Ed. 2018, 57, 1587–1591; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 1603–1607. [Google Scholar]
- 72. Winterwerber P., Harvey S., Ng D. Y. W., Weil T., Angew. Chem. Int. Ed. 2019, 58, doi: anie.201911249. [Google Scholar]
- 73. Garcia-Parajo M. F., Hernando J., Mosteiro G. S., Hoogenboom J. P., van Dijk E. M. H., van Hulst N. F., ChemPhysChem 2005, 6, 819–827. [DOI] [PubMed] [Google Scholar]
- 74. Ruiz-Carretero A., Janssen P. G., Stevens A. L., Surin M., Herz L. M., Schenning A. P. H. J., Chem. Commun. 2011, 47, 884–886. [DOI] [PubMed] [Google Scholar]
- 75. Stevens A. L., Janssen P. G. A., Ruiz-Carretero A., Surin M., Schenning A. P. H. J., Herz L. M., J. Phys. Chem. B 2011, 115, 10550–10560. [Google Scholar]
- 76. Ruiz-Carretero A., Janssen P. G., Stevens A. L., Surin M., Herz L. M., Schenning A. P., Chem. Commun. (Camb.) 2011, 47, 884–886. [DOI] [PubMed] [Google Scholar]
- 77. Su W., Schuster M., Bagshaw C. R., Rant U., Burley G. A., Angew. Chem. Int. Ed. 2011, 50, 2712–2715; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 2764–2767. [Google Scholar]
- 78. Ensslen P., Fritz Y., Wagenknecht H. A., Org. Biomol. Chem. 2015, 13, 487–492. [DOI] [PubMed] [Google Scholar]
- 79. Ensslen P., Wagenknecht H. A., Acc. Chem. Res. 2015, 48, 2724–2733. [DOI] [PubMed] [Google Scholar]
- 80. Ensslen P., Gartner S., Glaser K., Colsmann A., Wagenknecht H. A., Angew. Chem. Int. Ed. 2016, 55, 1904–1908; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 1936–1941. [Google Scholar]
- 81.
- 81a. Nickels P., Dittmer W. U., Beyer S., Kotthaus J. P., Simmel F. C., Nanotechnology 2004, 15, 1524–1529; [Google Scholar]
- 81b. Nagarajan R., Liu W., Kumar J., Tripathy S. K., Bruno F. F., Samuelson L. A., Macromolecules 2001, 34, 3921–3927. [Google Scholar]
- 82. Hassanien R., Al-Hinai M., Al-Said S. A. F., Little R., Siller L., Wright N. G., Houlton A., Horrocks B. R., ACS Nano 2010, 4, 2149–2159. [DOI] [PubMed] [Google Scholar]
- 83.
- 83a. Pruneanu S., Al-Said S. A. F., Dong L. Q., Hollis T. A., Galindo M. A., Wright N. G., Houlton A., Horrocks B. R., Adv. Funct. Mater. 2008, 18, 2444–2454; [Google Scholar]
- 83b. Dong L. Q., Hollis T., Fishwick S., Connolly B. A., Wright N. G., Horrocks B. R., Houlton A., Chem. Eur. J. 2007, 13, 822–828. [DOI] [PubMed] [Google Scholar]
- 84.
- 84a. Watson S. M. D., Hedley J. H., Galindo M. A., Al-Said S. A. F., Wright N. G., Connolly B. A., Horrocks B. R., Houlton A., Chem. Eur. J. 2012, 18, 12008–12019; [DOI] [PubMed] [Google Scholar]
- 84b. Chen W., Güler G., Kuruvilla E., Schuster G. B., Chiu H.-C., Riedo E., Macromolecules 2010, 43, 4032–4040. [Google Scholar]
- 85. Watson S. M., Pike A. R., Pate J., Houlton A., Horrocks B. R., Nanoscale 2014, 6, 4027–4037. [DOI] [PubMed] [Google Scholar]
- 86. Watson S. M. D., Galindo M. A., Horrocks B. R., Houlton A., J. Am. Chem. Soc. 2014, 136, 6649–6655. [DOI] [PubMed] [Google Scholar]
- 87.
- 87a. Moulin E., Armao J. J., Giuseppone N., Acc. Chem. Res. 2019, 52, 975–983; [DOI] [PubMed] [Google Scholar]
- 87b. Kocsis I., Rotaru A., Legrand Y. M., Grosu I., Barboiu M., Chem. Commun. 2016, 52, 386–389; [DOI] [PubMed] [Google Scholar]
- 87c. Armao J. J., Maaloum M., Ellis T., Fuks G., Rawiso M., Moulin E., Giuseppone N., J. Am. Chem. Soc. 2014, 136, 11382–11388; [DOI] [PubMed] [Google Scholar]
- 87d. Faramarzi V., Niess F., Moulin E., Maaloum M., Dayen J. F., Beaufrand J. B., Zanettini S., Doudin B., Giuseppone N., Nat. Chem. 2012, 4, 485–490. [DOI] [PubMed] [Google Scholar]
- 88.
- 88a. Gorska K., Winssinger N., Angew. Chem. Int. Ed. 2013, 52, 6820–6843; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 6956–6980; [Google Scholar]
- 88b. Silverman A. P., Kool E. T., Chem. Rev. 2006, 106, 3775–3789. [DOI] [PubMed] [Google Scholar]
- 89.
- 89a. Teo Y. N., Kool E. T., Chem. Rev. 2012, 112, 4221–4245; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89b. Malinovskii V. L., Wenger D., Haner R., Chem. Soc. Rev. 2010, 39, 410–422. [DOI] [PubMed] [Google Scholar]
- 90. Yang Y., Mao G., Jia X., He Z., J. Mater. Chem. B 2019, DOI 10.1039/C1039TB01870 K. [Google Scholar]
- 91. Morrone S. R., Matyszewski M., Yu X., Delannoy M., Egelman E. H., Sohn J., Nat. Commun. 2015, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.
- 92a. Rubio-Magnieto J., Azene E. G., Knoops J., Knippenberg S., Delcourt C., Thomas A., Richeter S., Mehdi A., Dubois P., Lazzaroni R., Beljonne D., Clément S., Surin M., Soft Matter 2015, 11, 6460–6471; [DOI] [PubMed] [Google Scholar]
- 92b. Rubio-Magnieto J., Thomas A., Richeter S., Mehdi A., Dubois P., Lazzaroni R., Clement S., Surin M., Chem. Commun. 2013, 49, 5483–5485. [DOI] [PubMed] [Google Scholar]
- 93. Fossépré M., Trévisan M. E., Cyriaque V., Wattiez R., Beljonne D., Richeter S., Clément S., Surin M., ACS Appl. Bio Mater. 2019, 2, 2125–2136. [DOI] [PubMed] [Google Scholar]
- 94. Duan X. R., Liu L. B., Feng F. D., Wang S., Acc. Chem. Res. 2010, 43, 260–270. [DOI] [PubMed] [Google Scholar]
- 95. Zhang Y., Petit E., Barboiu M., ChemPlusChem 2018, 83, 354–360. [DOI] [PubMed] [Google Scholar]
- 96. Wang Y., Chen J., Jiao H. P., Chen Y., Li W. Y., Zhang Q. F., Yu C., Chem. Eur. J. 2013, 19, 12846–12852. [DOI] [PubMed] [Google Scholar]
- 97. Kotras C., Fossépré M., Roger M., Gervais V., Richeter S., Gerbier P., Ulrich S., Surin M., Clément S., Front. Chem. 2019, 7, Article 493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Li M., Ehlers M., Schlesiger S., Zellermann E., Knauer S. K., Schmuck C., Angew. Chem. Int. Ed. 2016, 55, 598–601; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 608–611. [Google Scholar]
- 99.
- 99a. LaManna C. M., Lusic H., Camplo M., McIntosh T. J., Barthelemy P., Grinstaff M. W., Acc. Chem. Res. 2012, 45, 1026–1038; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99b. Patwa A., Gissot A., Bestel I., Barthelemy P., Chem. Soc. Rev. 2011, 40, 5844–5854; [DOI] [PubMed] [Google Scholar]
- 99c. Gissot A., Camplo M., Grinstaff M. W., Barthelemy P., Org. Biomol. Chem. 2008, 6, 1324–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Ruff Y., Moyer T., Newcomb C. J., Demeler B., Stupp S. I., J. Am. Chem. Soc. 2013, 135, 6211–6219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Atkins J. L., Patel M. B., Daschbach M. M., Meisel J. W., Gokel G. W., J. Am. Chem. Soc. 2012, 134, 13546–13549. [DOI] [PubMed] [Google Scholar]
- 102. Laroui N., Coste M., Lichon L., Bessin Y., Gary-Bobo M., Pratviel G., Bonduelle C., Bettache N., Ulrich S., Int. J. Pharm. 2019, 569. [DOI] [PubMed] [Google Scholar]
- 103.
- 103a. O′Reilly R. K., Turberfield A. J., Wilks T. R., Acc. Chem. Res. 2017, 50, 2496–2509; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103b. Kleiner R. E., Dumelin C. E., Liu D. R., Chem. Soc. Rev. 2011, 40, 5707–5717; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103c. Li X. Y., Liu D. R., Angew. Chem. Int. Ed. 2004, 43, 4848–4870; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004, 116, 4956–4979. [Google Scholar]
- 104. Usanov D. L., Chan A. I., Maianti J. P., Liu D. R., Nat. Chem. 2018, 10, 704–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.
- 105a. Meng W. J., Muscat R. A., Mckee M. L., Milnes P. J., El-Sagheer A. H., Bath J., Davis B. G., Brown T., O′Reilly R. K., Turberfield A. J., Nat. Chem. 2016, 8, 542–548; [DOI] [PubMed] [Google Scholar]
- 105b. McKee M. L., Milnes P. J., Bath J., Stulz E., O′Reilly R. K., Turberfield A. J., J. Am. Chem. Soc. 2012, 134, 1446–1449; [DOI] [PubMed] [Google Scholar]
- 105c. McKee M. L., Milnes P. J., Bath J., Stulz E., Turberfield A. J., O′Reilly R. K., Angew. Chem. Int. Ed. 2010, 49, 7948–7951; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 8120–8123; [Google Scholar]
- 105d. He Y., Liu D. R., Nat. Nanotechnol. 2010, 5, 778–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Roelfes G., Feringa B. L., Angew. Chem. Int. Ed. 2005, 44, 3230–3232; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 3294–3296. [Google Scholar]
- 107.
- 107a. Duchemin N., Heath-Apostolopoulos I., Smietana M., Arseniyadis S., Org. Biomol. Chem. 2017, 15, 7072–7087; [DOI] [PubMed] [Google Scholar]
- 107b. Boersma A. J., Megens R. P., Feringa B. L., Roelfes G., Chem. Soc. Rev. 2010, 39, 2083–2092. [DOI] [PubMed] [Google Scholar]
- 108. Amirbekyan K., Duchemin N., Benedetti E., Joseph R., Colon A., Markarian S. A., Bethge L., Vonhoff S., Klussmann S., Cossy J., Vasseur J. J., Arseniyadis S., Smietana M., ACS Catal. 2016, 6, 3096–3105. [Google Scholar]
- 109. Mansot J., Aubert S., Duchemin N., Vasseur J. J., Arseniyadis S., Smietana M., Chem. Sci. 2019, 10, 2875–2881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Wang J., Benedetti E., Bethge L., Vonhoff S., Klussmann S., Vasseur J. J., Cossy J., Smietana M., Arseniyadis S., Angew. Chem. Int. Ed. 2013, 52, 11546–11549; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 11760–11763. [Google Scholar]
- 111. Duchemin N., Benedetti E., Bethge L., Vonhoff S., Klussmann S., Vasseur J. J., Cossy J., Smietana M., Arseniyadis S., Chem. Commun. 2016, 52, 8604–8607. [DOI] [PubMed] [Google Scholar]
- 112. Duchemin N., Skiredj A., Mansot J., Leblanc K., Vasseur J. J., Beniddir M. A., Evanno L., Poupon E., Smietana M., Arseniyadis S., Angew. Chem. Int. Ed. 2018, 57, 11786–11791; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 11960–11965. [Google Scholar]
- 113. Naaman R., Paltiel Y., Waldeck D. H., Nat. Rev. Chem. 2019, 3, 250–260. [Google Scholar]
- 114.
- 114a. Kownacki M., Langenegger S. M., Liu S. X., Haner R., Angew. Chem. Int. Ed. 2019, 58, 751–755; [DOI] [PubMed] [Google Scholar]
- 114b. Engelen W., Wijnands S. P. W., Merkx M., J. Am. Chem. Soc. 2018, 140, 9758–9767; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114c. Bakker M. H., Lee C. C., Meijer E. W., Dankers P. Y., Albertazzi L., ACS Nano 2016, 10, 1845–1852. [DOI] [PubMed] [Google Scholar]
- 115. Holtzer L., Oleinich I., Anzola M., Lindberg E., Sadhu K. K., Gonzalez-Gaitan M., Winssinger N., ACS Cent. Sci. 2016, 2, 394–400. [DOI] [PMC free article] [PubMed] [Google Scholar]